

Abstract
The highly polymorphic genes of the major histocompatibility complex (MHC) play a key role in adaptive immunity. Divergent allele advantage, a mechanism of balancing selection, is proposed to contribute to their exceptional polymorphism. It assumes that MHC genotypes with more divergent alleles allow for broader antigen-presentation to immune effector cells, by that increasing immunocompetence. However, the direct correlation between pairwise sequence divergence and the corresponding repertoire of bound peptides has not been studied systematically across different MHC genes. Here, we investigated this relationship for five key classical human MHC genes (human leukocyte antigen; HLA-A, -B, -C, -DRB1, and -DQB1), using allele-specific computational binding prediction to 118,097 peptides derived from a broad range of human pathogens. For all five human MHC genes, the genetic distance between two alleles of a heterozygous genotype was positively correlated with the total number of peptides bound by these two alleles. In accordance with the major antigen-presentation pathway of MHC class I molecules, HLA-B and HLA-C alleles showed particularly strong correlations for peptides derived from intracellular pathogens. Intriguingly, this bias coincides with distinct protein compositions between intra- and extracellular pathogens, possibly suggesting adaptation of MHC I molecules to present specifically intracellular peptides. Eventually, we observed significant positive correlations between an allele’s average divergence and its population frequency. Overall, our results support the divergent allele advantage as a meaningful quantitative mechanism through which pathogen-mediated selection leads to the evolution of MHC diversity.
Keywords: HLA, balancing selection, heterozygote advantage, pathogen-mediated selection, human evolution
Introduction
Pathogens are suspected to be one of the strongest selective forces in human evolution and the constant exposure to parasites over evolutionary time has likely contributed to the genetic variation found at a large number of genes within and among present day populations (Fumagalli et al. 2011). In this light, the genes of the major histocompatibility complex (MHC) with their exceptional genetic diversity and immune function are a prime candidate to investigate the exact mechanisms through which pathogen-mediated selection has contributed to human evolution.
The MHC is a key component of the adaptive immune system common to all jawed vertebrates (Klein 1986). In humans, it is known as a gene-dense region that spans ∼4 Mb on the short arm of chromosome 6. It comprises over 200 genes, many of which are involved in immunity (Beck and Trowsdale 2000). Among these genes, the classical MHC genes (also called human leukocyte antigen, HLA) encode for cell-surface glycoproteins with a key role in adaptive immunity (Hughes and Yeager 1998; Trowsdale 2011). In cells infected by intracellular parasites, MHC class I molecules can present parasite-derived peptides to cytotoxic T lymphocytes (CTL). Upon recognition of these foreign peptides, the infected cells are destroyed. The MHC class II molecules present antigens, mainly derived from extracellular pathogens, on the surface of specialized antigen-presenting cells. The exposed peptides are recognized by helper T lymphocytes (TH cells), leading to a complex cascade of specific immune responses (Hughes and Yeager 1998; Jensen 2007; Neefjes et al. 2011).
The classical MHC genes are among the most polymorphic genes in the human genome and thousands of different alleles have been identified at some of these loci (Klein 1986; Trowsdale 2011). This polymorphism is characterized by a remarkable sequence variation in the peptide-binding grooves of MHC molecules (i.e., the pocket where antigens are bound) (Parham 1988; Reche and Reinherz 2003) as well as an enhanced rate of nonsynonymous substitutions (Hughes and Nei 1988). MHC polymorphisms are often ancient and allele lineages whose origin predates species divergence are retained across multiple species, an observation described as transspecies polymorphism (Klein 1987). The general action of balancing selection in enhancing both the rate of nonsynonymous substitutions in codons forming the peptide binding groove (Hughes and Nei 1988, 1989) and the persistence of allelic diversity over extremely long time periods is strongly supported (Klein et al. 1998, 2007). However, the exact mechanisms of balancing selection are still disputed. Accordingly, three main mechanisms of pathogen-mediated selection have been suggested (Spurgin and Richardson 2010) which are potentially not mutually exclusive and may interact with one another: heterozygote advantage (Doherty and Zinkernagel 1975), rare-allele advantage (Bodmer 1972), and fluctuating selection (Hill 1991).
The heterozygote advantage was first proposed by Doherty and Zinkernagel (1975). Heterozygous individuals at MHC loci are assumed to present a broader range of pathogen-derived peptides than homozygotes, thus increasing the probability of triggering a specific immune response. They show increased resistance to pathogens, and are more likely to have higher relative fitness, resulting in an increased persistence of different MHC alleles in the population (Hughes and Yeager 1998; Penn et al. 2002). The heterozygote advantage hypothesis has been further extended by taking into account the sequence level, leading to the idea of a divergent allele advantage (Potts and Wakeland 1990; Wakeland et al. 1990). The high sequence divergence observed at MHC genes results in structural polymorphism that may impact the functional properties of MHC molecules. Heterozygous individuals with more divergent MHC allele combinations (i.e., larger number of amino acid differences along the sequence of the antigen-binding domains) are thought to encode glycoproteins that differ more in the repertoire of antigens they can bind. Those individuals may thus be able to present a wider array of antigens to immune effector cells, conferring an advantage against pathogen infections. In contrast, alleles more similar at the sequence level presumably exhibit more similar peptide binding specificities, thus leading to recognition of a lower overall number of peptides when co-occurring in a heterozygous individual (Lenz 2011).
Because of the extremely high number of pathogen proteins to which each host might be exposed throughout its lifetime, comprehensively measuring the relevant repertoire of MHC-bound peptides is impractical in humans and impossible in nonmodel species. Consequently, the divergent allele advantage hypothesis has been difficult to test. However, different measures of MHC sequence divergence are increasingly being used as a proxy for the potential MHC-bound peptide repertoire diversity, leading to correlative evidence that highlights how selection has favored the evolution of multiple MHC loci with divergent alleles in natural populations (She et al. 1990; Landry et al. 2001; Richman et al. 2001; Forsberg et al. 2007; Neff et al. 2008; Lenz et al. 2009; Schwensow et al. 2010; Lenz, Eizaguirre, et al. 2013; Lenz, Mueller, et al. 2013). In humans, the development of computational MHC antigen-binding prediction algorithms has enabled a more direct test of the divergent allele advantage. With this approach it has been shown previously that more divergent HLA-DRB1 allele pairs experience less overlap in the antigenic peptides they can bind, that is, they are able to present a broader range of potential antigens (Lenz 2011), thus supporting the divergent allele advantage hypothesis at this locus. The investigation of the DRB1 locus has been further extended by considering two distinct phylogenetic groups of alleles, denoted as group A and B (Yasukochi and Satta 2014). The same pattern of increased pathogen recognition capacity was observed only for those alleles that in the phylogenetic tree cluster together with primate alleles forming a polyphyletic group (group B) (Lau et al. 2015). Recently, a mechanism of joint divergent asymmetric selection acting on HLA-A and B as a whole was suggested, which has potentially evolved to counter-balance the lack of diversity at individual HLA loci often found in small-sized and isolated human populations (Buhler et al. 2016).
In order to evaluate the divergent allele advantage hypothesis more systematically across all key classical human MHC genes, we here investigated the relationship between sequence divergence and peptide binding properties for three class I genes (HLA-A, -B, -C) and two class II genes (HLA-DRB1 and -DQB1). Focusing on “common” alleles for each locus, as defined by the CWD catalogue (Mack et al. 2013) (supplementary table S1, Supplementary Material online), we evaluated different estimates of amino acid sequence divergence as proxies for the functional divergence among different alleles. Functional divergence was characterized by allele-specific computational binding prediction for a broad range of representative human pathogens (supplementary table S2, Supplementary Material online). Considering a larger and more comprehensive data set of pathogen-derived peptides compared with the set of pathogenic peptides that has been used in previous studies, we were also able to describe the functional features of the divergent allele advantage, by investigating the differential pattern of antigenic presentation between MHC class I and class Il loci. Finally, the frequency distribution of HLA allele pools was investigated in several European populations in order to explore ongoing selection for divergent MHC alleles in modern humans.
Results
Functional Characterization of Common Human MHC Alleles
The set of 232 proteins from a broad collection of relevant human pathogens (N = 27), including macroparasites, bacteria, and viruses, resulted in a total of 118,097 unique pathogen-derived peptides. These peptides are meant to represent a comprehensive repertoire of potential antigens to which humans may have been exposed to throughout their evolutionary history, and which may thus have contributed to the exceptional MHC diversity that we see in present-day human populations.
The number of alleles defined as common varied among the different MHC loci: HLA-A: 63, HLA-B: 123, HLA-C: 40, HLA-DRB1: 73, HLA-DQB1: 21 (supplementary table S1, Supplementary Material online), reflecting general differences in allelic diversity among the loci (Trowsdale 2011). The proportion of peptides predicted to be bound by a given allele varied substantially within and among the different loci (table 1 and supplementary fig. S1, Supplementary Material online). For each locus, the proportions of common (shared among different alleles) and private (allele-specific) peptides bound by each allele are reported in supplementary figure S2, Supplementary Material online. The overlap in bound peptides between different loci was significantly higher for class I genes (A ∩ B = 8,253, B ∩ C = 5,930, A ∩ C = 5,692, A ∩ B ∩ C = 3,545) than for class II gene (DRB1 ∩ DQB1 = 133) (χ2 test, P <0.001) (supplementary fig. S3, Supplementary Material online). Interestingly, we also found a remarkable amount of bound peptides that was shared between class I and class II genes (A ∩ DRB1 = 1,377, A ∩ DQB1 = 385, B ∩ DRB1 = 933, B ∩ DQB1 = 403, C ∩ DRB1 = 689, C ∩ DQB1 = 251) (supplementary fig. S4, Supplementary Material online). However, since the binding prediction algorithms do not account for different antigen processing pathways, it remains to be explored whether in reality these shared peptides are presented by alleles from both classes of MHC. The number of combined peptides bound by any two alleles of a given locus (equivalent to a heterozygous genotype) differed significantly among the genes (Kruskal–Wallis test, P < 0.001) (supplementary fig. S5a, Supplementary Material online). Generally, class II genes showed a lower median number of peptides bound by allele pairs (HLA-DRB1 = 587, HLA-DQB1 = 446) compared with class I genes (HLA-A = 3,936, HLA-B = 3,753, HLA-C = 3,349).
Table 1.
Proportion of Bound Peptides across the Five Classical MHC Loci.
| Locus | Proportion of Peptides Bound By At Least One Allele | Proportion of Bound Peptides Per Allele |
||
|---|---|---|---|---|
| Min | Max | Median (95% CI) | ||
| HLA-A | 0.185 | 0.014 | 0.074 | 0.017 (0.0159, 0.0178) |
| HLA-B | 0.192 | 0.012 | 0.018 | 0.017 (0.0168, 0.0172) |
| HLA-C | 0.079 | 0.014 | 0.019 | 0.017 (0.0165, 0.0171) |
| HLA-DRB1 | 0.025 | 0.002 | 0.005 | 0.003 (0.0026, 0.0031) |
| HLA-DQB1 | 0.011 | 0.001 | 0.004 | 0.002 (0.0015, 0.0027) |
Note.—MHC allele-specific peptide binding was predicted computationally for each locus. Total number of peptides: 118,097.
Sequence-Based Divergence Parameters and Functional Differences among MHC Alleles
A growing number of studies are investigating the fitness consequences of MHC allele divergence both in natural populations and in model species by using some estimate of sequence dissimilarity as a proxy for functional divergence among alleles. These estimates range from simple nucleotide differences (only partially relevant at the protein level) to sophisticated methods that take into account different physicochemical properties of amino acids at the protein sequence level.
For instance, the pairwise amino acid p-distance simply counts the relative number of differences along the amino acid sequence, but information on amino acid properties and relationships are not incorporated, and all nonidentical amino acids are treated as equivalent (Henikoff 1996). However, as the substitution rate usually varies among amino acid site, methods including information about different mutation rates for each amino acid as well as scores that take into account residue-specific properties have been introduced (May 1999). DayHoff (Dayhoff et al. 1978) and JTT (Jones et al. 1992) are two examples of the most popular methods mainly used to investigate mutational trajectories and evolutionary distances between amino acids. Additionally, quantitative measures of pairwise distance have been developed, in which the physicochemical properties of the amino acids, and thus the functional similarity between sequences are considered. Among the different physicochemical features, the molecular volumes of amino acid residues might be particularly meaningful for the question whether a peptide fits into the various pockets of the peptide-binding groove of an MHC molecule. Grantham (Grantham 1974) and Sandberg (Sandberg et al. 1998) distances are two examples of sequence-based measures of sequence divergence where the molecular volume of the different amino acids is taken into account.
So far, a comprehensive evaluation has been lacking as to which sequence parameters are most suitable, that is, most strongly correlated with functional divergence. Thus, in order to identify the most relevant sequence-based parameter, the allele-specific functional binding properties at each MHC locus were correlated with five different, commonly used measures of sequence divergence: pairwise amino acid p-distance (Henikoff 1996), DayHoff (Dayhoff et al. 1978), JTT (Jones et al. 1992), Grantham (Grantham 1974) and Sandberg (Sandberg et al. 1998).
For each possible allele pair at a given HLA locus, we calculated the total number of unique pathogen-derived peptides (obtained by computational binding prediction as described earlier) bound by both alleles together (meant to reflect the MHC-presented antigen repertoire conferred by a heterozygote genotype). The pairwise number of bound peptides was then correlated with the sequence divergence of the two given alleles, estimated by the different measures. As expected, the five measures of genetic distance were highly correlated with each other (supplementary fig. S6 and table S3, Supplementary Material online). Accordingly, the correlation values between allele divergence and the combined number of bound peptides by all possible allele pairs at each locus under investigation were largely consistent across the different parameters of sequence divergence (supplementary table S4, Supplementary Material online). Nevertheless, despite similar correlation values, a rank analysis across the five human MHC gene (HLA-A, -B, -C, -DRB1, and -DQB1) revealed that the Grantham distance measure consistently ranked at the top, that is, showed the strongest correlation values (table 2). Therefore, for subsequent analyses, we focused on one parameter only, the Grantham distance.
Table 2.
Rank analysis between different measures of sequence divergence.
| Total |
Extracellular |
Intra-Extra |
Intracellular |
|||||
|---|---|---|---|---|---|---|---|---|
| Average Tau | Rank | Average Tau | Rank | Average Tau | Rank | Average Tau | Rank | |
| P-distance | 0.321 | 2 | 0.265 | 3 | 0.297 | 2 | 0.297 | 2 |
| Dayhoff | 0.313 | 3 | 0.257 | 4 | 0.285 | 5 | 0.297 | 2 |
| JTT | 0.309 | 5 | 0.256 | 5 | 0.286 | 4 | 0.287 | 4 |
| Grantham | 0.327 | 1 | 0.272 | 1 | 0.298 | 1 | 0.305 | 1 |
| Sandberg | 0.310 | 4 | 0.269 | 2 | 0.289 | 3 | 0.282 | 5 |
Note - Average correlation values (Kendall’s tau coefficient) across the five human MHC genes (HLA-A, -B, -C, -DRB1 and -DQB1) and rank analysis across the five parameters of sequence divergence.
Relation between Sequence Divergence and Functional Divergence
The median distance between allele pairs of a given locus differed significantly between the five genes (Kruskal–Wallis test, P < 0.001) with class II genes showing larger median Grantham distances (HLA-DRB1 = 9, HLA-DQB1 = 11) compared with class I genes (HLA-A = 7, HLA-B = 8, HLA-C = 5) (supplementary fig. S5b, Supplementary Material online).
For the sake of completeness, we first used our data to confirm the intuitive assumption of the heterozygote advantage hypothesis that allele pairs (representing heterozygous genotypes) together bind a larger number of peptides than single alleles (representing homozygous genotypes). This was generally true for all five loci (Kruskal–Wallis test, P < 0.001; supplementary fig. S7, Supplementary Material online), even though there were some rare cases where certain alleles alone bound more peptides than certain allele combinations, suggesting interesting variation in peptide promiscuity among alleles. This general result is in line with a large body of empirical studies showing higher pathogen resistance for MHC heterozygotes (Carrington et al. 1999; Penn et al. 2002).
Subsequently, we focus all our analyses on allele pairs with two different alleles (reflecting heterozygous genotypes). According to the divergent allele advantage hypothesis, we expect that the number of peptides bound by heterozygote genotypes increases with increasing sequence divergence between the two given HLA alleles. Following this expectation, all five HLA genes revealed a significant positive correlation between the pairwise genetic distance and the combined number of bound peptides across all possible allele pairs (fig. 1 and table 3). Interestingly, for HLA-A, -B, and -DRB1, the rate at which the number of bound peptides increases in response to larger allele divergence appears to slow down after a certain point, seemingly approaching a maximum. This can be explained by the fact that, for some loci, even allele pairs with only intermediate sequence divergence do not share any bound peptides anymore (supplementary fig. S8, Supplementary Material online). As the combined number of bound peptides cannot be larger than the sum of peptides bound be each allele, as soon as zero overlap is reached, any further sequence divergence cannot increase the combined number of bound peptides any further. This suggests that alleles at some loci can diverge functionally with only a small number of sequence changes, possibly at sites located in the peptide binding region.
Fig. 1.

In silico evidence for divergent allele advantage across five classical human MHC genes. Correlation between pairwise genetic distances reported as Grantham distance (x axes) and number of bound peptides (y axes) counted for all possible pairs of common HLA alleles. Each dot represents an allele pair. Binding prediction analyses performed on the complete data set of pathogen proteins (n = 232). Linear model (red line) and smoothed lowess curve (dashed blue line), describing the association between the combined number of bound peptides and pairwise Grantham sequence divergence. Note the different axis scales.
Table 3.
Divergent Allele Advantage and Different Pathogen Groups.
| HLA-A |
HLA-B |
HLA-C |
HLA-DRB1 |
HLA-DQB1 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pathogen Groups | Tau | Padj | Tau | Padj | Tau | Padj | Tau | Padj | Tau | Padj |
| Total | 0.361 | <0.001 | 0.397 | <0.001 | 0.507 | <0.001 | 0.157 | <0.001 | 0.210 | 0.001 |
| Extracellular | 0.345 | <0.001 | 0.192 | <0.001 | 0.392 | <0.001 | 0.130 | <0.001 | 0.303 | <0.001 |
| Intra–Extra | 0.351 | <0.001 | 0.289 | <0.001 | 0.475 | <0.001 | 0.166 | <0.001 | 0.210 | 0.024 |
| Intracellular | 0.293 | <0.001 | 0.377 | <0.001 | 0.544 | <0.001 | 0.137 | <0.001 | 0.172 | <0.001 |
Note.—Correlation values (Kendall’s tau) between combined number of bound peptide and Grantham genetic distance between all possible allele pairs across the five key classical MHC genes. Binding prediction was performed on the complete data set of pathogen proteins (n = 232) as well as considering proteins separately within three groups of pathogens: extracellular (n = 57), intracellular (n = 100), and intra-extracellular (n = 75).
Tau, Kendall’s tau coefficient; Padj, P value after Bonferroni-correction across multiple alleles tested at each locus and number of loci.
Furthermore, a significant negative correlation could be observed between pairwise genetic distance and the proportion of shared peptides (peptides bound by both alleles of a given combination; supplementary fig. S8, Supplementary Material online), revealing a decreasing proportion of peptides shared between more divergent alleles. Of note, for the two class II loci, the correlations between genetic distance and peptide sharing were stronger than between genetic distance and the total number of bound peptides. This is owing to the fact that the latter measure includes additional variation from differences in the size of the bound peptide repertoire among HLA class II alleles (supplementary fig. S2, Supplementary Material online), an allele-specific property that is independent of the divergence between alleles.
Phylogenetic analysis of the HLA-DRB1 gene has revealed two subgroups of allelic lineages: a human-specific monophyletic group (Group A), and a polyphyletic group with primates (Group B) (Yasukochi and Satta 2014). It has been proposed that group A and B allele lineages have evolved with contrasting binding capacity, and only the alleles from the polyphyletic group B showed increased presentation of pathogen peptides with increasing sequence divergence (Lau et al. 2015). In contrast with previous findings, our binding prediction analysis revealed a significant positive correlation for the whole set of DRB1 alleles (fig. 1 and table 3) as well as for both groups of alleles separately (supplementary fig. S9, Supplementary Material online). This discrepancy to the earlier results might be due to the much larger and more comprehensive data set of pathogen-derived peptides used in our analysis (here 118,097 peptides vs. 265 peptides in the previous study).
Antigen Processing and Different Origins of Pathogenic Peptides
Up to this point, our analysis treated all pathogen peptides as equally likely targets for each given MHC locus. However, in reality some of those peptides will never be in contact with certain MHC molecules, due to the different processing pathways by which eukaryotic cells degrade proteins: the proteasome and lysosomal proteases. Peptides resulting from proteasome degradation, generally derived from intracellular proteins, are presented by MHC class I molecules, whereas peptides presented by MHC class II molecules are usually of extracellular origin and processed through lysosomal protease degradation in antigen-presenting cells (Jensen 2007). If the exceptional sequence divergence among MHC alleles evolved at least partly as a consequence of pathogen-mediated selection for divergent alleles, we would expect to see a stronger signature of selection at a given MHC locus when focusing on biologically meaningful pathogens that are likely to actually be encountered by a given MHC molecule. That is, we expect a stronger correlation between sequence divergence and functional divergence when only focusing on peptides originating from the locus-specific antigen-processing pathway. We thus divided the pathogen proteins used for binding prediction analysis into three groups. The three groups were based on their agent’s lifestyle in the host: “extracellular” (n = 58 proteins), “intracellular” (n = 100) and a third group of pathogen proteins belonging to those agents whose life cycle involves both intracellular and extracellular stages inside the host (Silva 2012), here named as “intra-extracellular” (n = 75) (supplementary table S2, Supplementary Material online). Within each of the three groups of pathogen proteins, and for all five investigated HLA genes, we again observed a significant positive correlation between the pairwise sequence divergence and the combined number of bound peptides across all possible allele pairs. Interestingly however, in some cases, the strength of correlation differed among the three pathogen groups: at two of the MHC class I genes, HLA-B and HLA-C, a stronger positive correlation was observed for the group of intracellular pathogens, compared with extracellular pathogens, while this bias was not observed at HLA-A (fig. 2 and table 3). Conversely, a stronger correlation for peptides derived from extracellular pathogens was detected at one MHC class II locus, HLA-DQB1; while correlation values between pairwise sequence divergence and the combined number of bound peptides at the HLA-DRB1 locus were comparable across the three groups of pathogen proteins (fig. 2 and table 3). In the three cases where a bias across the groups of pathogen proteins was detected, the intra-extracellular proteins showed intermediate correlation values between the intra- and the extracellular proteins (table 3). However, permutation of the proteins among the three groups revealed that only for HLA-B and HLA-C, the observed difference between extracellular and intracellular correlation values were indeed larger than expected by chance (both P < 0.001; supplementary fig. S10, Supplementary Material online), while the other three loci did not show a statistically significant deviation from random expectations.
Fig. 2.

Different origins of pathogenic peptides. Correlation between pairwise genetic distances reported as Grantham distance (x axes) and number of bound peptides (y axes) calculated for all possible pairs of common HLA alleles. Each dot represents an allele pair. Binding prediction analyses performed considering proteins within three groups of pathogens: extracellular (orange, n = 57 proteins), intracellular (green, n = 100), and intra-extracellular (purple, n = 75).
Our main analyses were performed considering pathogen-derived peptides that are all of the same length (9 aa). However, MHC class II molecules allow binding longer peptides than class I. Thus, for alleles at the two class II loci, binding prediction of 15mer peptides from the same set of pathogen proteins were considered. This analysis showed stronger correlations for DRB1 and weaker correlations for DQB1, compared with the 9mer predictions, but overall support the main conclusions (supplementary table S5, Supplementary Material online). Furthermore, to test if our results were sensitive to the choice of the binding threshold, we additionally repeated the primary analysis using a different binding threshold (%rank of 0.5, indicating strong binding). The comparable results suggest that our main conclusions hold across a range of established binding thresholds (supplementary table S6, Supplementary Material online).
Distinct Amino Acid Composition among Pathogen Groups
The observed bias in the correlation between sequence divergence and functional divergence (peptide binding) toward a specific group of pathogens suggests distinct differences in the peptide repertoires among these groups. This could be due either to certain group-specific peptide sequences or to a more general difference in the amino acid composition of proteins among the pathogen groups. Amino acid usage has changed over evolutionary time in different species, and proteins have evolved in terms of physico-chemical and structural properties, reflecting adaptations to specific environmental conditions (Bogatyreva et al. 2006; Tekaia and Yeramian 2006). Intriguingly, intra- and extracellular environments exhibit significant differences, including different pH value (Casey et al. 2010) and availability of different nutrients (Goetz et al. 2001; O’Riordan and Portnoy 2002; Ross 2014). It thus appears plausible that intra- and extracellular pathogens may have evolved proteomes with distinct amino acid compositions.
In order to test if the observed bias in correlation values across the groups of pathogen proteins was the results of group-specific peptide sequences or due to more general differences in the amino acid composition within each group, we created four different data sets of artificial proteins. These four data sets were then analyzed in the same way as above, again assessing the strength of correlation between allele divergence and functional divergence (here based on bound peptides from the artificial proteins) for the three pathogen groups and across the five HLA loci. First, amino acids forming each pathogen protein sequence were randomly shuffled, maintaining the same amino acid composition of a given protein, but changing its actual sequence. If the stronger correlation with intracellular pathogen proteins by HLA-B and HLA-C were due to group-specific peptide sequences, we would expect this bias to disappear when reshuffling the protein sequences. However, correlation values resulting from shuffled proteins did not differ substantially from the true observed correlation values obtained with the real data set of pathogen proteins (supplementary table S7, Supplementary Material online). For the second set of artificial proteins, we created random protein sequences but maintained amino acid frequencies as they occurred within each group of pathogen proteins. Again, correlation values did not differ substantially from the observed true correlation values obtained in the initial test and the specific bias across the three groups was still observed (supplementary table S7, Supplementary Material online). For the third set of artificial proteins, we again created random protein sequences but this time maintained amino acid frequencies as they occurred in the whole data set of pathogen proteins. While a general positive correlation was also observed in this data set, the specific bias across the groups of pathogen protein was not detected anymore (supplementary table S7, Supplementary Material online). Finally, the amino acid composition computed from UniProtKB/Swiss-Prot data bank (Gasteiger et al. 2005; Boutet et al. 2016) was used to create the fourth set of artificial proteins. Again, the specific bias across the three groups of pathogen proteins was not detected anymore (supplementary table S7, Supplementary Material online). Thus, the observed bias in correlation values persisted only when amino acid frequencies mirrored the specific frequencies observed within each group of pathogen proteins. One of the possible explanations of our results could be that the observed stronger correlation of HLA-B and HLA-C alleles with peptides from intracellular pathogens is not due to specific peptides but is the results of adaptation of MHC alleles to the differences in amino acid composition between groups of pathogens. To further explore this hypothesis, a nonparametric multivariate analysis of variance was performed to quantify the similarity among proteins with regard to their amino-acid composition. The two groups of intracellular and extracellular pathogen proteins indeed differed significantly in their amino acid composition (PerMANOVA test, P < 0.001), with 9% of the total variance associated with the divisions in intracellular and extracellular proteins (R^2 estimate from PerMANOVA) (fig. 3). Accordingly, when average amino acid compositions were compared between the two groups of pathogen proteins, significant variations in the mean amino acid composition were observed for specific amino acids (one-way ANOVA, P < 0.05) (supplementary fig. S11 and table S8, Supplementary Material online). The observed differences in amino acid composition could be linked to glycosylation patterns, which differ between extracellular and intercellular peptides (Marshall 1972). N-glycosylation is one of the forms of protein glycosylation in eukaryotic organisms which is mainly targeting extracellular and secreted proteins. It has been shown that N-glycosylation sites are specific to the consensus sequence Asn-Xaa-Ser/Thr and that the presence of proline between Asn and Ser/Thr inhibits N-glycosylation (Bause 1983). Accordingly, in our analysis proteins of extracellular pathogens show low proline concentration which is instead prevalent in proteins of intracellular pathogens. These exploratory analyses suggest that the amino acid composition might be different between the two groups of intracellular and extracellular pathogen proteins and that MHC alleles might have potentially adapted their binding specificities accordingly, at least at the HLA-B and HLA-C loci. However, further research is necessary to validate this conclusion and to exclude other potential causes, such as taxonomy, driving the observed difference in amino acid composition.
Fig. 3.
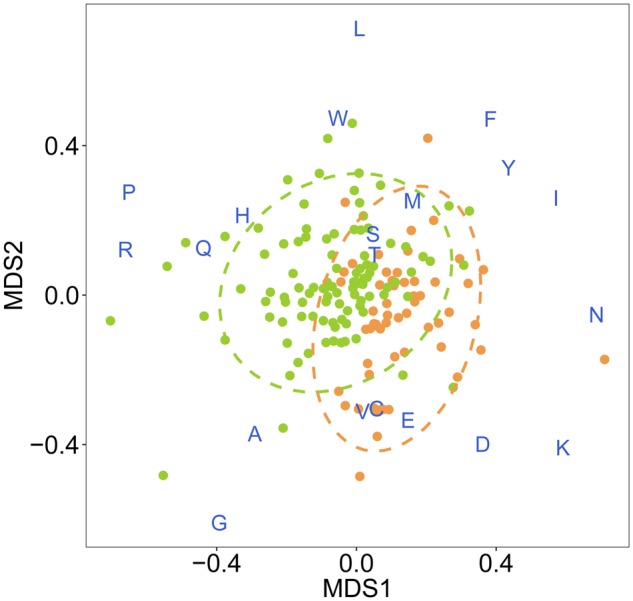
Multidimensional scaling plot of amino acid composition in intracellular and extracellular pathogen proteins. Multidimensional scaling (MDS) based on amino acid frequencies indicates similarity in amino acid composition among individual proteins (dots). Intracellular proteins (n = 100) are reported in green while extracellular proteins (n = 57) in orange. MDS enables a standardized unit-less representation of variation among data points in 2D space (along perpendicular axes MDS1 and MDS2): location of proteins within the plot is indicative of potential bias toward specific amino acids (blue characters in one letter code), proteins with more similar amino acid composition are displayed closer to each other. The dashed circles indicate 95% confidence intervals for each group. Stress for 2D representation: 0.21.
Population Frequency of Divergent HLA Alleles
The above-described results support historical pathogen-mediated selection through divergent allele advantage at the human MHC. However, we were also interested in exploring whether the divergent allele advantage was still maintaining diverse HLA allele pools in present day human populations. We hypothesized that, under the divergent allele advantage, alleles that on an average yield a more divergent genotype (conferring higher fitness) when paired with another allele in a heterozygote individual, would be selected for and thus exhibit higher frequencies in a given population. A similar observation had been made in the allele pool of a social marine mammal whose reproductive success is partly predicted by the divergence of its MHC genotype (Lenz, Mueller, et al. 2013). In order to test this hypothesis across the five classical HLA loci, we calculated for each allele of a given locus the average pairwise amino acid sequence divergence to the most common alleles at this locus (≥5% allele frequency, representing alleles most likely to be forming a heterozygote with the allele in question). This average sequence divergence was then correlated with the allele’s population frequency. For this analysis, we focused again on alleles defined as “common” in the CWD catalogue (Mack et al. 2013), assuming that very rare alleles are more susceptible to stochasticity and neutral demographic processes. In line with our expectation, a significant positive correlation between the average pairwise sequence divergence of an allele and its population frequency was observed for a number of HLA genes (HLA-B, -C, and -DRB1) across different European populations (USA European Caucasian, German, and Polish) (fig. 4 and supplementary table S9, Supplementary Material online).
Fig. 4.

Population frequency of divergent HLA alleles. Correlation between the average Grantham pairwise divergence to the most common alleles and the allele frequency in the USA NMDP European Caucasian (N = 1,242,890), German (N= 39,689), and Polish (N= 20,653) populations, for four classical HLA loci with available allele frequency data in AlleleFrequencies.net. Significant associations that persisted after Bonferroni correction across populations are reported with a solid line, while dashed lines indicate associations that are only nominally significant (P < 0.05 before Bonferroni correction; for exact values see supplementary table S9, Supplementary Material online).
Discussion
Here, we used computational antigen-binding prediction on a large data set of potentially antigenic pathogen peptides to investigate whether pairs of highly diverged MHC alleles together bind more different antigens than more similar alleles. Such an association is predicted by the divergent allele advantage hypothesis (Wakeland et al. 1990) and had previously been investigated only for the HLA-DRB1 gene in humans (Lenz 2011; Lau et al. 2015). The observed positive correlation between the genetic distance of two alleles and the combined number of peptides they bind together confirmed and extended the predictions of the divergent allele advantage for all five investigated human MHC genes. These results support the hypothesis that enhanced sequence diversity between alleles in a heterozygous MHC genotype increases the range of potential MHC-presented peptides, thus raising the chance to recognize specific antigens and consequently enhance immune surveillance.
Our observation that HLA-B and HLA-C (and potentially HLA-DQB1) exhibit the strongest associations when considering antigens originating from their most plausible targets is intriguing and lends further support to the biological relevance of this mechanism. It might indicate that the exceptional sequence divergence commonly observed among alleles of a given MHC locus has evolved specifically in response to selection by pathogens that are processed through the major protein degradation and antigen-presentation pathways that this given locus is associated with: alleles at HLA-B and -C loci have apparently evolved to bind specifically peptides derived from intracellular pathogens, while alleles at the HLA-DQB1 locus may have evolved to bind a wider array of peptides from extracellular pathogens. The fact that we did not observe such a pathogen group-specific bias at the HLA-A and HLA-DRB1 loci might indicate either that divergent allele advantage has not played a significant role in their evolution, or that they are less specific with regard to the pathogen origin of the peptides they present. It has indeed been shown that autophagy of intracellular components can promote the presentation of endogenous antigens by MHC class II molecules (Paludan et al. 2005; Levine and Deretic 2007; Münz 2012; Roche and Furuta 2015). Furthermore, several studies have reported a potential role for DRB1 molecules in viral infections (Martin and Carrington 2005). For instance, HLA-DR variants have been associated with spontaneous clearance of HBV and HCV infections (Thursz et al. 1995; McKiernan et al. 2004), with protective effect against dengue shock syndrome (DSS) development (Nguyen et al. 2008) and with HIV suppression (Malhotra et al. 2001). In this light, the lack of a particular bias by DRB1 alleles toward either of the pathogen groups may indicate that this locus evolves under selection by both intra- and extracellular pathogens.
In addressing the frequency of divergent HLA alleles in different human populations, we observed in some human populations, and for specific HLA loci, significant correlations between an allele’s population frequency and its average pairwise sequence divergence. These results might suggest still ongoing selection for divergent HLA genotypes, at least in some modern human populations, possibly depending on population-specific differences in historical pathogen communities. However, allelic age may also contribute to the observed pattern, as, in principle, older alleles are both more likely to have reached high frequencies, even under neutrality, and to have accumulated more point mutations (thus being more divergent). On the other hand, HLA genes are known to undergo frequent recombination and gene conversion events, yielding novel alleles with high divergence from their origin at the very start (i.e., at low frequency). It is thus unclear to what extent novel alleles contribute to the observed pattern, warranting further research to explore the effect of genetic drift on the frequency of divergent HLA alleles. Furthermore, HLA alleles that on an average form more divergent allele combination, and which have been maintained in the population because of their increased capacity in presenting pathogen-derived peptides, might also be advantageous in case of newly emerging and fast-evolving pathogens (i.e., HIV).
Humans share similar MHC allelic lineages with closely related species (Klein 1987; Lawlor et al. 1988). This observation is a typical feature of MHC genes, compatible with the theory of transspecies evolution: ancestral lineages present in the common ancestor are inherited through successive speciation events, persisting over long periods of time (Klein et al. 2007). The ancestral and highly diverged MHC variants are assumed to be adaptive and selectively maintained as a polymorphism by balancing selection (Hughes and Nei 1988; Lenz 2011). Recently, the role of adaptive introgression has been proposed to contribute to the exceptional level of polymorphism at the MHC (Abi-Rached et al. 2011; Wegner and Eizaguirre 2012). So far, explanations for the maintenance of introgressed MHC alleles have largely relied on the idea that such alleles were somehow locally adapted and thus beneficial. For instance, it has been suggested that modern humans might have maintained introgressed archaic HLA variants because they conferred an advantage against local pathogens (Abi-Rached et al. 2011). However, another explanation appears also plausible: MHC alleles from another species are, on average, likely to have diverged significantly from the species’ own allele pool. Thus, any allele that introgresses from another species is likely to lead to highly divergent MHC genotypes. Following the divergent allele advantage hypothesis (and our results), such introgressed alleles should then confer a significant advantage and should consequently be selected for in the new species. This scenario would easily explain the maintenance of introgressed MHC alleles, but further research is necessary to support this hypothesis.
While the present analysis focuses exclusively on the divergent allele advantage, in reality, selection at MHC genes is a dynamic process that involves additional mechanisms apart from the divergent allele advantage. Conceptually, the divergent allele advantage can be considered a quantitative mechanism, which works independent of specific pathogen species or strains. It can act over long evolutionary time scales, promoting the maintenance of ancient allelic lineages in natural populations (Lenz 2011) and facilitating immunity against the constant simultaneous barrage by many different pathogens. In contrast, negative frequency-dependent selection (NFDS) is a qualitative mechanism in which specific alleles can be selected by specific pathogens (Slade and McCallum 1992; Lenz 2018). This mechanism likely works on a shorter time scale, for instance affecting MHC evolution in humans in very recent history (Lindo et al. 2016; Krause-Kyora et al. 2018). Both mechanisms, the divergent allele advantage and NFDS might also act in parallel, but at different time scales, creating an intriguing combination of shared polymorphism but distinct allele pools among populations and possibly even species (Lighten et al. 2017). Local adaptation plays another significant role in MHC evolution and might modulate the effect of the above mechanisms (Eizaguirre and Lenz 2010). The simultaneous action of these additional mechanisms might occasionally mask the effect of the divergent allele advantage and potentially explain the only sporadic evidence for this mechanism in the population frequency analysis reported here. Nevertheless, our results strongly support the divergent allele advantage as a meaningful quantitative mechanism through which pathogen-mediated selection contributes to the evolution of MHC diversity.
Materials and Methods
MHC Loci and Alleles Included in Analyses
Five key classical human MHC genes (HLA-A, -B, -C, -DRB1, and -DQB1) were analyzed in this study. Alleles at each locus were defined at second field (four-digit) resolution and only alleles annotated as “common” in the CWD catalogue (Mack et al. 2013) were included in the analyses. The allele annotation “common” in the CWD catalogue does not specifically indicate a high population frequency but more the extent and quality of documentation available for the given allele. This category indicates that there is universal agreement about the identity of this allele because it has been observed in multiple populations and there is sufficient data for robust frequency estimation (Mack et al. 2013). These criteria resulted in the analysis of 63 alleles for HLA-A, 123 for HLA-B, 40 for HLA-C, 73 for HLA-DRB1, and 21 for HLA-DQB1 (supplementary table S1, Supplementary Material online).
Pathogen Proteins
Binding prediction analyses were performed on a data set of representative human pathogen proteins. Pathogens were selected from the Gideon database (Berger 2005) based on the following criteria: a global distribution, a potential for high mortality and/or morbidity, and a significant impact over the course of human history (Wolfe et al. 2007). The rational for these criteria was that such pathogens are likely to have contributed significantly to human evolution in general and to the evolution of MHC genes in particular. Wolfe et al. (2007) provided a comprehensive list of infectious diseases with the greatest evolutionary and historical significance. From that list, we have taken the majority of pathogens in our data set. However, to assess mortality and morbidity, epidemiological data were also collected from two published reports: the Annual report of the European Centre for Disease Prevention and Control (European Centre for Disease Prevention and Control 2013) and the WHO Global Health Estimates (World Health Organization 2016). First, pathogens with the highest current mortality were included. However, not just mortality, but also nonfatal morbidity can be historically and evolutionarily significant. Indeed, morbid pathogens can reduce the fitness of their host in different ways (e.g., by increasing the sterility), thus pathogens considered morbid were also included. Finally, eradicated pathogens known to be important in human history were taken into account. Here, we used protein sequences of present day pathogens to explore signatures of historical selection, even though ancient pathogen strains might have differed slightly in their antigen repertoires. While we do not expect an effect on the general patterns observed here, it might be interesting to explore subtle differences in future work. We further aimed for a balanced representation of different groups of pathogens (i.e., viruses, bacteria, parasites). Based on these criteria, we identified 27 pathogens (10 viruses, 10 bacteria, 7 macroparasites) that were classified into three groups: extracellular, intracellular, and intra-extracellular, based on their primary environment in the human body (supplementary table S2, Supplementary Material online). Then, for the selected pathogens, amino acid sequences of 232 pathogen proteins (8.5 ± 5.8 per pathogen) known to be antigenic (Vita et al. 2015) and/or likely exposed to the host immune system (mostly secreted and surface proteins) (Rana et al. 2016) were obtained from GenBank (for accession numbers see supplementary table S2, Supplementary Material online).
Peptide Binding Prediction Algorithms
Computational antigen-binding prediction algorithms for MHC molecules were used to determine pathogen peptides potentially bound by the MHC alleles under investigation. Binding prediction was computed for all alleles at each of the five human MHC genes. Furthermore, as prediction analysis are likely to be more accurate for the core of the binding groove, which is known to be nine residues long and contributes the most to the recognition of the antigens, binding prediction was performed considering all possible 9mer pathogen-derived peptides. The data set of 232 representative human pathogen proteins described above resulted in a total of 118,097 unique pathogen-derived 9mer peptides that were analyzed using two different algorithms: NetMHCpan (v2.8) (Hoof et al. 2009) for the alleles at class I loci (HLA-A, -B, -C) and NetMHCIIpan (v3.0) (Karosiene et al. 2013) for the alleles at class II loci (HLA-DRB1, -DQB1). For alleles at the two class II loci (HLA-DRB1 and HLA-DQB1), we repeated the binding prediction analysis considering all possible 15mer pathogen-derived peptides. The predicted binding affinity between pathogen peptides and MHC molecule variants (defined in nanomolar IC50, i.e., half maximal inhibitory concentration) are ranked by the respective software, based on comparison with a large pool of naturally occurring peptides, and a rank percentage score (%rank) is assigned to each peptide. To define “bound” peptides, we used the default %rank threshold of 2, which includes weak and strong binders. All analysis were also repeated using another established binding threshold (%rank of 0.5) which includes only strong binders. The allele HLA-A*30:04 was predicted to bind about four times as many peptides as the other 62 HLA-A alleles (supplementary fig. S2, Supplementary Material online) and was thus excluded as an outlier from subsequent analysis in order to prevent distortion of results. The binding prediction analyses were performed first on the complete data set of pathogen proteins (n = 232), and then considering proteins within three groups separately: extracellular (n = 58), intracellular (n = 100), and intra-extracellular (n = 75).
Sequence Divergence
Allele divergence was computed on the same set of alleles used in the binding prediction analysis reported in supplementary table S1, Supplementary Material online. Protein sequences of HLA alleles were obtained from IMGT/HLA database (Robinson et al. 2015). Exons forming the variable region in the peptide binding groove (i.e., exon 2 and 3 for class I alleles and exon 2 for class II alleles) were selected following the annotation obtained from Ensemble database (Aken et al. 2016). Amino-acid sequence alignments were performed using MUSCLE (Edgar 2004), and sites containing alignment gaps at the beginning or the end of sequences were removed. Genetic distances between alleles for all possible allele pairs at each locus were determined removing missing sites in pairwise comparisons and using five different pairwise parameters of allele divergence: p-distance (Henikoff 1996), DayHoff (Dayhoff et al. 1978), JTT (Jones et al. 1992), Grantham (Grantham 1974), and Sandberg (Sandberg et al. 1998). Pairwise amino acid p-distance, DayHoff and JTT distances were calculated in MEGA 7 (Kumar et al. 2016). Grantham and Sandberg sequence distances were calculated using a custom Perl script that required two input files: a FASTA file with aligned HLA alleles and a specific amino acid distance matrix. Grantham amino acid distance matrix was constructed from Grantham (1974). Sandberg amino acid distance matrix was calculated based on Euclidian distances between all 20 amino acids, using the Euclidian distance method in R version 3.4.1 (R Development Core Team 2017) according to the five physicochemical z-descriptors described in Sandberg et al. (1998): z1 (hydorphobicity), z2 (steric bulk), z3 (polarity), z4, and z5 (electronic effects). Our perl script (together with the Grantham amino acid similarity matrix) is freely available for download from SourceForge (https://granthamdist.sourceforge.io/). It can be used for calculation of pairwise Grantham divergence for any set of aligned MHC alleles of any species.
Allele Frequencies
Information about HLA allele frequencies in different human populations where obtained from the Allele Frequency Net Database (AFND) (Gonzalez-Galarza et al. 2015). We considered only populations of European ancestry with large sample sizes and for which frequencies of alleles at second field resolution were available: USA NMDP European Caucasian (N = 1,242,890), German (N= 39,689), and Polish (N= 20,653) populations. Furthermore, as with the analyses above, we focused on alleles defined as “common” in the CWD catalogue, which led to exclusion of some alleles with a frequency <1%. For each population, we first determined the most common alleles (allele frequency >= 5%) and for all the alleles under investigation in a given population, we calculated the average Grantham pairwise divergence to the most common alleles, considering all possible heterozygote genotypes.
Statistical Analyses
Correlation Tests
The Shapiro–Francia test was performed for all the parameters under investigation (i.e., measures of genetic distance, combined number of bound peptides and average Grantham pairwise amino acid divergence to the most common alleles) to explore samples’ distribution. As parameters were not normally distributed and tied ranks could be detected within our data, the nonparametric Kendall correlation was used to test for associations between parameters. When testing the association between sequence divergence and functional divergence, all P values were adjusted for multiple testing using a sequential Bonferroni correction across the number of alleles tested at each locus as well as across the number of different loci tested. When testing the association between the allele’s average divergence and its population frequency, P values were corrected across the number of populations tested. Correlations were performed in R version 3.4.1 (R Development Core Team 2017).
Permutation Tests
To test for significant differences in the strength of correlation between allele divergence and the binding to pathogen group-specific peptides, we performed permutation tests. For this analysis, the set of 232 representative human pathogen proteins were randomly shuffled among the three groups of pathogens, maintaining the same number of proteins as observed in the original data (extracellular n = 57, intracellular n = 100 and intra-extracellular n = 75). For each group of pathogens, permuted proteins were used to perform binding prediction analyses and compute correlation values between genetic distances and combined number of bound peptides counted for all possible allele pairs for the five HLA genes (analogous to original analysis). Each permutation was run 1,000 times, and the difference between correlation coefficients for intracellular and extracellular proteins for the five HLA genes was recorded. If there was no significant bias for intracellular or extracellular pathogens, on average this difference should be zero. The distribution of permuted differences was then used to infer the significance of our initial observations using a one-tailed test with a 0.05 cut-off.
Artificial Proteins
Four sets of artificial proteins were created and analyzed to test for potential differentiation of the amino acid composition (AAC) among the three groups of pathogens. The first set of artificial proteins was created by randomly shuffling amino acids within each pathogen protein by using the Shuffle Protein program (Stothard 2000), thus maintaining the AAC of each protein intact. Three more sets of artificial proteins were created in R version 3.4.1 (R Development Core Team 2017) by assembling random amino acids while maintaining several features as they occurred within each of the three pathogen groups used in the initial test (i.e., the number of proteins, the average length of sequences, the SD of the length and the minimum and maximum length). The second set of artificial proteins was created from random amino acids but maintaining the AAC as it occurred within each group of pathogen proteins. The third set of artificial proteins was created from random amino acids, while maintaining amino acid frequencies as they occur in the whole data set of pathogen proteins. Finally, amino acid composition computed from UniProtKB/Swiss-Prot data bank (Gasteiger et al. 2005; Boutet et al. 2016) was used to create the fourth set of artificial proteins.
Multivariate Analysis of Variance
Multidimensional scaling is a multivariate statistical technique that can be used to display and summarize a high-dimensional data set in 2D graphical form. The technique was here applied to explore associations between subsets of pathogen proteins and amino acids. A nonparametric, permutational multivariate analysis of variance (PerMANOVA) was used to test for differences in the amino acid composition between pathogen groups. The PerMANOVA, based on a Bray–Curtis dissimilarity distance matrix, was run with 999 permutations to tests for statistical significance. Both procedures are implemented in the vegan package (Oksanen et al. 2012) in R version 3.4.1 (R Development Core Team 2017).
Comparison of Average Amino Acid Compositions
Comparison of mean amino acid compositions between the two groups of pathogen proteins (extracellular and intracellular) were performed using one-way analysis of variance; all P values were adjusted for multiple testing using Bonferroni correction across the number of amino acids tested.
Supplementary Material
Acknowledgments
We thank Martin Kalbe and Jamie Winternitz for their precious input and suggestions. We also thank Matteo Fumagalli and an anonymous reviewer for very constructive comments on an earlier version of this article. This work was supported by the Max Planck Society and an Emmy Noether grant from the German Research Foundation (DFG; LE 2593/3-1 to T.L.L.).
References
- Abi-Rached L, Jobin MJ, Kulkarni S, McWhinnie A, Dalva K, Gragert L, Babrzadeh F, Gharizadeh B, Luo M, Plummer FA.. 2011. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 3346052:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, et al. 2016. The Ensembl gene annotation system. Database 2016:baw093.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bause E. 1983. Structural requirements of N-glycosylation of proteins. Studies with proline peptides as conformational probes. Biochem J. 2092:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S, Trowsdale J.. 2000. The human major histocompatibility complex: lessons from the DNA sequence. Annu Rev Genomics Hum Genet. 11:117–137. [DOI] [PubMed] [Google Scholar]
- Berger SA. 2005. GIDEON: a comprehensive Web-based resource for geographic medicine. Int J Health Geogr. 41:10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer WF. 1972. Evolutionary significance of the HL-A system. Nature 2375351:139–183. [DOI] [PubMed] [Google Scholar]
- Bogatyreva NS, Finkelstein AV, Galzitskaya OV.. 2006. Trend of amino acid composition of proteins of different taxa. J Bioinform Comput Biol. 42:597–608. [DOI] [PubMed] [Google Scholar]
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bansal P, Bridge AJ, Poux S, Bougueleret L, Xenarios I.. 2016. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. Methods Mol Biol. 1374:23–54. [DOI] [PubMed] [Google Scholar]
- Buhler S, Nunes JM, Sanchez-Mazas A.. 2016. HLA class I molecular variation and peptide-binding properties suggest a model of joint divergent asymmetric selection. Immunogenet. 68(6–7):401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington M, Nelson GW, Martin MP, Kissner T, Vlahov D, Goedert JJ, Kaslow R, Buchbinder S, Hoots K, O’Brien SJ.. 1999. HLA and HIV-1: heterozygote advantage and B*35-Cw*04 disadvantage. Science 2835408:1748–1752. [DOI] [PubMed] [Google Scholar]
- Casey JR, Grinstein S, Orlowski J.. 2010. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 11:50–61. [DOI] [PubMed] [Google Scholar]
- Dayhoff MO, Schwartz RM, Orcutt BC.. 1978. {A model of evolutionary change in proteins}. Atlas Protein Seq Struct. 5:345–351. [Google Scholar]
- Doherty PC, Zinkernagel RM.. 1975. Enhanced immunological surveillance in mice heterozygous at H-2 gene complex. Nature 256:50–52. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eizaguirre C, Lenz TL.. 2010. Major histocompatibility complex polymorphism: dynamics and consequences of parasite-mediated local adaptation in fishes. J Fish Biol. 77:2023–2047. [DOI] [PubMed] [Google Scholar]
- European Centre for Disease Prevention and Control. 2013. Annual Epidemiological Report 2013. Reporting on 2011 surveillance data and 2012 epidemic intelligence data. Stockholm: ECDC.
- Forsberg LA, Dannewitz J, Petersson E, Grahn M.. 2007. Influence of genetic dissimilarity in the reproductive success and mate choice of brown trout – females fishing for optimal MHC dissimilarity. J Evol Biol. 20:1859–1869. [DOI] [PubMed] [Google Scholar]
- Fumagalli M, Sironi M, Pozzoli U, Ferrer-Admettlla A, Pattini L, Nielsen R.. 2011. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 7:e1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A.. 2005. Protein identification and analysis tools on the ExPASy server In: Walker JM, editor. The proteomics protocols handbook. Totowa (NJ: ): Humana Press; p. 571–607. [Google Scholar]
- Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W.. 2001. Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci U S A. 98:12221–12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Galarza FF, Takeshita LY, Santos EJ, Kempson F, Maia MH, da Silva AL, Teles e Silva AL, Ghattaoraya GS, Alfirevic A, Jones AR.. 2015. Allele frequency net 2015 update: new features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 43:D784–D788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R. 1974. Amino acid difference formula to help explain protein evolution. Science 185:862–864. [DOI] [PubMed] [Google Scholar]
- Henikoff S. 1996. Scores for sequence searches and alignments. Curr Opin Struct Biol. 6:353–360. [DOI] [PubMed] [Google Scholar]
- Hill AVS. 1991. HLA associations with malaria in Africa: some implications for MHC evolution In: Klein J, Klein D, editors. Molecular evolution of the major histocompatibility complex. Heidelberg (Berlin: ): Springer Berlin Heidelberg; p. 403–420. [Google Scholar]
- Hoof I, Peters B, Sidney J, Pedersen L, Sette A, Lund O, Buus S, Nielsen M.. 2009. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Nei M.. 1988. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335:167–170. [DOI] [PubMed] [Google Scholar]
- Hughes AL, Nei M.. 1989. Nucleotide substitution at major histocompatibility complex class-II loci – evidence for overdominant selection. Proc Natl Acad Sci U S A. 86:958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Yeager M.. 1998. Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet. 32:415. [DOI] [PubMed] [Google Scholar]
- Jensen PE. 2007. Recent advances in antigen processing and presentation. Nat Immunol. 8:1041–1048. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM.. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 8:275–282. [DOI] [PubMed] [Google Scholar]
- Karosiene E, Rasmussen M, Blicher T, Lund O, Buus S, Nielsen M.. 2013. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65:711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J. 1986. Natural history of the major histocompatibility complex. New York: John Wiley and Sons. [Google Scholar]
- Klein J. 1987. Origin of major histocompatibility complex polymorphism: the trans-species hypothesis. Hum Immunol. 19:155–162. [DOI] [PubMed] [Google Scholar]
- Klein J, Sato A, Nagl S, O’hUigín C.. 1998. Molecular trans-species polymorphism. Annu Rev Ecol Syst. 29:1–21. [Google Scholar]
- Klein J, Sato A, Nikolaidis N.. 2007. MHC, TSP, and the origin of species: from immunogenetics to evolutionary genetics. Annu Rev Genet. 41:281–304. [DOI] [PubMed] [Google Scholar]
- Krause-Kyora B, Nutsua M, Boehme L, Pierini F, Pedersen DD, Kornell S-C, Drichel D, Bonazzi M, Möbus L, Tarp P, et al. 2018. Ancient DNA study reveals HLA susceptibility locus for leprosy in medieval Europeans. Nat Commun. 91:1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K.. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 337:1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry C, Garant D, Duchesne P, Bernatchez L.. 2001. ‘Good genes as heterozygosity’: the major histocompatibility complex and mate choice in Atlantic salmon (Salmo salar). Proc R Soc Lond B Biol Sci. 2681473:1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau Q, Yasukochi Y, Satta Y.. 2015. A limit to the divergent allele advantage model supported by variable pathogen recognition across HLA-DRB1 allele lineages. Tissue Antigens 865:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P.. 1988. Hla-a and Hla-B polymorphisms predate the divergence of humans and chimpanzees. Nature 3356187:268–271. [DOI] [PubMed] [Google Scholar]
- Lenz TL. 2011. Computational prediction of MHC II-antigen binding supports divergent allele advantage and explains trans-species polymorphism. Evolution 658:2380–2390. [DOI] [PubMed] [Google Scholar]
- Lenz TL. 2018. Adaptive value of novel MHC immune gene variants. Proc Natl Acad Sci U S A. 1157:1414.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz TL, Eizaguirre C, Kalbe M, Milinski M.. 2013. Evaluating patterns of convergent evolution and trans-species polymorphism at MHC immunogenes in two sympatric stickleback species. Evolution 678:2400–2412. [DOI] [PubMed] [Google Scholar]
- Lenz TL, Mueller B, Trillmich F, Wolf JBW.. 2013. Divergent allele advantage at MHC-DRB through direct and maternal genotypic effects and its consequences for allele pool composition and mating. Proc R Soc B Biol Sci. 2801762:20130714.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz TL, Wells K, Pfeiffer M, Sommer S.. 2009. Diverse MHC IIB allele repertoire increases parasite resistance and body condition in the Long-tailed giant rat (Leopoldamys sabanus). BMC Evol Biol. 9:269.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Deretic V.. 2007. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 710:767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighten J, Papadopulos AST, Mohammed RS, Ward BJ, Paterson G, I, Baillie L, Bradbury IR, Hendry AP, Bentzen P, van Oosterhout C.. 2017. Evolutionary genetics of immunological supertypes reveals two faces of the Red Queen. Nat Commun. 81:1294.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindo J, Huerta-Sánchez E, Nakagome S, Rasmussen M, Petzelt B, Mitchell J, Cybulski JS, Willerslev E, DeGiorgio M, Malhi RS.. 2016. A time transect of exomes from a Native American population before and after European contact. Nat Commun. 7:13175.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SJ, Cano P, Hollenbach JA, He J, Hurley CK, Middleton D, Moraes ME, Pereira SE, Kempenich JH, Reed EF, et al. 2013. Common and well-documented HLA alleles: 2012 update to the CWD catalogue. Tissue Antigens 814:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra U, Holte S, Dutta S, Berrey MM, Delpit E, Koelle DM, Sette A, Corey L, McElrath MJ.. 2001. Role for HLA class II molecules in HIV-1 suppression and cellular immunity following antiretroviral treatment. J Clin Investig. 1074:505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RD. 1972. Glycoproteins. Annu Rev Biochem. 41:673–702. [DOI] [PubMed] [Google Scholar]
- Martin MP, Carrington M.. 2005. Immunogenetics of viral infections. Curr Opin Immunol. 175:510–516. [DOI] [PubMed] [Google Scholar]
- May AC. 1999. Towards more meaningful hierarchical classification of amino acid scoring matrices. Protein Eng. 129:707–712. [DOI] [PubMed] [Google Scholar]
- McKiernan SM, Hagan R, Curry M, McDonald GS, Kelly A, Nolan N, Walsh A, Hegarty J, Lawlor E, Kelleher D.. 2004. Distinct MHC class I and II alleles are associated with hepatitis C viral clearance, originating from a single source. Hepatology 401:108–114. [DOI] [PubMed] [Google Scholar]
- Münz C. 2012. Antigen processing for MHC class II presentation via autophagy. Front Immunol. 3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neefjes J, Jongsma MLM, Paul P, Bakke O.. 2011. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 1112:823–836. [DOI] [PubMed] [Google Scholar]
- Neff BD, Garner SR, Heath JW, Heath DD.. 2008. The MHC and non-random mating in a captive population of Chinook salmon. Heredity 1012:175–185. [DOI] [PubMed] [Google Scholar]
- Nguyen TP, Kikuchi M, Vu TQ, Do QH, Tran TT, Vo DT, Ha MT, Vo VT, Cao TP, Tran VD, et al. 2008. Protective and enhancing HLA alleles, HLA-DRB1*0901 and HLA-A*24, for severe forms of dengue virus infection, dengue hemorrhagic fever and dengue shock syndrome. PLoS Negl Trop Dis. 210:e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, et al. 2012. vegan: community ecology package. Version R package version 2.0-3. https://CRAN.R-project.org/package=vegan.
- O’Riordan M, Portnoy DA.. 2002. The host cytosol: front-line or home front? Trends Microbiol. 108:361–364. [DOI] [PubMed] [Google Scholar]
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C.. 2005. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 3075709:593–596. [DOI] [PubMed] [Google Scholar]
- Parham P. 1988. Function and polymorphism of human leukocyte antigen-A, B, C molecules. Am J Med. 85(6A):2–5. [DOI] [PubMed] [Google Scholar]
- Penn DJ, Damjanovich K, Potts WK.. 2002. MHC heterozygosity confers a selective advantage against multiple-strain infections. Proc Natl Acad Sci U S A. 9917:11260–11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts WK, Wakeland EK.. 1990. Evolution of diversity at the major histocompatibility complex. Trends Ecol Evol. 56:181–187. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2017. R: a language and environment for statistical computing. Version Version 3.4.1. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Rana A, Thakur S, Bhardwaj N, Kumar D, Akhter Y.. 2016. Excavating the surface-associated and secretory proteome of Mycobacterium leprae for identifying vaccines and diagnostic markers relevant immunodominant epitopes. Pathog Dis. 74:ftw110.. [DOI] [PubMed] [Google Scholar]
- Reche PA, Reinherz EL.. 2003. Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J Mol Biol. 3313:623–641. [DOI] [PubMed] [Google Scholar]
- Richman AD, Herrera LG, Nash D.. 2001. MHC class II beta sequence diversity in the deer mouse (Peromyscus maniculatus): implications for models of balancing selection. Mol Ecol. 1012:2765–2773. [DOI] [PubMed] [Google Scholar]
- Robinson J, Halliwell JA, Hayhurst JD, Flicek P, Parham P, Marsh Steven GE.. 2015. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43(Database issue):D423–D431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche PA, Furuta K.. 2015. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol. 154:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross AC. 2014. Modern nutrition in health and disease. Philadelphia: Wolters Kluwer Health/Lippincott Williams and Wilkins. [Google Scholar]
- Sandberg M, Eriksson L, Jonsson J, Sjostrom M, Wold S.. 1998. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J Med Chem. 4114:2481–2491. [DOI] [PubMed] [Google Scholar]
- Schwensow N, Eberle M, Sommer S.. 2010. Are there ubiquitous parasite-driven major histocompatibility complex selection mechanisms in gray mouse lemurs? Int J Primatol. 314:519–537. [Google Scholar]
- She JX, Boehme S, Wang TW, Bonhomme F, Wakeland EK.. 1990. The generation of MHC class II gene polymorphism in the genus Mus. Biol J Linn Soc. 41(1–3):141–161. [Google Scholar]
- Silva MT. 2012. Classical labeling of bacterial pathogens according to their lifestyle in the host: inconsistencies and alternatives. Front Microbiol. 3:71.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade RW, McCallum HI.. 1992. Overdominant vs. frequency-dependent selection at MHC loci. Genetics 1323:861–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurgin LG, Richardson DS.. 2010. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. Proc R Soc B Biol Sci. 2771684:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stothard P. 2000. The sequence manipulation suite: javaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28:1102, 1104. [DOI] [PubMed] [Google Scholar]
- Tekaia F, Yeramian E.. 2006. Evolution of proteomes: fundamental signatures and global trends in amino acid compositions. BMC Genomics 7:307.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thursz MR, Kwiatkowski D, Allsopp CE, Greenwood BM, Thomas HC, Hill AV.. 1995. Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N Engl J Med. 33216:1065–1069. [DOI] [PubMed] [Google Scholar]
- Trowsdale J. 2011. The MHC, disease and selection. Immunol Lett. 137(1–2):1–8. [DOI] [PubMed] [Google Scholar]
- Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, et al. 2015. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43(Database issue):D405–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeland EK, Boehme S, She JX, Lu CC, McIndoe RA, Cheng I, Ye Y, Potts WK.. 1990. Ancestral polymorphisms of MHC class-II genes – divergent allele advantage. Immunol Res. 92:115–122. [DOI] [PubMed] [Google Scholar]
- Wegner KM, Eizaguirre C.. 2012. New(t)s and views from hybridizing MHC genes: introgression rather than trans-species polymorphism may shape allelic repertoires. Mol Ecol. 214:779–781. [DOI] [PubMed] [Google Scholar]
- Wolfe ND, Dunavan CP, Diamond J.. 2007. Origins of major human infectious diseases. Nature 4477142:279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. 2016. Global health estimates 2015: deaths by cause, age, sex, by country and by region, 2000-2015. Geneva: World Health Organization. [Google Scholar]
- Yasukochi Y, Satta Y.. 2014. A human-specific allelic group of the MHC DRB1 gene in primates. J Physiol Anthropol. 33:14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.