

Abstract
In this Opinion article, we aim to address how cells adapt to stress and the repercussions chronic stress has on cellular function. We consider acute and chronic stress-induced changes at the cellular level, with a focus on a regulator of cellular stress, the chaperome, which is a protein assembly that encompasses molecular chaperones, co-chaperones and other co-factors. We discuss how the chaperome takes on distinct functions under conditions of stress that are executed in ways that differ from the one-on-one cyclic, dynamic functions exhibited by distinct molecular chaperones. We argue that through the formation of multimeric stable chaperome complexes, a state of chaperome hyperconnectivity, or networking, is gained. The role of these chaperome networks is to act as multimolecular scaffolds, a particularly important function in cancer, where they increase the efficacy and functional diversity of several cellular processes. We predict that these concepts will change how we develop and implement drugs targeting the chaperome to treat cancer.
One of the key questions in cancer biology is how cells cope with and adapt to the proteotoxic stresses that accompany the malignant phenotype. The chaperome, an ensemble of molecular chaperones and their many partners, acts as a buffer to the myriad changes during malignancy and as a source of adaptation1,2. Yet, how chaperome actions are executed and whether this entity takes on distinct structures and functions remains debated.
Normal physiological conditions engage the cellular chaperome in a variety of housekeeping functions, including de novo protein folding during polypeptide synthesis, the transport of proteins to specific cellular locations and the assembly of protein complexes3–8. Cellular stress, as in cancer and other diseases, heightens the demand for these essential functions1,7,9,10. For example, the stresses experienced by cancer cells require the chaperome to buffer protein misfolding and reduce protein aggregation that may result from elevated protein synthesis and metabolic demands, the production of reactive oxygen species, growth under hypoxic and acidic conditions and the expression of altered protein stoichiometries that result from aneuploidy. To balance the resulting high level of protein misfolding in a cancer cell, a heat shock response is activated that can increase production of many chaperones. This view has its roots in the discovery of the heat shock response and heat shock proteins (HSPs)11–13. However, only a fraction (perhaps one-fifth) of the human chaperome is heat inducible14,15.
While the concept of the chaperome as a folding and disaggregase machine helps define how cells cope with stress and, in fact, was first used to define how a failure in the folding chaperome environment may contribute to the pathophysiology of cystic fibrosis16, we argue here that in cancer, this view has limitations. First, one expects a heightened folding capacity in cancer cells to be executed by an increase in the levels of chaperome members. This holds true for some cancers but not for all17. Second, the rationale that cancer is only a disease of improper protein folding — as posited above — and thus requires the chaperome to lessen proteotoxic stress may also be unsatisfactory. A simple proteome ʿfixʾ cannot fully explain the remarkable fitness of the cancer proteome despite the perceived defects, the harsh environment in which cancer cells thrive and an immune system that surveys and mediates cancer cell destruction18.
We provide a complementary view where chaperome function and structural organization, but not necessarily levels, are modulated by stress with the goal of augmenting cellular fitness and increasing cellular adaptation. We focus on the literature that describes the biochemical and functional relationships between chaperome members under normal conditions and in cells exposed to acute and chronic stress. We discuss changes in the chaperome that arise from a re-wiring of the chaperome capacity, which results in the formation of new entities with distinct functions and thermodynamic properties. We next reason that adaptation to cellular stress becomes maladaptive under chronic stress, leading to a global change in function and, in turn, to a malignant disease state. Finally, we outline how such stress-specific modifications of the chaperome impact how we perceive these proteins in cancer and, more pertinently, how we may develop anticancer therapeutics on the basis of chaperome modulators.
The chaperome
While the term chaperome was introduced in 2006 to denote an assembly of chaperones, co-chaperones and related factors16, a precise definition of this term is unresolved and will evolve as our understanding of these proteins develops. The first study to compile a list of the human chaperome was published in 2013 and described 147 bioinformatically predicted members14. This list included members of the HSP40 (also known as DNAJ proteins), HSP60, HSP70, HSP90 and HSP110 families, HSP10 and the small HSPs (sHSPs), as well as their co-chaperones and members of the folding peptidylprolyl isomerase and protein disulfide isomerase enzyme families. Later studies expanded the list to 332 members19, included tetratricopeptide repeat domain-containing proteins on the basis of their functional interactions with chaperones20 and provided a web-based chaperome analysis tool21. The function of the chaperome is fluid, and in several instances, it may function as a protective system that ensures proper folding of newly synthesized proteins (ʿfoldase activityʾ), prevents misfolding and aggregation of folded proteins and facilitates protein disaggregation (ʿ holdase activityʾ or ʿ disaggregase activityʾ)1,2,7–10. Besides folding and disaggregation, the chaperome assists in the formation of protein complexes22–25 (BOX 1). Owing to its complexity, the large number of chaperome members and their many functions, we limit our discussion to the major cytosolic chaperome machineries encompassing the HSP90 and the HSP70 chaperones and a variety of co-chaperones and co-factors.
Box 1 |. The chaperome in folding and complex assembly.
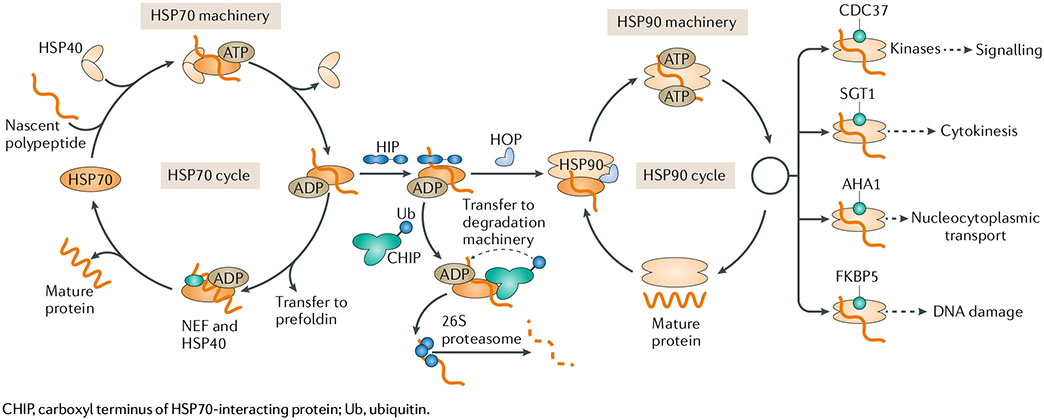
To rationalize how the chaperome participates in protein folding, a model was built in which nascent proteins are passed — from the time of synthesis to the final folded state — through several chaperone machineries, each with a specialized function and each supported by a major chaperone156,157. In the classic heat shock protein 70 (HSP70)-HSP90 chaperome folding cycle, a general model (see the figure) that has emerged comprises binding of a client protein to the HSP70 and HSP40 chaperones followed by transfer from HSP70 to HSP90 via HSP70-HSP90 organizing protein (HOP). HSP40s and nucleotide exchange factors (NEFs) then drive the HSP70 cycle of substrate binding and release, retaining the client in a folding-competent state. A role for heat shock cognate 70 interacting protein (HIP) was proposed at the stage of HSP70-HSP90 transfer, where it may attenuate HSP70 cycling by stabilizing the ADP state, thereby increasing the substrate holdase activity of HSP70 and diverting certain HSP70-ADP-substrate complexes to HSP90 or towards proteasomal degradation. Once transferred onto HSP90, cochaperones and other factors can lead to the formation of the mature HSP90 complex, which keeps the client in an activatable state136,156. Co-chaperones help regulate specific proteins or cellular functions: cell division cycle 37 (CDC37) assists in kinase regulation, suppressor of G2 allele of SKP1 (SGT1; also known as SUGT1) is involved in cytokinesis158, activator of HSP90 ATPase 1 (AHA1) plays a role in nucleocytoplasmic transport32 and FK-506-binding protein 5 (FKBP5) acts in DNA damage regulation33. In addition to assisting chaperone activity, many co-chaperones can also function independently.
Besides folding and disaggregation, the chaperome may assist in the formation of protein complexes22,23. In this case, chaperones help unite protein assemblies under normal cellular conditions but dissociate once the multicomponent assembly is formed23–25. In a classic example, nucleoplasmin is required only transiently for nucleosome assembly and is not a component of the nucleosomes22,23. Similar transitory involvement was also proposed for HSP90 during the assembly of several complexes such as small nucleolar ribonucleoprotein (snoRNP), RNA polymerase II, PI3K-related protein kinase (PIKK), the telomere complex, kinetochores, RNA-induced silencing complex (RISC) and the 26S proteasome25. Among the assembly chaperones are the histone chaperones, which play important roles in nucleosome assembly in addition to a variety of other functions related to histones in normal cells as well as in human diseases24,88,159. Finally, a variety of chaperones are involved in the many steps that lead to the assembly of the 34-subunit 26S proteasome160.
The HSP90-HSP70 chaperome
An analysis of protein expression in immortalized human cells (both nontransformed and cancer cells) identified members of the chaperome as some of the most abundant proteins, and they were, for example, 20-fold more abundant than non-chaperome proteins in HeLa cervical cancer cells14. The 147 chaperome members together contribute 7.6% of the total number of polypeptides and 10.3% of the total protein mass in HeLa cells. HSP90s are the most abundant members, averaging 2.8% of the total protein mass alone and up to 5.5% when combined with the HSP70s, whereas 1.5% of the total mass consisted mostly of substoichiometric regulatory co-chaperones of the HSP90 and HSP70 machineries14.
Connectivity between the HSP90-HSP70 chaperome.
Which factors influence the connectivity within the HSP90-HSP70 chaperome in human cells? (FIG. 1) While a reductionist approach is often used to study protein function, biological systems are characterized by immense diversity. Often, such complexity is better described by a network analysis, where network parameters define the centrality of a protein and provide information on its potential interaction spectrum, that is, the interactome. Several such interactomes and protein network analyses were reported in yeast26–32 and uncovered HSP90 and HSP70 not only as hubs but also as connectors of hubs, integrating and enabling distinct cellular processes.
Fig. 1 |. Chaperome connectivity from normal cellular states to conditions characterized by increasing stress.
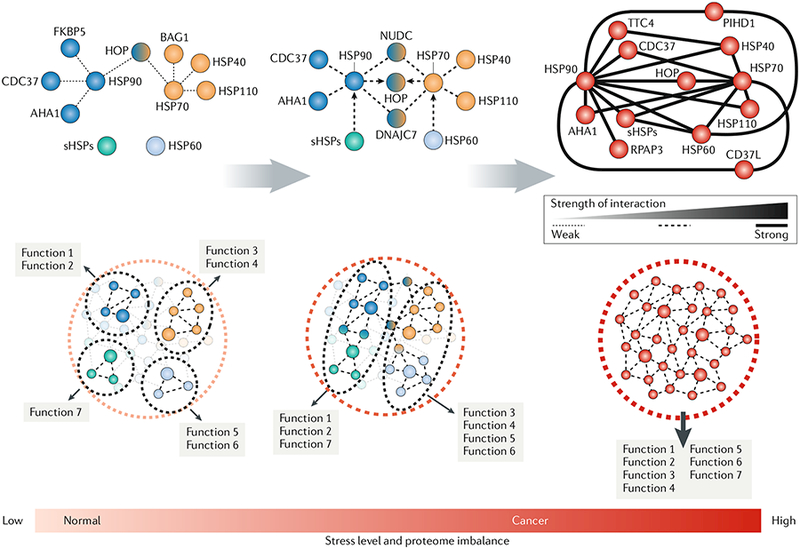
Under normal cellular conditions, the chaperome acts as a flexible and versatile connector of cellular pathways. Such flexibility is enabled by dynamic interactions among the chaperome members and between the major chaperome machineries. In these conditions, each chaperome machinery executes its set of dedicated functions with the use of individual sets of chaperones and co-chaperones. Such flexibility also permits a rapid chaperome rewiring when cells are exposed to acute stress (for example, heat) and certain chronic stresses to enable cellular stability and functionality. Networks are context-dependent, and the formation, re-wiring or restructuring of connectivities all depend on the applied molecular stress. Cellular stress increases the connectivity between distinct chaperome machineries, with the goal of increasing their functional diversity and competence. Certain stresses, such as MYC hyperactivation during oncogenesis (see the main text), can lead to a state of chaperome hyperconnectivity, where cellular demand requires maximal chaperome participation. Here, hyperconnectivity of the chaperome is executed by an increase in affinity between the chaperome machineries through their associated chaperones and co-chaperones. Thus, dynamic interactions between the chaperome members are characteristic of normal physiological conditions, but cellular stress alters the thermodynamics of such interactions, resulting in increased stability of chaperome complexes. Such a change also produces an altered functional interdependence among chaperome members. Most of the current data relates to the heat shock protein 90 (HSP90)-HSP70 chaperome, which we present as an example here, but the chaperome extends beyond these highly studied members. AHA1, activator of HSP90 ATPase 1; BAG1, BAG family molecular chaperone regulator 1; CD37L, HSP90 co-chaperone CDC37-ίike 1; CDC37, cell division cycle 37; FKBP5, FK-506-binding protein 5; HOP, HSP70-HSP90 organizing protein; NUDC, nuclear distribution protein C; PIHD1, PIH1 domain-containing protein 1; RPAP3, RNA polymerase Il-associated protein 3; sHSPs, small HSPs; TTC4, tetratricopeptide repeat domain 4.
To understand the connectivity between the HSP90 and HSP70 chaperome machineries in human cells, a large-scale study was conducted in human embryonic kidney 293T (HEK293T) cells in which several tagged chaperome members were introduced exogenously and their potential interactors identified33. Among the analysed chaperome components were cytosolic HSP90s and HSP70s and >50 co-factors and co-chaperones. The study found that while both HSP90 and HSP70 were hubs of protein networks, they functioned separately, each with its own co-chaperone subset and protein network. Specific co-chaperones connected each chaperone to distinct cellular networks. HSP90 was connected to kinases via cell division cycle 37 (CDC37), to DNA damage regulators through FK-506-binding protein 5 (FKBP5, also known as FKBP51) and to G protein signalling via FKBP8 (also known as FKBP38). By contrast, HSP70 was involved in mRNA-associated processes through BAG family molecular chaperone regulator 4 (BAG4). While the study proposed that the two chaperome machineries act mainly in isolation, with each designated for specific cellular functions, it also indicated that connectivity between them was possible and could be established through select co-chaperones, such as HSP70-HSP90 organizing protein (HOP; also known as STIP1), nuclear distribution protein C (NUDC), carboxy terminus of HSP70-interacting protein (CHIP; also known as STUB1), the HSP40 DNAJC7 (also known as TPR2 or TTC2), erythroid differentiation-related factor 1 (EDRF1) and HSP70-binding protein 1 (HSPBP1)33.
By performing network analyses on the HSP90 interactome in non-transformed and cancer cell lines, our group demonstrated that HSP90-HSP70 chaperome connectivity was influenced by stress, including proteome imbalance (which may arise from changes in protein levels, association or cellular location34), irrespective of the cellular levels of specific chaperome members35. Notably, there was little connectivity linking the HSP90 and HSP70 machineries in non-transformed cells. However, in a number of cancer cell lines, connectivity was increased, as reflected by increased participation of HOP and the inclusion of new co-chaperones in the HSP90-HSP70 chaperome networks. Connectivity was further increased in another subset of cancer cell lines, unified by at least transcriptionally active MYC expression, and was executed by over 40 co-chaperones, in addition to increased HOP participation35. As we discuss further, increased participation of chaperome members in the HSP90-HSP70 chaperome network is executed by increasing the interaction strength between HSP90 and network-participant chaperomes. Exogenous introduction and knockdown of MYC was sufficient to connect and disconnect, respectively, the network in these cancer cells. Conversely, transformation of cells with the viral oncogene v-src or mutant MET led to an increase in the cellular levels of several chaperones and co-chaperones (HSP70, heat shock cognate 70 kDa protein (HSC70; also known as HSPA8), HOP, HSP110, HSP40 and activator of HSP90 ATPase 1 (AHA1)) but, in this case, failed to substantially influence connectivity35.
Why does MYC hyperactivation augment HSP90-HSP70 chaperome connectivity in cancer cells? Among the direct transcriptional targets of MYC are components of the protein synthesis machinery, including translation initiation and elongation factors, tRNA synthetases and small and large ribosomal subunits, and these components function together to result in a global increase in cellular protein biogenesis that supports cell growth36. Beyond this, the transcriptional response of several MYC-responsive genes is further amplified by a coupled translation response37. The point at which an increase in protein production is balanced by an increase in protein degradation is unknown, but MYC activation is often associated with creating a state of proteome imbalance34. While this may require a ‘tune-up’ in the folding capacity of the chaperome, it also requires increased participation of the chaperome in processes that increase the fitness of transcription, translation and protein localization and that facilitate assembly of specific multiprotein complexes with roles in signalling, transcription, cell division and other cellular processes that lead to the aggressive behaviour of tumours associated with MYC hyperactivation. A proteome imbalance such as this (as we discuss below) is best sustained by a hyperconnected chaperome state in which the capacity of both the HSP90 and the HSP70 machineries are used most effectively to increase proteome activity and fitness. Interestingly, and supportive of the studies in human cancer cells35, recent studies in yeast found that deletion of genes involved in translation, transcriptional control and mitochondria-related processes rendered cells hypersensitive to protein overexpression — referred to as protein burden — and that an HSP90-HOP-HSP70 chaperome network was necessary to shape the response of yeast cells to protein burden38.
To date, the nature of the specific stresses that augment connectivity is ill-defined. Besides an increase in the ‘workload’ produced by specific pro-oncogenic proteins or by proteostasis inhibitors themselves35,39–43, it is also possible (and perhaps likely) that post-translational modifications of chaperome components, or even a change in pH or specific conditions in the tumour microenvironment44–47, may trigger this phenomenon. It is also possible that the restructuring of the network might come about as a result of a quantitative overload of distinct chaperome components owing to an increase in substrates1,13,21.
Physical chaperome connectivity during stress.
What is the biochemical basis for the low connectivity versus the hyperconnectivity of the HSP90-HSP70 chaperome networks? To address this question we need to analyse, where available, the experimentally defined biochemical and functional relationships among chaperome members in models ranging from simple organisms (BOX 2) to humans and determine how these relationships are modulated by cellular stress.
Box 2 |. Insights into chaperome networks from model organisms.
Chaperome connectivity during stress
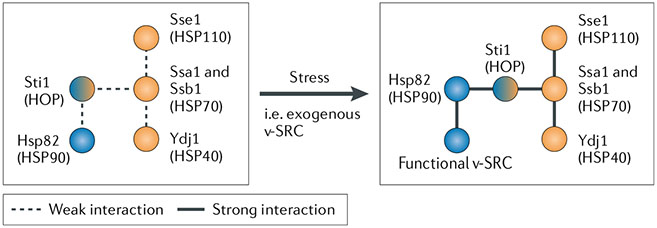
Studies in yeast investigated the relationship between the major chaperones, heat shock protein 90 (HSP90) (Hsp82 and Hsc82 in yeast) and HSP70 (Ssa1 and Ssb1 in yeast), and how this is influenced by HSP70-HSP90 organizing protein (HOP; Sti1 in yeast; see the figure). HSP90 does not form stable complexes with HOP and other chaperones unless cells are stressed, such as by the introduction of an exogenous protein. For example, HSP90 could be affinity purified with HSP70, HOP and an immunophilin only after viral oncogene v-src introduction161. Several HSP70 co-chaperones, such as an HSP40 (Ydj1 in yeast) and an HSP110 (Sse1 in yeast) were also required, and yeast with a defective form of HSP40 failed to maintain functional v-SRC. HSP110 overexpression could rescue this phenotype162–164. Tolerance to heat and antifungal agents also requires the HSP90-H0P-HSP70 link. While HSP90 is required for Aspergillus fumigatus resistance to the antifungal caspofungin, a mutation in HSP70 that impairs HOP binding, inhibition of HSP70 function with pifithrin-μ or deletion of the gene encoding HOP (sti1Δ) inhibited the ability of the fungi to adapt to and overcome caspofungin-induced cell wall stress165.
Chaperome redundancy
The relationship between inhibition of chaperome network components and redundancy was addressed in a large-scale investigation of proteome changes following deletion of SSA1 and SSB1 (two HSP70 paralogues) in yeast that were grown under optimal conditions30. In addition to being highly abundant (both proteins are among the top 5% of yeast proteins by mass), Ssa1 and Ssb1 contain the most connections among all hub proteins, with 3,269 and 2,489 links to client proteins, respectively, and interact with >40 other chaperones28. Surprisingly, no substantial changes in individual protein concentrations were associated with loss of SSA1 and SSB1, suggesting that the function of the chaperome network continues following their loss and is instead maintained by other chaperones, a process more efficient than regulating chaperone concentration. This ‘functional takeover’ can be achieved either by another HSP70 member or chaperome machinery.
Chaperome interconnectivity
Similar to tumour cells with a hyperconnected chaperome network, yeast under heat stress may also use chaperome interconnectivity to survive. For example, yeast harbouring mutations in HSC82 that cause temperature-sensitive growth166 were hypersensitive to the HSP90 inhibitors geldanamycin and radicicol167. HSP90 levels in the mutants were essentially unaltered compared to wild-type cells, and binding of inhibitors to HSP90 was unaffected by these mutations167. Defects in HOP and HSP110 function, but not in the HSP90 co-chaperones p23 (Sba1 in yeast), cyclophilin (Cpr6 in yeast), Hch1 or activator of HSP90 ATPase 1 (Aha1), led to increased sensitivity to HSP90 inhibitors164,167.
In the 1980s to the 1990s, pioneering studies on mammalian HSP90 found that its association with co-chaperones and client proteins was dynamic48–50. One feature of these multiprotein complexes that enabled their isolation and biochemical characterization was their stabilization by molybdate, vanadate and tungstate48. Such oxoanions induce a conformational state in the HSP90 chaperone that favours the capture of a client protein in a specific functional state49·50. The influence of cellular, proteome-mediated stress on the stability of chaperome complexes was not appreciated at that time.
A decade later, Kamal et al.51 investigated the interaction of HSP90 with its cochaperones p23 (also known as PTGES3) and HOP and discovered that HSP90 resided entirely in high-affinity complexes with p23 and HOP in tumour cells but not normal cells. These interactions were independent of the total levels of HSP90, p23 and HOP. However, later studies indicated that HSP90 was not entirely in a high-affinity, complexed state in cancer cells43. Instead, dynamic HSP90 complexes characteristic of normal cells were found alongside the high-affinity HSP90 complexes; these high-affinity complexes incorporated — in addition to HSP90 co-chaperones — factors normally associated with the HSP70 chaperome machinery, such as HSP40, HOP and HSC70 interacting protein (HIP). In addition to being biochemically distinct, with dynamic or strong interactions between partners in the dynamic complexes and high-affinity complexes, respectively, each of the two HSP90 pools exhibited distinct functions (see below). Unlike dynamic HSP90 complexes in normal conditions that required a stabilizing agent to capture co-chaperones, the high-affinity complexes remained stable under native PAGE35. They also had a distinct isoelectric focusing signature. In addition, unlike the dynamic HSP90 complexes, which dissociate under native PAGE and appear as dimers, these high-affinity complexes had distinct molecular masses, reflecting HSP90 incorporation into stable protein complexes with chaperones, co-chaperones and other proteins35. In addition, the more the chaperome participated in the formation of the stable, multimeric complexes, the more the HSP90-HSP70 chaperome networks became hyperconnected35. Because the formation of the hyperconnected chaperome is driven by proteome imbalance (in this case caused by MYC hyperactivation), and because hyperconnectivity creates an entity that is thermodynamically distinct from its constituent chaperome units, we coined this network the ʿepichaperomeʾ.
Together, these studies suggest that increased connectivity, via an increase in interaction strength among chaperome members, is important and more efficient in terms of energy expenditure and cellular fitness than an increase in expression levels following proteome alterations associated with chronic stress52.
Functional gain by increased connectivity under stress.
The formation of stable, multimeric chaperome complexes under stress may provide a template for the acquisition of new structures, activities and stabilities (FIG. 2). Oligomer formation can modulate ligand binding sites and thus alter affinity and specificity, increase the concentration of bound molecules, generate links between different cellular components and transmit signals, ions and other molecules across biological membranes53,54. More explicitly, oligomerization increases fitness and functionality, and components within the chaperome may assemble to acquire new competitive advantages and expand functional breadth55. Chaperome oligomerization to modulate function has been observed in bacteria, plants and other organisms in addition to human cells (BOX 3).
Fig. 2 |. Functional gains from formation of multimeric chaperome scaffolding platforms under cellular stress.
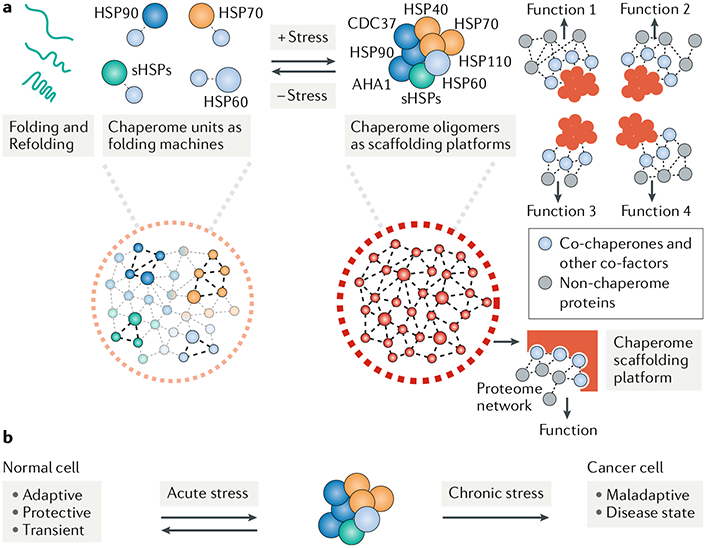
a | Cellular stress results in the formation of multimeric chaperome complexes of increased stability These structures, which we call chaperome scaffolding platforms, form after stress to execute folding-independent functions and provide a template for the acquisition of new activities. Chaperome scaffolding platforms have the advantage of increased quaternary structure diversity, where formation of new connections between chaperome units may increase the function and versatility of such platforms35,43. Stabilization of chaperome platforms by reducing the dynamic nature of chaperome-chaperome interactions may improve efficiency. These high-affinity chaperome complexes may help organize and maintain the function of the oncoprotein complement of a cancer cell in a tumour-specific and transformation-specific manner. In some cases, these context-dependent functions include the activation of signalling pathways in certain leukaemias73, the processing of key mRNAs of lymphoma subtypes78 and the maintenance of the viral oncoproteome in gammaherpesvirus-associated malignancies74. b | Formation of the chaperome platforms enables a dynamic adaptation to acute insults, providing a protective mechanism for cells under transient stress. When stresses become chronic, the adaptive functions meant to provide survival under acute stress become cemented and lead to maladaptive phenotypes associated with disease. AHA1, activator of HSP90 ATPase 1; CDC37, cell division cycle 37; HSP, heat shock protein; sHSPs, small HSPs.
Box 3 |. What are the functions gained through chaperome oligomerization?
In Arabidopsis spp., a thioredoxin-like protein (tetratricoredoxin (AtTDX)) can act as a disulfide reductase, a foldase and a holdase, each determined by its oligomeric status. AtTDX oligomerization is reversibly regulated by heat shock, which causes a transition from low to high molecular mass complexes with concomitant functional switching from a disulfide reductase and foldase chaperone to a holdase chaperone. Increased heat shock resistance in plants is conferred primarily via the holdase activity of AtTDX, that is, the high molecular mass oligomers. This is modulated via its tetratricopeptide repeat (TPR) domains, as their deletion results in increased disulfide reductase activity and loss of the holdase function168. Formation of the oligomeric form is associated with a conformational change in AtTDX structure169.
In Escherichia coli, oligomeric heat shock protein 70 (HSP70; DnaK in E. coli) is also more prevalent in cells exposed to heat shock. The oligomers retain ATPase and holdase activity but have a reduced ability to refold substrate and do not respond to stimulation by HSP40 (DnaJ in E. coli)170.
In human cells, stress conditions also induce chaperome oligomerization. The cytosolic HSP70s form oligomeric structures in response to heat, osmotic stress or hydrostatic pressure171,172, whereas the endoplasmic reticulum HSP70 glucose-regulated protein 78 (GRP78; also known as BIP and HSPA5) forms homo-oligomers in response to glucose deprivation173. Stable, ATP-independent, oligomeric forms of HSP70 and HSP110 were identified in a mouse C3H mammary carcinoma174. HSP27 (also known as HSPB1), a small HSP, forms small oligomers in exponentially growing cultured cells and large oligomers in tumour cells in vivo or in those grown to confluence in vitro175–178. The large oligomers represent the active form and are plausibly responsible for underlying oncogenic functions owing to their ability to inhibit apoptosis and dampen the effects of oxidative stress175–178. HSP90 also oligomerizes under conditions of thermal stress179–183. while all of the members of the HSP90 family are believed to exist primarily as homodimeric species, some reports suggest the presence of a monomeric form, while others have noted a tendency of HSP90 to self-associate when purified from cells under thermal stress58. These HSP90 oligomers were not aggregates but rather soluble high molecular mass species that retained the ability to bind co-chaperones, such as activator of HSP90 ATPase 1 (AHA1). In the absence of AHA1 binding, HSP90 ATPase activity is otherwise extremely low129. In fact, AHA1 preferentially binds oligomeric HSP90 over the dimeric species182.
A recent study by Drummond and colleagues56 elegantly described how heat stress induced global changes in the yeast proteome that led to the formation of reversible, large protein assemblies rather than irreversible, insoluble aggregates of misfolded proteins destined for degradation. Even severely aggregated endogenous proteins were recovered without degradation following heat shock, and certain heteromeric complexes remained active with unaltered fidelity, even during stress. The authors concluded that heat-induced ʿreversible aggregationʾ or oligomerization reflects an adaptive, autoregulatory process that aids cellular survival under conditions of thermal stress, and these ideas are in accordance with studies in human cancer35. Interestingly, exposing yeast to short-term thermal stress led to only a modest increase in the cellular levels of most chaperome components (~1.9-fold) but to a robust increase in members involved in oligomerization, such as several sHSPs and holdases57.
Because HSP90 is stable as a dimer even at high concentrations, a conformational change is most likely necessary to drive stress-induced oligomerization. This may lead to the unmasking of an oligomerization site that is absent in the dimer, giving rise to a new protein-protein interaction platform and a new quaternary structure. This postulate is supported by findings that the formation of oligomeric species is inhibited by ATP and the HSP90 inhibitor geldanamycin but favoured by agents such as vanadate and molybdate, which induce a substantial conformational change in HSP9049,58. Interestingly, binding of p23 shifted the HSP90 dimer-oligomer equilibrium towards the dimer, suggesting that a specific HSP90 conformation exists that is more permissive of the oligomerization process59.
HSP90 and other chaperome components may use oligomerization to trigger functions that are normally silent but become active during proteome imbalance58,60,61, and importantly, these specific functions may be executed more effectively than through chaperome units (FIG. 2). For example, a multimeric chaperome scaffold could facilitate oncogene addiction by allowing constitutive activation of signalling networks that contribute to malignancy39–41. Plants, which are constantly exposed to pathogenic stressors, maintain assembled heteromultimeric protein complexes of certain chaperomes even in the absence of pathogens. It is suggested that such complexes enable the indirect recognition of pathogen effector molecules during pathogen invasion and then interact with diverse proteins and protein complexes to activate the immune signalling pathway that protects against the pathogen62. The presence of stable chaperome complexes in cancer cells may be inferred to have similar ʿsensorʾ and ʿeffectorʾ mechanisms.
The functions of molecular scaffolds in general are to increase the efficacy of existing cellular processes and to facilitate the evolution of new functions, either by linking pre-existing components in a new way or by rewiring system thermodynamics63. Compared with molecular scaffolds, chaperome scaffolding platforms have the advantage of increased quaternary structure diversity, where formation of new connections between chaperome units may increase the function and versatility of platforms. Stabilization of chaperome platforms by reducing the dynamic nature of the chaperome-chaperome interactions may further increase efficiency. In cancer cells, where increased activity in a multitude of cellular activities is essential for survival, bringing proteins in proximity and posing them in the right orientation has obvious benefits. The idea of increased proximity for increased efficacy is well studied in enzymes, which align reactive chemical groups64. This reduces entropy, making the reaction more favourable. Therefore, we suggest that stable chaperome assemblies similarly drive unfavourable processes by reducing entropy, which may further increase the efficacy of such scaffolding platforms.
An indication of the function of chaperome platforms in cancer comes from the analysis of large interactome data sets derived mostly from yeast26–32. These analyses are challenging in human cells, and several studies have employed tagged proteins to isolate specific chaperomes and their interactomes33,38,65,66. Such cellular models in which a tagged protein is introduced into a cell line cannot recreate the environment in tumours. This may explain why some studies failed to isolate transcription factors along with kinases as HSP90 substrates33. Furthermore, transfection introduces cellular stress and could impose artificial chaperome interactions with exogenous proteins. A potential solution to this problem comes from the use of small molecule probes as baits for specific chaperome complexes. The use of probes with a binding preference for the multimeric chaperome complexes may enrich these species from a tumour lysate and in turn permit an analysis of the chaperome and its binding partners in native tumours35,43.
The use of a chaperome bait in a large-scale approach was used to investigate the interactome of multimeric, tumour-associated HSP90 complexes in K562 cells, a BCR-ABL fusion-driven chronic myeloid leukaemia (CML) cell line43. In these cells, dynamic HSP90 species retained their activity in regulating housekeeping protein functions and interacted with the non-oncogenic kinase ABL. By contrast, high-affinity, multimeric HSP90 species regulated the oncoprotein complement of the cell and interacted instead with the oncogenic BCR-ABL kinase. The interactome of multimeric HSP90 also contained multiple proteins as part of active signalling megacomplexes, as well as adaptor proteins such as growth factor receptor-bound protein 2 (GRB2), dedicator of cytokinesis (DOCK), CRK-like (CRKL) and epidermal growth factor receptor substrate 15 (EPS15). These factors link BCR-ABL to key effectors of multiple, aberrantly activated signalling pathways in K562 cells, indicating a clear presence of HSP90 in assembled — and active — signalling complexes. This was also true for HSP90 in B cell receptor complexes and their downstream signalling components in lymphomas67, for B cell receptor hyperactivity in chronic lymphocytic leukaemia (CLL)68 and for increased Janus kinase (JAK)-signal transducer and activator of transcription (STAT) activity in myeloproliferative neoplasms and acute lymphoblastic leukaemias (ALLs)69–71.
In the context of CML, the stable, multimeric HSP90 species were also found to regulate STAT5 activity43. Regulation occurred at multiple nodes in the STAT5 signalling pathway, indicating a role in increasing pathway (rather than just singular protein) activity. Specifically, multimeric HSP90 binding to STAT5 altered STAT5 phosphorylation and dephosphorylation kinetics and, moreover, maintained STAT5 in an active conformation in STAT5-containing transcription complexes43. Similar activities were also reported for STAT3, whereby HOP and HSP90 were found to form a complex required both for the maturation of JAK2 and as a scaffolding complex for the transduction of JAK2-STAT3 signalling in certain ovarian and endometrial cancer cells72. In acute myeloid leukaemia (AML), the activity of the STAT5 pathway is dependent on stable, multimeric HSP90, which is the basis for AML cell sensitivity to HSP90 inhibition73.
Survival of lymphoma cells transformed by Epstein-Barr virus (EBV) or Kaposi’s sarcoma-associated herpesvirus (KSHV) also requires the formation of the multimeric HSP90 species74,75. For example, viral FLICE inhibitory protein (vFLIP), a viral oncoprotein homologous to CASP8 and FADD-like apoptosis regulator (CFLAR; also called cFLIP) with nuclear factor-κB (NF-κB)-activating and anti-apoptotic activities, bound to the multimeric HSP90, but endogenous CFLAR did not74. In these lymphomas, multimeric HSP90 regulated several viral and cellular proteins, including many involved in NF-κB signalling, apoptosis and autophagy. Stable chaperome platforms may also be needed for the regulation of other viruses76,77. In another example, multimeric HSP90 sustained eukaryotic translation initiation factor 4E (eIF4E) activity in the nucleus and the cytoplasm by increasing the activity of eIF4E in driving nuclear export and translation of BCL6, MYC and BCL2 mRNA in diffuse large B cell lymphoma cell lines78. These data indicate a role for multimeric HSP90 in controlling the post-transcriptional dynamics of key mRNA species. In these and other B cell lymphoma cells, multimeric HSP90 also functions as a co-repressor for B cell lymphoma 6 protein (BCL-6) by maintaining it in a stable conformation within repressive complexes in the nucleus to block expression of BCL-6 target genes79.
Other evidence of the involvement of chaperome platforms in modulating the plasticity of signalling networks comes from studies with HSP90 inhibitors in cancer. For example, one of the effects of HSP90 inhibitors in cancer models, as well as in other disease models, is to disable the formation of signalling feedback loops that enable resistance to kinase inhibitors80. This concept is based on landmark studies in yeast and plants81. Signalling networks maintain a degree of dynamism and adapt to the ever-increasing changes induced by environmental conditions, such as extracellular and intracellular stimulatory or inhibitory signals. These signals can be derived from the tumour microenvironment or from other factors, such as a kinase inhibitor82. Crosstalk of signalling pathways and their dependence on the chaperome system for rapid remodelling are well-studied phenomena, and such work resulted in the use of HSP90 inhibitors in cancer in combination with kinase inhibitors83,84. If signalling network rewiring can be inhibited, the outgrowth of resistant clones should be reduced, and the duration of disease control would be prolonged. This phenomenon is evident when an HSP90 inhibitor is added to a kinase inhibitor, such as the JAK inhibitor ruxolitinib in myeloproliferative neoplasms and the human epidermal growth factor receptor 2 (HER2; also known as ERBB2) inhibitor trastuzumab in breast cancer85,86.
The scaffolding functions of the chaperome in cancer are likely to include other essential dynamic processes, such as transcription, translation, metabolism, immune adaptation, cell death and cell division pathways. One role may be to regulate the transcriptional and translational landscape, whose flexibility plays a key role in adaptation to the malignant state. Studies in yeast highlight the sophistication and fine-tuning of stress-regulated gene expression that is needed to promote balanced and coordinated protein production under each stress condition87. In transcription-related processes, the functions of chaperomes have been the subject of intensive study, but little is known about the relationships between chaperome entities, such as the histone chaperones and the HSP90 and HSP70 chaperone machineries. Relationships between the chaperones, co-chaperones and processes associated with nucleosome assembly and disassembly, chromatin remodelling and the integration and function of large protein machines involved in DNA transcription, replication and repair have been reported88–91. Yet in most cases, it is unknown whether the individual chaperone machineries function redundantly, represent independent activities at distinct stages during transcriptional regulation or demonstrate interconnectivity to increase activity and specificity.
Together, evidence suggests that the chaperome acts as a multimolecular scaffold in cancer, providing cellular components with a framework on which they can work more efficiently or differently than they would without chaperome participation92. Another benefit of such chaperome scaffolding platforms is reorganization of dynamic biological networks. To maintain robustness in the face of ever-changing genomic and environmental challenges, cells must maintain highly reconfigurable protein networks and reorganize macromolecular complexes93–98, and chaperome platforms can facilitate this. Dynamic adaptation-required remodelling of intricate cellular networks may then become stabilized when stress is chronically applied, such as in cancer, turning the adaptive function meant to provide survival under acute stress into maladaptive phenotypes associated with disease.
Redundancy and hyperconnectivity
Redundancy (or the lack thereof) is key to understanding the effect that inhibition of specific chaperome members has on cancer cells (FIG. 3). Recently, the cytotoxicity of PU-H71, an inhibitor with specificity for HSP90 when the chaperone is part of stable, multimeric chaperome complexes, was studied by our team in a large panel of cancer cell lines and primary tumour specimens encompassing pancreatic, gastric, lung and breast cancers, as well as lymphomas and leukaemias35. We observed that tumours could be classified into two types. In type 1, the drug induced substantial apoptosis, with little recovery after drug washout, whereas in the other subset, termed type 2, only growth inhibition was observed, with cancer cells recovering after drug washout. Intriguingly, in both type 1 and type 2 tumours, inhibition or small interfering RNA (siRNA) knockdown of HSP90 led to the depletion of select client proteins (for example, phosphorylated ribosomal protein S6 kinase (p-S6K), phosphorylated ERK (p-ERK) and epidermal growth factor receptor (EGFR)) and an identical HSP70 induction profile, indicating that the different responses were intrinsic to HSP90 rather than secondary effects induced in one type and not the other, such as impartial inhibition of HSP90 or apoptosis suppression by HSP70 induction (given that HSP70 has several anti-apoptotic functions99–103). Analysing network connectivity in type 1 and type 2 tumours revealed why HSP90 impairment was toxic to type 1 but not type 2 tumours. Type 1 tumours were characterized by highly interconnected or hyperconnected HSP90 and HSP70 chaperome machineries. By contrast, the network connectivity of type 2 tumours was similar to that observed in HEK293T cells33, where the HSP90 and HSP70 machineries behaved as insular chaperome communities, each utilizing a subset of its dedicated co-chaperones to execute designated cellular functions.
Fig. 3 |. Chaperome networks use redundancy to protect from temporary or partial impairment.
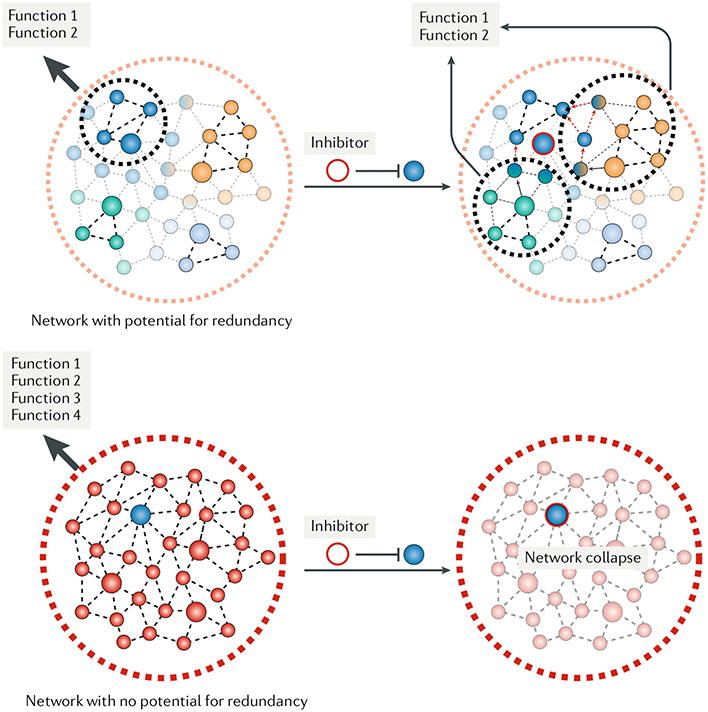
An easy way to understand redundancy is to think of a technical network (for example, a power grid). In this model, when a primary path is unavailable, an alternate path can be instantly deployed to ensure minimal downtime and continuity of network services. In a biological system, redundancy has a similar connotation and provides chaperome networks with a rapid response to insults, whether genetic or environmental stress, or to drugs targeting any of the chaperome network components. Under normal conditions or perhaps low stress (that is, type 2 tumours), the networking capacity of the chaperome is in place but executed via flexible interactions between network components31. Here, inhibition of chaperome units may be overcome by transfer of the workload to another chaperome or chaperome machinery In reality, networks cannot be infinitely redundant, and at some point, one may imagine that the entire network, including fail-safe systems, must be utilized; such is the case in conditions of high stress (such as with MYC hyperactivation), where the many connections between chaperome components become occupied and the network, which we call a hyperconnected chaperome network, reaches full capacity and may become ‘rigidʾ (REFS35,186). Here, the range of adaptive responses is diminished, and cancer cells are now vulnerable to therapeutic interventions that target critical hubs and nodes. In this sense, the stresses encountered by a cancer cell leads to depletion of the networking capacity of the chaperome and stabilization of network connections. Unlike dynamic chaperome networks, which can restructure under stress, this hyperconnected network lacks ʿrestructuring capacityʾ and collapses after inhibition of a key chaperome network unit.
Thus, the network in type 2 tumours enables redundancy. Studies in yeast (BOX 2) and human cells indicate that other chaperome members may take over the workload of the disabled chaperome and retain a robust cellular network30,35,104–106. When HSP90 is impaired, alternative use of the HSP70 machinery is a logical solution, and several lines of evidence support this hypothesis. First, proteome analyses of the interactome of cyclin-dependent kinase 4 (CDK4) indicated that CDK4 associates with HSP90α and HSP90β, CDC37 and two immunophilins (FKBP4 (also known as FKBP52) and FKBP5), but upon HSP90 inhibition with NVP-AUY922, the kinase becomes bound to HSC70, HSP70, HOP and HIP104. This was later confirmed by Taipale et al.33, who found that HSP90 inhibition by ganetespib led to stronger associations of some proteins — mainly kinases — with HSC70. Therefore, upon HSP90 inhibition, CDK4 and other client proteins may be transiently scaffolded by the HSC70-HIP complex to slow their degradation. This has also been observed for the microtubule-associated protein tau, where binding by HSC70 or HSP70 slowed or accelerated tau clearance, respectively105. Second, ʿworkload transferʾ by increased connectivity between the HSP90 and HSP70 machineries has been demonstrated in a study in which the HSP90 interactome was analysed in HEK293T cells in the presence of ATP, ADP and geldanamycin106. As discussed above for non-transformed cells, little connectivity was noted between HSP90 and the HSP70 machinery under native conditions because HSC70, the BAG proteins and HIP were mostly absent from the HSP90 isolates. However, upon treatment with geldanamycin, HSP90 association with these particular chaperome members increased and a number of other chaperome members also became associated. These other factors, including CDC37, FKBP4, tetratricopeptide repeat domain 9C (TTC9C), TTC4, DNAJC7, PIH1 domain-containing protein 1 (PIHD1), HSP90 co-chaperone CDC37-like 1 (CD37L) and RNA polymerase II-associated protein 3 (RPAP3), are also reported to be involved in forming the hyperconnected chaperome network in type 1 tumours35.
Collectively, these findings suggest that the chaperome network uses redundancy to protect itself from temporary or partial impairment of one of its hubs and is best seen when cells evade toxicity as a result of HSP90 inhibition (FIG. 3). Such workload transfer owing to redundancy in the chaperome network may also explain why inhibiting the direct interaction of CDC37 with HSP90 fails to compromise kinase function in some cancer cells107. However, this protective mechanism is absent in tumours in which the hyperconnected network is reinforced between the HSP90 and the HSP70 chaperome machineries. Indeed, in these cancer cells, but not in the type 2, low-connectivity tumours, knockdown of HSP110 (an HSP70 co-chaperone) or HOP was sufficient to diminish the activity or expression of HSP90-regulated kinases, such as p-S6K, p-ERK and EGFR, in a fashion similar to knockdown of HSP90α and HSP90β or AHA1 (REF.35). Also supporting the lack of redundancy in tumours with chaperome hyperconnectivity is the finding that the more the chaperome was integrated into the hyperconnected networks, that is, the epichaperome, the more vulnerable it became to hub impairment35. Consequently, pharmacological or genetic inhibition of several chaperome members (such as HSP90, HSP110, HOP or AHA1) was most toxic in type 1 cancer cells. Maintenance of the hyperconnected network was vital for the survival of these tumours and for the function of their oncogenic proteome networks35.
Chaperome inhibitors
Individual chaperomes have been actively sought as cancer targets. Nonetheless, clinical studies of chaperome inhibitors, such as those targeting HSP90, have had limited success in clinical trials108–111. In light of our discussion, we propose that therapies targeting chaperome members need to address the complexity of the networks in which they participate. HSP90 is routinely targeted without considering its context within the framework of the cancer chaperome. Instead, therapeutic strategies were based on a specific model: If an HSP90 client protein is important for the function of a particular tumour cell, then the tumour will respond to HSP90 therapy. Although this model has had success, for example, in HER2-positive breast cancers, it has largely failed for other ʿclient-dependentʾ tumours. This has led to a state of overt negativity towards the chaperome field. We propose a fresh look at the chaperome based on a novel mechanistic understanding of chaperome interconnectivity and interdependence.
The phenomenon of cooperation or interconnectivity for survival is not unique to the chaperome and has been studied in the context of genetic interactions via epistasis analysis and synthetic lethality112·113. HSP90 inhibition is lethal only when HSP90 is hyperconnected with the HSP70 machinery and other chaperomes, just as poly(ADP-ribose) polymerase (PARP) inhibition is lethal when breast or ovarian cancer cells harbour BRCA1 or BRCA2 mutations114,115. Therefore, we suggest that HSP90, per se, is not a target in cancer, in the sense that tumours become addicted to HSP90 only when HSP90 becomes integrated into epichaperome networks, potentially explaining the poor performance of HSP90 inhibitors in non-selected patient populations.
By analysing 95 cancer cell lines, 40 primary AMLs and 23 primary breast tumours ex vivo and 51 solid tumours and lymphomas in patients, we found that 50–60% express variable epichaperome levels but only ~10% are high expressors, as defined by the amount of HSP90 residing in hyperconnected chaperome networks35. We suggest that these cancers with high expression of the epichaperome are ideal for single-agent HSP90 therapy. Basket trials116 where epichaperome levels rather than genetics or cancer type are used for patient selection are more likely to capture potential responders to HSP90 therapies. Interestingly, most preclinical studies that reported substantial anticancer activity in xenografted human tumours (both patient-derived and cell line-derived) in mice with single-agent HSP90 therapy73,79,117–119 were later shown to have been performed with tumours harbouring high epichaperome levels35.
Realization that the HSP90 epichaperome is the optimal target for cancer therapy provides a new route for patient selection. Because of the unique presence of the epichaperome in tumours, and the distinct biochemical nature of HSP90-containing epichaperomes, chemical probes specific for the epichaperome can be developed35. These probes enable epichaperome detection in patients via positron emission tomography (PET) for solid tumours and flow cytometry for liquid tumours and are currently under clinical investigation as potential companion diagnostics for PU-H71 (incorporated into NCT01393509 (REF120), NCT01269593 (REF121) and NCT03166085 (REF122)).
Clearly, therapies built around chaperome inhibitors will need to consider the effect of combination therapies on chaperome networks. The sequence of therapy administration will also likely play an important role in optimizing potency and efficacy, as therapies may either increase or decrease chaperome connectivity and in turn the effectiveness of chaperome inhibitors123. Moreover, an optimal therapeutic index and an ability to be incorporated into combinatorial regimens, the mainstay for most cancer therapies, will also be crucial for chaperome inhibitors. As multimeric chaperome complexes support the oncogenic functions of the proteome, inhibitors that discriminate between a single chaperome and a chaperome incorporated into epichaperomes (that is, HSP90 versus HSP90-containing epichaperomes) are preferred as they may offer better target engagement and a safer profile. While acute chaperome member inhibition may not be toxic to normal cells, chronic suppression may have unwanted effects. This has been noted for HSP90 agents that persisted in the eye or in the gastrointestinal tract, resulting in visual disturbances and gastrointestinal toxicity, respectively124,125.
In the background of these discoveries, notable efforts over the past 2 decades have resulted in the development of a variety of HSP90 and HSP70 inhibitors, and although a few are selective for certain paralogues, most act as pan-HSP90 or pan-HSP70 inhibitors17,126–128. In the context of type 2 tumours35, the chaperome network concept would suggest that dual targeting of two hubs (for example, HSP90 and HSP70) will overcome redundancy and provide synthetic lethality. Nevertheless, there are a growing number of HSP70 inhibitors, which on their own have shown efficacy for select cancers101,127. Compounds that target the interaction of chaperones with specific co-chaperones have also been investigated, but these studies are still in their infancy107,129. The sensitivity of tumours to chaperome inhibitors may or may not necessarily overlap with the sensitivities exhibited with HSP90-specific inhibitors, as distinct chaperome networks may be evident among diverse tumours. In addition, inhibitors of chaperome-associated components in the proteostasis network, such as autophagy, the ubiquitin-proteasome system and endoplasmic reticulum (ER) and mitochondrial stress responses, may further increase vulnerability of tumours to chaperome inhibitors39. Because inhibitors of HSP90 are the most studied and the only ones so far translated to the clinic86,130, we focus the rest of our discussion on these small molecules.
One misconception is that all the small-molecule HSP90 inhibitors are identical. This concept derives from findings that most bind a similar pocket in HSP90. For agents that have moved into clinical investigation, this site is the nucleotide binding pocket located in the amino-terminal domain17,39–41. This view overlooks the reality that agents directed towards HSP90 in cancer cells are in fact being directed towards a multitude of HSP90 complexes, some of which may be dynamic and others that may be stable. Yet, HSP90 complexes are differentiated by their composition and possibly by post-translational modifications in resident components131–133. Ligands may also bind in the HSP90 pocket in unique orientations, and small-molecule binding may trigger pocket rearrangements134. Of note, HSP90 undergoes large conformational changes that may influence the pocket configuration as well as the residence time of the bound ligand135–137. These factors may all influence the association and dissociation kinetics of an HSP90-targeted drug35,134,138.
Evidence also indicates that not all inhibitors preferentially bind a common set of HSP90 conformations139. This is evident from studies using immobilized inhibitors to trap HSP90 complexes in cancer cells. For example, while geldanamycin efficiently captures HSP90, it binds poorly to HSP90 complexes containing client proteins43,140. This was not the case for other inhibitors, such as PU-H71, which instead preferred HSP90 species bound to co-chaperones and client proteins35,43. Compounds that target the HSP90 carboxyl terminus, such as celastrol and the novobiocin-derived compound KU174, prefer or perhaps even elicit a distinct HSP90 conformation that is different from that favoured by geldanamycin141. Importantly, studies comparing the protein partners of HSP90 inhibitors with those obtained after treatment with HSP90 antibodies (which instead capture the entire cellular pool of HSP90) indicate that each inhibitor prefers unique HSP90 pools35,43,68.
What are the consequences of such complexity in the binding preferences of chaperome inhibitors? First, equilibrium binding metrics are poorly suited for understanding the binding profiles of chaperome inhibitors. While the interaction of a small molecule with a protein or protein complex is often described using such parameters, there is an increased appreciation in the drug discovery field that such terms are unable to describe the complexities of a target in the cellular context and are even less apt to describe the many factors operating in vivo142,143. This realization has implications for the development of chaperome inhibitors and emphasizes the importance of considering factors such as the residence time, pharmacokinetics (PKs) and pharmacodynamics (PDs) to fully appreciate the distinctions between HSP90 agents17,144,145 (BOX 4).
Box 4 |. Factors that influence the biological activity and therapeutic index of chaperome inhibitors in the context of cancer.
We consider a scenario (see the figure) where a type 1 tumour with a hyperconnected chaperome network is treated with three heat shock protein 90 (HsP90) inhibitors with either a preference for the stable, multimeric HsP90 complexes (inhibitor A), a preference for the lower affinity, dynamic HsP90 complexes (inhibitor B) or no preference (inhibitor C). However, small molecules are not infinitely selective; an inhibitor with preference for epichaperomes may interact at much higher concentrations with non-complexed or low-affinity species. Drug selectivity comes not from static measurements performed at equilibrium but rather from a combination of factors that can be defined only in vivo. Indeed, in cancer cells under conditions of thermodynamic equilibrium, there is little difference in the phenotype observed with the three agents. During the time the compound resides inside the cell at an invariant concentration over the time course of the experiment and in the confined environment of tissue culture, the drug will occupy and inhibit the phenotype-determining species regardless of its binding preference.
However, in vivo, other factors will substantially influence the behaviour of the inhibitors17,142,144,146. For inhibitor A, we posit that Cmax, the maximum or peak concentration that a drug achieves in the tumour after administration, is most important. After compound administration and once the epichaperome is engaged, limited target turnover and slow target dissociation kinetics (K) assure extended epichaperome suppression. This is associated with a tumour pharmacokinetic (PK) profile, which mirrors the tumour pharmacodynamic (PD) profile and in turn leads to effective antitumour activity. If this drug also has a profile associated with rapid clearance from plasma and other sites in the body (with no obvious accumulation of toxic metabolites), inhibitor A can be dosed without overt toxicities. This is the case for an HsP90 inhibitor with a good therapeutic index and ability to suppress the target at safe doses.
For inhibitor B, the ability to engage the epichaperome in vivo is a challenge and is dependent upon maintaining extended systemic levels. The longer the systemic exposure, the more likely equilibrium is reached and the epichaperome is engaged. This becomes a considerable challenge in the body, as long systemic exposure also increases the chance for off-target effects and potential toxicity. As a result, the ability to use these drugs and obtain a favourable PD profile and antitumour effect is limited. This is a scenario where an extended tumour PK profile is indicative of binding to a specific HSP90 conformation142,184,185 and not to epichaperomes. Here, the tumour PK profile does not match the tumour PD profile, and limited antitumour activity is achieved144.
For inhibitor C, Cmax is important, but limitations in reaching it derive from factors related to off-target effects and possible toxicity owing to HsP90 inhibition in certain tissues and organs. These considerations greatly influence the ability to deliver sufficient drug to the target in a tumour at a safe dose144.
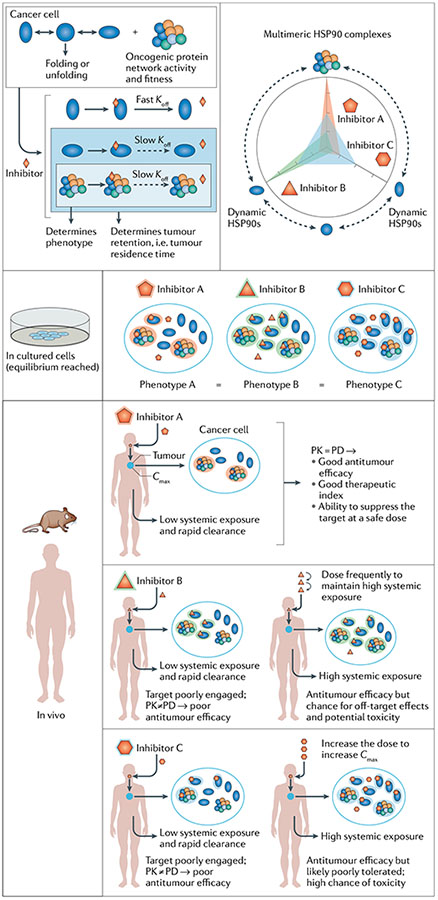
Overall, the biological activity and therapeutic index of chaperome inhibitors are influenced by many complex factors, and no single measure is sufficient to understand or predict behaviour. Ultimately, then, classical approaches to drug discovery are inappropriate when one targets the chaperome. Instead, attention must be paid during drug development to the ability of the compound to modulate relevant, disease-specific chaperome species.
Perspectives
With this Opinion article, we hope to catalyse a shift in our thinking of and the approach towards the study of the chaperome in cancer. We suggest that efforts must be directed to understand how individual chaperomes are integrated into intricate cellular networks and that only through a network approach will we be able to fully understand the implications of chaperome proteins as therapeutic targets. The previous rush to move the first class of chaperome inhibitors — the HSP90 inhibitors — into clinical studies without a deep understanding of the complex biology of the chaperome has led to disappointment in the medical community. This reticence to support future trials may simply neglect chaperome complexity. Instead, we believe that chaperome-based therapeutics have high potential for the treatment of cancer as well as a number of other diseases, such as neurodegenerative and infectious diseases.
This reality highlights the need to study the function of the chaperome in the context of native cellular states. Therefore, information derived from the use of cellular models must ultimately be validated in relevant biospecimens or other systems more closely related to the disease. Efforts should appreciate and distinguish the housekeeping and disease contributions of the chaperome to cellular function. Only through a better understanding of the biochemical factors that enable the formation of disease-associated chaperome networks and epichaperomes will we be able to design drugs that target such species.
In this context, several questions remain unanswered. First, how are the relatively stable oligomeric chaperomes formed? Changes in the quaternary structure of chaperomes may occur through conformational changes within individual subunits, which can be induced by post-translational modifications, changes in cellular location, alterations in the pH or ion composition or other unforeseen mechanisms. Future studies must address the mechanisms underlying this form of regulation but will be challenging considering the context-dependent formation of chaperome networks. Studies conducted in vitro with recombinant chaperome proteins suggest that formation of specific complexes is executed by conformational changes that render chaperones competent to bind a specific protein146. In cells, this is likely to be modulated, and perhaps facilitated, by the factors listed above, which may constrict the chaperone component to favour binding44,47,133,147. Second, is the hyperconnected HSP90-HSP70 network the only epichaperome that forms in cancer cells during chronic stress? Instead, we envision that chaperome networks are driven by specific stresses. In this scenario, each stress might potentially lead to the formation of distinct epichaperome networks. This has yet to be investigated. Third, might chaperome networks form within cellular compartments? One can imagine that specific networks in the mitochondria or ER might be linked to drug efficacy, as these compartments contain homologues of most cytoplasmic chaperome components148–150. In addition, altered cellular locations of chaperomes and changes in homeostasis or proteostasis in these organelles are tightly associated with some forms of cancer46,151,152. Therefore, these factors will need to be considered when we design and develop chaperome inhibitors for cancer.
It is also important to emphasize that the chaperome does not act in isolation in regulating the proteome. Instead, the chaperome partners with the proteostasis network that comprises cellular protein quality control processes. These processes include autophagy, the ubiquitin-proteasome pathway, the mitochondrial stress response and the ER-linked unfolded protein response153,154. Previously described studies associated changes in the expression levels of chaperome. members with the maintenance of the proteostasis network153,154. We propose a complementary view, where changes in the association strength between chaperome members endow the cell with new functions that enable robust proteostasis during conditions of chronic stress. In fact, proteostasis-targeting agents, such as proteasome inhibitors, represent examples of clinically successful agents that, in well-defined cancers and in combination with other agents, have provided substantial clinical benefit in select cancers, most notably multiple myeloma155. This provides precedent as well as additional impetus for the introduction of judicious patient selection and drug combination strategies for chaperome-based therapeutics.
In conclusion, while changes in the expression levels of chaperome members, the interaction strength between chaperomes, chaperome complex composition and cellular location of chaperome members are all hallmarks of cancer, we are only beginning to understand how these factors influence how chaperome networks form, how they are regulated and how they might be co-opted to develop hypothesis-driven, therapeutic approaches in cancer. In the future, we envision that the range of diseases linked to chaperome biology and new therapeutic targets will emerge from these efforts.
Acknowledgements
G.C. is supported by the US National Institutes of Health (NIH) (R01 CA172546, R01 CA155226, P01 CA186866, P30 CA08748 and P50 CA192937), the Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of the Memorial Sloan Kettering Cancer Center; T.W. is supported by the Lymphoma Research Foundation; T.L.S.A. is supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (17/01130–6); and J.L.B. is supported by the Cystic Fibrosis Foundation Therapeutics (BR0DSK13XX0) and by the NIH (grants GM75061 and DK79307).
Footnotes
Author contributions
S.J., T.W., T.L.S.A. and S.S. researched data for the article and contributed to the writing of the article and to the review of the manuscript. T.W., T.L.S.A. and G.C. designed the figures and their content. J.L.B. edited the manuscript and provided specific text. G.C. designed the content of the manuscript, researched data for the article and wrote, edited and reviewed the manuscript.
Competing interests
G.C. has partial ownership in Samus Therapeutics Inc., which develops chaperome inhibitors. S.J., T.W., T.L.S.A., S.S. and J.L.B. declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Cancer thanks R. Kaufman and the other anonymous reviewer(s) for their contribution to the peer review of this work.
References
- 1.Balch WE, Morimoto RI, Dillin A & Kelly JW Adapting proteostasis for disease intervention. Science 319, 916–919 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Lindquist S Protein folding sculpting evolutionary change. Cold Spring Harb. Symp. Quant. Biol 74, 103–108 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Hartl FU, Bracher A & Hayer-Hartl M Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Laskey RA, Honda BM, Mills AD & Finch JT Nucleosomes are assembled by an acidic protein which binds histones and transfers them to DNA. Nature 275, 416–420 (1978). [DOI] [PubMed] [Google Scholar]
- 5.Barraclough R & Ellis RJ Protein synthesis in chloroplasts. IX. Assembly of newly-synthesized large subunits into ribulose bisphosphate carboxylase in isolated intact pea chloroplasts. Biochim. Biophys. Acta 608, 19–31 (1980). [DOI] [PubMed] [Google Scholar]
- 6.Goloubinoff P, Gatenby AA & Lorimer GH GroE heat-shock proteins promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase oligomers in Escherichia coli. Nature 337, 44–47 (1989). [DOI] [PubMed] [Google Scholar]
- 7.Labbadia J & Morimoto RI The biology of proteostasis in aging and disease. Annu. Rev. Biochem 84, 435–464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horwich AL Molecular chaperones in cellular protein folding: the birth of a field. Cell 157, 285–288 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Miller SB, Mogk A & Bukau B Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J. Mol. Biol 427, 1564–1574 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Sontag EM, Samant RS & Frydman J Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem 86, 97–122 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ritossa F New puffing pattern induced by temperature shock and Dnp in Drosophila. Experientia 18, 571–573 (1962). [Google Scholar]
- 12.Ritossa FM Experimental activation of specific loci in polytene chromosomes of Drosophila. Exp. Cell Res, 601–607 (1964). [DOI] [PubMed] [Google Scholar]
- 13.Richter K, Haslbeck M & Buchner J The heat shock response: life on the verge of death. Mol. Cell 40, 253–266 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Finka A & Goloubinoff P Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 18, 591–605 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finka A, Mattoo RU & Goloubinoff P Meta-analysis of heat-and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperones 16, 15–31 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang X et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Taldone T, Ochiana SO, Patel PD & Chiosis G Selective targeting of the stress chaperome as a therapeutic strategy. Trends Pharmacol. Sci 35, 48–59 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Brehme M et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9, 1135–1150 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allan RK & Ratajczak T Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones 16, 353–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadizadeh Esfahani A, Sverchkova A, Saez-Rodriguez J, Schuppert AA & Brehme M A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLoS Comput. Biol 14, e1005890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellis RJ Molecular chaperones: assisting assembly in addition to folding. Trends Biochem. Sci 31, 395–401 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Ellis RJ Assembly chaperones: a perspective. Phil. Trans. R. Soc. B Biol. Sci 368, 20110398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burgess RJ & Zhang Z Histone chaperones in nucleosome assembly and human disease. Nat. Struct. Mol. Biol 20, 14–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makhnevych T & Houry WA The role of Hsp90 in protein complex assembly. Biochim. Biophys. Acta 1823, 674–682 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Palotai R, Szalay MS & Csermely P Chaperones as integrators of cellular networks: changes of cellular integrity in stress and diseases. IUBMB Life 60, 10–18 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Echtenkamp FJ et al. Global functional map of the p23 molecular chaperone reveals an extensive cellular network. Mol. Cell 43, 229–241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong Y et al. An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol. Syst. Biol 5, 275 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McClellan AJ et al. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Jamuczak AE, Eyers CE, Schwartz JM, Grant CM & Hubbard SJ Quantitative proteomics and network analysis of SSA1 and SSB1 deletion mutants reveals robustness of chaperone HSP70 network in Saccharomyces cerevisiae. Proteomics 15, 3126–3139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gyurko DM, Soti C, Stetak A & Csermely P System level mechanisms of adaptation, learning, memory formation and evolvability: the role of chaperone and other networks. Curr. Protein Pept. Sci, 171–188 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Echeverria PC, Bernthaler A, Dupuis P, Mayer B & Picard D An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS ONE 6, e26044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taipale M et al. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158, 434–448 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harper JW & Bennett EJ Proteome complexity and the forces that drive proteome imbalance. Nature 537, 328–338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodina A et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 538, 397–401 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barna M et al. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 456, 971–U979 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Elkon R et al. Myc coordinates transcription and translation to enhance transformation and suppress invasiveness. EMBO Rep 16, 1723–1736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farkas Z et al. Hsp70-associated chaperones have a critical role in buffering protein production costs. eLife, e29845 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trepel J, Mollapour M, Giaccone G & Neckers L Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrott JJ & Haystead TA Hsp90, an unlikely ally in the war on cancer. FEBS J 280, 1381–1396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whitesell L & Lindquist SL HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Ambati SR et al. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol 8, 323–336 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moulick K et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat. Chem. Biol 7, 818–826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunn DM et al. c-Abl mediated tyrosine phosphorylation of Aha1 activates its co-chaperone function in cancer cells. Cell Rep 12, 1006–1018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zuehlke A & Johnson JL Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 93, 211–217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong DS & Jay DG Emerging roles of extracellular Hsp90 in cancer. Adv. Cancer Res 129, 141–163 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Bachman AB et al. Phosphorylation induced cochaperone unfolding promotes kinase recruitment and client class-specific Hsp90 phosphorylation. Nat. Commun 9, 265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dahmer MK, Housley PR & Pratt WB Effects of molybdate and endogenous inhibitors on steroid-receptor inactivation, transformation, and translocation. Annu. Rev. Physiol 46, 67–81 (1984). [DOI] [PubMed] [Google Scholar]
- 49.Csermely P et al. ATP induces a conformational change of the 90-kDa heat shock protein (hsp90). J. Biol. Chem 268, 1901–1907 (1993). [PubMed] [Google Scholar]
- 50.Hutchison KA, Stancato LF, Jove R & Pratt WB The protein-protein complex between pp60v-src and hsp90 is stabilized by molybdate, vanadate, tungstate, and an endogenous cytosolic metal. J. Biol. Chem 267, 13952–13957 (1992). [PubMed] [Google Scholar]
- 51.Kamal A et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Shachrai I, Zaslaver A, Alon U & Dekel E Cost of unneeded proteins in E. coli is reduced after several generations in exponential growth. Mol. Cell 38, 758–767 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Matthews JM Protein Dimerization and Oligomerization in Biology Vol. 747 (Springer Science+Business Media, 2012). [Google Scholar]
- 54.Matthews JM & Sunde M Dimers, oligomers, everywhere. Adv. Exp. Med. Biol 747, 1–18 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Tai W, Guzman ML & Chiosis G The epichaperome: the power of many as the power of one. Oncoscience 3, 266–267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wallace EW et al. Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162, 1286–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mackenzie RJ et al. Absolute protein quantification of the yeast chaperome under conditions of heat shock. Proteomics 16, 2128–2140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chadli A, Ladjimi MM, Baulieu EE & Catelli MG Heat-induced oligomerization of the molecular chaperone Hsp90. Inhibition by ATP and geldanamycin and activation by transition metal oxyanions. J. Biol. Chem 274, 4133–4139 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Lepvrier E et al. Hsp90 oligomerization process: how can p23 drive the chaperone machineries? Biochim. Biophys. Acta 1854, 1412–1424 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Ehrnsperger M, Graber S, Gaestel M & Buchner J Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 16, 221–229 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Benaroudj N, Batelier G, Triniolles F & Ladjimi MM Self-association of the molecular chaperone Hsc70. Biochemistry 34, 15282–15290 (1995). [DOI] [PubMed] [Google Scholar]
- 62.Shirasu K & Schulze-Lefert P Complex formation, promiscuity and multi-functionality: protein interactions in disease-resistance pathways. Trends Plant Sci 8, 252–258 (2003). [DOI] [PubMed] [Google Scholar]
- 63.Good MC, Zalatan JG & Lim WA Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cooper MG The Cell: A Molecular Approach (Sinauer Associates, 2000). [Google Scholar]
- 65.Hartson SD & Matts RL Approaches for defining the Hsp90-dependent proteome. Biochim. Biophys. Acta 1823, 656–667 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weidenauer L, Wang T, Joshi S, Chiosis G & Quadroni M Proteomic interrogation of HSP90 and the insights for medical research. Expert Rev. Proteomics 14, 1105–1117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goldstein RL et al. Pharmacoproteomics identifies combinatorial therapy targets for diffuse large B cell lymphoma. J. Clin. Invest 125, 4559–4571 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo A et al. HSP90 stabilizes B cell receptor kinases in a multi-client interactome: PU-H71 induces CLL apoptosis in a cytoprotective microenvironment. Oncogene 36, 3441–3449 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kucine N et al. Tumor-specific HSP90 inhibition as a therapeutic approach in JAK-mutant acute lymphoblastic leukemias. Blood 126, 2479–2483 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koppikar P et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 489, 155–U222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marubayashi S et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Invest 120, 3578–3593 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsai CL et al. Stress-induced phosphoprotein-1 maintains the stability of JAK2 in cancer cells. Oncotarget 7, 50548–50563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zong H et al. A hyperactive signalosome in acute myeloid leukemia drives addiction to a tumor-specific Hsp90 species. Cell Rep 13, 2159–2173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nayar U et al. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood 122, 2837–2847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ojala PM Naughty chaperone as a target for viral cancer. Blood 122, 2767–2768 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Baquero-Perez B & Whitehouse A Hsp70 isoforms are essential for the formation of Kaposi’s sarcoma-associated herpesvirus replication and transcription compartments. PLoS Pathog 11, e1005274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anderson I et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl Acad. Sci. USA 111, E1528–E1537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Culjkovic-Kraljacic B et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B cell lymphomas. Blood 127, 858–868(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cerchietti LC et al. A purine scaffold Hsp90 inhibitor destabilizes BCL-6 and has specific antitumor activity in BCL-6-dependent B cell lymphomas. Nat. Med 15, 1369–1376 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schwartz H et al. Combined HSP90 and kinase inhibitor therapy: insights from The Cancer Genome Atlas. Cell Stress Chaperones 20, 729–741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jarosz DF, Taipale M & Lindquist S Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu. Rev. Genet 44, 189–216 (2010). [DOI] [PubMed] [Google Scholar]
- 82.Bernards RA Missing link in genotype-directed cancer therapy. Cell 151, 465–468 (2012). [DOI] [PubMed] [Google Scholar]
- 83.Lu X, Xiao L, Wang L & Ruden DM Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem. Pharmacol 83, 995–1004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Whitesell L & Lin NU HSP90 as a platform for the assembly of more effective cancer chemotherapy. Biochim. Biophys. Acta 1823, 756–766 (2012). [DOI] [PubMed] [Google Scholar]
- 85.Meyer SC Mechanisms of resistance to JAK2 inhibitors in myeloproliferative neoplasms. Hematol. Oncol. Clin. North Am 31, 627–642 (2017). [DOI] [PubMed] [Google Scholar]
- 86.Neckers L & Workman P Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res 18, 64–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lackner DH, Schmidt MW, Wu S, Wolf DA & Bahler J Regulation of transcriptome, translation, and proteome in response to environmental stress in fission yeast. Genome Biol 13, R25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hammond CM, Stromme CB, Huang H, Patel DJ & Groth A Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol 18, 141–158 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Khurana N & Bhattacharyya S Hsp90, the concertmaster: tuning transcription. Front. Oncol 5, 100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Isaacs JS Hsp90 as a “chaperone” of the epigenome: insights and opportunities for cancer therapy. Adv. Cancer Res 129, 107–140 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Echtenkamp FJ et al. Hsp90 and p23 molecular chaperones control chromatin architecture by maintaining the functional pool of the RSC chromatin remodeler. Mol. Cell 64, 888–899 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manjarrez JR, Sun L, Prince T & Matts RL Hsp90-dependent assembly of the DBC2/RhoBTB2-Cullin3 E3-ligase complex. PLoS ONE 9, e90054 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patra B et al. A genome wide dosage suppressor network reveals genomic robustness. Nucleic Acids Res 45, 255–270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Albert R, Jeong H & Barabasi AL Error and attack tolerance of complex networks. Nature 406, 378–382 (2000). [DOI] [PubMed] [Google Scholar]
- 95.Kitano H Biological robustness. Nat. Rev. Genet 5, 826–837 (2004). [DOI] [PubMed] [Google Scholar]
- 96.Felix MA & Barkoulas M Pervasive robustness in biological systems. Nat. Rev. Genet 16, 483–496 (2015). [DOI] [PubMed] [Google Scholar]
- 97.Bandyopadhyay S et al. Rewiring of genetic networks in response to DNA damage. Science 330, 1385–1389 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Navlakha S, He X, Faloutsos C & Bar-Joseph Z Topological properties of robust biological and computational networks. J. R. Soc. Interface 11, 20140283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Murphy ME The HSP70 family and cancer. Carcinogenesis 34, 1181–1188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sherman MY & Gabai VL Hsp70 in cancer: back to the future. Oncogene 34, 4153–4161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brodsky JL & Chiosis G Hsp70 molecular chaperones: emerging roles in human disease and identification of small molecule modulators. Curr. Top. Med. Chem 6, 1215–1225 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Assimon VA, Gillies AT, Rauch JN & Gestwicki JE Hsp70 protein complexes as drug targets. Curr. Pharm. Des 19, 404–417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rerole AL, Jego G & Garrido C Hsp70: anti-apoptotic and tumorigenic protein. Methods Mol. Biol 787, 205–230 (2011). [DOI] [PubMed] [Google Scholar]
- 104.Lambert JP et al. Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition. Nat. Methods 10, 1239–1245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jinwal UK et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J 27, 1450–1459 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gano JJ & Simon JA A proteomic investigation of ligand-dependent HSP90 complexes reveals CHORDC1 as a novel ADP-dependent HSP90-interacting protein. Mol. Cell. Proteomics 9, 255–270 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Smith JR et al. Restricting direct interaction of CDC37 with HSP90 does not compromise chaperoning of client proteins. Oncogene 34, 15–26 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Butler LM, Ferraldeschi R, Armstrong HK, Centenera MM & Workman P Maximizing the therapeutic potential of HSP90 inhibitors. Mol. Cancer Res 13, 1445–1451 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Garcia-Carbonero R, Carnero A & Paz-Ares L Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol 14, e358–e369 (2013). [DOI] [PubMed] [Google Scholar]
- 110.Alarcon SV et al. Tumor-intrinsic and tumor-extrinsic factors impacting hsp90-targeted therapy. Curr. Mol. Med 12, 1125–1141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jhaveri K, Taldone T, Modi S & Chiosis G Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 1823, 742–755 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lehner B Molecular mechanisms of epistasis within and between genes. Trends Genet 27, 323–331 (2011). [DOI] [PubMed] [Google Scholar]
- 113.Collins SR, Weissman JS & Krogan NJ From information to knowledge: new technologies for defining gene function. Nat. Methods 6, 721–723 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ashworth A, Lord CJ & Reis-Filho JS Genetic interactions in cancer progression and treatment. Cell 145, 30–38 (2011). [DOI] [PubMed] [Google Scholar]
- 115.Kim G et al. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res 21, 4257–4261 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Redig AJ & Janne PA Basket trials and the evolution of clinical trial design in an era of genomic medicine. J. Clin. Oncol 33, 975–977 (2015). [DOI] [PubMed] [Google Scholar]
- 117.Smith DL et al. The HSP90 inhibitor ganetespib potentiates the antitumor activity of EGFR tyrosine kinase inhibition in mutant and wild-type non-small cell lung cancer. Target. Oncol 10, 235–245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Caldas-Lopes E et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl Acad. Sci. USA 106, 8368–8373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Menezes DL et al. The novel oral Hsp90 inhibitor NVP-HSP990 exhibits potent and broad-spectrum antitumor activities in vitro and in vivo. Mol. Cancer Ther 11, 730–739 (2012). [DOI] [PubMed] [Google Scholar]
- 120.US National Library of Medicine. ClinicalTrials.govhttp://www.clinicaltrials.gov/ct2/show/NCT01393509 (2018). [DOI] [PubMed]
- 121.US National Library of Medicine. ClinicalTrials.govhttp://www.clinicaltrials.gov/ct2/show/NCT01269593 (2018). [DOI] [PubMed]
- 122.US National Library of Medicine. ClinicalTrials.govhttp://www.clinicaltrials.gov/ct2/show/NCT03166085 (2017). [DOI] [PubMed]
- 123.Lee MJ et al. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 149, 780–794 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kanamaru C et al. Retinal toxicity induced by small-molecule Hsp90 inhibitors in beagle dogs. J. Toxicol. Sci 39, 59–69 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Zhou D et al. A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicol. Appl. Pharmacol 273, 401–409 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Shrestha L & Young JC Function and chemotypes of human Hsp70 chaperones. Curr. Top. Med. Chem 16, 2812–2828 (2016). [DOI] [PubMed] [Google Scholar]
- 127.Li X, Shao H, Taylor IR & Gestwicki JE Targeting allosteric control mechanisms in heat shock protein 70 (Hsp70). Curr. Top. Med. Chem 16, 2729–2740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Goloudina AR, Demidov ON & Garrido C Inhibition of HSP70: a challenging anti-cancer strategy. Cancer Lett 325, 117–124 (2012). [DOI] [PubMed] [Google Scholar]
- 129.Stiegler SC et al. A chemical compound inhibiting the Aha1-Hsp90 chaperone complex. J. Biol. Chem 292, 17073–17083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jhaveri K et al. Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Expert Opin. Investig. Drugs 23, 611–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Prince TL et al. Client proteins and small molecule inhibitors display distinct binding preferences for constitutive and stress-induced HSP90 isoforms and their conformationally restricted mutants. PLoS ONE 10, e0141786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Woodford MR et al. Impact of posttranslational modifications on the anticancer activity of Hsp90 inhibitors. Adv. Cancer Res 129, 31–50 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Mollapour M & Neckers L Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 1823, 648–655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Taldone T et al. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J. Med. Chem 56, 6803–6818 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sattin S et al. Activation of Hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands. Chemistry 21, 13598–13608 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schopf FH, Biebl MM & Buchner J The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol 18, 345–360 (2017). [DOI] [PubMed] [Google Scholar]
- 137.Krukenberg KA, Street TO, Lavery LA & Agard DA Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys 44, 229–255 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gooljarsingh LT et al. A biochemical rationale for the anticancer effects of Hsp90 inhibitors: slow, tight binding inhibition by geldanamycin and its analogues. Proc. Natl Acad. Sci. USA 103, 7625–7630 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Beebe K et al. Posttranslational modification and conformational state of heat shock protein 90 differentially affect binding of chemically diverse small molecule inhibitors. Oncotarget 4, 1065–1074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsaytler PA, Krijgsveld J, Goerdayal SS, Rudiger S & Egmond MR Novel Hsp90 partners discovered using complementary proteomic approaches. Cell Stress Chaperones 14, 629–638 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Matts RL et al. A systematic protocol for the characterization of Hsp90 modulators. Bioorg. Med. Chem 19, 684–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Copeland RA The drug-target residence time model: a 10-year retrospective. Nat. Rev. Drug Discov 15, 87–95 (2016). [DOI] [PubMed] [Google Scholar]
- 143.de Witte WE, Danhof M, van der Graaf PH & de Lange EC In vivo target residence time and kinetic selectivity: the association rate constant as determinant. Trends Pharmacol. Sci 37, 831–842 (2016). [DOI] [PubMed] [Google Scholar]
- 144.Patel HJ, Modi S, Chiosis G & Taldone T Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin. DrugDis 6, 559–587 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shrestha L, Bolaender A, Patel HJ & Taldone T Heat shock protein (HSP) drug discovery and development: targeting heat shock proteins in disease. Curr. Top. Med. Chem 16, 2753–2764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ebong IO et al. Heterogeneity and dynamics in the assembly of the heat shock protein 90 chaperone complexes. Proc. Natl Acad. Sci. USA 108, 17939–17944 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zuehlke AD et al. An Hsp90 co-chaperone protein in yeast is functionally replaced by site-specific posttranslational modification in humans. Nat. Commun 8, 15328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Voos W Chaperone-protease networks in mitochondrial protein homeostasis. Biochim. Biophys. Acta 1833, 388–399 (2013). [DOI] [PubMed] [Google Scholar]
- 149.Sabnis AJ et al. Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc. Natl Acad. Sci. USA 113, 9015–9020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Altieri DC Mitochondrial HSP90s and tumor cell metabolism. Autophagy 9, 244–245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Patel PD et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat. Chem. Biol 9, 677–684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee AS Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 14, 263–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Carvalho AS, Rodriguez MS & Matthiesen R Review and literature mining on proteostasis factors and cancer. Methods Mol. Biol 1449, 71–84 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Brandvold KR & Morimoto RI The chemical biology of molecular chaperones — implications for modulation of proteostasis. J. Mol. Biol 427, 2931–2947 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gandolfi S et al. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev 36, 561–584 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Li Z, Hartl FU & Bracher A Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat. Struct. Mol. Biol 20, 929–935 (2013). [DOI] [PubMed] [Google Scholar]
- 157.Pratt WB & Toft DO Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev 18, 306–360 (1997). [DOI] [PubMed] [Google Scholar]
- 158.Davies AE & Kaplan KB Hsp90-Sgt1 and Skp1 target human Mis12 complexes to ensure efficient formation of kinetochore-microtubule binding sites. J. Cell Biol 189, 261–274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gurard-Levin ZA, Quivy JP & Almouzni G Histone chaperones: assisting histone traffic and nucleosome dynamics. Annu. Rev. Biochem 83, 487–517 (2014). [DOI] [PubMed] [Google Scholar]
- 160.Rosenzweig R & Glickman MH Chaperone-driven proteasome assembly. Biochem. Soc. T 36, 807–812 (2008). [DOI] [PubMed] [Google Scholar]
- 161.Chang HC & Lindquist S Conservation of Hsp90 macromolecular complexes in Saccharomyces cerevisiae. J. Biol. Chem 269, 24983–24988 (1994). [PubMed] [Google Scholar]
- 162.Meacham GC et al. Mutations in the yeast Hsp40 chaperone protein Ydj1 cause defects in Axl1 biogenesis and pro-a-factor processing. J. Biol. Chem 274, 34396–34402 (1999). [DOI] [PubMed] [Google Scholar]
- 163.Dey B, Caplan AJ & Boschelli F The Ydj1 molecular chaperone facilitates formation of active p60v-src in yeast. Mol. Biol. Cell 7, 91–100 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liu XD, Morano KA & Thiele DJ The yeast Hsp110 family member, SSE1, is an Hsp90 cochaperone. J. Biol. Chem 274, 26654–26660 (1999). [DOI] [PubMed] [Google Scholar]
- 165.Lamoth F, Juvvadi PR, Soderblom EJ, Moseley MA & Steinbach WJ Hsp70 and the cochaperone StiA (Hop) orchestrate Hsp90-mediated caspofungin tolerance in Aspergillus fumigatus. Antimicrob. Agents Chemother 59, 4727–4733 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nathan DF & Lindquist S Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol 15, 3917–3925 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Piper PW et al. Sensitivity to Hsp90-targeting drugs can arise with mutation to the Hsp90 chaperone, cochaperones and plasma membrane ATP binding cassette transporters of yeast. Eur. J. Biochem 270, 4689–4695 (2003). [DOI] [PubMed] [Google Scholar]
- 168.Lee JR et al. Heat-shock dependent oligomeric status alters the function of a plant-specific thioredoxin-like protein, AtTDX. Proc. Natl Acad. Sci. USA 106, 5978–5983 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee SS et al. Enhancement of chaperone activity of plant-specific thioredoxin through gamma-ray mediated conformational change. Int. J. Mol. Sci 16, 27302–27312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Thompson AD, Bernard SM, Skiniotis G & Gestwicki JE Visualization and functional analysis of the oligomeric states of Escherichia coli heat shock protein 70 (Hsp70/DnaK). Cell Stress Chaperones 17, 313–327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Angelidis CE, Lazaridis I & Pagoulatos GN Aggregation of hsp70 and hsc70 in vivo is distinct and temperature-dependent and their chaperone function is directly related to non-aggregated forms. Eur. J. Biochem 259, 505–512 (1999). [DOI] [PubMed] [Google Scholar]
- 172.Araujo TL et al. Conformational changes in human Hsp70 induced by high hydrostatic pressure produce oligomers with ATPase activity but without chaperone activity. Biochemistry 53, 2884–2889 (2014). [DOI] [PubMed] [Google Scholar]
- 173.Freiden PJ, Gaut JR & Hendershot LM Interconversion of three differentially modified and assembled forms of BiP EMBO J 11, 63–70 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hatayama T, Yasuda K & Yasuda K Association of HSP105 with HSC70 in high molecular mass complexes in mouse FM3A cells. Biochem. Biophys. Res. Commun 248, 395–401 (1998). [DOI] [PubMed] [Google Scholar]
- 175.Bruey JM et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol 2, 645–652 (2000). [DOI] [PubMed] [Google Scholar]
- 176.Bruey JM et al. Differential regulation of HSP27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 19, 4855–4863 (2000). [DOI] [PubMed] [Google Scholar]
- 177.Charette SJ, Lavoie JN, Lambert H & Landry J Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol. Cell. Biol 20, 7602–7612 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Garrido C Size matters: of the small HSP27 and its large oligomers. Cell Death Differ 9, 483–485 (2002). [DOI] [PubMed] [Google Scholar]
- 179.Yonehara M, Minami Y, Kawata Y, Nagai J & Yahara I Heat-induced chaperone activity of HSP90. J. Biol. Chem 271, 2641–2645 (1996). [DOI] [PubMed] [Google Scholar]
- 180.Nemoto T & Sato N Oligomeric forms of the 90-kDa heat shock protein. Biochem. J 330, 989–995 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nemoto TK, Ono T & Tanaka K Substrate-binding characteristics of proteins in the 90 kDa heat shock protein family. Biochem. J 354, 663–670 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Lepvrier E et al. Hsp90 oligomers interacting with the Aha1 cochaperone: an outlook for the Hsp90 chaperone machineries. Anal. Chem 87, 7043–7051 (2015). [DOI] [PubMed] [Google Scholar]
- 183.Lanks KW Temperature-dependent oligomerization of hsp85 in vitro. J. Cell. Physiol 140, 601–607 (1989). [DOI] [PubMed] [Google Scholar]
- 184.Song D et al. Antitumor activity and molecular effects of the novel heat shock protein 90 inhibitor, IPI-504, in pancreatic cancer. Mol. Cancer Ther 7, 3275–3284 (2008). [DOI] [PubMed] [Google Scholar]
- 185.Vogen S et al. Radicicol-sensitive peptide binding to the N-terminal portion of GRP94. J. Biol. Chem 277, 40742–40750 (2002). [DOI] [PubMed] [Google Scholar]
- 186.Gaspar ME & Csermely P Rigidity and flexibility of biological networks. Brief Funct. Genomics 11, 443–456 (2012). [DOI] [PubMed] [Google Scholar]