

Abstract
Here, we seek to summarize the current understanding of the biochemical and molecular events mediated by visual cycle molecules in the eye. The structures and functions of selected visual cycle proteins and their roles in human retinal diseases are also highlighted. Genetic mutations and malfunctions of these proteins provide etiological evidence that many ocular diseases arise from anomalies of retinoid (vitamin A) metabolism and related visual processes. Genetic retinal disorders such as retinitis pigmentosa, Leber's congenital amaurosis, and Stargardt's disease are linked to structural changes in visual cycle proteins. Moreover, recent reports suggest that visual cycle proteins may also play a role in the development of diabetic retinopathy. Basic science has laid the groundwork for finding a cure for many of these blindness-causing afflictions, but much work remains. Some translational research projects have advanced to the clinical trial stage, while many others are still in progress, and more are at the ideas stage and remain yet to be tested. Some examples of these studies are discussed. Recent and future progress in our understanding of the visual cycle will inform intervention strategies to preserve human vision and prevent blindness.
Keywords: protein isomerase, membrane protein, metabolic disease, genetic disease, vitamin A
Classic rod/cone visual cycle
The classic visual cycle is initiated by the conversion of a single photon of light energy into an electrical signal in the retina. This signal transduction occurs due to a G protein–coupled receptor (GPCR)2 called opsin, which contains an 11-cis-retinal chromophore. When activated by a photon, 11-cis-retinal undergoes photoisomerization to all-trans-retinal leading to a change in the conformation of opsin GPCR and a signal transduction cascade to close cGMP-gated cation channels resulting in hyperpolarization of the photoreceptor cell. The collective change in the receptor potentials of rods and cones triggers nerve impulses that our brain interprets as vision. Following isomerization and release from opsin, all-trans-retinal is reduced to all-trans-retinol and then transferred to the adjacent retinal pigment epithelium. It is esterified by lecithin–retinol acyltransferase to retinyl ester and then converted to 11-cis-retinol by the isomerohydrolase RPE65 (also known as isomerase I). It is oxidized to 11-cis-retinal before returning to the photoreceptors to combine with opsin to form rhodopsin (Fig. 1) (1–6).
Figure 1.
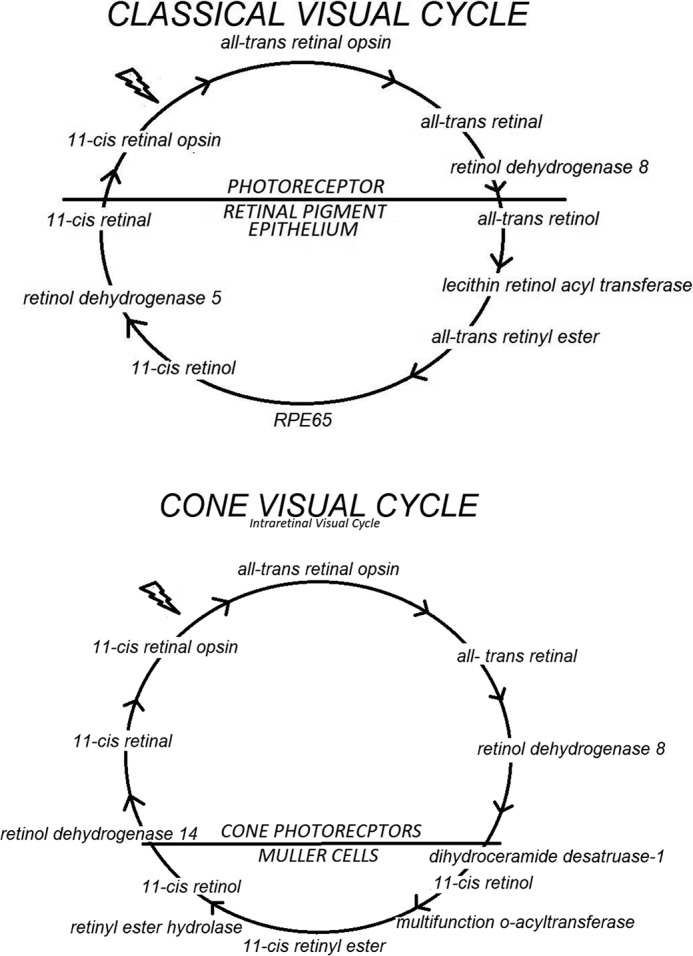
Classic (rod and cone photoreceptor) and the cone (cone photoreceptor) visual cycles.
Intraretinal cone visual cycle
Cones and their photopigments are responsible for daylight vision (photopic) and the perception of colors. There are three types of cones in the retina that respond to short-, medium-, and long-wavelength light, also called S-cones, M-cones, and L-cones, respectively. The intraretinal cone visual cycle begins after photoisomerization, and all-trans-retinal is released from cone pigments. After reduction to all-trans-retinol, it is transported from the cone outer segments to Müller cells in the retina where it is isomerized to become 11-cis-retinol and then esterified to retinyl ester. Upon hydrolysis, 11-cis-retinol is returned to the cone photoreceptors where it is oxidized to 11-cis-retinal to conjugate with cone opsins to form cone pigments. The cone visual cycle is supported by isomerase II, dihydroceramide desaturase 1 (DES1), and multifunction O-acyltransferase (MFAT) (Fig. 1) (7, 8).
Structure and function of selected visual cycle proteins
Rhodopsin structure and function
As mentioned above, rhodopsin consists of opsin and a covalently-bound retinal chromophore. It is a light-sensitive G protein–coupled receptor located in the lipid bilayer of outer segment disc membranes of rod cells. It has seven transmembrane α helices across the disc membrane. The photoreactive chromophore, 11-cis-retinal, is conjugated to a lysine residue of rhodopsin and oriented horizontally in the disc membrane to optimize interaction with photons. Isomerization of 11-cis-retinal into all-trans-retinal by light sets off a cascade of opsin conformational changes that leads to the formation of metarhodopsin II and activates the associated G proteins. This signal is transduced to a cGMP second messenger resulting in a change in the level of 5′GMP and the closure of cation channels (9, 10).
RPE 65 structure and function
RPE65 is the known isomerohydrolase (isomerase I) in the classic rod and cone visual cycle. RPE65 is a member of the carotenoid oxygenase family and is expressed in the retinal pigment epithelium bound to endoplasmic reticulum. It is a Fe2+-dependent isomerase enzyme that catalyzes the hydrolysis of all-trans-retinyl ester and the isomerization of all-trans-retinol to 11-cis-retinol (2). This is consistent with the observation in animal models where RPE65 KO resulted in a low level of 11-cis-retinol with an accumulation of retinyl esters in the eye. There are at least 60 known genetic mutations to RPE65 proteins that account for a variety of retinal and ocular diseases. Individuals afflicted with RPE65 gene mutations generally have early onset blindness (autosomal recessive LCA or RP).
IRBP structure and function
IRBP is secreted by rods and cones into the subretinal space where it constitutes the major protein component of the inter-photoreceptor matrix and interacts with the cone “matrix sheath” (11). IRBP is composed of multiple modules (four in tetrapods and humans and two to three modules in teleosts) with each harboring ∼300 amino acid residues (11).
IRBP has long been thought to play a role in the canonical visual cycle involving retinoid exchange between rods and RPE (12–14). However, understanding IRBP's function in this process has proved to be more challenging than initially anticipated (15–18). Recently, experimental evidence has implicated IRBP as having an important function in the cone visual cycle, involving retinoid exchange between cones and Müller glial cells (19–27).
The mechanism(s) involved in IRBP's role(s) in the cone visual cycle are only beginning to be elucidated. We recently reported that IRBP binds to the cone outer segment and Müller cell microvilli pericellular matrices (11). Such association could target and/or facilitate delivery/uptake of its retinol ligands. Furthermore, we reported that IRBP has free radical scavenging activity (28) and can protect all-trans- and 11-cis-retinol from photodegradation (29).
Gene mutations and retinal diseases
Retinal diseases from gene mutations
Based on all listings in the RetNet, there are 321 known gene mutations in human chromosomes 1–22, X, and mitochondria that result in retinal diseases. They consist of mutations in a wide range of proteins from ocular components such as rhodopsin to complexes I, II, and IV of the mitochondrial electron transfer chain. Table 1 provides a summary of all 321 gene mutations with an example of a mutated protein in each chromosome.
Table 1.
Genes and mapped loci causing retinal diseases
The data were accessed at https://sph.uth.edu/retnet/disease.htm on March 28, 2018. Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
| Chromosome | Mutations | Example of a disease with the mutated protein and gene mutation ID |
|---|---|---|
| 1 | 28 | Recessive Leber's congenital amaurosis; RPE65; candidate gene for LCA |
| 2 | 22 | Recessive retinitis pigmentosa; zinc finger protein; linkage mapping |
| 3 | 17 | Dominant retinitis pigmentosa; rhodopsin; linkage mapping |
| 4 | 21 | Recessive retinitis pigmentosa; LRAT; candidate gene |
| 5 | 9 | Dominant Wagner disease; versican; linkage mapping |
| 6 | 20 | Age-related macular degeneration; complement component 2; association study |
| 7 | 9 | Dominant tritanopia; blue cone opsin; candidate gene |
| 8 | 12 | Recessive Jobert syndrome; centrosome--spindle pole protein; whole-exome sequencing |
| 9 | 11 | Age-related macular degeneration; Toll-like receptor 4; link mapping; association study |
| 10 | 22 | Recessive retinitis pigmentosa; IRBP; homozygosity mapping; candidate gene |
| 11 | 19 | Recessive Usher syndrome; myosin VIIA; linkage mapping |
| 12 | 13 | Recessive fundus albipunctatus; RDH5; candidate gene |
| 13 | 5 | Somatic retinoblastoma; RB1; deletion mapping; candidate gene |
| 14 | 13 | Recessive Leber's congenital amaurosis; RDH 12; homozygosity mapping; linkage mapping |
| 15 | 9 | Recessive Usher syndrome; calcium- and integrin-binding protein; linkage mapping |
| 16 | 16 | Recessive Leber's congenital amaurosis; clusterin-associated protein 1; whole exome sequence |
| 17 | 16 | Dominant retinitis pigmentosa; carbonic anhydrase IV; linkage mapping |
| 18 | 3 | Recessive retinal dystrophy; α1-laminin; homozygosity mapping |
| 19 | 10 | Age-related macular degeneration; complement component 3; association study |
| 20 | 8 | Recessive retinitis pigmentosa; Kizuna centrosomal protein; whole-exome sequencing |
| 21 | 2 | Recessive cone-red dystrophy; chromosome 21 open reading frame 2; homozygosity Ma |
| 22 | 5 | Dominant Sorsby's fundus dystrophy; tissue inhibitor of MP3; linkage mapping |
| X | 24 | Protanopia; red cone opsin; candidate gene |
| Mitochondria | 7 | Leber's hereditary optic neuropathy; complex I, II or V; sequencing |
| Total | 321 |
Selected retinal diseases from gene mutations in visual cycle proteins
RP
This retinal disease is classified as a rod-cone dystrophy, meaning that rods are affected first and then the cones. Typically, patients initially notice difficulties with night vision, trouble adapting to dim illumination, or trouble with side vision in dim light. This can begin in childhood, but some patients may not notice it until in their teens or twenties. Most patients will have symptoms by the time they reach age 30. Eventually, only a small area of central vision remains, and this central vision may be maintained for years with 25% of patients maintaining vision well enough to be able to read for most of their lives (30). Of course, this means that 75% of patients will eventually lose enough central vision, in addition to their already lost peripheral vision, that they are unable to read.
Clinically, a classic “bone-spicule” pattern appears in the mid-peripheral retina; the retinal arterioles are narrowed, and the optic nerve has a pallor. Visual fields early on may exhibit what is described as a ring scotoma, but later the scotoma extends both directions to include all but the central vision. Electroretinography signal, a measurement of visual response to light, may be severely depressed even before retinal changes are visible.
RP is the most common (1 in 3000) genetically inherited retinal disease and can be either an autosomal recessive, autosomal dominant, or X-linked disorder of the eye that can evolve from mutations in more than 50 different genes. As many as half of the RP cases have no other affected family members and are designated as sporadic. Some of these cases designated as sporadic may turn out to have been recessive, but many may be due to new mutations occurring in the germline cells that lead to RP. The inherited genetic mutation causes a progressive loss of rod photoreceptor cells followed by loss of cone photoreceptor cells. One form of autosomal dominant RP is associated with a missense mutation, A346P, located in the rhodopsin gene. This mutation has been found to interfere with normal regeneration of photoreceptors. Mutations resulting in a truncated rhodopsin protein have been associated with autosomal recessive disease. The loss of these photoreceptor cells results in poor night and peripheral vision, and later central vision, which can and will ultimately lead to blindness (31, 32). An RPE65 mutation accounts for ∼2% of RP (32, 33).
LCA
This is an inherited autosomal recessive retinopathy. People born with LCA have greatly reduced vision at birth, although their retinas appear to be normal fundoscopically. Nystagmus is common with the eyes showing difficulty tracking. Sufferers are found to rub their eyes frequently stimulating their retinas to produce light-like impulses called pressure phospenes. Electoretinograms reveal very little retinal function. By the time patients reach puberty the retinal arterioles are constricted, and pigmentary changes of the retinal pigment epithelium occur similar to those with RP. Although the best corrected vision with glasses or contact lenses is limited to finger counting or light perception, it can remain fairly stable throughout early adulthood (34). A co-morbidity is often keratoconus. Speculation exists whether this stems from the associated eye rubbing or whether it is due to the genetics of LCA (35). LCA2 is the form of LCA linked to a mutation in the RPE65 gene (36). RPE65 is the isomerase responsible for converting all-trans-retinal to 11-cis-retinal, which is essential for proper functioning of the visual cycle (1). An RPE65 mutation results in the accumulation of all-trans-retinyl esters and the reduction of rhodopsin in the rod photoreceptor outer segment. The reduction of rhodopsin leads to major retinal abnormalities and dysfunction at birth (37, 38). The autosomal recessive mutations in RPE65 account for ∼6–16% of LCA instances (33, 39).
Stargardt disease (STGD)
This is the most common inherited (1:10,000) juvenile macular condition. Clinically, yellow flecks of lipofuscin pigment are found in the macula (40). Disease progression occurs at different rates among individuals, but usually when the vision decreases to 20/40, it descends rapidly toward a final vision of 20/200 to 20/400 (34). The patient retains peripheral vision because only the central vision is impaired. The loss of central vision stems from atrophy of the macular retinal pigment epithelium and neuroepithelium (40). STGD is a recessive hereditary condition (41). The causality of STGD disease is generally a mutation in the ABCA4 gene that codes for a transmembrane protein that moves all-trans-retinal from inside the photoreceptor disc into the cytoplasm where it is converted to retinol in the visual cycle (42). A multitude of STGD causative ABCA4 mutations have been described (43). The lack of ABCA4 protein function leads to a toxic accumulation of all-trans-retinal, which ultimately causes the death of photoreceptor cells. Currently, there is no treatment for Stargardt disease (44, 45).
Diabetic retinopathy (DR)
This represents the leading cause of blindness in working age adults. It is a reaction to the hyperglycemia associated with both type 1 and type 2 diabetes, but other factors such as lipid levels, blood pressure, and genetics also play a role (43, 46). Diabetic retinopathy begins with damage to retinal capillaries noted clinically as small dots of hemorrhage and microaneurysms and loss of retinal neurons. When the damage to the vasculature reaches a stage where oxygen supply and carbon dioxide removal are sufficiently reduced, the hypoxic retina responds with the development of new blood vessels. Unfortunately, it is this abnormal blood vessel development, referred to as proliferative diabetic retinopathy, that results in retinal detachment and possible blindness.
Currently, the treatments for diabetic retinopathy are only applicable at the later stages of the disease process (46). The pan-retinal photocoagulation entails a series of laser burns on the retina, which destroy some retinal elements thus decreasing oxygen demand while increasing oxygen flow from the underlying choroid. While preserving sight, the patient is left with reduced peripheral and night vision. More recently, anti-vascular endothelial growth factor injections have been used, resulting in constructive atrophy of aberrant vasculature and apparent stabilization of the diabetic retina, but many questions remain about the long-term impact of this treatment on the retina. Although control of the systemic factors in diabetes helps to prevent or delay the development of retinopathy, there is no regimen available to specifically treat or prevent disease development. Many agents, including inflammation, advanced glycation end products, protein kinase C, and oxidants, are thought to play a role in the induction of diabetic retinopathy. It has also been noted that patients with diabetes have reduced levels of IRBP, which have been linked to the progression of DR (43). Garcia-Ramirez et al. (58) found that the elevated levels of glucose and of the cytokines TNFα and IL-1β, associated with diabetes, lead to reduced IRBP expression. Recently, Malechka et al. (47) documented attenuated levels of 11-cis-retinal, IRBP, and rhodopsin in diabetic rats. How the lower amount of IRBP in patients may contribute toward the development of diabetic retinopathy is yet unclear (48).
Strategies to advance the cure of blinding afflictions
Pharmacological approach
Some have recommended the use of vitamin A and fish oils to help reduce progression of RP in adults, but its effectiveness remains controversial (30, 49, 50). Although the use of retinoid isomers and visual cycle inhibitors has been shown to be effective in research studies (9), no pharmaceutical treatments are available or currently approved for retinal diseases such as LCA or STGD. Targets for pharmaceuticals such as β-cyclodextrins are being investigated in an effort to enhance photoreceptor survival in individuals suffering from these maladies (43). Systemic treatments for DR are the intensive control of blood glucose, blood pressure, and lipid levels. Minocycline has been suggested as a possible treatment to prevent the development of DR due to its anti-inflammatory effects and the fact that it crosses the blood–brain barrier enabling it to reach the target inflammatory diabetic processes in the retina, but studies have not yet been completed to explore this possibility (48).
Gene therapy approach
Gene therapy represents one of the experimental strategies for the prevention and treatment of maladies associated with RPE65 mutation. Bainbridge et al. (51) used an rAAV vector to subretinally deliver human RPE65 cDNA under the control of the RPE65 native promoter. One of the three patients in the trial “showed evidence of improvement in retinal function by microperimetry, dark-adapted perimetry, and visual mobility” (51). Maguire et al. (52) subretinally injected rAAV harboring human RPE65 cDNA under the control of the chicken β-actin promoter. All three young adults in this trial “showed evidence of improvement in retinal function based on testing of visual acuity and pupillometry (pupillary light reflex).” Following treatment, the pupillary response to light was three times greater than the baseline. Visual acuity improved, and the visual field was enlarged 2 weeks after treatment” (52). A third group also tested rAAV-mediated delivery of human RPE65 cDNA with the RPE65 native promoter. Cideciyan et al. (53) also reported that one out of three of their trial subjects showed evidence of improved retinal function, including dark adaptation, increased light sensitivity, and visual field expansion (54).
December, 2017, marked the first approval by the Food and Drug Administration of an injection-based gene-delivery regimen to treat an eye disease. Voretigene neparovovec-ryzl (Luxturna) was approved to treat homozygous dysfunctional RPE65 retinal conditions such as LCA. Voretigene neparvovec-ryzl (Luxturna) uses an adeno-associated vector to deliver a complete copy of the RPE65 gene to treat retinal conditions such as LCA (55).
Other approaches
Stem cell therapy is being tested for the treatment of blinding disorders. Muniz et al. (5) utilized human pluripotent stem cells in vitro and derived functional RPE capable of all-trans-retinol uptake from the conditioned culture medium, processing it into 11-cis-retinal for secretion. This study provided the proof of principle of using pluripotent stem cells (iPS) from a patient to generate iPS-RPE for intraocular or retinal injection. Assawachananont et al. (56) were able to subretinally transplant both embryonic and induced pluripotent stem cell–derived three-dimensional retinal sheets into mice with advanced retinal degeneration and showed that the transplanted tissue developed an outer nuclear layer along with complete inner and outer photoreceptor segments. Thus, the authors of this study provided the “proof of concept” for transplantation therapy in the treatment of retinal degenerative diseases (56).
More recently, Kashani et al. (57) reported evidence for improved visual function in a small group of patients with non-neovascular age-related macular degeneration after implanting a sheet of RPE cells under the degenerated macula. None of the four patients receiving the implant have shown any further vision loss, and one patient demonstrated visual improvement by being able to view 17 more letters of the alphabet compared with before the treatment (57).
Discussion
Tremendous efforts are being expended in devising strategies to prevent and cure maladies of the eye. Basic science has laid the requisite foundation in this regard, and different laboratories are testing therapeutic strategies in cell and animal models, although a few groups have been conducting early clinical trials. Basic scientists, clinicians, and physicians from different countries are able to share research results and information freely over cross-sections of populations and to different countries around the globe. Given these favorable circumstances, one can be optimistic that a cure for the blinding disorders is forthcoming.
Acknowledgments
We thank the University of Texas Rio Grande Valley and University of Texas Rio Grande Valley School of Medicine for support of this work.
The authors declare that they have no conflicts of interest with the contents of this article.
- GPCR
- G protein–coupled receptor
- RP
- retinitis pigmentosa
- iPS
- induced pluripotent stem cell
- LCA
- Leber's congenital amaurosis
- IRBP
- interphotoreceptor retinoid-binding protein
- DR
- diabetic retinopathy
- STGD
- Stargardt disease
- RPE
- retinal pigment epithelium
- rAAV
- recombinant adeno-associated virus.
References
- 1. Jin M., Li S., Moghrabi W. N., Sun H., and Travis G. H. (2005) Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell 122, 449–459 10.1016/j.cell.2005.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moiseyev G., Chen Y., Takahashi Y., Wu B. X., and Ma J. X. (2005) RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc. Natl. Acad. Sci. U.S.A. 102, 12413–12418 10.1073/pnas.0503460102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muniz A., Villazana-Espinoza E. T., Hatch A. L., Trevino S. G., Allen D. M., and Tsin A. T. (2007) A novel cone visual cycle in the cone-dominated retina. Exp. Eye Res. 85, 175–184 10.1016/j.exer.2007.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang J. S., and Kefalov V. J. (2011) The cone-specific visual cycle. Prog. Retin. Eye Res. 30, 115–128 10.1016/j.preteyeres.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muñiz A., Greene W. A., Plamper M. L., Choi J. H., Johnson A. J., Tsin A. T., and Wang H. C. (2014) Retinoid uptake, processing, and secretion in human iPS-RPE support the visual cycle. Invest. Ophthalmol. Vis. Sci. 55, 198–209 10.1167/iovs.13-11740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Saari J. C. (2016) Vitamin A and vision. Subcell Biochem. 81, 231–259 10.1007/978-94-024-0945-1_9 [DOI] [PubMed] [Google Scholar]
- 7. Muniz A., Betts B. S., Trevino A. R., Buddavarapu K., Roman R., Ma J. X., and Tsin A. T. (2009) Evidence for two retinoid cycles in the cone-dominated chicken eye. Biochemistry 48, 6854–6863 10.1021/bi9002937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Travis G. H., Kaylor J., and Yuan Q. (2010) Analysis of the retinoid isomerase activities in the retinal pigment epithelium and retina. Methods Mol. Biol. 652, 329–339 10.1007/978-1-60327-325-1_19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiser P. D., and Palczewski K. (2016) Retinoids and retinal diseases. Annu. Rev. Vis. Sci. 2, 197–234 10.1146/annurev-vision-111815-114407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schnedermann C., Yang X., Liebel M., Spillane K. M., Lugtenburg J., Fernández I., Valentini A., Schapiro I., Olivucci M., Kukura P., and Mathies R. A. (2018) Evidence for a vibrational phase-dependent isotope effect on the photochemistry of vision. Nat. Chem. 10, 449–455 10.1038/s41557-018-0014-y [DOI] [PubMed] [Google Scholar]
- 11. Garlipp M. A., and Gonzalez-Fernandez F. (2013) Cone outer segment and Muller microvilli pericellular matrices provide binding domains for interphotoreceptor retinoid-binding protein (IRBP). Exp. Eye Res. 113, 192–202 10.1016/j.exer.2013.02.003 [DOI] [PubMed] [Google Scholar]
- 12. McBee J. K., Palczewski K., Baehr W., and Pepperberg D. R. (2001) Confronting complexity: the interlink of phototransduction and retinoid metabolism in the vertebrate retina. Prog. Retin. Eye Res. 20, 469–529 10.1016/S1350-9462(01)00002-7 [DOI] [PubMed] [Google Scholar]
- 13. Pepperberg D. R., Okajima T. L., Wiggert B., Ripps H., Crouch R. K., and Chader G. J. (1993) Interphotoreceptor retinoid-binding protein (IRBP). Molecular biology and physiological role in the visual cycle of rhodopsin. Mol. Neurobiol. 7, 61–85 10.1007/BF02780609 [DOI] [PubMed] [Google Scholar]
- 14. Gonzalez-Fernandez F. (2003) Interphotoreceptor retinoid-binding protein–an old gene for new eyes. Vision Res. 43, 3021–3036 10.1016/j.visres.2003.09.019 [DOI] [PubMed] [Google Scholar]
- 15. Gonzalez-Fernandez F. (2012) Interphotoreceptor retinoid binding protein; myths and mysteries. J. Ophthalmic. Vis. Res. 7, 100–104 [PMC free article] [PubMed] [Google Scholar]
- 16. Gonzalez-Fernandez F., and Ghosh D. (2008) Focus on molecules: interphotoreceptor retinoid-binding protein (IRBP). Exp. Eye Res. 86, 169–170 10.1016/j.exer.2006.09.003 [DOI] [PubMed] [Google Scholar]
- 17. Ho M. T., Massey J. B., Pownall H. J., Anderson R. E., and Hollyfield J. G. (1989) Mechanism of vitamin A movement between rod outer segments, interphotoreceptor retinoid-binding protein, and liposomes. J. Biol. Chem. 264, 928–935 [PubMed] [Google Scholar]
- 18. Palczewski K., Van Hooser J. P., Garwin G. G., Chen J., Liou G. I., and Saari J. C. (1999) Kinetics of visual pigment regeneration in excised mouse eyes and in mice with a targeted disruption of the gene encoding interphotoreceptor retinoid-binding protein or arrestin. Biochemistry 38, 12012–12019 10.1021/bi990504d [DOI] [PubMed] [Google Scholar]
- 19. Okajima T. I., Pepperberg D. R., Ripps H., Wiggert B., and Chader G. J. (1990) Interphotoreceptor retinoid-binding protein promotes rhodopsin regeneration in toad photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 87, 6907–6911 10.1073/pnas.87.17.6907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carlson A., and Bok D. (1992) Promotion of the release of 11-cis-retinal from cultured retinal pigment epithelium by interphotoreceptor retinoid-binding protein. Biochemistry 31, 9056–9062 10.1021/bi00152a049 [DOI] [PubMed] [Google Scholar]
- 21. Kolesnikov A. V., Ala-Laurila P., Shukolyukov S. A., Crouch R. K., Wiggert B., Estevez M. E., Govardovskii V. I., and Cornwall M. C. (2007) Visual cycle and its metabolic support in gecko photoreceptors. Vision Res. 47, 363–374 10.1016/j.visres.2006.08.024 [DOI] [PubMed] [Google Scholar]
- 22. Parker R. O., Fan J., Nickerson J. M., Liou G. I., and Crouch R. K. (2009) Normal cone function requires the interphotoreceptor retinoid binding protein. J. Neurosci. 29, 4616–4621 10.1523/JNEUROSCI.0063-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parker R. O., and Crouch R. K. (2010) The interphotoreceptor retinoid binding (IRBP) is essential for normal retinoid processing in cone photoreceptors. Adv. Exp. Med. Biol. 664, 141–149 10.1007/978-1-4419-1399-9_17 [DOI] [PubMed] [Google Scholar]
- 24. Parker R., Wang J. S., Kefalov V. J., and Crouch R. K. (2011) Interphotoreceptor retinoid-binding protein as the physiologically relevant carrier of 11-cis-retinol in the cone visual cycle. J. Neurosci. 31, 4714–4719 10.1523/JNEUROSCI.3722-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ala-Laurila P., Kolesnikov A. V., Crouch R. K., Tsina E., Shukolyukov S. A., Govardovskii V. I., Koutalos Y., Wiggert B., Estevez M. E., and Cornwall M. C. (2006) Visual cycle: dependence of retinol production and removal on photoproduct decay and cell morphology. J. Gen. Physiol. 128, 153–169 10.1085/jgp.200609557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Qtaishat N. M., Wiggert B., and Pepperberg D. R. (2005) Interphotoreceptor retinoid-binding protein (IRBP) promotes the release of all-trans-retinol from the isolated retina following rhodopsin bleaching illumination. Exp. Eye Res. 81, 455–463 10.1016/j.exer.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 27. Betts-Obregon B. S., Gonzalez-Fernandez F., and Tsin A. T. (2014) Interphotoreceptor retinoid-binding protein (IRBP) promotes retinol uptake and release by rat Muller cells (rMC-1) in vitro: implications for the cone visual cycle. Invest. Ophthalmol. Vis. Sci. 55, 6265–6271 10.1167/iovs.14-14721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez-Fernandez F., Sung D., Haswell K. M., Tsin A., and Ghosh D. (2014) Thiol-dependent antioxidant activity of interphotoreceptor retinoid-binding protein. Exp. Eye Res. 120, 167–174 10.1016/j.exer.2014.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonzalez-Fernandez F., Betts-Obregon B., Yust B., Mimun J., Sung D., Sardar D., and Tsin A. T. (2015) Interphotoreceptor retinoid-binding protein protects retinoids from photodegradation. Photochem Photobiol. 91, 371–378 10.1111/php.12416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sharma M. C., and Ho A. C. (2012) in Color Atlas and Synopsis of Clinical Ophthalmology (Fineman M. S., and Ho A. C., eds) Chapter 5, pg. 228, Wills Eye Institute, Lippincott Williams and Wilkins, Philadelphia [Google Scholar]
- 31. Gal A., Li Y., Thompson D. A., Weir J., Orth U., Jacobson S. G., Apfelstedt-Sylla E., and Vollrath D. (2000) Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 26, 270–271 10.1038/81555 [DOI] [PubMed] [Google Scholar]
- 32. Hartong D. T., Berson E. L., and Dryja T. P. (2006) Retinitis pigmentosa. Lancet 368, 1795–1809 10.1016/S0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- 33. Cai X., Conley S. M., and Naash M. I. (2009) RPE65: role in the visual cycle, human retinal disease, and gene therapy. Ophthalmic. Genet. 30, 57–62 10.1080/13816810802626399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumaran N., Ripamonti C., Kalitzeos A., Rubin G. S., Bainbridge J. W. B., and Michaelides M. (2018) RPE65 associated Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 59, 85–93 10.1167/iovs.17-22905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elder M. J. (1994) Leber congenital amaurosis and its association with keratoconus and keratoglobus. J. Pediatr. Ophthalmol. Strabismus. 31, 38–40 [DOI] [PubMed] [Google Scholar]
- 36. Morimura H., Fishman G. A., Grover S. A., Fulton A. B., Berson E. L., and Dryja T. P. (1998) Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc. Natl. Acad. Sci. U.S.A. 95, 3088–3093 10.1073/pnas.95.6.3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Redmond T. M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J. X., Crouch R. K., and Pfeifer K. (1998) Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 20, 344–351 10.1038/3813 [DOI] [PubMed] [Google Scholar]
- 38. Thompson D. A., Gyürüs P., Fleischer L. L., Bingham E. L., McHenry C. L., Apfelstedt-Sylla E., Zrenner E., Lorenz B., Richards J. E., Jacobson S. G., Sieving P. A., and Gal A. (2000) Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest. Ophthalmol. Vis. Sci. 41, 4293–4299 [PubMed] [Google Scholar]
- 39. Kiser P. D., Golczak M., Maeda A., and Palczewski K. (2012) Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta 1821, 137–151 10.1016/j.bbalip.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Allikmets R., Singh N., Sun H., Shroyer N. F., Hutchinson A., Chidambaram A., Gerrard B., Baird L., Stauffer D., Peiffer A., Rattner A., Smallwood P., Li Y., Anderson K. L., Lewis R. A., Nathans J., et al. (1997) A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 15, 236–246 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- 41. Wiggs J. L. (2014) in Molecular Genetics of Selected Ocular Disorders (Yanoff M., and Duker J. S., eds) 4th Ed., pp. 9–15, Elsevier-Saunders, Philadelphia [Google Scholar]
- 42. Maeda A., Maeda T., Golczak M., Chou S., Desai A., Hoppel C. L., Matsuyama S., and Palczewski K. (2009) Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J. Biol. Chem. 284, 15173–15183 10.1074/jbc.M900322200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen Y., Palczewska G., Mustafi D., Golczak M., Dong Z., Sawada O., Maeda T., Maeda A., and Palczewski K. (2013) Systems pharmacology identifies drug targets for Stargardt disease-associated retinal degeneration. J. Clin. Invest. 123, 5119–5134 10.1172/JCI69076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Westerfeld C., and Mukai S. (2008) Stargardt's disease and the ABCR gene. Semin. Ophthalmol. 23, 59–65 10.1080/08820530701745249 [DOI] [PubMed] [Google Scholar]
- 45. López-Rubio S., Chacon-Camacho O. F., Matsui R., Guadarrama-Vallejo D., Astiazarán M. C., and Zenteno J. C. (2018) Retinal phenotypic characterization of patients with ABCA4 retinopathydue to the homozygous p.Ala1773Val mutation. Mol. Vis. 24, 105–114 [PMC free article] [PubMed] [Google Scholar]
- 46. Grigsby J. G., and Tsin A. T. C. (eds) (2018) Early Events in Diabetic Retinopathy and Intervention Strategies, Chapter 1, pp. 3–6, IntechOpen, London [Google Scholar]
- 47. Malechka V. V., Moiseyev G., Takahashi Y., Shin Y., and Ma J. X. (2017) Impaired rhodopsin generation in the rat model of diabetic retinopathy. Am. J. Pathol. 187, 2222–2231 10.1016/j.ajpath.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Grigsby J. G., Cardona S. M., Pouw C. E., Muniz A., Mendiola A. S., Tsin A. T., Allen D. M., and Cardona A. E. (2014) The role of microglia in diabetic retinopathy. J. Ophthalmol. 2014, 705783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Berson E. L., Rosner B., Sandberg M. A., Hayes K. C., Nicholson B. W., Weigel-DiFranco C., and Willett W. (1993) A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch. Ophthalmol. 111, 761–772 10.1001/archopht.1993.01090060049022 [DOI] [PubMed] [Google Scholar]
- 50. Rayapudi S., Schwartz S. G., Wang X., and Chavis P. (2013) Vitamin A and fish oils for retinitis pigmentosa. Cochrane Database Syst. Rev. CD008428 10.1002/14651858.CD008428.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bainbridge J. W., Mehat M. S., Sundaram V., Robbie S. J., Barker S. E., Ripamonti C., Georgiadis A., Mowat F. M., Beattie S. G., Gardner P. J., Feathers K. L., Luong V. A., Yzer S., Balaggan K., Viswanathan A., et al. (2015) Long-term effect of gene therapy on Leber's congenital amaurosis. N. Engl. J. Med. 372, 1887–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Maguire A. M., High K. A., Auricchio A., Wright J. F., Pierce E. A., Testa F., Mingozzi F., Bennicelli J. L., Ying G. S., Rossi S., Fulton A., Marshall K. A., Banfi S., Chung D. C., Morgan J. I., et al. (2009) Age-dependent effects of RPE65 gene therapy for Leber's congenital amaurosis: a phase 1 dose-escalation trial. Lancet 374, 1597–1605 10.1016/S0140-6736(09)61836-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cideciyan A. V., Alerman T. S., Boye S. L., Schwartz S. B., Kaushal S., Roman A. J., Pang J.-J., Sumaroka A., Windsor E. A. M., Wilson J. M., Flotte T. R., Fishman G. A., Heon E., Stone E. M., Byrne B. J., Jacobson S. G., and Hauswirth W. H. (2008) Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. U.S.A. 105, 15112–15117 10.1073/pnas.0807027105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hareendran S., Balakrishnan B., Sen D., Kumar S., Srivastava A., and Jayandharan G. R. (2013) Adeno-associated virus (AAV) vectors in gene therapy: immune challenges and strategies to circumvent them. Rev. Med. Virol. 23, 399–413 10.1002/rmv.1762 [DOI] [PubMed] [Google Scholar]
- 55. Spark Therapeutics (2017) LuxturnaTM, an Injectable Drug, rAAV with a RPE65 Repair, Spark Therapeutics, Philadelphia [Google Scholar]
- 56. Assawachananont J., Mandai M., Okamoto S., Yamada C., Eiraku M., Yonemura S., Sasai Y., and Takahashi M. (2014) Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice. Stem Cell Reports 2, 662–674 10.1016/j.stemcr.2014.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kashani A. H., Lebkowski J. S., Rahhal F. M., Avery R. L., Salehi-Had H., Dang W., Lin C. M., Mitra D., Zhu D., Thomas B. B., Hikita S. T., Pennington B. O., Johnson L. V., Clegg D. O., Hinton D. R., and Humayun M. S. (2018) A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Transl. Med. 10, eeao4097 10.1126/scitranslmed.aao4097 [DOI] [PubMed] [Google Scholar]
- 58. García-Ramirez M., Hernández C., Villarroel M., Canals F., Alonso M. A., Fortuny R., Masmiquel L., Navarro A., García-Arumí J., and Simó R. (2009) Interphotoreceptor retinoid-binding protein (IRBP) is downregulated at early stages of diabetic retinopathy. Diabetologia 52, 2633–2641 10.1007/s00125-009-1548-8 [DOI] [PubMed] [Google Scholar]