

Abstract
Lytic polysaccharide monooxygenases (LPMOs) are copper-dependent enzymes that catalyze the oxidative cleavage of polysaccharides such as cellulose and chitin, a feature that makes them key tools in industrial biomass conversion processes. The catalytic domains of a considerable fraction of LPMOs and other carbohydrate-active enzymes (CAZymes) are tethered to carbohydrate-binding modules (CBMs) by flexible linkers. These linkers preclude X-ray crystallographic studies, and the functional implications of these modular assemblies remain partly unknown. Here, we used NMR spectroscopy to characterize structural and dynamic features of full-length modular ScLPMO10C from Streptomyces coelicolor. We observed that the linker is disordered and extended, creating distance between the CBM and the catalytic domain and allowing these domains to move independently of each other. Functional studies with cellulose nanofibrils revealed that most of the substrate-binding affinity of full-length ScLPMO10C resides in the CBM. Comparison of the catalytic performance of full-length ScLPMO10C and its isolated catalytic domain revealed that the CBM is beneficial for LPMO activity at lower substrate concentrations and promotes localized and repeated oxidation of the substrate. Taken together, these results provide a mechanistic basis for understanding the interplay between catalytic domains linked to CBMs in LPMOs and CAZymes in general.
Keywords: cellulose, copper monooxygenase, carbohydrate-binding module, carbohydrate-active enzyme, LPMO, lytic polysaccharide monooxygenase, nanofibril, protein linker, biotechnology, hydrolytic enzyme
Introduction
A biobased economy strives to derive biofuels, biomaterials, and commodity chemicals from biocatalytically processed biomass (1). Although cellulose is the most abundant form of biomass, its crystalline nature affects enzyme accessibility and hence the efficiency and costs of biochemical conversion processes (2). A major breakthrough came with the discovery of lytic polysaccharide monooxygenases (LPMOs)2 (3–6), which are monocopper enzymes that catalyze oxidative cleavage of glycosidic bonds in crystalline regions of polysaccharides such as chitin and cellulose (4, 7–11). By acting on crystalline regions, LPMOs generate new access points for hydrolytic enzymes such as endoglucanases and processive cellobiohydrolases (3–5, 11, 12). Furthermore, it is conceivable that LPMO action promotes the dissociation of cellobiohydrolases, which is known to be rate-limiting under many conditions, e.g. by removal of obstacles (13–17). The synergistic action between LPMOs and cellulases results in a significant enhancement in the efficiency of biomass saccharification and has led to the inclusion of LPMOs in commercial enzyme mixtures for degradation of lignocellulosic biomass (18, 19).
Like many other industrially important cellulose-degrading enzymes, a considerable fraction of LPMOs contain a carbohydrate-binding module (CBM). Such CBMs may be connected to the catalytic domain through a variety of linkers, differing in length, sequence, and flexibility (20). These linkers often contain regions of low sequence complexity and are predicted to be extended and flexible. The nature of these linkers hampers structural studies of the full-length proteins, although such studies could yield important insights into the interplay between the domains, substrate binding, and overall enzyme functionality. This lack of information exists for LPMOs and for carbohydrate-active enzymes in general. In an attempt to bridge this knowledge gap, we used NMR spectroscopy to characterize the structure and dynamics of a cellulose-active modular LPMO.
ScLPMO10C (previously known as CelS2), is an LPMO from Streptomyces coelicolor A3 (2) that cleaves cellulose by oxidizing C1 in the susceptible β-1,4-glycosidic bond, thus generating aldonic acids (7). Full-length ScLPMO10C is composed of an N-terminal catalytic domain (hereafter called ScAA10; residues 35–234) connected by a linker region (30 residues; residues 235–264) to a C-terminal family 2 carbohydrate-binding module (hereafter called ScCBM2; residues 265–364). The X-ray structure of the ScAA10 domain (Protein Data Bank (PDB) code 4OY7) depicts the characteristic LPMO β-sandwich fold with a densely packed hydrophobic core and a flat substrate-binding surface that includes the exposed catalytic copper site (21). The copper ion is coordinated by a histidine brace composed of the side chain (Nδ1) and the amino nitrogen of the N-terminal histidine (His35) and the side chain (Nϵ2) of His144. The linker region (Fig. 1A) consists of a Gly-rich part starting in the C-terminal end of the ScAA10 domain that is followed by a Pro-Thr-Asp–rich part and a Gly-Ser dyad just in front of the ScCBM2 domain. ScCBM2 belongs to the CBM2a subfamily whose members are known to bind cellulose and for which there is structural information (22–24). The presence of ScCBM2, the structure of which is not known, significantly enhances ScLPMO10C binding to cellulose (25) and results in higher yields of oxidized products compared with a truncated version comprising only the catalytic domain (21). Despite additional studies on LPMOs (26–28), the mechanism through which CBMs enhance product yields and the interactions among CBMs, linker regions, and catalytic domains are not well-understood.
Figure 1.
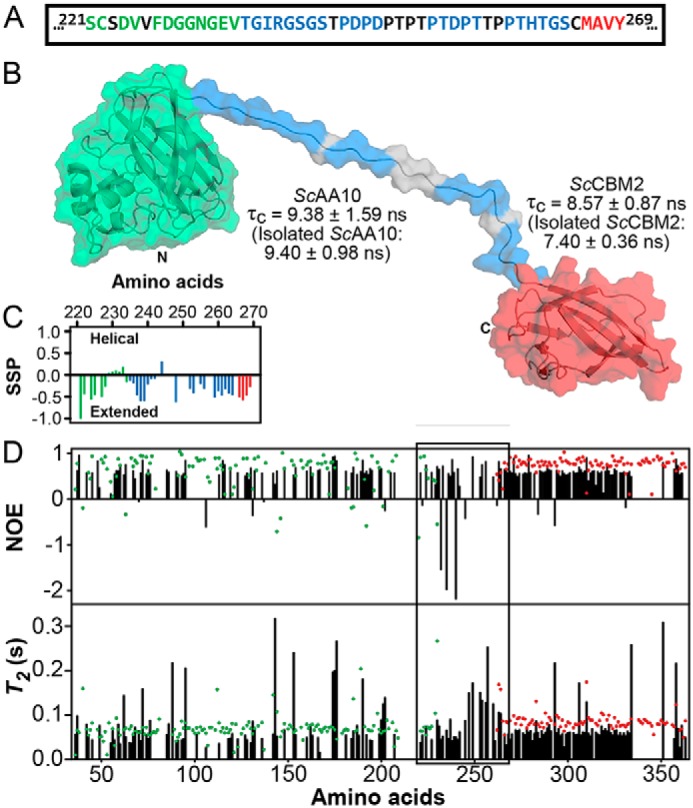
Dynamic features of ScLPMO10C. A shows the amino acid sequences of the C terminus of ScAA10 (green; residues 221–234), the linker region (blue; residues 235–264), and the N terminus of ScCBM2 (red; residues 265–269). Amino acids in black do not have their chemical shifts assigned. B shows a representative model of full-length ScLPMO10C. The N and C termini are labeled as are the ScAA10 (green) and ScCBM2 (red) domains with their respective τc. The blue colored patches on the linker region correspond to the residues for which NMR assignments are available. C shows the secondary structure propensity (SSP) of the linker region (blue) and adjacent C-terminal residues of ScAA10 (green) and N-terminal residues of ScCBM2 (red) derived from the secondary chemical shifts for ScLPMO10C. D shows 1H-15N NOEs and T2 relaxation times for full-length ScLPMO10C (black bars) and isolated ScAA10 (green dots) and ScCBM2 (red dots). The 221–269 region, including the linker, 235–264 region, is indicated by a box.
We have used NMR to investigate structural and dynamic aspects of full-length ScLPMO10C in solution. As a result, we have solved the first NMR structure of an LPMO-associated CBM (ScCBM2) and generated a structural model for full-length ScLPMO10C by combining the structures of ScCBM2 and ScAA10 with the dihedral angle constraints derived from the secondary chemical shift data for the linker region. This model was complemented with NMR relaxation data (T1, T2, and 1H-15N NOE) to provide a description of the dynamic features of ScLPMO10C. Furthermore, we have used NMR to map the substrate interaction surface of ScAA10 and ScCBM2 binding to cellulose nanofibrils and have compared the catalytic performance of full-length ScLPMO10C and isolated ScAA10. Taken together, the results provide a comprehensive, experimentally supported picture of the interplay among the catalytic domain, flexible linker, and CBM and its impact on LPMO functionality.
Results
Structure and dynamics of full-length ScLPMO10C
A model of full-length ScLPMO10C (Fig. 1B) was generated from X-ray crystal diffraction data (21) (for the ScAA10 domain; PDB code 4OY7) and NMR data (29) for the linker region (Biomagnetic Resonance Data Bank accession number 27078) and the ScCBM2 domain (Fig. S1). The result was an ensemble showing large conformational variation as a result of the low structural restriction in the linker region. The ensemble of conformers provides an experimentally supported picture of the overall conformational dynamics of ScLPMO10C (see Figs. 1 and 6 and Videos S1 and S2). Assignment of secondary structure based on chemical shifts (30) analyzed using TALOS-N (31) indicated that the linker has an extended conformation (Fig. 1C), which was confirmed by dynamic light scattering. Indeed, the average radius of gyration for the model ensemble calculated by YASARA (32) was 40 ± 10 Å, and the z-average hydrodynamic radius determined by dynamic light scattering was 35 ± 9.5 Å (Fig. S2); a hydrodynamic radius of about 25 Å would be expected for a spherical protein of similar mass. The linker region of ScLPMO10C is evidently disordered as its amide proton chemical shifts are distributed in a narrow region (8.0–8.6 ppm) of the 15N HSQC spectrum (Fig. S3B), showing narrower line widths (Fig. S3A) compared with signals from the ScAA10 and ScCBM2 domains.
Figure 6.

Illustration depicting cellulose oxidation by ScLPMO10C. ScCBM2 (orange) is docked on a cellulose surface (green) and tethered by the linker (cyan) to the copper-loaded LPMO domain (blue). The brown shadow indicates the theoretical area of ∼1300 Å2, corresponding to about 300 glucose residues, that the LPMO domain can sample. Random oxidation events that may have occurred are indicated by a yellow glow. See also Video S2.
To investigate the global mobility features of ScLPMO10C, rotational correlation times (τc) were calculated for isolated ScAA10 and ScCBM2 and full-length ScLPMO10C (Fig. 1B) based on the ratio between the T1 (Fig. S4) and T2 (Fig. 1D) relaxation times (33). τc is a measure of the overall tumbling of a protein, and it increases proportionally with molecular weight. We found that the τc values for the isolated domains (ScAA10, 9.40 ± 0.98 ns; ScCBM2, 7.40 ± 0.36 ns) are not significantly different from the values observed when the domains are linked together in full-length ScLPMO10C (ScAA10, 9.38 ± 1.59 ns; ScCBM2, 8.57 ± 0.87 ns). Notably, these τc values are significantly lower than the ∼17 ns that would be expected for a 34.5-kDa globular protein (34), suggesting that the domains tumble independently of each other when tethered together in ScLPMO10C by the flexible linker. NMR relaxation data support this by showing that the linker displays features of flexibility in the ps–ns timescale in the form of decreased 1H-15N NOEs and increased T2 values (Fig. 1D). Furthermore, the 1H-15N NOEs and the T2 values show that the dynamic features of the individual ScAA10 and ScCBM2 domains do not change when they are linked together.
Cellulose binding
Interactions of 15N-labeled ScAA10 and 15N-labeled ScCBM2 with cellulose nanofibrils were probed by measuring the changes in amide H and NH signal intensities in 15N HSQC spectra upon addition of cellulose nanofibrils to each of the protein samples (Fig. S5). The largest reduction of signal intensities is expected to occur at the cellulose-binding surface as substrate binding reduces protein mobility, resulting in signal broadening and decrease of intensity (35). Both proteins showed decreased signal intensities for residues clustered on their putative binding surfaces (Figs. 2 and S5), in particular around the active-site histidines (His35 and His144) and Tyr79 for ScAA10 and around the conserved tryptophans (Trp275 and Trp312) and His331 for ScCBM2. The decrease in signal intensity for all the affected residues was on average almost 10 times higher for ScCBM2 compared with ScAA10, indicating that cellulose binds more strongly to the CBM compared with the catalytic domain (Fig. S5).
Figure 2.
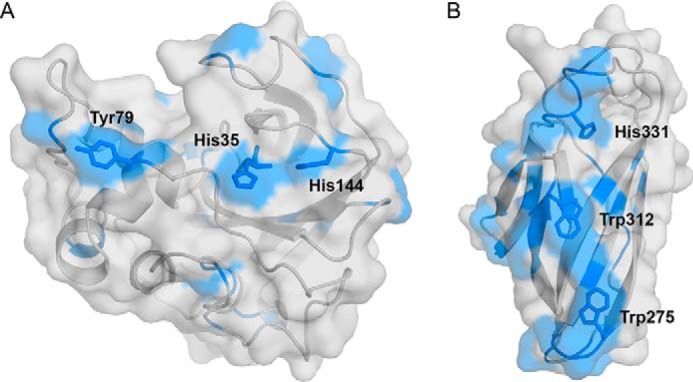
Experimental assessment of the interaction of ScAA10 (A) and ScCBM2 (B) with cellulose nanofibrils. Residues showing normalized amide signal intensities lower than 1 for ScAA10 and lower than 0.1 for ScCBM2 (see Fig. S5) in the presence of cellulose nanofibrils are colored blue. All residues meeting this criterium are visible in the figure, underpinning the clustering of these residues around the putative substrate-binding surfaces (ScAA10, His35, His144, and Tyr79; ScCBM2, Trp275, Trp312, and His331). These experiments were carried out at pH 5.5 and 25 °C.
Cellulose binding was also assessed by measuring the free–bound enzyme equilibrium in reactions with Avicel® (Fig. 3). The substrate-binding capacity (Bmax) for the full-length enzyme was 2.8 ± 0.2 μmol/g of Avicel, and the equilibrium dissociation constant (Kd) was 7.8 ± 1.0 μm. As only minimal binding was detected for the isolated ScAA10 domain (Fig. 3A), the obtained values primarily reflect the binding strength of ScCBM2. Indeed, the isolated CBM2 domain and the full-length protein showed similar binding properties (Fig. 3A).
Figure 3.

Binding of full-length ScLPMO10C, ScCBM2, and ScAA10 to Avicel. A shows binding of ScLPMO10C (solid line), ScCBM2 (long-dashed lines), and ScAA10 (short-dashed lines) to Avicel. B shows a plot for the binding data for ScLPMO10C. Pbound corresponds to bound protein (μmol/g of Avicel), and Pfree corresponds to nonbound protein (μm). These experiments were carried out at pH 7.0 and 22 °C. Each point represents the average of values obtained in three independent experiments. For some of the data points, the error bars (showing ±S.D.) are too small to be seen.
Cellulose degradation
To investigate the effect of the CBM2 on cellulose degradation, Cu(II)-loaded ScLPMO10C and ScAA10 were incubated with 10 g/liter Avicel in the presence of 1 mm ascorbic acid for 24 h (Fig. S6). As observed before (21), both enzyme variants yielded aldonic acids with a degree of polymerization ranging from 2 to 9 with yields after 24 h being almost 10 times higher for the full-length enzyme (Fig. S6).
It has recently been shown that, in the case of LPMOs, the effect of the CBM is not only due to the generally accepted targeting effect (i.e. increased substrate binding (36)). In LPMOs, substrate binding protects the enzyme from auto-oxidative inactivation, which implies that weaker substrate binding as a result of CBM removal leads to decreased enzyme stability (28, 37). For the same reasons, at nonsaturating substrate concentrations, the substrate concentration will not only affect LPMO activity but also stability. Another complicating factor in the study of CBM functionality resides in the payoff between CBM-mediated substrate affinity and possible negative effects of the CBM related to low off-rates and/or nonproductive binding (38). To overcome these complex issues and to gain true insight into the role of the CBM, reactions with ScLPMO10C and ScAA10 were set up with varying concentrations of Avicel (2–40 g/liter), and formation of soluble products was monitored over time (Fig. 4). At the last time point (60 min), we analyzed the total amounts of oxidized sites (i.e. soluble and insoluble) (Fig. 4, F and G) and the product distributions in the soluble fraction (Figs. 5 and S7) for each substrate concentration.
Figure 4.

Generation of oxidized products from Avicel by full-length ScLPMO10C and its catalytic domain. A–E show solubilized oxidized products released from 2 (A), 5 (B), 10 (C), 20 (D), or 40 (E) g/liter Avicel over a 60-min time period (solid lines, ScLPMO10C; dotted lines, ScAA10). F and G show the amounts of solubilized oxidized products (black bars) and total oxidized products (gray bars; i.e. the sum of oxidized products found in the insoluble and soluble fractions) after 60 min of incubation for ScLPMO10C (F) and ScAA10 (G). The error bars show ±S.D. (n = 3). Product quantification was achieved by converting all products to oxidized dimers and trimers with cellulases as described under “Experimental procedures.”
Figure 5.

Generation of solubilized oxidized products by full-length ScLPMO10C (A and C) and its catalytic domain, ScAA10 (B and D). The panels show the relative distribution (A and B) and the absolute values (C and D) of oxidized products with a degree of polymerization of 2–8, released after incubating the enzymes for 60 min with Avicel, at varying concentrations. To produce A and B, the peak areas for each of the eight monitored products were summed, and the sum was set to 100%. Note that the method used for product quantification differs from the method used to produce Fig. 4.
The action of a C1-oxidizing LPMO such as ScLPMO10C on crystalline cellulose will lead to release of soluble oxidized products if the same cellulose chain is cut twice at internal positions that are maximally ∼10 glycosidic bonds apart (longer fragments have too low solubility). Oxidized products may also result from a single cleavage near the nonreducing end of a cellulose chain, whereas a single cleavage close to the reducing end will yield a native (nonoxidized) cello-oligomer. Indeed, all reactions displayed in Figs. 4, 5, and S7 and discussed below yielded both oxidized and native products.
Fig. 4 shows that at the lower substrate concentrations ScAA10 is less effective than ScLPMO10C and that this is in part due to rapid enzyme inactivation (Fig. 4, A–C, nonlinear dotted progress curves). At these concentrations, the initial rate of ScAA10 seems to increase with the substrate concentration, which, thus, is nonsaturating. At higher substrate concentrations, ScAA10 is stable during the 60-min incubation time (Fig. 4, D and E), and measurements of the total amount of oxidized sites after 60 min (Fig. 4G) show that enzyme activity keeps increasing with substrate concentration up to 40 g/liter. Because a plateau in product formation is not reached, a substrate concentration of 40 g/liter is not a saturating substrate concentration for ScAA10. At the lower substrate concentrations, ∼30% of the oxidized sites are in the insoluble fraction. Interestingly, the proportion of nonsoluble oxidized sites increases at the higher substrate concentrations, reaching ∼60% at 40 g/liter (Fig. 4G).
For the CBM-containing full-length enzyme, stability issues are only observed at the lowest substrate concentration (2 g/liter; Fig. 4A, nonlinear solid curve). At all substrate concentrations from 5 g/liter and higher, stability issues are not observed (Fig. 4, B–E), and the total amount of oxidized sites after 60 min is the same (Fig. 4F). Thus, at 5 g/liter, the substrate concentration becomes saturating. In contrast with ScAA10, at lower substrate concentrations, almost all oxidized sites appear as soluble products (Fig. 4F). Interestingly, the absolute amount and the fraction (relative to the total) of soluble oxidized sites decrease at higher substrate concentrations, and this effect is more pronounced compared with ScAA10 (Fig. 4, F and G). It is also worth noting that, at the highest substrate concentration tested, the truncated enzyme generates more oxidized sites than the full-length enzyme (Fig. 4, E–G).
The fact that, at lower substrate concentrations, the CBM-containing enzyme produces a higher fraction of soluble products indicates a higher probability of the same cellulose chain being cut twice. This could be due to an immobilizing effect of the CBM that would keep the catalytic domain in the proximity of a previous cut for a prolonged time. Such an immobilizing effect would also increase the chances of two cuts in the same cellulose chain happening close to each other, meaning that, on average, one would expect shorter soluble oxidized products for the full-length enzyme. Fig. 5 shows that this is indeed the case. At the lower substrate concentrations, ∼60% of the oxidized products solubilized by full-length ScLPMO10C have degrees of polymerization of 2–4 as compared with ∼40% for ScAA10.
Most of the functional differences, including the difference in product distribution, were not visible at the highest of the tested substrate concentrations where the two enzyme forms show quite similar functionalities (Figs. 4 and 5). Furthermore, differences between the full-length and the truncated enzymes were largely absent when assessing the production of native soluble cello-oligomers (Fig. S7), which are the result of single cleavages near reducing chain ends.
Discussion
Taken together, the experiments described above yield a structural (Figs. 1, 2, S1, and S2) and a dynamic (Figs. 1, 6, and S3 and Videos S1 and S2) model of the interactions between the CBM-containing LPMO and its substrate, cellulose. Despite a lack of structural information for full-length proteins, models of full-length carbohydrate-active enzymes containing both a CBM and a flexible and extended linker have appeared in the literature (e.g. Ref. 39). To our knowledge, none of these models is supported by atomic-resolution structural data for the full-length protein molecule of the type we present here.
Comparative functional characterization of the full-length enzyme and its isolated catalytic domain revealed complexities that are unique for LPMOs and that relate to the multiple effects of substrate affinity and substrate binding on LPMO performance as discussed above. Although not being the core focus of this study, these complexities are of major importance and must be taken into account when interpreting existing functional data on the effect of CBMs on LPMO efficiency (21, 26–28) and when planning novel studies of the roles of CBMs in LPMOs. Clearly, characterization of LPMO variants by measuring product formation at one single time point and/or at one single substrate concentration is not sufficient to fully appreciate the interplay between an LPMO and its CBM.
Functional studies at low substrate concentrations revealed clear differences between the full-length and truncated LPMOs, which can largely be explained by the CBM promoting binding to internal positions on the substrate surface and by the immobilizing effect of the CBM, which promotes multiple cleavages in the same region and same cellulose chain. As a consequence, the full-length protein produces a higher fraction of soluble oxidized products (relative to insoluble products; Fig. 4F), and the soluble products are shorter (Fig. 5).
Native products can only emerge from cleavage at chain ends, which for Avicel, with an average degree of polymerization of 200 (40), are lower in concentration than the amount of internal LPMO-binding sites. Thus, one would expect the amount of native products to increase with substrate concentration all the way up to 40 g/liter as is indeed observed (Fig. S7). The dependence of native product formation on the substrate concentration differs between the two enzyme variants. The increase in product formation upon increasing the substrate concentration is less pronounced for the full-length enzyme (Fig. S7, compare B and C). This is likely because at high substrate concentrations the overall efficiency of the CBM-containing enzyme becomes lower than the efficiency of the truncated enzyme (for reasons discussed below). This efficiency difference is clearly apparent from the quantification of oxidized products shown in Fig. 4, F and G. Interestingly, at lower substrate concentrations, the ratio between native and oxidized products is considerably lower for the full-length LPMO than for the truncated version (Figs. S7, B and C). This shows that the CBM promotes binding of the LPMO at internal positions on the crystalline surface.
Although the distribution of oxidized products showed that the full-length enzyme generates shorter products than the truncated enzyme (Fig. 5), a similar trend is not observed for the production of native products, which are the result of one single cleavage. Substrate binding by the CBM to some extent immobilizes the LPMO, allowing it to carry out multiple oxidations in the same substrate region. It is thus not surprising that the effect of the CBM on the product distribution is only seen for oxidized products, which are primarily the result of two spatially close chain cleavages.
The effect of the substrate concentration on several of the analyzed functional features is remarkable. First, the data (Figs. 4, 5, and S7) show that removal of the CBM is beneficial for overall enzyme performance at the highest substrate concentrations, similar to what has been observed and discussed for CBMs in certain cellulases (38). The work by Várnai et al. (38) on cellobiohydrolases indicated that there may be a payoff between (beneficial) CBM-mediated substrate affinity and possible negative effects of the CBM related to low off-rates and/or nonproductive binding.
Second, the data show that the product mixtures generated by the full-length and the truncated enzymes become more similar as the substrate concentration increases. At the highest substrate concentration, the two enzyme forms behave similarly in terms of the ratio of soluble versus total oxidized products (Fig. 4), the length distributions of the oligomeric products (Figs. 5 and S7), and the ratio between native and oxidized soluble products (Fig. S7). Thus, at the highest substrate concentration, the tendency of the full-length enzyme to promote multiple cleavages in the same region of the substrate disappears, and the cleavage patterns become equally random for both enzyme forms. Although the similar overall activities can be explained by the compensatory effect of a high substrate concentration on weaker substrate affinity, this effect does not explain the changes in product profiles. The freely moving LPMO domain of a bound ScLPMO10C molecule could act on a cellulose chain in another fibril to which it is not directly bound. It is conceivable that this effectively more random, as opposed to local, mode of action, which is also expected for the truncated CBM-free enzyme, becomes more prominent as the substrate concentration increases. This would explain why the product profiles generated by the full-length and the truncated enzymes become more similar as the substrate concentration increases. Probing interactions between LPMOs and their polymeric substrate is challenging but has been accomplished by using a soluble polymeric substrate (xyloglucan (41)) and by using hydrogen/deuterium exchange to measure binding of insoluble chitin fibrils to a chitin-active LPMO (42). Here, we show that cellulose nanofibrils forming a stable suspension can be used to probe interactions between ScLPMO10C and its substrate. This approach allowed direct mapping of the cellulose-binding surfaces of ScCBM2 and ScAA10 and showed, together with classical binding studies, that ScCBM2 binds much more strongly to the insoluble cellulose substrate than ScAA10. The clear difference in binding strength implies that ScCBM2 prolongs the residence time of ScLPMO10C on cellulose, keeping ScAA10 proximal to the substrate.
Using chemical shifts as a probe of backbone conformation, we provide experimental evidence for an extended linker region. This was expected as proline residues in the linker confer rotational restriction around their peptide bonds, which has been observed previously for Pro-rich linkers (43, 44). Increased T2 values and decreased 1H-15N NOEs in the linker region (Fig. 1D) show that, overall, the linker is conformationally dynamic. As a consequence, the linker in ScLPMO10C decouples the motions of the tethered ScCBM2 and ScAA10 domains, which can move independently of each other (Video S1), as clearly indicated by the rotational correlation times (Fig. 1B).
The dynamic features described above should be regarded qualitatively. In particular, residue-specific relaxation data should not be evaluated individually (as “spikes” in the data may arise from integration errors or signal overlap) but in the context of clusters of amino acids displaying similar trends. Such trends can be clearly seen in the linker region but can also be observed for flexible loop regions in the structured domains (e.g. residues 174–176 and 199–202 as seen in Fig. 1D). In the case of ScLPMO10C, the dynamic nature of the linker will in part be due to the various glycine residues adjacent to the Pro-Thr-Asp–rich region (Fig. 1A). It should be noted, however, that NMR studies of a shorter (20-residue) Pro-Thr linker in Xyn10A, a xylanase from Cellulomonas fimi (43), also showed considerable conformational flexibility.
The linker in ScLPMO10C serves as a flexible spacer, maintaining a distance between the carbohydrate-binding and catalytic domains while simultaneously enabling these domains to move independently of each other. Due to the prolonged residence time on a local area of cellulose, caused by ScCBM2, the linker keeps the LPMO domain in proximity of the substrate. The consequences of these features are visible in Video S2, which is based on an ensemble of conformations calculated from amino acid–specific NMR data for full-length ScLPMO10C. The video, summarized in Fig. 6, shows that upon binding to the cellulose surface by ScCBM2 the ScAA10 domain moves around as a result of the flexibility conferred by the linker, sampling an area of ∼1300 Å2, equaling around 300 glucose residues. This dynamic model of ScLPMO10C explains the preferential release of shorter, soluble oxidized products by the full-length protein as discussed above. Of note, although Fig. 6 may give the impression that the CBM is statically bound to the substrate surface, lateral diffusion of the CBM on the cellulose surface cannot be excluded.
Despite the insights presented here, several questions related to the interplay of catalytic domains, flexible linkers, and CBMs remain. One issue concerns the events that trigger eventual desorption and relocation of the CBM. Another issue concerns the effect of glycosylation on linker structure and dynamics. Such glycosylation is known to happen in CAZymes from fungi (39, 45) and actinomycetes (46), and its impact is currently receiving considerable attention (39, 45). In their study on the C. fimi xylanase, Poon et al. (43) concluded that glycosylation of the 20-residue Pro-Thr linker had limited effects on linker structure and dynamics. Studies on the roles of glycosylated linkers in fungal modular CAZymes have revealed an impact of glycosylation on substrate binding and proteolytic resistance, but information on the impact of glycosylation on linker shape and dynamics is scarce. Interestingly, in a recent study, Amore et al. (45) concluded that glycosylation of the linker in a fungal cellobiohydrolase ensures the separation between the catalytic domain and the CBM. The present data show that the nonglycosylated linker of ScLPMO10C has an extended conformation that separates the domains. Another issue concerns the functional implications of variation in linker length and composition as exemplified by the contrast between the 30-residue linker of mixed nature in ScLPMO10C versus the 20-residue strict Pro-Thr linker in Xyn10A from C. fimi. A final question concerns the biological implications of the localized multiple substrate oxidations that are enabled by the CBM. One could envisage that such localized multiple action helps to create “weak spots” in an otherwise tough substrate, which could facilitate further degradation or, perhaps, in the case of an invading plant pathogen, easier penetration.
In conclusion, the present results provide a mechanistic description of the function, conformation, and dynamics of a modular carbohydrate-active enzyme with a flexible linker. Furthermore, we have unraveled some of the pitfalls in assessing the effect of a CBM on LPMO functionality, and by dealing with these pitfalls, we were able to generate functional data that contribute to a better understanding of the role of CBMs in LPMOs. Taking into account the abundance and diversity of CAZymes that have CBMs and flexible linkers (39, 43, 47), the present experimental insights will contribute to a greater understanding of CAZymes in general and lignocellulose-degrading enzymes in particular.
Experimental procedures
Sample preparation
The protocols used for the production of pure, isotopically labeled (15N and 13C) full-length ScLPMO10C; the catalytic domain, ScAA10; and the carbohydrate-binding domain, ScCBM2, as well as sample conditions for NMR measurements have been described previously (29). Briefly, ScLPMO10C was produced using Escherichia coli HI-ControlTM BL21 (DE3) and the ExpressoTM pETite N-His SUMO T7 expression vector (Lucigen). The SUMO-fused protein was subsequently purified on a nickel column, the SUMO tag was proteolytically cleaved using SUMO Express Protease, and pure ScLPMO10C was obtained by inverse nickel-affinity purification to remove the SUMO tag and SUMO Express Protease. ScAA10 was produced in E. coli RV308 using an LPMO expression cassette (48). The protein was isolated from the periplasm and purified by ion-exchange chromatography and size-exclusion chromatography. ScCBM2 was produced in E. coli BL21 (DE3) using the isopropyl 1-thio-β-d-galactopyranoside–inducible pNIC-CH vector (Addgene) with a C-terminal His tag and purified by immobilized metal affinity chromatography as described previously (29). The His tag was not removed after purification. Prior to NMR experiments, samples of isotopically labeled ScLPMO10C, ScAA10, and ScCBM2 were incubated in 10 mm Na-EDTA for 1 h after which the buffer was exchanged to 20 mm sodium phosphate pH 5.5 buffer with 10 mm NaCl.
Samples of ScLPMO10C and ScAA10 for cellulose degradation experiments were produced as described previously (25). Before use, these enzymes were saturated with copper, using Cu(II)SO4, prior to exchanging the buffer to 50 mm sodium phosphate, pH 7.0, using a PD MidiTrap G-25 (GE Healthcare) as described previously (49).
NMR spectroscopy
NMR spectra were recorded at 25 °C on a Bruker Ascend 800-MHz spectrometer Avance III HD equipped with a 5-mm Z-gradient CP-TCI (H/C/N) cryoprobe at the NV-NMR Center/Norwegian NMR Platform (NNP) in Trondheim, Norway. NMR data were processed and analyzed using Bruker TopSpin version 3.5 and Protein Dynamic Center software version 2.3.1 from Bruker BioSpin. The NMR assignments of ScLPMO10C, ScAA10, and ScCBM2 have been published elsewhere (29).
Nuclear spin relaxation times T1 and T2 and heteronuclear 1H-15N NOE measurements of amide 15N for all three proteins were carried out using transverse relaxation optimized spectroscopy (TROSY) experiments (50). T1 and T2 spectra were recorded as pseudo-3D spectra where two frequency dimensions correspond to the amide 1H and 15N chemical shifts, respectively, and the third dimension is made up of variable relaxation time delays. For T1, the time points were 0.1, 0.2, 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, and 4.5 s. For T2, the time points were 17, 34, 68, 136, 170, 204, 238, and 272 ms. The 1H-15N NOE spectra were composed of two 2D planes recorded with and without presaturation, respectively. Overall τc values were determined from the ratio between T1 and T2 (33).
Structure determination
For ScCBM2, NOE cross-peak intensities were converted into distance restraints using the CALIBA (51) subroutine in CYANA 3.97 (52, 53). Dihedral torsion angles (ϕ and ψ) calculated from chemical shift data (Cα, Cβ, HN, N, and C′) by TALOS-N (31) were included as conformational restraints as was one disulfide bridge (Cys265–Cys361). Based on this input, the structure was calculated using CYANA by generating 256 conformers that were optimized using 10,000 steps of simulated annealing to fit the NOE- and TALOS-N–derived distance restraints. The 20 conformers with lowest CYANA target function values were energy-minimized using YASARA (32) with the YASARA force field (54). The first minimization step, in vacuo, was followed by minimization in water and calculating electrostatics by applying the particle mesh Ewald method (55). The coordinates of the 20 ScCBM2 conformers with lowest energy (Table S1) have been deposited in the Protein Data Bank under code 6F7E.
The model of full-length ScLPMO10C was made in CYANA 3.97 by generating 256 conformers. These were then optimized using 10,000 steps of simulated annealing to simultaneously fit the following three conformational constraint inputs. Input I consisted of 1H–1H distances shorter than 5 Å that were extracted from the X-ray crystal diffraction structure of ScAA10 (PDB code 4OY7) (21) by using MOLMOL software (56). Input II was derived from the chemical shift assignment (29) of ScLPMO10C, which was used as an input for TALOS-N (31) to generate dihedral angle restraints for the linker region. Input III was the same 1H–1H distance constraints that were used for calculation of the structure of ScCBM2.
Videos S1 and S2 were produced from the experimentally determined ensemble of 32 conformers (calculated from amino acid–specific NMR data for full-length ScLPMO10C as described above). Video S1 was made by aligning conformers of ScLPMO10C with respect to all the α-carbons and using UCSF Chimera version 1.11.2 (57) to create the animation by using each conformer as a different frame. To make Video S2, the same 32 conformers of ScLPMO10C from the experimentally determined ensemble were aligned with respect to the CBM2 domain. A low-energy pathway between each of the conformers was calculated with YASARA (32) using simulated annealing with the YASARA force field (54) to generate a total of 3800 frames from the experimentally determined conformers. UCSF Chimera version 1.11.2 (57) was used to create the animation from the frames generated by YASARA. The cellulose fibril was generated using Cellulose-builder (58).
Binding to cellulose nanofibrils
Cellulose nanofibrils were produced from never-dried softwood bleached pulp fibers. A mechanical pretreatment, i.e. beating in a Claflin mill (1000 kilowatt h/ton for 1 h), was performed before fibrillation. Subsequently, the fibrillation was done by using a Rannie15 type 12.56X homogenizer (APV, SPX Flow Technology, Silkeborg, Denmark) with a pressure drop of 1000 bars in each pass. The cellulose nanofibrils were collected after three passes. The concentration of the cellulose dispersions used for homogenization was 1%, and the final product contained 0.98% dry weight (w/v).
Reference 15N HSQC spectra were recorded for 15N-labeled samples of ScAA10 (0.1 mm) and ScCBM2 (0.1 mm) in 20 mm sodium phosphate pH 5.5 buffer with 10 mm NaCl. Cellulose nanofibrils were added in a 20:1 ratio (w/w) to the protein samples, and new 15N HSQC spectra were recorded. The normalized signal intensity for each amino acid was estimated from the ratio between peak intensities in the 15N HSQC spectra recorded with and without cellulose nanofibrils.
Binding to Avicel
Binding to Avicel PH-101 (Fluka) was assessed in reaction mixtures containing 10 mg/ml substrate and 0.08 mg/ml protein (ScLPMO10C or ScAA10) in 50 mm sodium phosphate buffer, pH 7.0, that were incubated at 22 °C in an Eppendorf Comfort Thermomixer set to 1000 rpm. At various time points (2.5, 5, 15, 30, and 60 min), a sample was taken and filtered using a 96-well filter plate (Millipore) operated by a Millipore vacuum manifold to remove insoluble substrate and substrate-bound protein. The relative amount of protein in the supernatant was determined by measuring A280 (Eppendorf BioPhotometer, Eppendorf, Hamburg).
The Kd and Bmax for the full-length enzyme were determined by mixing protein solutions of varying concentrations (0, 10, 20, 50, 75, 150, 300, and 500 μg/ml) with 10 g/liter Avicel. Before adding Avicel, the A280 was measured for each of the prepared protein solutions (in 50 mm sodium phosphate buffer, pH 7.0) to create a standard curve. After addition of Avicel, the solutions were placed at 22 °C in an Eppendorf Comfort Thermomixer set to 1000 rpm for 60 min. Subsequently, samples were filtered using a 96-well filter plate (Millipore), and the concentration of free protein in the supernatant was determined by measuring A280. All assays were performed in triplicates and with blanks (buffer and 10 g/liter Avicel). The Kd (μm) and Bmax (μmol/g of Avicel) were determined by fitting the binding isotherms to a one-site binding equation where P represents protein: [Pbound] = Bmax [Pfree]/Kd + [Pfree]. The fitting was done by nonlinear regression using Prism 7 software (GraphPad, La Jolla, CA).
Cellulose degradation experiments
For all cellulose degradation experiments, 0.5 μm Cu(II)-loaded ScLPMO10C or ScAA10 was incubated with 2–40 g/liter Avicel PH-101, in 50 mm sodium phosphate buffer, pH 7.0, in the presence of 1 mm ascorbic acid. The reaction mixtures were incubated in an Eppendorf Thermomixer set to 40 °C and 1000 rpm. Samples were taken at 10, 20, 30, and 60 min, and solubilized products were separated from the insoluble fraction by filtration as described above. The soluble products were further degraded by incubation with a 0.5 μm concentration of the endoglucanase TfCel5A (59) at 37 °C for 16 h, yielding oxidized products with a degree of polymerization of 2 and 3 (GlcGlc1A and Glc2Glc1A), which were quantified to yield the total concentration of soluble oxidized sites. Cellobiose (98% purity; purchased from Sigma-Aldrich) and cellotriose (95% purity; purchased from Megazyme) were used as substrates for production of C1-oxidized dimer (cellobionic acid; GlcGlc1A) and trimer (cellotrionic acid; Glc2Glc1A) by incubation with 1.5 μm cellobiose dehydrogenase from Myriococcum thermophilum (MtCDH) (60). These in-house–made standards were used to quantify LPMO-generated products.
To determine the total concentration of oxidized products (soluble and insoluble) at the last time point (60 min), the LPMOs were inactivated by boiling the reaction mixture for 15 min at 100 °C. Subsequently, the reaction mixtures were diluted with 50 mm sodium phosphate buffer, pH 7.0, to a final concentration of 2 g/liter LPMO-treated Avicel and further hydrolyzed by adding a mixture of two endoglucanases, TfCel5A (1.2 μm) and TfCel6A (61) (5 μm), and incubation for 48 h at 50 °C with shaking at 1000 rpm. As a result of this procedure, all cellulose is solubilized, and oxidized products appear as dimers and trimers only.
Native and oxidized products were analyzed by high-performance anion-exchange chromatography using a DionexTM ICS-5000 system (Thermo Scientific, Sunnyvale, CA) set up with a disposable electrochemical gold electrode. Five-microliter samples were injected on a CarboPac PA1 (2 × 50-mm) column operated with 0.1 m NaOH (eluent A) at a flow rate of 0.25 ml/min and a column temperature of 30 °C. Elution was achieved using a stepwise gradient with increasing amounts of eluent B (0.1 m NaOH + 1 m NaOAc) as follows: 0–10% B over 10 min, 10–30% B over 25 min, 30–100% B over 5 min, 100–0% B over 1 min, and 0% B (reconditioning) for 9 min. For analysis of endoglucanase-treated samples containing only oxidized cellobiose and cellotriose, a steeper gradient of acetate was used as follows: 0–10% B over 10 min, 10–14% B over 5 min, 14–30% B over 1 min, 30–100% B over 2 min, 100–0% B over 0.1 min, and 0% B over 10.9 min. Eluted oligosaccharides were monitored using a pulsed detector, and chromatograms were recorded using Chromeleon 7.0 software (62).
Author contributions
G. C., Z. F., V. G. H. E., and F. L. A. conceptualization; G. C., Z. F., E. B. H., V. G. H. E., and F. L. A. data curation; G. C., Z. F., V. G. H. E., and F. L. A. formal analysis; G. C. and Z. F. investigation; G. C. and Z. F. methodology; G. C., Z. F., E. B. H., V. G. H. E., and F. L. A. writing-original draft; G. C., Z. F., V. G. H. E., and F. L. A. writing-review and editing; V. G. H. E. and F. L. A. supervision; F. L. A. funding acquisition; F. L. A. project administration.
Supplementary Material
Acknowledgment
We thank Gustav Vaaje-Kolstad for helpful discussions.
This work was supported by SO funds from NTNU Norwegian University of Science and Technology and by the OXYMOD project, the Norwegian NMR Platform, and an FRIPRO project, all funded by Research Council of Norway Grants 269408, 226244, and 262853, respectively. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7, Table S1, and Videos S1 and S2.
The atomic coordinates and structure factors (code 6F7E) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- LPMO
- lytic polysaccharide monooxygenase
- CAZyme
- carbohydrate-active enzyme
- CBM
- carbohydrate-binding module
- Sc
- S. coelicolor
- HSQC
- heteronuclear single quantum coherence
- τc
- rotational correlation times
- T
- relaxation time
- Bmax
- substrate-binding capacity
- Kd
- equilibrium dissociation constant
- SUMO
- small ubiquitin-like modifier
- Tf
- Thermobifida fusca.
References
- 1. Sheldon R. A. (2016) Green chemistry, catalysis and valorization of waste biomass. J. Mol. Catal. A Chem. 422, 3–12 10.1016/j.molcata.2016.01.013 [DOI] [Google Scholar]
- 2. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., and Foust T. D. (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–807 10.1126/science.1137016 [DOI] [PubMed] [Google Scholar]
- 3. Vaaje-Kolstad G., Horn S. J., van Aalten D. M., Synstad B., and Eijsink V. G. (2005) The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J. Biol. Chem. 280, 28492–28497 10.1074/jbc.M504468200 [DOI] [PubMed] [Google Scholar]
- 4. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., and Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 10.1126/science.1192231 [DOI] [PubMed] [Google Scholar]
- 5. Harris P. V., Welner D., McFarland K. C., Re E., Navarro Poulsen J.-C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., and Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 10.1021/bi100009p [DOI] [PubMed] [Google Scholar]
- 6. Horn S. J., Vaaje-Kolstad G., Westereng B., and Eijsink V. G. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45–56 10.1186/1754-6834-5-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunæs A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., and Eijsink V. G. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 10.1002/pro.689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J.-C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., et al. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 10.1073/pnas.1105776108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Phillips C. M., Beeson W. T., Cate J. H., and Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 10.1021/cb200351y [DOI] [PubMed] [Google Scholar]
- 10. Langston J. A., Shaghasi T., Abbate E., Xu F., Vlasenko E., and Sweeney M. D. (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77, 7007–7015 10.1128/AEM.05815-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eibinger M., Ganner T., Bubner P., Rošker S., Kracher D., Haltrich D., Ludwig R., Plank H., and Nidetzky B. (2014) Cellulose surface degradation by a lytic polysaccharide monooxygenase and its effect on cellulase hydrolytic efficiency. J. Biol. Chem. 289, 35929–35938 10.1074/jbc.M114.602227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vermaas J. V., Crowley M. F., Beckham G. T., and Payne C. M. (2015) Effects of lytic polysaccharide monooxygenase oxidation on cellulose structure and binding of oxidized cellulose oligomers to cellulases. J. Phys. Chem. B 119, 6129–6143 10.1021/acs.jpcb.5b00778 [DOI] [PubMed] [Google Scholar]
- 13. Igarashi K., Uchihashi T., Koivula A., Wada M., Kimura S., Okamoto T., Penttilä M., Ando T., and Samejima M. (2011) Traffic jams reduce hydrolytic efficiency of cellulase on cellulose surface. Science 333, 1279–1282 10.1126/science.1208386 [DOI] [PubMed] [Google Scholar]
- 14. Jalak J., Kurašin M., Teugjas H., and Väljamäe P. (2012) Endo-exo synergism in cellulose hydrolysis revisited. J. Biol. Chem. 287, 28802–28815 10.1074/jbc.M112.381624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurasin M., and Väljamäe P. (2011) Processivity of cellobiohydrolases is limited by the substrate. J. Biol. Chem. 286, 169–177 10.1074/jbc.M110.161059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kari J., Olsen J., Borch K., Cruys-Bagger N., Jensen K., and Westh P. (2014) Kinetics of cellobiohydrolase (Cel7A) variants with lowered substrate affinity. J. Biol. Chem. 289, 32459–32468 10.1074/jbc.M114.604264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cruys-Bagger N., Tatsumi H., Ren G. R., Borch K., and Westh P. (2013) Transient kinetics and rate-limiting steps for the processive cellobiohydrolase Cel7A: effects of substrate structure and carbohydrate binding domain. Biochemistry 52, 8938–8948 10.1021/bi401210n [DOI] [PubMed] [Google Scholar]
- 18. Harris P. V., Xu F., Kreel N. E., Kang C., and Fukuyama S. (2014) New enzyme insights drive advances in commercial ethanol production. Curr. Opin. Chem. Biol. 19, 162–170 10.1016/j.cbpa.2014.02.015 [DOI] [PubMed] [Google Scholar]
- 19. Johansen K. S. (2016) Discovery and industrial applications of lytic polysaccharide mono-oxygenases. Biochem. Soc. Trans. 44, 143–149 10.1042/BST20150204 [DOI] [PubMed] [Google Scholar]
- 20. Czjzek M., and Ficko-Blean E. (2017) Probing the complex architecture of multimodular carbohydrate-active enzymes using a combination of small angle X-ray scattering and X-ray crystallography, in Protein-Carbohydrate Interactions: Methods and Protocols (Abbott D. W., and van Bueren A., eds) pp. 239–253, Springer, New York: [DOI] [PubMed] [Google Scholar]
- 21. Forsberg Z., Mackenzie A. K., Sørlie M., Røhr Å. K., Helland R., Arvai A. S., Vaaje-Kolstad G., and Eijsink V. G. (2014) Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8446–8451 10.1073/pnas.1402771111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simpson P. J., Xie H., Bolam D. N., Gilbert H. J., and Williamson M. P. (2000) The structural basis for the ligand specificity of family 2 carbohydrate-binding modules. J. Biol. Chem. 275, 41137–41142 10.1074/jbc.M006948200 [DOI] [PubMed] [Google Scholar]
- 23. Brun E., Moriaud F., Gans P., Blackledge M. J., Barras F., and Marion D. (1997) Solution structure of the cellulose-binding domain of the endoglucanase Z secreted by Erwinia chrysanthemi. Biochemistry 36, 16074–16086 10.1021/bi9718494 [DOI] [PubMed] [Google Scholar]
- 24. Xu G. Y., Ong E., Gilkes N. R., Kilburn D. G., Muhandiram D. R., Harris-Brandts M., Carver J. P., Kay L. E., and Harvey T. S. (1995) Solution structure of a cellulose-binding domain from Cellulomonas fimi by nuclear magnetic resonance spectroscopy. Biochemistry 34, 6993–7009 10.1021/bi00021a011 [DOI] [PubMed] [Google Scholar]
- 25. Forsberg Z., Røhr A. K., Mekasha S., Andersson K. K., Eijsink V. G., Vaaje-Kolstad G., and Sørlie M. (2014) Comparative study of two chitin-active and two cellulose-active AA10-type lytic polysaccharide monooxygenases. Biochemistry 53, 1647–1656 10.1021/bi5000433 [DOI] [PubMed] [Google Scholar]
- 26. Crouch L. I., Labourel A., Walton P. H., Davies G. J., and Gilbert H. J. (2016) The contribution of non-catalytic carbohydrate binding modules to the activity lytic polysaccharide monooxygenases. J. Biol. Chem. 291, 7439–7449 10.1074/jbc.M115.702365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Forsberg Z., Nelson C. E., Dalhus B., Mekasha S., Loose J. S., Crouch L. I., Røhr Å. K., Gardner J. G., Eijsink V. G., and Vaaje-Kolstad G. (2016) Structural and functional analysis of a lytic polysaccharide monooxygenase important for efficient utilization of chitin in Cellvibrio japonicus. J. Biol. Chem. 291, 7300–7312 10.1074/jbc.M115.700161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Forsberg Z., Bissaro B., Gullesen J., Dalhus B., Vaaje-Kolstad G., and Eijsink V. G. H. (2018) Structural determinants of bacterial lytic polysaccharide monooxygenase functionality. J. Biol. Chem. 293, 1397–1412 10.1074/jbc.M117.817130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Courtade G., Forsberg Z., Vaaje-Kolstad G., Eijsink V. G. H., and Aachmann F. L. (2017) Chemical shift assignments for the apo-form of the catalytic domain, the linker region, and the carbohydrate-binding domain of the cellulose-active lytic polysaccharide monooxygenase ScLPMO10C. Biomol. NMR Assign. 11, 257–264 10.1007/s12104-017-9759-2 [DOI] [PubMed] [Google Scholar]
- 30. Wishart D. S., Sykes B. D., and Richards F. M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222, 311–333 10.1016/0022-2836(91)90214-Q [DOI] [PubMed] [Google Scholar]
- 31. Shen Y., and Bax A. (2013) Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241 10.1007/s10858-013-9741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krieger E., Koraimann G., and Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins 47, 393–402 10.1002/prot.10104 [DOI] [PubMed] [Google Scholar]
- 33. Fushman D., Weisemann R., Thüring H., and Rüterjans H. (1994) Backbone dynamics of ribonuclease T1 and its complex with 2′GMP Studied by two-dimensional heteronuclear NMR Spectroscopy. J. Biomol. NMR 4, 61–78 10.1007/BF00178336 [DOI] [PubMed] [Google Scholar]
- 34. Daragan V. A., and Mayo K. H. (1997) Motional model analyses of protein and peptide dynamics using 13C and 15N NMR relaxation. Prog. Nucl. Magn. Reson. Spectrosc. 31, 63–105 10.1016/S0079-6565(97)00006-X [DOI] [Google Scholar]
- 35. Takashima T., Ohnuma T., and Fukamizo T. (2018) NMR analysis of substrate binding to a two-domain chitinase: comparison between soluble and insoluble chitins. Carbohydr. Res. 458–459, 52–59 10.1016/j.carres.2018.02.004 [DOI] [PubMed] [Google Scholar]
- 36. Boraston A. B., Bolam D. N., Gilbert H. J., and Davies G. J. (2004) Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem. J. 382, 769–781 10.1042/BJ20040892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bissaro B., Røhr Å. K., Müller G., Chylenski P., Skaugen M., Forsberg Z., Horn S. J., Vaaje-Kolstad G., and Eijsink V. G. H. (2017) Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 13, 1123–1128 10.1038/nchembio.2470 [DOI] [PubMed] [Google Scholar]
- 38. Várnai A., Siika-Aho M., and Viikari L. (2013) Carbohydrate-binding modules (CBMs) revisited: reduced amount of water counterbalances the need for CBMs. Biotechnol. Biofuels 6, 30 10.1186/1754-6834-6-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Payne C. M., Resch M. G., Chen L., Crowley M. F., Himmel M. E., Taylor L. E. 2nd, Sandgren M., Ståhlberg J., Stals I., Tan Z., and Beckham G. T. (2013) Glycosylated linkers in multimodular lignocellulose-degrading enzymes dynamically bind to cellulose. Proc. Natl. Acad. Sci. U.S.A. 110, 14646–14651 10.1073/pnas.1309106110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Y.-H., and Lynd L. R. (2005) Determination of the number-average degree of polymerization of cellodextrins and cellulose with application to enzymatic hydrolysis. Biomacromolecules. 6, 1510–1515 10.1021/bm049235j [DOI] [PubMed] [Google Scholar]
- 41. Courtade G., Wimmer R., Røhr Å. K., Preims M., Felice A. K., Dimarogona M., Vaaje-Kolstad G., Sørlie M., Sandgren M., Ludwig R., Eijsink V. G., and Aachmann F. L. (2016) Interactions of a fungal lytic polysaccharide monooxygenase with β-glucan substrates and cellobiose dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 113, 5922–5927 10.1073/pnas.1602566113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aachmann F. L., Sørlie M., Skjåk-Bræk G., Eijsink V. G., and Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 10.1073/pnas.1208822109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Poon D. K., Withers S. G., and McIntosh L. P. (2007) Direct demonstration of the flexibility of the glycosylated proline-threonine linker in the Cellulomonas fimi xylanase Cex through NMR spectroscopic analysis. J. Biol. Chem. 282, 2091–2100 10.1074/jbc.M609670200 [DOI] [PubMed] [Google Scholar]
- 44. George R. A., and Heringa J. (2002) An analysis of protein domain linkers: their classification and role in protein folding. Protein Eng. 15, 871–879 10.1093/protein/15.11.871 [DOI] [PubMed] [Google Scholar]
- 45. Amore A., Knott B. C., Supekar N. T., Shajahan A., Azadi P., Zhao P., Wells L., Linger J. G., Hobdey S. E., Vander Wall T. A., Shollenberger T., Yarbrough J. M., Tan Z., Crowley M. F., Himmel M. E., et al. (2017) Distinct roles of N- and O-glycans in cellulase activity and stability. Proc. Natl. Acad. Sci. U.S.A. 114, 13667–13672 10.1073/pnas.1714249114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ong E., Kilburn D. G., Miller R. C. Jr., and Warren R. A. (1994) Streptomyces lividans glycosylates the linker region of a β-1,4-glycanase from Cellulomonas fimi. J. Bacteriol. 176, 999–1008 10.1128/jb.176.4.999-1008.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sammond D. W., Payne C. M., Brunecky R., Himmel M. E., Crowley M. F., and Beckham G. T. (2012) Cellulase linkers are optimized based on domain type and function: insights from sequence analysis, biophysical measurements, and molecular simulation. PLoS One 7, e48615 10.1371/journal.pone.0048615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Courtade G., Le S. B., Sætrom G. I., Brautaset T., and Aachmann F. L. (2017) A novel expression system for lytic polysaccharide monooxygenases. Carbohydr. Res. 448, 212–219 10.1016/j.carres.2017.02.003 [DOI] [PubMed] [Google Scholar]
- 49. Loose J. S., Forsberg Z., Fraaije M. W., Eijsink V. G., and Vaaje-Kolstad G. (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett. 588, 3435–3440 10.1016/j.febslet.2014.07.036 [DOI] [PubMed] [Google Scholar]
- 50. Zhu G., Xia Y., Nicholson L. K., and Sze K. H. (2000) Protein dynamics measurements by TROSY-based NMR experiments. J. Magn. Reson. 143, 423–426 10.1006/jmre.2000.2022 [DOI] [PubMed] [Google Scholar]
- 51. Güntert P., Braun W., and Wüthrich K. (1991) Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS and GLOMSA. J. Mol. Biol. 217, 517–530 10.1016/0022-2836(91)90754-T [DOI] [PubMed] [Google Scholar]
- 52. Güntert P., Mumenthaler C., and Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 10.1006/jmbi.1997.1284 [DOI] [PubMed] [Google Scholar]
- 53. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 10.1385/1-59259-809-9:353 [DOI] [PubMed] [Google Scholar]
- 54. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., and Karplus K. (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77, 114–122 10.1002/prot.22570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 10.1063/1.470117 [DOI] [Google Scholar]
- 56. Koradi R., Billeter M., and Wüthrich K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14, 51–55, 29–32 10.1016/0263-7855(96)00009-4 [DOI] [PubMed] [Google Scholar]
- 57. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 58. Gomes T. C., and Skaf M. S. (2012) Cellulose-builder: a toolkit for building crystalline structures of cellulose. J. Comput. Chem. 33, 1338–1346 10.1002/jcc.22959 [DOI] [PubMed] [Google Scholar]
- 59. McGinnis K., and Wilson D. B. (1993) Disulfide arrangement and functional domains of β-1,4-endoglucanse E5 from Thermomonospora fusca. Biochemistry 32, 8157–8161 10.1021/bi00083a015 [DOI] [PubMed] [Google Scholar]
- 60. Harreither W., Sygmund C., Augustin M., Narciso M., Rabinovich M. L., Gorton L., Haltrich D., and Ludwig R. (2011) Catalytic properties and classification of cellobiose dehydrogenases from ascomycetes. Appl. Environ. Microbiol. 77, 1804–1815 10.1128/AEM.02052-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Calza R. E., Irwin D. C., and Wilson D. B. (1985) Purification and characterization of two β-1,4-endoglucanases from Thermomonospora fusca. Biochemistry 24, 7797–7804 10.1021/bi00347a044 [DOI] [Google Scholar]
- 62. Westereng B., Agger J. W., Horn S. J., Vaaje-Kolstad G., Aachmann F. L., Stenstrøm Y. H., and Eijsink V. G. (2013) Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J. Chromatogr. A 1271, 144–152 10.1016/j.chroma.2012.11.048 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.