

Abstract
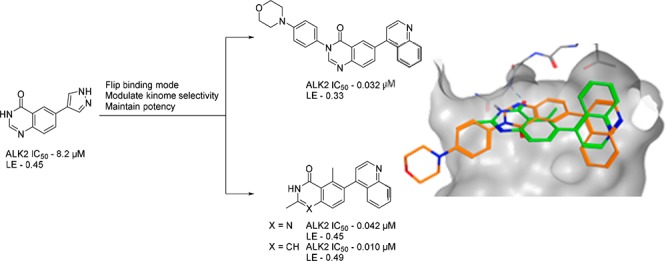
Structure–activity relationship and crystallographic data revealed that quinazolinone-containing fragments flip between two distinct modes of binding to activin receptor-like kinase-2 (ALK2). We explored both binding modes to discover potent inhibitors and characterized the chemical modifications that triggered the flip in binding mode. We report kinase selectivity and demonstrate that compounds of this series modulate ALK2 in cancer cells. These inhibitors are attractive starting points for the discovery of more advanced ALK2 inhibitors.
Introduction
ALK2 (gene: ACVR1) is a serine/threonine kinase in the bone morphogenetic protein (BMP) pathway and one of seven (ALK1–7) type-I receptors in the BMP and transforming growth factor beta (TGFβ) signaling pathways.1
Signaling through ALK2 is deregulated in two disease contexts. The first is fibrodysplasia ossificans progressiva (FOP), an extremely rare condition of ectopic bone formation resulting in a progressive loss of mobility.2 Heterozygous missense mutations in ACVR1 (most commonly R206H) lead to neofunction in response to activin A as well as increased BMP signaling through SMAD1/5/8—the driving force in FOP.3 The second disease is diffuse intrinsic pontine glioma (DIPG), a highly infiltrative tumor originating in the pons of the brainstem.4 DIPGs arise with a peak age of incidence of 6–7 years and have a fatality of 100%; median survival is 9–12 months.4 As DIPGs grow diffusely throughout the vital midline brain region, surgical resection is generally considered impossible, and the current treatment of radiotherapy, while providing short-term relief of symptoms, does not prevent rapid disease progression.5
ACVR1 mutations are observed in 24% of DIPG patients.4 These mutations occur in the cytoplasmic domains of ALK2 and modulate kinase activity though (i) destabilizing the inactive conformation of the kinase and (ii) disrupting the binding of a negative regulator protein, FKBP12.2 The high frequency of ALK2 mutations in DIPG strongly suggests a contribution to disease phenotype.
ALK2 inhibitors have been reported and fall into two series (Figure 1). The first contains a pyrazolo[1,5-a]pyrimidine core and derives from dorsomorphin. Dorsomorphin was initially reported as an adenosine monophosphate (AMP)-activated protein kinase inhibitor, but was found in a zebrafish embryo dorsalization assay to selectively inhibit BMP signaling through SMAD1/5/8.6 Further development led to LDN-193189,7LDN-212854,8 and ML347,9 which had improved microsomal stability, potency, and selectivity. In addition, these molecules demonstrated efficacy in mouse models of FOP.8 However, these compounds have a number of kinase off-targets and display dose-limiting toxicity with a 10% loss in body weight in animal models.10 A second series of inhibitor based on a pyridine core (e.g., K02288(11) and LDN-214117(12)), with equivalent biochemical potency and improved kinome selectivity, has also been reported (Figure 1).
Figure 1.

A selection of ALK2 and ALK5 inhibitors (the atom marked in red is the main anchor point to the kinase hinge residues).
Results
We identified 6-pyrazole quinazolinone, 1, as a ligand efficient inhibitor of ALK2 (IC50 = 8.2 μM; LE = 0.45) through cross-screening of a focused kinase fragment library, using Invitrogen’s LanthaScreen binding assay. 1 shares features with reported ALK5 inhibitors such as PF-03671148(13) and compound 19.14 These are known to bind to the hinge of ALK5 through a single polar contact at the N-1 position of the quinazolinone moiety, with the 2-methylpyridine directing toward the ALK5 Ser280 gatekeeper residue, forming a key water mediated hydrogen bond to Lys232 (Figure 2).14 Interestingly, PF-03671148 has been shown to be selective against ALK1 likely due a larger gate keeper (Thr) causing a clash with the pyridine substituent.13 Given that ALK2 also features a threonine gatekeeper residue (Thr283), it is also unlikely to be potently inhibited by PF-03671148.
Figure 2.
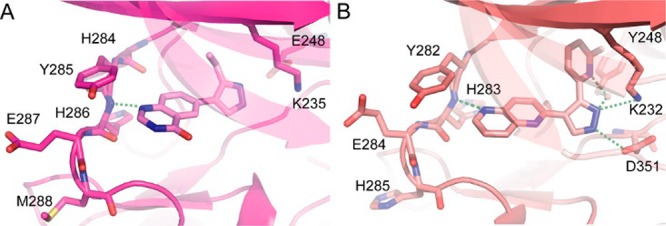
(A) Docking pose of 4 in ALK2 (structure used: 3Q4U). (B) X-ray structure of analogous naphthyridine-based inhibitor (compound 19) of ALK514 (PDB 1VJY).
Unfortunately, attempts to solve the structure of 1 bound to ALK2 were not successful. However, since 1 did not feature the pyridine substituent of the published ALK5 inhibitor (compound 19), we initially hypothesized that it explored a similar binding mode when binding to ALK5 (Figure 2).
We sought to investigate whether quinazolinone 1 could be optimized into an independent series of ALK2 inhibitor and characterize its binding mode.
We started our investigations at the quinazolinone 6-position, by modifying the pyrazole moiety (Table 1). N-Methylation of pyrazole (2) had a minor negative effect on ALK2 binding affinity, whereas substitution at the pyrazole 3-position was tolerated. Interestingly, increasing the size from methyl to cyclopropyl (4) was not only tolerated but led to a 21-fold potency increase. A 3,5-dimethylpyrazole (6) maintained sub-μM potency and was the most efficiently binding pyrazole derivative with an impressive LE of 0.51. This initial set of compounds, particularly compounds 2 and 6, suggested that the pyrazole was not the hinge binding motif and instead binds to Lys235 through a water bridge, as observed for compound 19 in Figures 1 and 2. This prompted us to replace the pyrazole for a bicycle (7–9). Since DIPG requires brain penetrable drugs, this had the added benefit of reducing the TPSA, a well-established predictor for passive permeability across the blood–brain barrier.15 Gratifyingly, this afforded a 40–80-fold improvement in potency over the initial hit. As the 4- and 5-quinoline examples (7 and 8) represented the most promising compounds so far, with sub-μM potency, good ligand efficiency and low TPSA, we decided to focus further modification on these isomers.
Table 1. SAR at Quinazolinone 6-Positiona.

IC50 data is an average of 2–4 measurements by LanthaScreen Eu kinase binding assay.
On the basis of the observed structure–activity relationship (SAR), we hypothesized that the compounds presented in Table 1 adopted a binding mode in which the variable heteroaromatic group occupied the ALK2 central pocket, extending toward the catalytic lysine (Lys235), similarly to reported ALK5 inhibitors (Figure 2).14 We also expected that this binding mode would be preserved between the pyrazoles and quinolines, which are present in several reported ALK2 inhibitors (Figure 1). However, it was still not clear how the quinazolinone core interacted with the hinge region. In an effort to shed light on this question, we positioned a methyl group at various positions around the quinazolinone ring (Table 2).
Table 2. Quinazolinone Methyl Scan SARa.

IC50 data is an average of 2–4 measurements by LanthaScreen Eu kinase binding assay.
When compared to the desmethyl analogues (7 and 8), a 5-methyl group (10 and 11) did not alter potency. This was consistent with the binding mode suggested in Figure 2, where the additional methyl group would direct toward solvent. 3-Methyl isomers 12 and 13 were also of equivalent potency, which again was consistent with a binding mode as in Figure 2 where the 3-methyl directs toward the solvent channel of ALK2. Surprisingly, analogous 2-methyl isomers (14 and 15) also displayed equal potency at ALK2. This was an unexpected result given that a 2-position substituent should clash with protein under the proposed binding model.
While this surprising tolerance to substitution at all three positions (Table 2) did not clearly suggest a preferred hinge binding motif, the 3-methyl isomer 12 stood out due to the low TPSA and closest analogy to published inhibitors.7
We decided to explore further substitution at the quinazolinone 3-position and incorporated solvent channel groups that had led to activity gains in other series, e.g., the pyrazolo[1,5-a]pyrimidine series. Introduction of a 4-morpholinophenyl group (16) was found to increase the potency 5-fold (Table 3) and yielded the most potent compound up to this point. However, the additional 12 heavy atoms from the 4-morpholinophenyl group did inflict a 0.11–0.19 LE unit penalty, suggesting that the solvent channel group was suboptimal.
Table 3. SAR at Quinazolinone 3-Positiona.

IC50 data is an average of 2–4 measurements by LanthaScreen Eu kinase binding assay.
To understand why only modest potency gains were afforded a series of truncated and modified analogues were prepared: phenyl (17), cyclohexyl (18), 3-morpholinopropyl (19), and 4-dimethyl aniline (20). Comparison of these analogues showed that the phenyl ring alone leads to a loss of activity compared to the methyl derivative, 12. Replacement by a cyclohexyl group did not drastically reduce binding further, suggesting that saturated or otherwise three-dimensional groups may be tolerated in this region of the ALK2 pocket. The 30-fold gain in potency achieved through the addition of morpholine (16) to phenyl-only compound 17 suggests that the phenyl ring is acting as a linker for this polar function. However, a simple propyl linker (19) to the morpholine unit does not afford a similar potency boost. In addition, maintaining the phenyl ring, but removing the ether functionality [of 16], leaving a dimethyl aniline (20) had a negligible effect on binding—strongly suggesting that the amine, and its precise position, are key to achieving potent inhibitors of ALK2.
With more potent compounds in hand, we reattempted cocrystallization and indeed managed to obtain the cocrystal structure of 16 with ALK2 to 2.2 Å resolution (Figure 3). The compound indeed displayed a binding mode similar to the reported inhibitor LDN-193189—quinazolinone N-1 interacts as an HBA for the ALK2 hinge residue His286, the 4-morpholino phenyl group directs through the solvent channel, and the 4-quinoline occupies the central pocket, with the quinoline-N-atom forming a water-bridged interaction to Lys235. Other similarities between the solved structure of 16 and LDN-193189 include the variable position of morpholine and methylpiperidine in the solvent channel—multiple conformations exist for both compounds in their respective asymmetric units suggesting that while the phenyl-bonded N atoms may have a significant role in binding the distal N-Me/O atom is unlikely to contribute heavily. We can only speculate on the reason why the morpholine amine contributes so strongly to binding. It is unlikely that this amine is charged due to the aniline character and electron withdrawing ether function. However, upon inspection of the electron density map for 16 a water bridged hydrogen bond to V214 is visible in one ALK2 chain in the asymmetric unit and may contribute to the gain in potency (Figure 3A). A notable difference versus available structures of pyrazolo[1,5-a]pyrimidine-based ALK2 inhibitors (PDB 3Q4U) is that the central pocket quinoline penetrates deeper toward Lys235 (Figure 3B), by virtue of the larger hinge-binding core.
Figure 3.
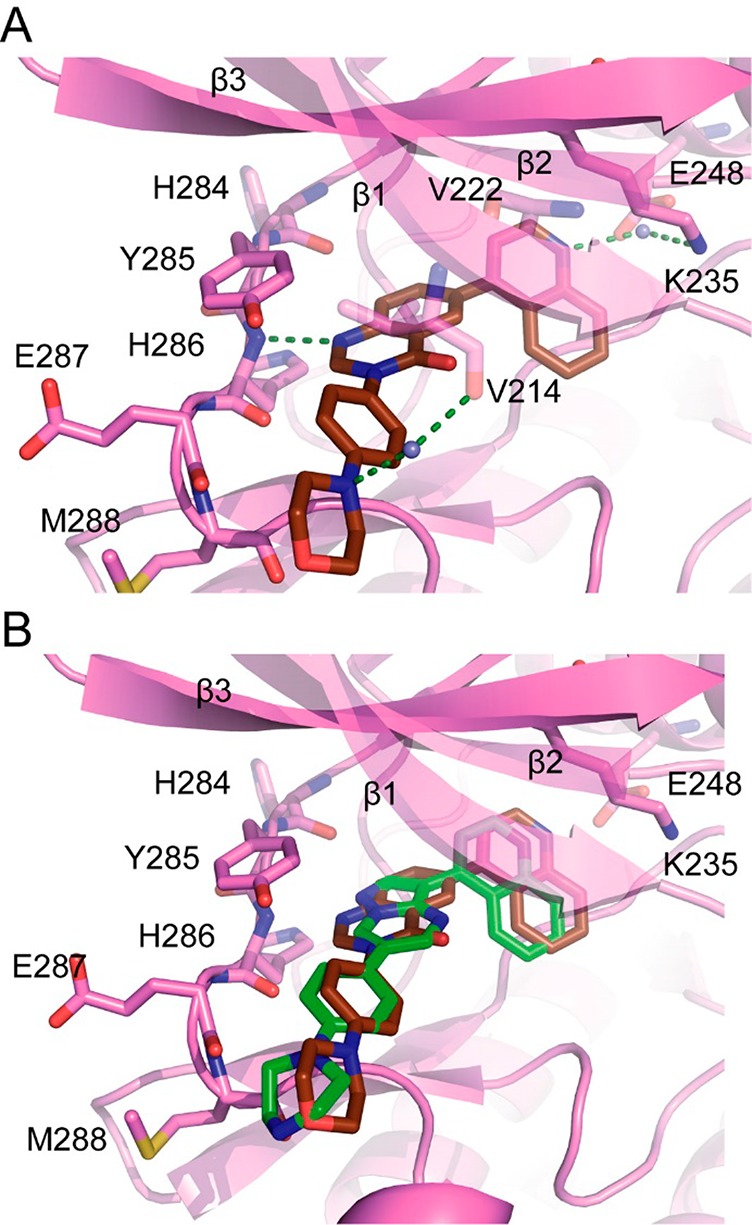
(A) Cocrystal structure of 16 in ALK2 showing three H-bond acceptors for H286, K235, and V214 (PDB 6GIN). (B) Overlay of 16 (brown) with LDN-193189 (green, PDB 3Q4U) showing deeper penetrance of 4-quinoline toward K235 for 16, and slight change in position of the solvent channel group.
The binding mode revealed by the crystal structure was consistent with our earlier hypothesis of preferred binding mode (cf. Figure 2 with the key hinge binding interaction between the quinazolinone N-1 and His286). An interesting observation was that some compounds (e.g., 14 and 15) maintained potency even though the additional 2-methylation would likely cause a clash with the hinge residues of ALK2 suggesting that they bind through an alternate binding mode. We therefore sought to solve additional structures and were able to obtain the structure of compound 11. Interestingly, 11 displayed a flipped binding mode (Figure 4A) in which the quinazolinone core bound to His286 through the amide, as a donor and acceptor. This also altered the vector from the quinazolinone 6-position such that the quinoline also flipped in order to fill the same volume in the ALK2 central pocket as for 16 (Figure 4B).
Figure 4.
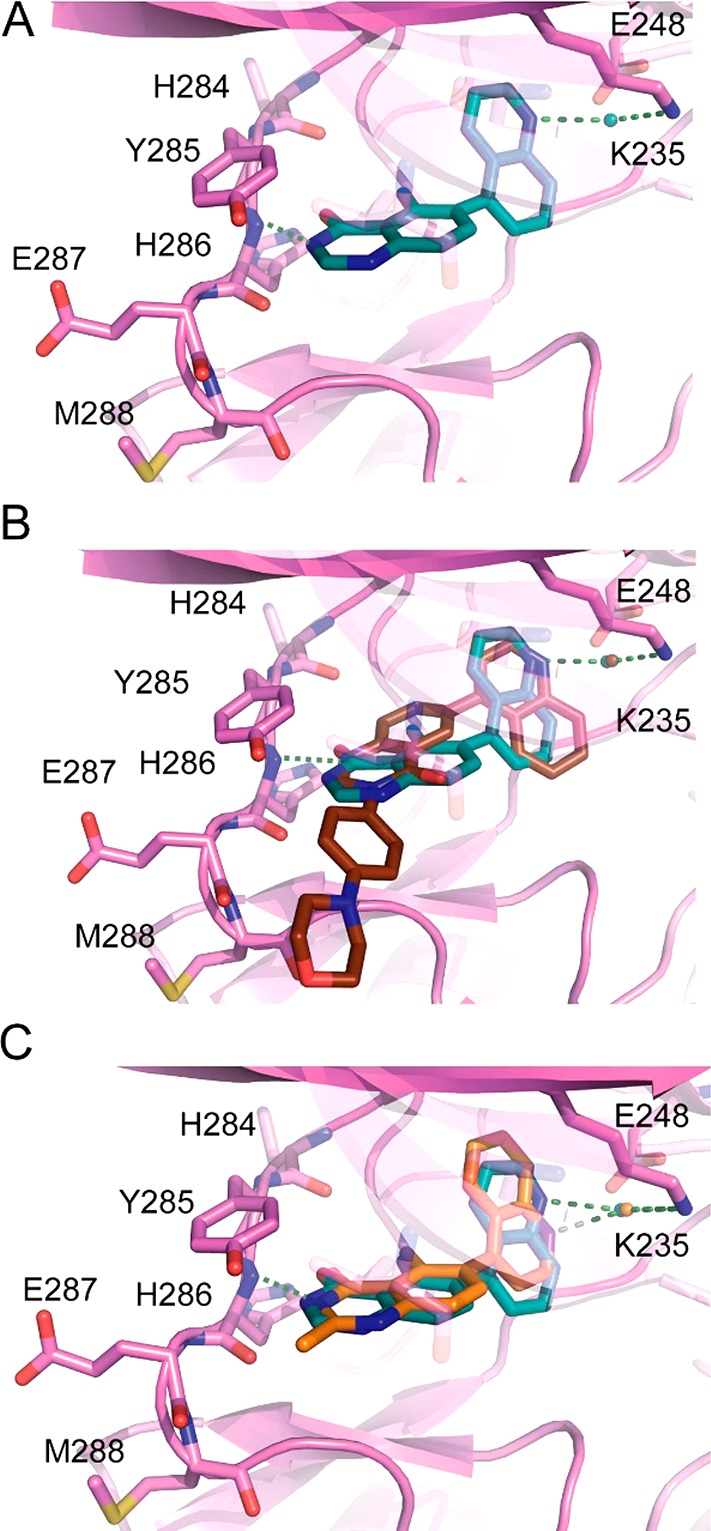
(A) Cocrystal structure of 11 with ALK2 (PDB 6GI6). (B) Superposition of costructures of 11 (blue) and 16 (brown). (C) Superposition of the costructures of 11 (blue) and 21 (orange, PDB 6GIP).
Inspection of the compound bound in the flipped binding mode (11) suggested that introduction of small hydrophobic groups in the 2-position should not only be tolerated but would lead to additional hydrophobic interaction (Table 4). Interestingly, while introduction of the 2- and 5-methyl groups individually (Table 2) was tolerated but did not lead to an increase in activity, introduction of both (21) afforded a 6-fold increase in potency (Table 4). We hypothesized that this cooperative increase in potency can be explained by the change in binding mode: 2-methylation forces adoption of the flipped, and presumably lower preference binding mode [for unsubstituted quinazolinones] (Figure 4C); however, there is no change in potency due to the addition of a methyl group in a relatively nonpolar part of the binding pocket. 5-Methylation fills the excluded volume created by flipping binding mode, which in the absence of 2-methylation is apparently enough to compensate for adopting the less energetically favored flipped binding mode. Once the flipped mode is induced by one methyl substituent, addition of the second does not incur a further penalty and reaps the benefit of the newly formed interaction.
Table 4. SAR of Flipped Binding Seriesa.

IC50 data is an average of 2–4 measurements by LanthaScreen Eu Kinase Binding Assay.
To assess the available space at the quinazolinone 2-position of the inhibitors binding in the flipped binding mode, we designed and prepared a few additional compounds. Crystallography suggested that the 2-methyl group could be replaced by larger groups. This was indeed the case. Increasing the size of the 2-substitutent to ethyl (22) or benzyl (23) was tolerated but did lead to a modest reduction in potency—mirroring the phenyl-only compound for the precedented binding mode (13), suggesting that there is scope to further investigate substituents at this position.
Furthermore, as the flipped binders do not appear to make use of N-1 for interaction with ALK2, we hypothesized that replacement with a C atom would result in additional hydrophobic interaction and loss of the desolvation penalty upon binding due to the polar heteroatom. The resulting isoquinolinone 24 indeed displayed 10 nM potency with a superb LE of 0.49.
At this stage, two divergent series with quinazolinone cores were developed to the stage of a having potency < 50 nM. We hypothesized that inhibitors of the normal and flipped modes would display distinct selectivity profiles. To test this hypothesis, the most potent normal and flipped binding quinazolinones (16 and 21 respectively) were assessed in DiscoveRx’s scanEDGE panel of 97 diverse kinases, plus all reported off-targets of the pyrazolo[1,5-a]pyrimidine and pyridine series (Table S1).8,9,12
Both quinazolinone based inhibitors displayed excellent selectivity in the kinome panel tested, though some notable differences were evident (Figure 5). At 1 μM the normal binder, 16, showed similarly potent inhibition of ALK6, platelet-derived growth factor receptor (PDGFR) A and B and Proto-Oncogene receptor tyrosine kinase KIT. While the flipped binder, 21, showed selectivity confined to the tyrosine kinase-like (TKL) family of kinases—a surprising result given its low molecular weight. Aside from the highly homologous ALKs 1, 4, 5, and 6, RAF1 and BRAF were also inhibited by 21. These results indicate that the two different compounds have distinct inhibition profiles within the kinome, versus each other as well as reported inhibitors and that both have a highly encouraging level of selectivity against the panel of kinases screened.
Figure 5.
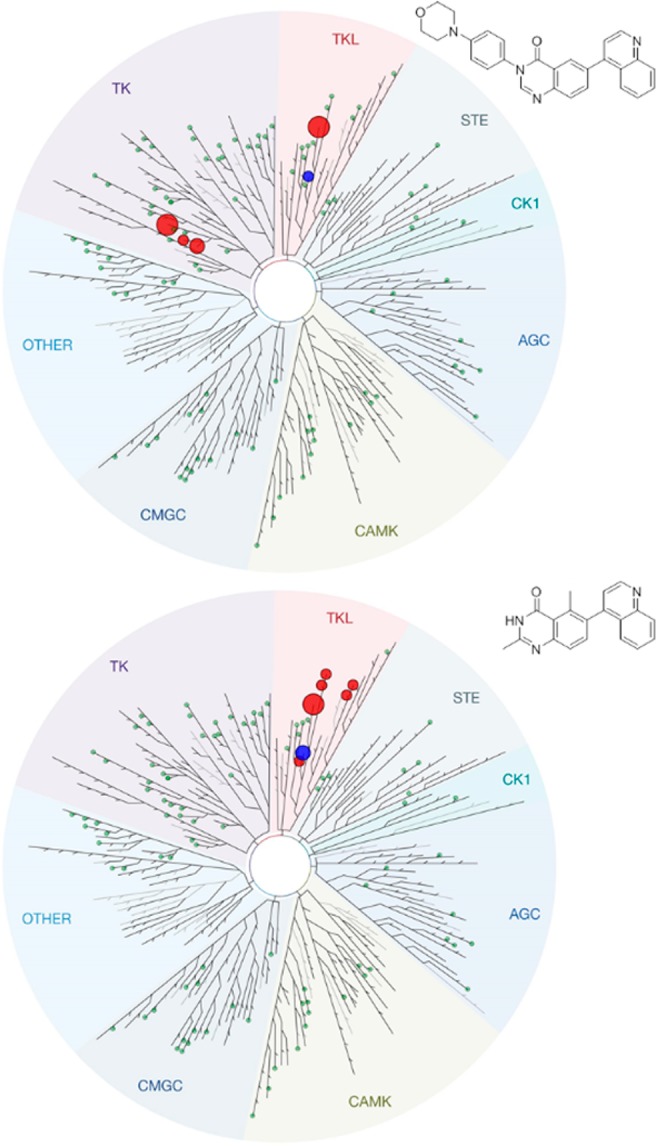
Kinase selectivity profile of 16 and 21 at 1 μM. Blue circle – ALK2; Green circles ≤ 65% probe displacement; small red circle = 65–90% probe displacement; intermediate circle = 90–95% probe displacement; larger red circle = 95–99% probe displacement.
KD values were obtained for ALKs 1–6 and all off-targets identified in Figure 5, for both 16 and 21 (Table 5) using DiscoverX’s KdELECT assay. 16 had similar affinity for ALKs 1, 2, 3, and 6 (which all signal through SMADs 1, 5, and 8) with moderate selectivity over ALKs 4 and 5 (which signal through SMADs 2 and 3). This profile is similar to reported ALK2 inhibitors.8,9,12 The smaller, flipped binder, 21, displayed an atypical profile—with lower selectivity over ALKs 4 and 5 and considerable variance in affinity for ALKs 1, 2, 3, and 6. The fact that 21 binds ALKs 1 and 6 16-fold more potently than ALK2 shows that 21 is not yet favorable for FOP and DIPG which harbor various activating mutations in ALK2. However, this is the first case, to our knowledge, of a small molecule with ALK1 selectivity over ALK2, which shares 79% sequence identity in their kinase domains.11 This is a potentially useful property if developed further to investigate the role of ALK1 in various disease contexts, particularly in regulating angiogenesis.16
Table 5. KD’s for ALKs 1–6 and All off-Targets Identified for Compounds 16 and 21a.
|
KD (nM) |
||
|---|---|---|
| kinase | 16 | 21 |
| ALK1 | 420 | 41 |
| ALK2 | 330 | 640 |
| ALK3 | 610 | 2700 |
| ALK4 | 11000 | 690 |
| ALK5 | 19000 | 1000 |
| ALK6 | 96 | 39 |
| BRAF | 65 | |
| RAF1 | 330 | |
| KIT | 54 | |
| PDGRFA | 43 | |
| PDGRFB | 250 | |
KD’s calculated from duplicate 11-point dose–response curves; KdELECT, DiscoverX
Finally, to achieve proof of concept that this class of compound shows activity in cells, we tested if 24 modulates the BMP pathway downstream of ALK2 (Figure 6) in HSJD-DIPG-007 patient derived cells (H3F3A K27M and ACVR1 R206H) at three different concentrations (0.1 μM, 1 μM, and 10 μM). Encouragingly, 24 displayed a dose-dependent reduction of pSMAD1/5/8 and ID1.
Figure 6.

Dose-dependent reduction in markers of ALK2 inhibition in HSJD-DIPG7 cells.
Compounds were prepared principally by a multicomponent reaction between orthoesters, amines, and anthranillic acids, followed by a Suzuki coupling (Scheme 1). For noncommercially available pyrazole boronic acids/esters, the parent pyrazole was brominated, then tosyl-protected (ix–x), which allowed for a one-pot borylation and Suzuki protocol.
Scheme 1. General Route to Quinazolinone Derivatives.

(a) 110 °C, 16–100%; (b) PdCl2(PPh3)2, NaOH(aq), 1,4-dioxane, 120–150 °C, 12–100%.
2-Benzyl quinazolinones were prepared by treatment of benzylcyanide with hydroxylamine, forming an intermediate amidoxime (Scheme 2).17 In the same pot, reaction with anthranillic acid, 25, afforded 6-bromo intermediate 26, which was functionalized by Suzuki coupling.
Scheme 2. Synthesis of 2-Benzyl Quinazolinones.

(a) (i) 50% NH2OH(aq), 120 °C, (ii) 25, 150 °C, 24%; (b) PdCl2(PPh3)2, NaOH(aq), 1,4-dioxane, 100 °C, 51–100%.
Isoquinolinones were prepared by a Doebner-modified Knoevenagel condensation with 4-bromo-3-methylbenzaldehyde, leading to 27 (Scheme 3).18 Preparation of a mixed anhydride with ethyl chloroformate followed by treatment with sodium azide gave 28, and subsequent Curtius rearrangement allowed for intramolecular trapping of the isocyanate to yield a 3:2 mixture of isoquinolinone regioisomers (29 and 30). After chromatographic separation, the major product (29) was functionalized by a Suzuki coupling.
Scheme 3. Synthesis of Isoquinolinones.

(a) Piperidine, 110 °C, 64%; (b) (i) NEt3, acetone, ethyl chloroformate, 0 °C – RT; (ii) NaN3, water, RT, 97%; (c) I2, 1,2-dichlorobenzene, 140–180 °C, 50% [3:2] 29:30; (d) PdCl2(PPh3)2, Na2CO3(aq), 1,4-dioxane, 150 °C, 61%.
Discussion and Conclusions
In conclusion, we identified a ligand efficient quinazolinone fragment (1) for inhibition of the ALK2 kinase domain through systematic cross screening. We found that the 6-pyrazole of 1 could be modified or replaced with bicyclic groups for gains in potency and that the activity was surprisingly tolerant to addition of methyl groups at the quinazolinone 2-, 3-, and 5-positions. Guided by crystallography, we were able to rationalize this tolerance through a flipped binding mode. We explored both binding modes to discover potent inhibitors. Our work shows that there is scope for further investigation of the SAR of both series and that the kinome selectivity profiles for example compounds (16 and 21) are not only distinct from one another but also do not hit off-targets for previously reported ALK2 inhibitors at the concentration tested. Finally we demonstrated that compound 24 modulates ALK2 in cells in a dose-dependent manner.
The compounds presented here thus represent attractive starting points to discover potent and selective inhibitors of activin receptor-like kinases.
Experimental Section
Unless otherwise stated, commercially available reagents and solvents were used without further purification. Yields were not optimized. NMR experiments were performed on a BrukerAvance 500 MHz spectrometer using an internal deuterium lock. Chemical shifts were measured in parts per million (ppm) relative to the residual signal of the deuterated solvent. Data are presented in the following format: chemical shift (multiplicity [s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, and br = broad], coupling constants (J in Hz), integration). Where mixed solvent systems were used residual solvent peaks to which the spectra are referenced are underlined. Assignment of 13C NMR data was achieved through analysis of 2D NMR spectra (HSQC, HMBC, COSY, and NOSEY). LCMS analyses and high resolution mass spectrometry were performed on an Agilent 1200 series HPLC and diode array detector coupled to a 6210 time-of-flight mass spectrometer with dual multimode APCI/ESI source (methods A and B) or a Waters Acquity UPLC and diode array detector coupled to a Waters G2 QToF mass spectrometer fitted with a multimode ESI/APCI source (method C); see Supporting Information for method details. All compounds described herein exhibited spectral data consistent with their proposed structures and, with the exception of 6 (purity >90%), had purities >95% as determined by HPLC.
General Procedure 1: Multicomponent Reaction
A mixture of an anthranillic acid or isatoic anhydride (1.0 equiv), amine (1.0 equiv), and orthoester (1.0 equiv) was stirred for 1–16 h at 110 °C, until LCMS indicated that reaction was complete. The mixture was cooled to RT, and the product was purified either by crystallization or trituration with hot EtOH unless otherwise indicated.
6-Bromo-3-(4-morpholinophenyl)quinazolin-4(3H)-one, i
Following general procedure 1, 6-bromo-2H-benzo[d][1,3]oxazine-2,4(1H)-dione (1.00 g; 4.15 mmol; 1.0 equiv), 4-morpholinoaniline (740 mg; 4.15 mmol; 1.0 equiv), and triethoxymethane (940 mg; 4.15 mmol; 1.0 equiv) were used to yield 6-bromo-3-(4-morpholinophenyl)quinazolin-4(3H)-one (1.59 g; 4.13 mmol; 100%) as a light purple solid. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 2.3 Hz, 1H), 8.08 (s, 1H), 7.84 (dd, J = 8.6, 2.3 Hz, 1H), 7.59 (d, J = 8.6 Hz, 1H), 7.26–7.23 (m, 2H), 7.00–6.97 (m, 2H), 3.86–3.82 (m, 4H), 3.21–3.18 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 160.07, 151.65, 147.00, 146.44, 137.80, 129.57, 129.04, 128.43, 127.52, 123.54, 121.30, 115.86, 66.64, 48.63. HRMS (ESI) m/z calc C18H17BrN3O2 [M + H]+ 386.0499; found = 386.0501.
6-Bromo-3-phenylquinazolin-4(3H)-one, ii
6-Bromo-2H-benzo[d][1,3]oxazine-2,4(1H)-dione (1.00 g; 4.15 mmol; 1.0 equiv), aniline (379 μL; 4.15 mmol; 1.0 equiv), and triethoxymethane (615 mg; 4.15 mmol; 1.0 equiv) were stirred at 110 °C for 18 h. The resulting tar was loaded onto silica with CH2Cl2 and purified by column chromatography in a solvent system of 0–5% MeOH in CH2Cl2. 6-Bromo-3-phenylquinazolin-4(3H)-one (387 mg; 1.29 mmol; 31%) was obtained as a beige solid. 1H NMR (500 MHz, CDCl3) δ 8.51 (d, J = 2.2 Hz, 1H), 8.15 (s, 1H), 7.90 (dd, J = 8.7, 2.3 Hz, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.60–7.56 (m, 2H), 7.55–7.51 (m, 1H), 7.45–7.42 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 159.61, 146.75, 146.41, 137.79, 137.19, 129.76, 129.74, 129.43, 129.34, 126.91, 123.79, 121.34. HRMS (ESI) m/z calc C14H10BrN2O [M + H]+ 300.9971; found = 300.9977.
6-Bromo-3-cyclohexylquinazolin-4(3H)-one, iii
6-Bromo-2H-benzo[d][1,3]oxazine-2,4(1H)-dione (1.00 g; 4.15 mmol; 1.0 equiv), cyclohexanamine (475 μL; 4.15 mmol; 1.0 equiv) and triethoxymethane (615 mg; 4.15 mmol; 1.0 equiv) were stirred at 110 °C for 18 h. The resulting tar was loaded onto silica with CH2Cl2 and purified by column chromatography in a solvent system of 0–5% MeOH in CH2Cl2. 6-Bromo-3-phenylquinazolin-4(3H)-one (429 mg; 1.40 mmol; 34%) was obtained as a light yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.45 (d, J = 2.2 Hz, 1H), 8.13 (s, 1H), 7.83 (dd, J = 8.7, 2.3 Hz, 1H), 7.58 (d, J = 8.7 Hz, 1H), 4.81 (tt, J = 12.3, 3.7 Hz, 1H), 2.05–1.99 (m, 2H), 1.99–1.93 (m, 2H), 1.81 (ddtd, J = 14.7, 4.9, 3.2, 1.4 Hz, 1H), 1.71–1.59 (m, 2H), 1.54 (qt, J = 13.2, 3.3 Hz, 2H), 1.28 (qt, J = 13.0, 3.7 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 159.55, 146.38, 144.22, 137.29, 129.52, 129.11, 123.33, 120.68, 53.65, 32.58, 25.86, 25.24. HRMS (ESI) m/z calc C14H16BrN2O [M + H]+ 307.0441; found = 307.0448.
6-Bromo-3-(3-morpholinopropyl)quinazolin-4(3H)-one, iv
6-Bromo-2H-benzo[d][1,3]oxazine-2,4(1H)-dione (1.0 g; 4.15 mmol; 1.0 equiv), 3-morpholinopropan-1-amine (598 mg; 4.15 mmol; 1.0 equiv), and triethoxymethane (940 mg; 6.35 mmol; 1.5 equiv) were stirred at 110 °C for 5 h. The resulting tar was loaded onto silica with CH2Cl2 and purified by column chromatography in a solvent system of 0–8% MeOH in CH2Cl2 to yield 6-bromo-3-(3-morpholinopropyl)quinazolin-4(3H)-one (1.06 g; 3.02 mmol; 73%) as a yellow oil. 1H NMR (500 MHz, DMSO) δ 8.43 (s, 1H), 8.22 (d, J = 2.4 Hz, 1H), 7.96 (dd, J = 8.7, 2.4 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H), 4.02 (t, J = 6.9 Hz, 2H), 3.44 (t, J = 4.7 Hz, 4H), 2.31 (t, J = 6.6 Hz, 2H), 2.27 (br-s, 4H), 1.86 (p, J = 6.7 Hz, 2H). 13C NMR (126 MHz, DMSO) δ 159.68, 149.44, 147.48, 137.47, 130.02, 128.52, 123.66, 119.77, 66.52, 55.58, 53.50, 45.53, 24.79. HRMS (ESI) m/z calc C15H19BrN3O2 [M + H]+ 352.0655; found = 352.0636.
6-Bromo-3-(4-(dimethylamino)phenyl)quinazolin-4(3H)-one, v
Following general procedure 1, 6-bromo-2H-benzo[d][1,3]oxazine-2,4(1H)-dione (1.0 g; 4.15 mmol; 1.0 equiv), N1,N1-dimethylbenzene-1,4-diamine (565 mg; 4.15 mmol; 1.0 equiv), and triethoxymethane (615 mg; 4.15 mmol; 1.0 equiv) were used to yield 6-bromo-3-(4-(dimethylamino)phenyl)quinazolin-4(3H)-one (833 mg; 2.43 mmol; 59%) as a glittery, dark-purple solid. 1H NMR (500 MHz, CDCl3) δ 8.50 (d, J = 2.3 Hz, 1H), 8.14 (s, 1H), 7.87 (dd, J = 8.7, 2.3 Hz, 1H), 7.64 (d, J = 8.6 Hz, 1H), 7.26–7.22 (m, 2H), 6.84–6.79 (m, 2H), 3.04 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 160.13, 150.74, 147.30, 146.84, 137.46, 129.70, 129.30, 127.41, 125.62, 123.88, 120.99, 112.49, 40.46. HRMS (ESI) m/z calc C16H15BrN3O [M + H]+ 344.0393; found = 344.0403.
6-Bromo-2-methylquinazolin-4(3H)-one, vi
6-Bromo-2H-benzo[d][1,3]oxazine-2,4(1H)-dione (1.00 g; 4.15 mmol; 1.0 equiv), NH4OAc (385 mg; 5.00 mmol; 1.2 equiv), and triethoxymethane (673 mg; 4.15 mmol; 1.0 equiv) were stirred at 110 °C for 18 h. The crude reaction product was loaded onto silica using CH2Cl2 and purified by column chromatography with a solvent system of 0–8% MeOH in CH2Cl2. 6-Bromo-2-methylquinazolin-4(3H)-one (212 mg; 0.891 mmol; 21%) was isolated as a cream solid. 1H NMR (500 MHz, CDCl3/MeOH) δ 8.28 (d, J = 2.3 Hz, 1H), 7.76 (dd, J = 8.7, 2.4 Hz, 1H), 7.45 (d, J = 8.7 Hz, 1H), 2.40 (s, 3H). 13C NMR (126 MHz, CDCl3/MeOH) δ 161.87, 154.36, 147.56, 137.82, 128.72, 128.20, 121.88, 119.82, 21.36. HRMS (ESI) m/z calc C9H8BrN2O [M + H]+ 238.9815; found = 238.9822.
6-Bromo-2,5-dimethylquinazolin-4(3H)-one, vii
6-Amino-3-bromo-2-methylbenzoic acid (950 mg; 4.15 mmol; 1.0 equiv), NH4OAc (385 mg; 5.00 mmol; 1.2 equiv) and triethoxymethane (673 mg; 4.15 mmol; 1.0 equiv) were stirred at 110 °C for 48 h. The crude reaction product was loaded onto silica using CH2Cl2 and purified by column chromatography with a solvent system of 0–8% MeOH in CH2Cl2. 6-Bromo-2,5-dimethylquinazolin-4(3H)-one (255 mg; 1.01 mmol; 24%) was isolated as a cream solid. 1H NMR (500 MHz, CDCl3/MeOH) δ 7.77 (dd, J = 8.8, 1.5 Hz, 1H), 7.25 (d, J = 8.3 Hz, 1H), 2.90 (s, 3H), 2.33 (s, 3H). 13C NMR (126 MHz, CDCl3/MeOH) δ 166.76, 157.95, 153.44, 143.71, 142.04, 129.43, 127.89, 124.22, 25.22, 24.73. HRMS (ESI) m/z calc C10H10BrN2O [M + H]+ 252.9971; found = 252.9980.
6-Bromo-2-ethyl-5-methylquinazolin-4(3H)-one, viii
6-Amino-3-bromo-2-methylbenzoic acid (950 mg; 4.15 mmol; 1.0 equiv), NH4OAc (385 mg; 5.00 mmol; 1.2 equiv), and triethyl orthopropionate (731 mg; 4.15 mmol; 1.0 equiv) were stirred at 110 °C for 48 h. The crude reaction product was loaded onto silica using CH2Cl2 and purified by column chromatography with a solvent system of 0–8% MeOH in CH2Cl2. 6-Bromo-2-ethyl-5-methylquinazolin-4(3H)-one (418 mg; 1.57 mmol; 38%) was isolated as an off-white solid. 1H NMR (500 MHz, DMSO) δ 12.17 (s, 1H), 7.91 (d, J = 8.7 Hz, 1H), 7.36 (d, J = 8.7 Hz, 1H), 2.92 (s, 3H), 2.58 (q, J = 7.5 Hz, 2H), 1.23 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 162.45, 159.18, 150.45, 138.98, 137.79, 127.17, 122.94, 121.18, 27.87, 21.54, 11.57. HRMS (ESI) m/z calc C11H12BrN2O [M + H]+ 267.0128; found = 267.0135.
4-Bromo-3-cyclopropyl-1-tosyl-1H-pyrazole, ix
To a solution of 4-bromo-3-cyclopropyl-1H-pyrazole (100 mg; 0.538 mmol; 1.0 equiv) in CH2Cl2 (10 mL; 0.054 M) was added NaOH(aq) (118 μL; 5 M; 0.591 mmol; 1.1 equiv) and 4-toluenesulfonyl chloride (113 mg; 0.591 mmol; 1.1 equiv). The mixture was heated to reflux and stirred for 72 h before being diluted with water (400 mL) and extracted with CH2Cl2 (2 × 200 mL). The combined organic portions were washed with brine, dried (Na2SO4), and filtered, and solvent was removed under reduced pressure. The crude product was washed through an SCX-II column with MeOH to yield 4-bromo-3-cyclopropyl-1-tosyl-1H-pyrazole (103 mg; 0.303 mmol; 56%) as a white solid after removal of solvent under reduced pressure. 1H NMR (500 MHz, DMSO) δ 8.68 (s, 1H), 7.86–7.81 (m, 2H), 7.48 (d, J = 8.1 Hz, 2H), 2.40 (s, 3H), 1.84 (tt, J = 8.3, 4.9 Hz, 1H), 0.99–0.93 (m, 2H), 0.78–0.73 (m, 2H). 13C NMR (126 MHz, DMSO) δ 158.61, 146.80, 133.46, 133.17, 130.88, 128.12, 99.30, 21.64, 8.83, 7.76. HRMS (ESI) m/z calc C13H13BrN2O2SNa [M + Na]+ 362.9779; found = 362.9771.
4-Bromo-3-ethyl-1-tosyl-1H-pyrazole, x
p-Toluene sulfonyl chloride (194 mg; 1.02 mmol; 1.1 equiv) was added to a solution of 4-bromo-3-ethyl-1H-pyrazole (162 mg; 0.930 mmol; 1.0 equiv) and [5 M] NaOH solution (0.2 mL; 1.02 mmol; 1.1 equiv) in CH2Cl2 (18.5 mL), and the mixture was stirred at 38 °C. After 16 h the reaction mixture was washed with water (15 mL) and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The combined organic fractions were washed with brine, dried over MgSO4, filtered, and concentrated under vacuum. The crude mixture was purified by normal phase flash chromatography in a solvent system of 0–5% EtOAc in cyclohexane to afford 4-bromo-3-ethyl-1-tosyl-1H-pyrazole (246 mg; 0.826 mmol; 81%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.03 (s, 1H), 7.85–7.93 (m, 2H), 7.30–7.38 (m, 2H), 2.62 (q, J = 7.6, 2H), 2.44 (s, 3H), 1.22 (t, J = 7.6, 3H). 13C NMR (126 MHz, CDCl3) δ 158.69, 145.92, 133.86, 128.13, 98.52, 21.74, 20.38, 12.23. HRMS (ESI) m/z calc C12H14BrN2O2S [M + H]+ 328.9954; found = 328.9954.
General Procedure 2: Suzuki Coupling
To a Biotage microwave vial was added the required bromo-aryl (1.0 equiv), boronic acid or ester (1.0–1.2 equiv), PdCl2(PPh3)2 (0.05 equiv), Na2CO3(aq) (0.5 M; 1.0 equiv), and 1,4-dioxane or DME. The vial was sealed and purged of air with 3 rounds of vacuum and N2 or Ar. The mixture was heated to 120–150 °C for 1 h under microwave irradiation. The reaction mixture was filtered through Celite, washed with water, and extracted twice with CH2Cl2. Combined organic portions were dried (Na2SO4), filtered, and had solvent removed under reduced pressure. The crude product was purified by column chromatography in a solvent system of MeOH in CH2Cl2.
6-(1H-Pyrazol-4-yl)quinazolin-4(3H)-one, 1
Following general procedure 2, 6-bromoquinazolin-4(3H)-one (44 mg; 0.20 mmol) was reacted with [1-(tert-butoxycarbonyl)-1H-pyrazol-4-yl]boronic acid pinacol ester (69 mg; 0.24 mmol) in DME to yield 6-(1H-pyrazol-4-yl)quinazolin-4(3H)-one (22 mg; 54%) as an off-white solid. Reaction temperature: 150 °C. Purification: 1–8% MeOH in CH2Cl2. 1H NMR (500 MHz, DMSO) 13.06 (br-s, 1H), 12.21 (br-s, 1H), 8.38 (br-s, 1H), 8.28 (d, J = 2.2 Hz, 1H), 8.08 (dd, J = 8.4, 2.2 Hz, 1H), 8.05 (s, 1H), 8.04 (br-s, 1H), 7.65 (d, J = 8.4 Hz, 1H). 13C NMR (126 MHz, DMSO) 161.2, 147.4, 145.0, 136.9, 132.1, 128.2, 126.6, 123.5, 121.3, 120.6. HRMS (ESI) m/z calc C11H9N4O [M + H]+ 213.0771; found = 213.0779.
6-(1-Methyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one, 2
Following general procedure 2, 6-bromoquinazolin-4(3H)-one (47 mg; 0.21 mmol) was reacted with 1-methylpyrazole-4-boronic acid pinacol ester (52 mg; 0.25 mmol) in DME to yield 6-(1-methyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (42 mg; 88%) as a white solid. Reaction temperature: 150 °C. Purification: 1–8% MeOH in CH2Cl2. 1H NMR (500 MHz, DMSO) 12.2 (br-s, 1H), 8.32 (d, J = 0.9 Hz, 1H), 8.23 (d, J = 2.1 Hz, 1H), 8.04 (br-s, 1H), 8.02 (dd, J = 8.5, 2.2 Hz, 1H), 7.99 (d, J = 0.9 Hz, 1H), 7.65 (d, J = 8.5 Hz, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, DMSO) 161.2, 147.4, 145.0, 136.8, 131.8, 128.9, 128.3, 123.6, 121.1, 121.1, 39.2. HRMS (ESI) m/z calc C12H11N4O [M + H]+ 227.0927; found = 227.0924.
6-(3-Methyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one, 3
Following general procedure 2, 6-bromoquinazolin-4(3H)-one (31 mg; 0.14 mmol) was reacted with 3-methyl-1H-pyrazole-4-boronic acid pinacol ester (35 mg; 0.17 mmol) and Pd(PPh3)4 (11 mg; 7 mol %) in DME. After 40 min, additional 3-methyl-1H-pyrazole-4-boronic acid pinacol ester (29 mg; 0.14 mmol) and Pd(PPh3)4 (8 mg; 0.05 equiv) were added, and the mixture was stirred under microwave irradiation at 150 °C for 30 min. After this time, the reaction was purified by SCX-2 chromatography (2 g, 30 mL MeOH then 30 mL 1 M ammonia in MeOH) and column chromatography (10 g SNAP, 3- 9% MeOH in CH2Cl2) to afford 6-(3-methyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (14 mg; 44%) as a cream solid. 1H NMR (500 MHz, DMSO) δ 12.75 (d, J = 41.8 Hz, 1H), 12.24 (s, 1H), 8.11 (s, 1H), 8.06 (d, J = 3.3 Hz, 1H), 7.94 (dd, J = 8.3, 2.2 Hz, 1H), 7.81 (s, 1H), 7.68 (d, J = 8.5 Hz, 1H), 2.42 (d, J = 30.9 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 161.19, 147.06, 145.11, 138.75, 135.75, 133.27, 132.88, 128.14, 123.32, 122.47, 117.15, 11.13. HRMS (ESI) m/z calc C12H11N4O [M + H]+ 227.0927; found = 227.0933.
6-(3-Ethyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one, 4
4-Bromo-3-ethyl-1-tosyl-1H-pyrazole (66 mg; 0.194 mmol; 1.0 equiv), KOAc (57 mg; 0.583 mmol; 1.1 equiv), bis(pinacolato)diboron (55 mg; 0.214 mmol; 1.1 equiv), and PdCl2(PPh3)2 (6.8 mg; 9.70 μmol; 0.05 equiv) were charged to a microwave vial. The vial was sealed and purged of air with avacuum and N2 five times. 1,4-Dioxane (4.4 mL; 0.044 M) was added, and the mixture was heated at 120 °C for 1 h under microwave irradiation. At RT, 6-bromoquinazolin-4(3H)-one (44 mg; 0.194 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (389 μL; 0.194 mmol; 1.0 equiv), and PdCl2(PPh3)2 (6.8 mg; 9.70 μmol; 0.05 equiv) were added to the reaction mixture before the vial was sealed and purged of air with vacuum and N2 five times. The mixture was heated to 120 °C for 1 h under microwave irradiation. The mixture was then diluted with water (100 mL) and extracted with CH2Cl2 (3 × 75 mL). Combined organic portions were washed with brine (100 mL), dried (Na2SO4), and filtered, and solvent was removed under reduced pressure. The crude product was purified by column chromatography in a solvent system of 2–15% MeOH in CH2Cl2 to yield 6-(3-ethyl-1-tosyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (13 mg; 0.03 mmol; 15%) as a white solid. A solution of the tosyl-protected intermediate was formed in EtOH (1.1 mL), to which was added [1 M] NaOH(aq) (0.8 mL; 0.82 mmol). The mixture was stirred at 50 °C for 14 h and then neutralized using 1 M HCl and purified by SCX-2 chromatography (2 g, 10 mL MeOH then 30 mL 1 M NH3 in MeOH). The crude mixture was further purified by Biotage column chromatography (5–12% MeOH in DCM) and concentrated under a vacuum. The resulting solid was triturated with n-hexane (3 × 1 mL) to afford 6-(3-ethyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (7 mg, 0.025 mmol, 82%) as an off-white solid. 1H NMR (500 MHz, DMSO) δ 12.77 (s, 1H), 12.25 (s, 1H), 8.09 (d, J = 2.1 Hz, 1H), 8.06 (d, J = 3.4 Hz, 1H), 7.90 (dd, J = 8.4, 2.2 Hz, 1H), 7.68 (d, J = 8.5 Hz, 1H), 2.82 (t, J = 7.6 Hz, 2H), 1.22 (t, J = 7.6 Hz, 4H). 13C NMR (126 MHz, DMSO) δ 161.18, 147.14, 145.16, 133.73, 132.92, 128.12, 123.32, 122.97, 18.25, 13.86. HRMS (ESI) m/z calc C13H13N4O [M + H]+ 241.1084; found = 241.1084.
6-(3-Cyclopropyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one, 5
4-Bromo-3-cyclopropyl-1-tosyl-1H-pyrazole (30 mg; 0.0882 mmol; 1.0 equiv), KOAc (26 mg; 0.265 mmol; 1.1 equiv), bis(pinacolato)diboron (25 mg; 0.0971 mmol; 1.1 equiv), and PdCl2(PPh3)2 (3.1 mg; 4.41 μmol; 0.05 equiv) were charged to a microwave vial. The vial was sealed and purged of air with a vacuum and N2 five times. 1,4-Dioxane (2 mL; 0.044 M) was added, and the mixture was heated at 120 °C for 1 h under microwave irradiation. 6-Bromoquinazolin-4(3H)-one (20 mg; 0.0882 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (176 μL; 0.0882 mmol; 1.0 equiv), and PdCl2(PPh3)2 (3.1 mg; 4.41 μmol; 0.05 equiv) were added to the reaction mixture (<40 °C) before the vial was sealed and purged of air with a vacuum and N2 five times. The mixture was heated to 120 °C for 1 h under microwave irradiation. The mixture was diluted with water (100 mL) and extracted with CH2Cl2 (3 × 75 mL). The combined organic portions were washed with brine (100 mL), dried (Na2SO4) and filtered and solvent was removed under reduced pressure. The crude intermediate was purified by column chromatography in a solvent system of 2–15% MeOH in CH2Cl2 to yield 6-(3-cyclopropyl-1-tosyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (31 mg; 0.0763 mmol; 87%) as a white solid. A mixture of 6-(3-cyclopropyl-1-tosyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (5 mg; 0.0123 mmol; 1.0 equiv), [1 M] NaOH(aq) (308 μL; 0.308 mmol; 25 equiv), and EtOH (1 mL; 0.012 M) was heated at 50 °C for 2 h before being cooled to RT and neutralized with [1 M] HCl(aq). The mixture had solvent removed under reduced pressure, and the residue was purified by column chromatography in a solvent system of 2–10% MeOH in CH2Cl2. 6-(3-Cyclopropyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (2.1 mg; 0.00833 mmol; 68%) was isolated as a white solid. 1H NMR (500 MHz, MeOD) δ 8.49 (q, J = 2.1 Hz, 1H), 8.12 (d, J = 8.6 Hz, 1H), 8.10 (s, 1H), 7.87 (s, 1H), 7.75 (d, J = 8.4 Hz, 1H), 2.13–2.06 (m, 1H), 1.12–0.82 (m, 4H). 13C NMR (126 MHz, MeOD) δ 161.90, 146.50, 144.37, 133.76, 133.02, 128.41, 126.83, 125.55, 123.26, 122.52, 118.86, 6.36, 6.35. HRMS (ESI) m/z calc C14H13N4O [M + H]+ 253.1084; found = 253.1089.
6-(3,5-Dimethyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one, 6
Following general procedure 2, 6-bromoquinazolin-4(3H)-one (37 mg; 0.165 mmol; 1 equiv) was reacted with 3,5-dimethyl-1H-pyrazole-4-boronic acid pinacol ester (43 mg; 0.194 mmol; 1.2 equiv) in DME (1.00 mL; 0.16 M) were reacted to yield 6-(3,5-dimethyl-1H-pyrazol-4-yl)quinazolin-4(3H)-one (19 mg; 0.0792 mmol; 50%) as a white solid. Reaction temperature: 150 °C. Purification: 4–11% MeOH in CH2Cl2. 1H NMR (500 MHz, DMSO) δ 12.17 (s, 1H), 8.11 (d, J = 8.4 Hz, 1H), 8.08 (d, J = 3.5 Hz, 1H), 7.77 (s, 1H), 7.64 (d, J = 1.7 Hz, 1H), 7.59 (dd, J = 8.2, 1.8 Hz, 1H), 3.81 (s, 3H), 2.45 (s, 3H). 13C NMR (126 MHz, DMSO) δ 160.95, 149.73, 146.12, 140.30, 137.33, 136.47, 126.77, 126.12, 124.50, 120.49, 118.82, 36.90, 10.91. HRMS (ESI) m/z calc C13H13N4O [M + H]+ 241.1084; found = 241.1081.
6-(Quinolin-4-yl)quinazolin-4(3H)-one, 7
Following general procedure 2, quinolin-4-ylboronic acid (44 mg; 0.255 mmol 1.0 equiv), 6-bromoquinazolin-4(3H)-one (57 mg; 0.255 mmol; 1.0 equiv), PdCl2(PPh3)2 (9 mg; 0.0128 mmol; 0.05 equiv), [0.5 M] Na2CO3 (510 μL; 0.255 mmol; 1.0 equiv) and 1,4-dioxane (1.28 mL; 0.20 M) were used to yield 6-(quinolin-4-yl)quinazolin-4(3H)-one (38 mg; 0.139 mmol; 55%) as a white solid. Reaction temperature: 120 °C. Purification: 1–8% MeOH in CH2Cl2. 1H NMR (500 MHz, MeOH) δ 8.91 (d, J = 4.5 Hz, 1H), 8.43 (d, J = 1.9 Hz, 1H), 8.18–8.12 (m, 1H), 8.12 (s, 1H), 7.97 (dd, J = 8.4, 2.1 Hz, 1H), 7.91 (dd, J = 8.4, 1.3 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.83–7.79 (m, 1H), 7.62–7.57 (m, 1H), 7.49 (d, J = 4.5 Hz, 1H). 13C NMR (126 MHz, MeOD/CDCl3) δ 161.70, 149.36, 148.78, 148.01, 147.77, 145.49, 136.86, 135.75, 130.05, 128.81, 127.59, 127.41, 127.22, 126.51, 125.43, 122.97, 121.65. HRMS (ESI) m/z calc C17H12N3O [M + H]+ 274.0975; found = 274.0985.
6-(Quinolin-5-yl)quinazolin-4(3H)-one, 8
Following general procedure 2, quinolin-5-ylboronic acid (44 mg; 0.255 mmol 1.0 equiv), 6-bromoquinazolin-4(3H)-one (57 mg; 0.255 mmol; 1.0 equiv), PdCl2(PPh3)2 (9 mg; 0.0128 mmol; 0.05 equiv), [0.5 M] Na2CO3 (510 μL; 0.255 mmol; 1.0 equiv) and 1,4-dioxane (1.28 mL; 0.20 M) were used to yield 6-(quinolin-5-yl)quinazolin-4(3H)-one (40 mg; 0.146 mmol; 57%) as a white solid. Reaction temperature: 120 °C. Purification: 1–8% MeOH in CH2Cl2. 1H NMR (500 MHz, MeOD/CDCl3) δ 8.88 (dd, J = 4.2, 1.6 Hz, 1H), 8.40–8.35 (m, 1H), 8.29 (ddd, J = 8.6, 1.7, 0.9 Hz, 1H), 8.13 (dt, J = 8.6, 1.1 Hz, 1H), 8.11 (s, 1H), 7.93 (dd, J = 8.4, 2.1 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.85 (dd, J = 8.5, 7.1 Hz, 1H), 7.63 (dd, J = 7.1, 1.2 Hz, 1H), 7.49 (dd, J = 8.6, 4.2 Hz, 1H). 13C NMR (126 MHz, MeOD/CDCl3) δ 162.26, 149.88, 148.16, 147.70, 145.15, 138.92, 138.09, 136.41, 134.74, 129.44, 128.38, 128.03, 127.58, 127.37, 126.44, 122.70, 121.56. HRMS (ESI) m/z calc C17H12N3O [M + H]+ 274.0975; found = 274.0984.
6-(Pyrazolo[1,5-a]pyridin-3-yl)quinazolin-4(3H)-one, 9
(4-Oxo-3,4-dihydroquinazolin-6-yl)boronic acid (30 mg; 0.158 mmol; 1.0 equiv), 3-bromopyrazolo[1,5-a]pyridine (31 mg; 0.158 mmol; 1.0 equiv), K2CO3 (66 mg; 4.74 mmol; 3.0 equiv), bis(di-tert-butyl(4-dimethylaminophenyl)phosphine)dichloropalladium(II) (5.6 mg; 0.00790 mmol; 5 mol %), DME (1 mL; 0.079 M), and water (1 mL; 0.079 M) were charged to a microwave vial. The vial was sealed and purged of air with five rounds of vacuum and N2. The mixture was stirred at 150 °C for 1 h under microwave irradiation. The reaction mixture was cooled to <40 °C and diluted with CH2Cl2 (50 mL) and passed through a syringe filter, and solvent was removed under a vacuum. The residue was purified initially by column chromatography using a solvent system of 0–8% MeOH in CH2Cl2, followed by purification by preparative HPLC (see Supporting Information for details). 6-(Pyrazolo[1,5-a]pyridin-3-yl)quinazolin-4(3H)-one (14 mg; 0.0537 mmol; 34%) was isolated as a white solid. 1H NMR (500 MHz, MeOD) δ 8.53 (dt, J = 7.0, 1.1 Hz, 1H), 8.47 (d, J = 2.2 Hz, 1H), 8.25 (s, 1H), 8.07 (dd, J = 8.4, 2.2 Hz, 1H), 8.01 (s, 1H), 7.99 (dt, J = 9.1, 1.2 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.34 (ddd, J = 9.0, 6.7, 1.1 Hz, 1H), 6.94 (td, J = 6.9, 1.3 Hz, 1H). 13C NMR (126 MHz, MeOD) δ 161.94, 146.63, 144.09, 140.21, 137.18, 133.48, 132.45, 128.82, 127.64, 125.37, 123.09, 123.04, 117.31, 113.02, 111.42. HRMS (ESI) m/z calc C15H11N4O [M + H]+ 263.0927; found = 263.0929.
5-Methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 10
Following general procedure 2, quinolin-4-ylboronic acid (22 mg; 0.126 mmol; 1.0 equiv), PdCl2(PPh3)2 (4.4 mg; 0.0063 mmol; 0.05 equiv), 6-bromo-5-methylquinazolin-4(3H)-one (30 mg; 0.126 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (252 μL; 0.126 mmol; 1.0 equiv) and 1,4-dioxane (2 mL; 0.063 M) were used to yield 5-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (18 mg; 0.0627 mmol; 50%) as a white solid. Reaction temperature: 120 °C. Purification: 3–20% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.87 (d, J = 4.4 Hz, 1H), 8.13–8.10 (m, 1H), 7.96 (d, J = 3.7 Hz, 1H), 7.70 (ddd, J = 8.4, 6.8, 1.6 Hz, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.53–7.51 (m, 1H), 7.43 (ddd, J = 8.1, 6.7, 1.2 Hz, 1H), 7.37 (dd, J = 8.4, 1.5 Hz, 1H), 7.30–7.25 (m, 1H), 2.49 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.63, 150.23, 149.54, 148.41, 147.68, 144.71, 139.31, 136.93, 135.28, 129.98, 129.03, 127.89, 127.22, 125.67, 125.05, 121.85, 121.58, 19.67. HRMS (ESI) m/z calc C18H14N3O [M + H]+ 288.1132; found = 288.1144.
5-Methyl-6-(quinolin-5-yl)quinazolin-4(3H)-one, 11
Following general procedure 2, quinolin-5-ylboronic acid (22 mg; 0.126 mmol; 1.0 equiv), PdCl2(PPh3)2 (4.4 mg; 0.0063 mmol; 0.05 equiv), 6-bromo-5-methylquinazolin-4(3H)-one (30 mg; 0.126 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (252 μL; 0.126 mmol; 1.0 equiv) and 1,4-dioxane (2 mL; 0.063 M) were used to yield 5-methyl-6-(quinolin-5-yl)quinazolin-4(3H)-one (21 mg; 0.0731 mmol; 58%) was isolated as a white solid. Reaction temperature: 120 °C. Purification: 3–20% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.86 (dd, J = 4.2, 1.7 Hz, 1H), 8.13 (dt, J = 8.6, 1.1 Hz, 1H), 7.97 (s, 1H), 7.79 (dd, J = 8.6, 7.0 Hz, 1H), 7.76–7.72 (m, 1H), 7.64 (d, J = 8.3 Hz, 1H), 7.58 (d, J = 8.3 Hz, 1H), 7.42 (dd, J = 7.0, 1.2 Hz, 1H), 7.33 (dd, J = 8.5, 4.2 Hz, 1H), 2.50 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.37, 150.09, 147.72, 144.30, 139.89, 139.20, 138.41, 137.77, 136.31, 134.56, 129.33, 128.66, 127.78, 127.18, 125.01, 121.43, 120.96, 19.70. HRMS (ESI) m/z calc C18H14N3O [M + H]+ 288.1132; found = 288.1138.
3-Methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 12
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-3-methylquinazolin-4(3H)-one (34 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 3-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (27 mg; 0.0940 mmol; 65%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3/MeOD) δ 8.77 (d, J = 4.6 Hz, 1H), 8.31 (d, J = 2.0 Hz, 1H), 8.09 (s, 1H), 8.04–7.99 (m, 1H), 7.79 (dd, J = 8.3, 2.0 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.64 (t, J = 7.6 Hz, 2H), 7.46–7.39 (m, 1H), 7.31 (d, J = 4.6 Hz, 1H), 3.52 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 161.50, 149.40, 149.35, 147.93, 147.87, 147.51, 136.90, 135.46, 129.96, 128.89, 127.45, 127.33, 127.27, 126.40, 125.33, 121.84, 121.57, 34.11. HRMS (ESI) m/z calc C18H14N3O [M + H]+ 288.1132; found = 288.1124.
3-Methyl-6-(quinolin-5-yl)quinazolin-4(3H)-one, 13
Following general procedure 2, quinolin-5-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-3-methylquinazolin-4(3H)-one (34 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 3-methyl-6-(quinolin-5-yl)quinazolin-4(3H)-one (23 mg; 0.0801 mmol; 56%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3/MeOD) δ 8.78 (dd, J = 4.2, 1.7 Hz, 1H), 8.30 (d, J = 1.8 Hz, 1H), 8.14 (ddd, J = 8.6, 1.8, 1.0 Hz, 1H), 8.10 (d, J = 1.4 Hz, 1H), 8.04 (dt, J = 8.5, 1.1 Hz, 1H), 7.78 (dd, J = 8.3, 2.0 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.72 (dd, J = 8.6, 7.1 Hz, 1H), 7.49 (dd, J = 7.1, 1.3 Hz, 1H), 7.34 (ddd, J = 8.7, 4.3, 1.3 Hz, 1H), 3.56 (d, J = 1.4 Hz, 3H).13C NMR (126 MHz, CDCl3/MeOD) δ 161.58, 149.90, 147.74, 147.49, 147.29, 138.73, 138.44, 136.10, 134.47, 129.30, 128.63, 127.92, 127.44, 127.29, 126.47, 121.74, 121.45, 34.11. HRMS (ESI) m/z calc C18H14N3O [M + H]+ 288.1132; found = 288.113.
2-Methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 14
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-2-methylquinazolin-4(3H)-one (34 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 2-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (29 mg; 0.101 mmol; 70%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, MeOD) δ 8.90 (d, J = 4.5 Hz, 1H), 8.36 (d, J = 2.1 Hz, 1H), 8.14 (dt, J = 8.5, 1.0 Hz, 1H), 7.95 (dd, J = 8.4, 2.2 Hz, 1H), 7.93–7.91 (m, 1H), 7.83–7.78 (m, 2H), 7.59 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 7.51 (d, J = 4.5 Hz, 1H), 2.53 (s, 3H). 13C NMR (126 MHz, MeOD) δ 162.60, 155.65, 149.36, 148.31, 147.93, 147.88, 135.76, 135.69, 129.94, 128.56, 127.29, 126.97, 126.52, 126.38, 125.41, 121.59, 120.73, 20.69. HRMS (ESI) m/z calc C18H14N3O [M + H]+ 288.1132; found = 288.1133.
2-Methyl-6-(quinolin-5-yl)quinazolin-4(3H)-one, 15
Following general procedure 2, quinolin-5-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-2-methylquinazolin-4(3H)-one (34 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 2-methyl-6-(quinolin-5-yl)quinazolin-4(3H)-one (24 mg; 0.0836 mmol; 58%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, MeOD) δ 8.86 (dd, J = 4.3, 1.7 Hz, 1H), 8.30 (d, J = 2.0 Hz, 1H), 8.27 (ddd, J = 8.6, 1.7, 0.9 Hz, 1H), 8.11 (dt, J = 8.5, 1.1 Hz, 1H), 7.86 (dd, J = 8.4, 2.1 Hz, 1H), 7.82 (dd, J = 8.5, 7.1 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.60 (dd, J = 7.1, 1.2 Hz, 1H), 7.46 (dd, J = 8.6, 4.3 Hz, 1H), 2.51 (s, 3H). 13C NMR (126 MHz, MeOD) δ 162.79, 154.99, 149.84, 148.15, 147.72, 139.04, 137.35, 136.41, 134.78, 129.43, 128.28, 127.96, 127.22, 126.62, 126.43, 121.49, 120.65, 20.96. HRMS (ESI) m/z calc C18H14N3O [M + H]+ 288.1132; found = 288.1144.
3-(4-Morpholinophenyl)-6-(quinolin-4-yl)quinazolin-4(3H)-one, 16
Following general procedure 2, 6-bromo-3-(4-morpholinophenyl)quinazolin-4(3H)-one (333 mg; 0.867 mmol; 1.0 equiv), quinolin-4-yl boronic acid (170 mg; 0.867 mmol; 1.0 equiv), PdCl2(PPh3)2 (30 mg; 0.0433 mmol; 0.05 equiv), [0.5 M] Na2CO3(aq) (1.73 mL; 0.867 mmol; 1.0 equiv) and 1,4-dioxane (10 mL; 0.087 M) were used to yield 3-(4-morpholinophenyl)-6-(quinolin-4-yl)quinazolin-4(3H)-one (180 mg; 0.414 mmol; 48%) as pale purple solid. Reaction temperature: 150 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 9.00 (d, J = 4.4 Hz, 1H), 8.54 (dd, J = 1.8, 0.9 Hz, 1H), 8.25–8.20 (m, 2H), 7.95–7.94 (m, 2H), 7.92–7.89 (m, 1H), 7.78 (ddd, J = 8.3, 6.8, 1.4 Hz, 1H), 7.55 (ddd, J = 8.3, 6.8, 1.3 Hz, 1H), 7.43 (d, J = 4.4 Hz, 1H), 7.37–7.34 (m, 2H), 7.08–7.02 (m, 2H), 3.92–3.88 (m, 4H), 3.29–3.23 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 160.95, 151.67, 149.96, 148.70, 147.91, 147.22, 146.86, 137.35, 136.15, 135.63, 130.04, 129.59, 128.03, 127.95, 127.67, 127.10, 125.37, 122.62, 121.57, 115.91, 115.87, 66.73, 48.74. HRMS (ESI) m/z calc C27H23N4O2 [M + H]+ 435.1816; found = 435.1814.
3-Phenyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 17
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-3-phenylquinazolin-4(3H)-one (42 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 3-phenyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (37 mg; 0.106 mmol; 74%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.98 (d, J = 4.4 Hz, 1H), 8.54 (dd, J = 1.8, 0.9 Hz, 1H), 8.23 (s, 1H), 8.21 (dt, J = 8.6, 0.8 Hz, 1H), 7.96–7.95 (m, 2H), 7.90 (dd, J = 8.5, 1.4 Hz, 1H), 7.77 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.63–7.50 (m, 4H), 7.48–7.45 (m, 2H), 7.43 (d, J = 4.4 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 160.63, 149.82, 149.73, 148.50, 147.80, 146.83, 137.47, 137.24, 135.81, 129.77, 129.77, 129.74, 129.37, 128.04, 127.97, 127.21, 126.96, 126.42, 125.34, 122.57, 121.59. HRMS (ESI) m/z calc C23H16N3O [M + H]+ 350.1288; found = 350.1303.
3-Cyclohexyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 18
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-3-cyclohexylquinazolin-4(3H)-one (44 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv) and 1,4-dioxane (2 mL; 0.072 M) were used to yield 3-cyclohexyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (42 mg; 0.118 mmol; 82%) as a pale yellow glass. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.97 (d, J = 4.4 Hz, 1H), 8.48 (dd, J = 2.0, 0.6 Hz, 1H), 8.22 (s, 1H), 7.90 (dd, J = 8.3, 2.0 Hz, 1H), 7.87 (d, J = 8.1 Hz, 1H), 7.77 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.66 (ddd, J = 12.0, 8.3, 1.4 Hz, 1H), 7.54 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 7.47 (td, J = 7.6, 3.0 Hz, 1H), 7.41 (d, J = 4.4 Hz, 1H), 4.85 (tt, J = 12.3, 3.7 Hz, 1H), 2.09–2.02 (m, 2H), 1.97 (dt, J = 13.8, 3.6 Hz, 2H), 1.82 (d, J = 13.5 Hz, 1H), 1.69 (qd, J = 12.4, 3.4 Hz, 2H), 1.55 (qt, J = 13.1, 3.4 Hz, 2H), 1.34–1.24 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 160.55, 149.83, 149.75, 148.51, 147.41, 144.68, 136.87, 135.35, 132.02, 129.76, 128.59, 127.87, 127.60, 127.13, 126.47, 122.08, 121.58, 53.67, 32.63, 25.88, 25.24. HRMS (ESI) m/z calc C23H22N3O [M + H]+ 356.1758; found = 356.1768.
3-(3-Morpholinopropyl)-6-(quinolin-4-yl)quinazolin-4(3H)-one, 19
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-3-(3-morpholinopropyl)quinazolin-4(3H)-one (51 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv) and 1,4-dioxane (2 mL; 0.072 M) were used to yield 3-(3-morpholinopropyl)-6-(quinolin-4-yl)quinazolin-4(3H)-one (21 mg; 0.0525 mmol; 36%) as a pale yellow glass. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 8.93 (d, J = 4.4 Hz, 1H), 8.44 (d, J = 2.0 Hz, 1H), 8.22 (s, 1H), 8.18 (dd, J = 8.6, 1.2 Hz, 1H), 7.90 (dd, J = 8.4, 2.0 Hz, 1H), 7.88–7.84 (m, 2H), 7.76 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.53 (ddd, J = 8.4, 6.9, 1.3 Hz, 1H), 7.40 (d, J = 4.4 Hz, 1H), 4.12 (t, J = 6.7 Hz, 2H), 3.68 (t, J = 4.6 Hz, 4H), 2.42 (q, J = 6.4 Hz, 6H), 2.02 (p, J = 6.7 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 161.04, 149.65, 148.28, 147.93, 147.90, 147.25, 136.97, 135.48, 129.82, 129.48, 127.65, 127.46, 127.24, 126.43, 125.36, 122.21, 121.59, 66.85, 54.91, 53.34, 45.39, 24.85. HRMS (ESI) m/z calc C24H25N4O2 [M + H]+ 401.1972; found = 401.1984.
3-(4-(Dimethylamino)phenyl)-6-(quinolin-4-yl)quinazolin-4(3H)-one, 20
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-3-(4-(dimethylamino)phenyl)quinazolin-4(3H)-one (49 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 3-(4-(dimethylamino)phenyl)-6-(quinolin-4-yl)quinazolin-4(3H)-one (45 mg; 0.115 mmol; 80%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 9.00 (d, J = 4.4 Hz, 1H), 8.54 (dd, J = 1.6, 1.0 Hz, 1H), 8.23–8.20 (m, 2H), 7.93–7.92 (m, 2H), 7.91 (ddd, J = 8.5, 1.4, 0.6 Hz, 1H), 7.76 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.54 (ddd, J = 8.3, 6.9, 1.3 Hz, 1H), 7.42 (d, J = 4.4 Hz, 1H), 7.29 (dd, J = 8.8, 2.0 Hz, 2H), 6.85–6.80 (m, 2H), 3.03 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 161.11, 150.73, 149.98, 148.71, 147.98, 147.65, 146.91, 137.15, 135.47, 130.03, 129.55, 128.03, 127.88, 127.48, 127.06, 126.44, 125.78, 125.42, 122.69, 121.58, 112.50, 40.47. HRMS (ESI) m/z calc C25H21N4O [M + H]+ 393.1710; found = 393.1712.
2,5-Dimethyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 21
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-2,5-dimethylquinazolin-4(3H)-one (36 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 2,5-dimethyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (23 mg; 0.0764 mmol; 53%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, MeOD) δ 8.89 (d, J = 4.4 Hz, 1H), 8.12 (dt, J = 8.4, 0.9 Hz, 1H), 7.75 (ddd, J = 8.4, 6.7, 1.5 Hz, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 8.4 Hz, 1H), 7.48 (ddd, J = 8.1, 6.7, 1.2 Hz, 1H), 7.44–7.41 (m, 1H), 7.33 (d, J = 4.4 Hz, 1H), 2.51 (s, 3H), 2.46 (s, 3H). 13C NMR (126 MHz, MeOD) δ 163.54, 154.82, 150.45, 149.45, 148.80, 147.55, 139.02, 136.03, 135.27, 130.05, 128.75, 127.39, 127.26, 125.78, 124.12, 121.97, 119.44, 20.65, 19.48. HRMS (ESI) m/z calc C19H16N3O [M + H]+ 302.1288; found = 302.1282.
2-Ethyl-5-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 22
Following general procedure 2, quinolin-4-ylboronic acid (25 mg; 0.144 mmol; 1.0 equiv), PdCl2(PPh3)2 (5.1 mg; 0.00721 mmol; 0.05 equiv), 6-bromo-2-ethyl-5-methylquinazolin-4(3H)-one (38 mg; 0.144 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (288 μL; 0.144 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.072 M) were used to yield 2-ethyl-5-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (28 mg; 0.0889 mmol; 62%) as a white solid. Reaction temperature: 130 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 11.93 (s, 1H), 9.04 (d, J = 4.3 Hz, 1H), 8.24 (dt, J = 8.6, 0.9 Hz, 1H), 7.77 (ddd, J = 8.4, 6.0, 2.2 Hz, 1H), 7.69 (dd, J = 8.3, 0.7 Hz, 1H), 7.59 (d, J = 8.3 Hz, 1H), 7.53–7.47 (m, 2H), 7.34 (d, J = 4.3 Hz, 1H), 2.83 (q, J = 7.6 Hz, 2H), 2.63 (s, 3H), 1.46 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.23, 158.07, 151.32, 150.00, 148.29, 148.05, 139.05, 136.32, 135.55, 129.88, 129.67, 127.36, 126.99, 125.78, 125.26, 121.92, 119.41, 28.83, 19.77, 11.49. HRMS (ESI) m/z calc C20H18N3O [M + H]+ 316.1445; found = 316.1449.
2-Benzyl-5-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one, 23
Following general procedure 2, quinolin-4-ylboronic acid (12 mg; 0.0692 mmol; 1.0 equiv), PdCl2(PPh3)2 (5 mg; 0.00712 mmol; 0.1 equiv), 2-benzyl-6-bromo-5-methylquinazolin-4(3H)-one (23 mg; 0.0692 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (138 μL; 0.0692 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.035 M) were used to yield 2-benzyl-5-methyl-6-(quinolin-4-yl)quinazolin-4(3H)-one (17 mg; 0.0451 mmol; 65%) as a white solid. Reaction temperature: 150 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, DMSO) δ 12.39 (s, 1H), 9.00 (d, J = 4.3 Hz, 1H), 8.13 (d, J = 8.4 Hz, 1H), 7.80 (ddd, J = 8.4, 6.8, 1.4 Hz, 1H), 7.58 (d, J = 0.7 Hz, 2H), 7.56–7.52 (m, 1H), 7.44–7.41 (m, 3H, 14), 7.39 (ddd, J = 8.5, 1.6, 0.7 Hz, 1H), 7.38–7.34 (m, 2H), 7.30–7.25 (m, 1H), 3.96 (s, 2H), 2.43 (s, 3H). 13C NMR (126 MHz, DMSO) δ 163.35, 156.81, 151.13, 150.75, 148.24, 147.58, 138.26, 137.06, 135.87, 135.32, 130.10, 130.00, 129.32, 128.99, 127.84, 127.29, 127.10, 125.95, 125.49, 122.53, 119.88, 40.31, 19.80. HRMS (ESI) m/z calc C25H20N3O [M + H]+ 378.1601; found = 378.1608.
3,8-Dimethyl-7-(quinolin-4-yl)isoquinolin-1(2H)-one, 24
Following general procedure 2, quinolin-4-ylboronic acid (38 mg; 0.218 mmol; 1.1 equiv), PdCl2(PPh3)2 (7.0 mg; 0.0100 mmol; 0.05 equiv), 7-bromo-3,8-dimethyl-2H-isoquinolin-1-one (50 mg; 0.198 mmol; 1.0 equiv), [0.5 M] Na2CO3(aq) (397 μL; 0.198 mmol; 1.0 equiv), and 1,4-dioxane (2 mL; 0.099 M) were used to yield 3,8-dimethyl-7-(quinolin-4-yl)isoquinolin-1(2H)-one (36 mg; 0.120 mmol; 61% yield) as a white solid. Reaction temperature: 150 °C. Purification: 0–8% MeOH in CH2Cl2. 1H NMR (500 MHz, CDCl3) δ 10.48 (s, 1H), 9.01 (d, J = 4.3 Hz, 1H), 8.21 (dt, J = 8.4, 0.9 Hz, 1H), 7.75 (ddd, J = 8.4, 6.3, 1.9 Hz, 1H), 7.53–7.45 (m, 2H), 7.41–7.40 (m, 2H), 7.32 (d, J = 4.3 Hz, 1H), 6.32 (d, J = 1.3 Hz, 1H), 2.65 (s, 3H), 2.37 (d, J = 1.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 165.32, 150.08, 148.72, 148.37, 140.67, 139.68, 138.01, 135.92, 133.52, 129.84, 129.50, 127.52, 126.79, 126.00, 123.58, 122.87, 121.94, 104.89, 20.03, 18.89. HRMS (ESI) m/z calc C20H17N2O [M + H]+ 301.1335, found = 301.1328.
2-Benzyl-6-bromo-5-methylquinazolin-4(3H)-one, 26
2-Phenylacetonitrile (500 mg; 4.27 mmol; 1.0 equiv) and hydroxylamine [50% in water] (424 μL; 6.41 mmol; 1.5 equiv) were stirred at 120 °C for 2 h. The mixture was cooled to <40 °C, and 6-amino-3-bromo-2-methylbenzoic acid (985 mg; 4.27 mmol; 1.0 equiv) was added. The mixture was heated to 150 °C, stirred for 2 h, and cooled to <40 °C. EtOH (20 mL) was added to the reaction mixture which induced precipitation. The solvent was decanted three times from EtOH, and the solid residue was dried under high vacuum. 2-Benzyl-6-bromo-5-methylquinazolin-4(3H)-one (341 mg; 1.04 mmol; 24%) was isolated at a white solid. 1H NMR (500 MHz, DMSO) δ 12.15 (s, 1H), 7.91 (d, J = 8.7 Hz, 1H), 7.38–7.34 (m, 3H), 7.34–7.30 (m, 2H), 7.26–7.22 (m, 1H), 3.89 (s, 2H), 2.90 (s, 3H). 13C NMR (126 MHz, DMSO) δ 157.94, 156.89, 150.36, 138.88, 137.89, 136.83, 129.32, 128.96, 127.27, 127.14, 122.99, 121.14, 40.81, 21.54. HRMS (ESI) m/z calc C16H14BrN2O [M + H]+ 329.0284; found = 329.0284.
3-(4-Bromo-3-methylphenyl)-2-methylacrylic acid, 27
A mixture of 4-bromo-3-methylbenzaldehyde (4.98 g, 25 mmol), methylmalonic acid (5.90 g, 50 mmol), and piperidine (4.94 mL, 50 mmol) in pyridine (30 mL) was heated at reflux for 24 h. The mixture was cooled and added to a mixture of ice (ca. 125 g) and conc. HCl (70 mL). The precipitate formed was filtered off and washed with cold water. The product was dried under vacuum to afford 3-(4-bromo-3-methylphenyl)-2-methylacrylic acid (6.1 g, 64.3% yield) as a waxy solid. 1H NMR (500 MHz, CDCl3) δ 11.77 (s, 1H), 7.74 (d, J = 1.6 Hz, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.30 (d, J = 2.1 Hz, 1H), 7.14 (dd, J = 8.3, 2.2 Hz, 1H), 2.45 (s, 3H), 2.14 (d, J = 1.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.72, 139.99, 138.10, 134.72, 132.44, 132.11, 128.49, 127.94, 125.48, 22.98, 13.79.
3-(4-Bromo-3-methylphenyl)-2-methylacryloyl azide, 28
To a suspension of 3-(4-bromo-3-methylphenyl)-2-methylacrylic acid (4.80 ;, 18.8 mmol; 1.0 equiv) in acetone (100 mL) at 0 °C was added triethylamine (3.41 mL; 24.5 mmol; 1.3 equiv). A clear solution was formed. Then, ethyl chloroformate (2.16 mL; 22.6 mmol; 1.2 equiv) was added, and a thick suspension was formed. The suspension was allowed to warm to room temperature. After 2 h, sodium azide (1.83 g; 28.2 mmol; 1.5 equiv) in water (15 mL) was added dropwise, and the suspension was stirred for a further 2 h. The suspension was cooled down and diluted with water (50 mL). Ethyl acetate (200 mL) was added, the layers were separated, and the aqueous layer was extracted with more ethyl acetate (2 × 50 mL). The organic were washed with sat. aq. NaHCO3 (2 × 50 mL), dried (MgSO4), filtered and concentrated (without heating) to yield the product as a white solid (5.1 g, 97% yield). 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 1.5 Hz, 1H), 7.58 (d, J = 8.3 Hz, 1H), 7.29–7.28 (m, 1H), 7.12 (dd, J = 8.3, 2.3 Hz, 1H), 2.44 (s, 3H), 2.12 (d, J = 1.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.36, 139.94, 138.23, 134.36, 132.54, 132.22, 129.99, 128.59, 125.92, 22.98, 13.87.
7-Bromo-3,8-dimethyl-2H-isoquinolin-1-one, 29 and 7-bromo-3,6-dimethyl-2H-isoquinolin-1-one, 30
A suspension of 3-(4-bromo-3-methylphenyl)-2-methylacryloyl azide (4.76 g; 17.0 mmol; 1.0 equiv) in 1,2-dichlorobenzene (70 mL) was heated to 140 °C for 1 h. Then, iodine (431.5 mg; 1.7 mmol; 0.1 equiv) was added, and the mixture was heated at reflux (ca. 180 °C) for 6 h and cooled to room temperature. The solution was stirred overnight at room temperature, and the solid formed was filtered and washed with 20 mL of cyclohexane. The solid was dried for 1 h at 50 °C and 20 Torr to give a mixture of 29 and 30 (2.16 g; 50% yield) in a ratio 3:2 (1H NMR). Normal phase flash chromatography of the mixture (0–25% EtOAc in cyclohexane) gave 116 mg of pure 29, 660 mg of mixture and 260 mg of pure 30. For 29: 1H NMR (500 MHz, DMSO) δ 11.24 (s, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.29 (d, J = 8.5 Hz, 1H), 6.27 (dd, J = 2.0, 1.1 Hz, 1H), 2.95 (s, 3H), 2.16 (d, J = 1.1 Hz, 3H). 13C NMR (126 MHz, DMSO) δ 163.21, 140.03, 139.80, 139.40, 136.12, 125.89, 124.19, 123.08, 103.48, 21.76, 18.77. HRMS (ESI) m/z calc for C11H1179BrNO [M + H]+ 252.0019, found = 252.0014. For 30: 1H NMR (DMSO, 500 MHz) δ 11.30 (br-s, 1H), 8.20 (s, 1H), 7.51 (a, 1H), 6.26 (s, 1H), 2.43 (s, 3H), 2.18 (s, 3H). 13C NMR (DMSO, 126 MHz) δ 161.2, 141.8, 139.5, 137.6, 129.6, 127.4, 123.8, 121.1, 102.0, 22.8, 18.8. HRMS (ESI) m/z calc for C11H1179BrNO [M + H]+ 252.0019, found = 252.0015.
Acknowledgments
L.H. and J.M. were funded by Wellcome Trust [Grant Number 090171/Z/09/Z]. We acknowledge NHS funding to the NIHR Biomedical Research Centre and funding from Cancer Research UK [Grant Number C309/A11566]. S.V. thanks the Spanish Ministerio de Educación, Cultura y Deporte for a “Salvador de Madariaga” mobility grant. C.J. and D.C. acknowledge support from Children with Cancer UK, Abbie’s Army and the DIPG Collaborative, the Lyla Nsouli Foundation and Lucas’ Legacy. The authors thank Diamond Light Source for beamtime (proposal mx10619), as well as the staff of beamlines I02, I03, and I04 for assistance with crystal testing and data collection. The SGC is a registered charity (Number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD Grant No. 115766], Janssen, MSD, Merck KGaA., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust [106169/ZZ14/Z].
Glossary
Abbreviations Used
- ALK
activin receptor-like kinase
- DIPG
diffuse intrinsic pontine glioma
- FKBP12
FK506 binding protein 12
- SMAD
small mothers against decapantaplegic
- FOP
fibrodysplasia ossificans progressiva
- RAF
rapidly accelerated fibrosarcoma
- PDGFR
platelet-derived growth factor receptor
- BMP
bone morphogenic protein
- ID1
inhibitor of DNA binding 1
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b00782.
Accession Codes
The PDB ID codes for 11, 16, and 21 bound to ALK2 are 6GI6, 6GIN and 6GIP, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
Supplementary Material
References
- Vogt J.; Traynor R.; Sapkota G. P. The specificities of small molecule inhibitors of the TGFß and BMP pathways. Cell. Signalling 2011, 23 (11), 1831–1842. 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- Chaikuad A.; Alfano I.; Kerr G.; Sanvitale C. E.; Boergermann J. H.; Triffitt J. T.; Von Delft F.; Knapp S.; Knaus P.; Bullock A. N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287 (44), 36990–36998. 10.1074/jbc.M112.365932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsell S. J.; Idone V.; Wolken D. M. A.; Huang L.; Kim H. J.; Wang L.; Wen X.; Nannuru K. C.; Jimenez J.; Xie L.; Das N.; Makhoul G.; Chernomorsky R.; D’Ambrosio D.; Corpina R. A.; Schoenherr C. J.; Feeley K.; Yu P. B.; Yancopoulos G. D.; Murphy A. J.; Economides A. N. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7 (303), 303ra137. 10.1126/scitranslmed.aac4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. R.; Vinci M.; Bullock A. N.; Jones C. ACVR1 mutations in DIPG: lessons learned from FOP. Cancer Res. 2014, 74 (17), 4565–4570. 10.1158/0008-5472.CAN-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren K. E. Diffuse intrinsic pontine glioma: poised for progress. Front. Oncol. 2012, 2 (December), 205. 10.3389/fonc.2012.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P. B.; Hong C. C.; Sachidanandan C.; Babitt J. L.; Deng D. Y.; Hoyng S. a; Lin H. Y.; Bloch K. D.; Peterson R. T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4 (1), 33–41. 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuny G. D.; Yu P. B.; Laha J. K.; Xing X.; Liu J.-F.; Lai C. S.; Deng D. Y.; Sachidanandan C.; Bloch K. D.; Peterson R. T. Structure-activity relationship study of bone morphogenetic protein (BMP) signaling inhibitors. Bioorg. Med. Chem. Lett. 2008, 18 (15), 4388–4392. 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohedas A. H.; Xing X.; Armstrong K. a.; Bullock A. N.; Cuny G. D.; Yu P. B. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem. Biol. 2013, 8 (6), 1291–1302. 10.1021/cb300655w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers D. W.; Frist A. Y.; Lindsley C. W.; Hong C. C.; Hopkins C. R. Synthesis and structure-activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of dorsomorphin: the discovery of ML347 as an ALK2 versus ALK3 selective MLPCN probe. Bioorg. Med. Chem. Lett. 2013, 23 (11), 3248–3252. 10.1016/j.bmcl.2013.03.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- i Vogt J.; Traynor R.; Sapkota G. P. The specificities of small molecule inhibitors of the TGFß and BMP pathways. Cell. Signalling 2011, 23 (11), 1831–1842. 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]; ii Sinha S.; Mundy C.; Bechtold T.; Sgariglia F.; Ibrahim M. M.; Billings P. C.; Carroll K.; Koyama E.; Jones K. B.; Pacifici M. Unsuspected osteochondroma-like outgrowths in the cranial base of Hereditary Multiple Exostoses patients and modeling and treatment with a BMP antagonist in mice. PLoS Genet. 2017, 13 (4), e1006742. 10.1371/journal.pgen.1006742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanvitale C. E.; Kerr G.; Chaikuad A.; Ramel M.-C.; Mohedas A. H.; Reichert S.; Wang Y.; Triffitt J. T.; Cuny G. D.; Yu P. B.; Hill C. S.; Bullock A. N. A new class of small molecule inhibitor of BMP signaling. PLoS One 2013, 8 (4), e62721. 10.1371/journal.pone.0062721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohedas A.; Wang Y.; Sanvitale C. E.; Canning P.; Choi S.; Xing X.; Bullock A. N.; Cuny G. D.; Yu P. B. Structure–activity relationship of 3,5-diaryl-2-aminopyridine ALK2 inhibitors reveals unaltered binding affinity for fibrodysplasia ossificans progressiva causing mutants. J. Med. Chem. 2014, 57, 7900–7915. 10.1021/jm501177w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boys M. L.; Bian F.; Kramer J. B.; Chio C. L.; Ren X. D.; Chen H.; Barrett S. D.; Sexton K. E.; Iula D. M.; Filzen G. F.; Nguyen M. N.; Angell P.; Downs V. L.; Wang Z.; Raheja N.; Ellsworth E. L.; Fakhoury S.; Bratton L. D.; Keller P. R.; Gowan R.; Drummond E. M.; Maiti S. N.; Hena M. a.; Lu L.; McConnell P.; Knafels J. D.; Thanabal V.; Sun F.; Alessi D.; McCarthy A.; Zhang E.; Finzel B. C.; Patel S.; Ciotti S. M.; Eisma R.; Payne N. a.; Gilbertsen R. B.; Kostlan C. R.; Pocalyko D. J.; Lala D. S. Discovery of a series of 2-(1H-pyrazol-1-yl)pyridines as ALK5 inhibitors with potential utility in the prevention of dermal scarring. Bioorg. Med. Chem. Lett. 2012, 22 (10), 3392–3397. 10.1016/j.bmcl.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Gellibert F.; Woolven J.; Fouchet M.-H.; Mathews N.; Goodland H.; Lovegrove V.; Laroze A.; Nguyen V.-L.; Sautet S.; Wang R.; Janson C.; Smith W.; Krysa G.; Boullay V.; De Gouville A.-C.; Huet S.; Hartley D. Identification of 1,5-naphthyridine derivatives as a novel series of potent and selective TGF-β type I receptor inhibitors. J. Med. Chem. 2004, 47, 4494–4506. 10.1021/jm0400247. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A.; Pennington L. D. Structure–brain exposure relationships. J. Med. Chem. 2006, 49 (26), 7559–7583. 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- Larrivée B.; Prahst C.; Gordon E.; del Toro R.; Mathivet T.; Duarte A.; Simons M.; Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev. Cell 2012, 22, 489–500. 10.1016/j.devcel.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adib M.; Ansari S.; Mohammadi A.; Bijanzadeh H. R. A novel, one-pot, solvent-, and catalyst-free synthesis of 2-aryl/alkyl-4 (3H)-quinazolinones. Tetrahedron Lett. 2010, 51 (1), 30–32. 10.1016/j.tetlet.2009.06.034. [DOI] [Google Scholar]
- Gensler W. J.; Berman E. Decarboxylative condensation. α-alkylcinnamic acids from aromatic aldehydes and alkylmalonic acids. J. Am. Chem. Soc. 1958, 80, 4949–4954. 10.1021/ja01551a048. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.