

Simulated shift work uncovers potential tissue mechanisms for metabolic effects attributed to sleep loss and circadian disruption.
Abstract
Curtailed sleep promotes weight gain and loss of lean mass in humans, although the underlying molecular mechanisms are poorly understood. We investigated the genomic and physiological impact of acute sleep loss in peripheral tissues by obtaining adipose tissue and skeletal muscle after one night of sleep loss and after one full night of sleep. We find that acute sleep loss alters genome-wide DNA methylation in adipose tissue, and unbiased transcriptome-, protein-, and metabolite-level analyses also reveal highly tissue-specific changes that are partially reflected by altered metabolite levels in blood. We observe transcriptomic signatures of inflammation in both tissues following acute sleep loss, but changes involving the circadian clock are evident only in skeletal muscle, and we uncover molecular signatures suggestive of muscle breakdown that contrast with an anabolic adipose tissue signature. Our findings provide insight into how disruption of sleep and circadian rhythms may promote weight gain and sarcopenia.
INTRODUCTION
Chronic sleep loss, social jet lag, and shift work—widespread in our modern 24/7 societies—are associated with an increased risk of numerous metabolic pathologies, including obesity, metabolic syndrome, and type 2 diabetes (1–4). Even minor weekly shifts in sleep timing, or as few as five consecutive nights of short sleep, have been associated with an increased risk of weight gain in healthy humans (4, 5).
Many of the adverse effects attributed to sleep loss and circadian misalignment might arise due to tissue-specific metabolic perturbations in peripheral tissues such as skeletal muscle and adipose tissue (6–9). Recurrent sleep loss combined with moderate calorie restriction in humans increases the loss of fat-free body mass, while decreasing the proportion of weight lost as fat (10), suggesting that sleep loss can promote adverse tissue-specific catabolism and anabolism. Human cohort studies and interventional sleep restriction studies in animals also suggest that sleep loss specifically promotes loss of muscle mass (11–13), but the underlying molecular mechanisms remain elusive.
Notably, sleep restriction studies controlling for caloric intake provide evidence that sleep loss reduces the respiratory exchange ratio (8, 14), indicating a shift toward non-glucose, that is, fatty acid, oxidation. Animal studies have elegantly shown that metabolic fuel selection and overall anabolic versus catabolic homeostasis are regulated by tissue-specific rhythms driven by the core circadian clock (15). Key metabolic processes, for example, glycolysis and mitochondrial oxidative metabolism, exhibit 24-hour rhythms in tissues such as skeletal muscle (16–18). This is, to a significant extent, orchestrated through circadian regulation of key transcription factors and enzymes such as pyruvate dehydrogenase kinase 4 (Pdk4), Ldhb, and phosphofructokinase 2 (Pfk2), which belong to some of the most highly rhythmic transcripts in skeletal muscle across circadian data sets in mice (19). Correspondingly, ablation of the core clock gene Bmal1 alters metabolic fuel utilization in mice (20, 21), and circadian desynchrony in humans results in decreased resting metabolic rate (22). Furthermore, even a single night of sleep loss has been shown to induce tissue-specific transcriptional and DNA methylation (an epigenetic modification that can regulate chromatin structure and gene expression) changes to core circadian clock genes in humans (23), but the downstream tissue-specific impact on metabolic pathways remains to be determined. Moreover, it is presently unknown to what extent DNA methylation may be modulated throughout the human genome in metabolic tissues in response to acute sleep loss, and whether metabolic tissues respond in a tissue-specific manner across multiple genomic and molecular levels.
On the basis of the above observations, and as a model of shift work that often entails overnight wakefulness, we hypothesized that acute sleep loss (that is, overnight wakefulness) would induce tissue-specific alterations at the genomic and physiological levels in pathways regulating metabolic substrate utilization and anabolic versus catabolic state. Specifically, we expected acute sleep loss to increase non-glycolytic oxidation and protein breakdown in skeletal muscle (12, 13), with the former favoring hyperglycemia. Since recurrent sleep loss has also been linked to adverse weight gain (2, 10, 24), we also hypothesized that acute sleep loss would promote signatures of increased adipogenesis and that some of these tissue-specific changes would be reflected at the DNA methylation level, indicating altered “metabolic memory.” To this end, we carried out a range of molecular analyses in subcutaneous adipose tissue and skeletal muscle samples, complemented by analyses in blood, in samples obtained from healthy young men both after a night of sleep loss and after a night of full sleep.
RESULTS
Acute sleep loss results in tissue-specific DNA methylation and transcriptomic changes
To examine whether acute sleep loss induces genome-wide alterations in epigenetic modifications, we used the Infinium HumanMethylation450 BeadChip (485,764 probes) to interrogate changes in DNA methylation in adipose tissue and skeletal muscle samples obtained from 15 healthy participants in the morning fasting state, both after one night of sleep loss and after a night of normal sleep (age, 22.3 ± 0.5 years; body mass index, 22.6 ± 0.5 kg/m2; further characteristics and sleep data are presented in table S1, and experimental design is shown in Fig. 1A). We found that sleep loss resulted in 148 significant differentially methylated regions (DMRs) [false discovery rate (FDR) < 0.05] in subcutaneous adipose tissue (Fig. 1B and table S2, A and B), most of which were hypermethylated (92 DMRs) and within 5 kilo–base pairs of the transcription start site (TSS) (129 DMRs or 87%). To investigate which gene pathways were associated with altered methylation status following sleep loss, we used gene ontology (GO) analyses to identify which biological pathways were enriched for genes close to our significant DMRs (Fig. 1C). When the directionality of DNA methylation was not considered, we found that pathways associated with, for example, lipid metabolism, cell differentiation, and DNA damage response were altered (table S2C). Next, to gain a better understanding of whether these pathways were driven by increased or decreased DNA methylation for specific genes in adipose tissue in response to sleep loss, we separately investigated pathways associated with the identified hypermethylated versus hypomethylated DMRs. Notably, hypermethylated genes were found to enrich for biological pathways such as lipid response and cell differentiation, whereas hypomethylated genes were related to pathways such as DNA damage response regulation and lipid metabolism (table S2, D and E). We found that sleep loss resulted in hypermethylation for DMRs near the TSS of genes that have been observed to be in a hypermethylated state before gastric bypass surgery, such as TNXB, TRIM2, and FOXP2 (Fig. 1D and table S2A) (25). We also observed altered methylation near the TSS of genes involved in adipogenesis: CD36, AKR1CL1 (an aldose reductase), and HOXA2, with the latter being hypermethylated. Specifically, we found that HOXA2, a homeobox transcription factor, was hypermethylated near its TSS, which, through altered DNA methylation and gene expression level, has been found to distinguish adipogenesis in subcutaneous white adipose tissue from that in, for example, brown or visceral adipose tissue (26–28). Notably, several of the 56 DMRs that were hypomethylated in response to sleep loss were in chromosomal regions or near the TSS of genes known to be genomically imprinted (such as TSPAN32, GNAS, INS, and GFI1; Fig. 1D and table S2B) and, through this or other mechanisms, have been associated with obesity (29, 30). Hypomethylated DMRs were also found at the TSS of genes implicated in insulin response or type 2 diabetes (for example, INS, CPT1A) as well as lipolysis or beiging (ADORA2A) (31–33). Whereas the average fold change in methylation for the significant DMRs did not exceed 3% in response to sleep loss (average, +2.8 ± 0.0% and −2.4 ± 0.0% for hyper- and hypomethylated DMRs, respectively), the most highly hypermethylated DMR (on average +6.9%) was found for CD36 (Fig. 1D and table S2A), which is involved in fatty acid import and whose expression is dysregulated in obese and type 2 diabetic patients (34).
Fig. 1. Acute sleep loss induces changes in DNA methylation in adipose tissue in healthy humans.
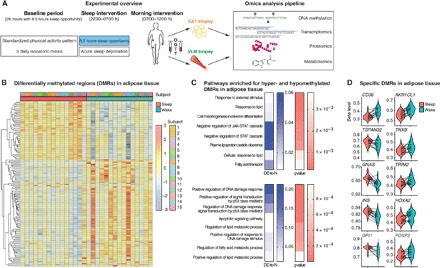
(A) Participants were investigated both after a night of sleep loss (that is, overnight wakefulness) and after a night of normal sleep, in each condition after an in-lab baseline day and night (26 hours in total, with an 8.5-hour baseline sleep opportunity) with standardized physical activity levels and isocaloric meals. Biopsies from the vastus lateralis muscle (VLM) and subcutaneous adipose tissue (SAT), as well as fasting blood sampling, preceded an oral glucose tolerance test (OGTT) and subsequent blood sampling. This was followed by a pipeline of omic analyses across tissues. (B) Differentially methylated regions (DMRs; FDR < 0.05) in adipose tissue showing DNA methylation (beta levels) after sleep and sleep loss (wake) across the 15 participants, with hierarchical clustering of DMR beta levels (z scores). (C) Significant gene ontology (GO) annotations based on hypermethylated (top) and hypomethylated DMRs (bottom) in adipose tissue in response to sleep loss, showing the ratio of differentially expressed gene-associated DMRs (DE) to the total number (N) of genes in a given pathway (“DE-to-N”) and adjusted P values (q values, FDR< 0.05). (D) Beta levels across some of the most significant DMRs in adipose tissue, in proximity to the specified genes, following sleep and sleep loss.
In contrast to adipose tissue, no significant DMRs were observed in skeletal muscle following sleep loss compared with sleep (table S2F). This finding could indicate that other epigenetic modifications—that may also respond to environmental changes (for example, at the chromatin level) regulate the transcriptional response to sleep loss in skeletal muscle or, alternatively, that DNA methylation changes occur at, for example, earlier or later time points in our intervention.
To assess genome-wide gene expression changes following acute sleep loss in humans in the morning hours, we next performed transcriptomic RNA sequencing (RNA-seq) analyses of total RNA isolated from the corresponding skeletal muscle and adipose tissue samples. We found that acute sleep loss altered expression of 117 (19 up-regulated, 98 down-regulated) mRNA transcripts in skeletal muscle, whereas 96 transcripts (59 up-regulated, 37 down-regulated) were significantly altered in subcutaneous adipose tissue (table S3, A to D). A comparison of transcripts that were significantly altered either in skeletal muscle or in subcutaneous adipose tissue revealed that many transcripts exhibited tissue-specific directionalities (that is, with regard to their fold change) in response to sleep loss compared with sleep (Fig. 2, A and B). In addition, almost no overlap was found between the two tissues for mRNA transcripts that were differentially expressed (Fig. 2C), further highlighting the tissue specificity of the response to acute sleep loss in human metabolic tissues. An untargeted analysis of all DNA methylation values versus all corresponding mRNA transcript levels confirmed that, overall, the degree of methylation was negatively correlated with the level of gene expression, a phenomenon observed in both adipose tissue (Spearman rs = −0.39) and skeletal muscle (Spearman rs=−0.41; fig. S1, A and B). Notably, however, when we next compared the changes in DNA methylation and mRNA transcript levels that were observed following acute sleep loss, neither tissue exhibited any significant correlation between DNA methylation and transcript expression levels (adipose tissue, Spearman rs = −0.01; skeletal muscle, Spearman rs = 0.00; fig. S1, C and D). Similarly, no overlap was found when comparing genes with altered mRNA expression with genes that had altered DNA methylation near the TSS in response to sleep loss in adipose tissue. Overall, this raises the possibility that these correlations in response to sleep loss may have been captured by more frequent biopsy sampling.
Fig. 2. Tissue-specific transcriptomic alterations in response to acute sleep loss in healthy humans.
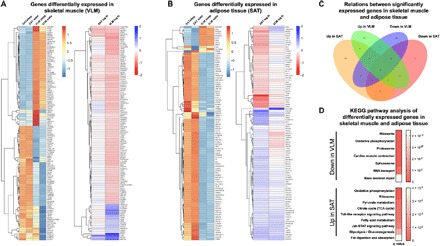
(A) Relative expression levels of differentially expressed genes (FDR < 0.05) in VLM showing levels across both VLM and SAT in both sleep and wake states (left; normalized by row, that is, all rows share the same mean and the same variance; the scale is truncated at −1 and 1). The fold changes for each tissue in response to sleep loss (that is, overnight wakefulness, wake) are also shown (right). (B) Corresponding analysis as shown in (A) for genes differentially expressed in adipose tissue in response to sleep loss. (C) Venn diagram displaying the number and overlap for significantly up- and down-regulated genes in each tissue following sleep loss. (D) GSEA using the R package GAGE against the KEGG ontology showing significant pathways (q values, with FDR < 0.05; scale shown to the right) that are down-regulated in VLM compared with pathways up-regulated in SAT in response to sleep loss (see table S4, A to D, for a complete list of all up- and down-regulated pathways in each tissue). fc, fold change.
In skeletal muscle, a gene set enrichment analysis (GSEA) using the KEGG (Kyoto Encyclopedia of Genes and Genomes) database (Fig. 2D and table S4, A and B) indicated that sleep loss down-regulated genes associated with ribosomes and oxidative phosphorylation (for example, NDUFS7 and ATP5D; table S4B). In line with down-regulation of ribosomal pathways, our analysis indicated that translational and cellular protein targeting processes—that is, energy-demanding and stress-sensitive biological processes—were down-regulated in skeletal muscle in response to sleep loss. We instead observed up-regulation of metabolic genes such as TXNIP and NNMT in response to sleep loss (Fig. 2A and table S3A); of these, TXNIP has consistently found to be up-regulated in prediabetic to diabetic patients and to be inversely correlated with glycolysis. Accordingly, its down-regulation in skeletal muscle has been positively correlated with insulin sensitivity during clamp measurements in nondiabetic patients (35). Further suggesting that sleep loss may promote catabolic stress, several immune-, injury-, and stress-related genes (such as NNMT, CXCR2, LRRK2, and FCGR3B), as well as several inflammation-related pathways, were up-regulated in skeletal muscle following acute sleep loss (tables S3A and S4A). LRRK2 is the most commonly mutated gene contributing to Parkinson’s disease, a gene known to be expressed in muscle and immune cells and with a putative role in autophagy (36).
A GSEA of RNA-seq data from adipose tissue revealed that sleep loss up-regulated KEGG pathways such as oxidative phosphorylation and ribosome pathways, that is, a direction opposite to that observed for skeletal muscle (Fig. 2D). Pathways related to glycolysis and Toll-like receptor (TLR) signaling were also up-regulated in adipose tissue following sleep loss (Fig. 2D and table S4C). Up-regulated TLR pathway components included CD14 and TLR4 (Fig. 2B and table S3C), both of which also possibly modulate adipose tissue insulin sensitivity, possibly through their role in response to bacterial lipopolysaccharide by adipocytes or present macrophages (37). Along with up-regulation of additional genes involved in inflammation (for example, IL1RAP) and protective cellular responses (such as TP53IP and GPX1), this suggests that increased inflammation occurs across tissues following sleep loss in humans. Furthermore, we observed a down-regulation of spliceosome and RNA transport pathways in GSEA-derived KEGG pathway analysis in adipose tissue, and BCL6, a gene that is markedly suppressed by insulin (35), was also down-regulated in response to sleep loss (table S3D). Compared with the observed up-regulation of TXNIP mRNA in skeletal muscle, this could suggest tissue-specific alterations of insulin signaling in these two tissues after sleep loss.
To determine what transcription factor pathways might be activated following sleep loss in each tissue, we also carried out chromatin immunoprecipitation enrichment analysis against the ChEA database, based on our RNA-seq data of genes that were up-regulated in either skeletal muscle or adipose tissue in response to sleep loss. For skeletal muscle, this analysis revealed enrichment of targets of the transcription factors PPARG and LXR (such as CPT1a, NNMT, PFKB3, PDK4, and TXNIP), which regulate, for example, fatty acid uptake in skeletal muscle (table S4E) (38). In adipose tissue, we found increased enrichment of targets—for example, THBD, GLYCTK, and GPCPD1—of transcription factors that promote adipogenesis or adipose tissue inflammation, such as CEBPD, FOXA1, and the nuclear factor κB subunit p65 (RELA) (table S4F).
Acute sleep loss induces tissue-specific changes within substrate-utilizing and anabolic-versus-catabolic pathways
Given that our transcriptomic analyses revealed changes in pathways regulating metabolic state and fuel-determining pathways, we next carried out label-free mass spectrometry to quantify the tissue-specific impact on relative protein concentrations following our intervention of sleep loss versus normal sleep. We detected 1264 proteins in the skeletal muscle samples, and of these, 23 were down-regulated, whereas 9 were up-regulated (Table 1, A and B). Several of the down-regulated proteins were involved in or downstream of glycolysis, such as phosphofructokinase-1 (PFK1), phosphoglycerate kinase 1 (PGK1), and pyruvate kinase (Table 1B). A subsequent KEGG pathway analysis of pathways that were up- and down-regulated in response to sleep loss substantiated that glycolysis (FDR-corrected, P < 10−5) was among the most significantly down-regulated pathways in skeletal muscle (Fig. 3A and table S5, A and B); this was further supported by independent validation of down-regulated PFK1 and other glycolytic targets [Western blot for PFK1: P = 0.009 and quantitative polymerase chain reaction (qPCR) analyses; Fig. 3, B and C].
Table 1. Proteins with significantly changed abundance in skeletal muscle and adipose tissue in response to acute sleep loss in humans.
Proteins (A) up-regulated and (B) down-regulated in skeletal muscle (vastus lateralis muscle) and up-regulated in (C) subcutaneous adipose tissue in response to sleep loss compared with normal sleep. No proteins were found to be down-regulated in subcutaneous adipose tissue. Protein IDs shown are limited to 3. n = 15 pairs for each tissue; two-sided t tests.
| Protein IDs | Protein names (gene name in parentheses) |
Log2 ratio (sleep loss/sleep) |
SEM | P |
| A. Up-regulated proteins in skeletal muscle | ||||
| O14558 | Heat shock protein beta-6 (HSPB6) | 0.33 | 0.43 | 0.012 |
| P07195 | l-Lactate dehydrogenase B chain; l-lactate dehydrogenase (LDHB) | 0.37 | 0.51 | 0.017 |
| P08238 | Heat shock protein HSP 90-beta (HSP90AB1) | 0.12 | 0.16 | 0.018 |
| P35609; P35609-2 | Alpha-actinin-2 (ACTN2) | 0.09 | 0.13 | 0.033 |
| Q2TBA0; Q2TBA0-2 | Kelch-like protein 40 (KLHL40) | 0.38 | 0.55 | 0.035 |
| Q9GZV1 | Ankyrin repeat domain-containing protein 2 (ANKRD2) | 0.44 | 0.72 | 0.038 |
| Q5XKP0 | Protein QIL1 (QIL1) | 0.22 | 0.26 | 0.040 |
| C9JFR7; P99999 | Cytochrome c (CYCS) | 0.21 | 0.32 | 0.042 |
| O14949 | Cytochrome b-c1 complex subunit 8 (UQCRQ) | 0.23 | 0.37 | 0.049 |
| B. Down-regulated proteins in skeletal muscle | ||||
| O00757 | Fructose-1.6-bisphosphatase isozyme 2 (FBP2) | −0.27 | 0.28 | 0.003 |
| Q9NR12-6 | PDZ and LIM domain protein 7 (PDLIM7) | −0.31 | 0.35 | 0.005 |
| P00558; P00558-2 | Phosphoglycerate kinase 1 (PGK1) | −0.22 | 0.22 | 0.006 |
| Q9UKS6 | Protein kinase C and casein kinase substrate in neurons protein 3 (PACSIN3) | −0.18 | 0.19 | 0.011 |
| P02585 | Troponin C, skeletal muscle (TNNC2) | −0.27 | 0.32 | 0.012 |
| P14543-2; P14543 | Nidogen-1 (NID1) | −0.31 | 0.35 | 0.013 |
| P14618-2 | Pyruvate kinase (PKM) | −0.21 | 0.26 | 0.015 |
| Q08043 | Alpha-actinin-3 (ACTN3) | −0.40 | 0.47 | 0.017 |
| Q5T7C4; Q5T7C6; P09429 | High mobility group protein B1 (HMGB1) | −0.19 | 0.23 | 0.019 |
| Q8N142; Q8N142-2 | Adenylosuccinate synthetase isozyme 1 (ADSSL1) | −0.14 | 0.20 | 0.022 |
| P61586; Q5JR08; P08134 | Transforming protein RhoA; Rho-related guanosine 5′-triphosphate–binding protein RhoC (RHOA; RHOC) |
−0.17 | 0.22 | 0.024 |
| P55786; P55786-2 | Puromycin-sensitive aminopeptidase (NPEPPS) | −0.08 | 0.11 | 0.026 |
| Q14324 | Myosin-binding protein C, fast-type (MYBPC2) | −0.37 | 0.56 | 0.026 |
| P04075 | Fructose-bisphosphate aldolase A; Fructose-bisphosphate aldolase (ALDOA) | −0.14 | 0.20 | 0.027 |
| P50995-2; P50995 | Annexin A11 (ANXA11) | −0.33 | 0.28 | 0.028 |
| P62942; Q5W0X3 | Peptidyl-prolyl cis-trans isomerase FKBP1A; peptidyl-prolyl cis-trans isomerase (FKBP1A; FKBP12-Exip2) |
−0.12 | 0.17 | 0.030 |
| P17612; P17612-2; P22694-4 | cAMP-dependent protein kinase catalytic subunit alpha; cAMP-dependent protein kinase catalytic subunit beta (PRKACA; PRKACB; KIN27) |
−0.13 | 0.19 | 0.032 |
| P00338; P00338-3; P00338-4 | l-Lactate dehydrogenase A chain (LDHA) | −0.21 | 0.31 | 0.036 |
| P07108; P07108-3; P07108-2 | Acyl-CoA–binding protein (DBI) | −0.17 | 0.26 | 0.037 |
| P12882 | Myosin-1 (MYH1) | −0.93 | 1.38 | 0.038 |
| P08237; P08237-3; P08237-2 | 6-Phosphofructokinase, muscle type (PFKM) | −0.16 | 0.26 | 0.040 |
| P05976 | Myosin light chain 1/3, skeletal muscle isoform (MYL1) | −0.16 | 0.24 | 0.041 |
| Q0VAK6; Q0VAK6-2 | Leiomodin-3 (LMOD3) | −0.23 | 0.31 | 0.043 |
| C. Up-regulated proteins in subcutaneous adipose tissue | ||||
| P09211 | Glutathione S-transferase (GSTP) | 0.23 | 0.06 | 0.004 |
| P02788 | Lactotransferrin (LTF) | 0.95 | 0.26 | 0.004 |
| P00558; P00558-2 | Phosphoglycerate kinase 1 (PGK1) | 0.23 | 0.08 | 0.011 |
Fig. 3. Acute sleep loss down-regulates protein levels in the glycolysis pathway in skeletal muscle of healthy young men.
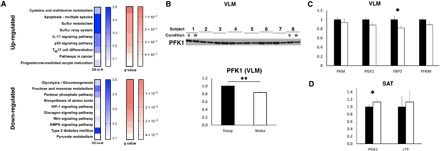
(A) KEGG pathway analysis of significantly altered VLM proteins (via mass spectrometry) in the morning following sleep loss compared with after a night of normal sleep (n = 15 pairs; see also Table 1 and table S5). Shown as ratio of differentially expressed proteins in relation to total number of proteins in pathway (DE-to-N), and as adjusted P values (q values; FDR < 0.05) for pathways based on up-regulated (top) and down-regulated (bottom) proteins. (B) Immunoblot analysis of PFK1 in VLM (P = 0.009), normalized to loading control (loading control shown in fig. S4A; showing 8 representative pairs out of a total of 13 analyzed pairs); quantified in the bottom for sleep loss [wake (w)] compared with normal sleep (s). qPCR analyses of significant proteomic hits in response to sleep loss in (C) VLM and in (D) SAT (P = 0.027 for FBP2 in VLM; P = 0.031 for PGK1 in adipose tissue for hypothesized contrasts between sleep versus sleep loss). Solid black bars represent values after sleep (set to 1); white bars indicate values obtained after sleep loss (n = 15 pairs for both tissues). FBP2, fructose-bisphosphatase 2; LTF, lactotransferrin; PFKM, 6-phosphofructokinase, muscle type; PKM, pyruvate kinase muscle isozyme. *P < 0.05 and **P < 0.01; two-sided t tests. TH17, T helper 17; IL-17, interleukin 17.
Our mass spectrometry data instead indicated that levels of proteins involved in mitochondrial energy metabolism were up-regulated in skeletal muscle after our intervention, further suggesting a shift in metabolic fuel utilization toward non-glycolytic oxidation after a night of sleep loss (Table 1A). Given that components of oxidative phosphorylation exhibit circadian rhythms (17), this finding may represent a circadian phase misalignment compared with the aforementioned transcriptomic changes. However, no differences were found in Western blot analyses of the major mitochondrial complexes, suggesting a subtle impact on overall mitochondrial function after acute sleep loss (fig. S2, A and B).
To quantify whether the shift in glycolytic protein levels correlated with altered systemic insulin sensitivity, we assessed fasting and postprandial systemic insulin sensitivity in our participants in samples obtained on the same day as the biopsies and found increased morning fasting insulin resistance after sleep loss (P = 0.006; fig. S2, C and D). A subsequent OGTT revealed significantly higher postprandial levels of glucose, but not of insulin, following sleep loss, and postprandial insulin sensitivity was reduced by ~15% (P = 0.033; fig. S2, D and E). Together with unaltered fasting and postprandial insulin levels, this suggests that the decreased postprandial insulin sensitivity after sleep loss was primarily driven by altered glucose handling in peripheral tissues, possibly primarily in skeletal muscle.
Our mass spectrometry analysis also revealed decreased levels of several structural proteins in skeletal muscle, such as myosin-1 (encoding myosin light chain IIx) and troponin C, following sleep loss (Table 1B). This provides molecular support for earlier indirect evidence that sleep loss enhances skeletal muscle catabolism, especially of fast (type II) fibers (that express myosin light chain IIx) (9, 10, 12). Catabolic stress can up-regulate levels of heat shock proteins (HSPs), in part to protect against muscle breakdown (39, 40). Our mass spectrometry analysis of skeletal muscle demonstrated increased levels of HSP beta-6 and HSP 90-beta after sleep loss (Table 1A), indicating that sleep loss acts as a cellular catabolic stressor in skeletal muscle. Levels of HSF1 were instead down-regulated at the mRNA level in our skeletal muscle transcriptomic data (table S3B), consistent with negative feedback due to the up-regulation of HSP protein levels. Mechanistically, HSP 90-beta stabilizes, for example, glucocorticoid receptors (41), and glucocorticoids have been implicated in skeletal muscle atrophy in, for example, starvation and diabetes, particularly affecting fast type II fibers (42). Suggesting that glucocorticoids may be involved in acute catabolic effects on skeletal muscle following sleep loss, we observed significantly elevated cortisol levels during our morning blood sampling interval [analysis of variance (ANOVA) wake and time effects: P = 0.029 and P = 0.003, respectively; fig. S2F].
To investigate whether adipose tissue also exhibits signs of altered metabolic fuel utilization in response to sleep loss, we also carried out mass spectrometry–based proteomics on subcutaneous adipose tissue obtained at the same time as the skeletal muscle biopsies. In an analysis of the absolute (“static”) levels of proteins compared with mRNA expression levels (based on our RNA-seq data), we found that mRNA transcript levels overall correlated positively with protein levels in both adipose tissue (Spearman rs = 0.29) and skeletal muscle (Spearman rs = 0.55; fig. S3, A and B). However, no correlations were observed when sleep loss–induced changes in mRNA transcript levels were compared with the corresponding changes in protein levels (adipose tissue, Spearman rs = 0.09; skeletal muscle, Spearman rs = 0.08; fig. S3, C and D), which could be due to a lack of temporally separated biopsies required to identify the delay between changes in gene versus protein expression following sleep loss.
Of the 1358 identified proteins or protein groups identified in adipose tissue, we found 3 significantly up-regulated proteins but no down-regulated proteins after sleep loss compared with sleep (Table 1C). Specifically, levels of PGK1 were significantly increased at the protein and mRNA level (Fig. 3D and Table 1C) and, thus, consistent with the observed up-regulation (KEGG-based) of the glycolysis pathway in our concurrent transcriptomic data set of adipose tissue (Fig. 2D). This provides further evidence that at least metabolic pathways are altered in a directionality opposite to those observed in skeletal muscle, in response to sleep loss. Enhanced glycolysis in adipose tissue could possibly be indicative of increased triglyceride synthesis, a process for which glycolysis can enable greater availability of glycerol as the triglyceride backbone (43).
Our proteomics data further indicated that protein levels of glutathione S-transferase (GSTP) and lactotransferrin increased in subcutaneous adipose tissue following sleep loss. These proteins have been linked not only to adipogenesis and adipocyte differentiation (44, 45) but also to protection from oxidative stress in adipose tissue (46). Altered expression of GSTP in adipose tissue has been associated specifically with insulin resistance and obesity, also in humans (47). Collectively, our findings indicate that subcutaneous adipose tissue exhibits a state promoting increased glucose utilization and triglyceride synthesis in the morning following sleep loss, whereas skeletal muscle concurrently decreases glucose utilization and promotes muscle protein breakdown, possibly to increase amino acid efflux to the liver for gluconeogenesis and ketone body synthesis. These changes further support earlier findings that forgoing sleep favors the retention of adipose tissue over skeletal muscle mass (10).
Evidence for muscle-specific alterations in the core circadian clock after acute sleep loss
The core molecular clock—specifically through the component BMAL1—affects metabolic fuel utilization in the liver and skeletal muscle in mice (19–21), and several glycolytic genes exhibit circadian expression patterns (17, 19). As we have previously demonstrated, tissue-specific changes in DNA methylation and transcription that are indicative of circadian misalignment occur following one night of sleep loss in humans (23). Herein, we found that protein levels of the core clock component BMAL1 were significantly higher in skeletal muscle (P = 0.017) but were unaltered in adipose tissue (P = 0.51) in response to sleep loss (Fig. 4, A to C). Furthermore, for several clock genes, fold changes at the descriptive transcriptomic level were opposite in skeletal muscle versus adipose tissue (these, however, did not survive FDR correction), providing further preliminary support for tissue-specific circadian misalignment after sleep loss in humans (Fig. 4D).
Fig. 4. Acute sleep loss induces tissue-specific changes in clock genes and downstream pathways in healthy young men.
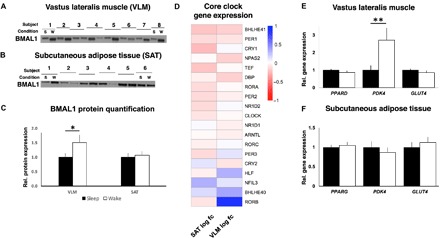
Representative blots for protein abundance of BMAL1 in (A) skeletal muscle (VLM; P = 0.017; showing 8 representative pairs out of a total of 13 analyzed pairs) and in (B) SAT (P = 0.51; 6 representative pairs out of 11 analyzed pairs shown), (C) with quantification, after a night of sleep (s) and a night of sleep loss (wake or w). Western blots were normalized to loading control (see fig. S4, B and C; expression shown relative to controls that were set to 1). (D) Transcriptomic changes in core circadian clock genes, with log2 fold change for each of the investigated tissues (VLM and SAT, n = 15 pairs for each tissue), after sleep loss (wake) compared with after normal sleep (all FDR > 0.05). (E and F) Relative gene expression of targeted genes based on qPCR (PDK4: P = 0.007; all other P > 0.10, n = 15 pairs for each tissue). BMAL1, brain and muscle Arnt-like protein-1; GLUT4, glucose transporter 4; PDK4, pyruvate dehydrogenase kinase isozyme 4; PPARD/PPARG, peroxisome proliferator–activated receptor delta (PPARD)/gamma (PPARG); s, sleep; w, wake (sleep loss). *P < 0.05 and **P < 0.05; two-sided t tests.
Notably, in our skeletal muscle RNA-seq data set, the clock gene–regulated (19, 48) and substrate-determining gene PDK4 was the most up-regulated transcript following sleep loss (table S3A; also confirmed by qPCR, Fig. 4E). Mechanistically, PDK4 directs glucose utilization away from glycolysis toward beta oxidation: Fasting glucose is thus reduced in mice lacking the Pdk4 gene (49), whereas PDK4 expression is increased in states of insulin resistance and in mouse models of type 2 diabetes (50, 51). Our RNA-seq in skeletal muscle also revealed up-regulation of PFKFB3, which is also involved in regulation of glucose metabolism in skeletal muscle. Across time course transcriptomic data sets in murine skeletal muscle, PDK4 and PFKFB3 have been found to be among the top five most highly regulated transcripts by the circadian clock and are also among the only transcripts associated with metabolism that show a high circadian amplitude in skeletal muscle (19). Altogether, these findings hint at the potential involvement of the molecular circadian clock in dysregulation of glucose metabolism in skeletal muscle following acute sleep loss.
In contrast to PDK4, qPCR-assessed levels of PPARD, which can regulate PDK4 expression, and the glucose transporter GLUT4 (also assessed at the protein level) were unaltered in skeletal muscle after sleep loss (Fig. 4E and fig. S3E), suggesting that other components that are regulated by the circadian clock in animal models and human myotubes (18, 19, 48) are at least not as acutely affected in skeletal muscle by sleep loss in humans. Corresponding analyses in subcutaneous adipose tissue, that is, overall RNA-seq, as well as qPCR of PDK4, PPARG (the corresponding major isoform of PPARD in adipose tissue) (Fig. 4F), and GLUT4 (qPCR and protein level), demonstrated no similar changes in the morning after sleep loss (Fig. 4F and fig. S3, F and G).
Metabolomic changes indicate altered metabolic substrate utilization following acute sleep loss
To assess whether the transcriptomic and proteomic changes due to acute sleep loss are reflected in altered metabolic flux, we also carried out metabolomic analyses by gas chromatography coupled to mass spectrometry (GCMS) in the previously analyzed skeletal muscle, subcutaneous adipose tissue, and venous blood samples, to also allow us to assess how tissue-specific changes were reflected by systemic changes.
Fasting compared with post-OGTT blood sampling demonstrated that most serum metabolites changed significantly in response to an OGTT (ANOVA time effect) following both sleep loss and sleep (table S6A). Further suggesting that sleep loss alters amino acid metabolism, possibly to promote skeletal muscle protein breakdown, we observed decreased fasting serum levels of several (some essential) amino acids such as arginine, asparagine, and threonine (table S6B) as well as lower levels of glycine in skeletal muscle (table S6C). Levels of glutamic acid and aspartic acid were instead significantly increased in subcutaneous adipose tissue (table S6D). In both the fasting and post-OGTT state, serum levels of ornithine and urea were decreased, coupled with trends for lower levels of muscle urea and post-OGTT serum uric acid (table S6, B and C). Together with altered levels of structural muscle proteins, these changes support the notion that sleep loss promotes skeletal muscle breakdown by increasing amino acid flux.
In adipose tissue, we observed increased levels of both malic acid and glyceric acid–3-phosphate in response to sleep loss compared with sleep (table S6D), possibly indicative of increased fatty acid synthesis via glycerol synthesis (52). Levels of the ketone 3-hydroxybutyric acid were significantly increased in subcutaneous adipose following sleep loss, supported by a similar directionality for ketone bodies in serum (rs = 0.703, P = 0.007) and a decrease in levels of the ketogenic amino acid threonine in skeletal muscle (P = 0.051) and fasting blood serum (table S6, B and C). We also observed lower fasting serum levels of 1,5-anhydro-d-glucitol (table S6B), a marker that decreases in response to hyperglycemia in, for example, diabetes, altogether further arguing for impaired glucose handling and altered metabolic substrate utilization after sleep loss.
Through hierarchical clustering, we found tissue-specific changes in shared metabolite levels in skeletal muscle and adipose tissue to be reflected by overlapping changes in blood serum in the fasting state in response to sleep loss (Fig. 5A). Shared overall changes in metabolite levels were significant at the correlational level between skeletal muscle and serum (r = 0.606, P < 0.001; fig. S3H), and a metabolite enrichment analysis combining data from serum and skeletal muscle indicated that changes in protein biosynthesis and in the urea cycle occurred following sleep loss (Fig. 5B). In contrast, correlations in GCMS-based metabolites following sleep loss in subcutaneous adipose tissue appeared to be reflected to a much lesser extent by serum (rs = 0.023, P = 0.91; fig. S3I). A subsequent pathway analysis in adipose tissue revealed significant changes in the malate-aspartate shuttle pathway in response to sleep loss (Fig. 5C), a pathway that can be used to produce NADH (reduced form of nicotinamide adenine dinucleotide) for glyceroneogenesis and lipid synthesis (43).
Fig. 5. Hierarchical clustering analyses reveal close relationship between changes in serum and skeletal muscle metabolite levels in response to acute sleep loss.
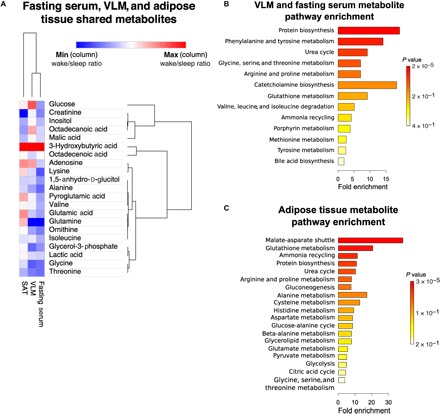
(A) Shared metabolites across subcutaneous adipose tissue, skeletal muscle, and fasting serum. Rows indicate metabolites—based on gas chromatography mass spectrometry (GCMS) metabolomic data—and have been ranked according to relatedness in terms of (i) changes across tissues (column-wise ranking) and (ii) fold changes across metabolites, following sleep loss (wake) (using log2 values for the sleep loss/sleep ratio). The degree of changes in metabolites following sleep loss is color-coded, with red indicating increased levels and blue indicating decreased levels (sleep loss/sleep). Metabolite set enrichment analysis for (B) skeletal muscle and serum metabolites and for (C) subcutaneous adipose tissue metabolites. n = 13 for each tissue.
DISCUSSION
Shift work often entails overnight work, forgoing sleep and concomitantly incurring acute circadian misalignment, both of which are associated with a range of metabolic disruptions. Sleep loss can promote both catabolism (9, 12) and anabolism—for example, via increased risk of weight gain (2, 5)—yet few studies have focused on the underlying tissue-specific molecular responses to acute sleep loss. Here, by parallel sampling of skeletal muscle and adipose tissue, we provide insight into the tissue-specific mechanisms by which acute sleep loss affects key metabolic tissues in humans, demonstrating critical differences in how these tissues respond at a number of molecular levels to acute sleep loss. Our results indicate that sleep loss is associated with down-regulation of the glycolytic pathway in skeletal muscle, whereas this pathway instead is up-regulated in subcutaneous adipose tissue. Our analyses further suggest that these changes may be due to acute tissue-specific circadian misalignment and provide evidence that acute sleep loss may reprogram DNA methylation in adipose tissue to promote increased adiposity. Our observations indicate that levels of structural proteins in skeletal muscle decrease in response to sleep loss, contrasting with increased levels of proteins linked to adipogenesis in adipose tissue. These observations are thus the first to offer an explanation at the tissue level for two seemingly contrasting clinical phenotypes seen following experimental sleep loss in humans: gain of fat mass occurring concomitantly with loss of lean mass (10).
Our findings support the notion that curtailed sleep can promote a catabolic state in skeletal muscle. Recent studies have demonstrated that rats exposed to extended rapid eye movement sleep deprivation exhibit atrophy in glycolytic and mixed but not in oxidative muscles (13, 53). Notably, a similar down-regulation at the genetic transcript levels for fast muscle fibers is observed in isolated human myotubes when the circadian clock is genetically disrupted (18), possibly linking our alterations in structural muscle protein levels to our observed muscle-specific changes in BMAL1 protein levels. Together with increased levels of adipogenesis-promoting proteins and a DNA methylation profile that shares features of the obese state, these observations provide molecular-level observations for the altered body composition that was previously first observed by Nedeltcheva et al. (10) via whole-body dual-energy x-ray absorptiometry, in subjects exposed to several days of partial sleep loss compared with normal sleep. Metabolomic studies have also indicated that sleep loss promotes a catabolic state in blood and urine (9, 54), and two recent cohort studies of middle-age and older community-dwelling adults have indeed found insufficient sleep to be associated with lower skeletal muscle mass (11, 12). Increased catabolism in response to acute sleep loss may be driven by hormonal disruptions that regulate the anabolic versus catabolic state in skeletal muscle and adipose tissue. As found herein, acute sleep loss can increase levels of the catabolic hormone cortisol. Concomitantly, sleep loss can reduce testosterone levels (55) and abolish nocturnal growth hormone release, which normally occurs during slow-wave sleep (56).
In response to one night of sleep loss, we observed DNA methylation changes for genes that have previously been demonstrated to be differentially methylated in adipose tissue of obese and type 2 diabetes patients, including several imprinted genes. Recent evidence suggests that differences in imprinted genes can distinguish obese from nonobese subjects (57), and hence, recurrent disruption of sleep and circadian rhythms may promote obesity development by reprogramming DNA methylation or other environmentally plastic epigenomic modifications in adipose tissue to favor adipogenesis and lipogenesis. The modulation and duration of these changes following additional stressful or protective stimuli such as shifts in diet and physical exercise remain to be determined.
It should furthermore be noted that the present study was a short-term acute intervention that was restricted to young male Caucasians, with a limited number of analytical time points. Hence, it is presently unknown whether the observed tissue-specific changes in response to acute sleep loss also extend to, for example, different age groups, females, or other ethnicities. It is also not known whether the observed short-term effects might differ from the effects of chronic sleep restriction.
In summary, our results indicate that acute sleep loss results in a tissue-specific switch in metabolic fuel utilization, which may be associated with changes in the core circadian clock due to acute circadian misalignment. Furthermore, we find that sleep loss induces a molecular catabolic signature in skeletal muscle, mirrored by changes in blood, and that this contrasts with an adiposity-promoting molecular and DNA methylation signature in adipose tissue. These tissue-specific findings thus provide novel insight into why chronic sleep loss and circadian misalignment may promote adverse weight gain in humans.
MATERIALS AND METHODS
Experimental design
This was a randomized, two-session, within-subject crossover design study in which participants took part in two experimental conditions—acute sleep loss (that is, overnight wakefulness) versus sleep. Written informed consent was obtained from a total of 17 male Caucasian participants who were deemed eligible and, thus, were included in the study; out of these, 16 partook in both sessions. Fifteen participants successfully adhered to the sleep protocol of the study (one subject was unable to sleep during the sleep session). All participants received financial reimbursement for participating in the study.
Participants were screened by a medical doctor (J.C.) who assessed questionnaires about sleep and general health and recorded anthropomorphic data. Extreme morningness or eveningness was excluded with the morningness-eveningness questionnaire (see table S1), and only participants with normal self-reported sleeping habits (7 to 9 hours of sleep per night) and normal sleep quality (as assessed by the Pittsburgh Sleep Quality Index; score ≤5) were included in the study. All participants were of self-reported good health and were free from chronic medical conditions and chronic medications as assessed by a medical interview by J.C. No participant drank more than 5 U of alcohol on average per week, and all were nonsmokers. Participants were furthermore screened for normal blood cell counts and fasting glucose levels.
Before final participation, the participants underwent a separate electroencephalography (EEG)–monitored adaptation night, whereby they were habituated to the sleep environment. Participants also completed sleep, food, and activity diaries as part of their screening, as well as during the week before each study session. There was no significant difference in the average nighttime, self-reported sleep duration in the week leading up to each intervention (8 hours 3 min ± 10 min for the wake condition versus 8 hours 7 min ± 10 min for the sleep condition; P = 0.71).
Study design and procedure
Two evenings before each session’s final experimental morning during which biopsies were collected, participants came to the laboratory (day 0), where they remained under constant supervision until the end of the experimental session (total time in the laboratory for each session, ~42 hours).
At 1930 h on the evening of day 0, participants were provided with a dinner that provided a third of their total daily energy requirement (the Harris-Benedict equation was factored 1.2 for light physical activity to calculate each individual’s daily energy requirements). All served meals were of low sugar and fat content. Furthermore, while meals varied between breakfast, lunch, and dinner, meals were isocaloric and identical for a given recurring time point (that is, breakfast, lunch, and dinner), and meals were kept identical between sessions (that is, sleep loss and normal sleep).
During the first (baseline) night in each condition, participants had an 8.5-hour-long sleep opportunity (2230 to 0700 h); baseline sleep characteristics were typical for laboratory conditions and were not significantly different between the two conditions (P = 0.21 for total sleep duration). The following day, the participants were provided three isocaloric meals—each had to be consumed within 20 min after being served—and participants were taken on two standardized and supervised 15-min long walks.
Throughout each session, participants were restricted to their rooms but were free to engage in sedentary-level activities and were instructed not to exert themselves physically in any way. Participants were furthermore blinded to the experimental condition (sleep or sleep loss) during the second night, until 90 min in advance of the intervention onset at 2230 h, and the nighttime intervention lasted until 0700 h the following morning. Under the sleep loss (overnight wakefulness) condition, room lights were kept on (250 to 300 lux at eye level) and participants were confined to their beds from 2230 to 0700 h to approximate sleep-like activity levels, while continuing to be constantly monitored to ensure complete wakefulness. Every 2 hours, participants were provided with water (1.5 dl; with the possibility to obtain more if requested) to avoid dehydration throughout the nocturnal wakefulness; however, no food intake was allowed. Room lights were kept off during the nighttime intervention in the sleep condition, and EEG, electrooculography, and electromyography were used to record sleep with Embla A10 recorders (Flaga hf, Reykjavik). For sleep stage assessment, standard criteria were used by a scorer blinded to the study hypothesis (58). Before biopsy collection in the morning after sleep or sleep loss, participants completed a short computer task to assess cognitive function, the results of which have been previously published (59).
Participants continued to be closely monitored and were instructed to remain sedentary throughout the intervention morning following sleep or sleep loss and were not allowed to support their body weight using their legs, but rather to primarily recline with leg support. Biopsies were collected after an initial blood sample in the fasting state. The subcutaneous adipose tissue biopsy collection preceded the muscle biopsy collection; the timing of each type of biopsy collection was kept the same for each participant for both sessions (±15 min). Subcutaneous adipose tissue and skeletal muscle were collected from the left or right subcutaneous fat and vastus lateralis muscle, respectively, in a randomized counterbalanced order for the first session. During the second session, the biopsies were obtained from the contralateral side of the abdomen and leg. For the subcutaneous adipose tissue biopsy, the skin of the left or right umbilical region was first anesthetized using lidocaine (10 mg/ml) without epinephrine. A millimeter-large incision was made through the skin, after which a large-caliber needle (14 gauge) was used to collect subcutaneous fat [as described in (23)]. For the skeletal muscle biopsy, the skin and fascia overlying the left or right vastus lateralis were anesthetized, and a conchotome was used to obtain skeletal muscle biopsies (23). Collected subcutaneous adipose tissue and skeletal muscle samples were quickly washed in phosphate-buffered saline to remove visible blood and connective tissue before snap-freezing the samples in liquid nitrogen. All biopsy specimens were thereafter stored at −80°C.
After biopsy sampling, participants underwent an OGTT before leaving the facility. Before the OGTT, fasting blood samples were obtained around 1030 h using an indwelling venous catheter. Participants then consumed a glucose solution (75 g of glucose in 300 ml of water) within 2 min. To ensure equal initial physiological distribution of the consumed OGTT solution, participants were instructed to lie on their right side for the next 5 min and were not allowed to walk around until after the final post-OGTT blood sample had been collected. Additional blood samples were obtained 30, 60, 90, and 120 min after having consumed the solution.
To ensure full recovery following each session, at least 4 weeks elapsed between the two different sessions (sleep loss versus normal sleep) that the 15 included subjects participated in. The study was approved by the Ethical Review Board in Uppsala (EPN 2012/477/1) and was conducted in accordance with the Helsinki Declaration.
Genomic and molecular analyses
More detailed descriptions of genomic and molecular analyses are provided in the Supplementary Materials. For all biochemical runs and tissue extractions, as well as for all subsequent runs (for example, for DNA/RNA extractions, mass spectrometry, and GCMS), samples from both conditions (sleep loss and sleep) were always extracted in the same batch for a given individual to avoid interbatch effects.
Genome-wide DNA methylation
For genome-wide DNA methylation analysis, the samples were analyzed via Uppsala SciLifeLab core facility services (Uppsala). The HumanMethylation450 BeadChip (Illumina; examines 485,764 CpG dinucleotides) was run, and changes in DNA methylation levels were assessed using differences in mean beta values (range, 0 to 1 corresponding to 0 to 100% methylation) for the sleep loss (overnight wakefulness) versus sleep condition (wake-sleep). P values are presented as FDR-corrected (Benjamini-Hochberg–adjusted). All processing and statistical analyses of the DNA methylation 450K BeadChip data were carried out using the statistical software R (version 3.1.1; www.r-project.org), with software and statistical packages detailed in the Supplementary Materials.
qPCR and RNA-sequencing
Applied Biosystem’s 7500 Fast Real-Time PCR System (Applied Biosystems) was used to analyze gene expression by qPCR of the collected skeletal muscle and adipose tissue biopsies, and RNA-seq analysis was carried out for genome-wide analysis of transcription. The TruSeq stranded total RNA library preparation kit with Ribo-Zero Gold treatment (Illumina) was used to prepare libraries for sequencing. FastQ files generated from RNA-seq were run through the RNA-seq pipeline [National Genomics Infrastructure (NGI) Sweden; https://github.com/ewels/NGI-RNAseq] for basic processing of RNA-seq data, as detailed in the Supplementary Materials. Data from adipose and muscle were analyzed separately. Following initial read count analysis with featureCounts, the R packages edgeR and GAGE (Generally Applicable Gene-set Enrichment) were used for differential gene expression and GSEA, respectively (see Supplementary Materials).
Mass spectrometry–based protein quantification
For label-free quantification of relative protein expression in skeletal muscle and adipose tissue samples, protein identification was performed following mass spectrometry analysis against a FASTA database, which contained proteins from Homo sapiens extracted from the UniProtKB/Swiss-Prot database (December 2014). A decoy search database including common contaminants and a reverse database were used to estimate the identification FDR. The search parameters included the following: maximum 10 parts per million and 0.6-Da error tolerances for the survey scan and tandem mass spectrometry analysis, respectively; enzyme specificity was trypsin; maximum one missed cleavage site allowed; cysteine carbamidomethylation was set as static modification; and oxidation (M) was set as variable modification. A total label-free intensity analysis was performed for each individual sample, followed by bioinformatic analyses of the generated results. Proteins that were present in at least 12 of the 15 subject pairs for each tissue type were included in the subsequent analyses of group-level expression level differences. For each protein, a log2 ratio between the sample obtained after sleep loss and the sample obtained after normal sleep was calculated for the individual participants, followed by one-sample Student’s t test.
Western blot
Aliquots of the skeletal muscle biopsies (20 to 30 mg) were freeze-dried, followed by microscopy-assisted fine dissection to remove any visible blood and connective tissue. The aliquots were homogenized in homogenization buffer [500 μl of ice-cold buffer, containing 2.7 mM KCl, 1 mM MgCl2, 137 mM NaCl, 20 mM tris (pH 7.8), 1 mM EDTA, 10 mM NaF, 5 mM Na pyrophosphate, 10% (v/v) glycerol, 1% Triton X-100, 0.2 mM phenylmethylsulfonyl fluoride, 0.5 mM Na3VO4, and protease inhibitor cocktail; Set I, 1×; Calbiochem, EMD Biosciences]. Samples were then rotated at 4°C for 1 hour, followed by centrifugation at 12,000g for 10 min, both at 4°C.
For the adipose tissue, the aliquoted (20 to 35 mg) biopsies were first ground in liquid nitrogen to a fine powder using a mortar and pestle, after which the samples were immersed in roughly 400 μl of homogenization buffer. The samples were then rapidly homogenized using a motor-driven pestle while maintained on ice and were centrifuged at 12,000g for 10 min at 4°C. Following centrifugation, insoluble material was removed from processed muscle and adipose tissue protein lysates, and protein concentration for each sample was determined with duplicate samples run with the Bradford Protein Assay Kit (Thermo Scientific).
Protein aliquots were suspended in equal amounts in Laemmli sample buffer, followed by SDS–polyacrylamide gel electrophoresis separation using precast gels (Criterion XT; Bio-Rad). Following protein separation, proteins were transferred to polyvinylidene difluoride membranes and Ponceau staining was used to verify equal protein loading. Protein extraction was unsuccessful for two muscle samples (from two separate individuals). Therefore, these two subjects were not included in Western blot analyses, yielding a total of 13 paired skeletal muscle samples used for Western blot analysis. Subcutaneous adipose tissue samples were prepared and run similarly to the muscle samples but on separate gels. Because of the low starting material, a total of 11 paired subcutaneous adipose tissue protein samples (that is, including both the sleep loss and sleep condition biopsy samples from a given subject) were available for Western blot analysis. Furthermore, because of the low total protein content, we were unable to further validate the mass spectrometry proteomic hits in adipose tissue protein lysates with immunoblotting.
Following loading verification, transferred membranes were blocked with 5% nonfat dry milk for 90 min at room temperature. Skeletal muscle and subcutaneous adipose tissue membranes were then incubated overnight at 4°C with primary antibodies against BMAL1 (1:2000; Santa Cruz Biotechnology), GLUT4 (1:1000; 07-741, Millipore), mitochondrial complexes I-V (1:1000; ab110411, Abcam), and phosphofructokinase 1 (PFK1; 1:1000; sc-67028, Santa Cruz Biotechnology). After repeated washing in Tris Buffered Saline, with Tween 20 (TBST), the membranes were incubated with appropriate secondary antibodies (anti-goat, anti-mouse, or anti-rabbit) for 60 min. Enhanced chemiluminescence (Amersham) was used to visualize detected proteins.
Quantification of protein densitometry was done with the software Fiji/ImageJ (v2.0.0) (60); normalization of protein content was done to Ponceau staining to normalize to total protein content and avoid bias from variation in housekeeping protein (for example, due to circadian variation) content in individual samples (61). Because the number of analyzed samples per tissue exceeded the amount permitted by a single gel, protein quantification for each tissue was done by running two gels, which together contained all samples for each tissue at the same time. Samples from both conditions (that is, the wake and sleep samples) from each individual were always run together on the same gel, but to account for possible intergel variation, ratios (sleep loss/sleep) were calculated for each subject and assessed using one-sample t tests.
Metabolomic analyses
For metabolite analyses in sampled tissues and serum, following mass spectrometry and initial analysis (see Supplementary Materials), identified retention indices and mass spectra were compared with libraries of retention time indices and mass spectra to identify the extracted mass spectra. Compound identification was based on comparison with mass spectra libraries (in-house database) and the retention index. All metabolomic data were normalized to internal standards, and muscle and adipose tissue samples were also normalized to the mass of each individual sample, determined with 0.1-mg resolution in the frozen state. Metabolomic data were analyzed following log2 transformation with one-sample Student’s t tests of protein and metabolite ratios (sleep loss/sleep). Correlational analyses of metabolomic data were carried out with the Morpheus tool (https://software.broadinstitute.org/morpheus/) using hierarchical clustering (1 − Pearson correlation) for both metabolites and subjects; pathway enrichment analyses were performed with MetaboAnalyst using pathway-associated metabolite sets (http://www.metaboanalyst.ca/). A total of 15 pairs of subcutaneous adipose tissue samples were included in the metabolomic analyses of sleep loss compared with normal sleep; however, because of two outlier samples for two separate subjects (>2 SDs for multiple metabolites), the metabolomic analysis in skeletal muscle included only 13 paired samples.
Serum insulin and cortisol values were analyzed with commercial enzyme-linked immunosorbent assay (ELISA) kits (Human Insulin ELISA, Mercodia AB, Uppsala; Cortisol Parameter Assay Kit, R&D Systems). Plasma glucose levels were analyzed with a chemistry analyzer (Architect C16000, Abbott Laboratories). Serum aliquots (100 μl) from the fasting pre-OGTT and 120-min post-OGTT were used for serum GCMS analyses as described above.
Statistics
Normally distributed data (Kolmogorov-Smirnov’s test, P > 0.05) were analyzed with paired Student t tests (qPCR data), one-sample t tests (Western blot data), or Pearson’s correlation; nonparametric variables were analyzed with the Wilcoxon signed-rank test or Spearman’s rank test. Repeatedly measured biochemical and metabolomic parameters were analyzed with ANOVA with the factors “wake” (reflecting sleep condition) and “time” (reflecting time point, that is, before and up until 120 min after the OGTT). ANOVA sphericity deviations were corrected with the Greenhouse-Geisser method; post hoc comparisons were carried out with the paired Student’s t test. For RNA-seq analyses, genes with >1 count per million in at least five samples were included in the analysis. For genome-wide DNA methylation and transcriptomic analyses, only hits with FDR < 0.05 were considered significantly different; in GSEA and classical pathway analysis, only gene sets with FDR < 0.05 were reported. Unless otherwise specified, values are reported as mean ± SEM and P values < 0.05 were considered significant. Biochemical and metabolomic data were analyzed with SPSS (v.23, SPSS Inc.). Additional details on statistical methods are described for individual methods in the Supplementary Materials.
Supplementary Material
Acknowledgments
We acknowledge J. Brandell, O. Ros, and J.E. Broman for helping with the experiments. We are grateful to K. M. Ramsey and C. B. Peek for input regarding the manuscript, to B. Egan for fruitful discussions, and to B. Marcheva and E. Cedernaes for help with the illustrations. We also thank the participants for their central contribution to this study. Funding: Work from the authors’ laboratory was supported by AFA Försäkring (140006 to C.B.), Avtal om Läkarutbildning och Forskning (ALFBGB-723681 to S.L.D.), the Bissen Brainwalk Foundation (to J.C.), the Carl Trygger Foundation (to S.B.L.), the Erik, Karin and Gösta Selander Foundation (to J.C.), the Fredrik och Ingrid Thurings Foundation (to J.C.), the Lars Hiertas Minne Foundation (to J.C.), the Mats Kleberg Foundation (to J.C.), the Magnus Bergvalls Foundation (to S.B.L.), the Novo Nordisk Foundation (to C.B. and J.R.Z.), the Tore Nilson Foundation (to J.C.), the Swedish Medical Research Society (to J.C.), the Swedish Society for Medicine (SLS-694111 to J.C.), the Swedish Brain Foundation (to J.C. and C.B.), the Swedish Research Council (2015-03100 to C.B., 2014-6888 to J.C., 2016-01088 to H.B.S., 2016-02195 to S.L.D., and 2015-4870 to J.B.), the Åke Wiberg Foundation (to J.C. and S.B.L.), and the National Natural Science Foundation of China (grant no. 31671139 to J.M.). The funding sources had no role in the design, conduct, or reporting of this study, or in any aspect of manuscript writing and submission. This work was supported by the Science for Life Laboratory Mass Spectrometry Based Proteomics Facility in Uppsala. J.O.W. was supported by the Knut and Alice Wallenberg Foundation as part of the National Bioinformatics Infrastructure Sweden at SciLifeLab. The computations were performed on resources provided by Swedish National Infrastructure for Computing (SNIC) through the Uppsala Multidisciplinary Center for Advanced Computational Science. Sequencing was performed by the SNP&SEQ Technology Platform in Uppsala. The facility is part of NGI Sweden and Science for Life Laboratory. The SNP&SEQ Platform was also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation. Author contributions: J.C. designed the study. J.C. wrote the protocol with input from C.B. J.C. collected the data. J.C., M.S., J.O.W., J.M., A.C., S.V., H.V., and C.B. conducted the analyses. J.C., M.S., J.O.W., J.M., A.C., S.V., M.O., H.V., K.H., S.L.D., S.B.L., J.B., H.B.S., J.R.Z., and C.B. interpreted the data. J.C. wrote the original draft, and all authors contributed to review and editing. J.C. and C.B. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The utilized code for data processing can be accessed at https://github.com/orzechoj/cedernaes2018_analysis/tree/master. Processed and raw genomic data can be accessed through ArrayExpress [accession numbers E-MTAB-6908 (DNA methylation data) and E-MTAB-6903 (RNA-seq data)].
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaar8590/DC1
Fig. S1. Correlations between methylation and gene expression levels in subcutaneous adipose tissue and skeletal muscle at baseline and in response to acute sleep loss in humans.
Fig. S2. Insulin sensitivity is adversely affected, and cortisol levels are significantly elevated following acute sleep loss in healthy young men without changes to protein levels of mitochondrial complexes.
Fig. S3. Gene expression and protein levels correlate at baseline but not in response to sleep loss in subcutaneous adipose tissue and skeletal muscle in humans.
Fig. S4. Loading controls for Western blots used throughout the manuscript.
Table S1. Data for the 15 participants that were included in the study.
Table S2. DMRs and enriched biological pathways based on methylation changes in subcutaneous adipose tissue in response to acute sleep loss.
Table S3. Differentially expressed genes in skeletal muscle and subcutaneous adipose tissue in response to acute sleep loss.
Table S4. Altered pathways and transcription factors based on RNA-seq data from skeletal muscle and subcutaneous adipose tissue in response to acute sleep loss compared with normal sleep.
Table S5. Enriched pathways based on proteomic analyses of skeletal muscle tissue in response to acute sleep loss compared with normal sleep.
Table S6. Changes in serum, skeletal muscle, and subcutaneous adipose tissue metabolites in response to acute sleep loss.
Supplementary Methods
REFERENCES AND NOTES
- 1.Cappuccio F. P., D’Elia L., Strazzullo P., Miller M. A., Quantity and quality of sleep and incidence of type 2 diabetes: A systematic review and meta-analysis. Diabetes Care 33, 414–420 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cappuccio F. P., Taggart F. M., Kandala N. B., Currie A., Peile E., Stranges S., Miller M. A., Meta-analysis of short sleep duration and obesity in children and adults. Sleep 31, 619–626 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong P. M., Hasler B. P., Kamarck T. W., Muldoon M. F., Manuck S. B., Social jetlag, chronotype, and cardiometabolic risk. J. Clin. Endocrinol. Metab. 100, 4612–4620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roenneberg T., Allebrandt K. V., Merrow M., Vetter C., Social jetlag and obesity. Curr. Biol. 22, 939–943 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Spaeth A. M., Dinges D. F., Goel N., Effects of experimental sleep restriction on weight gain, caloric intake, and meal timing in healthy adults. Sleep 36, 981–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeFronzo R. A., Tripathy D., Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32 (suppl. 2), S157–S163 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hotamisligil G. S., Arner P., Caro J. F., Atkinson R. L., Spiegelman B. M., Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Invest. 95, 2409–2415 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao M. N., Neylan T. C., Grunfeld C., Mulligan K., Schambelan M., Schwarz J.-M., Subchronic sleep restriction causes tissue-specific insulin resistance. J. Clin. Endocrinol. Metab. 100, 1664–1671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bell L. N., Kilkus J. M., Booth J. N. III, Bromley L. E., Imperial J. G., Penev P. D., Effects of sleep restriction on the human plasma metabolome. Physiol. Behav. 122, 25–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nedeltcheva A. V., Kilkus J. M., Imperial J., Schoeller D. A., Penev P. D., Insufficient sleep undermines dietary efforts to reduce adiposity. Ann. Intern. Med. 153, 435–441 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchmann N., Spira D., Norman K., Demuth I., Eckardt R., Steinhagen-Thiessen E., Sleep, muscle mass and muscle function in older people. Dtsch. Arztebl. Int. 113, 253–260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien M.-Y., Wang L.-Y., Chen H.-C., The relationship of sleep duration with obesity and sarcopenia in community-dwelling older adults. Gerontology 61, 399–406 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Monico-Neto M., Giampá S. Q., Lee K. S., de Melo C. M., de Sá Souza H., Dáttilo M., Minali P. A., Santos Prado P. H., Tufik S., de Mello M. T., Antunes H. K. M., Negative energy balance induced by paradoxical sleep deprivation causes multicompartmental changes in adipose tissue and skeletal muscle. Int. J. Endocrinol. 2015, 908159 ( 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shechter A., Rising R., Wolfe S., Albu J. B., St-Onge M.-P., Postprandial thermogenesis and substrate oxidation are unaffected by sleep restriction. Int. J. Obes. 38, 1153–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bass J., Lazar M. A., Circadian time signatures of fitness and disease. Science 354, 994–999 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Hodge B. A., Wen Y., Riley L. A., Zhang X., England J. H., Harfmann B. D., Schroder E. A., Esser K. A., The endogenous molecular clock orchestrates the temporal separation of substrate metabolism in skeletal muscle. Skeletal Muscle 5, 17 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neufeld-Cohen A., Robles M. S., Aviram R., Manella G., Adamovich Y., Ladeuix B., Nir D., Rousso-Noori L., Kuperman Y., Golik M., Mann M., Asher G., Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proc. Natl. Acad. Sci. U.S.A. 113, E1673–E1682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perrin L., Loizides-Mangold U., Chanon S., Gobet C., Hulo N., Isenegger L., Weger B. D., Migliavacca E., Charpagne A., Betts J. A., Walhin J.-P., Templeman I., Stokes K., Thompson D., Tsintzas K., Robert M., Howald C., Riezman H., Feige J. N., Karagounis L. G., Johnston J. D., Dermitzakis E. T., Gachon F., Lefai E., Dibner C., Transcriptomic analyses reveal rhythmic and CLOCK-driven pathways in human skeletal muscle. eLife 7, e34114 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thurley K., Herbst C., Wesener F., Koller B., Wallach T., Maier B., Kramer A., Westermark P. O., Principles for circadian orchestration of metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 114, 1572–1577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brager A. J., Heemstra L., Bhambra R., Ehlen J. C., Esser K. A., Paul K. N., Novak C. M., Homeostatic effects of exercise and sleep on metabolic processes in mice with an overexpressed skeletal muscle clock. Biochimie 132, 161–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peek C. B., Levine D. C., Cedernaes J., Taguchi A., Kobayashi Y., Tsai S. J., Bonar N. A., McNulty M. R., Ramsey K. M., Bass J., Circadian clock interaction with HIF1α mediates oxygenic metabolism and anaerobic glycolysis in skeletal muscle. Cell Metab. 25, 86–92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buxton O. M., Cain S. W., O’Connor S. P., Porter J. H., Duffy J. F., Wang W., Czeisler C. A., Shea S. A., Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Sci. Transl. Med. 4, 129ra43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cedernaes J., Osler M. E., Voisin S., Broman J. E., Vogel H., Dickson S. L., Zierath J. R., Schiöth H. B., Benedict C., Acute sleep loss induces tissue-specific epigenetic and transcriptional alterations to circadian clock genes in men. J. Clin. Endocrinol. Metab. 100, E1255–E1261 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Hall M. H., Muldoon M. F., Jennings J. R., Buysse D. J., Flory J. D., Manuck S. B., Self-reported sleep duration is associated with the metabolic syndrome in midlife adults. Sleep 31, 635–643 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benton M. C., Johnstone A., Eccles D., Harmon B., Hayes M. T., Lea R. A., Griffiths L., Hoffman E. P., Stubbs R. S., Macartney-Coxson D., An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 16, 8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim Y. C., Chia S. Y., Jin S., Han W., Ding C., Sun L., Dynamic DNA methylation landscape defines brown and white cell specificity during adipogenesis. Mol. Metab. 5, 1033–1041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karastergiou K., Fried S. K., Xie H., Lee M.-J., Divoux A., Rosencrantz M. A., Chang R. J., Smith S. R., Distinct developmental signatures of human abdominal and gluteal subcutaneous adipose tissue depots. J. Clin. Endocrinol. Metab. 98, 362–371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Billon N., Kolde R., Reimand J., Monteiro M. C., Kull M., Peterson H., Tretyakov K., Adler P., Wdziekonski B., Vilo J., Dani C., Comprehensive transcriptome analysis of mouse embryonic stem cell adipogenesis unravels new processes of adipocyte development. Genome Biol. 11, R80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinstein L. S., Xie T., Qasem A., Wang J., Chen M., The role of GNAS and other imprinted genes in the development of obesity. Int. J. Obes. 34, 6–17 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalgaard K., Landgraf K., Heyne S., Lempradl A., Longinotto J., Gossens K., Ruf M., Orthofer M., Strogantsev R., Selvaraj M., Lu T. T., Casas E., Teperino R., Surani M. A., Zvetkova I., Rimmington D., Tung Y. C., Lam B., Larder R., Yeo G. S., O’Rahilly S., Vavouri T., Whitelaw E., Penninger J. M., Jenuwein T., Cheung C. L., Ferguson-Smith A. C., Coll A. P., Körner A., Pospisilik J. A., Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell 164, 353–364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gnad T., Scheibler S., von Kügelgen I., Scheele C., Kilić A., Glöde A., Hoffmann L. S., Reverte-Salisa L., Horn P., Mutlu S., El-Tayeb A., Kranz M., Deuther-Conrad W., Brust P., Lidell M. E., Betz M. J., Enerbäck S., Schrader J., Yegutkin G. G., Müller C. E., Pfeifer A., Adenosine activates brown adipose tissue and recruits beige adipocytes via A2A receptors. Nature 516, 395–399 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Kojima H., Fujimiya M., Matsumura K., Nakahara T., Hara M., Chan L., Extrapancreatic insulin-producing cells in multiple organs in diabetes. Proc. Natl. Acad. Sci. U.S.A. 101, 2458–2463 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Warfel J. D., Vandanmagsar B., Dubuisson O. S., Hodgeson S. M., Elks C. M., Ravussin E., Mynatt R. L., Examination of carnitine palmitoyltransferase 1 abundance in white adipose tissue: Implications in obesity research. Am. J. Physiol. Regul. Integr. Comp. Physiol. 312, R816–R820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonen A., Tandon N. N., Glatz J. F. C., Luiken J. J. F. P., Heigenhauser G. J. F., The fatty acid transporter FAT/CD36 is upregulated in subcutaneous and visceral adipose tissues in human obesity and type 2 diabetes. Int. J. Obes. 30, 877–883 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Parikh H., Carlsson E., Chutkow W. A., Johansson L. E., Storgaard H., Poulsen P., Saxena R., Ladd C., Christian Schulze P., Mazzini M. J., Jensen C. B., Krook A., Björnholm M., Tornqvist H., Zierath J. R., Ridderstråle M., Altshuler D., Lee R. T., Vaag A., Groop L. C., Mootha V. K., TXNIP regulates peripheral glucose metabolism in humans. PLOS Med. 4, 868–879 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alessi D. R., Sammler E., LRRK2 kinase in Parkinson’s disease. Science 360, 36–37 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Fernandez-Real J. M., Pérez del Pulgar S., Luche E., Moreno-Navarrete J. M., Waget A., Serino M., Sorianello E., Sánchez-Pla A., Pontaque F. C., Vendrell J., Chacón M. R., Ricart W., Burcelin R., Zorzano A., CD14 modulates inflammation-driven insulin resistance. Diabetes 60, 2179–2186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu S., Yao J., Howe A. A., Menke B. M., Sivitz W. I., Spector A. A., Norris A. W., Peroxisome proliferator-activated receptor γ decouples fatty acid uptake from lipid inhibition of insulin signaling in skeletal muscle. Mol. Endocrinol. 26, 977–988 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senf S. M., Skeletal muscle heat shock protein 70: Diverse functions and therapeutic potential for wasting disorders. Front. Physiol. 4, 330 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tupling A. R., Bombardier E., Stewart R. D., Vigna C., Aqui A. E., Muscle fiber type-specific response of Hsp70 expression in human quadriceps following acute isometric exercise. J. Appl. Physiol. 103, 2105–2111 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Kirschke E., Goswami D., Southworth D., Griffin P. R., Agard D. A., Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Balkom R. H. H., Zhan W.-Z., Prakash Y. S., Dekhuijzen P. N., Sieck G. C., Corticosteroid effects on isotonic contractile properties of rat diaphragm muscle. J. Appl. Physiol. 83, 1062–1067 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Nye C., Kim J., Kalhan S. C., Hanson R. W., Reassessing triglyceride synthesis in adipose tissue. Trends Endocrinol. Metab. 19, 356–361 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Jowsey I. R., Smith S. A., Hayes J. D., Expression of the murine glutathione S-transferase α3 (GSTA3) subunit is markedly induced during adipocyte differentiation: Activation of the GSTA3 gene promoter by the pro-adipogenic eicosanoid 15-deoxy-Δ12,14-prostaglandin J2. Biochem. Biophys. Res. Commun. 312, 1226–1235 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Moreno-Navarrete J. M., Ortega F., Moreno M., Serrano M., Ricart W., Fernández-Real J. M., Lactoferrin gene knockdown leads to similar effects to iron chelation in human adipocytes. J. Cell. Mol. Med. 18, 391–395 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salgado-Somoza A., Teijeira-Fernandez E., Fernandez A. L., Gonzalez-Juanatey J. R., Eiras S., Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 299, H202–H209 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Boden G., Duan X., Homko C., Molina E. J., Song W., Perez O., Cheung P., Merali S., Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 57, 2438–2444 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dyar K. A., Ciciliot S., Wright L. E., Biensø R. S., Tagliazucchi G. M., Patel V. R., Forcato M., Paz M. I., Gudiksen A., Solagna F., Albiero M., Moretti I., Eckel-Mahan K. L., Baldi P., Sassone-Corsi P., Rizzuto R., Bicciato S., Pilegaard H., Blaauw B., Schiaffino S., Muscle insulin sensitivity and glucose metabolism are controlled by the intrinsic muscle clock. Mol. Metab. 3, 29–41 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeoung N. H., Wu P., Joshi M. A., Jaskiewicz J., Bock C. B., Depaoli-Roach A. A., Harris R. A., Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochem. J. 397, 417–425 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Majer M., Popov K. M., Harris R. A., Bogardus C., Prochazka M., Insulin downregulates pyruvate dehydrogenase kinase (PDK) mRNA: Potential mechanism contributing to increased lipid oxidation in insulin-resistant subjects. Mol. Genet. Metab. 65, 181–186 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Rosa G., Di Rocco P., Manco M., Greco A. V., Castagneto M., Vidal H., Mingrone G., Reduced PDK4 expression associates with increased insulin sensitivity in postobese patients. Obes. Res. 11, 176–182 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Fell D. A., Small J. R., Fat synthesis in adipose tissue. An examination of stoichiometric constraints. Biochem. J. 238, 781–786 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Sá Souza H., Antunes H. K., Dáttilo M., Lee K. S., Mônico-Neto M., de Campos Giampa S. Q., Phillips S. M., Tufik S., de Mello M. T., Leucine supplementation is anti-atrophic during paradoxical sleep deprivation in rats. Amino Acids 48, 949–957 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Giskeødegård G. F., Davies S. K., Revell V. L., Keun H., Skene D. J., Diurnal rhythms in the human urine metabolome during sleep and total sleep deprivation. Sci. Rep. 5, 14843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leproult R., Van Cauter E., Effect of 1 week of sleep restriction on testosterone levels in young healthy men. JAMA 305, 2173–2174 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sassin J. F., Parker D. C., Mace J. W., Gotlin R. W., Johnson L. C., Rossman L. G., Human growth hormone release: Relation to slow-wave sleep and sleep-walking cycles. Science 165, 513–515 (1969). [DOI] [PubMed] [Google Scholar]
- 57.Soubry A., Murphy S. K., Wang F., Huang Z., Vidal A. C., Fuemmeler B. F., Kurtzberg J., Murtha A., Jirtle R. L., Schildkraut J. M., Hoyo C., Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int. J. Obes. 39, 650–657 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.A. Rechtschafen, A. Kales, A Manual of Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects (BIS/BRI, UCLA, 1968). [DOI] [PubMed] [Google Scholar]
- 59.Cedernaes J., Brandell J., Ros O., Broman J.-E., Hogenkamp P. S., Schiöth H. B., Benedict C., Increased impulsivity in response to food cues after sleep loss in healthy young men. Obesity 22, 1786–1791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A., Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romero-Calvo I., Ocón B., Martínez-Moya P., Suárez M. D., Zarzuelo A., Martínez-Augustin O., de Medina F. S., Reversible Ponceau staining as a loading control alternative to actin in Western blots. Anal. Biochem. 401, 318–320 (2010). [DOI] [PubMed] [Google Scholar]
- 62.Matsuda M., DeFronzo R. A., Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care 22, 1462–1470 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Lachmann A., Xu H., Krishnan J., Berger S. I., Mazloom A. R., Ma’ayan A., ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Almén M. S., Nilsson E. K., Jacobsson J. A., Kalnina I., Klovins J., Fredriksson R., Schiöth H. B., Genome-wide analysis reveals DNA methylation markers that vary with both age and obesity. Gene 548, 61–67 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Du P., Zhang X., Huang C.-C., Jafari N., Kibbe W. A., Hou L., Lin S. M., Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aryee M. J., Jaffe A. E., Corrada-Bravo H., Ladd-Acosta C., Feinberg A. P., Hansen K. D., Irizarry R. A., Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peters T. J., Buckley M. J., Statham A. L., Pidsley R., Samaras K., Lord R. V., Clark S. J., Molloy P. L., De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin 8, 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barrès R., Yan J., Egan B., Treebak J. T., Rasmussen M., Fritz T., Caidahl K., Krook A., O’Gorman D. J., Zierath J. R., Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 15, 405–411 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Dokas J., Chadt A., Nolden T., Himmelbauer H., Zierath J. R., Joost H.-G., Al-Hasani H., Conventional knockout of Tbc1d1 in mice impairs insulin- and AICAR-stimulated glucose uptake in skeletal muscle. Endocrinology 154, 3502–3514 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Broad Institute, Picard Tools, GitHub repository; http://broadinstitute.github.io/picard/ [accessed 28 Feb 2017].
- 73.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luo W., Friedman M. S., Shedden K., Hankenson K. D., Woolf P. J., GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., Ma’ayan A., Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Kanehisa M., Goto S., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.A J., Trygg J., Gullberg J., Johansson A. I., Jonsson P., Antti H., Marklund S. L., Moritz T., Extraction and GC/MS analysis of the human blood plasma metabolome. Anal. Chem. 77, 8086–8094 (2005). [DOI] [PubMed] [Google Scholar]
- 80.Gullberg J., Jonsson P., Nordström A., Sjöström M., Moritz T., Design of experiments: An efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 331, 283–295 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Jonsson P., Johansson A. I., Gullberg J., Trygg J., A J., Grung B., Marklund S., Sjöström M., Antti H., Moritz T., High-throughput data analysis for detecting and identifying differences between samples in GC/MS-based metabolomic analyses. Anal. Chem. 77, 5635–5642 (2005). [DOI] [PubMed] [Google Scholar]
- 82.Matthews D. R., Hosker J. P., Rudenski A. S., Naylor B. A., Treacher D. F., Turner R. C., Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28, 412–419 (1985). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaar8590/DC1
Fig. S1. Correlations between methylation and gene expression levels in subcutaneous adipose tissue and skeletal muscle at baseline and in response to acute sleep loss in humans.
Fig. S2. Insulin sensitivity is adversely affected, and cortisol levels are significantly elevated following acute sleep loss in healthy young men without changes to protein levels of mitochondrial complexes.
Fig. S3. Gene expression and protein levels correlate at baseline but not in response to sleep loss in subcutaneous adipose tissue and skeletal muscle in humans.
Fig. S4. Loading controls for Western blots used throughout the manuscript.
Table S1. Data for the 15 participants that were included in the study.
Table S2. DMRs and enriched biological pathways based on methylation changes in subcutaneous adipose tissue in response to acute sleep loss.
Table S3. Differentially expressed genes in skeletal muscle and subcutaneous adipose tissue in response to acute sleep loss.
Table S4. Altered pathways and transcription factors based on RNA-seq data from skeletal muscle and subcutaneous adipose tissue in response to acute sleep loss compared with normal sleep.
Table S5. Enriched pathways based on proteomic analyses of skeletal muscle tissue in response to acute sleep loss compared with normal sleep.
Table S6. Changes in serum, skeletal muscle, and subcutaneous adipose tissue metabolites in response to acute sleep loss.
Supplementary Methods