

Abstract
We report the synthesis of SAHAquines and related primaquine (PQ) derivatives. SAHAquines are novel hybrid compounds that combine moieties of suberoylanilide hydroxamic acid (SAHA), an anticancer agent with weak antiplasmodial activity, and PQ, an antimalarial drug with low antiproliferative activity. The preparation of SAHAquines is simple, cheap, and high yielding. It includes the following steps: coupling reaction between primaquine and a dicarboxylic acid monoester, hydrolysis, a new coupling reaction with O‐protected hydroxylamine, and deprotection. SAHAquines 5 a–d showed significant reduction in cell viability. Among the three human cancer cell lines (U2OS, HepG2, and MCF‐7), the most responsive were the MCF‐7 cells. The antibodies against acetylated histone H3K9/H3K14 in MCF‐7 cells revealed a significant enhancement following treatment with N‐hydroxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}pentanediamide (5 b). Ethyl (2E)‐3‐({4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)prop‐2‐enoate (2 b) and SAHAquines were the most active compounds against both the hepatic and erythrocytic stages of Plasmodium parasites, some of them at sub‐micromolar concentrations. The results of our research suggest that SAHAquines are promising leads for new anticancer and antimalarial agents.
Keywords: acetylation, anticancer agents, antiplasmodial activity, cytostatic activity, drug design
1. Introduction
Despite extensive efforts and significant progress in cancer treatment, therapeutic interventions are not yet satisfactory. Almost all available cytostatic drugs cause undesirable side effects, whereas drug resistance presents an additional problem.1, 2, 3 In another field of medicine, an ongoing battle is being fought against malaria, a parasitic disease caused by Plasmodium species.4, 5 Malaria still poses a great health and economic burden to the highly populated countries in the tropical and subtropical parts of the world. The need for new, effective antimalarials arises from several factors, including the absence of an effective vaccine, insufficient vector control, and the emergence of multidrug‐resistant Plasmodium strains.6, 7, 8 The currently adopted approaches to the design of antiplasmodial compounds include the development of analogues of existing drugs, resistance reversers, and novel compounds with new mechanisms of action.6, 9
A number of studies have shown a relationship between cancer and malaria in regard to diagnostics, drug research, treatment, prevention, and epidemiology.10, 11, 12, 13 Different classes of antimalarial drugs display direct or adjuvant activity against cancer cell lines, are known as sensitivity reversers of resistant tumor cell lines or inhibitors of drug resistance development, or have synergistic action with known anticancer drugs.14, 15, 16, 17, 18, 19, 20 Although their exact mode of action against cancer is still not completely understood, various mechanisms have been proposed.21, 22, 23
Such observations prompted us, and others, to design and prepare novel derivatives of known antimalarial drugs and evaluate their cytostatic potential.24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 Our efforts have been focused on primaquine (PQ), which is an old drug with many flaws (e.g., induction of hemolytic anemia in individuals lacking glucose‐6‐phosphate dehydrogenase, quick metabolism, degradation to inactive carboxyprimaquine, altering the treatment outcome in dependence of CYP 2D6 enzyme activity)35 but is currently the only available Plasmodium hypnozoitocide.36 We previously showed that various PQ derivatives of amides, ureas, bis‐ureas, semicarbazides, and acylsemicarbazide‐type derivatives possessed significant cytostatic activity against a panel of cancer cell lines or high selectivity towards the breast adenocarcinoma cell line (MCF‐7).24, 25, 26, 27, 28, 29, 30 On the other hand, cytostatic agents of different classes, including histone deacetylase (HDAC) inhibitors, have been shown to exert antiplasmodial activity.6, 37, 38, 39, 40, 41, 42 Suberoylanilide hydroxamic acid (SAHA, vorinostat) was the first marketed pan‐HDAC inhibitor, and it was followed by other drugs from the same class (e.g., romidepsin, belinostat, and panobinostat.43, 44 Their hydroxamic group chelates zinc ion found in the active site of Zn‐dependent HDACs (classes I, II, and IV), and this leads to the accumulation of acetylated histones and other proteins.45 HDACs were also identified as transcription regulators in P. falciparum.46, 47 Out of five P. falciparum HDACs, three are Zn‐dependent enzymes, prone to inhibition by SAHA, and thus represent viable targets for drug development.48 Several studies have shown the antimalarial activity of SAHA and related HDAC inhibitors.49, 50
In this study, we employed one of the classical medicinal chemists’ tools—the combination of two pharmacophores in one molecule.7, 51, 52, 53, 54 The concept of hybrid drugs is also a valuable strategy to overcome the limitations of a combined therapy, as the resulting molecules could exhibit inhibitory activities on multiple targets.55 The hybrid compounds described here, SAHAquines, combine motifs of SAHA, an anticancer agent with weak antiplasmodial activity, and PQ, an antimalarial drug with low antiproliferative activity. Other here‐reported PQ derivatives differ in the linker length/type and/or functional groups: compounds 2 are esters, compounds 3 are carboxylic acids, and compounds 4 and 6 are O‐benzyl‐ and O‐methyl‐substituted hydroxamic acids. Similar to the known HDAC inhibitors, hydroxamic acids 5 consist of a capping group (quinoline ring), a linker, and a Zn‐binding group (hydroxamic acid) (Figure 1). Given that hydroxamic acid is a strong binding group for metal ions that might lead to poor selectivity and confer undesired properties, such as poor pharmacokinetics,56, 57 we introduced other functional groups instead: an ester, carboxylic acid, or O‐protected hydroxamic group.
Figure 1.
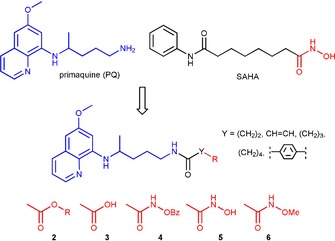
Design of SAHAquines 5 and other PQ derivatives.
Herein, we report the synthesis of SAHAquines 5 a–d as well as 20 novel PQ derivatives, their chemical characterization, the assessment of their cytostatic activity, and the evaluation of their activity against the erythrocytic and hepatic stages of Plasmodium.
2. Results and Discussion
2.1. Chemistry
The objective of our research was to prepare SAHAquines 5 and related PQ derivatives, which differ in the type/length of the spacer and/or functional groups: compounds 2 are esters and compounds 3 are carboxylic acids, whereas compounds 4 and 6 are O‐protected hydroxamic acids. Scheme 1 shows the synthetic pathway leading to derivatives 2–6. In the first reaction step, dicarboxylic acid monoesters 1 a–e were coupled with PQ to give derivatives 2 a–e by using 1‐[bis(dimethylamino)methylene]‐1H‐1,2,3‐triazolo[4,5‐b]pyridinium 3‐oxid hexafluorophosphate (HATU) as the coupling reagent, along with Hünig's base (N,N‐diisopropylethylamine, DIEA). The following dicarboxylic acid monoesters were used: 4‐methoxy‐4‐oxobutanoic acid (monomethyl hydrogen succinate) (1 a), (E)‐4‐ethoxy‐4‐oxobut‐2‐enoic acid (monoethyl fumarate) (1 b), 5‐methoxy‐5‐oxopentanoic acid (monomethyl glutarate) (1 c), 6‐methoxy‐6‐oxohexanoic acid (monomethyl adipate) (1 d), and 4‐(methoxycarbonyl)benzoic acid (monomethyl terephtalate) (1 e). Notably, an analogous coupling reaction between PQ and monomethyl malonate failed in our hands. Amide formation between these two compounds by using thionyl chloride or benzotriazolide58 was also unsuccessful and gave a mixture of products. Classical activation of the carboxylic group with thionyl chloride worked well with other dicarboxylic acid monoesters. Hydrolysis of 2 a–e with lithium hydroxide afforded corresponding acids 3 a–e, which were transformed into O‐benzylhydroxamic acids 4 a–e and O‐methylhydroxamic acids 6 a–e by means of O‐benzyl‐ and O‐methylhydroxylamine, respectively. Again, HATU/DIEA was used as the coupling system. Free hydroxamic acids 5 a–d were obtained by catalytic hydrogenation of O‐benzyl derivatives 4 a–e. Selective deprotection of the benzyl group in fumaric derivative 4 b failed. As a result of double‐bond hydrogenation, succinylhydroxamic acid 5 a was obtained instead of fumarylhydroxamic acid.
Scheme 1.
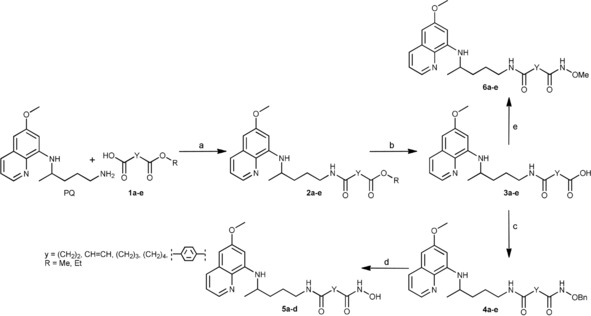
Synthesis of PQ derivatives 2–6. Reagents and conditions: a) HATU, DIEA, CH2Cl2, RT, 1 h; b) LiOH, MeOH, H2O, RT, 1 h; c) H2NOBn, HATU, DIEA, CH2Cl2, RT, 2 h; d) H2, Pd/C, MeOH, RT, 2–4 h; NH2OMe, HATU, DIEA, CH2Cl2, RT, 2 h.
All new compounds were fully characterized by conventional spectroscopy and analytical methods (IR, 1H NMR, and 13C NMR spectroscopy; MS; and elemental analyses). The data were consistent with the proposed structures and are given in short in the Experimental Section and in detail in the Supporting Information.
To evaluate the drug‐like properties of our novel compounds, a common set of physicochemical parameters were calculated: topological polar surface area (TPSA), number of atoms, molecular weight (MW), partition coefficient (log P), number of H‐bond donors (HBDs), number of H‐bond acceptors (HBAs), and molar refractivity (MR). The parameters were calculated with Chemicalize.org software and are presented in Table 1.59 All compounds 2–6 (except 4 e, which showed minimal aberration) are fully in agreement with Lipinski's and Gelovani's rules for prospective small‐molecule drugs (MW≤500, log P≤5, number of H‐bond donors ≤5, number of H‐bond acceptors ≤10, TPSA<140 Å2, MR within the range of 40 and 130 cm3 mol−1, number of atoms 20–70).60
Table 1.
Properties of compounds 2–6 calculated with the Chemicalize.org program.59 The Lipinski's and Gelovani's parameters.
| Compd | Molecular formula | Number of atoms | MW | log P | HBD[a] | HBA[b] | Lipinski score[c] | MR[d] [cm3 mol−1] | TPSA[e] [Å2] |
|---|---|---|---|---|---|---|---|---|---|
| 2 a | C20H27N3O4 | 54 | 373.453 | 1.43 | 2 | 5 | 4 | 103.62 | 89.55 |
| 2 b | C21H27N3O4 | 55 | 385.464 | 2.38 | 2 | 5 | 4 | 109.44 | 89.55 |
| 2 c | C21H29N3O4 | 57 | 387.480 | 1.88 | 2 | 5 | 4 | 108.22 | 89.55 |
| 2 d | C22H31N3O4 | 60 | 401.507 | 2.32 | 2 | 5 | 4 | 112.82 | 89.55 |
| 2 e | C24H27N3O4 | 58 | 421.497 | 3.32 | 2 | 5 | 4 | 120.65 | 89.55 |
| 3 a | C19H25N3O4 | 51 | 359.426 | 0.80 | 3 | 6 | 4 | 98.85 | 100.55 |
| 3 b | C19H23N3O4 | 49 | 357.410 | 0.90 | 3 | 6 | 4 | 99.92 | 100.55 |
| 3 c | C20H27N3O4 | 54 | 373.453 | 1.29 | 3 | 6 | 4 | 103.45 | 100.55 |
| 3 d | C21H29N3O4 | 57 | 387.480 | 1.74 | 3 | 6 | 4 | 108.05 | 100.55 |
| 3 e | C23H25N3O4 | 55 | 407.470 | 2.23 | 3 | 6 | 4 | 115.89 | 100.55 |
| 4 a | C26H32N4O4 | 66 | 464.566 | 2.58 | 3 | 6 | 4 | 131.53 | 101.58 |
| 4 b | C26H30N4O4 | 64 | 462.515 | 2.94 | 3 | 6 | 4 | 132.60 | 101.58 |
| 4 c | C27H34N4O4 | 69 | 478.580 | 3.02 | 3 | 6 | 4 | 136.13 | 101.58 |
| 4 d | C28H36N4O4 | 72 | 492.620 | 3.47 | 3 | 6 | 4 | 140.74 | 101.58 |
| 4 e | C30H32N4O4 | 70 | 512.600[b] | 4.27 | 3 | 6 | 3[f] | 148.57 | 101.58 |
| 5 a | C19H26N4O4 | 53 | 374.441 | 0.48 | 4 | 6 | 4 | 102.44 | 112.58 |
| 5 b | C20H28N4O4 | 56 | 388.468 | 0.92 | 4 | 6 | 4 | 107.04 | 112.58 |
| 5 c | C21H30N4O4 | 59 | 402.495 | 1.37 | 4 | 6 | 4 | 111.64 | 112.58 |
| 5 d | C23H26N4O4 | 57 | 422.485 | 2.16 | 4 | 6 | 4 | 119.47 | 112.58 |
| 6 a | C20H28N4O4 | 56 | 388.468 | 0.85 | 3 | 6 | 4 | 106.92 | 101.58 |
| 6 b | C20H26N4O4 | 54 | 386.452 | 1.21 | 3 | 6 | 4 | 107.99 | 101.58 |
| 6 c | C21H30N4O4 | 59 | 402.495 | 1.30 | 3 | 6 | 4 | 111.52 | 101.58 |
| 6 d | C22H32N4O4 | 62 | 416.522 | 1.74 | 3 | 6 | 4 | 116.12 | 101.58 |
| 6 e | C24H28N4O4 | 60 | 436.512 | 2.54 | 3 | 6 | 4 | 123.95 | 101.58 |
[a] H‐bond donor. [b] H‐bond acceptor. [c] Out of four. [d] Molar refractivity. [e] Topological polar surface area. [f] Minimal aberrations of the rules.
2.2. Biological Evaluation
Synthesized compounds 2–6 were tested for their anticancer activity on three human cancer cell lines (bone osteosarcoma U2OS, hepatocellular carcinoma HepG2, and breast adenocarcinoma MCF‐7) and human embryonic kidney (Hek293) cells. The cells were treated with the different compounds at different concentrations, and the median inhibitory concentration (IC50) values were determined (Table 2).
Table 2.
Antiproliferative screening of novel compounds 2–6 towards human cancer cell lines (U2OS, HepG2, MCF‐7) and human embryonic kidney cell line (Hek293) in vitro.
| Compd | Structure | IC50 [a] [μm] | |||
|---|---|---|---|---|---|
| U2OS | HepG2 | MCF‐7 | Hek293 | ||
| 2 a |
|
6.7±3.1 | >50 | >50 | >50 |
| 2 b |
|
14.9±2.2 | >50 | >50 | 17.0±1.7 |
| 2 c |
|
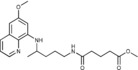
>50 | >50 | >50 | >50 |
| 2 d |
|
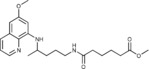
24.3±1.5 | >50 | 5.3±3.2 | >50 |
| 2 e |
|
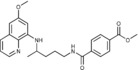
>50 | >50 | 7.1±3.0 | >50 |
| 3 a |
|
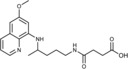
>50 | >50 | >50 | >50 |
| 3 b |
|
>50 | >50 | >50 | >50 |
| 3 c |
|
33.8±2.9 | >50 | >50 | >50 |
| 3 d |
|
>50 | >50 | >50 | >50 |
| 3 e |
|
>50 | >50 | 15.2±0.5 | >50 |
| 4 a |
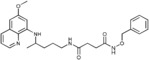
|
>50 | >50 | 12.0±1.4 | >50 |
| 4 b |
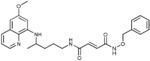
|
>50 | >50 | >50 | >50 |
| 4 c |
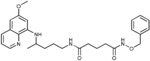
|
>50 | >50 | 16.6±0.8 | >50 |
| 4 d |
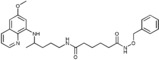
|
32.8±3.0 | 9.9±1.1 | 13.9±1.5 | >50 |
| 4 e |
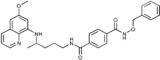
|
7.1±1.2 | >50 | 11.5±0.1 | 13.2±2.6 |
| 5 a |
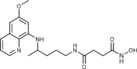
|
16.9±2.2 | 18.0±3.3 | 5.4±0.1 | 25.5±0.8 |
| 5 b |
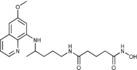
|
3.3±0.05 | 20.0±0.2 | 1.6±0.8 | 8.5±0.5 |
| 5 c |
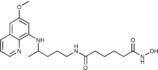
|
11.6±2.0 | 28.4±2.1 | 5.0±0.4 | 13.7±3.4 |
| 5 d |
|
11.6±2.1 | 21.7±0.2 | 4.7±0.1 | 6.1±1.9 |
| 6 a |
|
6.6±1.2 | >50 | 5.9±0.6 | 8.6±2.0 |
| 6 b |
|
>50 | >50 | 10.0±2.8 | >50 |
| 6 c |
|
>50 | >50 | 7.1±0.4 | >50 |
| 6 d |
|
>50 | >50 | 9.8±0.4 | >50 |
| 6 e |
|
>50 | >50 | >50 | >50 |
| PQ[b] | 12.0±0.7 | 37.7±5.8 | 13.8±1.4 | 8.1±1.3 | |
| SAHA[c] | 5.7±0.8 | 4.0±0.1 | 2.8±0.7 | 7.4±0.9 | |
| Cis[d] | 2.5±0.7 | – | 3.2±1.0 | 2.0±0.6 | |
[a] The concentration required to decrease viability by 50 %. [b] Primaquine. [c] Suberoylanilide hydroxamic acid. [d] Cisplatin.
The data from Table 2 clearly indicate that the SAHAquines were the most potent in the selected cancer cell lines. The IC50 values towards MCF‐7 cells were in the low micromolar concentrations (1.6–5.4 μm). Compounds from the other subclasses were, in general, less active than compounds 5. Compounds 4 d and 4 e were the most effective O‐benzylhydroxamic acids. In contrast, O‐methylhydroxamic acids 6 exhibited weak activity, except for succinic acid derivative 6 a. All compounds from the ester series, except 2 c, showed moderate activity, whereas carboxylic acid derivatives 3 a–e were practically inactive.
MCF‐7 cells were sensitive to 15 of the 24 tested compounds, namely, 5 a–d, 2 d, 2 e, 4 a, 4 c–e, and 6 a–d. Similar results were obtained in our previous studies with various PQ derivatives.24, 25, 26, 27, 28, 29, 30 On the other hand, HepG2 cells were very robust and only responded towards 4 d and compounds from subclass 5. The non‐cancer human cell line Hek293 was sensitive to 2 b, 4 e, 5 a–d, and 6 a but to a lesser extent than the cancer cell lines. Selectivity indices ranged from 2 to 12, depending on the tested compound and the cancer cell line employed.
To show the loss of MCF‐7 cells following their exposure to the most potent compounds from subclasses 4 and 5, that is, 4 e and 5 b, we labeled the cell nuclei with Hoechst 33342 and assessed the number of cells (Figure 2). The results from these studies complement the data from the MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide] assay (Table 2), showing a correlation between significant loss of mitochondrial metabolic activity and a concentration‐dependent decrease in the number of cells. A reduction in the number of MCF‐7 cells was found for treatment over periods of 24 and 72 h. The IC50 values for 5 b and 4 e obtained from these assays were within a comparable micromolar concentration range (5 b: 0.5 and 1.6 μm; 4 e: 9.0 and 11.5 μm). Determination of the number of cells is a more direct way of showing cell loss.
Figure 2.
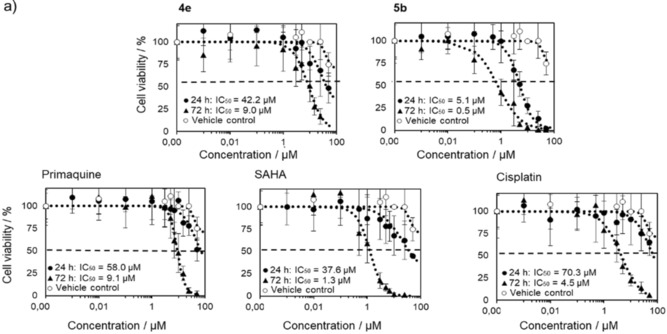
MCF‐7 cell viability following treatment with 4 e, 5 b, primaquine, SAHA, and cisplatin for 24 or 72 h at various concentrations ranging from 50 to 0.001 μm. Cell viability was measured by counting Hoechst 33342 labeled nuclei imaged by using a fluorescence microscope. Shown are average percentage cell viability compared to the untreated controls ± SD from two independent experiments.
As SAHAquines 5 with a free hydroxamic acid have the potential to chelate metal ions in Zn‐dependent HDACs and to increase the content of acetylated histones, we performed a immunocytochemical assay for histone acetylation. MCF‐7 cells were treated with 5 b and acetylated H3K9/H3K14 was measured. SAHA, PQ, and 4 e were used as controls in the histone acetylation experiments. Indeed, the results show that 5 b caused a significant accumulation of acetylated H3 histone (Figure 3), which suggested that HDAC inhibition was a possible contributing mechanism for SAHAquines 5 but not for the other PQ derivatives. Given that HDAC inhibition correlated with cellular histone acetylation but not with cell loss, we did not extensively study the kinetics of HDAC inhibition.61 An in‐depth mechanistic study should be performed to explain the mode of cell death caused by the SAHAquines.
Figure 3.
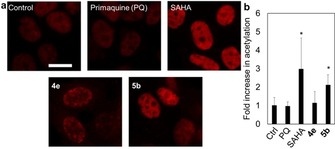
MCF‐7 cells were treated with PQ, SAHA, 4 e, or 5 b (1 μm) for 24 h. a) Representative fluorescence micrographs of MCF‐7 histone H3 acetylation (red) at lysine 9/lysine 14 in response to treatments. Cells were incubated with primary antibody (rabbit anti‐acetyl‐Histone‐H3 polyclonal antibody, 500× dilution) for 24 h and then with secondary antibody (Alexa Fluor 647 goat anti‐rabbit IgG −2 mg mL−1; 500× dilution) for 1 h in the dark. Cells were imaged by using a fluorescence microscope with a CY5 filter. Fluorescence was analyzed in ImageJ. Scale bar represents 20 μm. b) Averages of fluorescence per cell±SD (as fold increase in untreated control=1) from at least two independent experiments. * p<0.05, t‐test.
Literature data on the antimalarial activity of SAHA and related HDAC inhibitors49, 50 prompted us to evaluate SAHAquines and their synthetic precursors for their in vitro activity against P. falciparum erythrocytic stages. The IC50 values of SAHAquines 2–6 against the erythrocytic stage of two P. falciparum strains, 3D7 and Dd2, were determined (Table 3). In general, the 3D7 strain was more sensitive than the Dd2 strain. Again, the most active compounds were SAHAquines 5, the hydroxamic acid subclass. Compound 5 b had the lowest IC50 value (0.4 μm for the Pf3D7 strain and 1.9 μm for the PfDd2 strain), followed by 5 d (IC50=0.6 and 1.2 μm, respectively), 5 c (IC50=3.7 and 13.6 μm, respectively), and finally 5 a (IC50=15.8 and 27.1 μm, respectively). Derivatives 2 b and 2 d were the most active compounds from the ester subclass, and 4 e was the most active from the O‐benzylhydroxamic acids. Carboxylic acids 3 a–e were inactive.
Table 3.
IC50 values for compounds 2–6 against erythrocytic stage of two P. falciparum strains.
| Compd | IC50 [μm] | Compd | IC50 [μm] | ||
|---|---|---|---|---|---|
| Pf3D7 | Pf3Dd2 | Pf3D7 | Pf3Dd2 | ||
| 2 a | 100.0±11.0 | 74.1±5.3 | 4 d | >27.7 | >27.7 |
| 2 b | 2.9±0.2 | 7.2±1.9 | 4 e | 6.1±0.1 | >27.7 |
| 2 c | 81.1±3.4 | 80.1±31.0 | 5 a | 15.8±1.7 | 27.1±0.8 |
| 2 d | 6.6±0.2 | 50.4±4.6 | 5 b | 0.4±0.1 | 1.9±0.8 |
| 2 e | 27.1±0.6 | 25.0±2.8 | 5 c | 3.7±1.3 | 13.6±0.2 |
| 3 a | >111 | >111 | 5 d | 0.6±0.2 | 1.2±0.04 |
| 3 b | >111 | >111 | 6 a | >111 | 94.4±8.6 |
| 3 c | >111 | >111 | 6 b | 8.3±0.9 | 14.8±0.1 |
| 3 d | >111 | >111 | 6 c | >111 | >111 |
| 3 e | >111 | >111 | 6 d | >111 | >111 |
| 4 a | >55 | 50.0±5.0 | 6 e | >25 | >27.7 |
| 4 b | >27.7 | >27.7 | PQ[a] | 1.5±0.02 | 4.3±1.5 |
| 4 c | 39.5±15.5 | 35.5±1.2 | CQ[b] | 1.6×10−3 | 0.265±0.003 |
[a] Primaquine. [b] Chloroquine.
We further evaluated the in vitro activity of compounds 2–6 against the hepatic stages of P. berghei. Two concentrations of SAHAquines were tested: 1 and 10 μm. For comparison, the same concentrations of PQ were included in the assay as a positive control (IC50=8.4±3.4 μm), and DMSO was used as a negative control (Figure 4).
Figure 4.
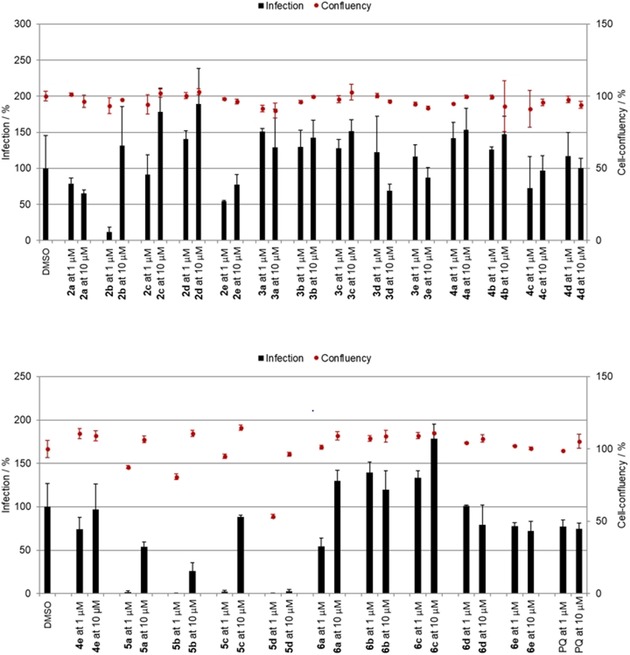
Activity of compounds 2–6 against P. berghei hepatic stages at concentrations of 1 and 10 μm. Anti‐infective activity (infections scale, bars) are shown.
As shown in Figure 4, only ester 2 b and SAHAquines 5 a–d were active against the P. berghei hepatic stages, whereas the other tested compounds were completely inactive. All compounds from hydroxamic acid subclass 5 exhibited strong antiplasmodial activity at both concentrations tested, with IC50 values ranging from 0.3 to 1.25 μm and without any noticeable effects on host‐cell confluency (Figure 5). Our results show that the free hydroxamic acid moiety was crucial for antiplasmodial activity, as activity was lost if this group was protected or replaced by a carboxylic acid. The activity of compound 2 b from the ester subclass was probably due to the α,β‐unsaturated carbonyl group (Michael acceptor moiety), which is capable of conjugate addition.
Figure 5.
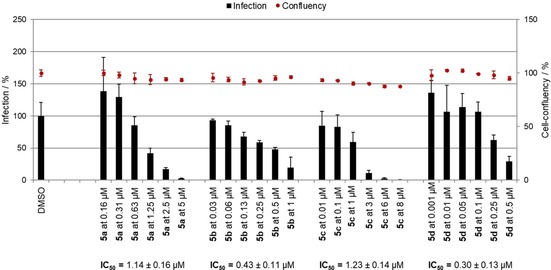
IC50 of SAHAquines 5 a–d against P. berghei hepatic stages.
The relationship between antimalarial and anticancer activity is complex and is discussed in detail in a review by Duffy, Wade, and Chang.62 Both main groups of existing antimalarial agents, the quinolines and the sesquiterpene lactones, have a range of known anticancer properties that could either be distinct from or overlap with their antimalarial properties. Although our results show a similar pattern of activity, it should be noted that the activities of 5 b and 5 d are an order of magnitude higher towards the P. falciparum erythrocytic stage than towards the tested cancer cell lines and the human embryonic kidney cells (Hek293).
Having in mind that very good safety profiles are needed for antimalarial drugs, as they should be delivered, amongst others, to two particularly vulnerable populations, small children and pregnant women,63 we are aware that our finding can only be a starting point for the development of a clinically applicable antimalarial drug. As the antiplasmodial activities of compounds 2 b and 5 a–d were comparable to the activity of PQ, they might potentially replace PQ against PQ‐resistant Plasmodium strains or in the treatment of glucose‐6‐phosphate dehydrogenase deficient patients and poor metabolizers, but this hypothesis still remains to be evaluated. Notably, the synthesis of SAHAquines is cheap and short (1–4 h) with good to excellent yields (43–97 %), which is beneficial for the development of antimalarial agents.64
3. Conclusions
Four SAHAquines based on SAHA and PQ motifs and 20 other PQ derivatives were synthesized and evaluated in vitro against three human cancer cell lines and a human embryonic kidney cell line. SAHAquines 5 a–d displayed cytostatic activity at low micromolar concentrations. A few compounds from other subclasses were also effective, but less so than 5 a–d. We showed that the most active hydroxamic acid, that is, 5 b, caused a significant accumulation of acetylated histone H3K9/H3K14, a downstream target of class I HDACs. The results from the antiplasmodial activity of SAHAquines against the erythrocytic stages of the 3D7 and Dd2 P. falciparum strains and against P. berghei hepatic stages correlated with their cytostatic activity. The in vitro cytostatic and dual‐stage antiplasmodial activity of SAHAquines suggest that these novel compounds could constitute a basis for the development of effective anticancer or malaria prophylactic/curative agents with improved potency and selectivity.
Experimental Section
General Methods
Melting points were measured with a Stuart Melting Point (SMP3) apparatus (Barloworld Scientific, UK) in open capillaries. IR spectra were recorded with a FTIR PerkinElmer Spectrum One, and UV/Vis spectra were recorded with a Lambda 20 double‐beam spectrophotometer (PerkinElmer, UK). All NMR (1H and 13C) spectra were obtained at 25 °C by using an NMR Avance 600 spectrometer (Bruker, Germany) at 300 and 150 MHz for 1H and 13C nuclei, respectively. Chemical shifts (δ) are reported in parts per million (ppm) relative to tetramethylsilane in the 1H spectra and relative to [D6]DMSO in the 13C spectra (δ=39.51 ppm). Coupling constants (J) are reported in hertz. Mass spectra were collected with a HPLC‐MS/MS instrument (HPLC, Agilent Technologies 1200 Series; MS, Agilent Technologies 6410 Triple Quad). Mass determination was realized by using electrospray ionization (ESI) in the positive mode. Elemental analyses were performed with a CHNS LECO analyzer (LECO Corporation, USA). All compounds were routinely checked by TLC with Merck silica gel 60F‐254 glass plates by using the following solvent systems: dichloromethane/methanol 9:1, 9.5:0.5, and 9.6:0.4; cyclohexane/ethyl acetate 1:1; and cyclohexane/ethyl acetate/methanol 3:1:0.5. Spots were visualized by short‐wave UV light and iodine vapor. Column chromatography was performed on silica gel 0.063–0.200 mm. All chemicals and solvents were of analytical grade and were purchased from commercial sources. PQ diphosphate, 4‐methoxy‐4‐oxobutanoic acid (monomethyl succinate), (E)‐4‐ethoxy‐4‐oxobut‐2‐enoic acid (monoethyl fumarate), 5‐methoxy‐5‐oxopentanoic acid (monomethyl glutarate), 6‐methoxy‐6‐oxohexanoic acid (monomethyl adipate), 4‐(methoxycarbonyl)benzoic acid (monomethyl terephtalate), DIEA, HATU, O‐benzylhydroxylamine, O‐methylhydroxylamine, triethylamine, MTT, cisplatin (Cis), and Hoechst 33342 were purchased from Sigma–Aldrich; rabbit anti‐acetyl‐Histone‐H3 polyclonal antibody was purchased from Millipore, Alexa Fluor 647 goat anti‐rabbit IgG was purchased from Life Technologies, and SAHA was acquired from Cayman Chemicals. PQ was prepared from PQ diphosphate prior to use. All reactions with PQ were performed under light‐protected conditions.
Synthesis
General Procedure for the Preparation of Esters 2 a–e
Method A: A solution of dicarboxylic acid monoester 1 a–e (1.4 mmol), HATU (0.532 g, 1.4 mmol), and DIEA (0.362 g, 2.8 mmol) in dichloromethane (8 mL) was stirred at room temperature. After 10 min, a solution of PQ (0.401 g, 1.5 mmol) in dichloromethane (7 mL) was added. The mixture was stirred at room temperature for 1 h and was then concentrated under reduced pressure. The residue was dissolved in ethyl acetate and extracted with brine (3×). The organic layer was dried with sodium sulfate, filtered, and concentrated under reduced pressure.
Method B: A solution of dicarboxylic acid monoester 1 a–e (1.8 mmol) in thionyl chloride (7 mL) was kept overnight and concentrated under reduced pressure. The residue was triturated with dichloromethane (2×), and the solvent was evaporated again. A solution of PQ (0.401 g, 1.5 mmol) and Et3N (0.152 g, 1.5 mmol) in dichloromethane (8 mL) was added dropwise to the carboxylic acid chloride dissolved in dichloromethane (10 mL). The mixture was stirred at room temperature for 1 h and extracted with brine (3×). The organic layer was dried with sodium sulfate, filtered, and concentrated under reduced pressure.
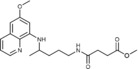
Methyl 3‐({4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)pro‐ panoate (2 a): Method A, from the reaction of monomethyl succinate (1 a, 0.185 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether), 2 a was obtained as a pale‐yellow solid (0.460 g, 88 %): m.p. 71–73 °C; 1H NMR ([D6]DMSO): δ=8.55–8.53 (dd, J=1.6, 4.2 Hz, 1 H, 10), 8.09–8.06 (dd, J=1.5, 8.3 Hz, 1 H, 12), 7.86 (t, J=5.3 Hz, 1 H, 1), 7.45–7.40 (m, 1 H, 11), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.12 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.66–3.57 (m, 1 H, 5), 3.56 (s, 1 H, 5′), 3.08–3.02 (m, 2 H, 2), 2.48 (t, J=6.6 Hz, 2 H, 2′), 2.33 (t, J=6.8 Hz, 2 H, 3′), 1.70–1.58, 1.58–1.42 (2 m, 4 H, 3,4), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=173.30, 170.87, 159.00, 144.62, 144.23, 134.79, 134.52, 129.57, 122.09, 96.09, 91.58, 54.97, 51.21, 46.97, 38.46, 33.37, 29.82, 28.82, 25.95, 20.19 ppm; IR (ATR): =2278, 3306, 3060, 2959, 2927, 2857, 1732, 1636, 1619, 1552, 1518, 1454, 1387, 1224, 1200, 1170, 1052, 1031, 825, 793, 679 cm−1; MS (ESI): m/z: 374.2 [M+1]+; elemental analysis calcd (%) for C20H27N3O4 (373.45): C 64.32, H 7.21, N 11.25; found: C 64.02, H 7.47, N 11.00.
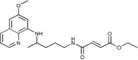
Ethyl (2E)‐3‐({4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)‐ prop‐2‐enoate (2 b): Method B, from the reaction of monoethyl fumarate (1 b, 0.202 g) and after purification by column chromatography (dichloromethane/methanol 9.6:0.4) and crystallization (ether), 2 b was obtained as a pale‐yellow solid (0.378 g, 70 %): m.p. 71–73 °C; 1H NMR ([D6]DMSO): δ=8.54–8.53 (dd, J=1.6, 4.2 Hz, 1 H, 10), 8.51 (t, J=5.5 Hz, 1 H, 1), 8.08–8.06 (dd, J=1.5, 8.3 Hz, 1 H, 12), 7.43–7.41 (m, 1 H, 11), 6.98 (d, J=15.5 Hz, 1 H, 2′), 6.55 (d, J=15.5 Hz, 1 H, 3′), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.27 (d, J=2.4 Hz, 1 H, 14), 6.13 (d, J=8.8 Hz, 1 H, 7), 4.20–4.16 (q, J=7.1 Hz, 2 H, 5′), 3.82 (s, 3 H, 17), 3.66–3.61 (m, 1 H, 5), 3.20–3.17 (dd, J=6.4, 12.3 Hz, 2 H, 2), 1.71–1.65, 1.62–1.51 (2 m, 4 H, 3,4), 1.23 (t, J=7.1 Hz, 3 H, 6′), 1.22 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=165.05, 162.63, 158.98, 144.60, 144.20, 137.60, 134.76, 134.51, 129.55, 128.08, 122.06, 96.11, 91.51, 60.60, 54.94, 46.93, 38.81, 33.36, 25.63, 20.18, 13.97 ppm; IR (ATR): =3391, 3325, 3079, 2969, 2939, 1716, 1645, 1616, 1554, 1520, 1457, 1424, 1388, 1365, 1334, 1302, 1224, 1205, 1163, 1159, 1038, 999, 831, 819, 790, 681, 660 cm−1; MS (ESI): m/z: 386.2 [M+1]+; elemental analysis calcd (%) for C21H27N3O4 (385.46): C 65.44, H 7.06, N 10.90; found: C 65.25, H 7.09, N 10.70.
Methyl 4‐({4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)but‐ anoate (2 c): Method A, from the reaction of monomethyl glutarate (1 c, 0.206 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5), 2 c was obtained as an oil (0.439 g, 81 %): 1H NMR ([D6]DMSO): δ=8.53 (d, J=2.6 Hz, 1 H, 10), 8.07 (d, J=8.2 Hz, 1 H, 12), 7.78 (t, J=3 Hz, 1 H, 1), 7.43–7.41 (m, 1 H, 11), 6.47 (d, J=2.2 Hz, 1 H, 16), 6.26 (d, J=2.2 Hz, 1 H, 14), 6.11 (d, J=8.8 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.64–3.60 (m, 1 H, 5), 3.57 (s, 3 H, 6′), 3.05 (t, J=6.0 Hz, 2 H, 2), 2.27 (t, J=3.0 Hz, 2 H, 2′), 2.07 (t, J=7.3 Hz, 2 H, 4′), 1.74–1.69 (m, 2 H, 3′), 1.67–1.62, 1.54–1.44 (2 m, 4 H, 3, 4), 1.21 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=173.00, 171.18, 158.98, 144.60, 144.20, 134.76, 134.50, 129.55, 122.05, 96.08, 91.59, 54.95, 51.14, 46.97, 38.36, 34.28, 33.40, 32.64, 25.93, 20.62, 20.17 ppm; IR (ATR): =3376, 3246, 3086, 2993, 2961, 2927, 1736, 1632, 1612, 1574, 1520, 1458, 1423, 1386, 1219, 1203, 1172, 1161, 1052, 992, 817, 788, 751, 720, 676 cm−1; MS (ESI): m/z: 388.3 [M+1]+; elemental analysis calcd (%) for C21H29N3O4 (387.47): C 65.09, H 7.54, N 10.84; found: C 65.35, H 7.33, N 10.99.
Methyl 5‐({4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)pent‐ anoate (2 d): Method A, from the reaction of monomethyl adipate (1 d, 0.224 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether/petroleum ether), 2 d was obtained as a pale‐yellow solid (0.483 g, 86 %): m.p. 56–58 °C; 1H NMR ([D6]DMSO): δ=8.54–8.52 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.5 Hz, 1 H, 12), 7.78 (t, J=5.4 Hz, 1 H, 1), 7.45–7.40 (m, 1 H, 11), 6.48 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.12 (d, J=8.8 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.66–3.59 (m, 1 H, 5), 3.57 (s, 3 H, 7′), 3.08–3.04 (m, 2 H, 2), 2.28 (t, J=6.9 Hz, 2 H, 2′), 2.04 (t, J=6.9 Hz, 2 H, 5′), 1.68–1.57, 1.57–1.42 (2 m, 8 H, 3, 4, 3′, 4′), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=173.22, 171.60, 159.00, 144.63, 144.23, 134.79, 134.53, 129.57, 122.09, 96.09, 91.58, 54.97, 51.15, 46.98, 38.36, 35.00, 33.43, 32.98, 25.99, 24.73, 24.03, 20.18 ppm; IR (ATR): =3373, 3311, 2943, 2876, 1730, 1640, 1618, 1518, 1508, 1461, 1425, 1389, 1265, 1222, 1169, 1055, 821, 680, 625 cm−1; MS (ESI): m/z: 402.3 [M+1]+; elemental analysis calcd (%) for C22H31N3O4 (401.50): C 65.81, H 7.78, N 10.47; found: C 65.66, H, 7.66, N 10.31.
Methyl 4‐({4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)ben‐ zoate (2 e): Method A, from the reaction of monomethyl terephtalate (1 e, 0.252 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether), 2 e was obtained as a pale‐yellow solid (0.489 g, 83 %): m.p. 112–113 °C; 1H NMR ([D6]DMSO): δ=8.65 (t, J=5.5 Hz, 1 H, 1), 8.54–8.52 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.08–8.06 (dd, J=8.2, 1.1 Hz, 1 H, 12), 8.01 (d, J=8.3 Hz, 2 H, 3′, 7′), 7.94 (d, J=8.3 Hz, 2 H, 4′, 6′), 7.43–7.41 (m, 1 H, 11), 6.47 (d, J=2.2 Hz, 1 H, 16), 6.28 (d, J=2.1 Hz, 1 H, 14), 6.15 (d, J=8.7 Hz, 1 H, 7), 3.88 (s, 3 H, 9′), 3.81 (s, 3 H, 17), 3.69–3.65 (m, 1 H, 5), 3.32–3.29 (m, 2 H, 2), 1.76–1.65, 1.65–1.57 (2 m, 4 H, 3, 4), 1.23 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=165.69, 165.24, 158.97, 144.59, 144.18, 138.76, 134.73. 134.51, 131.54, 129.53, 128.99, 127.47, 122.03, 96.09, 91.59, 54.91, 52.26, 47.00, 39.27, 33.39, 25.79, 20.15 ppm; IR (ATR): =3403, 3318, 3060, 2936, 1714, 1634, 1615, 1519, 1425, 1386, 1278, 1225, 1169, 1111, 820, 791, 736, 703 cm−1; MS (ESI): m/z: 422.2 [M+1]+; elemental analysis calcd (%) for C24H27N3O4 (421.49): C 68.39, H 6.46, N 9.97; found: C 68.25, H 6.76, N 10.08.
General procedure for the preparation of carboxylic acids 3 a–e: A solution of lithium hydroxide monohydrate (0.126 g, 3 mmol) in water (10 mL) was added to a solution of ester 2 (6 mmol) in methanol (10 mL). The mixture was stirred at room temperature for 1 h. Methanol was evaporated under reduced pressure, and the aqueous residue was neutralized with 10 % HCl and extracted with dichloromethane (3×). The organic layer was dried with sodium sulfate, filtered, and concentrated under reduced pressure.
3‐({4‐[(6‐Methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)propanoic acid (3 a): From the reaction of ester 2 a (0.224 g) and after crystallization (ether), 3 a was obtained as a pale‐yellow solid (0.209 g, 97 %): m.p. 146–148 °C; 1H NMR ([D6]DMSO): δ=12.06 (s, 1 H, 5′), 8.55–8.53 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.6 Hz, 1 H, 12), 7.85–7.82 (t, J=5.4 Hz, 1 H, 1), 7.45–7.41(m, 1 H, 11), 6.48 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.12 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.66–3.57 (m, 1 H, 5), 3.08–3.03 (m, 2 H, 2), 2.41 (t, J=6.5 Hz, 2 H, 2′), 2.29 (t, J=6.7 Hz, 2 H, 3′), 1.70–1.58, 1.58–1.38 (2 m, 4 H, 3, 4), 1.2 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=173.87, 170.73, 159.00, 144.63, 144.24, 134.80, 134.52, 129.58, 122.10, 96.10, 91.59, 54.97, 46.99, 38.48, 33.38, 30.04, 29.21, 25.96, 20.19 ppm; IR (ATR): =3454, 3268, 3094, 3009, 2962, 2928, 2863, 1713, 1649, 1615,1584, 1562, 1524, 1452, 1386, 1346, 1227, 1203, 1156, 1161, 824, 785, 746, 674 cm−1; MS (ESI): m/z: 360.2 [M+1]+; elemental analysis calcd (%) for C19H25N3O4 (359.42): C 63.49, H 7.01, N 11.69; found: C 63.22, H 7.30, N 11.45.
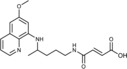
(2E)‐3‐({4‐[(6‐Methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl) prop‐2‐enoic acid (3 b): From the reaction of ester 2 b (0.231 g) and after crystallization (ether), 3 b was obtained as a pale‐yellow solid (0.178 g, 83 %): m.p. 147–149 °C; 1H NMR ([D6]DMSO): δ=12.83 (s, 1 H, 5′), 8.55–8.53 (dd, J=1.6, 4.2 Hz, 1 H, 10), 8.49 (t, J=5.5 Hz, 1 H, 1), 8.09–8.06 (dd, J=1.5, 8.3 Hz, 1 H, 12), 7.45–7.40 (m, 1 H, 11), 6.91 (d, J=15.5 Hz, 1 H, 2′), 6.50 (d, J=15.5 Hz, 1 H, 3′), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.27 (d, J=2.4 Hz, 1 H, 14), 6.14 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.68–3.58 (m, 1 H, 5), 3.22–3.16 (m, 2 H, 2), 1.71–1.49 (m, 4 H, 3, 4), 1.21 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=166.49, 162.95, 159.00, 144.62, 144.22, 137.11, 134.79, 134.52, 129.57, 129.33, 122.09, 96.13, 91.62, 54.96, 46.96, 38.80, 33.40, 25.71, 20.19 ppm; IR (ATR): =3453, 3271, 3087, 2937, 1716, 1654, 1613, 1564, 1527, 1380, 1337, 1269, 1236, 1213, 1198, 1171, 981, 909, 820, 785, 767, 673 cm−1; MS (ESI): m/z: 358.2 [M+1]+; elemental analysis calcd (%) for C19H23N3O4 (357.40): C 63.85, H 6.49, N 11.76; found: C 63.77, H 6.72, N 11.99.
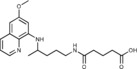
4‐({4‐[(6‐Methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)butanoic acid (3 c): From the reaction of ester 2 c (0.232 g) and after crystallization (ether), 3 c was obtained as a pale‐yellow solid (0.186 g, 88 %): m.p. 95–96 °C; 1H NMR ([D6]DMSO): δ=12.00 (s, 1 H, 6′), 8.55–8.53 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.10–8.07 (dd, J=8.3, 1.5 Hz, 1 H, 12), 7.80 (t, J=5.4 Hz, 1 H, 1), 7.45–7.41 (m, 1 H, 11), 6.48 (d, J=2.4 Hz, 1 H, 16), 6.27 (d, J=2.4 Hz, 1 H, 14), 6.13 (d, J=8.7 Hz, 1 H, 7), 3.83 (s, 3 H, 17), 3.69–3.55 (m, 1 H, 5), 3.09–3.05 (m, 2 H, 2), 2.20 (t, J=7.4 Hz, 2 H, 2′), 2.08 (t, J=7.4 Hz, 2 H, 4′), 1.75–1.43 (m, 6 H, 3′, 3, 4), 1.21 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=147.15, 171.36, 159.00, 144.63, 144.23, 134.79, 134.53, 129.57, 122.09, 96.11, 91.60, 54.97, 46.98, 38.38, 34.46, 33.41, 33.04, 25.97, 20.71, 20.19 ppm; IR (ATR): =3453, 3324, 2933, 1720, 1643, 1613, 1580, 1524, 1386, 1227, 1204, 1161, 1139, 1058, 822, 786, 675 cm−1; MS (ESI): m/z: 374.2 [M+1]+; elemental analysis calcd (%) for C20H27N3O4 (373.45): C 64.32, H 7.29, N 11.25; found: C 64.21, H 7.15, N 11.48.
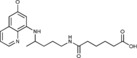
5‐({4‐[(6‐Methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)pentanoic acid (3 d): From the reaction of ester 2 d (0.241 g) and after crystallization (ether), 3 d was obtained as a pale‐yellow solid (0.198 g, 85 %): m.p. 68–69 °C; 1H NMR ([D6]DMSO): δ=11.99 (s, 1 H, 7′), 8.54 (d, J=2.9 Hz, 1 H, 10), 8.08 (d, J=8.1 Hz, 1 H, 12), 7.78 (t, J=6.0 Hz, 2 H, 1), 7.45–7.40 (m, 1 H, 11), 6.48 (d, J=3.0 Hz, 1 H, 16), 6.26 (d, J=3.0 Hz, 1 H, 14), 6.12 (d, J=8.6 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.69–3.56 (m, 1 H, 5), 3.09–3.0 (m, 2 H, 2), 2.18 (t, J=6.0 Hz, 2 H, 5′), 2.04 (t, J=6.0 Hz, 2 H, 2′), 1.70–1.57, 1.57–1.42 (2 m, 8 H, 3, 4, 3′, 4′), 1.20 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=174.37, 171.67, 159.00, 144.63, 144.23, 134.79, 134.52, 129.57, 122.09, 96.10, 91.59, 54.97, 46.98, 38.36, 35.11, 33.38, 25.99, 24.84, 24.13, 20.19 ppm; IR (ATR): =3453, 3370, 3294, 2943, 2863, 1731, 1609, 1562, 1519, 1455, 1424, 1385, 1224, 1203, 1166, 1156, 1133, 1053, 819, 790, 765, 677 cm−1; MS (ESI): m/z: 388.2 [M+1]+; elemental analysis calcd (%) for C21H29N3O4 (387.47): C 65.10, H 7.54, N 10.84; found: C 65.44, H 7.79, N 10.63.
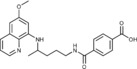
4‐({4‐[(6‐Methoxyquinolin‐8‐yl)amino]pentyl}carbamoyl)benzoic acid (3 e): From the reaction of ester 2 e (0.253 g) and after crystallization (ether), 3 e was obtained as a pale‐yellow solid (0.210 g, 86 %): m.p. 142–144 °C; 1H NMR ([D6]DMSO): δ=13.12 (s, 1 H, 9′), 8.64 (t, J=5.5 Hz, 1 H, 1), 8.54–8.53 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.5 Hz, 1 H, 12), 8.00 (d, J=8.4 Hz, 2 H, 3′, 7′), 7.92 (d, J=8.4 Hz, 2 H, 4′, 6′), 7.45–7.41 (m, 1 H, 11), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.28 (d, J=2.4 Hz, 1 H, 14), 6.16 (d, J=8.4 Hz, 1 H, 7), 3.81 (s, 3 H, 17), 3.71–3.63 (m, 1 H, 5), 3.34–3.28 (m, 2 H, 2), 1.76–1.57 (m, 4 H, 3, 4), 1.22 ppm (d, J=7.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=166.80, 165.42, 159.00, 144.61, 144.22, 138.45, 134.80, 134.52, 132.79, 129.58, 129.16, 127.36, 122.09, 96.14, 91.59, 54.96, 47.02, 39.28, 33.40, 25.86, 20.19 ppm; IR (ATR): =3413, 3308, 2941, 2864 1727, 1691, 1636, 1613, 1575, 1519, 1454, 1422, 1385, 1276, 1220, 1201, 1156, 818, 786, 674 cm−1; MS (ESI): m/z: 408.2 [M+1]+; elemental analysis calcd (%) for C23H25N3O4 (407.46): C 67.80, H 6.18, N 10.31; found: C 67.93, H 6.02, N 10.68.
General procedure for the preparation of O‐benzylhydroxamic acids 4 a–e: A solution of acid 3 (0.6 mmol), DIEA (0.155 g, 1.2 mol), and HATU (0.228 g, 0.6 mmol) in dichloromethane (6 mL) was stirred at room temperature. After 10 min, O‐benzylhydroxylamine hydrochloride (0.112 g, 0.7 mmol) and Et3N (0.071 g, 0.7 mmol) were added. The mixture was stirred at room temperature for 2 h and concentrated under reduced pressure. The residue was dissolved in ethyl acetate (20 mL) and extracted with brine (3×). The organic layer was dried with sodium sulfate, filtered, and concentrated under reduced pressure.
N‐(Benzyloxy)‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}butanediamide (4 a): From the reaction of 3 a (0.216 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether), 4 a was obtained as a pale‐yellow solid (0.111 g, 40 %): m.p. 121–123 °C; 1H NMR ([D6]DMSO): δ=10.99 (s, 1 H, 5′), 8.54 (d, J=2.7 Hz, 1 H, 10), 8.08 (d, J=7.2 Hz, 1 H, 12), 7.85 (s, 1 H, 1), 7.44‐7‐35 (m, 6 H, 11, 8′‐12′), 6.47 (d, J=2.2 Hz, 1 H, 16), 6.26 (d, J=2.0 Hz, 1 H, 14), 6.12 (d, J=8.6 Hz, 1 H, 7), 4.76 (s, 2 H, 6′), 3.82 (s, 3 H, 17), 3.66–3.57 (m, 1 H, 5), 3.08–3.02 (m, 2 H, 2), 2.30 (t, J=6.9 Hz, 2 H, 3′), 2.18 (t, J=6.8 Hz, 2 H, 2′), 1.70–1.57, 1.57–1.43 (2 m, 4 H, 3, 4), 1.20 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=170.71, 168.80, 159.00, 144.62, 144.23, 136.06, 134.79, 134.52, 129.57, 128.72, 128.26, 128.16, 122.09, 96.09, 91.59, 76.75, 54.97, 46.99, 38.49, 33.39, 30.37, 27.91, 25.97, 20.19 ppm; IR (ATR): =3367, 3311, 3060, 2936, 2862, 1728, 1636, 1615, 1546, 1519, 1457, 1424, 1387, 1223, 1200, 1159, 1052, 820, 790, 679 cm−1; MS (ESI): m/z: 465.3 [M+1]+; elemental analysis calcd (%) for C26H32N4O4 (464.56): C 67.22, H 6.94, N 12.06; found: C 67.46, H 6.69, N, 12.31.
(2E)‐N‐(Benzyloxy)‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}‐but‐2‐ene‐diamide (4 b): From the reaction of 3 b (0.215 g) and after purification by column chromatography (dichloromethane/methanol/ethyl acetate/cyclohexane 9.5:0.5:10:10) and crystallization (ether), 4 b was obtained as a pale‐yellow solid (0.119 g, 43 %): m.p. 157–158 °C; 1H NMR ([D6]DMSO): δ=11.52 (s, 1 H, 5′), 8.55–8.53 (dd, J=1.5, 4.2 Hz, 1 H, 10), 8.45 (t, J=5.5 Hz, 1 H, 1), 8.09–8.06 (dd, J=1.4, 8.3 Hz, 1 H, 12), 7.45–7.37 (m, 1 H, 11, 8′‐12′), 6.90 (d, J=15.1 Hz, 1 H, 2′), 6.62 (d, J=15.2 Hz, 1 H, 3′), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.27 (d, J=2.3 Hz, 1 H, 14), 6.15 (d, J=8.7 Hz, 1 H, 7), 4.86 (s, 2 H, 6′), 3.82 (s, 3 H, 17), 3.68–3.58 (m, 2 H, 5), 3.20–3.15 (m, 2 H, 2), 1.70–1.49 (m, 4 H, 3,4), 1.21 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=163.17, 161.22, 158.99, 144.60, 144.22, 134.82, 134.49, 133.58, 129.58, 129.07, 122.09, 96.16, 91.64, 63.35, 54.97, 46.97, 38.95, 33.42, 25.76, 20.19 ppm; IR (ATR): =3383, 3283, 3213, 3008, 2936, 2865, 1738, 1626, 1575, 1556, 1518, 1454, 1386, 1337, 1219, 1203, 1157,1051, 974, 818, 790, 738, 695, 675 cm−1; MS (ESI): m/z: 463.1 [M+1]+; elemental analysis calcd (%) for C26H30N4O4 (462.54): C 67.51, H 6.54, N 12.11; found: C 67.79, H 6.81, N 11.95.
N‐(Benzyloxy)‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}pentanediamide (4 c): From the reaction of 3 c (0.224 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether/petroleum ether), 4 c was obtained as a pale‐yellow solid (0.144 g, 50 %): m.p. 84–85 °C; 1H NMR ([D6]DMSO): δ=10.95 (s, 1 H, 6′), 8.54–8.53 (dd, J=4.1, 1.5 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.4 Hz, 1 H, 12), 7.78 (t, J=5.4 Hz, 1 H, 1), 7.44–7.33 (m, 6 H, 11, 9′‐13′), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.3 Hz, 1 H, 14), 6.13 (d, J=8.8 Hz, 1 H, 7), 4.77 (s, 2 H, 7′), 3.82 (s, 3 H, 17), 3.62–3.56 (m, 1 H, 5), 3.08–3.04 (m, 2 H, 2), 2.04 (t, J=7.4 Hz, 2 H, 4′), 1.95 (t, J=7.3 Hz, 2 H, 2′), 1.74–1.43 (m, 6 H, 3′, 3, 4), 1.20 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=171.32, 169.03, 159.00, 144.62, 144.23, 136.09, 134.79, 134.52, 129.57, 128.72, 128.26, 128.16, 122.09, 96.10, 91.59, 76.77, 54.97, 46.98, 38.41, 34.57, 33.43, 31.70, 25.99, 21.21, 20.19 ppm; IR (ATR): =3367, 3325, 3187, 2960, 2931, 2857, 1738, 1677, 1634, 1614, 1518, 1455, 1387, 1219, 1203, 1170, 1157, 1052, 1033, 821, 790, 740, 696, 676 cm−1; MS (ESI): m/z: 479.3 [M+1]+; elemental analysis calcd (%) for C27H34N4O4 (478.58): C 67.76, H 7.16, N 11.71; found: C 67.59, H 6.98, N, 12.04.
N‐(Benzyloxy)‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}hexanediamide (4 d): From the reaction of 3 d (0.232 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether), 4 d was obtained as a pale‐yellow solid (0.151 g, 51 %): m.p. 102–105 °C; 1H NMR ([D6]DMSO): δ=10.91 (s, 1 H, 7′), 8.53 (dd, J=4.0, 1.4 Hz, 1 H, 10), 8.06 (dd, J=8.2 Hz, 1 H, 12), 7.74 (s, 1 H, 1), 7.43–7.41 (m, 1 H, 11), 7.37–7.34 (m, 5 H, 10′‐14′), 6.47 (d, J=2.3 Hz, 1 H, 16), 6.26 (d, J=2.2 Hz, 1 H, 14), 6.11 (d, J=8.7 Hz, 1 H, 7), 4.77 (s, 2 H, 8′), 3.82 (s, 3 H, 17), 3.65–3.59 (m, 1 H, 5), 3.08–3.02 (m, 2 H, 2), 2.02 (s, 2 H, 2′), 1.93 (s, 2 H, 5′), 1.68–1.62, 1.56–1.41 (2 m, 8 H, 3, 4, 3′, 4′), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=171.61, 169.19, 158.96, 144.59, 144.17, 136.03, 134.72, 134.48, 129.52, 128.66, 128.19, 129.09, 122.02, 96.06, 91.60, 76.70, 54.93, 53.57, 46.97, 38.34, 35.13, 33.42, 32.04, 25.94, 24.83, 24.62, 20.15 ppm; IR (ATR): =3383, 3287, 2934, 2865, 1737, 1635, 1578, 1519, 1457, 1423, 1387, 1224, 1202, 1168, 1052, 820, 791, 743, 696 cm−1; MS (ESI): m/z: 493.3 [M+1]+; elemental analysis calcd (%) for C28H36N4O4 (492.61): C 68.27, H 7.37, N 11.37; found: C 68.47, H 7.25, N, 11.49.
N 1‐(Benzyloxy)‐N 4‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}‐benzene‐1,4‐dicarboxamide (4 e): From the reaction of 3 e (0.245 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether), 4 e was obtained as a pale‐yellow solid (0.188 g, 61 %): m.p. 183–184 °C; 1H NMR ([D6]DMSO): δ=11.88 (s, 1 H, 9′), 8.59 (t, J=5.5 Hz, 1 H, 1), 8.54–8.52 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.6 Hz, 1 H, 12), 7.89 (d, J=8.4 Hz, 2 H, 3′, 7′), 7.80 (d, J=8.4 Hz, 2 H, 4′, 6′), 7.45–7.34 (m, 6 H, 11, 12′‐16′), 6.47 (d, J=2.5 Hz, 1 H, 16), 6.28 (d, J=2.4 Hz, 1 H, 14), 6.15 (d, J=8.6 Hz, 1 H, 7), 4.94 (s, 2 H, 10′), 3.81 (s, 3 H, 17), 3.71–3.63 (m, 1 H, 5), 3.33–3.27 (m, 2 H, 2), 1.75–1.58 (m, 4 H, 3, 4), 1.23 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=165.35, 163.70, 158.99, 144.61, 144.22, 137.30, 135.82, 134.80, 134.52, 134.38, 129.57, 128.93, 128.32, 127.32, 127.02, 122.09, 96.13, 91.59, 77.03, 54.96, 47.02, 39.23, 33.39, 25.88, 20.19 ppm; IR (ATR): =3384, 3287, 2969, 2935, 1738, 1630, 1577, 1519, 1492, 1387, 1320, 1224, 1159, 1053, 863, 820, 790, 747, 718, 69 cm−1; MS (ESI): 513.3 [M+1]+; elemental analysis calcd (%) for C30H32N4O4 (512.60): C 70.29, H 6.29, N 10.93; found: C 70.55, H 6.03, N, 11.21.
General procedure for the preparation of SAHAquines 5 a–d: A suspension of O‐benzylhydroxamic acid 4 (0.27 mmol) and 10 % Pd/C (20 mg) in methanol (7 mL) was stirred at room temperature for 2–4 h under a hydrogen atmosphere. The catalyst was filtered off, and the mother liquor was concentrated under reduced pressure.
N‐Hydroxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}butane‐ diamide (5 a): From the reaction of 4 a (0.125 g) and after crystallization (ether), 5 a was obtained as a pale‐yellow solid (0.077 g, 76 %): m.p. 109–110 °C; 1H NMR ([D6]DMSO): δ=10.37 (s, 1 H, 5′), 8.67 (s, 1 H, 6′), 8.54–8.53 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.5 Hz, 1 H, 12), 7.85 (t, J=5.3 Hz, 1 H, 1), 7.45–7.41 (m, 1 H, 11), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.12 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.67–3.56 (m, 1 H, 5), 3.09–3.00 (m, 2 H, 2), 2.29 (t, J=6.5 Hz, 2 H, 2′), 2.17 (t, J=6.5 Hz, 2 H, 3′), 1.72–1.56, 1.56–1.39 (2 m, 4 H, 3, 4), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=170.84, 168.46, 159.00, 144.63, 144.24, 134.80, 134.52, 129.58, 122.11, 96.10, 91.60, 54.98, 47.00, 38.49, 33.42, 30.64, 27.92, 25.97, 20.20 ppm; IR (ATR): =3471, 3375, 3283, 3204, 3008, 2964, 2934, 1744, 1647, 1612, 1556, 1521, 1455, 1385, 1366, 1226, 1205, 1172, 1157, 1056, 821, 789, 677 cm−1; MS (ESI): m/z: 375.2 [M+1]+; elemental analysis calcd (%) for C19H26N4O4 (374.43): C 60.95, H 7.00, N 14.96; found: C 60.84, H 6.81, N, 15.17.
N‐Hydroxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}pentanediamide (5 b): From the reaction of 4 c (0.129 g) and after crystallization (ether), 5 b was obtained as a pale‐yellow solid (0.077 g, 73 %): m.p. 99–102 °C; 1H NMR ([D6]DMSO): δ=10.32 (s, 1 H, 6′), 8.63 (s, 1 H, 7′), 8.55–8.52 (dd, J=4.2, 1.6 Hz, 1 H, 12), 8.09–8.05 (dd, J=8.3, 1.6 Hz, 1 H, 12), 7.76 (t, J=5.3 Hz, 1 H, 1), 7.45–7.39 (m, 1 H, 11), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.11 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.68–3.56 (m, 1 H, 5), 3.05 (d, J=5.6 Hz, 2 H, 2), 2.04 (t, J=7.4 Hz, 2 H, 2′), 1.94 (t, J=7.4 Hz, 2 H, 4′), 1.76–1.57, 1.57–1.40 (2 m, 6 H, 3, 4, 3′), 1.21 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=171.37, 168.74, 158.98, 144.60, 144.20, 134.75, 134.50, 129.54, 122.05, 96.08, 91.61, 54.95, 46.98, 38.39, 34.73, 33.43, 31.75, 25.95, 21.42, 20.17 ppm; IR (ATR): =3468, 3375, 3283, 3210, 3008, 2963, 2933, 1744, 1645, 1613, 1520, 1455, 1386, 1204, 1171, 1158, 1056, 821, 789, 677 cm−1; MS (ESI): m/z: 389.2 [M+1]+; elemental analysis calcd (%) for C20H28N4O4 (388.46): C 61.84, H 7.27, N 14.42; found: C 61.69, H 7.03, N, 14.77.
N‐Hydroxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}hexanediamide (5 c): From the reaction of 4 d (0.133 g) and after crystallization (ether), 5 c was obtained as a pale‐yellow solid (0.082 g, 75 %): m.p. 109–111 °C; 1H NMR ([D6]DMSO): δ=10.34 (s, 1 H, 7′), 8.66 (s, 1 H, 8′), 8.55–8.52 (dd, J=4.2, 1.5 Hz, 1 H, 10), 8.10–8.05 (dd, J=8.3, 1.4 Hz, 1 H, 12), 7.77 (t, J=5.3 Hz, 1 H, 1), 7.45–7.40 (m, 1 H, 11), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.12 (d, J=8.8 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.67–3.57 (m, 1 H, 5), 3.04 (d, J=5.5 Hz, 2 H, 2), 2.03 (s, 2 H, 2′), 1.92 (s, 2 H, 5′), 1.72–1.58, 1.58–1.41 (2 m, 8 H, 3, 4, 3′, 4′), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=171.70, 168.95, 159.00, 144.62, 144.24, 134.80, 134.52, 129.57, 122.10, 96.10, 91.60, 54.98, 46.98, 38.38, 35.20, 33.44, 32.11, 26.01, 24.97, 24.87, 20.20 ppm; IR (ATR): =3428, 3283, 3191, 3088, 3044, 2943, 2923, 2856, 1738, 1621, 1578,1556, 1523, 1385, 1367, 1204, 1170, 1160, 954, 822, 788, 676 cm−1; MS (ESI): m/z: 403.3 [M+1]+; elemental analysis calcd (%) for C21H30N4O4 (402.49): C 62.67, H 7.51, N 13.92; found: C 62.98, H 7.36, N, 14.09.
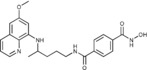
N 1‐Hydroxy‐N 4‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}benzene‐1,4‐dicarboxamide (5 d): From the reaction of 4 e (0.138 g) and after purification by column chromatography (dichloromethane/methanol 9:1) and crystallization (ether), 5 d was obtained as a pale‐yellow solid (0.048 g, 42 %): m.p. 156–158 °C; 1H NMR ([D6]DMSO): δ=11.32 (s, 1 H, 9′), 9.12 (s, 1 H, 10′), 8.57 (t, J=5.5 Hz, 1 H, 1), 8.54–8.53 (dd, J=4.2, 1.6 Hz, 1 H, 10), 8.09–8.06 (d, J=8.3, 1.6 Hz, 1 H, 12), 7.88 (d, J=8.3 Hz, 2 H, 3′, 7′), 7.80 (d, J=8.3 Hz, 2 H, 4′, 6′), 7.44–7.40 (m, 1 H, 11), 6.47 (d, J=2.2 Hz, 1 H, 16), 6.28 (d, J=2.1 Hz, 1 H, 14), 6.15 (d, J=8.7 Hz, 1 H, 7), 3.81 (s, 3 H, 17), 3.71–3.62 (m, 1 H, 5), 3.33–3.27 (m, 2 H, 2), 1.75–1.58 (m, 4 H, 3, 4), 1.23 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=165.44, 163.49, 159.00, 144.62, 144.20, 136.91, 134.86, 134.79, 134.53, 129.57, 127.17, 126.78, 122.09, 96.12, 91.60, 54.96, 47.02, 39.23, 33.41, 25.89, 20.19 ppm; IR (ATR): =3427, 3303, 3199, 2969, 2938, 1738, 1672, 1636, 1616, 1521, 1458, 1388, 1223, 1203, 1172, 1015, 898, 858, 821, 789, 677 cm−1; MS (ESI): m/z: 423.1 [M+1]+; elemental analysis calcd (%) for C23H26N4O4 (422.48): C 65.39, H 6.20, N 13.26; found: C 65.47, H 6.12, N, 12.99.
General procedure for the preparation of O‐methylhydroxamic acids 6 a–e: A solution of corresponding acid 3 (0.2 mmol), DIEA (0.052 g, 0.4 mmol), and HATU (0.076 g, 0.2 mmol) in dichloromethane (5 mL) was stirred at room temperature. After 10 min, O‐methylhydroxylamine hydrochloride (0.020 g, 0.24 mmol) and Et3N (0.024 g, 0.24 mmol) were added. The mixture was stirred at room temperature for 2 h and was then concentrated under reduced pressure. The residue was dissolved in ethyl acetate (8 mL) and extracted with water (3×). The organic layer was dried with sodium sulfate, filtered, and concentrated under reduced pressure.
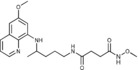
N‐Methoxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}butanediamide (6 a): From the reaction of 3 a (0.072 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether/petroleum ether), 6 a was obtained as a pale‐yellow solid (0.055 g, 71 %): m.p. 121–122 °C; 1H NMR ([D6]DMSO): δ=10.93 (s, 1 H, 5′), 8.54–8.53 (dd, J=3.0 Hz, 1 H, 10), 8.06 (d, J=8.2 Hz, 1 H, 12), 7.82 (s, 1 H, 1), 7.43–7.41 (m, 1 H, 11), 6.47 (d, J=2.2 Hz, 1 H, 16), 6.26 (d, J=2.1 Hz, 1 H, 14), 6.11 (d, J=8.7 Hz, 1 H, 7), 3.83 (s, 3 H, 17), 3.63–3.58 (m, 1 H, 5), 3.54 (s, 3 H, 6′), 3.08–3.02 (m, 2 H, 2), 2.29 (t, J=7.4 Hz, 2 H, 2′), 2.16 (t, J=7.0 Hz, 2 H, 3′), 1.67–1.63, 1.55–1.45 (2 m, 4 H, 3, 4), 1.21 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=170.64, 168.44, 158.96, 144.59, 144.17, 134.71, 134.48, 129.52, 122.01, 96.04, 91.60, 62.97, 54.92, 46.97, 38.44, 33.37, 30.27, 27.84, 25.90, 20.14 ppm; IR (ATR): =3302, 3234, 3001, 2969, 2933, 1739, 1658, 1634, 1556, 1519, 1457, 1424, 1388, 1336, 1224, 1204, 1168, 1053, 820, 791, 679 cm−1; MS (ESI): m/z: 389.2 [M+1]+; elemental analysis calcd (%) for C20H28N4O4 (388.46): C 61.84, H 7.27, N 14.42; found: C 62.05, H 7.01, N 14.65.
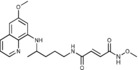
(2E)‐N‐Methoxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pent‐yl}but‐2‐enediamide (6 b): From the reaction of 3 b (0.072 g) and after purification by column chromatography (cyclohexane/ethyl acetate/methanol 3:1:0.5) and crystallization (ether), 6 b was obtained as a pale‐yellow solid (0.038 g, 49 %): m.p. 177–179 °C; 1H NMR ([D6]DMSO): δ=11.53 (s, 1 H, 5′), 8.55–8.53 (dd, J=1.6, 4.2 Hz, 1 H, 10), 8.44 (t, J=5.5 Hz, 1 H, 1), 8.09–8.06 (dd, J=1.5, 8.3 Hz, 1 H, 12), 7.45–7.41 (m, 6 H, 11), 6.89 (d, J=15.1 Hz, 1 H, 2′), 6.60 (d, J=15.1 Hz, 1 H, 3′), 6.47 (d, J=2.2 Hz, 1 H, 16), 6.27 (d, J=2.2 Hz, 1 H, 14), 6.14 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.65 (s, 4 H, 5, 6′), 3.20–3.14 (m, 2 H, 2), 1.70–1.49 (m, 4 H, 3, 4), 1.21 ppm (d, J=6.2 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=163.16, 160.97, 159.00, 144.60, 144.22, 135.72, 134.82, 134.49, 133.58, 129.58, 129.12, 128.85, 129.34, 122.09, 96.16, 91.63, 76.97, 54.97, 46.97, 38.77, 33.42, 25.77, 20.19 ppm; IR (ATR): =3285, 3223, 3097, 3007, 2969,2935, 1738, 1630, 1570, 1519, 1457, 1388, 1338, 1224, 1168, 1066, 1053, 991, 821, 792, 659 cm−1; MS (ESI): m/z: 387.1 [M+1]+; elemental analysis calcd (%) for C20H26N4O4 (386.44): C 62.16, H 6.78, N 14.50; found: C 61.94, H 6.55, N 14.87.
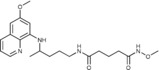
N‐Methoxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}pentanediamide (6 c): From the reaction of 3 c (0.075 g) and after crystallization (ether), 6 c was obtained as a pale‐yellow solid (0.045 g, 56 %): m.p. 112–113 °C; 1H NMR ([D6]DMSO): δ=10.94 (s, 1 H, 6′), 8.55–8.53 (dd, J=4.2, 1.5 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.4 Hz, 1 H, 12), 7.89 (t, J=5.3 Hz, 1 H, 1), 7.45–7.41 (m, 1 H, 11), 6.48 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.3 Hz, 1 H, 14), 6.12 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.62 (m, 1 H, 5), 3.56 (s, 3 H, 7′), 3.06–3.04 (m, 2 H, 2), 2.04 (t, J=7.3 Hz, 2 H, 2′), 1.93 (t, J=7.3 Hz, 2 H, 4′), 1.74–1.40 (m, 6 H, 3′, 3,4′), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=171.31, 168.72, 159.00, 144.62, 144.23, 134.79, 134.52, 129.57, 122.10, 96.10, 91.59, 63.10, 54.98, 46.98, 38.41, 34.54, 33.43, 31.68, 25.98, 21.12, 20.19 ppm; IR (ATR): =3396, 3285, 3162, 3097, 2969, 2936, 1737, 1670, 1630, 1564, 1521, 1456, 1424, 1388, 1227, 1206, 1170,1161, 1030, 820, 790, 679 cm−1; MS (ESI): m/z: 403.3 [M+1]+; elemental analysis calcd (%) for C21H30N4O4 (402.49): C 62.67, H 7.51, N 13.92; found: C 62.46, H 7.31, N 13.95.
N‐Methoxy‐N′‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}hexanediamide (6 d): From the reaction of 3 d (0.075 g) and after purification by column chromatography (dichloromethane/methanol 9.5:0.5) and crystallization (ether), 6 d was obtained as a pale‐yellow solid (0.042 g, 51 %): m.p. 108–109 °C; 1H NMR ([D6]DMSO): δ=10.91 (s, 1 H, 7′), 8.55–8.52 (dd, J=4.1, 1.6 Hz, 1 H, 10), 8.09–8.06 (dd, J=8.3, 1.5 Hz, 1 H, 12), 7.75 (t, J=5.4 Hz, 1 H, 1), 7.44–7.41 (m, 1 H, 11), 6.47 (d, J=2.4 Hz, 1 H, 16), 6.26 (d, J=2.4 Hz, 1 H, 14), 6.11 (d, J=8.7 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.65–3.55 (m, 4 H, 5, 8′), 3.08–3.00 (m, 2 H, 2), 2.03 (s, 2 H, 2′), 1.92 (s, 2 H, 5′), 1.55–1.49, 1.49–1.40 (m, 8 H, 3, 4, 3′, 4′), 1.20 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=171.64, 168.91, 158.98, 144.61, 144.20, 134.76, 134.50, 129.55, 122.06, 96.08, 91.60, 63.05, 54.95, 46.97, 38.36, 35.15, 33.43, 32.07, 25.97, 24.86, 24.60, 20.17 ppm; IR (ATR): =3395, 3287, 3166, 3090, 2927, 2861, 1739, 1674, 1631, 1577, 1563, 1521, 1457, 1423, 1388, 1227, 1206, 1170, 1161, 1053, 821, 790, 679 cm−1; MS (ESI): m/z: 417.3 [M+1]+; elemental analysis calcd (%) for C22H32N4O4 (416.51): C 63.44, H 7.74, N 13.45; found: C 63.26, H 7.53, N 13.71.
N 1‐Methoxy‐N 4‐{4‐[(6‐methoxyquinolin‐8‐yl)amino]pentyl}benzene‐1,4‐dicarboxamide (6 e): From the reaction of 3 e (0.082 g) and after crystallization (ether), 6 e was obtained as a pale‐yellow solid (0.040 g, 45 %): m.p. 142–145 °C; 1H NMR ([D6]DMSO): δ=11.84 (s, 1 H, 9′), 8.58 (t, J=5.56 Hz, 1 H, 1), 8.54–8.53 (dd, J=1.6, 4.2 Hz, 1 H, 10), 8.08–8.06 (dd, J=1.6, 8.3 Hz, 1 H, 12), 7.89 (d, J=8.4 Hz, 2 H, 3′, 7′), 7.80 (d, J=8.4 Hz, 2 H, 4′, 6′), 7.43–7.41 (m, 1 H, 11), 6.47 (d, J=2.5 Hz, 1 H, 16), 6.28 (d, J=2.4 Hz, 1 H, 14), 6.15 (d, J=8.8 Hz, 1 H, 7), 3.82 (s, 3 H, 17), 3.72 (s, 2 H, 10′), 3.69–3.65 (m, 1 H, 5), 3.31–3.28 (m, 2 H, 2), 1.76–1.66, 1.65–1.56 (m, 4 H, 3, 4), 1.23 ppm (d, J=6.3 Hz, 3 H, 6); 13C NMR ([D6]DMSO): δ=165.32, 163.36, 158.98, 144.61, 144.20, 137.28, 134.76, 134.52, 134.31, 129.55, 127.21, 126.93), 122.06, 96.10, 91.60, 63.26, 54.94, 47.01, 39.23, 33.41, 25.85, 20.17 ppm; IR (ATR): =3394, 3309, 3176, 3060, 2970, 2937, 1738, 1670, 1624, 1557, 1520, 1456, 1389, 1221, 1205, 1161, 1039, 867, 821, 791, 678 cm−1; MS (ESI): m/z: 437.3 [M+1]+; elemental analysis calcd (%) for C24H28N4O4 (436.50): C 66.04, H 6.47, N 12.84; found: C 66.33, H 6.31, N 13.04.
Biological Evaluation
Cell viability: All synthesized compounds were first screened for their inhibition of mitochondrial metabolic activity by using the MTT assay.65 Mitochondrial metabolic activity correlates with cell viability. The following cell lines were tested: U2OS, HepG2, MCF‐7, and Hek293 cells. The compounds were dissolved in DMSO (3×10−3 m) and stored at −20 °C. The cells were seeded at a density of 3000 cells per 200 μL in a 96‐well plate (Sarsted) in triplicate and grown in Dulbecco's modified Eagle medium with 4500 mg L−1 glucose (DMEM‐high glucose) (Lonza) containing 10 % fetal bovine serum (FBS) (Gibco), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (Sigma–Aldrich). The next day, the medium was aspirated and cells were treated for 72 h. Only the compounds that led to more than a 40 % reduction in mitochondrial metabolic activity at a concentration of 5×10−5 m were selected for further analysis. The following concentrations of selected compounds were used: 5×10−5, 1×10−5, 1×10−6, 1×10−7, and 1×10−8 m. All working concentrations were freshly prepared in DMEM on the day of the testing. A fresh growth medium was added to untreated control cells, which were defined as 100 % viable. DMSO (1.67, 0.33, 0.03, 0.3×10−2, and 0.3×10−3 %) in DMEM was considered as a negative control. Cisplatin, PQ, and SAHA were used as positive controls. After 72 h of incubation in the presence of selected compounds, media were aspirated and MTT (0.5 mg mL−1; 40 μL per well) was added, and the cells were incubated for 4 h 37 °C. Media were aspirated and formazan crystals were dissolved in DMSO (170 μL per well). The absorbance was measured on a microplate reader (Promega) at λ=560 nm. The IC50 values (concentration required to decrease viability by 50 %) were calculated by using nonlinear regression on the sigmoidal dose–response plots and are expressed as mean±SD of two independent experiments.
MCF‐7 cell labeling with Hoechst 33342: Fluorescent dye Hoechst 33342 was used to determine the total number of cells. MCF‐7 cells were seeded at a density of 3000 cells per 100 μL (72 h treatment) or 6000 cells per 100 μL (24 h treatment) in a 96‐well plate (Corning Inc.). Cells were grown in triplicate in DMEM containing 10 % FBS (Wisent), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin (Thermo Fischer). The next day, cells were treated with SAHAquines 4 e and 5 b, SAHA, PQ, and cisplatin in ten different concentrations ranging from 5×10−5 to 1×10−9 m. After 72 h, cells were stained with Hoechst 33342 (10 μm, 10 min). Cells were then imaged by using a fluorescence microscope (Leica, DMI4000B). Results were calculated by using nonlinear regression on the sigmoidal dose–response plot and are expressed as mean±SD of two independent experiments.
In vitro drug sensitivity assay against erythrocytic stages of P. falciparum: The antiplasmodial activity of compounds 2–6 was tested in a drug‐sensitivity assay against two laboratory P. falciparum strains (3D7—chloroquine sensitive, Dd2—chloroquine‐resistant) as described before by using the histidine‐rich protein 2 (HRP2) assay.66, 67 In brief, 96‐well plates were precoated with the tested compounds in a threefold dilution before ring‐stage parasites were added in complete culture medium at a hematocrit of 1.5 % and a parasitaemia of 0.05 %. After 3 days of incubation at 37 °C, 5 % CO2 and 5 % oxygen, plates were frozen until analyzed by HRP2‐ELISA. All compounds were evaluated in duplicate in at least two independent experiments. The IC50 was determined by analyzing the nonlinear regression of log concentration–response curves using the drc‐package v0.9.0 of R v2.6.1.68
In vitro activity against P. berghei hepatic stages: Compound activity on P. berghei infection of a human hepatoma cell line (HuH7) was assessed employing the luminescence‐based method, as previously described.69 Briefly, hepatic infection was determined by measuring the luminescence intensity of lysates of HuH7 cells infected with a firefly luciferase‐expressing P. berghei line. HuH7 cells (1.0×104 per well) were seeded in 96‐well plates the day before infection. One hour prior to infection, the medium was replaced by medium containing the appropriate drug concentrations. The addition of 1.0×104 sporozoites was followed by centrifugation at 1800×g for 5 min and parasite infection load was measured 48 h after parasite addition by a bioluminescence assay (Biotium, USA) using a multiplate reader Infinite M200 (Tecan, Switzerland). The effect of the different treatments on the viability of HuH7 cells was assessed by the CellTiter‐Blue assay (Promega, USA) according to the manufacturer's protocol. Nonlinear regression analysis was employed to fit the normalized results of the dose–response curves, and IC50 values were determined using GraphPad Prism V5.0.
Immunocytochemistry for histone acetylation: MCF‐7 cells were seeded at a density of 5000 cells per coverslip in DMEM containing 10 % FBS, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin and were left for 24 h to adhere. Cells were treated with 4 e, 5 b, SAHA, or PQ (at 1 μm concentration) for 24 h. Following treatment, cells were fixed with 4 % paraformaldehyde (10 min), then permeabilized using 0.1 % Triton X‐100 (10 min). Blocking was performed using 10 % goat serum in phosphate buffer saline (PBS) (1 h), followed by incubation with primary antibody (rabbit anti‐acetyl‐Histone‐H3 polyclonal antibody 1/500; Millipore 06–559) for 24 h in a humidified chamber at 4 °C. Samples were washed with PBS (3×5 min). Samples were then incubated with secondary antibody (Alexa Fluor 647 goat anti‐rabbit IgG −2 mg mL−1 1/500; Life Technologies A21244) for 1 h in the dark, after which they were washed with PBS (3×5 min). Nuclei were labeled with Hoechst 33342 (10 μm, 10 min). Samples were mounted on microscope slides using Poly Aqua Mount (PolySciences) and were dried overnight before imaging with a fluorescence microscope (Leica). Fluorescence intensity was quantified using ImageJ software, and the results are presented as means of fluorescence per cell±SD (as fold increase in the untreated control=1).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support for this study by Croatian Science Foundation (project number IP‐09–2014‐1501), Canadian Institute of Health Research (CIHR; project number MOP‐119425), and National Sciences and Engineering Research Council of Canada (NSERC; project number RGPIN 04994‐15) is acknowledged.
M. Beus, Z. Rajić, D. Maysinger, Z. Mlinarić, M. Antunović, I. Marijanović, D. Fontinha, M. Prudêncio, J. Held, S. Olgen, B. Zorc, ChemistryOpen 2018, 7, 624.
Contributor Information
Prof. Zrinka Rajić, Email: zrajic@pharma.hr.
Prof. Branka Zorc, Email: bzorc@pharma.hr.
References
- 1. Remesh A., Int. J. Basic Clin. Pharmacol. 2012, 1, 2–12. [Google Scholar]
- 2. Longley D. B., Johnston P. G., J. Pathol. 2005, 205, 275–292. [DOI] [PubMed] [Google Scholar]
- 3. Curtin N. J., Nat. Rev. 2012, 12, 801–817. [DOI] [PubMed] [Google Scholar]
- 4. Azad C., Saxena M., Siddiqui A. J., Bhardwaj J., Puri S. K., Dutta G. P., Anand N., Saxena A. K., Chem. Biol. Drug Des. 2017, 90, 254–261. [DOI] [PubMed] [Google Scholar]
- 5. Teixeira C., Vale N., Pérez B., Gomes A., Gomes J. R., Gomes P., Chem. Rev. 2014, 114, 11164–11220. [DOI] [PubMed] [Google Scholar]
- 6. Shahinas D., Folefoc A., Pillai D. R., Pathogens 2013, 2, 33–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Agarwal D., Gupta R. D., Awasthi S. K., Antimicrob. Agents Chemother. 2017, 61, e00249-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takasu K., Shomogama T., Saiin C., Kim H.-S., Wataya Y., Brun R., Ihara M., Chem. Pharm. Bull. 2005, 53, 653–661. [DOI] [PubMed] [Google Scholar]
- 9. Gomes A., Machado M., Lobo L., Nogueira F., Prudencio M., Teixeira C., Gomes P., ChemMedChem 2015, 10, 1344–1349. [DOI] [PubMed] [Google Scholar]
- 10. Higginbotham S., Wong W. R., Linington R. G., Spadafora C., Iturrado L., Arnold A. E., PLoS One 2014, 9, e84549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnston W. T., Mutalima N., Sun D., Emmanuel B., Bhatia K., Aka P., Wu X., Borgstein E., Liomba G. N., Kamiza S., Mkandawire N., Batumba M., Carpenter L. M., Jaffe H., Molyneux E. M., Goedert J. J., Soppet D., Newton R., Mbulaiteye S. M., Sci. Rep. 2014, 4, 3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Khan K. H., Germs 2013, 3, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fedosov D. A., Dao M., Karniadakis G. E., Suresh S., Ann. Biomed. Eng. 2014, 42, 368–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hooft van Huijsduijnen R., Guy R. K., Chibale K., Haynes R. K., Peitz I., Kelter G., Phillips M. A., Vennerstrom J. L., Yuthavong Y., Wells T. N., PLoS One 2013, 8, e82962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.K.-C. Keum, N.-C. Yoo, W.-M. Yoo, K. K. Chang, Y. N. Choon, Y. W. Min, WO 2002013826 A1, 2002.
- 16. Liu F., Shang Y., Chen S-Z., Acta Pharmacol. Sin. 2014, 35, 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kamal A., Aziz A., Shouman S., El-Demerdash E., Elgendy M., Abdel-Naim A. B., Sci. Proc. 2014, 1, e384. [Google Scholar]
- 18. Ganguli A., Choudhury D., Datta S., Bhattacharya S., Chakrabarti G., Biochimie 2014, 107, 338–349. [DOI] [PubMed] [Google Scholar]
- 19. Soo G. W., Law J. H., Kan E., Tan S. Y., Lim W. Y., Chay G., Bukhari N. I., Segarra I., Anticancer Drugs 2010, 21, 695–703. [PubMed] [Google Scholar]
- 20. Wong Y. K., Xu C., Kalesh K. A., He Y., Lin Q., Wong W. S. F., Shen H. M., Wang J., Med. Res. Rev. 2017, 37, 1492–1517. [DOI] [PubMed] [Google Scholar]
- 21. Nakase I., Lai H., Singh N. P., Sasaki T., Int. J. Pharm. 2008, 354, 28–33. [DOI] [PubMed] [Google Scholar]
- 22. Cheng C., Wang T., Song Z., Peng L., Gao M., Hermine O., Rousseaux S., Khochbin S., Mi J. Q., Wang J., Cancer Med. 2018, 7, 380–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi T., Yu X. X., Yan L. J., Xiao H. T., Cancer Chemother. Pharmacol. 2017, 79, 287–294. [DOI] [PubMed] [Google Scholar]
- 24. Džimbeg G., Zorc B., Kralj M., Ester K., Pavelić K., Balzarini J., De Clercq E., Mintas M., Eur. J. Med. Chem. 2008, 43, 1180–1187. [DOI] [PubMed] [Google Scholar]
- 25. Šimunović M., Perković I., Zorc B., Ester K., Kralj M., Hadjipavlou-Litina D., Pontiki E., Bioorg. Med. Chem. 2009, 17, 5605–5613. [DOI] [PubMed] [Google Scholar]
- 26. Perković I., Tršinar S., Žanetić J., Kralj M., Martin-Kleiner I., Balzarini J., Hadjipavlou-Litina D., Katsori A. M., Zorc B., J. Enzyme Inhib. Med. Chem. 2013, 28, 601–610. [DOI] [PubMed] [Google Scholar]
- 27. Pavić K., Perković I., Cindrić M., Pranjić M., Martin-Kleiner I., Kralj M., Schols D., Hadjipavlou-Litina D., Katsori A.-M., Zorc B., Eur. J. Med. Chem. 2014, 86, 502–514. [DOI] [PubMed] [Google Scholar]
- 28. Perković I., Antunović M., Marijanović I., Pavić K., Ester K., Kralj M., Vlainić J., Kosalec I., Schols D., Hadjipavlou-Litina D., Pontiki E., Zorc B., Eur. J. Med. Chem. 2016, 124, 622–636. [DOI] [PubMed] [Google Scholar]
- 29. Pavić K., Perković I., Gilja P., Kozlina F., Ester K., Kralj M., Schols D., Hadjipavlou-Litina D., Pontiki E., Zorc B., Molecules 2016, 21, 1629–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pavić K., Perković I., Pospíšilová Š., Machado M., Fontinha D., Prudêncio M., Jampilek J., Coffey A., Endersen L., Rimac H., Zorc B., Eur. J. Med. Chem. 2018, 143, 769–779. [DOI] [PubMed] [Google Scholar]
- 31. Gomes A., Fernandes I., Teixeira C., Mateus N., Sottomayor M. J., Gomes P., ChemMedChem 2016, 11, 2703–2712. [DOI] [PubMed] [Google Scholar]
- 32. Pérez C., Fernandes I., Mateus N., Teixeira C., Gomes P., Bioorg. Med. Chem. Lett. 2013, 23, 6769–6772. [DOI] [PubMed] [Google Scholar]
- 33. Slezakova S., Ruda-Kucerova J., Anticancer Res. 2017, 37, 5995–6003. [DOI] [PubMed] [Google Scholar]
- 34. Sun Q., Wang J., Li Y., Zhuang J., Zhang Q., Sun X., Sun D., Chem. Biol. Drug Des. 2017, 90, 1019–1028. [DOI] [PubMed] [Google Scholar]
- 35. Pybus B. S., Marcsisin S. R., Jin X., Deye G., Sousa J. C., Li Q., Caridha D., Zeng Q., Reichard G. A., Ockenhouse C., Bennett J., Walker L. A., Ohrt C., Melendez V., Malar. J. 2013, 12, e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vale N., Moreira R., Gomes P., Eur. J. Med. Chem. 2009, 44, 937–953. [DOI] [PubMed] [Google Scholar]
- 37. Tcherniuk S. O., Chesnokova O., Oleinikov I. V., Potopalsky A. I., Oleinikov A. V., Malar. J. 2015, 14, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wildbolz A., Ther. Umsch. 1973, 30, 218–222. [PubMed] [Google Scholar]
- 39. Pouvelle B., Farley P. J., Long C. A., Taraschi T. F., J. Clin. Invest. 1994, 94, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Usanga E. A., FEBS Lett. 1986, 209, 23–27. [DOI] [PubMed] [Google Scholar]
- 41. Nair L., Bhasin V. K., Jpn. J. Med. Sci. Biol. 1994, 47, 241–252. [DOI] [PubMed] [Google Scholar]
- 42. Kreidenweiss A., Kremsner P. G., Mordmüller B., Malar. J. 2008, 7, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grant S., Easley C., Kirkpatrick P., Nat. Rev. Drug. Discov. 2007, 6, 21–22. [DOI] [PubMed] [Google Scholar]
- 44. Chua M. J., Arnold M. S. J., Xu W., Lancelot J., Lamotte S., Späth G. F., Prina E., Pierce R. J., Fairlie D. P., Skinner-Adams T. S., Andrews K. T., Int. J. Parasitol. 2017, 7, 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andrews K. T., Haque A., Jones M. K., Immunol. Cell Biol. 2012, 90, 66–77. [DOI] [PubMed] [Google Scholar]
- 46. Chaal B. K., Gupta A. P., Wastuwidyaningtyas B. D., Luah Y. H., Bozdech Z., PLoS Pathog. 2010, 6, e1000737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu H., Cabrera A., Kono M., Mok S., Chaal B. K., Haase S., Engelberg K., Cheemadan S., Spielmann T., Preiser P. R., Gillberger T.-W., Bozdech Z., Nat. Biotechnol. 2010, 28, 91–98. [DOI] [PubMed] [Google Scholar]
- 48. Andrews K. T., Tran T. N., Wheatley N. C., Fairlie D. P., Curr. Top. Med. Chem. 2009, 9, 292–308. [DOI] [PubMed] [Google Scholar]
- 49. Engel J. A., Jones A. J., Avery V. M., Sumanadasa S. D., Ng S. S., Fairlie D. P., Skinner-Adams T., Andrews K. T., Int. J. Parasitol. 2015, 5, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trenholme K., Marek L., Duffy S., Pradel G., Fisher G., Hansen F. K., Skinner-Adams T. S., Butterworth A., Ngwa C. J., Moecking J., Goodman C. D., McFadden G. I., Sumanadasa S. D. M., Fairlie D. P., Avery V. M., Kurz T., Andrews K. T., Antimicrob. Agents Chemother. 2014, 58, 3666–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muregi F. W., Ishih A., Drug Dev. Res. 2010, 71, 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oliveira R., Miranda D., Magalhaes J., Capela R., Perry M. J., O'Neill P. M., Moreira R., Lopes F., Bioorg. Med. Chem. 2015, 23, 5120–5130. [DOI] [PubMed] [Google Scholar]
- 53. Shaveta S., Mishra S., Singh P., Eur. J. Med. Chem. 2016, 124, 500–536. [DOI] [PubMed] [Google Scholar]
- 54. Mishra M., Mishra V. K., Kashaw V., Iyer A. K., Kashaw S. K., Eur. J. Med. Chem. 2017, 125, 1300–1320. [DOI] [PubMed] [Google Scholar]
- 55. Zhang X., Zhang J., Tong L., Luo Y., Mingbo S., Zang Y., Li J., Lu W., Chen Y., Bioorg. Med. Chem. 2013, 21, 3240–3244. [DOI] [PubMed] [Google Scholar]
- 56. Payne J. E., Bonnefous C., Hassig C. A., Symons K. T., Guo X., Nguyen P. M., Annable T., Wash P. L., Hoffman T. Z., Rao T. S., Shiau A. K., Malecha J. W., Noble S. A., Hager J. F., Smith N. D., Bioorg. Med. Chem. Lett. 2008, 18, 6093–6096. [DOI] [PubMed] [Google Scholar]
- 57. Madsen A. S., Kristensen H. M., Lanz G., Olsen C. A., ChemMedChem 2014, 9, 614–626. [DOI] [PubMed] [Google Scholar]
- 58. Zorc B., Rajić Džolić Z., Butula I., Croat. Chem. Acta 2012, 85, 595–602. [Google Scholar]
- 59.Chemicalize, 2017, ChemAxon Ltd. Available from: http://www.chemicalize.org.
- 60. Luzina E. L., Popov A. V., Eur. J. Med. Chem. 2012, 53, 364–373. [DOI] [PubMed] [Google Scholar]
- 61. Lauffer B. E., Mintzer R., Fong R., Mukund S., Tam C., Zilberleyb I., Flicke B., Ritscher A., Fedorowicz G., Vallero R., Ortwine D. F., Gunzner J., Modrusan Z., Neumann L., Koth C. M., Lupardus P. J., Kaminker J. S., Heise C. E., Steiner P., J. Biol. Chem. 2013, 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Duffy R., Wade C., Chang R., Drug Discovery Today 2012, 17, 942–953. [DOI] [PubMed] [Google Scholar]
- 63. Held J., Kreidenweiss A., Mordmüller B., Expert Opin. Drug Discovery 2013, 8, 1325–1337. [DOI] [PubMed] [Google Scholar]
- 64. Barbaras D., Kaiser M., Brun R., Gademann K., Bioorg. Med. Chem. Lett. 2008, 18, 4413–4415. [DOI] [PubMed] [Google Scholar]
- 65. Boyd M. R., Paull K. D., Drug Dev. Res. 1995, 34, 91–109. [Google Scholar]
- 66. Held J., Gebru T., Kalesse M., Jansen R., Gerth K., Müller R., Mordmüller B., Antimicrob. Agents Chemother. 2014, 58, 6378–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Noedl H., Bronnert J., Yingyuen K., Attlmayr B., Kollaritsch H., Fukuda M., Antimicrob. Agents Chemother. 2005, 49, 3575–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.R Core Team (2015). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/.
- 69. Mendes A. M., Albuquerque I. S., Machado M., Pissarra J., Meireles P., Prudêncio M., Antimicrob. Agents Chemother. 2017, 61, e02005-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary