

Abstract
Benzoyl-CoA reductase (BCR) from the bacterium Thauera aromatica catalyzes the two-electron reduction of benzoyl-CoA (BCoA) to a nonaromatic cyclic diene. In a process analogous to enzymatic nitrogen reduction, BCR couples the electron transfer to the aromatic ring to a stoichiometric hydrolysis of 2 ATP/2e−. Reduced but not oxidized BCR hydrolyzes ATP to ADP. In this work, purified BCR was shown to catalyze an isotope exchange from [14C]ADP to [14C]ATP, which was ≈15% of the ATPase activity in the presence of equimolar amounts of ADP and ATP. In accordance, BCR (αβγδ-composition) autophosphorylated its γ-subunit when incubated with [γ-32P]ATP. Formation of the enzyme-phosphate was independent of the redox state, whereas only dithionite-reduced BCR catalyzed a dephosphorylation associated with the ATPase activity. This finding suggests that the ATPase- and autophosphatase-partial activities of BCR exhibit identical redox dependencies. BCoA or the nonphysiological electron-accepting substrate hydroxylamine stimulated the redox-dependent effects; the rates of both the overall ATPase and the autophosphatase activities of reduced BCR were increased 6-fold. In contrast, BCoA and hydroxylamine had no effect on oxidized and phosphorylated BCR. The reactivity of the phosphoamino acid fits best with a phosphoamidate or acylphosphate linkage. The results obtained suggest a mechanism of ATP hydrolysis-driven electron transfer, which differs from that of nitrogenase by the transient formation of a phosphorylated enzyme.
Benzoyl-CoA (BCoA) is a central intermediate in the metabolism of many aromatic compounds in anaerobic bacteria. This compound becomes reduced by benzoyl-CoA reductase (BCR), a key enzyme in anaerobic aromatic metabolism (1, 2). It catalyzes the reductive dearomatization of BCoA to cyclohexa-1,5-diene-1-carbonyl-CoA (Fig. 1; ref. 3). In this reaction, the hydrolysis of two ATP to ADP and Pi is stoichiometrically coupled to the transfer of two single electrons from reduced ferredoxin to the aromatic substrate. Notably, BCR also catalyzes the ATP-dependent reduction of the nonphysiological substrate hydroxylamine (2). The stoichiometric coupling of ATP hydrolysis to an electron transfer process has long been considered as a unique feature of nitrogenases (4, 5). However, there are no amino acid sequence similarities between BCR and nitrogenases.
Figure 1.

Reaction catalyzed by BCoA reductase from T. aromatica. As artificial substrate hydroxylamine is reduced ATP-dependently to ammonia and water.
The reduction of the aromatic ring is a mechanistically and energetically difficult process that in organic synthesis requires solvated electrons, which are generated by dissolving alkali metals in liquid ammonia. In a so-called Birch mechanism, the reduction of the aromatic ring proceeds in single electron and proton transfer steps by means of radical intermediates (6). A similar mechanism has been proposed for enzymatic benzene ring reduction (7). The crucial step is the first electron transfer to the aromatic ring yielding a nonaromatic radical anion, which requires a redox potential of −1.9 V (3). BCR overcomes this high redox barrier by coupling a stoichiometric ATP hydrolysis to the electron activation process and by an appropriate solvation of the substrate. Recent results gave preliminary evidence that a Birch-like mechanism involving radical species may operate in BCR as suggested by kinetic (H. Moblitz and M.B., unpublished data) and spectroscopic (8) studies.
The biochemistry of BCR has been studied only so far in the purified enzyme from the denitrifying bacterium Thauera aromatica, although gene sequence comparisons and immunological studies indicated that similar enzymes are distributed among other anaerobic bacteria (9). The extremely oxygen-labile 170-kDa enzyme from T. aromatica consists of 4 subunits with an αβγδ-composition. BCR contains three cysteine-ligated [4Fe-4S]+1/+2 clusters with midpoint potentials more negative than −500 mV (10). The four subunits of BCR in T. aromatica (11) and in the phototrophic bacterium Rhodopseudomonas palustris (12) show ≈70% identity to each other. Furthermore, the subunits of BCR show 38–52% similarities to the two-component enzyme system activase (Hgd C-homodimer)/2-hydroxyglutaryl-CoA dehydratase (Hgd A/B-heterodimer) of Acidaminococcus fermentans (11, 13). The monodimeric activase of this enzyme system catalyzes a nonstoichiometric ATP-dependent activation of electrons that are essential for the initiation of water elimination from the α-position of 2-hydroxyglutaryl-CoA (7, 14). Based on conserved amino acid motifs (11, 13) and on the recently solved structure of activase from A. fermentans (15), BCR is postulated to contain two functionally distinct modules (Fig. 2): (i) Bcr A and Bcr D (α- and δ-subunits) both contain one ATP-binding site. A [4Fe-4S] cluster is symmetrically coordinated by both subunits resulting in a structure that bears resemblance to the architecture of the Fe-protein of nitrogenase (4). This module is considered as the site of ATP-dependent electron activation. (ii) Bcr B and C (β- and γ-subunits) harbor the two other [4Fe-4S] clusters; together they form the substrate reduction module.
Figure 2.

Proposed molecular architecture of BCoA reductase. The model is based on spectroscopic studies and on amino acid sequence comparisons with the two-component system 2-hydroxygluaryl-CoA dehydratase/activase of A. fermentans. The iron-sulfur clusters of the Bcr A/D module are interfacially ligated by two cysteines of each subunit.
Reduced BCR catalyzes ATP hydrolysis to ADP + Pi coupled to substrate reduction (100% rate) and when the substrate is missing (15% rate) (2, 16). This ATPase activity was present only in the reduced state of BCR, suggesting that it is coupled to an electron activation process. EPR and Mössbauer spectroscopic studies further showed that ATP-hydrolysis induces a switch from the low-spin (S = 1/2) to a high-spin (S = 7/2) state of a [4Fe-4S]+1 cluster, thereby possibly lowering its redox potential. In single BCoA turnover experiments, the reversible low-spin/high-spin switch has been shown to be an event in the catalytic cycle of BCR (9).
In this work, two possible scenarios for the ATP-dependent electron transfer process, with and without the transient formation of an energy rich enzyme-phosphate linkage, were tested. The data obtained indicate that BCR catalyzes a stoichiometric ATP hydrolysis-driven electron transfer by means of an energy-rich enzyme-phosphate intermediate. This process is unique so far and differs from that of nitrogenases (17).
Experimental Procedures
Growth of Bacterial Cells.
T. aromatica (Deutsche Sammlung für Mikroorganismen, Braunschweig, Germany; DSM 6984) was grown anoxically in a 200-liter fermenter at 28°C in a mineral salt medium. 4-Hydroxybenzoate and nitrate in a ratio of 1:3.5 or acetate and nitrate in a ratio of 1:1 served as sole sources of energy and cell carbon. Continuous feeding of the substrates, cell harvesting, storage, and preparation of cell extracts were carried out as described (18).
Protein Purification, Activity Determination, and Sample Storage.
Purification of BCR from T. aromatica (wet cell mass 200 g) was performed under strictly anaerobic conditions in a glove box under an N2/H2 (95:5, by volume) atmosphere as described (2). The enzyme used was ≈95% pure. Protein samples were immediately frozen in liquid nitrogen and stored anaerobically at −196°C for several months. BCR as isolated was in 20 mM triethanolamine, pH 7.8/4 mM MgCl2/10% glycerol/0.5 mM sodium dithionite. The MgATP- and BCoA-dependent oxidation of methyl viologen catalyzed by BCR was determined in a spectrophotometric assay at 730 nm at 37°C as described (2). The specific BCoA reduction activity of the samples used was 0.3 μmol min−1 mg−1.
ATPase Activity of BCR.
The ATP hydrolyzing activity was tested in an anaerobic continuous spectrophotometric assay in gas-tight sealed glass cuvettes at 37°C as described (2). The formation of ADP was coupled to NADH oxidation by the use of pyruvate kinase and lactate dehydrogenase.
Isotope Exchange Reaction.
The isotope exchange from [8-14C]ADP to [14C]ATP catalyzed by BCR was performed at 37°C under aerobic and anaerobic conditions. The 0.5-ml reaction mixture contained 100 μM ATP/5 mM MgCl2/100 μM [8-14C]ADP [18.5 kBq(1 dps; 1 Ci = 37 GBq)]/1.3 or 2.7 μM BCR; the reaction was started by enzyme addition. For the aerobic assay, BCR (13.5 μM stock solution) was preincubated on air at 4°C for 30 min. The anaerobic assay was performed in gas-tight sealed glass tubes under a nitrogen/hydrogen atmosphere (95:5 by volume) and contained 2 mM sodium dithionite. To test the effect of substrates, 0.3 mM BCoA or hydroxylamine was added. Samples (50 μl) taken at different time points were added to 5 μl of 25% H2SO4. After centrifugation (12,000 × g), the radioactive samples were analyzed by HPLC with a Mono Q anion exchange column (5 cm × 5 mm, Amersham Pharmacia) as described (16). Detection of labeled compounds was carried out with a radioactivity monitoring analyzer.
Phosphorylation/Dephosphorylation Assays.
The routine reaction mixture (80 μl) contained 20 mM triethanolamine (TEA), pH 7.8/4 mM MgCl2/10 μM BCR/10 μM [γ-32P]ATP (150 kBq), prepared from a 222 GBq/mmol stock solution. [γ-32P]GTP or [α-32P]ATP was added in the same amounts and specific radioactivities when substituted for [γ-32P]ATP. When cell extracts from T. aromatica were used, the protein concentration was ≈25 mg ml−1, resulting in a BCR concentration of ≈9 μM; cells grown anaerobically on acetate were used as controls, in which BCR expression was only 5% of benzoate grown cells. Experiments were repeated with three different cell batches. The reaction mixtures were incubated at 22°C. Samples (15 μl) were taken at different time points and were subsequently added to the same amount of double-concentrated SDS/PAGE loading buffer. Before SDS-gel analysis, samples were incubated for 1 min at 80°C. After SDS/PAGE analysis, radioactive-labeled protein bands were visualized on SDS gels with a phosphorimaging system (Bio-Rad) after exposure for 24 h. Relative quantification of the labeled protein band was performed with the densitometric quantification software of the phosphorimaging system. Absolute quantification was carried out by cutting the radioactive-labeled band from the SDS gel. The cut gel piece was incubated with 1 M HCl at room temperature for 2 h and subsequently centrifuged (12,000 × g). The supernatant containing radioactivity was analyzed by Cerenkov counting. When air-oxidized BCR was used, BCR was preincubated for 30 min on air at 4°C. For testing the dithionite-reduced enzyme (2 mM sodium dithionite), the phosphorylation reaction was performed in anaerobic 0.5-ml glass tubes sealed with gas-tight stoppers under a nitrogen/hydrogen atmosphere (95:5, by volume). The thionine-oxidized samples were prepared in an anaerobic glove box by adding an anaerobically prepared saturated thionine stock solution (≈10 mM) to a 10 μM enzyme sample until the color remained blue, indicating that the enzyme sample was fully oxidized. Excess of thionine was removed by passing BCR (≈1 ml) over a Sephadex G-25 desalting column (volume ≈5 ml, Amersham Pharmacia). In the anaerobic assays, additions of the substrates were made with gas-tight glass syringes from anaerobic stock solutions.
The formation of free inorganic phosphate during the phosphorylation/dephosphorylation reactions was followed by TLC analysis with polyethyleneimine-cellulose plates (Merck), using a solvent of 0.52 M phosphate buffer (pH 7.8).
Chemical Stability Assay of Enzyme-Phospho-Linkage.
For testing the chemical stability of the phosphoamino acid of BCR, air-oxidized BCR was phosphorylated as described previously. After SDS/PAGE analysis, protein bands were blotted on a poly(vinylidene difluoride) (PVDF) membrane (Millipore). Cut membrane strips containing the phosphorylated protein band were incubated for 2 h at 22°C in 20 mM triethanolamine, pH 7.8/3 M NaOH/1 M HCl or 1 M hydroxylamine, pH 7.5. Strips were dried and exposed to a phosphorimaging system for 16 h. In a control experiment, strips were stained with amido black to ensure the removal of label was not the result of a loss of the protein band.
Further Determinations.
BCoA was synthesized from CoA and benzoic acid anhydride (19). Purification and purity control of the thioester was carried out by HPLC as described (3). The N-terminal amino acid sequence of the phosphorylated protein band was determined according to an automatized Edman degradation by H. Schägger (University of Frankfurt, Germany). Protein concentration was determined according to Bradford (20), using BSA as standard. SDS gels (10%) were prepared according to Laemmli (21), and protein bands were visualized by Coomassie staining (22).
Results
ATPase Activity of BCR.
The intrinsic (substrate-independent) rate of the ATPase activity was reinvestigated in this work (a summary is presented in Table 2). In accordance to previous results, dithionite-reduced but not thionine- or air-oxidized BCR catalyzed the hydrolysis of ATP to ADP and Pi, which could be stimulated by addition of BCoA or hydroxylamine by a factor of 6–7 (2, 17). Note that the nonphysiologic substrate hydroxylamine becomes ATP dependently reduced by BCR (2).
Table 2.
Rates of partial and overall activities of BCR under different conditions
| Condition | Substrate reduction* | ATP hydrolysis† | Auto-phosphorylation‡ | Auto-dephosphorylation | Isotope exchange¶ |
|---|---|---|---|---|---|
| Dithionite reduced | — | 15% | +++ | + | 100%‖ |
| Dithionite reduced + BCoA | 100% | 100% | +++§ | +++ | <1% |
| Dithionite reduced + hydroxylamine | 125% | ∼100% | +++§ | +++ | <1% |
| Air-oxidized | — | <1% | +++ | ≪+ | 100% |
| Air-oxidized + BCoA | — | <1% | +++ | <1% | 100% |
| Air-oxidized + hydroxylamine | — | <1% | +++ | <1% | 100% |
| Thionine-oxidized | — | <1% | +++ | <1% | ND |
100% refers to 0.3 μmol BCoA reduced min−1 mg−1.
100% refers to 0.6 μmol ADP formed from ATP min−1 mg−1.
+++ refers to maximal phosphorylation after 30-s incubation.
The rate of phosphorylation cannot be determined precisely as a result of the rapid dephosphorylation induced by BCoA and hydroxylamine.
Determined in the presence of equimolar amounts of ATP and ADP (100 μM). 100% refers to the formation of 1.4 nmol mg−1 min−1 [8-14C]ATP from [8-14C]ADP.
Initial rate after 5-min incubation. After prolonged incubation this rate decreased continuously.
Isotope Exchange from [8-14C]ADP to [8-14C]ATP.
In the absence of a reducible substrate, BCR catalyzed an isotope exchange between [8-14C]ADP and ATP. The experiments were performed at two different catalytic enzyme concentrations (1.3–2.7 μM). BCR was added to unlabeled ATP and [8-14C]ADP (100 μM each) and the time- and BCR-dependent formation of [8-14C]ATP from unlabeled ATP in the absence of an electron-accepting substrate was followed (Fig. 3). The isotope exchange reflects the following reactions catalyzed by BCR:
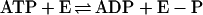 |
1 |
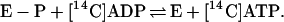 |
2 |
The initial rate of the isotope exchange rate was 1.4 nmol min−1 mg−1, as determined for the air-oxidized enzyme (Fig. 3A); doubling of the enzyme concentration resulted in a doubling of the exchange rate. Under the conditions used, the rate of the isotope exchange of BCR was ≈15% of the overall ATPase activity. The low rates can be rationalized by two special properties of BCR. (i) In the absence of a reducible substrate, the ATP hydrolyzing activity of BCR is only 15% of the rate coupled to electron transfer. (ii) ADP is competitive for ATP; both compounds bind with low affinity to BCR [KM(ATP) = 0.6 mM and Ki(ADP) = 1.1 mM (2, 17)]. The initial isotope exchange rate was hardly effected by the redox state of BCR. However, after long incubation, it continuously decreased in reduced BCR, whereas it remained stable in the oxidized state of the enzyme (Fig. 3A). As will be shown in detail below, the enzyme-phosphate formed in reaction 1 becomes continuously hydrolyzed by reduced but not by oxidized BCR. Thus, reactions 1 and 2 are pulled toward ATP/[14C]ATP hydrolysis in the reduced state.
Figure 3.

Isotope exchange from [8-14C] ADP to [8-14C]ATP catalyzed by BCR. BCR was incubated with [8-14C] ADP and unlabeled ATP (100 μM each) at 37°C under aerobic and anaerobic conditions. Samples were analyzed by HPLC, using a Mono Q column and a radioactivity monitoring analyzer. (A) [14C]ATP formed from [8-14C]ADP catalyzed by 1.3 μM (▾) and 2.7 μM (●) air-oxidized BCR and by 2.7 μM (○) dithionite-reduced BCR. (B) [8-14C]ATP formed from [8-14C]ADP catalyzed by 2.7 μM BCR; in the presence of 300 μM BCoA with air-oxidized (●) and dithionite-reduced (○) BCR; in the presence of 300 μM hydroxylamine with air-oxidized (▾) and dithionite-reduced (▿) BCR.
In the presence of excess of BCoA or of the nonphysiological electron-accepting substrate hydroxylamine (0.3 mM each), no isotope exchange of reduced BCR was observed (Fig. 3B). As the rate of ATP hydrolysis is stimulated 6-fold when coupled to substrate reduction (Table 2), the equilibrium of the isotope exchange reaction will be shifted further toward hydrolysis of enzyme-phosphate and of ATP/[14C]ATP, finally abolishing the isotope exchange activity. In contrast, the isotope exchange reaction of oxidized BCR was not affected in the presence of either of the electron-accepting substrates BCoA or hydroxylamine.
Autokinase/Autophosphatase Partial Activities of Oxidized and Reduced BCR in the Absence of an Electron-Accepting Substrate.
The isotope exchange experiments indicated the formation of an enzyme-phosphate intermediate. This prediction was tested by using [γ-32P]ATP, which should also enable the identification of the phosphorylated enzyme subunit. In the routine phosphorylation assay we mixed equimolar amounts of purified BCR with [γ-32P]ATP (10 μM each) for 1 min at room temperature and quenched the reaction by adding SDS gel loading buffer. In the air-oxidized state of BCR, a radioactive-labeled protein band was detected on SDS gels that comigrated with the 44-kDa γ-subunit of BCR (Fig. 4 A and B). The N-terminal amino acid sequence of the labeled protein band was determined, which was identical to that of the γ-subunit of BCR (not shown). When BCR was incubated under conditions as described with equimolar amounts of [α-32P]ATP, no radioactive-labeled protein band was detectable on SDS gels excluding the presence of a tightly bound nucleotide (Fig. 4B). In another control experiment, in which [γ-32P]GTP substituted for [γ-32P]ATP, again no labeled protein band was found on SDS gels fitting to the high specificity of BCR for ATP (Fig. 4B; ref. 2).
Figure 4.

Reaction of purified BCR and cell extracts of T. aromatica with radioactively labeled nucleotides. BCR (10 μM) or cell extracts (25 mg ml−1) were incubated with the nucleotide for 1 min at 20°C on air, and reaction products were separated by SDS/PAGE. (A) Coomassie-stained gel of BCR. The 30- and 49-kDa subunits contain one ATP-binding site each. (B) Phosphorimaging of an SDS gel of BCR after reaction with [α-32P]ATP (lane 1), [γ-32P]ATP (lane 2), and [γ-32P]GTP (lane 3). (C) Phosphorimaging of an SDS gel of extracts from T. aromatica grown on acetate (lane 4) and 4-hydroxybenzoate (lane 5) after reaction with [γ-32P]ATP. In some BCR preparations, a second labeled protein band at ≈80 kDa was observed (as showed in lane 5). It was either assigned to an impurity or a protein aggregate containing the γ-32P-labeled γ-subunit of BCR.
In further experiments, we incubated extracts of T. aromatica cells grown on benzoate and acetate, respectively, with [γ-32P]ATP under aerobic conditions. In cells grown anaerobically on an aromatic substrate, BCR made up to 6% of the soluble protein fraction (referred to as 100% expression), which should enable the detection of the phosphorylated subunit on SDS gels of cell extracts (2). In contrast, BCR is hardly expressed in cells grown on acetate (<5% expression). In extracts prepared from cells grown on benzoate, a labeled protein band was found that comigrated with the phosphorylated band of the purified enzyme (Fig. 4C). In extracts of cells grown on acetate, this labeled band was only ≈5% of the one observed in benzoate-grown cells. This finding fits perfectly with the differential expression of BCR when grown on different substrates (23).
To test the effect of the redox state, we carried out time-dependent enzyme phosphorylation studies in the dithionite-reduced, air-, and thionine-oxidized state of BCR. In the air- and thionine-oxidized states, the enzyme-phosphate was rapidly formed after 30 s (Fig. 5 A and D; only the experiment with air-oxidized BCR is shown). As a result of the high enzyme concentration used (10 μM) and the difficult sample handling, the kinase reaction was too fast to be followed in a time-dependent manner. In the air- and thionine-oxidized states, the degree of enzyme phosphorylation (referred to as 100%) remained stable in the course of the experiment (3 min). In the dithionite-reduced state of BCR, the enzyme-phosphate was also rapidly formed to a similar extent. In contrast to the oxidized sample, it decreased continuously after prolonged incubation (Fig. 5 A and D); in parallel, 32Pi was continuously formed (not shown). This result provides evidence that oxidized BCR shows autokinase (enzyme-phosphate + ADP forming) but no or low autophosphatase (enzyme + Pi forming)-partial activity, whereas reduced BCR exhibits both partial activities. The time-dependent decrease of the formed enzyme-phosphate of reduced BCR can be rationalized as a result of continuous enzyme-phosphate hydrolysis, ATP depletion, and a concomitant accumulation of ADP. The latter compound is competitive to ATP.
Figure 5.

Effects of dithionite, oxygen, and substrates on autokinase/autophosphatase reactions of BCR. BCR (10 μM) was mixed with [γ-32P]ATP (10 μM) at 22°C. After various time points samples were taken that were quenched by adding SDS loading buffer. The phosphorylated γ-subunit of BCR was visualized by phosphorimaging of SDS gels. 100% refers to maximal phosphorylation. A–C show the radioactively labeled phosphorylated γ-subunit of BCR as detected on SDS gels. r, anaerobic assay (2 mM dithionite); o, aerobic assay with air-oxidized BCR. Note that anaerobic oxidation of BCR with thionine resulted in identical effects as air oxidation (not shown). Effect of the redox state in the absence of a reducible substrate (A), in the presence of BCoA (30 μM) (B), and in the presence of hydroxylamine (30 μM) (C) on enzyme phosphorylation. (D) Quantitative analysis of A and B: air-oxidized BCR (●), dithionite-reduced BCR (■), air-oxidized BCR + 30 μM BCoA (○), and dithionite-reduced BCR + 30 μM BCoA (▾). (E) Quantitative analysis of C: air-oxidized BCR + 30 μM hydroxylamine (●) and dithionite-reduced BCR + 30 μM hydroxylamine (○).
Quantification of the formed enzyme-phosphate of oxidized BCR on SDS gels indicated that in the steady-state of the autokinase reaction, 0.05% of the γ-subunits of BCR were phosphorylated. This low value can be rationalized as follows. First, as will be shown below, the chemical properties of the enzyme-phospho-linkage indicated the presence of an energy-rich phosphoamidate or acyl-phosphate during the ATPase activity. Therefore, the autokinase partial reaction should rather favor ATP formation from ADP and enzyme-phosphate. As an example, the free energy of acetyl-phosphate formation from ATP and acetate is +13 kJ mol−1 (24), giving a ratio of enzyme:enzyme-phosphate of ≈200:1 in the equilibrium (assuming equimolar concentrations of all substrates/products). Thus, under the conditions used (10 μM of ATP and BCR each), maximally 0.5% of phosphorylated BCR can be expected if the reaction is solely thermodynamically driven. Second, as the formed enzyme-phospho-linkage is highly sensitive even under moderate conditions (see below), it is likely that considerable amounts of the formed phosphoamino acid are lost during sample preparation (80°C, high SDS concentration).
Effect of Substrates on the Autokinase/Autophosphatase Partial Activities of BCR.
To give further evidence that the observed autokinase/autophosphatase activities represent partial reactions in the catalytic cycle of BCR, the effects of BCoA and the nonphysiological substrate hydroxylamine on the two partial activities were investigated. Formation of the enzyme-phosphate was independent of BCoA with oxidized and reduced enzyme (Fig. 5 B and D). However, in the dithionite-reduced state of BCR, addition of 3-fold amounts of BCoA resulted in an immediate stimulation of the autophosphatase rate (Fig. 5 B and D). The initial rate could not be determined precisely because of the sample handling, but it is estimated to be >6-times increased in the presence of BCoA compared with the substrate-independent rate. Most importantly, BCoA did not induce the dephosphorylation of oxidized and phosphorylated BCR (Fig. 5 B and D). In a further experiment, BCoA was substituted by hydroxylamine under the same conditions. As shown in Fig. 5 C and E, hydroxylamine had the same effect on phosphorylated BCR as BCoA; the hydrolysis rate was similarly stimulated by 30 μM hydroxylamine, whereas the same concentration had no effect on the oxidized enzyme. The latter result clearly rules out a nonenzymatic reaction of hydroxylamine, which should be independent of the redox state of BCR.
Chemical Stability of the Phospho-Linkage.
To investigate the nature of the phosphorylated amino acid, BCR was autophosphorylated under aerobic conditions as described previously. Phosphorylated BCR was incubated in 50 mM Tris-buffer, pH 7.5/1 M HCl/3 M NaOH/1 M hydroxylamine (pH 7.5) for 2 h at room temperature. For this purpose, the [γ-32P]-labeled BCR was subjected to SDS/PAGE analysis, blotted on a poly(vinylidene difluoride) membrane, and quantitatively analyzed by phosphorimaging (Table 1). The results indicate that the phospho-linkage is highly sensitive to acid and hydroxylamine treatment, whereas it was considerably resistant to hydrolysis under alkaline conditions. However, even in Tris buffer (pH 7.5), one-third of the phosphorylated enzyme band was lost compared with the air-dried sample. The data favor the presence of a phosphoamidate (e.g., histidine or lysine phosphate) rather than an acyl-phosphate as the latter is usually more labile at alkaline conditions; the presence of O-phosphates can be excluded as they are generally resistant to both acidic conditions and hydroxylamine treatment (25).
Table 1.
Chemical stability of the enzyme-phospho-linkage
| Condition | Phosphorylated BCR, % |
|---|---|
| Air-dried | 100 |
| 20 mM Tris, pH 7.5 | 65 |
| 3 M NaOH | 35 |
| 1 M HCl | <1 |
| 1 M hydroxylamine, pH 7.5 | <1 |
Air-oxidized BCR (10 μM) was incubated for 1 min with 10 μM [γ-32P]ATP, analyzed by SDS/PAGE, and blotted on a poly(vinylidene difluoride) (PVDF) membrane. The cut membrane strips containing the phosphorylated γ-subunit were incubated for 2 h at 21°C in the solutions as indicated. As a control, one strip was kept air-dried for 2 h.
Discussion
In this work, two unique partial activities of BCR have been discovered, which indicate that a phosphorylated enzyme intermediate is formed during enzymatic benzene ring reduction. The assignment of the autokinase/autophosphatase partial activities to the catalytic cycle of BCR is based on the effects of the redox state/substrates on these partial activities and on the [8-14C]ADP/ATP isotope exchange activity. The properties of the partial/overall activities under different conditions are summarized in Table 2.
In Fig. 6, a simplified scheme for a catalytic cycle of BCR considering the partial reactions discovered in this work is presented. For a simpler presentation, the single-electron transfer events are not included; instead, the presentation focuses on the ATP-dependent events. We distinguish between two catalytic cycles in the absence (cycle I) and presence (cycle II) of a reducible substrate. Both cycles include the redox-independent reversible formation of an energy-rich enzyme-phosphate catalyzed by the autokinase partial activity of BCR. Assuming an energy-rich enzyme-phospho-linkage is formed, only a small fraction (possibly less than 0.5%) of BCR is considered to be in the phosphorylated state under turnover conditions. The next events in the catalytic cycle are a dephosphorylation and the low-spin/high-spin switch of a [4Fe-4S]+1 cluster. Both events are driven solely by the presence of one or more electrons on BCR as they do not depend on the presence of BCoA. Enzyme-phosphate hydrolysis will largely pull the kinase reaction toward ATP hydrolysis as evidenced by the isotope exchange studies. Although it has not been shown yet, it is conceivable that enzyme-phosphate hydrolysis induces conformational changes finally resulting in the low-spin/high spin switch of a [4Fe-4S]+1 cluster. In the futile cycle I, the energized enzyme switches back to the ground state (S = 1/2) without electron transfer, and the energy of enzyme-phosphate hydrolysis is dissipated as heat. Exchange of ADP by ATP will close the cycle and initiate a second turn. In cycle II, enzyme-phosphate hydrolysis is stimulated 6-fold when electrons are transferred to BCoA or hydroxylamine, indicating a coupling of the electron transfer and dephosphorylation events. The cycle will be closed by re-reduction of BCR by ferredoxin and by an exchange of ADP by ATP. Note that for the transfer of two electrons the cycle has to run twice. However, it is not clear yet if one ATP is hydrolyzed for each electron transferred or if for the thermodynamically more difficult first electron transfer step (possibly yielding a radical anion) two ATP are hydrolyzed.
Figure 6.

Proposed role of the autokinase and autophosphatase partial activities in the catalytic cycle of BCR. Cycle I operates in reduced BCR in the absence of a reducible substrate, and cycle II operates in reduced BCR in the presence of BCoA. Note, that for a simpler presentation, neither the individual electron transfer steps to the substrates nor the enzyme-bound nucleotides and the ADP/ATP exchanges are shown.
The stoichiometric ATP-dependent electron transfer catalyzed by BCR represents a unique energy transduction process so far. Despite several analogies between BCR and nitrogenase (e.g., the type of the overall reaction or the similar overall architecture of nitrogenase Fe protein and the ATP-binding module of BCR), there are no homologies between these two enzymes. In contrast to nitrogenase (a member of the nucleotide switch family including nitrogenases and G proteins; ref. 17), the fold of the ATP-binding sites of BCR is predicted to be of the so-called ASKHA superfamily (acetate and sugar kinases, heat shock protein cognate Hsc70, actin; refs. 26 and 27) as evidenced by amino acid sequence comparisons (11, 13). The only member of this family for which an enzyme-phosphate intermediate involving mechanism has been reported so far is acetate kinase (28, 29). In acetate kinase, a highly conserved glutamate is discussed as the site of phosphorylation although it is ≥6 Å apart from the γ-phosphate of ATP (27). Thus, the conserved ATP-binding motifs of the α- and δ-subunits of BCR can hardly be used for speculations on the phosphorylation site of the γ-subunit. The N-terminal region of the γ-subunit of BCR contains a motif, 16DLDFT20, which would fit to a recently described common N-terminal DXDX(T/V) motif of a phosphotransferase class (30). The first aspartate in this motif has been identified as the phosphorylated amino acid in phosphomannomutase and is suggested as the site of phosphorylation in other members of this class, e.g., in phosphoserine phosphatases, sugar phosphate phosphatases, or some ATPases. Although the chemical stability rather favors the presence of phosphoamidate linkage in BCR, it is tempting to speculate Asp-16 as the phosphorylated amino acid. Unfortunately, the low degree of phosphorylation and the high sensitivity of the phosphoamino acid exclude a straightforward identification, e.g., by mass spectrometry. As BCR, several members of the ASKHA family (Hsc70, F-actin, and hexokinase) catalyze a futile ATP hydrolysis at a low rate, which is greatly stimulated by addition of the natural substrate (26). In this family, a substrate-dependent cleft closing/opening process is considered to minimize the futile ATP hydrolysis rate (26). A similar situation is conceivable for BCR.
Next to BCR, there is one other example of an electron-activating enzyme for which a phosphorylated enzyme-phosphate has been described: the methyl transferase activating protein (MAP) of Methanosarcina barkeri activates a methyl transferase by an ATP-dependent reduction of the corrinoid center to the Co(I) state (31). The MAP protein was shown to catalyze a stoichiometric autophosphorylation, and the phosphorylated enzyme has been shown to substitute for ATP (32). However, an electron transfer-dependent dephosphorylation, as it is demonstrated for BCR in this work, has not been shown for this system.
Acknowledgments
We are grateful to Prof. G. Fuchs for helpful suggestions and for carefully reading the manuscript. We thank Prof. Schägger for amino acid sequence analysis. This work was supported by grants from the Deutsche Forschungsgemeinschaft.
Abbreviations
- BCR
benzoyl-CoA reductase
- BCoA
benzoyl-CoA
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Heider J, Fuchs G. Eur J Biochem. 1997;243:577–596. doi: 10.1111/j.1432-1033.1997.00577.x. [DOI] [PubMed] [Google Scholar]
- 2.Boll M, Fuchs G. Eur J Biochem. 1995;234:921–933. doi: 10.1111/j.1432-1033.1995.921_a.x. [DOI] [PubMed] [Google Scholar]
- 3.Boll M, Laempe D, Eisenreich W, Bacher A, Mittelberger T, Heinze J, Fuchs G. J Biol Chem. 2000;275:21889–21895. doi: 10.1074/jbc.M001833200. [DOI] [PubMed] [Google Scholar]
- 4.Howard J B, Rees D C. Chem Rev. 1996;96:2965–2982. doi: 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]
- 5.Jang S B, Seefeldt L C, Peters J W. Biochemistry. 2000;39:14745–14752. doi: 10.1021/bi001705g. [DOI] [PubMed] [Google Scholar]
- 6.Birch A J, Rao G S. Adv Org Chem. 1972;8:1–65. [Google Scholar]
- 7.Buckel W, Keese R. Angew Chem. 1995;107:1595–1598. [Google Scholar]
- 8.Boll M, Fuchs G, Lowe D J. Biochemistry. 2001;40:7612–7620. doi: 10.1021/bi002771l. [DOI] [PubMed] [Google Scholar]
- 9.Harwood C S, Burchhardt G, Herrmann H, Fuchs G. FEMS Microbiol Rev. 1999;22:439–458. [Google Scholar]
- 10.Boll M, Fuchs G, Meier C, Trautwein A, Lowe D J. J Biol Chem. 2000;275:31857–31868. doi: 10.1074/jbc.M001508200. [DOI] [PubMed] [Google Scholar]
- 11.Breese K, Boll M, Alt-Mörbe J, Schägger H, Fuchs G. Eur J Biochem. 1998;256:916–923. doi: 10.1046/j.1432-1327.1998.2560148.x. [DOI] [PubMed] [Google Scholar]
- 12.Egland P G, Pelletier D A, Dispensa M, Gibson J, Harwood C S. Proc Natl Acad Sci USA. 1997;94:6484–6489. doi: 10.1073/pnas.94.12.6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hans M, Sievers J, Müller U, Bill E, Vorholt J A, Linder D, Buckel W. Eur J Biochem. 1999;265:404–414. doi: 10.1046/j.1432-1327.1999.00748.x. [DOI] [PubMed] [Google Scholar]
- 14.Buckel W. FEBS Lett. 1996;389:20–24. doi: 10.1016/0014-5793(96)00530-3. [DOI] [PubMed] [Google Scholar]
- 15.Locher K P, Hans M, Yeh A P, Schmid B, Buckel W, Rees D C. J Mol Biol. 2001;307:297–308. doi: 10.1006/jmbi.2000.4496. [DOI] [PubMed] [Google Scholar]
- 16.Boll M, Albracht S J P, Fuchs G. Eur J Biochem. 1997;244:840–851. doi: 10.1111/j.1432-1033.1997.00840.x. [DOI] [PubMed] [Google Scholar]
- 17.Rees D C, Howard J B. J Mol Biol. 1999;293:343–350. doi: 10.1006/jmbi.1999.3005. [DOI] [PubMed] [Google Scholar]
- 18.Anders H J, Kaetzke A, Kämpfer P, Ludwig W, Fuchs G. Int J Syst Bacteriol. 1995;45:327–333. doi: 10.1099/00207713-45-2-327. [DOI] [PubMed] [Google Scholar]
- 19.Gross G G, Zenk Z H. Z Naturforsch, B: Anorg Chem, Org Chem. 1966;21:683–690. [Google Scholar]
- 20.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Laemmli U K. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 22.Zehr B D, Savin T J, Hall R E. Anal Biochem. 1989;182:157–159. doi: 10.1016/0003-2697(89)90734-3. [DOI] [PubMed] [Google Scholar]
- 23.Heider J, Boll M, Breese K, Breinig S, Ebenau-Jehle C, Feil U, Gad'on N, Laempe D, Leuthner B, Mohamed M, et al. Arch Microbiol. 1998;170:120–131. doi: 10.1007/s002030050623. [DOI] [PubMed] [Google Scholar]
- 24.Thauer R K, Jungermann K, Decker K. Bacteriol Rev. 1977;41:100–180. doi: 10.1128/br.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duclos B D, Mascardier S, Cozzone A J. In: Methods of Enzymology. Abelson J N, Simon M I, editors. Vol. 21. San Diego: Academic; 1981. pp. 10–21. [Google Scholar]
- 26.Hurley J H. Annu Rev Biophys Biomol Struct. 1996;25:137–162. doi: 10.1146/annurev.bb.25.060196.001033. [DOI] [PubMed] [Google Scholar]
- 27.Buss K A, Cooper D R, Ingram-Smith C, Ferry J G, Sanders D A, Hasson M S. J Bacteriol. 2001;183:680–686. doi: 10.1128/JB.183.2.680-686.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anthony R S, Spector L B. J Biol Chem. 1970;246:6730–6741. [Google Scholar]
- 29.Anthony R S, Spector L B. J Biol Chem. 1972;247:2120–2125. [PubMed] [Google Scholar]
- 30.Collet J-F, Strooban V, Pirard M, Delpierre G, Van Schaftingen E. J Biol Chem. 1998;273:14107–14112. doi: 10.1074/jbc.273.23.14107. [DOI] [PubMed] [Google Scholar]
- 31.Wassenaar R W, Daas P J H, Geerts W J, Keltjens J T, van der Drift C. J Bacteriol. 1996;178:6937–6944. doi: 10.1128/jb.178.23.6937-6944.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daas P J, Wassenaar R W, Willemsen P, Theunissen R J, Keltjens J T, van der Drift C, Vogels G D. J Biol Chem. 1996;271:22339–22345. doi: 10.1074/jbc.271.37.22339. [DOI] [PubMed] [Google Scholar]