

Abstract
The pyruvate dehydrogenase multienzyme complex (PDHc) connects glycolysis to the tricarboxylic acid cycle by producing acetyl-CoA via the decarboxylation of pyruvate. Because of its pivotal role in glucose metabolism, this complex is closely regulated in mammals by reversible phosphorylation, the modulation of which is of interest in treating cancer, diabetes, and obesity. Mutations such as that leading to the αV138M variant in pyruvate dehydrogenase, the pyruvate-decarboxylating PDHc E1 component, can result in PDHc deficiency, an inborn error of metabolism that results in an array of symptoms such as lactic acidosis, progressive cognitive and neuromuscular deficits, and even death in infancy or childhood. Here we present an analysis of two X-ray crystal structures at 2.7-Å resolution, the first of the disease-associated human αV138M E1 variant and the second of human wildtype (WT) E1 with a bound adduct of its coenzyme thiamin diphosphate and the substrate analogue acetylphosphinate. The structures provide support for the role of regulatory loop disorder in E1 inactivation, and the αV138M variant structure also reveals that altered coenzyme binding can result in such disorder even in the absence of phosphorylation. Specifically, both E1 phosphorylation at αSer-264 and the αV138M substitution result in disordered loops that are not optimally oriented or available to efficiently bind the lipoyl domain of PDHc E2. Combined with an analysis of αV138M activity, these results underscore the general connection between regulatory loop disorder and loss of E1 catalytic efficiency.
Keywords: pyruvate dehydrogenase complex (PDC), protein structure, protein conformation, decarboxylase, mutant, crystal structures, inborn error of metabolism, phosphorylation, pyruvate dehydrogenase deficiency, regulation
Introduction
During glycolysis, a molecule of glucose is converted into two molecules of pyruvate that subsequently undergo conversion to acetyl-CoA for entry into the tricarboxylic acid cycle. Pyruvate is thus a key intermediate in aerobic respiration. The production of acetyl-CoA from pyruvate is accomplished in several steps by a large protein complex known as the pyruvate dehydrogenase multienzyme complex (PDHc).3 Three different enzymes that are present in multiple copies within the complex provide the required enzymatic activities. Pyruvate dehydrogenase (E1) decarboxylates pyruvate and reductively acetylates a lipoyl group covalently linked to dihydrolipoamide acetyltransferase (E2). In an E2 active site, the acetyl group is transferred to CoA to produce acetyl-CoA, leaving the lipoyl group in a reduced state. The E3 component, dihydrolipoamide dehydrogenase, which is connected to the complex core via the E3 binding protein (E3BP), oxidizes the lipoyl group on E2, returning it to its initial state for further rounds of catalysis, and concomitantly reduces NAD+ to NADH. The overall activity of the complex is regulated by kinase/phosphatase action on the PDHc E1 component (1). The interactions between PDHc E1 and E2 relevant to the present work are shown schematically in Fig. 1A.
Figure 1.

A, schematic overview of the interaction between human PDHc E1 and E2. In E2, the subunit binding domain (labeled S) that anchors E2 to E1 is colored green; the corresponding domain is called PSBD in some species such as E. coli. E2's two lipoyl domains (L1 and L2) are each colored pink. For E1, the two αβ heterodimers that make up the α2β2 heterotetramer are colored yellow and blue. In this drawing, one active site entrance in the E1 heterotetramer is empty, whereas the other is occupied by a lipoyl domain from E2. No interactions of catalytic or structural consequence are known to exist between E1 and E3, although both are assumed to reside close to each other on the complex periphery. Each, however, has its own independent tethering linkage to a complex core domain. B, the structure from the present work of WT human E1 with bound ThDP:AcPhi adducts. In each α-subunit the two phosphorylatable regulatory loops are colored orange and labeled. The ThDP:AcPhi adducts are shown as space-filling representations.
Because of its central role in energy production, malfunction of PDHc has major consequences during development and the immediate postnatal period, in the form of PDHc deficiency. Before birth, the fetus may fail to gain weight appropriately. Shortly after birth, one or several metabolic and neurological complications become evident. These include lactic acidosis, a dangerous accumulation in the blood of lactate derived from the reduction of pyruvate in the absence of sufficient PDHc function, as well as neurological problems ranging from lethargy, poor muscle tone, and poor feeding to intellectual development deficits, seizures, and death (2–4). The severity of the symptoms is correlated with the degree of reduction in PDHc activity. Severe lactic acidosis is frequently fatal in affected newborns, and neurological problems related to PDHc deficiency are progressive.
Mutations resulting in PDHc deficiency can be found in the genes coding for the three main PDHc enzymatic components, but most are found in the pyruvate-decarboxylating E1 component. Human PDHc E1 is an α2β2 heterotetramer that has a molecular mass of 150 kDa. Its function requires both the coenzyme ThDP and magnesium ions. In the present work we examine the structural basis for impaired function in the pathogenic αV138M variant of human PDHc E1 (5, 6). The αV138M E1 structure, along with the structure of WT–AcPhi E1, underscores the connection between regulatory loop disorder and reduced function in this enzyme.
Results
The structure of WT E1 with bound ThDP and substrate analogue acetylphosphinate is similar to that of WT E1 with bound cofactor ThDP
We reported earlier that AcPhi is a tight slow binding inhibitor of WT E1. Kinetic analysis of the progress curves of the overall PDHc assay using reconstituted PDHc containing WT E1 enabled calculation of a Ki,AcPhi of 0.014 μm. The calculated rate constants for AcPhi association (k1) and dissociation (k−1) were 2.18 μm−1 min−1 and 0.0305 min−1, respectively (7). Also, CD spectra of WT E1 treated with AcPhi were unaffected by overnight dialysis, indicating tight binding; however, AcPhi could be replaced in the WT E1 active centers by pyruvate (5 mm) (7). The covalent adduct of ThDP and AcPhi is a mimic of the physiological pre-decarboxylation intermediate adduct of ThDP and pyruvate (Fig. 2A). Although the components of the adduct are covalently bonded to each other, the adduct itself is bound noncovalently to E1.
Figure 2.

A, the chemical structure of the native coenzyme thiamin diphosphate (top) and the pre-decarboxylation reaction intermediate adducts (bottom). Pyruvate is the native decarboxylation substrate, whereas AcPhi is the inhibitory substrate analogue used in this study. B, representative omit mFo − DFc electron density contoured at 3σ for the ThDP:AcPhi adduct in one binding site of WT human E1. The presence of acetylphosphinate covalently linked to ThDP is clearly observable in the density. The position of αVal-138 is indicated.
We have now determined and analyzed the structure of WT human E1 with the bound covalent adduct of ThDP and AcPhi. To our knowledge, this is the first structure of human E1 with a covalent ThDP:substrate or ThDP:analogue adduct in its active site. The crystals belong to the primitive orthorhombic space group P212121, and there is a single heterotetramer in the asymmetric unit. The structure was determined by molecular replacement using the native structure (PDB code 1NI4) as the search model. Detailed data collection and refinement statistics are given in Table 1. The overall structure of WT–AcPhi E1 is shown in Fig. 1B. There are no major structural changes compared with holo E1 (the E1 enzyme with cofactors ThDP and Mg2+, PDB code 1NI4). A superposition of the adduct-bound structure with holo E1 yields an r.m.s. deviation of 0.38 Å over 1384 aligned Cα atoms. All three active site histidine residues, namely αHis-63, αHis-263, and β′His-128, are involved in interactions with the substrate analogue. These contacts are shown in Fig. 8 and will be discussed later when regulatory issues are addressed. Of these, β′His-128 is hydrogen-bonded to the C2-hydroxyl oxygen, and αHis-63 and αHis-263 are hydrogen-bonded to one of the phosphonyl oxygens in the analogue. αHis-263, which is located in phosphorylation loop A, is also hydrogen-bonded to the diphosphate tail of ThDP. As will become apparent below, the conserved interaction of αHis-263 with diphosphate and the substrate analogue in the active site may play an important role in stabilizing phosphorylation loop A, residues α259–α284. This substrate analogue interaction mimics the interaction very likely present with the physiological pre-decarboxylation intermediate, 2-(1-carboxyl-1-hydroxyethyl)-ThDP, in the reaction of pyruvate with ThDP. The chemical structures of ThDP and the ThDP:pyruvate and ThDP:AcPhi adducts are given in Fig. 2A. Fig. 2B depicts omit electron density for one of the two ThDP:AcPhi adducts in the WT–AcPhi E1 structure's crystallographic asymmetric unit. The substrate analogue intermediate adduct was generated by soaking AcPhi into holo WT E1 crystals, and the density clearly confirms that AcPhi is covalently bound to ThDP. Although only one binding site is displayed, the density for ThDP:AcPhi in both binding sites within the heterotetramer is unambiguous.
Table 1.
X-ray data collection and refinement statistics
Values in parentheses refer to the highest-resolution shell. For each structure, 5% of the reflections were randomly assigned to the free set, and these reflections were not used at any point during structure refinement.
| WT–AcPhi E1 | αV138M E1 | |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.0 | 1.0 |
| Resolution range (Å) | 47.58–2.70 | 35.69–2.69 |
| Space group | P212121 | P21 |
| Unit cell lengths (Å) | 102.27, 127.73, 129.78 | 121.76, 123.92, 128.28 |
| Unit cell angles (°) | 90, 90, 90 | 90, 118.12, 90 |
| Total reflections | 307,019 | 247,251 |
| Unique reflections | 44,072 | 89,293 |
| Multiplicity | 7.0 (7.2) | 2.8 (2.7) |
| Completeness (%) | 99.2 (98.3) | 95.6 (96.8) |
| Mean I/σI | 14.9 (5.3) | 8.4 (1.6) |
| Rmerge | 0.096 (0.275) | 0.114 (0.684) |
| Rmeas | 0.105 (0.297) | 0.138 (0.826) |
| CC1/2 | 0.996 (0.969) | 0.993 (0.726) |
| Refinement | ||
| Rwork | 0.186 (0.254) | 0.192 (0.319) |
| Rfree | 0.235 (0.321) | 0.234 (0.369) |
| Number of tetramers in ASU | 1 | 2 |
| Number of non-hydrogen atoms | 11,139 | 20,948 |
| Macromolecules | 10,698 | 20,540 |
| Ligands | 66 | 106 |
| Solvent | 375 | 302 |
| Protein residues | 1,384 | 2,661 |
| R.m.s. bonds (Å) | 0.005 | 0.006 |
| R.m.s. angles (°) | 0.92 | 1.09 |
| Ramachandran favored (%) | 96.51 | 96.31 |
| Ramachandran allowed (%) | 3.12 | 3.50 |
| Ramachandran outliers (%) | 0.36 | 0.19 |
| Clashscore (percentile) | 3.27 (100) | 3.45 (100) |
| Average B-factor | 46.9 | 55.6 |
| Macromolecules | 47.0 | 55.7 |
| Ligands | 55.9 | 85.4 |
| Solvent | 45.9 | 39.3 |
| PDB code | 6CFO | 6CER |
Figure 8.
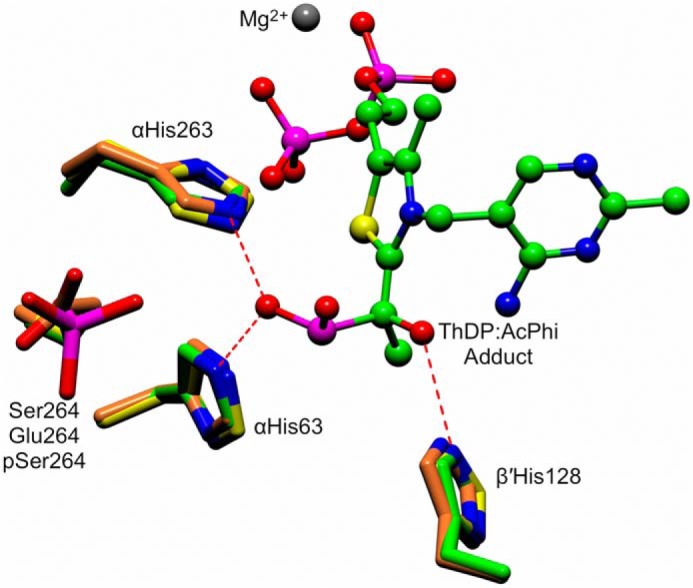
An active site superposition of the WT–AcPhi E1 structure with the phosphorylation-mimicking αS264E substitution (PDB code 2OZL) and the structure of the WT E1 enzyme phosphorylated at αSer-264 (PDB 3EXH). The ThDP:AcPhi adduct from the WT–AcPhi E1 structure is shown as a ball-and-stick representation. Conserved histidines that interact with and stabilize the substrate analogue are shown as green cylinders (WT-AcPhi), orange cylinders (αS264E substitution), and yellow cylinders (pSer-264). The position of Ser-264, the site of phosphorylation, is indicated.
The αV138M variant alters ThDP's torsion angles and displaces its diphosphate tail, resulting in disordered regulatory loops
The αV138M substitution in E1 is a naturally occurring mutation causing impairment in PDHc function (5). Previous X-ray structural studies predicted that Val-138 in the E1 active centers could provide hydrophobic interactions to the aminopyrimidine and thiazolium moieties of ThDP and could be important for maintaining ThDP in the V-conformation and for catalysis (8). We have determined and analyzed the X-ray crystal structure of human αV138M E1 to examine the structural etiology of the mutation's association with PDHc deficiency. Full data collection and refinement statistics can be found in Table 1. The variant crystallized in the primitive monoclinic space group P21 with two heterotetramers in the asymmetric unit; these are essentially identical to each other, having an r.m.s. deviation of 0.26 Å over 1312 Cα atoms. The global structure is also similar to the WT holo E1 (PDB code 1NI4) (8) and WT–AcPhi (this work) structures, yielding r.m.s. deviations of 0.41 and 0.47 Å for 1323 and 1313 aligned Cα atoms, respectively.
Examination of the structures reveals that the largest differences from WT are found in the active sites, specifically in the conformation of the ThDP coenzymes and nearby loops. Fig. 3A shows an active site after superposition of the WT–AcPhi E1 and αV138M E1 structures calculated using only protein and no coenzyme atoms. The mutation resulting in Met-138 substantially alters the conformation of ThDP, with the diphosphate moiety showing the largest changes, the thiazolium ring modest differences, and the 4′-aminopyrimidine ring only small differences. Fig. 3A reveals that the presence of the longer side chain upon mutation changing Val-138 to Met pushes the thiazolium ring away due to steric interactions between the Met Cϵ atom and the C4, C4α, and C5 atoms of the thiazolium ring. These atoms, if as in the ThDP conformation found in WT E1, would be only 2.2, 2.4, and 2.5 Å from the variant Met's Cϵ atom, respectively, i.e. well short of accepted van der Waals contact distances (Fig. S1). To analyze the conformations of the thiazolium and 4′-aminopyrimidine rings more quantitatively, we calculated the classic dihedral angles, φT and φP, defined by the atoms C5′-C(3,5′)-N3-C2 and N3-C(3,5′)-C5′-C4′ (9), respectively, for the ThDP molecules in the αV138M and WT–AcPhi E1 structures, as well as for those found in published structures of WT holo E1 (8, 10), the phosphorylation-mimicking substitution αS264E (11), and WT E1 actually phosphorylated at αSer-264 (10). The atoms defining these dihedral angles are labeled on the ThDP structure in Fig. 2A, and the results of the calculations are shown in Table 2. The structures reveal that the thiazolium dihedral angle φT is more variable than the 4′-aminopyrimidine dihedral angle φP over all structures examined. In WT E1, the average φT is ∼92–96°, values that are reduced only slightly to 91–93° upon introduction of a negative charge at Ser-264 via either the S264E substitution or phosphorylation at this position. In contrast, φT shows its smallest average value, 86.5°, in the αV138M variant, although the range of measured values is larger than for the other structures. Changes in this dihedral angle may be related to the ability of the coenzyme to sample multiple tautomeric states while bound to the enzyme (see below). Formation of an adduct between ThDP and the substrate analogue AcPhi results in an increase in this dihedral angle to ∼103°. This angle is also subject to a noticeable variation between homologues in different species, as φT for ThDP molecules in the homodimeric Escherichia coli pyruvate dehydrogenase E1 is ∼110°, larger than any value measured for ThDP in its human enzyme counterparts. In contrast, there is much less variation in φP, which has values of approximately −67° to −73° across all structures in Table 2.
Figure 3.
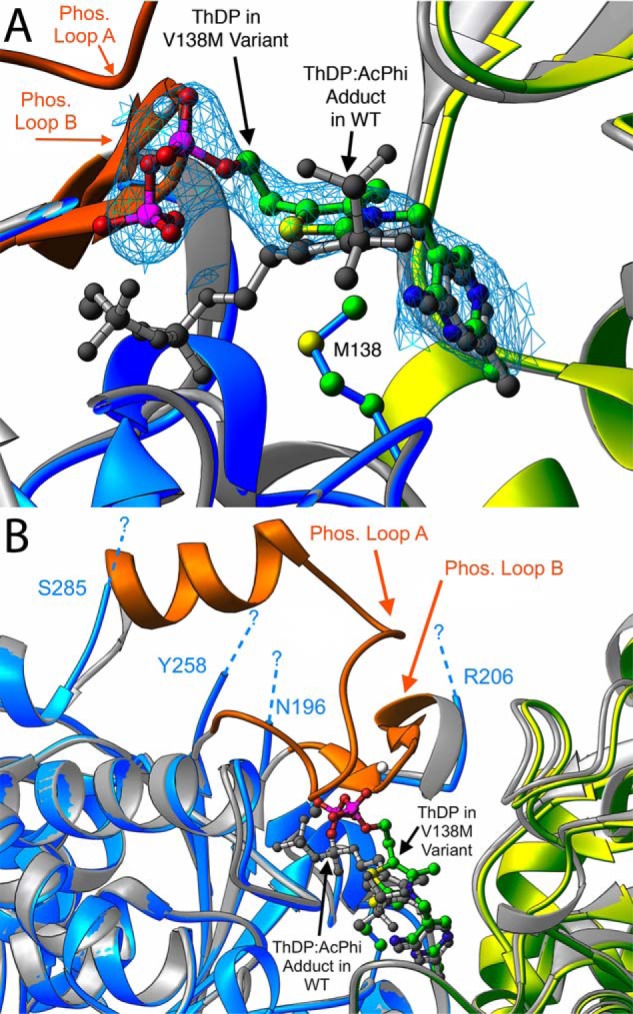
A comparison of the active sites in the αV138M and WT–AcPhi E1 structures. A, a magnified view showing the differences in ThDP binding. The blue and yellow ribbon diagrams correspond to the α′ and β chains of αV138M E1, whereas the gray ribbons indicate the WT–AcPhi E1 structure. ThDP bound to αV138M E1 is shown as a ball-and-stick representation colored by atom type, whereas the ThDP:AcPhi adduct from the WT structure is colored gray. The blue mesh is the 2mFo − DFc electron density contoured at 1σ for the ThDP bound to αV138M. The stretches of sequences that are observable in the WT–AcPhi E1 structure but disordered and not seen in the αV138M E1 structure, which correspond to phosphorylation loops A and B, are colored orange. The dashed lines indicate the limits of the disordered regions. B, a different view of the active site that makes clear the extent and location of disordering in the αV138M E1 variant.
Table 2.
Dihedral angles for the thiazolium (φT) and 4′-aminopyrimidine (φP) rings of ThDP coenzymes found in select human pyruvate dehydrogenase (E1) structures
Values are given as the mean ± S.D. over n ThDP molecules. The value of n depends on the number of heterotetramers (or homodimers, in the case of the E. coli structure) found in the given structure's crystallographic asymmetric unit.
| PDB code | Structure | φT (°) | φP (°) | n |
|---|---|---|---|---|
| 1NI4 | WT holo E1 | 95.7 ± 1.0 | −67.9 ± 2.6 | 2 |
| 3EXE | WT holo E1 | 92.9 ± 2.9 | −73.2 ± 0.5 | 4 |
| 2OZL | Phosphomimic αS264E E1 | 93.1 ± 0.3 | −71.8 ± 0.1 | 2 |
| 3EXH | Phosphorylated αSer-264 E1 | 91.1 ± 1.3 | −73.9 ± 0.7 | 4 |
| 6CER | αV138M E1 | 86.5 ± 5.9 | −68.5 ± 4.0 | 4 |
| 6CFO | WT–AcPhi E1 adduct | 103.4 ± 0.3 | −67.3 ± 0.1 | 2 |
| 2IEA | WT E. coli holo E1 | 110.3 ± 0.3 | −68.6 ± 0.5 | 2 |
By far the largest structural consequence in the αV138M variant is a substantial displacement of ThDP's diphosphate tail compared with its position in WT structures (Fig. 3A). In the variant structure, ThDP phosphate atoms P1 and P2 are displaced 6.6 and 3.4 Å, respectively, from their positions in the WT structure. We note that, with the exception of the thiazolium ring in one of the four ThDP molecules within the αV138M asymmetric unit, the electron density for the variant's cofactor molecules is clear, and the shifted positions of the ThDP diphosphate tails are unambiguous. Nevertheless, comparison of ThDP conformations in all four active sites does reveal appreciable variation among the diphosphate tails, as seen in Fig. 4.
Figure 4.
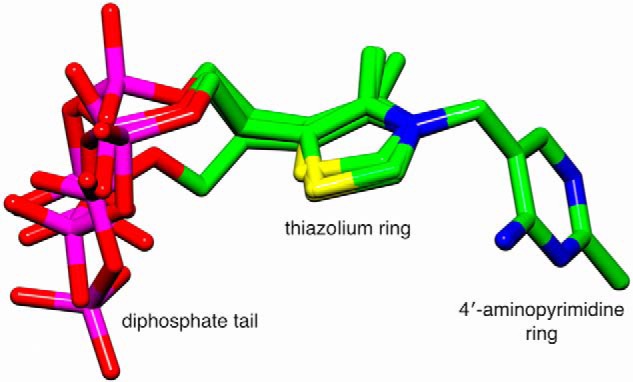
Shown is a superposition of the four ThDP coenzymes from the two heterotetramers of the new structure of human αV138M pyruvate dehydrogenase. The superposition was calculated using only the N1′, C2′, N3′, C4′, C5′, and C6′ atoms of the 4′-aminopyrimidine ring.
The altered locations of the diphosphate tails have two consequences. The first is that the conserved magnesium binding site is disrupted. Fig. 5 shows the canonical octahedral coordination of Mg2+ commonly found in WT E1 structures such as WT–AcPhi. In WT E1, Mg2+ is coordinated by two oxygen atoms from the ThDP diphosphate tail, as well as by a water molecule, the side chains of αAsp-167 and αAsn-196, and the backbone carbonyl oxygen of αTyr-198. In the αV138M variant, only the side chain of αAsp-167 retains its native position; both phosphates in the ThDP tail are located far from their WT position, the coordinating water molecule is absent, the backbone at αAsn-196 diverges from its WT position, and αTyr-198 is totally disordered and unobservable. As a consequence, we were unable to locate any reasonable density for Mg2+ in either binding site for one of the two tetramers in the αV138M asymmetric unit; in the other tetramer, we were able to model and successfully refine a magnesium ion within interaction distance of each ThDP diphosphate in density too strong to be a water molecule, although these magnesium ions have nothing resembling the octahedral coordination sphere found in the WT structure (Fig. 5). In the αV138M variant, then, magnesium binding is substantially altered, and possibly nearly abolished. This binding alteration contradicts the generally held belief that magnesium is required for E1 to bind ThDP; our structural observations indicate that the 6-coordinate magnesium binding site is probably necessary to anchor ThDP in its canonical bound conformation, but not strictly necessary for E1 to bind ThDP in general.
Figure 5.
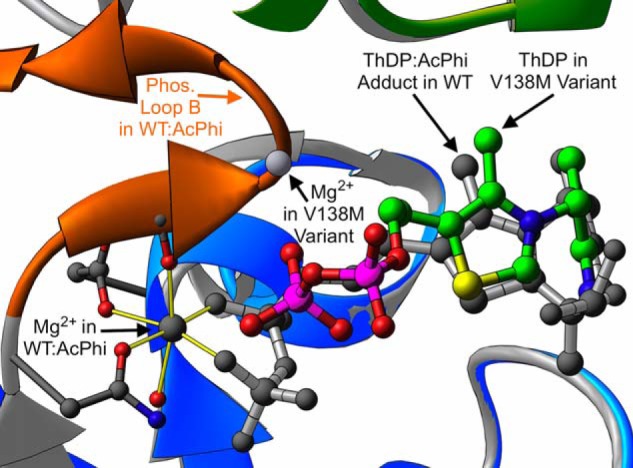
A depiction of magnesium binding in WT–AcPhi and the αV138M variant. The color coding is the same as described in the legend to Fig. 3. The yellow cylinders indicate the canonical octahedral coordination of the magnesium ion commonly found in ThDP-dependent enzymes. In contrast, in the αV138M variant this binding site is abolished due to the displacement of ThDP's diphosphate tail and the disordering of phosphorylation loop B.
Residues αAsn-196 and αTyr-198 from the magnesium binding site are part of phosphorylation loop B, which comprises residues Asn-196–Arg-206 and results in E1 inactivation when phosphorylated at αSer-203. In αV138M E1, this loop is completely disordered and unobservable from residues α197 to α205. The observed, displaced ThDP diphosphate tail would sterically clash with the backbone of residues αGly-199 and αMet-200 in this loop, if it had the same conformation as in the WT and intermediate WT–AcPhi structures. The disordering of phosphorylation loop B in the variant therefore likely results from the necessity of avoiding steric overlap, combined with the reduction/loss of magnesium coordination. Phosphorylation loop B is labeled and colored orange in Figs. 1, 3, and 5. The ThDP diphosphate tail in the variant structure also would directly overlap the side chain of residue αHis-263 in the WT–AcPhi structure. In fact, the entire stretch from residues α259 to α284 is also disordered and unobservable in the variant; this stretch corresponds to phosphorylation loop A. E1 is also inactivated when phosphorylated at αSer-264 and/or αSer-271 in this loop. The disordering of phosphorylation loop A in the variant therefore likely results from the steric clash between ThDP and residue αHis-263 as well as from the disruption of interactions with disordered phosphorylation loop B. The extent of disordering of these two loops is demonstrated in Fig. 3B.
In summary, the new structural results demonstrate that both the WT–AcPhi and αV138M E1 structures are globally very similar to the WT holo E1 structure. In WT–AcPhi E1, the active centers are also unchanged compared with WT holo E1. However, the active centers in αV138M E1 are altered compared with the WT structures: the ThDP thiazolium ring is modestly displaced from its WT position, the thiazolium ring dihedral angle is decreased, magnesium binding is altered or disrupted, and the diphosphate tail is substantially displaced from its native position, resulting in complete disordering of the regulatory phosphorylation loops A and B. We next relate these observed structural differences to functional changes.
PDHc activity is deficient in the presence of αV138M E1
We have assayed the overall activity of human PDHc by assembling αV138M E1 with E2·E3BP and E3. Overall complex activity was measured by monitoring the production of NADH. When WT E1 is replaced by αV138M E1, production of NADH falls to only 2–4% of the WT level (data not shown). This implies that either the decarboxylation or reductive acetylation functions of E1 (or both) are compromised by the mutation. Previous work examining αV138M E1 decarboxylation activity by monitoring the production of 14CO2 found the variant to have only 33% of the WT activity (6); this is lower, but probably not low enough to explain the greatly reduced NADH production that we observed for the overall reaction, which suggests that both E1 functions are compromised by the mutation. To determine whether reduced affinity for ThDP underlies the loss of enzymatic activities, we used fluorescence quenching experiments to measure the affinity of apo-WT and apo-αV138M E1 for ThDP. On excitation at 295 nm, the spectrum of WT E1 displayed an emission maximum at 338 nm that was quenched on addition of 0.10–100 μm ThDP (Fig. 6A). About 17.8% quenching of the WT E1 fluorescence was reached, which allowed us to calculate a Kd,ThDP of 5.8 μm and a Hill coefficient of 1.0, which implies no cooperativity between the two active centers of WT E1. In contrast, quenching of the αV138M E1 fluorescence did not reach a steady-state maximum even at 100 μm ThDP; its titration curve suggests ThDP can saturate only one of two active centers (Fig. 6B). The value of Kd,ThDP calculated for the first active site was 4.4 μm, but the dissociation constant for the second active site must be weaker because it was not saturated over the range of ThDP concentrations tested. This suggests again that ThDP must bind αV138M E1 differently than it does WT E1, which agrees with our structural results.
Figure 6.
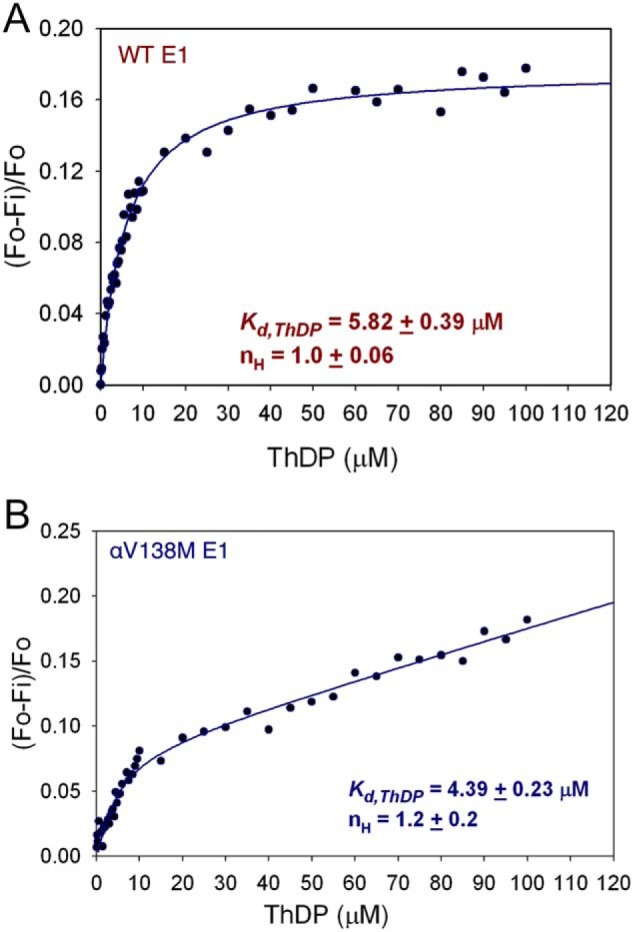
Fluorescence titration data used to calculate the dissociation constant for ThDP in WT E1 (A) and the first binding site in αV138M E1 (B). Apo-WT E1 (0.034 mg/ml) or apo-αV138M E1 (0.10 mg/ml) in 20 mm KH2PO4 (pH 7.0) containing 1 mm MgCl2 was titrated with 0.2–100 μm ThDP.
CD reveals that ThDP does not access the same tautomers when bound to αV138M E1 instead of WT E1
As reported previously (12–15), the binding of ThDP to WT E1 is accompanied by the appearance of two characteristic CD bands in the near-UV spectrum, both of which are shown in Fig. 7: (a) a negative CD band at 330 nm, corresponding to a charge-transfer transition between the thiazolium and 4′-aminopyrimidine rings of ThDP, the signature for its 4′-aminopyrimidine (AP) tautomer, and (b) a positive CD band at 305 nm assigned to its 1′,4′-iminopyrimidine (IP) tautomer (12, 16–18). The negative CD band at 320–330 nm has been observed with several different ThDP-dependent enzymes (15, 19). Importantly, both the AP and IP ThDP tautomers could simultaneously be detected in WT E1, suggesting an asymmetry of the two active centers (12).
Figure 7.
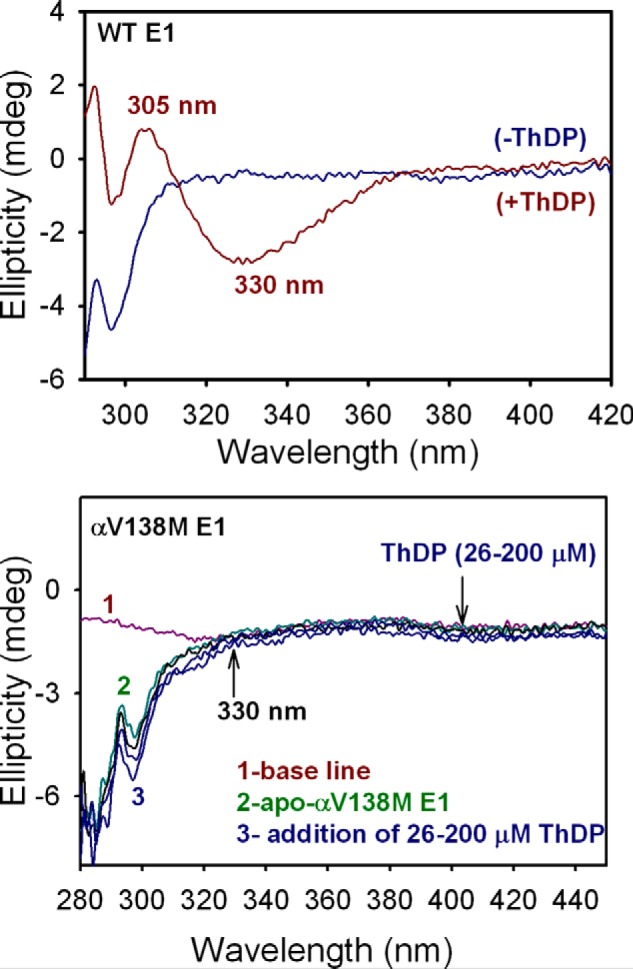
Near-UV CD spectra of WT E1 and αV138M E1 titrated with ThDP. Top, CD spectra of apo-WT E1 (25 μm concentration of active centers) in 10 mm KH2PO4 (pH 7.0) containing 1 mm MgCl2 were recorded in the absence (blue) and presence of 0.20 mm ThDP (red). Upon addition of ThDP to WT E1, a positive and a negative CD band (at 305 and 330 nm, respectively) developed, corresponding to the IP and AP tautomers of ThDP, respectively, when bound to WT E1. Bottom, apo-αV138M E1 (26 μm active centers, green) in 20 mm KH2PO4 (pH 7.0) containing 1 mm MgCl2 was titrated with ThDP (26–200 μm, blue). The spectra do not display the CD bands at 305 and 330 nm, indicating that ThDP binding in the active centers of αV138M E1 is altered compared with WT E1.
We have recorded a series of CD spectra in which αV138M E1 was titrated with ThDP over the range of 26–200 μm; the results are shown in Fig. 7. Notably, neither of the aforementioned bands at 305 or 330 nm appears over the course of the titration. In the general sense, this indicates that the conformation of ThDP when bound to αV138M E1 is different from when bound to WT E1, a result confirmed by its structure, which reveals modest displacement of the thiazolium ring and substantial displacement of the diphosphate tail. The characteristic CD bands that appear upon titration of the WT E1 enzyme with ThDP, however, report on specific tautomeric states of the ThDP coenzyme. The absence of these CD bands upon titration of αV138M E1 with ThDP therefore suggests that the conformation of ThDP when bound to αV138M E1 does not readily allow access to the AP and IP tautomers that are sampled by ThDP when bound to WT E1. Noting that the V-conformation (9) of the thiazolium and 4′-aminopyrimidine rings is more open in the αV138M E1 structure than in the WT structure and that the thiazolium dihedral angle φT is smaller than in any other examined structure, we suggest that these structural changes are related to the altered tautomeric sampling in ThDP when bound to the variant E1 enzyme.
Discussion
Regarding the WT–AcPhi E1 structure, there have been no similar ligand complex structures reported for any other PDHc, regardless of species, apart from our own earlier report of the E. coli PDHc E1-phosphonolactylthiamin diphosphate complex (20). That and the new work reported here represent the only structural examples of pre-decarboxylation intermediates in PDH, although there have been reports of post-decarboxylation intermediates (21). The pre/post distinction is important, because in the E. coli work it is only the pre-decarboxylation intermediate that triggers conformational changes affecting loop ordering at the active site entrance, likely facilitating interaction with E2's lipoyl domains. We also note that the E1 components involved in the two cases are quite different, with the E. coli and human E1s having differing assemblies (homodimers versus heterotetramers), little sequence homology, and only the human versions being subject to regulation via phosphorylation.
Implications of the WT–AcPhi and αV138M structures for E1 inactivation
Human PDHc E1 undergoes reversible inactivation by phosphorylation of its α-subunit at Ser-203, Ser-264, or Ser-271, with Ser-264, located in phosphorylation loop A, being the primary phosphorylation site. Different mechanisms of inactivation by Ser-264 phosphorylation have been proposed. Functional and structural analyses of two Ser-264 substitutions, S264E that mimics the length and negative charge of a phosphorylated serine, and S264Q that mimics the length but lacks the negative charge of a phosphorylated serine, revealed that phosphorylation loops A and B were ordered in both of these structures but that both exhibited little or no acetylation activity (11). The fact that the phosphorylation loops remained ordered in both structures and that S264Q E1 showed deficient activity in the absence of a negative charge, led to the conclusion that E1 inactivation by Ser-264 phosphorylation primarily results from steric occlusion of the substrate channel leading to the active site by the increased length of the Ser-264 side chain upon phosphorylation (11, 22).
Kato et al. (10) have analyzed the structure of WT E1 phosphorylated at Ser-264 and suggested that, rather than inactivation by steric occlusion of the active site channel, a phosphoryl group attached to Ser-264 sterically clashes with its neighboring residue Ser-266, resulting in the disruption of a hydrogen bonding network responsible for maintaining phosphorylation loops A and B in an ordered state. Accordingly, they attribute the reduction in E1 acetylation activity to a lack of binding between the disordered loops and the lipoyl domain of E2. Note that the two proposed inactivation mechanisms are not fundamentally incompatible, because both posit that reduced binding between E1 and the E2 lipoyl domains is the ultimate cause of reduced acetylation activity, regardless of how this reduced binding comes about.
The phosphorylated and substituted structures reported previously were analyzed in the absence of a substrate or analogue. The newly reported structure of WT–AcPhi E1 reported here is to our knowledge, the first structure of human E1 with a bound intermediate adduct, and the situation may differ. To assess this possibility, we have superimposed the phosphomimic and phosphorylated structures onto the WT–AcPhi E1 structure (Fig. 8) to determine whether the phosphomimic or phosphoryl group might disrupt any important active site interactions with the intermediate that could explain these modifications' effect on catalysis. αHis-63, αHis-263, and β′His-128 are the three key residues that interact with and stabilize the bound intermediate analogue in the active site and probably play a role in the reaction mechanism. Functional studies have indicated that substitutions at αHis-63 and αHis-263 E1 lead to near-zero overall PDHc activity (7, 23). In the WT–AcPhi E1 structure, these two histidine residues are hydrogen-bonded to one of the phosphonyl oxygens of the bound substrate analogue. The structural result clearly provides very strong support for the involvement of residues αHis-63 and αHis-263 in substrate interaction and later, in reductive acetylation of the E2 subunit. The residue β′His-128 forms a hydrogen bond with the C2α-hydroxyl oxygen atom, and may play an important role in properly positioning the substrate pyruvate in the enzyme active site. From the superposition, it is clear that modifications at αSer-264 do not alter the arrangement of these key histidine side chains stabilizing the substrate analogue. This suggests inactivation by phosphorylation does not fundamentally proceed by abolishing the ability of the enzyme to interact with the incoming substrate and provides a structural confirmation for the published observation that, even when phosphorylated at αSer-264, WT E1 is still able to decarboxylate pyruvate in the presence of saturating amounts of ThDP (10).
The αV138M E1 variant also retains appreciable decarboxylation activity, but only 2–4% overall PDHc activity, which implicates reduced reductive acetylation of E2 by E1 as the primary source of PDHc deficiency in patients harboring the associated mutation. Position α138 in E1 is located beneath bound ThDP toward the protein interior; it does not protrude into the substrate channel where it could potentially abolish E1's acetylation activity by blocking the entrance of the lipoylated lysine of E2. Rather, the results of the mutation are virtually identical to the consequences observed upon phosphorylation of αSer-264: phosphorylation loops A and B become disordered, and E1 reductive acetylation activity plummets because the disordered loops are not optimally oriented or available to bind E2's lipoyl domain efficiently. Although both phosphorylation and the αV138M variant result in disordered phosphorylation loops, the physical mechanisms inducing loop disorder are different. Although phosphorylation is proposed to disrupt an interaction between αSer-264 and αSer-266 (10), the αV138M methionine side chain displaces the diphosphate tail of ThDP, which in turn physically impacts both phosphorylation loops, resulting in their disorder. Until now, the only phenomena demonstrated structurally to modulate the order/disorder state of phosphorylation loops A and B were (i) the disorder-to-order shift upon transitioning from apo to holo E1 and (ii) the order-to-disorder transition induced by phosphorylation at αSer-264. The αV138M E1 structure reported here now demonstrates unambiguously that the loop order-to-disorder transition can also be induced by mutations associated with pyruvate dehydrogenase complex deficiency. In contrast, the previously reported WT holo E1 and new WT–AcPhi E1 reaction intermediate analogue structures indicate no such disorder throughout the catalytic cycle. Reduction of E1 acetylation activity by inducing phosphorylation loop disorder now appears to be a general phenomenon, not strictly limited to modulation via the evolved regulatory phosphorylation/dephosphorylation system. Given that more than 20 E1 variants associated with PDHc deficiency have been found in human patients (5, 6, 24), we expect that future disease-related variant structures may well reveal additional examples of reducing acetylation function via induction of phosphorylation loop disorder.
Experimental procedures
Expression and purification of human PDHc proteins
Recombinant WT E1 and its αV138M E1 variant were expressed and purified as previously reported (25). Briefly, the pQE-9–6HE1α/E1β plasmid for the simultaneous expression of the E1β subunit and either the WT or V138M E1α subunit was transformed into either E. coli M15 or E. coli JRG1342 cells for protein expression. In a typical preparative purification, 5 ml of an overnight culture in LB medium were used to inoculate 1 liter of LB medium containing 100 μg/ml of ampicillin and 25 μg/ml of kanamycin. The cells were grown at 37 °C to A600 = 0.7, at which point isopropyl β-d-1-thiogalactopyranoside was added to a final concentration of 200 μg/ml, and protein was expressed overnight at 25 °C. The harvested cells were resuspended in buffer containing 50 mm KH2PO4 (pH 7.5), 1 mm EDTA, and 5 mm β-mercaptoethanol, and were treated with lysozyme (1 mg/ml) for 30 min at 4 °C. Cells were then lysed by repeated passages in a French press. KCl was added to the supernatant to a final concentration of 300 mm, and the protein solution was loaded onto a nickel-nitrilotriacetic acid-agarose column equilibrated with 50 mm KH2PO4 (pH 7.0) containing 300 mm KCl. Protein was eluted with a linear gradient of 0.2 m imidazole in 50 mm KH2PO4 (pH 7.0) containing 300 mm KCl. The fractions containing E1 were pooled and concentrated to a final volume of 3 ml, and the concentrated protein was dialyzed against 4 liters of 50 mm KH2PO4 (pH 7.0). Recombinant E2·E3BP subcomplex (human dihydrolipoamide acetyltransferase–E3 binding protein) and recombinant human E3 were overexpressed in E. coli and purified as described previously (22, 26).
Crystallization of human PDHc E1 proteins
A 17 mg ml−1 solution of human αV138M E1 in 50 mm potassium phosphate buffer (pH 7.0), 0.2 mm ThDP, 2 mm MgCl2, and 1 mm DTT was used for crystallization trials via vapor diffusion in sitting drops at 22 °C. The best crystals were obtained with a reservoir solution containing 12–14% (w/v) PEG 3350, 0.2% (w/v) NaN3, 100 mm ammonium sulfate, and 50 mm potassium phosphate (pH 6.0). Drops had a total volume of 3–5 μl consisting of equal parts protein and reservoir solutions. Crystals were flash cooled in liquid nitrogen without additional cryoprotection.
Crystals of the holo form of human WT E1 (18 mg/ml in 50 mm potassium phosphate buffer, 0.2 mm ThDP, 0.2 mm MgCl2, and 1 mm DTT, pH 7.0) were obtained with a reservoir solution containing 12–14% (w/v) PEG 3350, 0.2% (w/v) NaN3, 10% (v/v) 1,2-propanediol, 200 mm NaSCN, and 50 mm potassium phosphate (pH 8.0). The WT E1–acetylphosphinate (WT–AcPhi) complex crystals were then obtained by soaking holo E1 crystals in reservoir solution containing the substrate analogue AcPhi at 2 mm for 1–4 min. All soaking experiments were performed at room temperature. Soaked crystals were flash cooled in liquid nitrogen without additional cryoprotection.
X-ray data collection and structure solution
Diffraction data were collected at SER-CAT beamlines 22-ID (for WT–AcPhi E1) and 22-BM (for αV138M E1) of the Advanced Photon Source (Argonne National Laboratory, Lemont, IL). For αV138M E1, 251 images of 0.5° oscillation width were recorded, and for WT–AcPhi E1, 180 images were collected at an oscillation width of 1°. Indexing, integration, and scaling were performed with XDS (27). WT–AcPhi E1 crystallized in the primitive orthorhombic space group P212121 with a single heterotetramer in the asymmetric unit. The αV138M variant crystallized in the primitive monoclinic space group P21 with two heterotetramers in the asymmetric unit. For both structures, phasing was accomplished via molecular replacement in Phaser (28) using the WT human E1 structure (PDB code 1NI4) as the search model (8). Each structure was subjected to several rounds of manual model building in Coot (29) and automated refinement in Phenix (30). The refined models were deposited in the Protein Data Bank under accession codes 6CFO (WT–AcPhi E1) and 6CER (αV138M E1 variant). Data collection and refinement statistics for both structures are provided in Table 1.
Enzyme activity measurements
The overall activity of PDHc containing either WT or αV138M E1 was measured by monitoring the formation of NADH (and H+) at 340 nm, as reported previously (22). PDHc was reconstituted by premixing the E1, E2·E3BP, and E3 proteins at a microgram mass ratio of 1:3:3 in a buffer of 50 mm KH2PO4 (pH 7.0) and 0.15 m NaCl for 60 min at 25 °C (31).
Fluorescence analysis of ThDP binding to E1
Apo-E1 was prepared using a PD-10 desalting column equilibrated with 50 mm KH2PO4 (pH 7.5) containing 0.15 m NaCl, 0.5 mm DTT, and 1.0 mm benzamidine-HCl, followed by dialysis for 15 h at 4 °C against the same buffer (31).
Apo-WT E1 (0.034 mg/ml) and apo-αV138M E1 (0.10 mg/ml) in 20 mm KH2PO4 (pH 7.0) containing 1 mm MgCl2 were titrated with ThDP (0.2–100 μm) in 3-ml quartz cuvettes. Fluorescence spectra were recorded at 25 °C using a Cary Eclipse spectrophotometer according to a procedure reported previously (32). The excitation wavelength was 295 nm, and the emission spectra were recorded in the range of 300–450 nm. The inner filter effect was corrected with the absorption of ThDP at the excitation wavelength (295 nm), whereas the emission maximum (334 nm) was corrected with a dilution factor calculated according to the following equation,
| (Eq. 1) |
where Fi,corr is the corrected value of the fluorescence intensity at a given point of titration, Fi,obs is the experimentally measured fluorescence intensity at the emission maximum, V0 is the initial volume of the sample, Vi is the volume at a given point of titration (Vi/Vo is the dilution factor), Aex is the absorbance of the sample at the excitation wavelength, and Aem is the absorbance of the sample at the emission maximum. The value of Kd,ThDP was calculated according to the modified Hill equation,
| (Eq. 2) |
where (Fo − Fi)/Fo is the relative fluorescence quenching following the addition of ThDP, n is the Hill coefficient, and S0.5 is the concentration of ThDP at half-saturation.
CD
For titration of apo-WT E1 with ThDP, the CD spectra of apo-WT E1 (25 μm concentration of active centers) in 10 mm KH2PO4 (pH 7.0) containing 1 mm MgCl2 were recorded (i) in the absence of ThDP and (ii) in the presence of a saturating concentration of ThDP (0.20 mm). For titration of apo-αV138M E1 with ThDP, the CD spectra of apo-αV138M E1 (26 μm active centers) in 20 mm KH2PO4 (pH 7.0) containing 1 mm MgCl2 were recorded (i) in the absence of ThDP and (ii) upon titration of 26–200 μm ThDP. CD spectra were recorded using a Chirascan CD instrument (Applied Photophysics, Leatherhead, UK) at 25 °C in a cuvette with a path length of 1 cm. The baseline represents the CD spectrum of the reaction mixture in the absence of protein. CD measurements are expressed in millidegrees, the unit of molecular ellipticity (ϴ), where ϴ (millidegrees) = 32,982 × ΔA (Δ absorbance).
Author contributions
M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. conceptualization; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. data curation; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. formal analysis; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. supervision; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. funding acquisition; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. validation; M. J. W., P. A., N. S. N., L. G. K., Y.-H. P., M. P., F. J., and W. F. F. investigation; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. visualization; M. J. W., P. A., N. S. N., L. G. K., Y.-H. P., M. P., F. J., and W. F. F. methodology; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. writing-original draft; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. project administration; M. J. W., P. A., N. S. N., L. G. K., M. P., F. J., and W. F. F. writing-review and editing; Y.-H. P. resources.
Supplementary Material
Acknowledgments
This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract DE-AC02-06CH11357.
This work was supported by National Institutes of Health Grants DK080748 (to M. S. P.), GM050380 (to F. J. and M. S. P.), GM116077 (to F. J.), GM061791 and GM121469 (to W. F.), and a Veterans Affairs Merit Review (to W. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 6CFO and 6CER) have been deposited in the Protein Data Bank (http://wwpdb.org/).
This article contains Fig. S1.
- PDHc
- pyruvate dehydrogenase multienzyme complex
- AcPhi
- acetylphosphinate
- E1
- pyruvate dehydrogenase from PDHc
- ThDP
- thiamin diphosphate
- E2
- dihydrolipoamide acetyltransferase from PDHc
- E3
- dihydrolipoamide dehydrogenase from PDHc
- r.m.s. deviation
- root mean square deviation
- PDB
- Protein Data Bank
- AP
- 4′-aminopyrimidine
- IP
- 1′,4′-iminopyrimidine.
References
- 1. Linn T. C., Pettit F. H., and Reed L. J. (1969) α-Keto acid dehydrogenase complexes: X. regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. U.S.A. 62, 234–241 10.1073/pnas.62.1.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown G. K., Otero L. J., LeGris M., and Brown R. M. (1994) Pyruvate dehydrogenase deficiency. J. Med. Genet. 31, 875–879 10.1136/jmg.31.11.875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patel K. P., O'Brien T. W., Subramony S. H., Shuster J., and Stacpoole P. W. (2012) The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 106, 385–394 10.1016/j.ymgme.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byron O., and Lindsay J. G. (2017) The pyruvate dehydrogenase complex and related assemblies in health and disease. Subcell. Biochem. 83, 523–550 10.1007/978-3-319-46503-6_19 [DOI] [PubMed] [Google Scholar]
- 5. Chun K., MacKay N., Petrova-Benedict R., and Robinson B. H. (1993) Mutations in the X-linked E1 α subunit of pyruvate dehydrogenase leading to deficiency of the pyruvate dehydrogenase complex. Hum. Mol. Genet. 2, 449–454 10.1093/hmg/2.4.449 [DOI] [PubMed] [Google Scholar]
- 6. Ferriero R., Manco G., Lamantea E., Nusco E., Ferrante M. I., Sordino P., Stacpoole P. W., Lee B., Zeviani M., and Brunetti-Pierri N. (2013) Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci. Transl. Med. 5, 175ra131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nemeria N. S., Korotchkina L. G., Chakraborty S., Patel M. S., and Jordan F. (2006) Acetylphosphinate is the most potent mechanism-based substrate-like inhibitor of both the human and Escherichia coli pyruvate dehydrogenase components of the pyruvate dehydrogenase complex. Bioorg. Chem. 34, 362–379 10.1016/j.bioorg.2006.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ciszak E. M., Korotchkina L. G., Dominiak P. M., Sidhu S., and Patel M. S. (2003) Structural basis for flip-flop action of thiamin pyrophosphate-dependent enzymes revealed by human pyruvate dehydrogenase. J. Biol. Chem. 278, 21240–21246 10.1074/jbc.M300339200 [DOI] [PubMed] [Google Scholar]
- 9. Pletcher J., Sax M., Turano A., and Chang C.-H. (1982) Effects of structural variations in thiamin, its derivatives and analogues. in Thiamin: Twenty Years of Progress (Sable H. Z., and Gubler C. J., eds) pp. 454–458, The New York Academy of Sciences, New: York [Google Scholar]
- 10. Kato M., Wynn R. M., Chuang J. L., Tso S. C., Machius M., Li J., and Chuang D. T. (2008) Structural basis for inactivation of the human pyruvate dehydrogenase complex by phosphorylation: role of disordered phosphorylation loops. Structure 16, 1849–1859 10.1016/j.str.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seifert F., Ciszak E., Korotchkina L., Golbik R., Spinka M., Dominiak P., Sidhu S., Brauer J., Patel M. S., and Tittmann K. (2007) Phosphorylation of serine 264 impedes active site accessibility in the E1 component of the human pyruvate dehydrogenase multienzyme complex. Biochemistry 46, 6277–6287 10.1021/bi700083z [DOI] [PubMed] [Google Scholar]
- 12. Nemeria N., Chakraborty S., Baykal A., Korotchkina L. G., Patel M. S., and Jordan F. (2007) The 1′,4′-iminopyrimidine tautomer of thiamin diphosphate is poised for catalysis in asymmetric active centers on enzymes. Proc. Natl. Acad. Sci. U.S.A. 104, 78–82 10.1073/pnas.0609973104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nemeria N., Korotchkina L., McLeish M. J., Kenyon G. L., Patel M. S., and Jordan F. (2007) Elucidation of the chemistry of enzyme-bound thiamin diphosphate prior to substrate binding: defining internal equilibria among tautomeric and ionization states. Biochemistry 46, 10739–10744 10.1021/bi700838q [DOI] [PubMed] [Google Scholar]
- 14. Nemeria N. S., Chakraborty S., Balakrishnan A., and Jordan F. (2009) Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS J. 276, 2432–2446 10.1111/j.1742-4658.2009.06964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Paulikat M., Wechsler C., Tittmann K., and Mata R. A. (2017) Theoretical studies of the electronic absorption spectra of thiamin diphosphate in pyruvate decarboxylase. Biochemistry 56, 1854–1864 10.1021/acs.biochem.6b00984 [DOI] [PubMed] [Google Scholar]
- 16. Jordan F., Zhang Z., and Sergienko E. (2002) Spectroscopic evidence for participation of the 1′,4′-imino tautomer of thiamin diphosphate in catalysis by yeast pyruvate decarboxylase. Bioorg. Chem. 30, 188–198 10.1006/bioo.2002.1249 [DOI] [PubMed] [Google Scholar]
- 17. Jordan F. (2003) Current mechanistic understanding of thiamin diphosphate-dependent enzymatic reactions. Nat. Prod. Rep. 20, 184–201 10.1039/b111348h [DOI] [PubMed] [Google Scholar]
- 18. Baykal A. T., Kakalis L., and Jordan F. (2006) Electronic and nuclear magnetic resonance spectroscopic features of the 1′,4′-iminopyrimidine tautomeric form of thiamin diphosphate, a novel intermediate on enzymes requiring this coenzyme. Biochemistry 45, 7522–7528 10.1021/bi060395k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jordan F., and Nemeria N. S. (2014) Progress in the experimental observation of thiamin diphosphate-bound intermediates on enzymes and mechanistic information derived from these observations. Bioorg. Chem. 57, 251–262 10.1016/j.bioorg.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 20. Arjunan P., Sax M., Brunskill A., Chandrasekhar K., Nemeria N., Zhang S., Jordan F., and Furey W. (2006) A thiamin-bound, pre-decarboxylation reaction intermediate analogue in the pyruvate dehydrogenase E1 subunit induces large scale disorder-to-order transformations in the enzyme and reveals novel structural features in the covalently bound adduct. J. Biol. Chem. 281, 15296–15303 10.1074/jbc.M600656200 [DOI] [PubMed] [Google Scholar]
- 21. Pei X. Y., Titman C. M., Frank R. A., Leeper F. J., and Luisi B. F. (2008) Snapshots of catalysis in the E1 subunit of the pyruvate dehydrogenase multienzyme complex. Structure 16, 1860–1872 10.1016/j.str.2008.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Korotchkina L. G., and Patel M. S. (2001) Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J. Biol. Chem. 276, 5731–5738 10.1074/jbc.M007558200 [DOI] [PubMed] [Google Scholar]
- 23. Seyda A., and Robinson B. H. (2000) Functional expression of four PDH-E(1)α recombinant histidine mutants in a human fibroblast cell line with zero endogenous PDH complex activity. Biochem. Biophys. Res. Commun. 270, 1068–1073 10.1006/bbrc.2000.2551 [DOI] [PubMed] [Google Scholar]
- 24. Kerr D. S., Wexler I. D., Tripatara A., and Patel M. S. (1996) Human defects of the pyruvate dehydrogenase complex. in Alpha-Keto Acid Dehydrogenase Complexes (Patel M. S., Roche T. E., and Harris R. A., eds) pp. 249–269, Birkhäuser Verlag, Basel [Google Scholar]
- 25. Korotchkina L. G., Tucker M. M., Thekkumkara T. J., Madhusudhan K. T., Pons G., Kim H., and Patel M. S. (1995) Overexpression and characterization of human tetrameric pyruvate dehydrogenase and its individual subunits. Protein Expr. Purif. 6, 79–90 10.1006/prep.1995.1011 [DOI] [PubMed] [Google Scholar]
- 26. Liu T. C., Korotchkina L. G., Hyatt S. L., Vettakkorumakankav N. N., and Patel M. S. (1995) Spectroscopic studies of the characterization of recombinant human dihydrolipoamide dehydrogenase and its site-directed mutants. J. Biol. Chem. 270, 15545–15550 10.1074/jbc.270.26.15545 [DOI] [PubMed] [Google Scholar]
- 27. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nemeria N. S., Shome B., DeColli A. A., Heflin K., Begley T. P., Meyers C. F., and Jordan F. (2016) Competence of thiamin diphosphate-dependent enzymes with 2′-methoxythiamin diphosphate derived from bacimethrin, a naturally occurring thiamin anti-vitamin. Biochemistry 55, 1135–1148 10.1021/acs.biochem.5b01300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nemeria N., Yan Y., Zhang Z., Brown A. M., Arjunan P., Furey W., Guest J. R., and Jordan F. (2001) Inhibition of the Escherichia coli pyruvate dehydrogenase complex E1 subunit and its tyrosine 177 variants by thiamin 2-thiazolone and thiamin 2-thiothiazolone diphosphates: evidence for reversible tight-binding inhibition. J. Biol. Chem. 276, 45969–45978 10.1074/jbc.M104116200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.