

MET-2/SETDB1 and interactors LIN-65/ATF7IP and ARLE-14/ARL14EP initiate heterochromatin formation during embryogenesis.
Abstract
Heterochromatin formation during early embryogenesis is timed precisely, but how this process is regulated remains elusive. We report the discovery of a histone methyltransferase complex whose nuclear accumulation and activation establish the onset of heterochromatin formation in Caenorhabditis elegans embryos. We find that the inception of heterochromatin generation coincides with the accumulation of the histone H3 lysine 9 (H3K9) methyltransferase MET-2 (SETDB) into nuclear hubs. The absence of MET-2 results in delayed and disturbed heterochromatin formation, whereas accelerated nuclear localization of the methyltransferase leads to precocious H3K9 methylation. We identify two factors that bind to and function with MET-2: LIN-65, which resembles activating transcription factor 7–interacting protein (ATF7IP) and localizes MET-2 into nuclear hubs, and ARLE-14, which is orthologous to adenosine 5′-diphosphate–ribosylation factor-like 14 effector protein (ARL14EP) and promotes stable association of MET-2 with chromatin. These data reveal that nuclear accumulation of MET-2 in conjunction with LIN-65 and ARLE-14 regulates timing of heterochromatin domains during embryogenesis.
INTRODUCTION
The nucleus of a young embryo undergoes major reorganization as it transitions from a fertilized egg to a multicellular embryo. As cells acquire specific fates and zygotic transcription commences, the nucleus is segregated into distinct domains of euchromatin and heterochromatin (1). While much has been learned about the mechanisms that control cell-fate specification and the onset of zygotic transcription, little is understood about the processes that establish chromatin domains de novo during embryogenesis.
Heterochromatin domains are generated de novo during development (1). In early mouse embryos, embryonic stem cells, and planarian neoblasts, higher-order heterochromatin is lacking but becomes established as these cells differentiate. Their nuclei shrink, chromatin compacts, and the nucleoplasm becomes divided into visible domains of euchromatin and heterochromatin (1). In Caenorhabditis elegans, the dynamics of embryonic chromatin has been tracked with artificial chromosomes. These appear large and distended at the earliest stages of embryogenesis and undergo compaction during gastrulation (2, 3). These examples suggest that the segregation of heterochromatin domains is a conserved feature of the differentiated state. Little is known, however, what drives formation of heterochromatin during embryogenesis or what determines the timing of chromatin reorganization.
Here, we characterize the regulation of MET-2 protein and its relation to heterochromatin formation during embryogenesis. We identify partner proteins that mediate MET-2 subcellular localization and chromatin association. We find that MET-2 and its partners are critical for the timely generation of heterochromatin during embryogenesis.
RESULTS
We began our analysis with a survey of wild-type (WT) embryos using two assays for heterochromatin: First, we used transmission electron microscopy (TEM). TEM studies since the 1960s have shown that heterochromatin domains can be detected as electron-dense regions (EDRs) and that heterochromatin-associated histone modifications such as histone H3 lysine 9 methylation (H3K9me) are located within EDRs (4, 5). Second, we surveyed histone modifications by antibody stains to track their abundance and distribution in early embryos. Histone modifications are associated with active or silent regions of the genome and can be a hallmark of heterochromatin (6).
As viewed by TEM, nuclei in the youngest embryos appeared relatively homogeneous, with light speckling in the nucleoplasm and a nuclear envelope free of electron-dense material (Fig. 1, A and B). Upon initiation of gastrulation (~21- to 50-cell stage), embryos gained more electron-dense puncta throughout their nuclei. By mid-gastrulation (51- to 100-cell stage), we observed dark material abutting the nuclear envelope, and the nucleoplasmic puncta coalesced into larger but fewer electron-dense compartments. These features became more pronounced over time, with large EDRs that spanned the nucleus and bordered the nuclear periphery (Fig. 1B, >200 cells). We note that EDRs appeared throughout the embryo, suggesting that cells destined to produce different cell types nevertheless generated heterochromatin domains at about the same time in development (fig. S1A).
Fig. 1. Heterochromatin and H3K9me domains are established during embryogenesis.
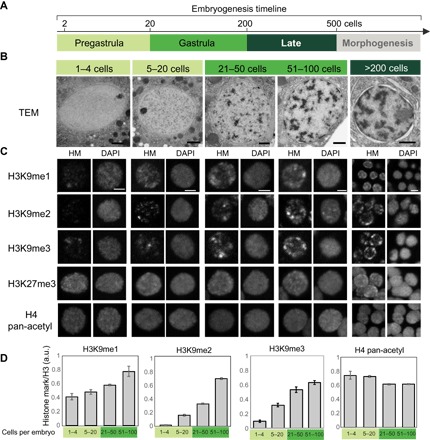
(A) Timeline of C. elegans embryogenesis. Stages are color-coded: light green (<20 cells, pregastrula), green (21 to 200 cells, gastrula), and dark green (200 to 500 cells, late stage). Morphogenesis starts after the 500-cell stage and was not analyzed in this study. (B) Transmission electron micrographs of representative nuclei from WT embryos. Scale bars, 1 μm. (C) Survey of histone modifications (HMs). Representative single nuclei at designated embryonic stages stained for histones and DNA. Scale bars, 2 μm. DAPI, 4′,6-diamidino-2-phenylindole. (D) Quantitation of histone modifications normalized to total histone H3. Error bars denote SEM. a.u., arbitrary units.
We surveyed histone modifications to determine whether they were dynamic during development. There was a dramatic increase in H3K9me from fertilization to the mid-gastrula in interphase cells. We quantified the effect, using antibodies against histone H3 for normalization (see Materials and Methods). H3K9me2 increased 10-fold (P = 4 × 10−19), while H3K9me1 increased 2-fold (P = 0.001) and H3K9me3 increased 6-fold (P = 4 × 10−7; Fig. 1, C and D). H3K9me2 was barely detectable at fertilization, consistent with previous studies that showed that H3K9me2 is erased in the germ line and undetectable in oocytes (7). Bright H3K9me2 puncta became apparent by the 20-cell stage throughout embryonic nuclei. The signal intensified during gastrulation, with more puncta and brighter staining within puncta (~51- to 100-cell stage, mid-stage). A time series of whole embryo stains indicated that most interphase nuclei behaved similarly (fig. S1B) and two different antibodies against H3K9me2 gave identical results (fig. S1C). Controls indicated that the signal was specific because mutants lacking H3K9me2 (7, 8) failed to stain with any of the H3K9me2 antibodies (fig. S1, D and E). Both puncta of histone modifications and EDRs arose during gastrulation, suggesting that H3K9me stains are a useful proxy to visualize heterochromatin domains.
Not all histone modifications were developmentally regulated (Fig. 1C and fig. S1B). The Polycomb-mediated modification H3K27me3 was abundant in early embryos and did not increase further, as observed previously (9). This result indicates that H3K27me3 is not sufficient for higher-order heterochromatin. H4 pan-acetylation (H4Ac), a modification associated with active chromatin, was present in early embryos before the onset of zygotic transcription and increased slightly over time. This observation reveals that bulk loss of H4Ac is not required for higher-order heterochromatin. In short, only some modifications were regulated dynamically during embryogenesis.
To identify the molecular basis of heterochromatin formation, we focused on the methyltransferase MET-2. MET-2 is homologous to vertebrate SETDB1 (10, 11) and required for virtually all H3K9me1 and H3K9me2, as well as for some H3K9me3 (7, 8). We focused on H3K9me2 and MET-2 because H3K9me2 was regulated most dynamically during embryogenesis, because it promotes H3K9me3 (8), and because its location in the genome, by chromatin immunoprecipitation (ChIP), tracks well with canonical heterochromatin proteins such as HPL-2/HP1 (12).
We asked whether loss of MET-2 affected heterochromatin domains visible by TEM. Pregastrula met-2 embryos matched their WT counterparts, with homogeneous, translucent nuclei (Fig. 2A). However, the speckles observed in WT nuclei at the 20-cell stage were dimmer in met-2 mutants, and they failed to coalesce into EDRs by the 50- to 100-cell stage (Fig. 2A). EDRs emerged in older embryos, but they occupied less nuclear volume and were reproducibly paler than the WT (Fig. 2A). We performed line-scan analysis to quantify the appearance of nuclear EDRs. The standard deviation (SD) of line-scan values is higher in nuclei with EDRs compared to homogeneous nuclei because the dark EDRs contrast with the pale nucleoplasm. met-2 nuclei had a more homogeneous signal distribution and a smaller SD at every stage (Fig. 2B). To assess the amount of residual electron density in met-2 late embryonic nuclei (>200-cell stage), we used an intensity threshold to define EDRs in WT and met-2 embryos (Fig. 2C). The percentage of pixels in EDRs was reduced almost threefold in met-2 mutants compared to the WT (Fig. 2C). To control for TEM fixation and sectioning, we examined cytoplasmic organelles and yolk droplets. Intensity thresholding revealed that electron-dense cytoplasmic structures were present in met-2 mutants (Fig. 2D), and they resembled those in WT embryos (fig. S2, A and B). These results indicate that met-2 is critical for the timely formation of segregated heterochromatin domains.
Fig. 2. H3K9 methyltransferase MET-2/SETDB1 is required for heterochromatin formation.
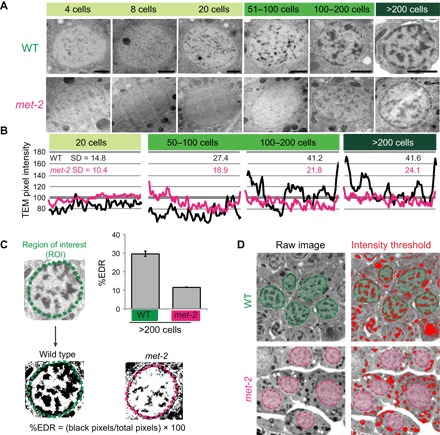
(A) TEM of representative nuclei from WT or met-2 embryos. Scale bars, 1 μm. (B) TEM line-scan analysis for WT (black) or met-2 (magenta) nuclei and the SD. Line scan for a single nucleus is shown as an example. (C) Proportion of pixels within EDRs at the 200-cell stage. Error bars denote SEM. (D) Contrast of the cytosol in WT versus met-2 mutants and intensity thresholding (red) to define electron-dense structures. WT nuclei are in green and met-2 nuclei are in magenta.
Given the dependence of H3K9me2 on MET-2, we asked whether expression of MET-2 tracked with the onset of H3K9me2. MET-2 protein gradually shifted from the cytosol to the nucleus, from the two-cell stage to the onset of gastrulation. We observed this change for both endogenous MET-2 and single-copy MET-2 reporters (Fig. 3, A to C, and fig. S3, B and C). In one- to eight-cell embryos, MET-2 was distributed throughout the nuclei and cytoplasm with little nuclear accumulation. As embryos aged, the concentration of MET-2 within nuclei increased approximately fivefold (Fig. 3B; P = 0.0002), whereas the absolute level of MET-2 protein did not change significantly over time (Fig. 3C). We note that early embryos had increased nuclear MET-2 and H3K9me2 transiently during prophase (fig. S3, C and F), but we focus on interphase nuclei here.
Fig. 3. Nuclear accumulation of MET-2 determines the onset of H3K9 dimethylation.
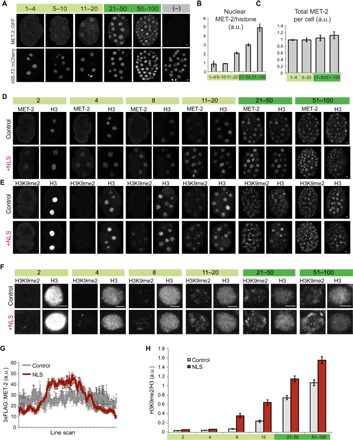
(A) Embryos stained for MET-2::GFP (top) and HIS-72::mCherry (bottom) at designated stages of embryogenesis. Scale bars, 2 μm. (B and C) Quantitation of nuclear (B) and total (C) MET-2 at designated stages, normalized to HIS-72::mCherry. Error bars denote SEM. (D and E) Localization of 3xFLAG::MET-2 with a c-Myc NLS compared to 3xFLAG::MET-2 control (D) and corresponding H3K9me2 levels (E). We used histone H3 as a staining control. Note that H3 appears to be released during mitosis, but we focused on interphase cells. (F) Interphase nuclei showing H3K9me2 levels for the c-myc NLS construct compared to on-slide control embryos. (G) 3xFLAG::MET-2 line scans in pregastrula embryos (one- to four-cell stage) with (red) or without (gray) the c-myc NLS. Average of line scans across multiple nuclei is shown, and error bars denote SEM. (H) H3K9me2 levels for the NLS construct (red), normalized to H3 and to WT embryos at the 51- to 100-cell stage (gray). Error bars denote SEM.
These data may help resolve a controversy regarding where MET-2 is localized within cells (8, 13–15). A previous study had concluded that MET-2 was cytosolic (8), whereas other work had suggested that MET-2 associated with chromatin (14) in the nucleus (15). Our data reveal that MET-2 transitions from the cytosol to the nucleus (Fig. 3A), a process that is regulated by its binding partner LIN-65 (see below and Discussion).
Given that met-2 is necessary for heterochromatin domains and that H3K9me2 accumulated dynamically in early embryos, we asked whether regulation of MET-2 constituted part of the embryonic timer for heterochromatin establishment. We hypothesized that if nuclear MET-2 was rate-limiting, then premature accumulation of MET-2 in nuclei would lead to precocious H3K9me2 and initiate heterochromatin. We added a nuclear localization signal (NLS) from c-Myc to a FLAG-tagged copy of endogenous MET-2 using CRISPR-Cas9 and examined embryos for H3K9me2 and histone H3 (a control for staining). We note that H3 appears to be released during mitosis, but our quantitation focused only on interphase cells.
Addition of an NLS resulted in a twofold increase in nuclear MET-2 in pregastrula embryos (Fig. 3, D and G). Increased nuclear MET-2 led to precocious accumulation of H3K9me2, beginning at least one cell division earlier than WT embryos (Fig. 3, E, F, and H; P < 0.05 at all stages). These results suggest that gradual accumulation of MET-2 within nuclei initiates H3K9me2.
To understand how MET-2 is regulated, we searched for binding partners using immunoprecipitation, followed by multidimensional protein identification technology mass spectrometry (MudPIT), using MET-2::GFP and 3xFLAG::MET-2. WT C. elegans bearing no tagged proteins and strains bearing an unrelated green fluorescent protein (GFP) or FLAG reporter served as negative controls. Results of our MET-2::GFP proteomics analysis can be found in table S1. We chose a candidate list of interacting partners based on the specificity of MET-2 binding, on the peptide counts, and on protein coverage. We undertook a secondary screen for the effects of these candidates on H3K9me2. From this survey, two proteins emerged as likely MET-2 partners (fig. S4A): LIN-65 is a 100-kDa protein and the most abundant interactor of MET-2 (fig. S4B); B0336.5 codes for a smaller protein (~30 kDa) and had lower spectral counts but similar protein coverage to LIN-65 (fig. S4A). We renamed B0336.5 as arle-14 for ARL14 effector protein, as explained below.
To test the role of the MET-2 binding partners in H3K9me deposition, we analyzed loss-of-function mutants. lin-65 mutants had markedly reduced levels of H3K9me1 and H3K9me2, as well as low H3K9me3 (Fig. 4, A and B). WT embryos had puncta of H3K9me3 and a large SD of line-scan values, whereas met-2 and lin-65 mutants each had dispersed H3K9me3 with a more uniform distribution in the nucleus (Fig. 4, C and D). Thus, lin-65 mutants resembled met-2 mutants, suggesting that it is an essential cofactor. On the other hand, arle-14 mutants had reduced H3K9me1/H3K9me2 levels and largely normal H3K9me3 (Fig. 4, A and B). These mutants were similar to a partial loss of met-2 activity, suggesting that arle-14 is less critical than lin-65.
Fig. 4. MET-2 and two conserved binding partners form concentrated nuclear hubs at gastrulation.
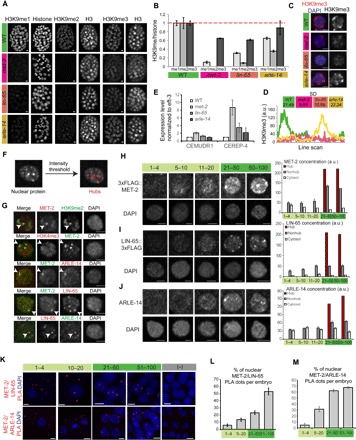
(A) Whole embryos stained with antibodies against methylated H3K9 and pan-histones in WT versus met-2, lin-65, or arle-14 mutants. We used H3K9me2 (Kimura 6D11) antibodies. Scale bars, 2 μm. (B) H3K9me levels in mutant embryos normalized to on-slide control embryos and to pan-histone. Error bars denote SEM. (C) Representative single nuclei showing H3K9me3 (red) and DAPI (blue) in WT versus met-2, lin-65, or arle-14 mutants. Scale bars, 2 μm. (D) H3K9me3 line-scan analysis for WT (green), met-2 (magenta), lin-65 (orange), arle-14 (yellow) nuclei, and the SD. Line scan for a single nucleus is shown as an example. (E) Expression of CEREP4 and CEMUDR1 repeat RNAs by real-time quantitative polymerase chain reaction (qPCR) in WT versus met-2, lin-65, or arle-14 mutant embryos. (F) Definition of hubs by intensity thresholding. (G) Single nuclei showing 3xFLAG::MET-2, LIN-65::3xFLAG, and ARLE-14 colocalization in concentrated protein hubs and exclusion of activating mark H3K4me3. Scale bars, 2 μm. (H to J) Representative single nuclei showing 3xFLAG::MET-2 (H), LIN-65::3xFLAG (I), and ARLE-14 (J) localization at different embryonic stages. Scale bars, 2 μm. Quantitation of signal intensity in nuclear hubs (brown), nuclear regions excluding hubs (“nonhub,” dark gray), and cytosol (light gray). Error bars denote SEM. (K) PLA signal showing interactions between MET-2/LIN-65 and MET-2/ARLE-14. (L and M) Percentage of nuclear PLA dots per total dots in embryos for MET-2/LIN-65 (L) and MET-2/ARLE-14 (M) interactions. Error bars denote SEM.
lin-65 and arle-14 resembled met-2 mutants in two additional assays. First, an important role of MET-2 is to silence repetitive DNA (14). arle-14 mutations and, to a greater degree, lin-65 derepressed RNAs for two repeats (Fig. 4E). Second, derepressed repeats led to a mortal germline phenotype for met-2 mutants at 26°C (11). Similarly, lin-65 mutants became sterile after a single generation and arle-14 mutants became sterile after two generations (n = 80 worms; generation 1, 20% sterility; generation 2, 100% sterility). These results indicate that LIN-65 and ARLE-14 are bona fide binding partners for MET-2 and contribute to its functions.
To address whether LIN-65 and ARLE-14 contribute to the onset of heterochromatin formation, we examined their expression during embryogenesis. We generated an antibody against bacterially produced ARLE-14 and inserted a 3xFLAG tag at the C terminus of endogenous LIN-65 by CRISPR. Both proteins behaved similarly to MET-2: They were enriched in the cytoplasm from the one-cell stage through the eight-cell stage but gradually moved into nuclei thereafter; total levels did not change (fig. S4, C and D). During gastrulation, we observed that concentrated hubs of MET-2, LIN-65, and ARLE-14 emerge within nuclei (Fig. 4, F and H to J). The hubs were visible by eye within nuclei and defined more rigorously with an intensity threshold (Fig. 4F). H3K9me2, MET-2, LIN-65, and ARLE-14 colocalized in hubs, and these excluded the activating mark H3K4me3 (Fig. 4G). MET-2 and its binding partners never excluded each other in the nucleus, but we frequently observed intense hubs of one protein that did not overlap with each other (Fig. 4G). This result suggests that although these proteins bind to each other, they are not always colocalized in the cell and that binding interactions may be specific to certain regions or functions.
The antibody stains suggested that MET-2 could bind LIN-65 and ARLE-14 in either the cytoplasm or the nucleus, but imaging of proteins under the light microscope lacks the resolution to define where binding occurs. We took advantage of the proximity ligation assay (PLA), which detects pairs of proteins when they are within ~30 nm of each other, to investigate MET-2 binding to its partners. Positive and negative controls demonstrated that our PLA signals were specific (fig. S4, E and F). At the earliest stages of embryogenesis, we observed a MET-2 PLA+ signal with LIN-65 and ARLE-14 in the cytoplasm but rarely in the nucleus (~5% nuclear; Fig. 4, K to M). As embryos matured, we not only continued to detect PLA signals in the cytoplasm but also observed a signal within nuclei for MET-2 with both LIN-65 (~53%) and ARLE-14 (~67%). These results reveal that MET-2 interacts closely with LIN-65 and ARLE-14 and that the interacting proteins accumulate in nuclear hubs over time.
To begin to address how ARLE-14 contributes to the deposition of H3K9me2 by MET-2, we examined MET-2 in WT and arle-14 embryos. Neither the localization nor the level of MET-2 changed in arle-14 mutants (Fig. 5, A and B). On the other hand, expression of ARLE-14 tracked extremely well with MET-2 (Fig. 5, C and D). In WT embryos, ARLE-14 was predominantly nuclear, but in met-2 mutants lacking MET-2 protein, ARLE-14 was lost, suggesting that MET-2 is required for ARLE-14 stability or accumulation. These data indicate that arle-14 is not required to localize MET-2 into nuclei.
Fig. 5. ARLE-14 promotes chromatin association of MET-2.
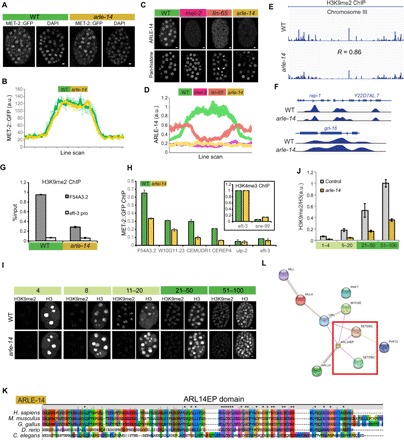
(A) Whole embryos showing the distribution of MET-2::GFP in WT versus arle-14 mutants. Scale bars, 2 μm. (B) Line-scan analysis showing the mean MET-2::GFP intensity across embryonic cells in WT versus arle-14 mutant embryos. Average of line scans across multiple nuclei is shown, and error bars denote SEM. (C) ARLE-14 antibody staining in WT versus met-2, lin-65, and arle-14 mutants. (D) ARLE-14 line-scan quantitation. Average of line scans across multiple nuclei is shown, and error bars denote SEM. (E and F) H3K9me2 ChIP-seq (logLR) track in WT versus arle-14 mutant embryos (chromosome III and loci: rep-1, Y22D7AL.7, and grl-16). Five WT ChIP experiments and 10 arle-14 mutant ChIP experiments were pooled in for sequencing, and results are not quantitative. (G) H3K9me2 ChIP-qPCR in WT versus arle-14 mutants before combining parallel ChIP experiments together for sequencing. Error bars denote SEM. (H) MET-2::GFP ChIP-qPCR for WT (green) versus arle-14 (yellow) mutant embryos. Inset shows H3K4me3 ChIP-qPCR as a control. The y axis shows normalized %input values for both WT and mutants, where the %input value at the H3K4me3-positive region was set to 1 for both genotypes. %input for MET-2::GFP ChIP was adjusted accordingly. Error bars denote SEM for n = 3 experiments. (I and J) Acquisition of H3K9me2 in WT versus arle-14 mutants during embryogenesis, with an H3 costain (I) and quantitation normalized to H3 (J). Error bars denote SEM. (K) Amino acid sequence alignment of worm ARLE-14 to the human ARL14EP domain using ClustalX2. Color scheme: blue, hydrophobic; red, positive charge; magenta, negative charge; green, polar; pink, cysteines; orange, glycines; yellow, prolines; cyan, aromatic. (L) Protein interaction map for human ARL14EP from STRING database (https://string-db.org/). The red box highlights interactions with human SETDB1/2.
Next, we used ChIP sequencing (ChIP-seq) to determine whether arle-14 affects the genome-wide location of H3K9me2 or MET-2. The distribution of H3K9me2 was normal in arle-14 mutants (WT versus arle-14 gave a genome-wide correlation of 0.86 and 0.66 for two independent experiments; Fig. 5, E and F). We conclude that ARLE-14 is not required to target MET-2 in the genome. We also quantitatively examined the level of H3K9me2. ChIP-seq experiments are rarely quantitative owing to the methods of sample isolation and library preparation (see Materials and Methods). Thus, while ChIP-seq can reveal where H3K9me2 is located in the genome, it does not reveal how much. To quantify the level of H3K9me2, we used ChIP-qPCR on material before DNA amplification and normalized to input DNA. H3K9me2 ChIP-qPCR confirmed the decrease in H3K9me2 levels in arle-14 mutants (Fig. 5G), similar to immunostaining results (Fig. 4A). These findings indicate that H3K9me2 is in the right places in the genome but at a reduced level.
Quantitative analysis by ChIP-qPCR revealed a twofold decrease in MET-2::GFP binding to known genomic targets in arle-14 mutants (Fig. 5H). These data show that arle-14 is required for robust association of MET-2 with chromatin. The reduction in chromatin-associated MET-2 may explain the low level of H3K9me2 in arle-14 mutants.
The reduction in chromatin-associated MET-2 and H3K9me2 suggested that arle-14 would be important for timing H3K9me2 in early embryos. We tested this idea with stains of arle-14 mutants and quantified the level of H3K9me2 with histone H3 normalization. From the 5- to 20-cell to the 51- to 100-cell stage, we observed a linear accumulation of H3K9me2 with a slope of 0.41 (R2 = 0.99) for the WT and a slope of 0.15 (R2 = 0.98) for arle-14 (Fig. 5, I and J). From the 1- to 4-cell to the 5- to 20-cell stage, H3K9me2 appeared to accumulate more slowly with a slope of 0.11 for the WT and 0.02 for arle-14. These data indicate that arle-14 is required for accumulation of H3K9me2 at every stage analyzed and suggest that the rate of deposition is slowest during the first few cell divisions.
Before this study, ARLE-14 was an uncharacterized C. elegans protein with homology to adenosine 5′-diphosphate ribosylation factor-like 14 effector protein (ARL14EP; Fig. 5K). In vertebrates, the only published function for ARL14EP is in the cytoplasm (16). However, the Human Protein Atlas shows ARL14EP in the nuclei of many human tissues (www.proteinatlas.org). We surveyed large-scale interaction databases (17–19) and uncovered an interaction between ARL14EP and SETDB proteins in both humans and Drosophila (Fig. 5L). We suggest that, in addition to its cytoplasmic function, ARLE-14 and its orthologs function in nuclei with SETDB methyltransferases.
Next, we examined LIN-65. In lin-65 mutants, MET-2 remained cytoplasmic (Fig. 6A and fig. S5A), as did ARLE-14 (Fig. 5C). MET-2 levels did not decrease in lin-65 mutants (fig. S5B), indicating that LIN-65 affected the subcellular distribution of MET-2 but not its stability. These data show that LIN-65 is required for the timely accumulation of MET-2 within nuclei. MET-2, LIN-65, and ARLE-14 likely bind in a complex together because MET-2 requires LIN-65 for nuclear localization, whereas ARLE-14 requires MET-2 for both nuclear localization and cellular accumulation.
Fig. 6. LIN-65 is rate-limiting for H3K9me2 and nuclear MET-2 during embryogenesis.
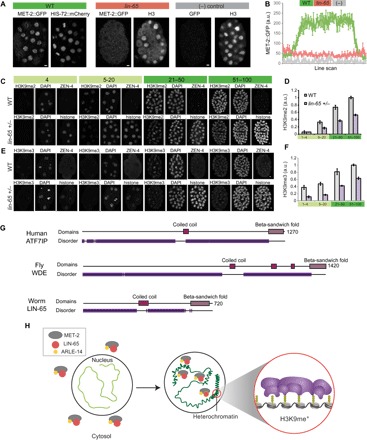
(A) Distribution of MET-2::GFP in WT versus lin-65 mutants versus no-GFP WT strain. Scale bars, 2 μm. Note that this H3 antibody detects mostly cytosolic histone H3 during mitosis. (B) Line-scan analysis across embryonic nuclei shows mean MET-2::GFP intensity in WT (green) versus lin-65 (pink) mutants versus no-GFP control (“−,” gray). Average of line scans across multiple nuclei is shown, and error bars denote SEM. (C and E) H3K9me2 and H3K9me3 levels in embryos with a single-copy ZEN-4::GFP (“WT,” on-slide control) or progeny of lin-65(+/−) heterozygous mothers identified by HIS-72::mCherry at designated stages of embryogenesis. (D and F) Quantitation of H3K9me2 and H3K9me3 levels from WT (gray) or lin-65(+/−) (purple) offspring. Error bars denote SEM. (G) Domain architecture from HMMER (www.ebi.ac.uk/Tools/hmmer/) for human ATF7IP, fly Windei (WDE), and worm LIN-65 showing disordered (purple), coiled-coil (pink), and β-sandwich fold (violet) regions. (H) In early embryos, MET-2 (gray), LIN-65 (red), and ARLE-14 (yellow) are enriched in the cytosol, and there is little H3K9me2 or heterochromatin (light green). As embryos mature, MET-2 and interactors gradually accumulate in nuclei, form concentrated nuclear hubs, and deposit H3K9me2. MET-2–dependent H3K9me is required to generate heterochromatin domains (compacted, dark green).
MET-2 is required for nuclear accumulation of LIN-65 in adult worms upon mitochondrial stress (13). We wondered whether a similar relationship between MET-2 and LIN-65 might exist in embryos in the absence of mitochondrial stress. LIN-65::3xFLAG was nuclear in WT embryos but cytoplasmic in met-2 mutants (fig. S5C). These results show that LIN-65 and MET-2 are dependent on each other for nuclear localization.
The NLS experiment suggested that nuclear accumulation of MET-2 might be rate-limiting for H3K9me2 and heterochromatin formation. To test this idea further, we reduced the dose of MET-2 or LIN-65 by examining embryos from met-2/+ or lin-65/+ heterozygotes. Embryos from met-2/+ mothers behaved like WT, with normal levels and distribution of MET-2 and H3K9me2 (fig. S5, D and E), suggesting that MET-2 is dosage-compensated. A half-dose of lin-65 led to reduced accumulation of MET-2::GFP within nuclei and an increase of cytoplasmic MET-2 (fig. S5F). The slope of H3K9me2 was reduced by half, from 0.32 in the WT (R2 = 0.99) to 0.16 in the lin-65 heterozygotes (R2 = 0.99). H3K9me3 was also reduced but less markedly from a slope of 0.22 (R2 = 0.96) to 0.18 (R2 = 0.95). Gastrula embryos had approximately half the level of age-matched controls (Fig. 6, C to F; P = 1.5 × 10−14 for H3K9me2 and P = 8.8 × 10−13 for H3K9me3). These results indicate that LIN-65 is rate-limiting for nuclear accumulation of MET-2 and H3K9me.
lin-65 belongs to the synMuv B subclass of regulators, which are involved in chromatin regulation and transcriptional repression. Although lin-65 had been annotated as a novel protein (13, 20), we found similarities between LIN-65 and the cofactor activating transcription factor 7–interacting protein (ATF7IP; Fig. 6G). Like ATF7IP, LIN-65 has a high-probability coiled-coil region predicted by PCOILS and a high-confidence β-sandwich in the C terminus embedded within extensive disordered sequences (fig. S5, G and H). Like LIN-65, ATF7IP binds and localizes SETDB1 to nuclei (21). Despite these similarities, LIN-65 is not an obvious ortholog of ATF7IP and may be an example of convergent evolution.
DISCUSSION
De novo generation of heterochromatin domains is a conserved feature of embryogenesis, but it was previously unclear how timing was established. This study revealed that heterochromatin onset depends on the gradual accumulation of MET-2 within nuclei. Relocation depends on two MET-2 binding partners: LIN-65 is critical for MET-2 nuclear localization, and ARLE-14 is important for MET-2 to associate with chromatin. Naturally, these activities do not rule out additional roles for MET-2 regulation by LIN-65 and ARLE-14.
Similar to C. elegans, mammals and Drosophila rebuild heterochromatin domains during embryogenesis (1, 22). More generally, lack of heterochromatin domains appears to be a feature of undifferentiated cells, including embryonic stem cells and planarian neuroblasts, and differentiation involves reestablishing heterochromatin (1). Examination of previous studies suggests that murine SETDB1 is cytoplasmically enriched in early embryos, but its function has been difficult to address owing to early lethality (23). An intriguing idea is that nuclear localization of SETDB with ATF7IP and ARL14EP initiates heterochromatin formation in other animals as well.
The localization of MET-2 in C. elegans cells had previously been controversial. One study argued that MET-2 was cytoplasmic (8), while another demonstrated that MET-2 was required for nuclear localization of LIN-65 under mitochondrial stress conditions, but MET-2 itself was assumed to be cytosolic (13). On the other hand, ChIP experiments had suggested that MET-2 associated with chromatin, implying a nuclear focus (14, 15). One possibility is that excess MET-2 may overwhelm LIN-65 and accumulate in the cytosol under conditions where native MET-2 would be nuclear.
What is the function of de novo H3K9me and heterochromatin formation during embryogenesis? It has been proposed that generation of heterochromatin is critical for loss of pluripotency and restriction of cell fate during gastrulation. In support of this notion, H3K9me can act as an epigenetic barrier against reprogramming cells into a pluripotent state (24). In addition, absence of heterochromatin in early embryos may provide a brief window where repetitive sequences are transcribed, which is a prerequisite for initiating silencing (25).
Alterations in H3K9me have been observed in polycystic ovary syndrome (PCOS), a frequent cause of infertility in women (26, 27). PCOS leads to a range of defects including aberrant oocyte morphology and an increased incidence of miscarriage (28). ARL14EP was identified by genome-wide association studies as a promising candidate for PCOS (29). It is intriguing to speculate that there may be a link between ARL14EP and H3K9me in PCOS.
MATERIALS AND METHODS
Number of experiments and embryos surveyed
For imaging experiments, first, many embryos were surveyed under the microscope through the eye piece and general trends were noted. Then, a random subset of embryos was imaged and analyzed more deeply, with quantitation. Details of analysis are described separately in the “Microscopy and image analysis” section, and the analysis gave the same qualitative result as the trends observed in the initial survey.
In Fig. 1C, >50 embryos were surveyed for each histone mark. At least five WT embryos from each developmental stage were imaged and analyzed in n = 3 experiments.
TEM in met-2 mutants was performed in n = 2 experiments in Fig. 2A. For the latest stages of embryogenesis (>200 cells), in the first experiment, 49 nuclei across 8 embryos were surveyed, and in the second experiment, 71 nuclei across 13 embryos were surveyed.
In Fig. 3A, >100 embryos were surveyed. A total of 37 embryos were imaged and analyzed in n = 3 experiments. In Fig. 3D, >50 embryos were surveyed for each strain. Fourteen WT and 14 NLS embryos were imaged and analyzed in n = 3 experiments. In Fig. 3E, >50 additional embryos for each strain were surveyed. Twenty-five WT and 34 NLS embryos were imaged and analyzed in n = 3 experiments.
We analyzed the following number of embryos in n = 3 experiments in Fig. 4A: H3K9me1: WT versus met-2(ok2307) (5, 7), WT versus lin-65(n3441) (8, 12), and WT versus arle-14(tm6845) (6, 7). H3K9me2: WT versus met-2(ok2307) (11, 20), WT versus lin-65(n3441) (10, 13), and WT versus arle-14(tm6845) (22, 21). H3K9me3: WT versus met-2(ok2307) (14, 12), WT versus lin-65(n3441) (6, 6), and WT versus arle-14(tm6845) (5, 6). In Fig. 4I, 41 embryos were analyzed in n = 3 experiments. In Fig. 4J, 30 embryos were analyzed in n = 3 experiments. In Fig. 4K, for MET-2/ARLE-14 PLA, a total of 20 embryos were analyzed in n = 3 experiments. For MET-2/LIN-65 PLA, a total of 28 embryos were analyzed in n = 3 experiments.
More than 30 mutant embryos were surveyed in Fig. 5A. Thirty-one WT embryos and 21 arle-14(tm6845) mutant embryos were imaged and analyzed in n = 3 experiments. In Fig. 5C, >50 embryos were surveyed for each strain. Eleven WT versus 11 lin-65(n3441) mutant embryos and 8 WT versus 4 met-2(ok2307) mutant embryos were imaged and analyzed in n = 3 experiments. In Fig. 5E, two biological replicates were processed in parallel. In Fig. 5I, >40 embryos were surveyed for each strain. Ten WT and 19 arle-14(tm6845) mutant embryos were imaged and analyzed in n = 3 experiments.
In Fig. 6A, >100 lin-65 embryos were surveyed. Fourteen WT and 31 lin-65 embryos were imaged and analyzed in n = 3 experiments. In Fig. 6C, >50 lin-65 +/− embryos were surveyed. Twenty-two WT and 25 lin-65 +/− embryos were analyzed in n = 3 experiments. In Fig. 6E, >50 lin-65 +/− embryos were surveyed. Fourteen WT and 16 lin-65 +/− embryos were analyzed in n = 3 experiments.
Strains
Worms were maintained at 20°C according to Brenner (30), unless stated otherwise:
(1) N2 (WT, Bristol).
(2) RB1789 met-2(ok2307) III, provided by the C. elegans Gene Knockout Project at Oklahoma Medical Research Foundation.
(3) MT13232 lin-65(n3441) I (20).
(4) SM2078 stIs10389 (pha-4::gfp::3xflag); pha-4 (q500) rol-9 (sc148) (31), integrated array strain.
(5) SM2333 pxSi01 (zen-4::gfp, unc-119+) II; unc-119(ed3) III, generated by Mos1-Mediated Single-Copy Insertion (MosSCI).
(6) SM2529 arle-14/B0336.5(tm6845) III, provided by the Japanese National Bioresource Project.
(7) JAC500 his-72(csb43[his-72::mCherry]) III, provided by Norris et al. (32).
(8) EL597 omIs 1 [Cb-unc-119 (+) met-2::gfp II], generated by MosSCI.
(9) SM2491 omIs 1 [Cb-unc-119 (+) met-2::gfp II]; met-2(ok2307) unc-119 (ed3) III.
(10) SM2533 omIs1 [Cb-unc-119 (+) met-2::gfp II]; arle-14(tm6845) III.
(11) SM2536 omIs1 [Cb-unc-119 (+) met-2::gfp II]; lin-65 (n3441) I.
(12) SM2532 [Cb-unc-119 (+) met-2::gfp II]; his-72(csb43[his-72::mCherry]) III.
(13) EL634 3xflag::met-2 III. This study, generated by CRISPR.
(14) SM2575 lin-65::3xflag I. This study, generated by CRISPR.
(15) SM2576 arle-14(tm6845) III, lin-65::3xflag I. This study.
(16) SM2578 met-2(ok2307) III, lin-65::3xflag I. This study.
(17) SM2580 NLS::3xflag::met-2 III. This study, generated by CRISPR.
Antibody staining
Antibody staining was performed as described previously (33). Antibodies were used for immunostaining after fixation with 2% paraformaldehyde (PFA) for 5 min and cold methanol for 3 min (for all except ARLE-14 and MET-2 antibodies, which was fixed with 2% PFA for 10 min and methanol for 3 min). For mutant configurations, an on-slide WT sample was included, marked with a single-copy ZEN-4::GFP tag. On-slide controls allowed better quantitation between different genotypes or stages. A description of how antibodies against endogenous MET-2 or ARLE-14 were generated can be found in a separate section.
List of antibodies and dilution used
The following were the antibodies (dilution) used in this study:
(1) H3K9me1 (1:200) Abcam ab8896
(2) H3K9me2 (1:200) Abcam ab1220, Kimura 6D11–MABI0307
(3) H3K9me3 (1:200) Kimura 2F3–MABI0308
(4) H3K27me3 (1:200) Active Motif 61017
(5) H4-pan acetyl (1:500) Active Motif 39925
(6) Pan-histone (1:500) Chemicon/Millipore MAB052
(7) Histone H3 (1:500) Abcam ab1791
(8) FLAG M2 (1:100) Sigma-Aldrich F1804
(9) GFP (1:500) MilliporeSigma MAB3580
(10) GFP (1:500) Thermo Fisher Scientific A11122
(11) MET-2 (1:500) rabbit polyclonal, raised against the first 17 amino acids of MET-2 and affinity-purified.
(12) ARLE-14 (1:500) rabbit polyclonal, raised by Covance.
Generation of MET-2, LIN-65, ARLE-14, ZEN-4, and HIS-72 reagents
MET-2
To generate EL634, we inserted the 3xFLAG tag (DYKDHDGDYKDHDIDYKDDDDK) at the endogenous met-2 locus using CRISPR (34). The construct places the tag at the N terminus of MET-2 and is inserted immediately after the start codon without any linker sequences. To generate EL597, we inserted a single copy of MET-2::GFP with its endogenous promoter and upstream gene R05D3.2 at the ttTi5605 locus on chromosome II by MosSCI. The GFP tag was placed at the C terminus of MET-2 and was inserted immediately before the stop codon without any linker sequences. Both the CRISPR construct and the MosSCI construct could rescue H3K9me2 deposition, although the CRISPR allele did so better than the MosSCI allele. The MET-2 antibody was generated against the first 17 amino acids of endogenous MET-2 and affinity-purified.
To generate SM2580 (NLS::3xFLAG::MET-2), the c-Myc NLS sequence (CCAGCCGCCAAGCGTGTCAAGCTCGAC) was added directly upstream of 3xFLAG::MET-2 without any linker sequence by CRISPR (34). For the insertion, the 3xFlag sequence in EL634 was targeted by the following guide RNA: ATGGACTACAAAGACCATGA(CGG). The dpy-10 locus was used as a phenotypic marker. Because it segregated independently from the met-2 locus, nonroller non-dpy worms were isolated for further analysis by single worm PCR and genotyping. The edit was confirmed by sequencing the 200–base pair (bp) region around the insertion. CRISPR RNA (crRNA), transactivating crRNA (tracrRNA), and CRISPR-associated protein 9 (Cas9) protein were ordered from the Integrated DNA Technologies (IDT) Alt-R genome editing system. The 97-bp repair template was synthesized and polyacrylamide gel electrophoresis (PAGE)–purified by IDT.
LIN-65
We inserted a 3xFLAG at the endogenous lin-65 locus using CRISPR (34). The sequence of the guide RNA was TCATTCGAGAGTGATGAAGG(TGG). The 3xFLAG tag was located at the C terminus of LIN-65 and was inserted directly before the stop codon without any linker sequences. The dpy-10 locus was used as a phenotypic marker. Because it segregated independently from the lin-65 locus, nonroller non-dpy worms were isolated for further analysis by single worm PCR and genotyping. The edit was confirmed by sequencing the 200-bp region around the insertion. crRNA, tracrRNA, and Cas9 protein were ordered from the IDT Alt-R genome editing system. The 136-bp repair template was synthesized and PAGE-purified by IDT.
ARLE-14
An antibody against endogenous ARLE-14 was generated. Bacteria containing arle-14 complementary DNA (cDNA) in a pET-47b(+) (#71461, Novagen) plasmid backbone were grown at 30°C for 19 hours in LB + kanamycin (50 μg/ml). The culture was diluted 1:5 in LB + kanamycin, and protein expression was induced with 0.25 mM isopropyl-β-d-thiogalactopyranoside at 30°C for 3 hours. Bacteria were pelleted and flash-frozen at −80°C. The pellet was resuspended in 20 ml of lysis buffer [50 mM tris (pH 7.2), 300 mM NaCl, 5 mM β-ME, and 10% glycerol] and digested with 200 μl of lysozyme (50 mg/ml; #90082, Thermo Fisher Scientific) for 30 min on ice. Following lysozyme digestion and sonication on ice (four cycles, 30 s ON, 1 min OFF; output control 3, duty cycle 50%, pulsed), the protein was purified from inclusion bodies as follows: The sample was centrifuged at 4000 rpm at 4°C for 15 min and the supernatant was discarded. The pellet was resuspended in 20 ml of lysis buffer with 1% Triton X-100 and 200 μl of TURBO DNase (deoxyribonuclease; Thermo Fisher Scientific AM2239) and incubated for 20 min at room temperature. The sample was sonicated and centrifuged again with the same settings as before, and the supernatant was discarded. The pellet was rinsed once with dilution buffer [10 mM tris-Cl (pH 7.5),150 mM NaCl, and 0.5 mM EDTA] and resuspended in denaturation buffer [50 mM tris-HCl (pH 8), 300 mM NaCl, 2 mM β-ME, 5 mM MgCl2, and 6 M urea] by gentle rocking on a shaker at room temperature for 1 hour. The solution was dialyzed against 50 mM tris-Cl (pH 8), 150 mM NaCl, 5 mM MgCl2, and protein (1 mg/ml) and was sent to Covance for injections into rabbits. Total immunoglobulin G purification was performed by Covance after the final bleed. For in vivo imaging, the antibody solution was precleared overnight with arle-14(tm6845) mutant embryos before use. Protocol described in the “Antibody staining” section was followed to prepare arle-14(tm6845) embryos and to stain them with the ARLE-14 antibody. The resulting precleared antibody solution was transferred to a fresh tube, stored at 4°C for <1 week, and used in staining experiments.
ZEN-4
ZEN-4::GFP was amplified from bsem1129 (35) with zen-4_uni_5′_nested_2_attB1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTGCAAAAAGTCGCATCTGGGAA; attB1 underlined) and unc-54_3′UTR_Hobert_nested_3′_attB2 (GGAAACAGTTATGTTTGGTATATTGGGACCCAGCTTTCTTGTACAAAGTGGTCCCC; attB2 underlined) primers using TaKaRa PrimeSTAR (35). The resulting attB-flanked PCR product was recombined into pCFJ151 (Addgene) using Gateway BP Clonase II (Invitrogen/Thermo Fisher Scientific) to create bsem1267. SM2333 was generated by injecting bsem1267 along with pCFJ601, pMA122, pGH8, pCFJ90, and pCFJ104 (all available from Addgene) into SM2288 [ttTiS605 II; unc-119(ed3)III]. The mosSCI protocol on www.wormbuilder.org was used to generate single integrants. SM2333 was used as an on-slide WT control in antibody stains.
HIS-72
The mCherry tag was inserted at the C terminus of the endogenous his-72 locus by CRISPR. Briefly, JAC499 was injected with Cre recombinase to remove the selection cassette and produce a functional HIS-72::mCherry protein. JAC500 his-72(csb43[his-72::mCherry]) III was used as a histone control in MET-2::GFP stains (Fig. 2C) and to mark cross-progeny after mating (Fig. 4H and fig. S4E). The mCherry tag did not interfere with H3K9me2 (fig. S2C).
Proximity ligation assay
Sigma Duolink In Situ Kit (DUO92101) was used for this assay. Embryos were fixed as for regular antibody staining. After overnight staining with primary antibodies at 15°C, the sample was stained at 37°C for 1 hour with secondary antibodies that have oligonucleotide probes attached. Connector oligos were hybridized to the probes and served as templates for circularization by enzymatic ligation when in close proximity. The ligation reaction was incubated at 37°C for 30 min. The circularized DNA strands were used for rolling circle amplification (RCA), and the RCA product was detected by hybridizing fluorescently labeled oligos. The RCA reaction was incubated at 37°C for 100 min. After each step, slides were washed with tris-buffered saline (TBS) + 0.2% Triton X-100 for 5 min. Slides were mounted in DAPI.
Microscopy and image analysis
Histone modifications
Stacks of optical sections were collected with a ZEISS LSM700 or LSM880 Confocal Microscope and analyzed using Volocity software. Signal intensity for histone modifications was normalized to signal intensity for unmodified histones for each nucleus. Normalized values were averaged at given embryonic stages and plotted (Figs. 1D, 2, D, E, and J, 3B, and 4E, and figs. S1D and S3, C and D).
Quantitation of histone modifications by Volocity
Nuclei were identified in three dimensions using the DAPI channel, and sum signal intensity of histone modification was calculated for each nucleus in interphase. Mitotic nuclei were excluded from the analysis manually based on DAPI morphology. Mean cytoplasmic background was measured at a random point for each embryo and was used to extrapolate the sum background signal for each nucleus, which depends on nuclear volume. To calculate the total amount of background signal in the nucleus, mean cytosolic signal was multiplied by the voxel counts for each nucleus. Sum background intensity was subtracted from the sum signal intensity for that nucleus. For each nucleus, signal intensity of the histone modification was normalized to signal intensity of histones. Unmodified histones were similar between different genotypes and served as a staining control. Note that sum signal intensity for any mark positively correlates with the volume of the nucleus (36), and normalizing the sum intensity of marks to histone H3 controls for changes in nuclear volume at different stages of embryogenesis.
Transmission electron microscopy (TEM)
Adult hermaphrodites were treated by high-pressure freeze fixation and freeze substitution into 2% osmium tetroxide and 0.1% uranyl acetate in acetone, rinsed in 4% distilled H2O in acetone, and then rinsed in propylene oxide before embedding in plastic resin. The adults were thin-sectioned, lengthwise onto Pioloform-coated slot grids on a diamond knife. The thin sections were counterstained with uranyl acetate and then with lead citrate (4) before examination in a Philips CM10 electron microscope. Fertilized early embryos could be found in a row within the uterus of each adult, and sample images of nuclei from each embryo were collected at high resolution with an Olympus Morada digital camera. Images were collected at intervals across serial sections of each embryo to sample many different nuclei. The digital camera offers a much greater range of contrast levels compared to EM film images, and it becomes possible to detect weakly staining objects with minimal poststaining, just by digital contrast enhancement. However, we applied similar amounts of counterstain to each sample to avoid artifacts, spurious results, or undue contrast enhancement by digital manipulations (37).
TEM image processing
Raw images were processed with Photoshop as 8-bit grayscale images. Images from different preparations were standardized for accurate comparison. The cytoplasm was used to adjust signal intensity range for each image, but image contrast was not altered. Adjusted images were then saved and quantified with ImageJ by line-scan analysis.
TEM/H3K9me3 line-scan analysis
Random lines were drawn across the center of different nuclei, and the intensity was measured. SD was calculated for each individual line. The SD of 30 lines was averaged for each strain and listed on the plot. A randomly selected line profile for a nucleus is shown as an example (Fig. 2B for TEM and Fig. 4D for H3K9me3). The SD describes the morphology of the nucleus; that is, a higher SD stems from a more punctate staining pattern that alternates between high and low values (that is, electron-dense heterochromatin and electron-lucent nucleoplasm or H3K9me3-positive and -negative regions in the nucleus).
TEM percentage of EDR
A black-and-white image was created for each nucleus in ImageJ using the thresholding function. The threshold was manually chosen to best reflect EDRs in >200-cell WT embryos and was used to quantify all nuclei (threshold, 39 to 150). The inner nuclear membrane was manually traced to define an outermost ring, and the number of black pixels versus total pixels within the ring was used to quantify %EDRs.
MET-2/ARLE-14/LIN-65 line-scan analysis
Lines that go through the center of the nucleus were drawn across the cell, and the intensity was measured. Each line had 100 bins. The intensity in each bin was averaged for 30 lines, and the average line was plotted. Error bars denote the SEM at each bin (Figs. 3G, 5, B and D, 6B, and fig. S5, C and F).
Definition and quantitation of hubs
Nuclear hubs were defined by intensity thresholding in ImageJ. The threshold was selected manually in WT nuclei at the 51- to 100-cell stage, and the same threshold was applied to all the images in a given data set. The intensity measurements for each defined hub were averaged to yield the intensity of “hubs” at given embryonic stages. “Nonhub” was defined as nuclear areas that were below the intensity threshold. The mean intensity in nonhub areas was measured for each nucleus and averaged across 30 nuclei. Noninterphase nuclei were discarded manually. For intensity measurements in the cytosol, at least four random areas in the cytosol were chosen for every cell that was in interphase. The measured intensities were averaged.
Half-dose LIN-65 experiments
lin-65 (n3441) moms were crossed with JAC500 his-72::mCherry males. Progeny of lin-65 +/− heterozygotes marked by mCherry were analyzed. On the same slide, SM2333 containing a single copy of zen-4::gfp was used as a WT staining control. Mean H3K9me2 or H3K9me3 intensity in each nucleus was quantified using Volocity, and the average H3K9me2 or H3K9me3 intensity per nucleus was plotted and normalized as described in the “Quantitation of histone modifications by Volocity” section.
Biochemistry and bioinformatics
Harvesting embryos
Mix-staged embryos were collected from adult worms by bleaching. Embryos were shaken at 200 rpm at 20°C in Complete S Medium [100 mM NaCl, 5.6 mM K2HPO4, 4.4 mM KH2PO4, cholesterol (10 μg/ml), 10 mM potassium citrate, 2 mM CaCl2, 2 mM MgSO4, and 1× trace metals] without food (38). Once the embryos hatched and became L1s, concentrated NA22 bacteria were added to the culture. Synchronized embryos were harvested by bleaching after 62 to 66 hours when most worms carried one to eight embryos, frozen in liquid nitrogen, and stored at −80°C.
Immunoprecipitation
Frozen embryo pellets were resuspended in lysis buffer [50 mM Hepes (pH 7.4), 1 mM EGTA, 1 mM MgCl2, 100 mM KCl, 10% glycerol, and 0.05% NP-40] with protease inhibitors (Calbiochem Cocktail Set I, #539131) and incubated on ice for 10 min. Using the QSonica Q800 Sonicator, samples were sonicated at 40% amplitude, 10 s ON and 50 s OFF, for 3 cycles. After sonication, samples were centrifuged for 10 min at 10,000g at 4°C. The supernatant was transferred to a new tube and diluted in dilution buffer [10 mM tris-Cl (pH 7.5), 150 mM NaCl, and 0.5 mM EDTA]. Total lysate (1.5 mg) was precleared for 1 hour at 4°C with 25 μl of Chromotek magnetic agarose beads before immunoprecipitation. The precleared lysate was then used for immunoprecipitating MET-2::GFP with Chromotek GFP-Trap magnetic agarose beads or Sigma FLAG M2 antibody coupled to magnetic agarose beads for 5 hours at 4°C. Beads were rinsed with dilution buffer three times, washed with dilution buffer twice for 5 min, and eluted with 50 μl of 0.2 M glycine (pH 2.5) for 30 s under constant mixing or with 3xFLAG peptide (200 μg/ml) in TBS for 1 hour at 4°C. Five microliters of 1 M tris base (pH 10.4) was added for neutralization after glycine elution. Samples were boiled in 2× Laemmli sample buffer (#161-0737, Bio-Rad) with 50 mM dithiothreitol and analyzed by Western blotting or silver staining (SilverQuest Silver Staining Kit, LC6070, Thermo Fisher Scientific).
Mass spectrometry
Reagents and chemicals
Deionized water (18.2 megohms; Barnstead) was used for all preparations. Buffer A consists of 5% acetonitrile and 0.1% formic acid, buffer B consists of 80% acetonitrile and 0.1% formic acid, and buffer C consists of 500 mM ammonium acetate and 5% acetonitrile.
Sample preparation
Proteins were precipitated in 23% trichloroacetic acid (product number T-0699, Sigma-Aldrich) at 4°C overnight. After 30-min centrifugation at 18,000g, protein pellets were washed two times with 500 μl of ice-cold acetone. Air-dried pellets were dissolved in 8 M urea/100 mM tris (pH 8.5). Proteins were reduced with 1 M tris(2-carboxyethyl)phosphine hydrochloride (product number C4706, Sigma-Aldrich) and alkylated with 500 mM 2-chloroacetamide (product number 22790-250G-F, Sigma-Aldrich). Proteins were digested for 18 hours at 37°C in 2 M urea, 100 mM tris (pH 8.5), and 1 mM CaCl2 with 2 μg of trypsin (product number V5111, Promega). Digestion was stopped with formic acid, 5% final concentration. Debris was removed by centrifugation at 18,000g for 30 min.
Multidimensional protein identification technology microcolumn
A multidimensional protein identification technology (MudPIT) microcolumn (39) was prepared by first creating a Kasil frit at one end of an undeactivated 250-μm–inner diameter (ID)/360-μm–outer diameter (OD) capillary (Agilent Technologies Inc.). The Kasil frit was prepared by briefly dipping a 20- to 30-cm capillary in well-mixed 300 μl of Kasil 1624 (PQ Corporation) and 100 μl of formamide, curing at 100°C for 4 hours, and cutting the frit to ~2 mm in length. Strong cation exchange particles (SCX Partisphere, 5 μm diameter, 125 Å pores; Phenomenex) were packed in-house from particle slurries in methanol (2.5 cm). Additional 2.5-cm reversed-phase particles (C18 Aqua, 3 μm diameter, 125 Å pores; Phenomenex) were then similarly packed into the capillary using the same method as SCX loading to create a biphasic column. An analytical reversed-phase liquid chromatography (LC) column was generated by pulling a 100-μm-ID/360-μm-OD capillary (Polymicro Technologies Inc.) to a 5-μm-ID tip. Reversed-phase particles (Aqua C18, 3-μm diameter, 125 Å pores; Phenomenex) were packed directly into the pulled column at 5.5 MPa until 12 cm long. The MudPIT microcolumn was connected to an analytical column using a zero-dead volume union [P-720-01, Upchurch Scientific (IDEX Health & Science)]. LC–tandem mass spectrometry (MS/MS) analysis was performed using an Agilent Technologies 1200 high-performance LC pump and a Thermo Orbitrap Velos using an in-house built electrospray stage. MudPIT experiments were performed with steps of 0% buffer C, 30% buffer C, 50% buffer C, 90/10% buffer C/B, and 100% buffer C, being run for 3 min at the beginning of each gradient of buffer B. Electrospray was performed directly from the analytical column by applying the electrospray ionization voltage at a tee (150 μm ID; Upchurch Scientific) (39). Electrospray directly from the LC column was performed at 2.5 kV with an inlet capillary temperature of 325°C. Data-dependent acquisition of MS/MS spectra with the Orbitrap Velos was performed with the following settings: MS/MS on the 10 most intense ions per precursor scan; 1 microscan; reject unassigned charge state and charge state 1; dynamic exclusion repeat count, 1; repeat duration, 30 s; exclusion list size, 200; and exclusion duration, 30 s.
Data analysis
Protein and peptide identification and protein quantitation were carried out with Integrated Proteomics Pipeline (Integrated Proteomics Applications Inc.; www.integratedproteomics.com/). Tandem mass spectra were extracted from raw files using RawConverter (40) with monoisotopic peak option and were searched against WormBase protein database (WB257) with reversed sequences using ProLuCID (41, 42). The search space included all half and fully tryptic peptide candidates. Carbamidomethylation (+57.02146) of cysteine was considered as a static modification. Peptide candidates were filtered using DTASelect with the parameters -p 2 -y 1 --trypstat --pfp 0.01 --extra --pI -DM 10 --DB --dm -in -m 1 -t 2 --brief --quiet (43). Mass spectrometry identified a total of 602 proteins in the MET-2::GFP sample. Five hundred proteins were eliminated because of their presence in controls, and 43 proteins were eliminated because they had low sequence coverage (<6%). The remaining 59 proteins were further analyzed based on suggested function and localization from literature. Promising candidates were chosen for a loss-of-function screen.
RNA expression analysis
Embryos frozen in liquid nitrogen were partially thawed. An equal volume of glass beads (G8772, Sigma) was added, and samples were vortexed for 1 min. For TRIzol-chloroform extraction, 1 ml of TRIzol (#15596026, Thermo Fisher Scientific) was added to the sample, vortexed for 30 s, and incubated at room temperature for 5 min. Three hundred microliters of chloroform (Omnipur #3155, Calbiochem) was added, shaken by hand, and incubated at room temperature for 3 min. The sample was centrifuged at 14,000 rpm at 4°C for 15 min. Upper aqueous phase was transferred to a fresh tube, and RNA was precipitated using 500 μl of isopropanol and 1 μl of GlycoBlue (AM9516, Thermo Fisher Scientific). The pellet was air-dried and resuspended in nuclease-free water at room temperature for 10 min. Samples were treated with DNase at 37°C for 30 min using the TURBO DNA-free kit (AM1907, Thermo Fisher Scientific), and 500 ng of RNA was used for reverse transcription with a random primer mix (ProtoScript First Strand cDNA Synthesis Kit, E6300, NEB). The synthesis reaction was diluted in water to yield a total volume of 50 μl, and 3 μl of the cDNA was analyzed by qPCR (KAPA SYBR FAST qPCR Master Mix, KK4601). The following program was used for qPCR: 3 min at 95°C (3 s at 95°C, 15 s at 55°C, and 30 s at 72°C) for 40 cycles, followed by a melting curve. Reverse transcription reactions without the enzyme and water served as negative controls.
Chromatin immunoprecipitation
We used twice as much material for arle-14 mutants compared to WT because of the lower level of H3K9me2 in arle-14 mutants. H3K9me2 ChIP was performed as described previously (31) with the following changes: Embryos frozen in liquid nitrogen were thawed on ice and fixed in 1.5% formaldehyde (#15686, Electron Microscopy Sciences) for 15 min at room temperature. Using the QSonica Q800 Sonicator, samples were sonicated at 30% amplitude, 30 s ON and 30 s OFF, for a total of 15 min at 4°C, yielding 100- to 300-bp fragments. Six microliters of Kimura 6D11 antibody was coupled to 25 μl of beads (Pierce Protein A/G magnetic beads, #88802) for 8 hours at 4°C before ChIP. Forty micrograms of chromatin was used per ChIP reaction, and chromatin was precleared for 2 hours at 4°C using uncoupled magnetic beads (Pierce Protein A/G magnetic beads, #88802). To elute the bound immunocomplexes, 150 μl of elution buffer (50 mM NaHCO3, 140 mM NaCl, and 1% SDS) was added to each tube and heated at 65°C for 15 min. For MET-2::GFP ChIP, embryos were fixed with 1.5 mM ethylene glycol bis(succinimidyl succinate) (EGS) (Pierce Protein A/G magnetic beads, #21565) for 10 min and with 1% formaldehyde for another 10 min. Six microliters of GFP antibody (Abcam5665) was coupled to 25 μl of beads for 8 hours at 4°C before ChIP. Two microliters of H3K4me3 (Abcam8580) antibody was coupled to 25 μl of beads for 8 hours at 4°C before ChIP.
Library preparation
The ChIP-seq libraries were generated by using the Apollo 324 System and the PrepX ILM DNA Library Kit from IntegenX. After adaptor ligation, the input and ChIP DNA were enriched by PCR amplification using the NEBNext High-Fidelity 2X PCR Master Mix with Q5 polymerase and PrepX PCR primer with the following PCR conditions: 30 s at 98°C (10 s at 98°C, 30 s at 60°C, and 30 s at 72°C) for 8 cycles for input libraries and 11 cycles for ChIP libraries, following 5 min at 72°C (~15 μl of adaptor-ligated DNA, 25 μl of NEBNext High-Fidelity 2X PCR Master Mix, and 2 μl of universal PCR primer, brought to 50 μl with nuclease-free water). The enriched DNA was then purified using 50 μl (1:1 ratio of DNA volume and beads) of PCRClean DX Beads (Aline) and size-selected by Pippin Prep (180 to 600 bp). One microliter of each library was applied to measure the concentration using a Qubit dsDNA assay kit (Invitrogen). One nanogram of DNA from each library was checked by a TapeStation (Agilent Technologies). Input and ChIP libraries were pooled such that they each had the same amount of molecules expected for obtaining a similar number of reads. The Illumina sequencing was performed with 75-nucleotide (nt) paired-end reads.
Sequencing analysis
DNA fragments were sequenced on an Illumina HiSeq machine, yielding 64 to 95 million 75-nt paired-end reads per sample. Reads were processed as described at www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113841. Briefly, reads were aligned to the C. elegans reference genome (ce10) with Bowtie2, version 2.3.1, and default parameters except for “--very-sensitive.” MACS2, version 2.1.1.20160309, was used to call peaks in the ChIP samples, using input DNA as the control and analyzing only properly paired fragments (-f BAMPE). Log-likelihood ratio tracks were calculated using the MACS2 module “bdgcmp,” and correlations were calculated using the University of California Santa Cruz tools “bedGraphToBigWig” and “wigCorrelate.” The computations were run on the Odyssey cluster supported by the Faculty of Arts and Sciences Division of Science, Research Computing Group at Harvard University.
Data access
The ChIP-seq data from this study have been submitted to the National Center for Biotechnology Information Gene Expression Omnibus database with accession number GSE113841.
Supplementary Material
Acknowledgments
We thank A. Schier, D. Moazed, and the Mango Lab for comments on the manuscript; S. Von Stetina for zen-4::gfp; J. Calarco and A. Norris for his-72::mCherry; the Harvard Center for Biological Imaging, K. Nguyen, and B. Raja for their help on TEM procedures; and Maine Lab members, especially X. Xu, for their helpful input. Funding: We acknowledge support from the NIH (R37GM056264 to S.E.M., RO1 GM089818 to E.M.M., and R24OD010943 to D.H.H.), the John D. and Catherine T. MacArthur Foundation and Harvard University (to S.E.M.), and the American Association of University Women International Fellowship (to B.M.). B.D.O., J.J.M., and J.R.Y. were supported by the National Institute of General Medical Sciences (8 P41 GM103533). Some strains were provided by the Caenorhabditis Genetics Center, funded by NIH P40OD010440. Author contributions: B.M. and S.E.M. designed and conducted the study and wrote the manuscript. D.H.H. performed TEM. H.-M.C. performed TEM image processing. J.J.M., B.D.O., and J.R.Y. performed proteomics. B.Y., S.K.-R., and E.M.M. generated the MET-2 antibody and met-2 CRISPR and MosSCI strains. J.M.G. analyzed H3K9me2 ChIP-seq data. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Reagents can be provided by S.E.M. pending scientific review and a completed material transfer agreement. Requests should be submitted to S.E.M. (smango@mcb.harvard.edu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat6224/DC1
Fig. S1. Heterochromatin formation and establishment of H3K9me in WT embryos.
Fig. S2. Control for TEM conditions in WT versus met-2 mutants.
Fig. S3. MET-2 localization with additional reagents and during cell cycle.
Fig. S4. Specificity controls for MET-2 coimmunoprecipitation and PLA.
Fig. S5. Further analysis of LIN-65 and dosage compensation for MET-2 protein.
Table S1. Spectral counts and sequence coverage for proteins identified in the GFP immunoprecipitation and mass spectrometry experiment.
REFERENCES AND NOTES
- 1.Politz J. C. R., Scalzo D., Groudine M., Something silent this way forms: The functional organization of the repressive nuclear compartment. Annu. Rev. Cell Dev. Biol. 29, 241–270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yuzyuk T., Fakhouri T. H. I., Kiefer J., Mango S. E., The polycomb complex protein mes-2/E(z) promotes the transition from developmental plasticity to differentiation in C. elegans embryos. Dev. Cell 16, 699–710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fakhouri T. H. I., Stevenson J., Chisholm A. D., Mango S. E., Dynamic chromatin organization during foregut development mediated by the organ selector gene PHA-4/FoxA. PLOS Genet. 6, e1001060 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hall D. H., Hartwieg E., Nguyen K. C. Q., Modern electron microscopy methods for C. elegans. Methods Cell Biol. 107, 93–149 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Wirth M., Paap F., Fischle W., Wenzel D., Agafonov D. E., Samatov T. R., Wisniewski J. R., Jedrusik-Bode M., HIS-24 linker histone and SIR-2.1 deacetylase induce H3K27me3 in the Caenorhabditis elegans germ line. Mol. Cell. Biol. 29, 3700–3709 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rice J. C., Briggs S. D., Ueberheide B., Barber C. M., Shabanowitz J., Hunt D. F., Shinkai Y., Allis C. D., Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol. Cell 12, 1591–1598 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Bessler J. B., Andersen E. C., Villeneuve A. M., Differential localization and independent acquisition of the H3K9me2 and H3K9me3 chromatin modifications in the Caenorhabditis elegans adult germ line. PLOS Genet. 6, e1000830 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Towbin B. D., González-Aguilera C., Sack R., Gaidatzis D., Kalck V., Meister P., Askjaer P., Gasser S. M., Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150, 934–947 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Bender L. B., Cao R., Zhang Y., Strome S., The MES-2/MES-3/MES-6 complex and regulation of histone H3 methylation in C. elegans. Curr. Biol. 14, 1639–1643 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Poulin G., Dong Y., Fraser A. G., Hopper N. A., Ahringer J., Chromatin regulation and sumoylation in the inhibition of Ras-induced vulval development in Caenorhabditis elegans. EMBO J. 24, 2613–2623 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andersen E. C., Horvitz H. R., Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development 134, 2991–2999 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Garrigues J. M., Sidoli S., Garcia B. A., Strome S., Defining heterochromatin in C. elegans through genome-wide analysis of the heterochromatin protein 1 homolog HPL-2. Genome Res. 25, 76–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tian Y., Garcia G., Bian Q., Steffen K. K., Joe L., Wolff S., Meyer B. J., Dillin A., Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McMurchy A. N., Stempor P., Gaarenstroom T., Wysolmerski B., Dong Y., Aussianikava D., Appert A., Huang N., Kolasinska-Zwierz P., Sapetschnig A., Miska E. A., Ahringer J., A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. eLife 6, e21666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel T., Hobert O., Coordinated control of terminal differentiation and restriction of cellular plasticity. eLife 6, e24100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul P., van den Hoorn T., Jongsma M. L. M., Bakker M., Hengeveld R. C. C., Janssen L., Cresswell P., Egan D. A., van Ham M. S., ten Brinke A., Ovaa H., Beijersbergen R. L., Kuijl C. P., Neefjes J., A genome-wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell 145, 268–283 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Giot L., Bader J. S., Brouwer C., Chaudhuri A., Kuang B., Li Y., Hao Y. L., Ooi C. E., Godwin B., Vitols E., Vijayadamodar G., Pochart P., Machineni H., Welsh M., Kong Y., Zerhusen B., Malcolm R., Varrone Z., Collis A., Minto M., Burgess S., McDaniel L., Stimpson E., Spriggs F., Williams J., Neurath K., Ioime N., Agee M., Voss E., Furtak K., Renzulli R., Aanensen N., Carrolla S., Bickelhaupt E., Lazovatsky Y., DaSilva A., Zhong J., Stanyon C. A., Finley R. L. Jr., White K. P., Braverman M., Jarvie T., Gold S., Leach M., Knight J., Shimkets R. A., McKenna M. P., Chant J., Rothberg J. M., A protein interaction map of Drosophila melanogaster. Science 302, 1727–1736 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Guruharsha K. G., Rual J.-F., Zhai B., Mintseris J., Vaidya P., Vaidya N., Beekman C., Wong C., Rhee D. Y., Cenaj O., McKillip E., Shah S., Stapleton M., Wan K. H., Yu C., Parsa B., Carlson J. W., Chen X., Kapadia B., VijayRaghavan K., Gygi S. P., Celniker S. E., Obar R. A., Artavanis-Tsakonas S., A protein complex network of Drosophila melanogaster. Cell 147, 690–703 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rolland T., Taşan M., Charloteaux B., Pevzner S. J., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., Kamburov A., Ghiassian S. D., Yang X., Ghamsari L., Balcha D., Begg B. E., Braun P., Brehme M., Broly M. P., Carvunis A.-R., Convery-Zupan D., Corominas R., Coulombe-Huntington J., Dann E., Dreze M., Dricot A., Fan C., Franzosa E., Gebreab F., Gutierrez B. J., Hardy M. F., Jin M., Kang S., Kiros R., Lin G. N., Luck K., MacWilliams A., Menche J., Murray R. R., Palagi A., Poulin M. M., Rambout X., Rasla J., Reichert P., Romero V., Ruyssinck E., Sahalie J. M., Scholz A., Shah A. A., Sharma A., Shen Y., Spirohn K., Tam S., Tejeda A. O., Trigg S. A., Twizere J.-C., Vega K., Walsh J., Cusick M. E., Xia Y., Barabási A.-L., Iakoucheva L. M., Aloy P., De Las Rivas J., Tavernier J., Calderwood M. A., Hill D. E., Hao T., Roth F. P., Vidal M., A proteome-scale map of the human interactome network. Cell 159, 1212–1226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ceol C. J., Stegmeier F., Harrison M. M., Horvitz H. R., Identification and classification of genes that act antagonistically to let-60 Ras signaling in Caenorhabditis elegans vulval development. Genetics 173, 709–726 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herz H.-M., Garruss A., Shilatifard A., SET for life: Biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem. Sci. 38, 621–639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan K., O’Farrell P. H., TALE-light imaging reveals maternally guided, H3K9me2/3-independent emergence of functional heterochromatin in Drosophila embryos. Genes Dev. 30, 579–593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dodge J. E., Kang Y.-K., Beppu H., Lei H., Li E., Histone H3-K9 methyltransferase ESET is essential for early development. Mol. Cell. Biol. 24, 2478–2486 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Becker J. S., Nicetto D., Zaret K. S., H3K9me3-dependent heterochromatin: Barrier to cell fate changes. Trends Genet. 32, 29–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moazed D., Small RNAs in transcriptional gene silencing and genome defence. Nature 457, 413–420 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L., Baek K.-H., Molecular genetics of polycystic ovary syndrome: An update. Curr. Mol. Med. 15, 331–342 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Eini F., Novin M. G., Joharchi K., Hosseini A. K., Nazarian H., Piryaei A., Bidadkosh A., Intracytoplasmic oxidative stress reverses epigenetic modifications in polycystic ovary syndrome. Reprod. Fertil. Dev. 29, 2313–2323 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Palomba S., Daolio J., La Sala G. B., Oocyte competence in women with polycystic ovary syndrome. Trends Endocrinol. Metab. 28, 186–198 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Hayes M. G., Urbanek M., Ehrmann D. A., Armstrong L. L., Lee J. Y., Sisk R., Karaderi T., Barber T. M., McCarthy M. I., Franks S., Lindgren C. M., Welt C. K., Diamanti-Kandarakis E., Panidis D., Goodarzi M. O., Azziz R., Zhang Y., James R. G., Olivier M., Kissebah; A. H. Reproductive Medicine Network, Stener-Victorin E., Legro R. S., Dunaif A., Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat. Commun. 6, 7502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brenner S., The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu H.-T., Chen H.-M., Yang Z., Wang J., Lee N. K., Burger A., Zaret K., Liu T., Levine E., Mango S. E., Recruitment of RNA polymerase II by the pioneer transcription factor PHA-4. Science 348, 1372–1376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Norris A. D., Kim H.-M., Colaiácovo M. P., Calarco J. A., Efficient genome editing in Caenorhabditis elegans with a toolkit of dual-marker selection cassettes. Genetics 201, 449–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiefer J. C., Smith P. A., Mango S. E., PHA-4/FoxA cooperates with TAM-1/TRIM to regulate cell fate restriction in the C. elegans foregut. Dev. Biol. 303, 611–624 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paix A., Folkmann A., Seydoux G., Precision genome editing using CRISPR-Cas9 and linear repair templates in C. elegans. Methods 121–122, 86–93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Von Stetina S. E., Liang J., Marnellos G., Mango S. E., Temporal regulation of epithelium formation mediated by FoxA, MKLP1, MgcRacGAP, and PAR-6. Mol. Biol. Cell 28, 2042–2065 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weber S. C., Brangwynne C. P., Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr. Biol. 25, 641–646 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.J. C. Russ, J. C. Russ, Introduction to Image Processing and Analysis (CRC Press, 2007). [Google Scholar]
- 38.Stiernagle T., Maintenance of C. elegans. WormBook 2006, 1–11 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolters D. A., Washburn M. P., Yates J. R., An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 73, 5683–5690 (2001). [DOI] [PubMed] [Google Scholar]
- 40.He L., Diedrich J., Chu Y.-Y., Yates J. R. III, Extracting accurate precursor information for tandem mass spectra by RawConverter. Anal. Chem. 87, 11361–11367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P., Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC−MS/MS) for large-scale protein analysis: The yeast proteome. J. Proteome Res. 2, 43–50 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Xu T., Park S. K., Venable J. D., Wohlschlegel J. A., Diedrich J. K., Cociorva D., Lu B., Liao L., Hewel J., Han X., Wong C. C. L., Fonslow B., Delahunty C., Gao Y., Shah H., Yates J. R. III, ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tabb D. L., McDonald W. H., Yates J. R. III, DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/8/eaat6224/DC1
Fig. S1. Heterochromatin formation and establishment of H3K9me in WT embryos.
Fig. S2. Control for TEM conditions in WT versus met-2 mutants.
Fig. S3. MET-2 localization with additional reagents and during cell cycle.
Fig. S4. Specificity controls for MET-2 coimmunoprecipitation and PLA.
Fig. S5. Further analysis of LIN-65 and dosage compensation for MET-2 protein.
Table S1. Spectral counts and sequence coverage for proteins identified in the GFP immunoprecipitation and mass spectrometry experiment.