

ABSTRACT
The genetic code sectored via tRNA charging errors, and the code progressed toward closure and universality because of evolution of aminoacyl-tRNA synthetase (aaRS) fidelity and translational fidelity mechanisms. Class I and class II aaRS folds are identified as homologs. From sequence alignments, a structurally conserved Zn-binding domain common to class I and class II aaRS was identified. A model for the class I and class II aaRS alternate folding pathways is posited. Five mechanisms toward code closure are highlighted: 1) aaRS proofreading to remove mischarged amino acids from tRNA; 2) accurate aaRS active site specification of amino acid substrates; 3) aaRS-tRNA anticodon recognition; 4) conformational coupling proofreading of the anticodon-codon interaction; and 5) deamination of tRNA wobble adenine to inosine. In tRNA anticodons there is strong wobble sequence preference that results in a broader spectrum of contacts to synonymous mRNA codon wobble bases. Adenine is excluded from the anticodon wobble position of tRNA unless it is modified to inosine. Uracil is generally preferred to cytosine in the tRNA anticodon wobble position. Because of wobble ambiguity when tRNA reads mRNA, the maximal coding capacity of the three nucleotide code read by tRNA is 31 amino acids + stops.
KEYWORDS: The last universal common cellular ancestor, cloverleaf tRNA, standard genetic code, aminoacyl-tRNA synthetases, class I and class II aaRS homology, anticodon wobble preference, synonymous anticodons, tRNA wobble inosine
Abbreviations
- aminoacyl-tRNA synthetase enzymes(aaRS)
(i.e. glycine aminoacyl-tRNA synthetase (GlyRS))
- Homo sapiens
(Hs)
- inosine
(I)
- the last universal common cellular ancestor
(LUCA)
- Pyrobaculum aerophilum
(Pae)
- Pyrococcus furiosis
(Pfu)
- Staphylothermus marinus
(Sma)
- Sulfolobus solfataricus
(Sso)
- Thermus thermophilus
(Tth)
Introduction
Aminoacyl-tRNA synthetases (aaRS; i.e. GlyRS) accurately add amino acids to the 3’-CCA ends of tRNAs. Two distinct protein folds are identified for aaRS, described as class I and class II [1,2]. Class I aaRS have an active site that adenylates an amino acid at a “Rossmannoid” fold of parallel β-sheets before transferring the amino acid to tRNA. Class II aaRS, by strong contrast, have an active site of antiparallel β-sheets. The evolutionary relationship of class I and class II enzymes has not been clearly demonstrated, although the interesting suggestion has been made that class I and class II aaRS enzymes were encoded on opposing strands of a bi-directional ancestral gene [3–8]. We provide a simpler explanation. We show amino acid sequence similarity in archaea that indicates that class I and class II aaRS enzymes arose from unidirectional in-frame translation starting from different N-termini. The longer N-terminal region of class I aaRS enzymes forces the class I fold and prevents the class II fold. To detect class I and class II aaRS sequence similarity, one only has to gaze toward LUCA (the last universal common cellular ancestor; ∼3.85 billion years ago) by comparing sequences in ancient archaea [9].
All 64 codons are utilized in mRNA, but only a subset of matching anticodons is utilized in tRNA. A subset of tRNA anticodons is possible because of degeneracy of the genetic code and ambiguity in tRNA anticodon wobble bases reading mRNA codons, allowing (and limiting) a tRNA anticodon to read multiple synonymous mRNA codons. Potentially, therefore, ambiguity in tRNA anticodons reading mRNA codons could be positively selected in evolution, which might be reflected in anticodon wobble base preferences. It appears that tRNAomes (the collection of tRNAs for an organism) are generally selected to be small (even in complex eukaryotes) without skipping recognition of mRNA codons [9]. Specialization of tRNAs may occur [10], but this was not the major driving force in early evolution.
In a recent review, it was suggested that evidence was sparse for the error minimization hypothesis in standard genetic code evolution [11]. The error minimization theory describes sectoring of the code to minimize the impacts of random mutations in tRNAs and of tRNA charging errors. Clustering similar amino acids in the codon-anticodon table might be selected in order to reduce the impact of translation errors. Massey has argued, however, that the code likely did not sector strongly to minimize errors in translation and coding, but, rather, that clustering of similar amino acids occurred through the evolutionary sectoring mechanism [12]. Here, we also argue against error minimization as a strong selection pressure in building the genetic code. Rather, we argue that the sectoring of the code was largely driven by tRNA charging errors, and, therefore, error minimization resulted from the pathway of code evolution, essentially as proposed by Massey. Specifically, we show that minimization of translation errors via aaRS proofreading appears to have limited sectoring of the genetic code, indicating that tRNA charging errors led to reassignments of tRNAs during early code evolution. Reassignments of tRNAs could result in subdividing a 4-codon sector of the codon-anticodon table into two 2-codon sectors and adding a newly encoded amino acid. Mutations in the anticodon loop of tRNAs can also initiate invasion of neighboring genetic code sectors, but this process moves amino acids in the table without introducing new amino acids into the code. Because tRNA charging errors drove code evolution, mechanisms ensuring tRNA charging accuracy brought the code to closure and universality. The dominant model to analyze genetic code evolution, therefore, should be that tRNA charging errors induced sectoring of the code, and evolution of accuracy mechanisms brought the code to universality and closure. The coevolution hypothesis posits that tRNAs, amino acids, the genetic code and aaRS enzymes are coevolved, an idea that we support in this paper.
In recent work, we describe how a cloverleaf tRNA evolution model [13,14] is highly predictive for models of genetic code evolution [9]. Further, we show that evolution of the genetic code is centered more on tRNA than on mRNA or the ribosome. Primitive archaea have 46 tRNAs and 3 stop codons. Translation termination signals are recognized by proteins (not tRNA) that bind to an mRNA stop codon in the ribosome decoding center and reach into the ribosome peptidyl transferase center to terminate translation [15]. Included in sets of 46 tRNAs are encoded 44 unique tRNA anticodons. There are 3 tRNAMet (CAU anticodon) including 1 initiator tRNAiMet and 2 elongator tRNAMet [16,17]. Generally, in ancient archaea and bacteria, only a single tRNAIle (GAU) is utilized. All other permitted anticodons are found in tRNAs except for three potential anticodon sequences corresponding to stop codons. Because only 44 unique anticodons and 3 stop codons need to be considered in early code evolution, but all 64 codons are utilized in mRNA, tRNA anticodon structure and presentation appears to have placed the greatest restrictions on expansions of the genetic code [9].
In archaea, little or no tRNA wobble position adenine is found [9,18]. In bacteria, only tRNAArg (ACG) generally has adenine encoded in the anticodon wobble position [18,19]. In bacteria and eukarya, tRNA wobble adenine is modified to inosine (A→I) by a tRNA adenosine deaminase. Wobble A in tRNA specifies U in mRNA codons, but wobble inosine pairs A, C and U, indicating that increasing ambiguity in mRNA codon interpretation was positively selected as long as the specificity of coding remained unchanged [18,19]. Because tRNA wobble A is negatively selected, according to a tRNA-centric view, only 44 unique anticodons and 3 stop codons need to be considered in earliest standard genetic code evolution rather than 64 [9]. Because of tRNA wobble ambiguity reading mRNA, however, the maximum number of amino acids that can be encoded by a genetic code read by tRNA is 31 aas with stops.
Although the initial evolution of the genetic code may have involved ribozyme-catalyzed tRNA aminoacylation [20–23], at later stages, tRNAs coevolved with aaRS enzymes that attach amino acids at the tRNA 3’-CCA ends [1,2]. Some aaRS enzymes have the capability to proofread tRNA-aa attachments by moving an improperly joined amino acid from the aaRS synthetic site, where the amino acid is linked to the tRNA 3’-CCA end, to a separate aaRS editing or proofreading site, where the non-cognate amino acid is removed [1,2]. We make observations about aaRS editing that are not noted in reviews nor, as far as we can discover, in the literature. We make the observation that aaRS editing appears to inhibit continued sectoring of the code utilizing the anticodon wobble position, giving insight into the roles of tRNA charging errors in evolution of the code. Furthermore, in eukaryotes, left half and mostly 4-codon sectors of the genetic code, for which the aaRS enzymes have proofreading capacity, are also the sectors that have introduced the adenine→inosine anticodon wobble position modification. A→I modification blocks subdivision of a 4-codon sector to two 2-codon sectors because sectoring would result in translation errors. A→I conversions and U vs C wobble preference increase the ambiguity of the tRNA wobble base to allow broader sequence contacts to synonymous mRNA codons.
Results
Evolution and homology of class I and class II aaRS enzymes
The evolution of Pyrococcus furiosis (Pfu) aaRS enzymes is described in Figure 1. Interestingly, the apparent pathways for Pfu aaRS divergence show similarities to the proposed pathways for LUCA tRNA evolution [9]. AaRS enzymes, amino acids and tRNAs are coevolved, as predicted by the coevolution hypothesis [11]. To construct the pathway for aaRS evolution, NCBI (National Center for Biotechnology Information) Blast tools were used with relaxed search metrics to identify the closest apparent relationships between aaRS proteins. Pfu was selected as an example of an ancient archaea with a similar translation system to LUCA [9]. Separate structural comparisons (structural dendograms) of class I and class II aaRS enzymes have been published [24–26], and our analysis is consistent with these. Surprisingly, however, we identify similarities in protein sequences comparing class I and class II Pfu aaRS enzymes, which, to our knowledge, have not previously been reported (however, see Figure 13 of reference [26]). Specifically, GlyRS-IIA and ValRS-IA are similar in amino acid sequence (e-value = 2.1). AspRS-IIB and IleRS-IA are similar (e-value = 1.4 or 1.5; depending on the alignment). HisRS-IIA and TyrRS-IC are also similar (e-value = 3.7). TyrRS-1C is more similar in sequence to HisRS-IIA (e-value = 3.7) than it is to other class I aaRS enzymes, with the sole exception of closely related TrpRS-IC (e-value = 1e-4). ThrRS-IIA is similar in sequence to IleRS-IA (e-value = 4.2). The e-value scores are for the best local alignments, but Pfu GlyRS-IIA and ValRS-IA are similar in sequence over nearly the entire length of GlyRS-IIA.
Figure 1.
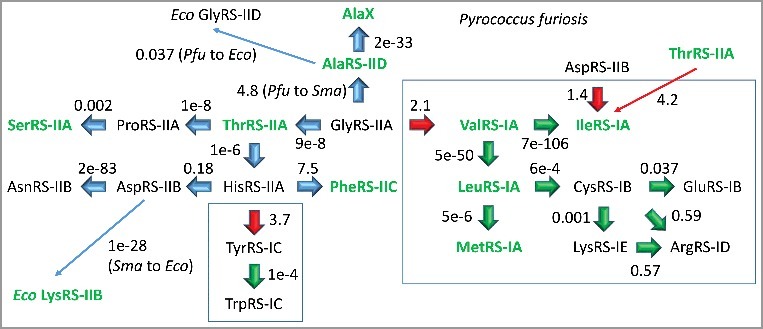
Pyrococcus furiosis (Pfu) aaRS enzymes were searched using NCBI Blast tools for nearest homologs in Pfu. In some cases, Staphylothermus marinus (Sma) (archaea) and Escherichia coli (Eco) (bacteria) homologs are identified. AlaX is one of a set of tRNAAla editing enzymes in Pfu.
Interestingly, Pfu ValRS-IA, LeuRS-IA, IleRS-IA and MetRS-IA are all very similar enzymes by aaRS structural class (IA) and e-value, and Val, Leu, Ile and Met are similar neutral and hydrophobic amino acids within the first column of the codon-anticodon table. For Pfu, therefore, amino acids, tRNAs and aaRS enzymes are coevolved for the first column of the code, as expected from the coevolution hypothesis. ThrRS-IIA, ProRS-IIA and SerRS-IIA are found in the second column of the code, and these are related enzymes by aaRS class, e-value and apparent lineage. Gly, Asp, Val and Ala have been proposed to be the first four amino acids in the code [9]. Interestingly, GlyRS-IIA, AspRS-IIB, ValRS-IA and AlaRS-IID are very different enzymes, indicating that, at the base of code evolution across rows, discrimination of tRNAs by distinct aaRS enzymes was strongly selected. Apparently, there is a greater tendency for amino acid, tRNA and aaRS coevolution within columns than across rows of the genetic code, particularly at the base of the code and at the earliest stage of code evolution. These observations appear to partly explain the distributions of similar amino acids within codon-anticodon table columns.
Figure 2 and Supplementary Figure 1 show the alignment of GlyRS-IIA, ValRS-IA and IleRS-IA enzymes from ancient archaea. The alignment in Figure 2 includes a Zn-binding motif that is shared among class I and class II aaRS enzymes. Some other features of class I and class II aaRS enzymes also appear to be conserved. The entire alignment is shown in Supplementary Figure 1. A summary of the alignment data is shown in the schematic in Figure 2. Relative to Pfu GlyRS-IIA, Staphylothermus marinus (Sma) IleRS-IA has an N-terminal extension that includes essential active site β-sheets, the HIGH active site motif and a Zn-binding motif, all of which are missing in GlyRS-IIA. These unique N-terminal determinants of class I aaRS enzymes, are likely to ensure the class I fold and to block the C-terminus of the protein from assuming the class II fold. The shorter GlyRS-IIA aligns to IleRS-IA and ValRS-IA over its entire length, and the C-terminus of these proteins is reasonably conserved across aaRS classes. GlyRS-IIA, IleRS-IA and ValRS-IA share: 1) a β-sheet in the Motif 1 region of GlyRS-IIA aligning with an active site β-sheet in IleRS-IA; 2) a Zn-binding domain including 3 similarly positioned β-sheets; 3) a β-sheet of GlyRS-IIA just C-terminal to the shared Zn-binding domain aligns with an active site β-sheet of IleRS-IA; and 4) the active site Motif 2 of GlyRS-IIA and the active site KMSKS of IleRS-IA align, including a shared active site β-sheet and loop. The quality of the amino acid sequence alignment is probably sufficient to demonstrate GlyRS-IIA, ValRS-IA and IleRS-IA homology. The structural similarities, such as the shared Zn-binding motif, strongly reinforce this conclusion. Local alignments with e-values as low as 0.001-0.002 have been obtained for GlyRS-IIA and IleRS-IA (i.e. a 1:500 to 1:1000 chance that the alignment is due to a random event).
Figure 2.
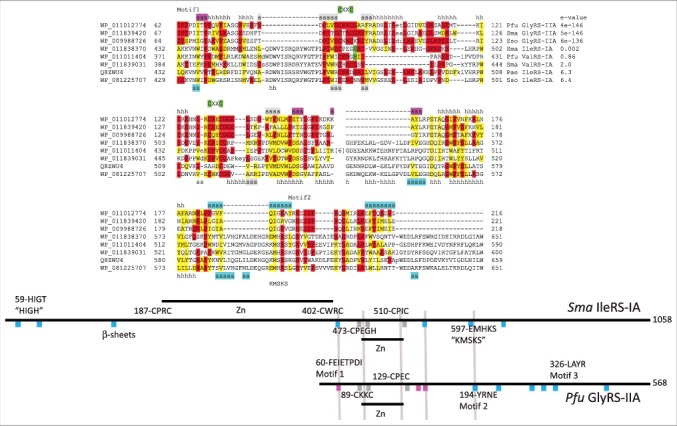
Similarity of class I and class II aaRS enzymes is indicated. A partial sequence alignment of GlyRS-IIA, ValRS-IA and IleRS-IA enzymes is shown demonstrating sequence similarity of a shared Zn-binding motif and GlyRS-IIA Motif 2 with IleRS-IA KMSKS motif. Red shading indicates identity comparing class I and class II aaRS. Yellow shading indicates similarity comparing class I and class II aaRS. Green shading is used to highlight Zn-binding motifs. Cyan shading indicates active site β-sheets (sss). Magenta shading indicates 3 β-sheets in GlyRS-IIA expected to block class I folding by a class II aaRS. Gray shading indicates β1-β3 of the shared Zn-binding domain. The entire alignment is shown in Supplementary Figure 1. The schematic diagram shows how Pfu GlyRS-IIA and Sma IleRS-IA align (gray lines highlight some similarities). Pae) Pyrobaculum aerophilum; Sso) Sulfolobus solfataricus.
In order to analyze the shared Zn-binding motif, a homology model for Pfu GlyRS-IIA was generated (Figure 3). The closest structure identified using the Phyre2 server was human GlyRS-IIA (i.e. PDB 4KQE and 4QEI) [27,28]. Although human GlyRS-IIA lacks cysteine ligands for Zn binding, the fold of the homologous region of the protein is maintained, so a model of the conserved Pfu GlyRS-IIA Zn-binding domain was obtained. Thermus thermophilus (Tth) ValRS-IA (PDB 1GAX) includes a shortened version of the shared Zn-binding domain [29]. In Figure 4, the shared Zn-binding regions are compared. Within the Zn-binding region, three similarly arranged β-sheets are identified (β1-β3) comparing Tth ValRS-IA, Pfu GlyRS-IIA and human GlyRS-IIA structures. We conclude from this structural comparison that class I and class II aaRS enzymes are homologous.
Figure 3.
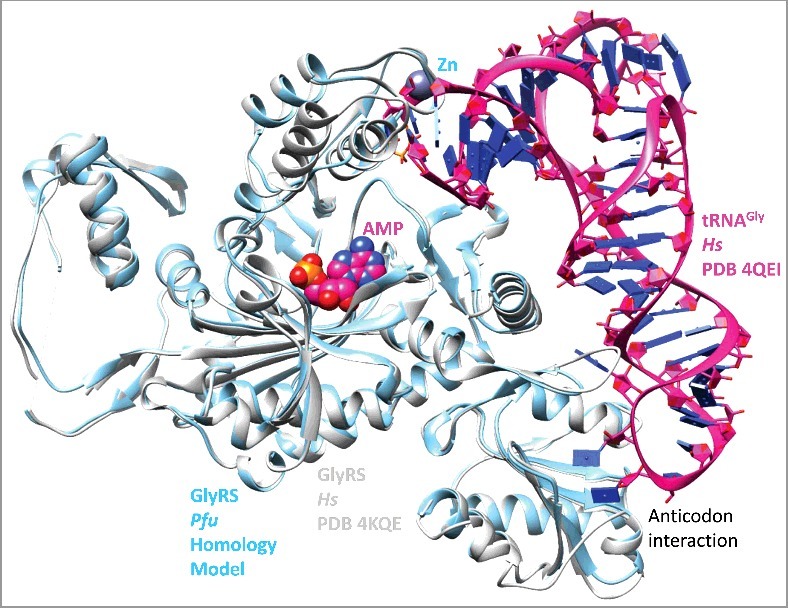
A homology model (Supplementary File 1) of Pyrococcus furiosis GlyRS-IIA was constructed by homology threading to human GlyRS-IIA (PDB 4KQE). The homology model (powder blue), PDB 4KQE [28] (white) and related PDB 4QEI [27] (magenta) were overlaid. Although human (Hs) GlyRS-IIA lacks Zn binding, the shape of the loops is maintained.
Figure 4.
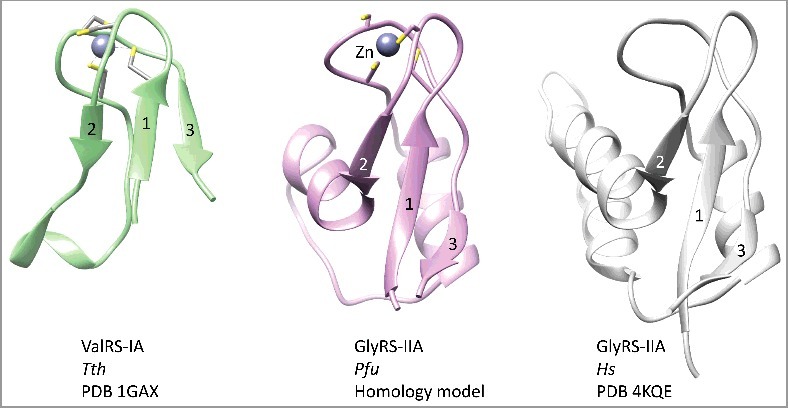
A structurally conserved Zn-binding motif among class I and class II aaRS enzymes. Similar orientations of Tth ValRS-IA (green), Pfu GlyRS-IIA (magenta) and human GlyRS-IIA (white) are shown.
Incompatibility of class I and class II aaRS folds
ValRS-IA and GlyRS-IIA folds are incompatible (Figure 5). The N-terminal extension of ValRS-IA helps to form the class I aaRS active site (i.e. the HIGH motif and essential active site β-sheets), and the N-terminal Zn-binding region of class I aaRS enzymes blocks class II aaRS folding. By comparison of structures, three antiparallel β-sheets in GlyRS-IIA that surround the shared Zn-binding motif establish a clash with the ValRS-IA N-terminal Zn-binding domain. The most C-terminal β-sheet of the Pfu GlyRS-IIA β-sheets (157-KAYL) corresponds to an active site β-sheet in Tth ValRS-IA (485-LVTG; Sma IleRS-IA 553-FIVEG), so formation of the β-sheets in GlyRS-IIA is incompatible with formation of the ValRS-IA active site. Formation of the ValRS-IA active site, therefore, is dependent on the N-terminal domain of Tth ValRS-IA, which includes the “HIGH” active site motif, parts of the active site parallel β-sheets (β-sheets 35-PFVIF, 73-EAVWL(P)GT, 137-DWSREAF) and the class I-specific Zn-binding domain. The more N-terminal class I-specific Zn-binding domain blocks class II aaRS folding.
Figure 5.
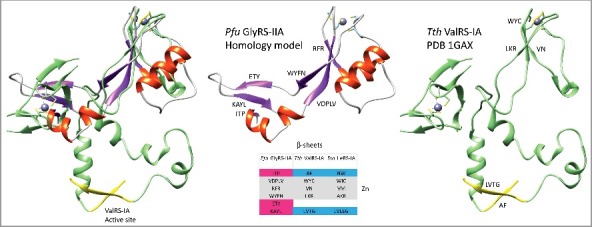
Incompatibility of class I and class II aaRS folding patterns. An overlay of the shared Zn-binding motif of GlyRS-IIA (secondary structure representation) and ValRS-IA (green) demonstrates a clash by three antiparallel GlyRS-IIA β-sheets with the N-terminal ValRS-IA Zn-binding domain. A ValRS-IA active site β-sheet (LVLEG) is yellow. LVLEG corresponds to Pfu GlyRS-IIA β-sheet KAYL in the 3 antiparallel β-sheet cluster surrounding the shared Zn-binding motif.
The standard genetic code
The initial standard genetic code, which is found in many ancient archaea, is shown in Figure 6 as a codon-anticodon table (top table, for archaea). Because of the central importance of tRNA in genetic code evolution, codon-anticodon tables are more informative than simpler representations. When the standard genetic code was established (i.e. in the RNA-protein world before LUCA), the anticodons shaded in red in the top chart were disallowed, because adenine was negatively selected in the tRNA anticodon wobble position [9,18]. Adenine in the wobble position can destabilize the anticodon loop. Also, because wobble A pairs with U much better than with C in mRNA, adenine in the tRNA wobble position supports an inflexible code that was negatively selected [9]. In addition, only one tRNAIle (GAU) is generally utilized. Therefore, only 44 unique tRNA anticodons and 3 stop codons need to be considered in early genetic code evolution [9].
Figure 6.
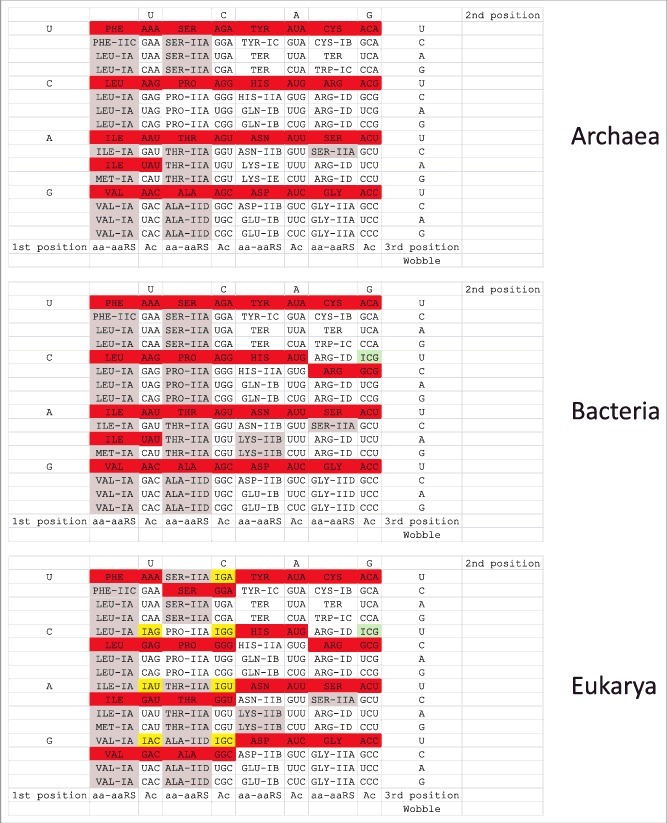
Codon-anticodon tables. Proofreading by aaRS enzymes in archaea is confined to the left half of the codon-anticodon table. Gray shading indicates editing by aaRS enzymes. Red shading indicates anticodons that are disallowed or strongly underrepresented. Green shading indicates adenosine→inosine conversion in bacteria and eukaryotes (tRNAArg (ACG→ICG)). Yellow shading indicates adenosine→inosine conversion in eukaryotes (very rarely, these modifications are found in some bacteria) [19].
AaRS enzymes that proofread
In the course of studies of genetic code evolution, we analyzed archaeal, bacterial and eukaryotic aaRS enzymes with proofreading active sites versus the standard genetic code (Figure 6) [1,2]. The figure also accounts for the tRNA wobble adenine→inosine modification lacking in archaea but found in bacteria (tRNAArg (ACG)) and eukarya (tRNALeu (AAG), tRNAIle (AAU), tRNAVal (AAC), tRNASer (AGA), tRNAPro (AGG), tRNAThr (AGU), tRNAAla (AGC) and tRNAArg (ACG)) [18,19]. Remarkably, the aaRS enzymes that proofread in archaea are restricted to the left half of the codon-anticodon table, and, in eukaryotes, aaRS enzymes that proofread correlate strongly with the wobble A→I modification. SerRS-IIA proofreads, but Ser is split between the left and right halves of the table. In bacteria and eukaryotes, LysRS-IIB proofreads, but, in archaea, LysRS-IE does not [1,2]. To our knowledge, near restriction of aaRS editing to the left half of the codon-anticodon chart is not recorded in recent reviews or in the literature on aaRS enzymes, tRNAs or the genetic code, although this observation is informative about code structure and evolution. Because the A→I wobble modification strongly correlates with aaRS enzymes that proofread, this added structure of the code requires explanation.
Synonymous anticodon preferences
I>>G>>A
In part because of strong G>>A anticodon wobble preference in archaea (Figure 6), we considered wobble preference more generally in tRNA (Figures 7–9). Unlike codon preference, anticodon wobble preference does not appear to be largely driven by gene regulation (i.e. to match codon bias). Inspection of anticodon wobble base frequencies indicated that, for each synonymous ANN and GNN pair (encoding the same amino acid) (Figure 7), there was a strong preference for wobble G>>A, unless A was deaminated to inosine, in which case, interestingly, the preference was strongly I>>G. In archaea, A is largely excluded in the wobble position. In bacteria, only tRNAArg (ACG→ICG) is strongly favored over tRNAArg (GCG). In all other cases in bacteria, G is strongly favored over A, as in archaea. In eukaryotes, tRNALeu (AAG vs GAG), tRNAIle (AAU vs GAU), tRNAVal (AAC vs GAC), tRNASer (AGA vs GGA), tRNAPro (AGG vs GGG), tRNAAla (AGC vs GGC) and tRNAArg (ACG vs GCG), for which wobble anticodons encoding A modify A→I, inosine is strongly favored over G. The A→I conversion is expected to increase the encoding of tRNAs with ANN anticodons but is not necessarily expected to so strongly suppress the use of synonymous GNN anticodons, which are functional in archaea. In this regard, tRNA wobble G can pair with mRNA codon C or U, but tRNA wobble I can pair with A, C or U. Apparently, I>>G preference in the anticodon wobble position reflects strong positive selection for broader recognition by tRNA of synonymous mRNA codons.
Figure 7.
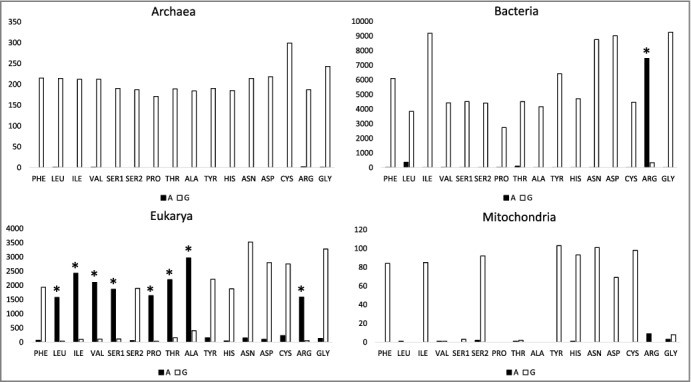
Anticodon wobble preferences comparing synonymous ANN and GNN tRNA anticodons. * indicates A→I conversion. Synonymous anticodons: Ser1 (AAA vs GAA), Ser2 (ACU vs GCU).
Figure 9.
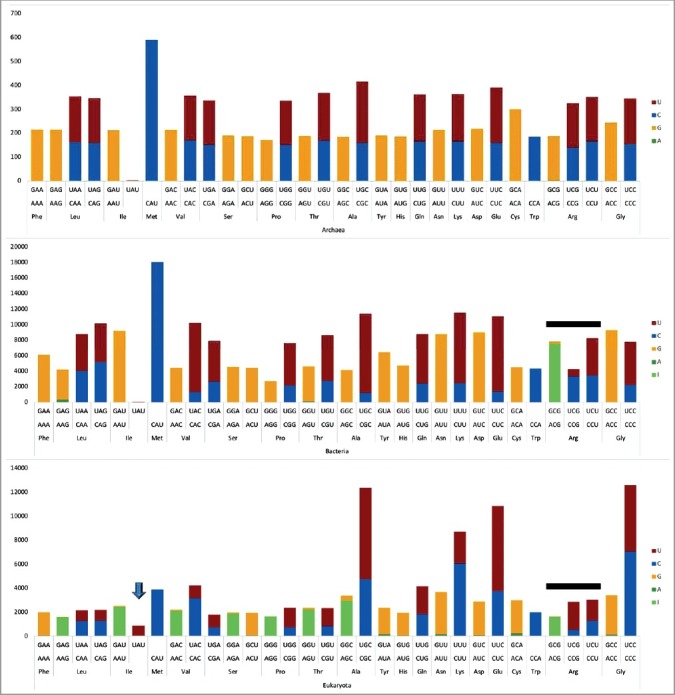
A representation that combines purine and pyrimidine anticodon wobble preference data. The down arrow indicates Ile (UAU) utilization in eukaryotes. The black lines indicate interesting differences comparing Arg anticodons in bacteria and eukarya.
We note that in eukaryotes tRNASer (ACU<<GCU) shows G preference over A in the anticodon wobble position. For tRNASer (ACU), A is not converted to I. If tRNASer (ACU) converted A→I, this would cause recognition by tRNASer (ICU) of AGA Arg codons in mRNA, causing translation errors (Ser replacement of Arg in proteins). The A→I conversion, therefore, only occurs in 4-codon sectors to prevent spillover of tRNA specificity to a 2-codon sector encoding a different amino acid. In eukaryotes, the only 4-codon sector of the genetic code for which there is no A→I conversion is tRNAGly (ACC) [18].
U>C
In Figure 8, pyrimidine wobble preferences are analyzed. Generally, U is preferred over C. In archaea, U is slightly preferred over C, for all synonymous anticodon pairs. Trp (CCA) is a special case, because anticodon UCA represents a UGA stop codon that is read in mRNA by a protein. In bacteria, U is generally preferred over C, except for Leu2 (UAG<CAG) and Arg1 (UCG<<CCG). For Val (UAC>>CAC), Ser (UGA>>CGA), Pro (UGG>>CGG), Thr (UGU>>CGG), Ala (UGC>>CGC), Gln (UUG>>CUG), Lys (UUU>>CUU), Glu (UUC>>CUC) and Gly (UCC>>CCC), wobble U is strongly preferred over wobble C. In eukaryotes, the U>C tRNA anticodon wobble preference is apparent for sets of synonymous anticodons for Ser (UGA>CGA), Pro (UGG>>CGG), Thr (UGU>>CGU), Ala (UGC>>CGC), Gln (UUG>CUG), Glu (UUC>>CUC), Arg1 (UCG>>CCG) and Arg2 (UCU>CCU). Interestingly, U versus C bias is opposite for eukaryotic Arg1 (UCG>>CCG) and bacterial Arg1 (UCG<<CCG). Also, U<C anticodon wobble preference is observed in eukaryotes for Leu1 (UAA<CAA), Leu2 (UAG<CAG), Val (UAC<<CAC), Lys (UUU<<CUU) and Gly (UCC<CCC).
Figure 8.
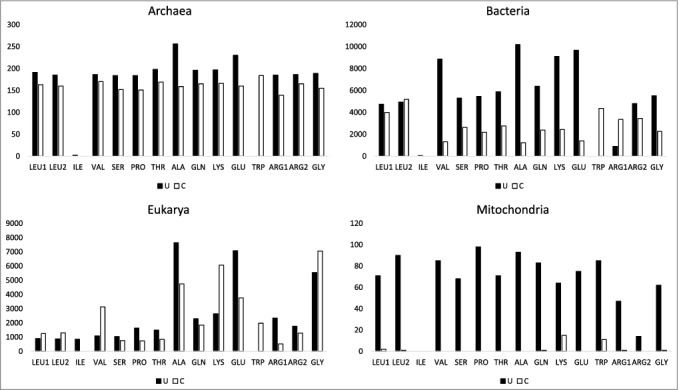
Anticodon wobble preferences comparing synonymous UNN and CNN tRNA anticodons. Synonymous anticodons: Leu1 (UAA vs CAA), Leu2 (UAG vs CAG), Arg1 (UCG vs CCG), Arg2 (UCU vs CCU).
Ile, Met and Trp are special cases. Ile and Met occupy the same 4-codon sector in the codon-anticodon table. Anticodon preference for Ile is shown in Figure 9. For archaea and bacteria, tRNAIle (GAU) is highly used and tRNAIle (AAU and UAU) are very rarely used. Generally, archaea and bacteria utilize a single tRNAIle (GAU). In eukaryotes tRNAIle (AAU→IAU) is strongly favored and tRNAIle (GAU) is suppressed, as expected. Interestingly, tRNAIle (UAU) is commonly utilized in eukaryotes, although tRNAIle (UAU) can potentially be ambiguous with tRNAMet (CAU). Trp (CCA) shares a 2-codon sector with a stop codon, which is read in mRNA by a protein rather than a tRNA [15].
Synonymous anticodon wobble preference in mitochondria
To maintain a small organelle genome, mitochondria encode a subset of tRNAs, potentially limiting available anticodons. Mitochondrial anticodon wobble preference, indeed, is strange and limited in coding capacity (Figures 7 and 8). In particular, Leu, Val, Pro, Thr, Ala, Arg and Gly tRNAs have scant or no mitochondria-encoded wobble anticodon A or G (Figure 7). In terms of codon usage and preference, however, mitochondria utilize mRNA codons with wobble C and U encoding these amino acids. Because mitochondria import cytosolic tRNAs [30–32], deficiencies in mitochondrial coding can be compensated, and, perhaps, all of the apparent mitochondrial anticodon wobble deficiencies are compensated by imported cytosolic tRNAs. We note that import of tRNAs with inosine in the anticodon wobble position (encoding Leu, Val, Pro, Thr, Ala and Arg) would be almost sufficient to compensate for limiting mitochondrial tRNAs. Import of tRNAIle (IAU) is less important, because tRNAIle (GAU) is encoded in the mitochondria and can suffice to read mRNA codons AUC and AUU. Import of a cytosolic tRNAGly (GCC) also appears necessary, and cytosolic tRNAGly (GCC) is imported into mitochondria [31]. Furthermore, mitochondrial-encoded tRNAs are heavily biased toward wobble U rather than C (Figure 8). Interestingly, tRNATrp (UCA>>CCA) in mitochondria utilizes the UCA anticodon corresponding to the UGA stop codon in place of the CCA anticodon, which is utilized to encode Trp in archaea, bacteria and eukaryotes. Of course, tRNA wobble U reads a broader spectrum of synonymous mRNA codons than tRNA wobble C because wobble U can pair with mRNA wobble A or G but wobble C strongly prefers to pair with mRNA wobble G.
Arg coding
Figure 9 shows interesting features of tRNAArg distributions in eukarya and bacteria. Notably, tRNAArg (CCG) is somewhat limiting in eukaryotes and tRNAArg (UCG) is limiting or absent in bacteria. In Figure 10, the consequences of these tRNA limitations are reviewed. It appears that eukaryotes primarily use tRNAArg (UCG) to read CGG codons. Bacteria primarily use tRNAArg (CCG) to read CGG codons. Eukaryotes read CGA codons using tRNAArg (ICG and UCG). Bacteria read CGA codons primarily using tRNAArg (ICG). Absence of tRNAArg (UCG) in some bacteria, therefore, appears to explain the evolution of the A→I tRNA wobble modification, which, in the case of missing tRNAArg (UCG), is required to read CGA codons.
Figure 10.
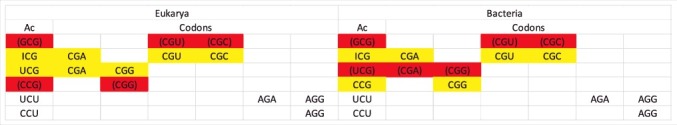
Consequences of apparent limiting of tRNAArg (CCG) in eukarya and tRNAArg (UCG) in bacteria. Ac for anticodon.
The editing hypothesis
Based on the genetic code table left side biased distribution of 4-codon sectors correlating with proofreading aaRS enzymes (Figure 6), sectoring of the genetic code utilizing the tRNA anticodon wobble position was likely inhibited by aaRS proofreading. Furthermore, editing appears to be limited to hydrophobic and neutral amino acids with limited charge and absent or limited side chain hydrogen bonding potential. Proofreading generally occurs for amino acids that are smaller than, or very similar to, the cognate amino acid [1,2]. Smaller amino acids may attach to a non-cognate tRNA and require editing because they can fit the synthetic aaRS active site and, therefore, can be linked to a non-cognate tRNA [1,2]. Aminoacylation errors are less likely for amino acids with charged side chains and/or with more hydrogen bonding groups, because more readily distinguished amino acids are more fully specified in their cognate aaRS synthetic active site. Interestingly, the densest sectoring employing the tRNA anticodon wobble position is observed for the third column (Glu, Asp, Lys, Asn, Gln, His, Ter (stop), Tyr) and the uppermost 4-codon sector of the fourth column (Trp, Ter, Cys). None of the corresponding aaRS enzymes proofread in archaea (Figure 6). In bacteria, LysRS-IIB proofreads to reject amino acids that are from outside the code (homocysteine, homoserine and ornithine). Lys and Arg are readily discriminated in the LysRS and ArgRS active sites. Lys has a flexible side chain with a localized positive charge. Arg, by contrast, has a much stiffer side chain with a distributed positive charge and hydrogen bonding potential. We posit that, as the code evolved, right half tRNAs initially did not require editing by an aaRS because encoded amino acids with more identifying functional groups were easier to specify through interactions in the aaRS synthetic active site. Because accurate specification of a cognate amino acid in the aaRS synthetic site limits tRNA charging errors, it is likely that the right half of the genetic code sectored more completely to 2-codon sectors prior to full evolution of accurate amino acid selectivity by aaRS enzymes. A slightly different but related view might be that amino acids with more identifying characteristics were more aggressive at invading 4-codon sectors compared to neutral amino acids with limited hydrogen bond forming potential. Evolution of aaRS anticodon recognition domains (in all aaRS except for AlaRS and SerRS) also enhanced the accuracy of tRNA charging and brought the code to universality. Fidelity mechanisms, therefore, continued to evolve and potentially take precedence over one another as the code continued to sector.
Because aaRS proofreading appears to inhibit sectoring, and because archaeal aaRS enzymes from the right half of the code do not edit, sectoring is more innovative on the right half of the codon-anticodon table than the left half. We posit the editing hypothesis that aaRS proofreading inhibited genetic code sectoring. Invasion to reassign a 4-codon sector encoding a single amino acid to two 2-codon sectors, each encoding a distinct amino acid, for instance, was initiated by aminoacylation errors on existing tRNAs. During code evolution, invasion could be by an amino acid that was not yet encoded, resulting in an increase in the complexity of the code. Note that accuracy of translation and tRNA charging continued to improve as the code evolved, and editing and specificity, therefore, became ever more important later in evolution as additional amino acids became encoded. Also, metabolism generates amino acids that are not encoded but could be charged to tRNAs in error and could be removed by aaRS proofreading. Alternatively, amino acids could be attached to tRNAs in error, and, then, through selection, could be added to the genetic code by dividing a 4-codon sector into two 2-codon sectors. With the exceptions of Met and Trp, only a stop codon (UGA), which is recognized in mRNA by a protein not a tRNA, can occupy a 1-codon sector. Only in eukaryotes does Ile strongly occupy the UAU anticodon (adjacent to Met (CAU)) (Figure 9), by which time in evolution, mechanisms were developed for tRNA modifications to support accurate tRNAIle (UAU) and tRNAMet (CAU) discrimination [33,34].
Met, which appears to have invaded a partially occupied 4-codon Ile sector, may be an apparent exception to the rule that proofreading aaRS enzymes resist sectoring around the wobble anticodon position. MetRS proofreads to remove homocysteine, which is not part of the genetic code [1,2]. The Ile-Met 4-codon sector, however, appears to be a special case (see Discussion). In evolution, Phe may have invaded a 4-codon Leu sector (AAA, GAA, UAA, CAA), perhaps being recruited from outside the code. Arg appears to have invaded a 4-codon Ser sector (ACU, GCU, UCU, CCU) (Figure 6), apparently demonstrating movement of amino acids within the code.
Coevolution of aaRS enzymes and tRNAs and the editing hypothesis
Figure 11 shows how aaRS enzymes may have coevolved with tRNAs [1,2]. Before the code was substantially evolved, it is difficult to imagine tRNA recognition by proteins, and initial aminoacyl transfers may have been catalyzed by ribozymes [20,22,23]. A proposed sequence of events was developed according to the aaRS mechanisms now used to discriminate different archaeal tRNAs. As aaRS enzymes evolved, acceptor stems of tRNAs and the discriminator base (position 76; 73 in historic numbering) [9,13] may have been the most important initial determinants for discrimination. In archaea, most discriminators are A, so the discriminator base is only used for a subset of amino acids (i.e. generally A in archaea except: G) Asp, Ser, Arg, Asn; U) Thr, Cys; and C) His) [16]. We posit that recognition of the anticodon of tRNAs by aaRS enzymes subsequently became a mechanism for tRNA specification that restricted further sectoring of the code. Only AlaRS and SerRS lack anticodon recognition domains. Later, the aaRS enzyme class (i.e. class I versus class II aaRS) became a determinant [1,2]. Without knowing the exact order of events, at some stage, longer V loops in tRNALeu and tRNASer became important as determinants and anti-determinants for tRNA charging. From Figure 1 of a recent paper [9], many tRNAs appear to be derived from tRNALeu and tRNASer, which could have driven tRNALeu and tRNASer V loop expansions in the evolving code in order to discriminate partially radiated tRNAs that may attach related amino acids. In archaea, generally, other tRNAs do not have V loop expansions (i.e. tRNATyr and tRNASec (Sec for selenocysteine) in bacteria). Along the pathway, active sites of aaRS enzymes continued to evolve to exclude attachment of incorrect amino acids. This exclusion is more difficult for amino acid side chains that are uncharged and that form only one hydrogen bond (i.e. the left half of the codon-anticodon table), explaining why aaRS proofreading became such a dominant mechanism for the left half of the code. At a late stage, therefore, proofreading by aaRS enzymes is posited to have been recruited as a mechanism for discrimination mostly restricted to the left half of the codon-anticodon table (Figure 6), consistent with the editing hypothesis that aaRS proofreading maintained 4-codon sectors in the genetic code by suppressing further sectoring. For bacteria, only LysRS-IIB from the right half of the table is capable of proofreading. AaRS enzyme classes are structurally related enzymes for archaea, bacteria and eukaryotes, except for LysRS, which is typically structural class IE in archaea and class IIB in bacteria and eukarya [1,2]. GlyRS is class IIA in archaea and eukaryotes but class IID (or historically classified IIC) in many bacteria [24].
Figure 11.
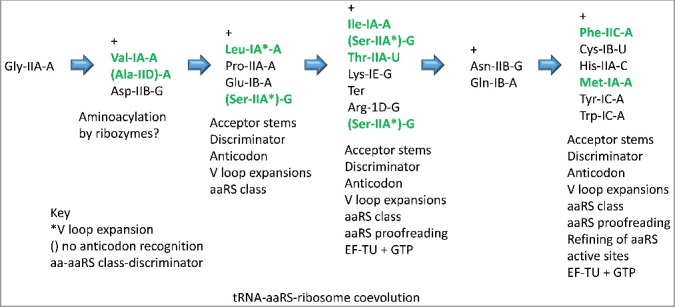
An approximate sequence of events for the requirement of different mechanisms for discrimination of tRNA identities by aaRS enzymes and the evolution of ribosome fidelity. Green text indicates aaRS proofreading (in archaea).
Proofreading is not utilized for GlyRS, ArgRS and archaeal/eukaryotic ProRS (Figure 6). Glycine is the smallest amino acid, so the GlyRS synthetic active site is constrained to block loading of larger amino acids (PDB 4KR2) [35]. Arginine is a large amino acid that is much less flexible than lysine. As with lysine, arginine is charged (+1), and arginine has significant hydrogen bonding potential. These distinguishing features of arginine are utilized in the ArgRS synthetic active site to exclude incorrect amino acids (PDB 1F7U) [36]. Proline is the only encoded imino acid, so proline is readily distinguished in the ProRS active site from other encoded amino acids. ProRS proofreads in bacteria because of addition of a bacterial-specific editing domain to ProRS-IIA that is missing in ProRS-IIA of archaea and eukaryotes. Of course, aaRS editing and accurate cognate amino acid specification also suppress inaccurate charging of tRNAs with amino acids that are generated from metabolism but are not encoded. When the code was evolving, aaRS enzymes were likely more error-prone in attaching amino acids, supporting sectoring of the code via tRNA charging errors.
Interestingly, the tRNAs added by eukaryotes (compared to bacteria) with adenine→inosine in the anticodon wobble position are mostly proofread by aaRS enzymes and, also, generally occupy 4-codon sectors on the left half of the genetic code table (Figure 6) [18,19]. In eukaryotes, all tRNAs on the left half of the chart utilize the A→I conversion except tRNAMet, which lacks an anticodon with encoded wobble A, and tRNAPhe, which occupies a 2-codon sector and, therefore, cannot adopt the A→I modification without substituting Phe for Leu in proteins. It appears that eukaryotes adopted a mechanism evolved in bacteria for tRNAArg (ACG→ICG) in order to modify and stabilize the left half of the eukaryotic genetic code table. Perhaps, most interestingly, when wobble inosine is utilized, the synonymous GNN anticodon is suppressed (Figures 7 and 9), indicating that the broader mRNA synonymous codon recognition of inosine compared to G is positively selected. The adenine→inosine modification has only invaded sectors with 4 codons because inosine pairs A, C and U in mRNA codons. In eukaryotes, the only 4-codon sector that is not altered with the adenine→inosine modification is the Gly sector [18].
In mitochondria, it appears that tRNAs encoding Leu, Val, Pro, Thr, Ala and Arg, which all convert wobble A→I in eukaryotes, must be imported from the cytosol, indicating a strong preference for utilizing tRNAs encoded in the cell nucleus with inosine in the wobble anticodon position. Because the mitochondrion was derived from a α-proteobacterial endosymbiont, mitochondria would encode tRNAs with wobble G to specify these amino acids. It appears that the mitochondria prefer to import nuclear-encoded eukaryotic tRNAs with inosine in the wobble position rather than to utilize mitochondrial tRNAs with G in the wobble position, indicating once again the importance of increasing ambiguity in tRNA reading synonymous mRNA codons. Import of tRNAs is a fascinating process supporting the mitochondria-eukaryote symbiotic relationship. Without a eukaryotic host to supply missing tRNAs, mitochondria would not be able to translate mitochondria-encoded mRNA.
Discussion
Alternate class I and class II aaRS folding
Class I and class II aaRS enzymes are related by amino acid sequence homology identified in archaeal species (Figures 1–2; Supplementary Figure S1). A shared Zn-binding domain in GlyRS-IIA and ValRS-IA is identified (Figure 4). Class I aaRS enzymes have a N-terminal extension that can include a second Zn-binding domain, which may have been a determinant in distinct class I aaRS folding. Additionally, class I aaRS active site β-sheets and the active site “HIGH” motif are found within the class I-specific N-terminus, so the class I aaRS active site cannot be assembled without the N-terminal domain. Because class I and class II enzymes have largely incompatible protein folds and bind to opposite faces of tRNA, we posit that an ancestral aaRS enzyme folded in distinct class I and class II conformations for three reasons. First, the N-terminal extension in class I aaRS enzymes that includes the HIGH active site motif and active site β-sheets and that can include a Zn-binding domain helped to enforce the class I fold. Second, a set of three antiparallel β-sheets in class II aaRS enzymes would clash with the N-terminal Zn motif found in class I aaRS and block assembly of the class I aaRS active site (Figure 5). Third, opposite faces of cloverleaf tRNA bind class I and class II aaRS enzymes, and tRNA binding may have helped direct the alternate aaRS folds. As domains evolved to take on the appropriate fold, Zn-binding disappears from some domains that initially evolved around Zn binding (Figure 4). The more complex model that class I and class II aaRS enzymes arose from transcription and translation of an ancestral bi-directional gene [3–6] we find less likely. Early in evolution, Zn-binding appears to have directed the stability and folding conformations of large proteins such as aaRS enzymes and RNA polymerase. Over time, some Zn domains hardened in conformation so that Zn binding was no longer necessary (Figure 4). Based on the determinants for class I and class II aaRS folding identified here, domain swap experiments can likely switch the folding of the two aaRS structural forms.
The maximal size of the genetic code
The standard genetic code is generally considered to potentially encode 64 amino acids. Because adenine is not utilized in the anticodon wobble position in archaea, however, this reduces the number of utilized anticodons to 48 at the base of code evolution [9]. When wobble A is encoded in tRNA, A→I modification occurs, and wobble G is suppressed in the synonymous anticodon (Figures 7 and 9). The most heavily divided 4-codon sectors of the standard genetic code that encode amino acids, and not stop codons, or Met, are divided into two 2-codon sectors. The reason that 2-codon sectors resisted further subdivision into 1-codon sectors encoding two different amino acids is that tRNA anticodons with wobble U and wobble C are read ambiguously to recognize mRNA codons with both wobble A and G. Anticodon wobble C is thought to mostly recognize codon wobble G but may have recognized mRNA codon A well enough to have supported ambiguous reading of mRNAs during the early evolution of the code. Interestingly, anticodon wobble C was not excluded from tRNA as strictly as was anticodon wobble A, and this observation requires further explanation. Because of ambiguous coding, reading tRNAs, the largest number of amino acids that could be encoded using a triplet tRNA code is 32 (or 31 aas with stops). Because division of 4-codon sectors was limited by aaRS proofreading, evolutionary refinement of aaRS active sites, aaRS anticodon recognition and the A→I modification, the standard genetic code has only 20 aas with stops, and, with minor partial exceptions, the code has remained universal in the three domains of life.
Coevolution of aaRS accuracy and genetic code universality
There is a “chicken and egg” problem to consider in terms of aaRS evolution. Notably, there is no known mechanism to generate aaRS proteins until the code has evolved, and, to our knowledge, there is no clear model for making functional proteins with subsets of amino acids. At this time, we offer no simple solution to this problem. Ribozymes as small as 5 nt created in vitro can aminoacylate tRNAs [20,22,23], but these ribozyme functions appear now to be fully replaced by aaRS enzymes, so a natural record of aminoacylating ribozymes may not now exist.
Because tRNA and aaRS enzymes must be coevolved [1,2], however, aaRS enzymes and proofreading by aaRS enzymes are considered with regard to evolution of the code. We note that aaRS proofreading, in archaea, is limited to the left half of the codon-anticodon table, which encodes only hydrophobic and neutral amino acids (Figure 6). ProRS, from the left half of the code, does not edit in archaea and eukarya, but ProRS edits in bacteria (Figure 6). Another partial exception to the left half rule is tRNASer (GCU). Ser is the only amino acid that is split into both the left and right halves of the table, and SerRS-IIA edits. We posit that a 4-codon sector encoding Ser (anticodons ACU, GCU, UCU, CCU) may have been invaded by Arg probably before SerRS proofreading evolved to adequately resist sectoring. Also, because Ser is encoded within separated genetic code sectors, SerRS did not recognize the tRNASer anticodon for discrimination in accurate Ser attachment, which may have increased tRNASer charging errors, leading to Ser sensitivity to invasion by Arg [1,2]. In bacteria, LysRS (class IIB (editing) in most bacteria; class IE (non-editing) in most archaea) is also a partial exception (Figure 6) [1]. we observe that the third column and also the uppermost 4-codon sector of the fourth column of the codon-anticodon table are the most heavily innovated, indicating that evolution of aaRS proofreading inhibited code sectoring, limiting the expansion of the code. Based on this observation, we posit that errors in amino acid attachment to tRNA were important to continue sectoring the code by utilizing the wobble anticodon position. As errors become more difficult to make or to sustain, i.e. because of aaRS synthetic site specificity (mostly the right half of the code) or because of aaRS editing (left half of the code), the code evolved toward closure and universality. Also, the tRNA cloverleaf structure and rugged RNA evolution may limit the potential size of the code. The advent of specific tRNA modifications (i.e. in bacteria and eukaryotes) can be assessed in expanding permitted anticodon contacts to mRNA [33,37,38]. Rugged evolution occurs when many or most substitutions are disruptive for structure, as expected for tRNA [39–41]. Expanding the code beyond 20 amino acids, therefore, may strain the capacity of tRNAs and aaRS enzymes to coevolve for adequate accuracy and discrimination. Evolution of tRNA covalent modifications supported innovation and refinement of the code (i.e. discrimination of tRNAIle (UAU) and tRNAMet (CAU) in eukaryotes) [33,37,42].
Positive selection of tRNA anticodon wobble ambiguity
Because tRNA wobble bases make ambiguous contacts with mRNA, a single tRNA can recognize multiple synonymous mRNA codons, but tRNA wobble ambiguity also limited the capacity for code expansions to encode new amino acids. Although there may be selection for tRNAs with specific purposes, particularly in complex eukaryotes [10], generally, selection was for increased ambiguity in reading tRNA anticodons. In evolution of the genetic code, tRNA anticodon wobble inosine is strongly preferred to guanine, which is strongly preferred to adenine (Figures 7 and 9). Anticodon wobble inosine recognizes A, C and U in mRNA. Anticodon wobble G recognizes C and U. Anticodon wobble A recognizes U, but tRNA wobble A recognizes mRNA wobble C poorly. We posit that the I>>G>>A preference reflects positive selection of increasing ambiguity in the tRNA anticodon wobble position without affecting the reading of synonymous mRNA codons. Because inosine recognizes A, C and U in mRNA codons, the A→I substitution strongly selects for, and can only occur in, 4-codon sectors. Similarly, anticodon wobble U can pair with both mRNA codon A and G. Anticodon wobble C pairs much more strongly with G than with A. We posit that U is generally preferred to C in the anticodon wobble position because tRNA wobble U recognizes synonymous mRNA wobble A and G more rapidly and readily than tRNA wobble C recognizes mRNA wobble A. It is also possible that G = C wobble pairs are (or were) too stable to be optimal for translation (i.e. gave slow tRNA release on the ribosome). The selection pressures at the inception of the code were different than subsequent selection pressures.
Resistance to forming 1-codon sectors
The reason that 4-codon sectors of the genetic code split into two 2-codon sectors around purine and pyrimidine wobble bases is that tRNA wobble bases are read ambiguously. The only 1-codon sectors are for tRNAMet (CAU) and tRNATrp (CCA). The Ile-Met 4-codon sector is a special case (see below). In the Cys-Ter-Trp 4-codon sector, tRNATrp (CCA) shares a 2-codon sector with a stop codon UGA (anticodon UCA), which is recognized in mRNA by a protein, not a tRNA. In mitochondria, however, anticodon UCA (corresponding to stop codon UGA) is utilized to encode Trp (Figure 8). Because of tRNA wobble ambiguity, the maximum coding potential of the standard genetic code is for 32 letters: 31 aas + stops.
The Ile-Met sector
Questions remain with regard to early evolution of the Ile-Met 4-codon sector of the standard genetic code. In archaea, typically only a single tRNAIle (GAU) (Figure 9), two elongator tRNAMet (CAU) and one tRNAiMet (CAU) are found. The sectoring and early proliferation of tRNAMet (CAU) is unusual so near the base of code evolution and requires explanation. From analysis of archaeal tRNA radiations from the primordial cloverleaf tRNAPri, it appears that tRNAMet and tRNAiMet may be derived from tRNAIle, as might be expected from code structure [9]. Furthermore, one tRNAMet and tRNAiMet appear to radiate further and further from tRNAIle in more derived archaeal species. Perhaps the 4-codon Ile-Met sector can be viewed as a partially occupied 4-codon Ile sector, partly invaded by Met. Invasion of the Ile 4-codon sector by Met probably involved recruitment of Met from outside the code via inaccurate tRNAIle (i.e. CAU) charging. Met invasion of Ile and tRNAMet proliferation were partly driven to establish the start signal for translation. Because Met (CAU) evolved at LUCA to discriminate three tRNAMet (CAU; 2 elongator and 1 initiator), at eukaryogenesis, discrimination of potentially synonymous Met (CAU) and Ile (UAU) could be supported by previously evolved tRNA modifications [33,37,42].
Evolution of the standard genetic code
Three main hypotheses for evolution of the standard genetic code include: 1) variations on the Gamow hypothesis (the stereochemical hypothesis: that amino acids interact directly with RNAs, i.e. codons or anticodons, leading to matching of codons and anticodons with amino acids and evolution of the code); 2) the coevolution theory (that code complexity coevolved with advances in amino acid metabolism); and 3) the error minimization theory (that the code evolved to minimize tRNA charging and translation errors) [11]. Recently, it was pointed out that these long-standing hypotheses may have limitations for furthering our understanding of code evolution [11]. Here, we give a simple hypothesis partly relating to, and slightly at odds with, the error minimization theory. We posit that the standard genetic code evolved through mechanisms of inaccurate tRNA charging, tRNA anticodon mutation and tRNA diversification. Mechanisms that enforced tRNA charging accuracy, therefore, brought the code to universality. We posit that similar amino acids are encoded in neighboring sectors and often in the same column of the codon-anticodon table because sectoring was driven by two mechanisms. First, errors in aaRS-catalyzed amino acid attachments to tRNAs induced the division of sectors, generally involving recruitment of similar amino acids, from outside the code, that attached to initially similar tRNAs. Secondly, tRNA anticodon mutations could result in local migrations to a neighboring sector, moving similar amino acids to nearby positions within the code. Selection for incorporation of a new amino acid into proteins drove tRNAs to diverge and discriminate amino acid attachments, leading to a more complex code with an increased number of sectors encoding different amino acids. We posit that the code was built by sectoring in a series of stages described in a recent paper [9].
Koonin and Novozhilov ask why the code is a triplet code [11]. The code is triplet because of the structure of the tRNA anticodon loop, which forces a triplet register for two adjacent tRNAs bound to adjacent mRNA codons [13]. In strict terms of coding, however, the code is almost a 2-nucleotide code, because of degeneracy in the anticodon wobble position, explaining why there are 20 amino acids + stops in the standard genetic code rather than a larger number (up to 31 aas + stops).
Koonin and Novozhilov suggest that translation systems should be analyzed to understand code evolution [11]. We identify two features of translation systems that are relevant. First of all, in the decoding center of the ribosome, proofreading of anticodon base pair attachments to mRNA codons, involving small ribosomal subunit conformational closure enabling EF-Tu and GTP hydrolysis, applies to the second and third anticodon positions only, not the first (wobble) position [43]. For most amino acids, the tRNA wobble position was selected to broaden recognition of mRNA codons, supporting code degeneracy and making tRNAs more readily available for insertion of the encoded amino acid. Secondly, translation systems evolved around tRNA, so a focus on tRNA evolution helps to interpret genetic code evolution. The tRNA-centric view significantly simplifies the problem of standard genetic code evolution, i.e. by shrinking the relevant number of anticodons. Because the genetic code is degenerate, analyzing code evolution from the point of view of mRNA is deceptive, because all 64 codons are utilized in mRNA, but only 44 unique tRNA anticodons and 3 stop codons were utilized at the inception of the standard genetic code (LUCA and ancient archaea). Furthermore, because of tRNA wobble ambiguity, the maximal capacity of the genetic code only expands to 31 amino acids + stops, but aaRS proofreading, accurate aaRS synthetic site specification of amino acid substrates, aaRS anticodon recognition, ribosome conformational proofreading of the anticodon-codon interaction and perhaps the A→I modification limited code expansions to 20 amino acids by preserving 4-codon sectors. Evolving 1-codon sectors of the genetic code was strongly resisted particularly via aaRS and ribosome fidelity mechanisms.
Ribosome proofreading the anticodon-codon interaction
In a recent paper, we posit that the genetic code sectored from a 1→4→8→16→21 letter code (20 aas + stops) [9]. The initial code evolved to utilize any mRNA sequence to synthesize polyglycine, used to stabilize protocells. According to this view, conformational tightening and EF-Tu and GTP proofreading of Watson-Crick base pairing between the anticodon and the codon in the second and third anticodon positions [43] became necessary at the 8→16 letter stage. The 8 letter stage is characterized by resolution of purines and pyrimidines only, but not individual bases, in the first mRNA codon position and the corresponding third tRNA anticodon position. At the 8 letter stage of code evolution, reading the third anticodon position is similar to the sectoring of the wobble position of the standard code, indicating that ribosome proofreading was not yet evolved at this stage. In order to fully resolve A, G, C and U in the first codon position and the corresponding third anticodon position, conformational tightening and EF-Tu and GTP hydrolysis proofreading was essential. The model for sectoring of the genetic code, therefore, makes a prediction about the evolution of translational fidelity mechanisms that brought the code to universality.
Correlation of aaRS proofreading and A→I modification
In eukaryotes, there is strong correlation between aaRS editing and tRNA wobble A→I modification (Figure 6; bottom panel). Primarily, we attribute this correlation to 4-codon sectors. Proofreading by aaRS enzymes maintains 4-codon sectors by inhibiting tRNA charging errors that could lead to further sectoring. A→I modification is most utilized by eukaryotes, which are about 2.2 billion years old. The standard genetic code, by vast contrast, is probably >3.8 billion years old. Because the code is ancient and universal, eukaryotic innovations do not bear on the birth of the code, although eukaryotic innovations may have stabilized the eukaryotic code to prevent further sectoring and a possible escape by eukaryotes from code universality. A→I conversion is limited to 4-codon sectors, because tRNA wobble inosine recognizes mRNA wobble A, C and U. A→I modification in a 2-codon sector, therefore, spills into a neighboring 2-codon sector, causing translation errors. Much earlier in code evolution, tRNA charging errors induced sectoring, adding amino acids to the code. Now such errors are lethal because they induce translation errors. In bacteria, the Arg (ACG, GCG, UCG, CCG) 4-codon sector was protected by the A→I modification, but, in archaea, the Arg 4-codon sector was faithfully preserved without the A→I modification, perhaps because of the high specificity of the ArgRS synthetic active site, ArgRS anticodon recognition and EF-TU proofreading on the ribosome.
Because Gly occupies a 4-codon sector of the code, this raises the question of why tRNAGly (ACC) is not modified A→I in eukaryotes [18]. GlyRS resists charging errors because of the small size of the synthetic active site, which made the Gly (ACC, GCC, UCC, CCC) sector resistant to subdivision. A similar argument can be made for the Arg (ACG, GCG, UCG, CCG) sector. ArgRS does not have a proofreading active site. The ArgRS synthetic active site accurately specifies Arg, however, because of the distinctive Arg side chain. Specificity of charging is enhanced because ArgRS recognizes the tRNAArg anticodon. The Arg 4-codon sector resists further division in bacteria and eukarya because ArgRS charging is accurate and because the A→I modification limits sectoring. It is possible that the standard genetic code is universal (i.e. in archaea, bacteria and eukaryotes), in part, because aaRS proofreading, high aaRS synthetic site specificity, anticodon recognition by aaRS, EF-TU proofreading and A→I modification prevented introduction of new 2-codon sectors in the bacterial and eukaryotic genetic codes. Because tRNA charging errors resulted in code sectoring, evolving mechanisms that enhanced the accuracy of amino acid attachments to tRNAs led to closure and universality of the genetic code.
The tRNA-centric view
We advocate a tRNA-centric view of genetic code and ribosome evolution [9,13]. The complexity of the genetic code was limited by tRNA anticodon loop structure and tRNA wobble degeneracy reading mRNA. The primitive ribosome might have been a decoding scaffold and a mobile peptidyl transferase center. According to our view, cloverleaf tRNA was the essential biological intellectual property leading to the evolution of the code and to the encoding of proteins including aaRS enzymes. According to this view, cloverleaf tRNAPri was a prerequisite to the coevolution of tRNAomes, aaRS enzymes, ribosomes and the genetic code. It appears to us that a small collection of ribozymes, most of which have been generated in vitro, is sufficient to convert a strange polymer and minihelix world into a cloverleaf tRNA world that leads inevitably to a RNA-protein world and cellular life. As described previously, tRNAPri evolved initially as an improved mechanism to synthesize polyglycine to stabilize protocells (as in bacterial cell walls) before the coevolution of tRNAomes, aaRS enzymes, ribosomes and the genetic code. Alternate views have been expressed by others [3,4,7,8,44].
Methods
NCBI Blast
NCBI Blast tools (https://blast.ncbi.nlm.nih.gov/Blast.cgi#alnHdr_317113484) were used to analyze the relatedness of Pfu aaRS enzymes (Figure 1) and to obtain alignments (Figure 2; Supplementary Figure S1).
Anticodon wobble preference
Sequences for tRNAs were collected from the tRNA database (http://trna.bioinf.uni-leipzig.de/) and the genomic tRNA database (http://gtrnadb.ucsc.edu /) [16,17]. Anticodon wobble position preference was analyzed for synonymous anticodons with A and G (ANN vs GNN) or U and C (UNN vs CNN).
Homology modeling
Pfu GlyRS-IIA was modeled to human GlyRS-IIA (PDB 4KQE) [28] using the program Phyre2 [45,46]. Atomic coordinates were refined using the YASARA energy minimization server (http://www.yasara.org/minimizationserver.htm). The PDB file for Pfu GlyRS-IIA is Supplementary File 1 for this paper. UCSF Chimera was used to visualize molecules [47,48]. Zn was oriented to ligands as previously described [46]. Because of low sequence similarity in shared Zn fingers, Pfu GlyRS-IIA and Tth ValRS-IA Zn fingers were aligned manually using Chimera.
Statistical methods
Anticodon wobble preference data sets were analyzed using a chi-square goodness of fit test (http://www.stat.yale.edu/Courses/1997-98/101/chigf.htm). Because of the large datasets used and the differences observed, all comparisons were judged to be significant (p-value<0.0001).
Supplementary Material
Acknowledgments
We thank Bruce Kowiatek (Blue Ridge Community and Technical College, WV) and Robert Root-Bernstein, Michigan State University, MI) for encouragement and helpful suggestions. Kristopher Opron (University of Michigan, Bioinformatics Core) helped with sequence alignments.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Perona JJ, Gruic-Sovulj I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top Curr Chem. 2014;344:1–41. PMID:23852030 [DOI] [PubMed] [Google Scholar]
- [2].Giege R, Eriani G. Transfer RNA recognition and aminoacylation by synthetases. John Wiley & Sons, Ltd; 2014. [Google Scholar]
- [3].Carter CW., Jr. Coding of class I and II aminoacyl-tRNA synthetases. Adv Exp Med Biol. 2017;966:103–148. doi: 10.1007/5584_2017_93. PMID:28828732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rodin AS, Rodin SN, Carter CW., Jr On primordial sense-antisense coding. J Mol Evol. 2009;69:555–567. doi: 10.1007/s00239-009-9288-4. PMID:19956936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pham Y, Li L, Kim A, et al. A minimal TrpRS catalytic domain supports sense/antisense ancestry of class I and II aminoacyl-tRNA synthetases. Mol Cell. 2007;25:851–862. doi: 10.1016/j.molcel.2007.02.010. PMID:17386262 [DOI] [PubMed] [Google Scholar]
- [6].Rodin SN, Ohno S. Two types of aminoacyl-tRNA synthetases could be originally encoded by complementary strands of the same nucleic acid. Orig Life Evol Biosph. 1995;25:565–589. doi: 10.1007/BF01582025. PMID:7494636 [DOI] [PubMed] [Google Scholar]
- [7].Carter CW, Jr., Wills PR. Interdependence, reflexivity, fidelity, impedance matching, and the evolution of genetic coding. Mol Biol Evol. 2018;35:269–286. doi: 10.1093/molbev/msx265. PMID:29077934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wills PR, Carter CW., Jr. Insuperable problems of the genetic code initially emerging in an RNA world. Biosystems. 2018;164:155–166. doi: 10.1016/j.biosystems.2017.09.006. PMID:28903058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pak D, Du N, Kim Y, et al. Rooted tRNAomes and evolu tion of the genetic code. Transcription. 2018. doi: 10.1080/21541264.2018.1429837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Percudani R. Restricted wobble rules for eukaryotic genomes. Trends Genet. 2001;17:133–135. doi: 10.1016/S0168-9525(00)02208-3. PMID:11314654 [DOI] [PubMed] [Google Scholar]
- [11].Koonin EV, Novozhilov AS. Origin and evolution of the universal g enetic code. Annu Rev Genet. 2017. doi: 10.1146/annurev-genet-120116-024713. PMID:28853922 [DOI] [PubMed] [Google Scholar]
- [12].Massey SE. A neutral origin for error minimization in the genetic code. J Mol Evol. 2008;67:510–516. doi: 10.1007/s00239-008-9167-4. PMID:18855039 [DOI] [PubMed] [Google Scholar]
- [13].Pak D, Root-Bernstein R, Burton ZF. tRNA structure and evolution and standardization to the three nucleotide genetic code. Transcription. 2017;8:205–219. doi: 10.1080/21541264.2017.1318811. PMID:28632998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Root-Bernstein R, Kim Y, Sanjay A, et al. tRNA evolution from the proto-tRNA minihelix world. Transcription. 2016;7:153–163. doi: 10.1080/21541264.2016.1235527. PMID:27636862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bertram G, Innes S, Minella O, et al. Endless possibilities: translation termination and stop codon recognition. Microbiology. 2001;147:255–269. doi: 10.1099/00221287-147-2-255. PMID:11158343 [DOI] [PubMed] [Google Scholar]
- [16].Juhling F, Morl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. PMID:18957446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016;44:D184–D189. doi: 10.1093/nar/gkv1309. PMID:26673694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Saint-Leger A, Bello C, Dans PD, et al. Saturation of recognition elements blocks evolution of new tRNA identities. Sci Adv. 2016;2:e1501860. doi: 10.1126/sciadv.1501860. PMID:27386510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rafels-Ybern A, Torres AG, Grau-Bove X, et al. Codon adaptation to tRNAs with I nosine modification at position 34 is widespread among Eukaryotes and present in two Bacterial phyla. RNA Biol. 2017:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xiao H, Murakami H, Suga H, et al. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature. 2008;454:358–361. doi: 10.1038/nature07033. PMID:18548004 [DOI] [PubMed] [Google Scholar]
- [21].Rodin SN, Rodin AS. Origin of the genetic code: first aminoacyl-tRNA synthetases could replace isofunctional ribozymes when only the second base of codons was established. DNA Cell Biol. 2006;25:365–375. doi: 10.1089/dna.2006.25.365. PMID:16792507 [DOI] [PubMed] [Google Scholar]
- [22].Lee N, Bessho Y, Wei K, et al. Ribozyme-catalyzed tRNA aminoacylation. Nat Struct Biol. 2000;7:28–33. doi: 10.1038/71225. PMID:10625423 [DOI] [PubMed] [Google Scholar]
- [23].Turk RM, Chumachenko NV, Yarus M. Multiple translational products from a five-nucleotide ribozyme. Proc Natl Acad Sci U S A. 2010;107:4585–4589. doi: 10.1073/pnas.0912895107. PMID:20176971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Valencia-Sanchez MI, Rodriguez-Hernandez A, Ferreira R, et al. Structural insights into the polyphyletic origins of glycyl tRNA synthetases. J Biol Chem. 2016;291:14430–14446. doi: 10.1074/jbc.M116.730382. PMID:27226617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Smith TF, Hartman H. The evolution of class II aminoacyl-tRNA synthetases and the first code. FEBS Lett. 2015;589:3499–3507. doi: 10.1016/j.febslet.2015.10.006. PMID:26472323 [DOI] [PubMed] [Google Scholar]
- [26].O'Donoghue P, Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67:550–573. doi: 10.1128/MMBR.67.4.550-573.2003. PMID:14665676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Qin X, Deng X, Chen L, et al. Crystal structure of the wild-type human GlyRS Bound with tRNA(Gly) in a productive conformation. J Mol Biol. 2016;428:3603–3614. doi: 10.1016/j.jmb.2016.05.018. PMID:27261259 [DOI] [PubMed] [Google Scholar]
- [28].Deng X, Qin X, Chen L, et al. Large conformational changes of insertion 3 in human glycyl-tRNA synthetase (hGlyRS) during catalysis. J Biol Chem. 2016;291:5740–5752. doi: 10.1074/jbc.M115.679126. PMID:26797133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fukai S, Nureki O, Sekine S, et al. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi: 10.1016/S0092-8674(00)00182-3. PMID:11114335 [DOI] [PubMed] [Google Scholar]
- [30].Salinas-Giege T, Giege R, Giege P. tRNA biology in mitochondria. Int J Mol Sci. 2015;16:4518–4559. doi: 10.3390/ijms16034518. PMID:25734984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Salinas T, Duby F, Larosa V, et al. Co-evolution of mitochondrial tRNA import and codon usage determines translational efficiency in the green alga Chlamydomonas. PLoS Genet. 2012;8:e1002946. doi: 10.1371/journal.pgen.1002946. PMID:23028354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schneider A. Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu Rev Biochem. 2011;80:1033–1053. doi: 10.1146/annurev-biochem-060109-092838. PMID:21417719 [DOI] [PubMed] [Google Scholar]
- [33].Agris PF, Narendran A, Sarachan K, et al. The importance of being modified: the role of RNA modifications in translational fidelity. Enzymes. 2017;41:1–50. doi: 10.1016/bs.enz.2017.03.005. PMID:28601219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Agris PF, Vendeix FA, Graham WD. tRNA's wobble decoding of the genome: 40 years of modification. J Mol Biol. 2007;366:1–13. doi: 10.1016/j.jmb.2006.11.046. PMID:17187822 [DOI] [PubMed] [Google Scholar]
- [35].Qin X, Hao Z, Tian Q, et al. Cocrystal structures of glycyl-tRNA synthetase in complex with tRNA suggest multiple conformational states in glycylation. J Biol Chem. 2014;289:20359–20369. doi: 10.1074/jbc.M114.557249. PMID:24898252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Delagoutte B, Moras D, Cavarelli J. tRNA aminoacylation by arginyl-tRNA synthetase: induced conformations during substrates binding. EMBO J. 2000;19:5599–5610. doi: 10.1093/emboj/19.21.5599. PMID:11060012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Agris PF, Eruysal ER, Narendran A, et al. Celebrating wobble decoding: half a century and still much is new. RNA Biol. 2017:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vare VY, Eruysal ER, Narendran A, et al. Chemical and conforma tional diversity of modified nucleosides affects tRNA structure and function. Biomolecules. 2017;7. doi: 10.3390/biom7010029. PMID:28300792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Novozhilov AS, Wolf YI, Koonin EV. Evolution of the genetic code: partial optimization of a random code for robustness to translation error in a rugged fitness landscape. Biol Direct. 2007;2:24. doi: 10.1186/1745-6150-2-24. PMID:17956616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Curtis EA, Bartel DP. Synthetic shuffling and in vitro selection reveal the rugged adaptive fitness landscape of a kinase ribozyme. RNA. 2013;19:1116–1128. doi: 10.1261/rna.037572.112. PMID:23798664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kun A, Szathmary E. Fitness landscapes of functional RNAs. Life (Basel). 2015;5:1497–1517. PMID:26308059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vendeix FA, Dziergowska A, Gustilo EM, et al. Anticodon domain modifications contribute order to tRNA for ribosome-mediated codon binding. Biochemistry. 2008;47:6117–6129. doi: 10.1021/bi702356j. PMID:18473483 [DOI] [PubMed] [Google Scholar]
- [43].Demeshkina N, Jenner L, Westhof E, et al. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–259. doi: 10.1038/nature10913. PMID:22437501 [DOI] [PubMed] [Google Scholar]
- [44].Zamudio GS, Jose MV. Phenotypic graphs and evolution unfold the standard genetic code as the optimal. Orig Life Evol Biosph. 2018;48:83–91. doi: 10.1007/s11084-017-9552-3. PMID:29082465 [DOI] [PubMed] [Google Scholar]
- [45].Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. PMID:25950237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kim Y, Benning N, Pham K, et al. Homology threading to generate RNA polymerase structures. Protein Expr Purif. 2018;147:13–16. doi: 10.1016/j.pep.2018.02.002. PMID:29444461 [DOI] [PubMed] [Google Scholar]
- [47].Yang Z, Lasker K, Schneidman-Duhovny D, et al. UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J Struct Biol. 2012;179:269–278. doi: 10.1016/j.jsb.2011.09.006. PMID:21963794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. PMID:15264254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.