

Abstract
Enteropathogenic bacteria elicit mucosal innate and adaptive immune responses. We investigated whether gut epithelial cells played a role in triggering an adaptive immune response by recruiting dendritic cells (DCs). Immature DCs are selectively attracted by the CCL20 chemokine. The expression of the CCL20 gene in human intestinal epithelial cell lines was up-regulated by pathogenic bacteria, including Salmonella species, but not by indigenous bacteria of the intestinal flora. The Salmonella machinery for epithelial cell invasion was not required for CCL20 gene activation. Flagellin but not the lipopolysaccharide was found to be the Salmonella factor responsible for stimulation of epithelial CCL20 production. CCL20 in turn triggered a specific migration of immature DCs. Our data show that crosstalk between bacterial flagellin and epithelial cells is essential for the recruitment of DCs, a mechanism that could be instrumental to initiate adaptive immune responses in the gut.
The gut represents a unique niche for bacteria from the normal flora and eventually for enteropathogenic microbes. The intestinal epithelium constitutes not only the physical barrier that separates the luminal environment from the host milieu, but it also acts as a sentinel sensing injuries in the intestinal tract. Enteropathogenic bacteria colonize the epithelium, and their intimate interaction with the epithelial cells activates proinflammatory signaling pathways (1–3). This innate response is essential for rapid clearance of bacteria. Adaptive immunity is also stimulated to prevent reinfection, but the mechanisms initiating this response in the gut epithelium have not been identified yet.
Dendritic cells (DCs) are bone marrow-derived antigen-presenting cells with the unique ability to induce primary immune responses. The recruitment of DCs into the epithelium is therefore a prerequisite to initiate an adaptive response. The trafficking of DCs depends on differential expression of CCR6 and CCR7 chemokine receptors (4–7). The CCL20 chemokine also known as LARC, MIP-3 alpha, and Exodus is the ligand of CCR6 receptor (8). Immature DCs express CCR6 and efficiently take up soluble and particulate antigens (for review, see ref. 9). Maturation of DCs is induced by danger signals, i.e., bacterial, viral, or cellular components, and is characterized by the up-regulation of antigen presentation, costimulatory molecules, and of the CCR7 chemokine receptor that mediates migration of activated DCs to the draining lymph nodes. The CCL20 gene is expressed in mouse colon and the epithelium over Peyer's patches and in human colon, appendix, tonsils, and skin keratinocytes (4, 6–8, 10, 11). CCR6-expressing DCs are found in tissues close to CCL20-expressing epithelial cells or keratinocytes (6, 7, 10). In CCR6 knockout mice, subepithelial myeloid DCs are absent in the dome of Peyer's patches, and mucosal immune responses are impaired (7). These findings have emphasized the instrumental role of CCL20-dependent DC trafficking in induction of adaptive responses in the gut.
Enteropathogens compete with the normal flora and produce specific virulence factors to overcome innate defenses. Enteroinvasive bacteria (e.g., Salmonella, Shigella, Yersinia, and Listeria) adhere and invade the epithelium via M cells of Peyer's patches (12). After subepithelial translocation, they invade enterocytes or phagocytes and/or replicate. Invasive bacteria divert cellular signaling by interacting with cell surface receptors or with cytosolic targets by using toxins injected into the cell cytoplasm via a type III secretion system (3, 13). Salmonella enterica of various serotypes provoke gastroenteritis in mammals characterized by mucosal inflammation and diarrhea. Salmonella are the only bacteria that can invade apically enterocytes along the crypt to villus axis of the small intestine (14). In human intestinal epithelial cells, the Salmonella-induced inflammatory response is characterized by basal secretion of IL-8 (CXCL8) and of various proinflammatory chemokines that recruit neutrophils in the subepithelial compartment (1, 15, 16). The induction of IL-8 secretion depends on virulence factors of Salmonella and on epithelial NF-κB signaling (1, 2, 17, 18).
In this study, we have investigated whether the release of intestinal epithelial chemokines in response to bacteria is able to recruit immune cells that initiate adaptive immunity. We show that Salmonella typhimurium flagellins stimulate in epithelial cells the secretion of the CCL20 chemokine, which triggers DC chemotaxis.
Methods
Bacterial Strains and Culture Conditions.
The bacterial strains are listed in Table 1. SIN strains were obtained by phage P22 HT105/int-1 transduction. Salmonella or Escherichia coli were grown in LB broth for 24 h at 37°C then diluted 1/1′000 in LB broth and grown in standing conditions for 18 h at 37°C (19). Bacterial concentration was estimated to 109 bacteria per milliliter per OD unit at 600 nm and calculated by plating. Ampicillin and kanamycin were added at 100 and 40 μg/ml, respectively. Listeria monocytogenes was grown in brain heart infusion (BHI) medium at 37°C, and Bifidobacterium bifidum and Bacteroides vulgatus were grown in BHI at 37°C in an anaerobic GasPak (Becton Dickinson) jar by using a glycerol frozen inoculum. Supernatants were filtered to remove residual bacteria, and proteolysis was performed at 37°C for 30 min with 10 μg/ml of trypsin (Worthington). When specified, bacteria or supernatants were heat-treated for 20 min at 65°C. For complementation, the ampicillin-resistant plasmid pRP2, harboring an EcoRI fragment with S. typhimurium fliC genes (gift of K. Hughes, University of Washington, Seattle), was introduced in Salmonella. Flagellin expression was checked by (i) agglutination with rabbit Salmonella H antiserum poly(A)-z (Difco), (ii) motility in 0.35% agar, and (iii) by SDS/PAGE analysis of supernatants and immunoblot with poly(A)-z serum and peroxidase-conjugated anti-rabbit serum (Sigma).
Table 1.
Bacterial strains
| Strains | Relevant characteristics‡‡ |
|---|---|
| DH5α | Laboratory strain derived from E. coli K-12, NM |
| EMO* | Plasmid-free E. coli from human colon flora, M |
| ATCC14028† | Virulent S. typhimurium strain, M |
| SB856‡ | sopE∷aphT mutation in S. typhimurium SL1344, M |
| TH714§ | fljB5001∷MudJ mutation in S. typhimurium LT2, M |
| VV341¶ | hilA∷kan-339 mutation in SL1344, M |
| SIN14 | hilA∷kan-339 in ATCC14028, M |
| SIN18 | sopE∷aphT in ATCC14028, M |
| SIN20 | fliC∷aphT from SEFK32 strain in ATCC14028, M |
| SIN22 | fljB5001∷MudJ in ATCC14028, M |
| SE857‖ | Virulent S. enteritidis strain, M |
| SEFK32‖ | fliC∷aphT mutation in SE857, NM |
| B. vulgatus* | Gram− anaerobe from human colon, NM |
| B. bifidum* | Gram+ anaerobe from human colon, NM |
| LO28** | Virulent L. monocytogenes strain, M |
Strains were obtained from
V. Gaboriau-Routhiau,
American Type Culture Collection,
J. Galan,
K. Hughes,
C. Lee,
F. Van Asten, and
P. Berche.
kan, MudJ, and aphT encode cassettes for resistance to kanamycin; M, motile; NM, nonmotile.
Cell Culture and Stimulation.
The human colon adenocarcinoma cell line Caco-2 clone 1 was grown in DMEM with glutamax, 10% FCS, 1% nonessential amino acids, and 4 μg/ml of transferrin (cell culture products from GIBCO/BRL). The T-84 intestinal epithelial cell line was grown in 50% DMEM/50% Ham's F-12 medium/10% FCS/2 mM l-glutamine. Cells were grown for 10 days at 37°C under 5% CO2 on Transwells (6 mm in diameter, 3 μm pore; Corning). The average of transepithelial electrical resistance was 450 Ω cm2 and 1,000 Ω cm2 for Caco-2 and T-84 cells, respectively. Differentiation was also checked by the presence of apical microvilli and by up-regulation of apical sucrase isomaltase with specific Abs (gift from A. Zweibaum, Institut National de la Santé et de la Recherche Médicale, U178, Villejuif, France), using electron and confocal microscopy. Bacteria or bioactive materials were suspended in complete DMEM and added either apically (300 μl) or basally (1 ml). For infection, cells were incubated for 45 min with 108 bacteria, i.e., a multiplicity of infection (moi) of ≈100, washed with PBS, and incubated with medium containing 50 μg/ml of gentamicin (5 μg/ml for Listeria) to kill extracellular bacteria. Alternatively, cells were exposed to supernatant, lipopolysaccharide (LPS), or flagellin for the duration of experiment. At indicated times, total RNA was prepared and/or culture medium was recovered.
Real-Time Quantitative PCR for Analysis of mRNA Levels.
Total RNA was isolated from cells of three Transwell filters (RNeasy; Qiagen, Switzerland), and reverse transcription (RT) was performed on 100 ng by using Superscript II (GIBCO/BRL). Resulting cDNA (1 ng) was amplified in triplicates by the SYBR-Green PCR assay, and products were detected on a Prism 5700 detection system (SDS, ABI/Perkin–Elmer). PCR reactions were incubated for 2 min at 50°C and for 10 min at 95°C, followed by 40 amplification cycles with 1 min annealing/extension at 60°C and 15-s denaturation at 95°C. The 18S ribosomal RNA was used to standardize the total amount of cDNA. The primers for CCL20 (CCAAGAGTTTGCTCCTGGCT and TGCTTGCTGCTTCTGATTCG), IL-8 (CACCGGAAGGAACCATCTCA and GGAAGGCTGCCAAGAGAGC), and 18S (ACATCCAAGG AAGGCAGCAG and TTTTCGTCACTACCTCCCCG) designed from sequences (NM004591, Y00787, and X03205) yielded PCR products of 75, 72, and 65 bp, respectively. Specificity of PCR was checked by analyzing melting curves and sequencing. Relative mRNA levels (2ΔΔC) were determined by comparing (i) the PCR cycle threshold (C) between cDNA of the gene of interest and of 18S rRNA (ΔC), and (ii) ΔC values between treated and untreated conditions (ΔΔC). SDs of relative mRNA levels were calculated as 2(ΔΔC ± √{SD[ΔCtreated]2 + SD[ΔCuntreated]2}). Increase of RNA levels lower than 2-fold were not considered as significant.
CCL20-Specific ELISA.
Microplates coated with 3 μg/ml of human CCL20-specific mAb (clone 67310.111; R & D Systems) were used to capture CCL20 in culture medium. Goat anti-human CCL20 (R & D Systems) diluted at 1 μg/ml was used as the detection Ab, and development was performed with peroxidase-conjugated rabbit anti-goat Ab (Sigma) diluted 1/2′000. CCL20 concentration was calculated from a standard curve by using recombinant human (rh) CCL20 (R & D Systems). The detection threshold was 0.5 ng/ml.
LPS and Flagellin Purification.
LPS was purified by hot phenol extraction as described (20). Alternatively, commercial S. typhimurium LPS was used (L-6511, Sigma). Flagellin was prepared from Salmonella strain SEFK32(pRP2) grown for 16 h at 37°C with agitation in LB as described (21). Briefly, flagella were sheared from surface, pelleted by ultracentrifugation at 100,000 × g, and acidified to release flagellin monomers. Flagellin was concentrated in PBS and stored at −80°C.
Generation of CD34+-Derived DCs.
Progenitors were isolated from umbilical cord blood by positive selection, using anti-CD34 mAb (Immu-133.3; Immunotech, Luminy, France), goat anti-mouse IgG-coated microbeads, and MidiMacs columns (Miltenyi Biotec, Bergisch Gladbach, Germany). CD34+ cells were grown in RPMI medium 1640/10% FCS/200 units/ml rhGM-CSF (Schering-Plough)/50 units/ml rhTNFα (PeproTech, Rocky Hill, NJ)/10 units/ml rhSCF (R & D Systems). After 7 days, the cells (30–50% CD1a+ DCs, 25–35% CD1a−CD14+ DC precursors, and undifferentiated CD34+ cells) were collected.
Chemotaxis Assay.
Supernatants from Caco-2 cells cultured in complete DMEM (2% FCS) or rhCCL20 were added to 24-well plates and 5 × 105 DCs to Transwell inserts (5 μm pores, Corning). Plates were incubated for 1.5 h at 37°C. Migrated cells were stained with FITC-labeled anti-CD1a mAb and phycoerythrin (PE)-labeled anti-CD14 mAb and counted by flow cytometry. For neutralization, samples were incubated for 30 min at 37°C with 10 μg/ml of goat anti-CCL20 Ab.
Results
S. typhimurium Induces Expression of CCL20 Gene in Intestinal Epithelial Cells.
We studied the expression of the CCL20 chemokine gene in the human intestinal epithelial Caco-2 cell line grown on permeable filters in response to various stimuli by real-time RT-PCR and by ELISA. In untreated cells about 1.8 ± 1.0 × 106 CCL20 copies per microgram of total mRNA were detected, which corresponds to ≈10 copies per cell. The concentration of CCL20 in the apical and basal medium never exceeded 0.5 ng/ml. These observations confirmed the constitutive CCL20 gene expression in Caco-2 cells reported by others (11).
Apical exposure of Caco-2 cells to virulent S. typhimurium ATCC14028 resulted in efficient infection, because 0.25% bacteria were internalized within 2 h (Table 2, which is published as supporting information on the PNAS web site, www.pnas.org). The transcription of CCL20 was maximally increased between 2 and 3.5 h after infection [15.2 ± 6.9-fold (n = 20)] (Fig. 1a). Under these conditions, IL-8 transcription was increased 27.6 ± 7.8-fold as reported (1, 15). CCL20 secretion increased significantly 2 h after infection and reached a plateau at 6 h (Fig. 1b). CCL20 secretion was partially polarized because 20 h after infection, 64.8 ± 6.9% (n = 7) were recovered in the basal compartment.
Figure 1.

S. typhimurium-regulated expression of CCL20 gene in epithelial cells. Caco-2 cells in Transwell cultures were infected apically for 45 min with S. typhimurium ATCC14028 (moi = 100), washed, and incubated for the indicated times in gentamicin-supplemented medium. (a) Transcriptional activation of CCL20 gene: Total RNA was extracted and reverse transcribed. CCL20 mRNA levels were quantified by using real-time PCR and 18S rRNA amplicons as standards. Values were expressed as relative increase of CCL20 mRNA quantity compared with noninfected Caco-2 cells. (b) Secretion of CCL20 chemokine in basal culture medium. CCL20 concentration was measured by CCL20-specific ELISA on cell culture medium of Caco-2 cells.
The CCL20 Response Is Induced by Pathogens.
The specificity of CCL20 induction was analyzed in response to various bacteria encountered in the gut. The E. coli strain DH5-α and the commensal bacteria E. coli EMO, B. bifidum, or B. vulgatus were unable to induce CCL20 expression (Fig. 2 a and b). In contrast, pathogenic bacteria including Salmonella enteritidis and L. monocytogenes activated CCL20 transcription as efficiently as S. typhimurium (Fig. 2c).
Figure 2.

Pathogen-specific induction of CCL20 transcription in epithelial cells. Monolayers of Caco-2 cells were exposed apically for 45 min to bacterial strains (moi = 100) and incubated for 2.5 h in gentamicin-containing medium. CCL20 expression was quantified by real-time RT-PCR. ATCC14028 was used as a positive control of CCL20 induction. Results are representative of at least 2 independent experiments. CCL20 transcription was analyzed after exposure to laboratory E. coli DH5α (a); bacteria from human colon flora, E. coli EMO, B. vulgatus, and B. bifidum (b); and enteroinvasive bacteria, S. enteritidis SE857 and L. monocytogenes LO28 (c).
CCL20 Induction Does Not Require Epithelial Cell Invasion.
Invasion of epithelial cell depends on a type III secretion system encoded by Salmonella pathogenicity island 1 (SPI-1) that injects toxins, such as SopE, in the cytoplasm of epithelial cells (13, 22). These toxins induce membrane ruffles resulting in bacterial internalization and disturb signaling pathways. Inactivation of the hilA gene that encodes an activator of SPI-1 genes impairs invasion. The S. typhimurium hilA mutant SIN14 and the sopE-inactivated strain SIN18 were found as efficient as ATCC14028 to induce CCL20 expression in epithelial Caco-2 cells (Fig. 3a). CCL20 induction by heat-killed and live bacteria was not significantly different (Fig. 3a), thus ruling out a role of bacterial invasion. Therefore, our experiments indicated that CCL20 stimulation does not require epithelial cell invasion nor the injection of SopE toxin.
Figure 3.

Salmonella induction factor for CCL20 expression is a heat-stable secreted protein. Polarized Caco-2 cells were exposed apically for 45 min to bacteria (moi = 100) (a). Then, cells were incubated for 2.5 h in gentamicin-supplemented medium. Alternatively, cells were exposed for 3.25 h to bacterial products (b and c). Activation of CCL20 gene transcription was quantified by real-time RT-PCR. Results are representative of at least 3 independent experiments. (a) Induction of CCL20 transcription does not depend on Salmonella-mediated invasion. (b) LPS-independent CCL20 transcription. Epithelial cells were treated apically or basally with 10 μg/ml of LPS from S. typhimurium. (c) Induction factor is a Salmonella-secreted protein. Cells were exposed apically to 100 μl of supernatants from S. typhimurium, heat-treated supernatant, or trypsin-digested and heat-treated supernatant. LB broth treated in the same conditions was used as control.
CCL20-Inducing Factor Is a Heat-Stable Secreted Protein.
S. typhimurium supernatant strongly induced CCL20 expression when applied apically on epithelial cells (Fig. 3c). LPS is a heat-resistant molecule of outer membrane from Gram-negative bacteria involved in cell signaling. Apical or basal treatment of Caco-2 cells with commercial S. typhimurium LPS or LPS purified from ATCC14028 did not activate CCL20 gene transcription (Fig. 3b). Thus, LPS per se is not the induction factor for CCL20 stimulation.
As observed with whole bacteria, heat treatment did not abolish the supernatant activity (Fig. 3c). Trypsin digestion of the supernatant, however, totally abrogated CCL20 induction. Altogether, these experiments indicated that the CCL20-specific induction factor is a heat-stable secreted protein.
Flagellin Is the CCL20 Induction Factor.
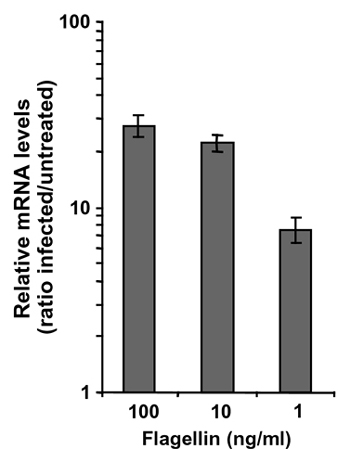
Flagellin, the subunit constituting the flagellar filament, is the major protein recovered from S. typhimurium or S. enteritidis supernatants (Fig. 4c; ref. 23). S. typhimurium produces two 52-kDa flagellins, FliC or FljB, whereas S. enteritidis produces a single 56-kDa flagellin, FliC. The fliC-deleted S. enteritidis SEFK32 was unable to induce CCL20 gene expression in contrast to the parental strain SE857 (Fig. 4a). Complementation of fliC mutant with fliC gene from S. typhimurium fully restored CCL20 induction. In addition, S. typhimurium mutants SIN20 or SIN22, producing either FljB or FliC, stimulated CCL20 transcription to similar levels as wild-type bacteria (data not shown). Finally, purified S. typhimurium FliC flagellin activated CCL20 transcription in Caco-2 cells (ED50 ≈ 20 pM and Fig. 4b). Similar results were obtained by using the T-84 epithelial cell line (Fig. 6, which is published as supporting information on the PNAS web site). As recently reported (21, 24), flagellin was found to induce transcription of the IL-8 gene in Caco-2 cells (Fig. 4 a and b). Our experiments demonstrated that flagellin is required for the induction of CCL20 and IL-8 gene expression in epithelial intestinal cells.
Figure 4.

Salmonella flagellins are inducing factors of CCL20 and IL-8 transcription in epithelial cells. Polarized Caco-2 cells were treated apically with bacteria (moi = 100) or flagellin. CCL20 and IL-8 gene transcription was quantified by real-time RT-PCR (a and b). Results are representative of at least 3 independent experiments. (a) Cells were infected for 45 min with S. enteritidis, the fliC mutant SEFK32, or SEFK32(pRP2) (complemented with the FliC flagellin of S. typhimurium) and incubated for 2.5 h in gentamicin-containing medium. (b) Dose-dependent induction of CCL20 and IL-8 expression by flagellin. Cells were exposed apically for 3.25 h to purified S. typhimurium FliC flagellin at the indicated concentrations. (c) Flagellin expression in Salmonella strains. Supernatants (0.5 ml) from Salmonella cultures or purified S. typhimurium FliC flagellin (1 μg) were analyzed after SDS/PAGE by Coomassie blue staining (Upper) and by immunoblotting (Lower) with flagellin-specific Ab. Arrow and asterisk indicate the position of flagellins from ATCC14028 (52 kDa) and SE857 (56 kDa), respectively. Agglutination with flagellin-specific Ab was performed on bacteria grown in the same conditions.
Medium from Flagellin-Treated Cells Induces Migration of Immature DCs.
Human immature DCs were able to migrate in response to rhCCL20 (Fig. 5). The migration was inhibited by incubation with CCL20-specific Abs. Low migration of DCs was observed with basal medium from untreated cells, probably reflecting the constitutive secretion of CCL20 by Caco-2 cells. The basal medium from flagellin-treated Caco-2 monolayers was as chemotactic as rhCCL20 at equivalent concentrations. Moreover, incubation of medium with CCL20-specific mAb fully abrogated chemotaxis. In conclusion, the migration of immature DC medium from flagellin-stimulated Caco-2 specifically depends on CCL20 activity.
Figure 5.

Immature DCs migrate in response to medium from flagellin-treated epithelial cells. rhCCL20 (7 ng/ml), control medium, or basal medium of untreated or of flagellin-treated Caco-2 cells (7 ng/ml of CCL20) were used in migration assays of immature DCs. When specified, CCL20-specific mAb was mixed with medium 30 min before assay to neutralize CCL20. Results are representative of 2 independent experiments.
Discussion
In this study, we provide the first evidence that Salmonella flagellins specifically stimulate CCL20 chemokine expression and secretion by epithelial intestinal cells, resulting in chemotaxis of immature DCs. Such DC migration could be essential for uptake of flagellated enteropathogens followed by antigen processing and presentation necessary for the induction of an adaptive immune response in the gut.
Flagellin is widely distributed and conserved among distant bacterial species (25). The domain involved in cell signaling is shared by S. typhimurium FliC and FljB and S. enteritidis FliC molecules, suggesting that it is located in conserved regions, i.e., 170 amino- and 90 carboxyl-terminal residues. Flagellins from various Gram-negative or -positive bacteria, including L. monocytogenes, are proinflammatory in the picomolar range (this study and refs. 21, 24, and 26–28). Therefore, flagellin presents all features of pathogen-associated molecular patterns (PAMPs). Toll-like receptors (TLRs) are involved in signal transduction of mammalian, plant, and insect PAMPs (29). Recently, TLR5 has been shown to mediate flagellin-dependent signaling in transfected mammalian cells (28). We and others have shown that TLR5 is expressed in Caco-2 cells (Fig. 7, which is published as supporting information on the PNAS web site, and ref. 30), suggesting that in the gut, flagellin could trigger chemokine expression by means of TLR5. Moreover, in human intestine, TLR5 is detected on the apical and basal surfaces of enterocytes (30). LPS, which signals injury in peripheral tissues or in sterile mucosal tissues, is inactive in the gut lumen where the Gram-negative bacteria are abundant. The gut has developed a detection system for danger by using other PAMPs. Flagellin is one PAMP candidate, but other bacterial factors are involved in mucosal cell signaling; for instance, E. coli P fimbriae in urinary epithelia triggers inflammation by means of TLR4 (31).
In our study, various enteropathogenic but not commensal bacteria stimulated CCL20 and IL-8 gene expression. In pathogens, flagella are expressed during infection, and the associated motility is crucial for virulence (3). Pathogenic bacteria also produce virulence factors for specific adhesion, and/or invasion, and/or injury of epithelial cells (3). Commensal bacteria, including the E. coli EMO used in this study, can also be equipped with flagella. However, even if expressed in vivo, the flagella of commensal bacteria are probably not contacting the epithelial cells. The microbial flora is confined to the luminal compartment and mucus layer (32). We propose that in vivo, only enteropathogenic bacteria could bring flagellin in close contact to the epithelial cell surface, resulting in induction of cell signaling. Alternatively, nonpathogenic bacteria have been shown to down-regulate the proinflammatory cascade in epithelium (33), a mechanism that could also result in the absence of flagellin-mediated signaling.
The gut is tolerant to most luminal material including resident bacteria. Under steady-state conditions, immature DCs are continually entering the gut probably via a constitutive CCL20-dependent mechanism and are sampling antigens (4, 7). The absence of injury and/or the anti-inflammatory environment of the gut have been proposed to induce tolerance (for review, see ref. 34) because antigen presentation by DCs occurs in the absence of costimulation. The coupling of CCL20 and IL-8 transcriptional activation could be crucial for induction of protective immune responses in the gut. Flagellin was already known to induce the proinflammatory IL-8 chemokine expression in epithelial cells (21, 24, 27). The resulting inflammation provides danger signals, especially tumor necrosis factor (TNF)-α and IL-1 cytokines, required for DC maturation. Thus, DCs attracted upon flagellin-stimulation may be fully activated and potent stimulators for adaptive responses. The recruitment of memory CD4 and B lymphocytes by CCL20 could also contribute to immunity in the gut (35).
Transcriptional activation of IL-8 and CCL20 genes is mediated by NF-κB (p65/p65 and p50/p65; refs. 11 and 18). A p65 binding site is present in the regulatory sequences of both IL-8 (18) and CCL20 genes (contig NT022115.2, −150 bp from ATG). These data are consistent with the flagellin-dependent TLR5-mediated NF-κB signaling (28). The coupling of IL-8 and CCL20 expression is, however, not absolute. IL-8 gene transcription is significantly higher in epithelial cells exposed to live Salmonella compared with heat-killed bacteria or to flagellin, whereas CCL20 mRNA levels remain the same. Therefore, activation of CCL20 expression seems to depend uniquely on flagellin, whereas IL-8 transcription is modulated by other components delivered by live bacteria as described (1, 2, 33).
Immature DCs recruited after interaction of enteropathogenic microbes with epithelial cells could constitute an appropriate niche for bacterial survival and dissemination. S. typhimurium are taken up in Peyer's patches by subepithelial DCs (36). The survival of S. typhimurium in DCs does not depend on virulence factors required for intracellular survival in macrophages (37). Therefore, the subepithelial immature DCs are the most potent candidates to carry the bacteria from the intestine to deeper organs such as mesenteric lymph nodes, spleen, or liver, where it is transferred to macrophages. The chemokine-stimulating activity of flagellin could be essential to enhance migration of DCs into subepithelial areas of Peyer's patches and villi. Dissemination by means of DCs has been documented for L. monocytogenes (38). Like Salmonella, L. monocytogenes produces flagella that are coordinately expressed with other virulence factors. Whether Listeria flagella are induction factors for CCL20 and whether immature DCs are vehicles for these bacteria are important questions to address for pathogenicity. Recently, Rescigno et al. (39) reported that, both in vitro and in vivo, mouse DCs penetrate intestinal epithelium to sample luminal bacteria. It remains to be tested whether this process is flagellin- and CCL20-mediated because the rapid migration of DCs does not parallel the CCL20 induction observed in Caco-2 cells.
Understanding the mechanisms of flagellin signaling in innate and adaptive immunity could provide new therapeutic approaches for preventing gut inflammation and new prospects in mucosal vaccination.
Supplementary Material
Acknowledgments
We thank A. Didierlaurent for bioinformatic analysis of the CCL20 gene, F. Niedergang and D. Schifferli for critical reading of the manuscript, and M. Reinhardt for excellent technical assistance. We are grateful to Drs. P. Berche (Institut National de la Santé et de la Recherche Médicale U411, Paris), K. Hughes, V. Gaboriau-Routhiau (Institut National de la Recherche Agronomique, Jouy-en-Josas, France), J. Galan (Yale School of Medicine, New Haven, CT), C. Lee (Harvard Medical School, Boston), and F. Van Asten (University of Utrecht, Utrecht, The Netherlands) for the generous gift of plasmid and strains. This work was supported by Swiss National Science Foundation Grant 31-56936-99, Swiss League Against Cancer Grant SKL635-2-1998 (to J.P.K.), and by European Union Grants QLRT-PL1999-01321 and OFES 99.041-1 and -2 (to J.P.K. and J.C.S.).
Abbreviations
- DC
dendritic cell
- moi
multiplicity of infection
- LPS
lipopolysaccharide
- RT
reverse transcription
- rh
recombinant human
- TLR
Toll-like receptor
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Eckmann L, Kagnoff M F, Fierer J. Infect Immun. 1993;61:4569–4574. doi: 10.1128/iai.61.11.4569-4574.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCormick B A, Miller S I, Carnes D, Madara J L. Infect Immun. 1995;63:2302–2309. doi: 10.1128/iai.63.6.2302-2309.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finlay B B, Falkow S. Microbiol Mol Biol Rev. 1997;61:136–169. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dieu M C, Vanbervliet B, Vicari A, Bridon J M, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, Caux C. J Exp Med. 1998;188:373–386. doi: 10.1084/jem.188.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charbonnier A S, Kohrgruber N, Kriehuber E, Stingl G, Rot A, Maurer D. J Exp Med. 1999;190:1755–1768. doi: 10.1084/jem.190.12.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwasaki A, Kelsall B L. J Exp Med. 2000;191:1381–1394. doi: 10.1084/jem.191.8.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook D N, Prosser D M, Forster R, Zhang J, Kuklin N A, Abbondanzo S J, Niu X D, Chen S C, Manfra D J, Wiekowski M T, et al. Immunity. 2000;12:495–503. doi: 10.1016/s1074-7613(00)80201-0. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka Y, Imai T, Baba M, Ishikawa I, Uehira M, Nomiyama H, Yoshie O. Eur J Immunol. 1999;29:633–642. doi: 10.1002/(SICI)1521-4141(199902)29:02<633::AID-IMMU633>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 9.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu Y J, Pulendran B, Palucka K. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 10.Dieu-Nosjean M C, Massacrier C, Homey B, Vanbervliet B, Pin J J, Vicari A, Lebecque S, Dezutter-Dambuyant C, Schmitt D, Zlotnik A, Caux C. J Exp Med. 2000;192:705–718. doi: 10.1084/jem.192.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Izadpanah A, Dwinell M B, Eckmann L, Varki N M, Kagnoff M F. Am J Physiol. 2001;280:G710–G719. doi: 10.1152/ajpgi.2001.280.4.G710. [DOI] [PubMed] [Google Scholar]
- 12.Kraehenbuhl J P, Neutra M R. Annu Rev Cell Dev Biol. 2000;16:301–332. doi: 10.1146/annurev.cellbio.16.1.301. [DOI] [PubMed] [Google Scholar]
- 13.Hueck C J. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Worton K J, Candy D C, Wallis T S, Clarke G J, Osborne M P, Haddon S J, Stephen J. J Med Microbiol. 1989;29:283–294. doi: 10.1099/00222615-29-4-283. [DOI] [PubMed] [Google Scholar]
- 15.McCormick B A, Colgan S P, Delp-Archer C, Miller S I, Madara J L. J Cell Biol. 1993;123:895–907. doi: 10.1083/jcb.123.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S K, Eckmann L, Panja A, Kagnoff M F. Gastroenterology. 1997;113:1214–1223. doi: 10.1053/gast.1997.v113.pm9322516. [DOI] [PubMed] [Google Scholar]
- 17.Gewirtz A T, Siber A M, Madara J L, McCormick B A. Infect Immun. 1999;67:608–617. doi: 10.1128/iai.67.2.608-617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elewaut D, DiDonato J A, Kim J M, Truong F, Eckmann L, Kagnoff M F. J Immunol. 1999;163:1457–1466. [PubMed] [Google Scholar]
- 19.Lee C A, Falkow S. Proc Natl Acad Sci USA. 1990;87:4304–4308. doi: 10.1073/pnas.87.11.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slauch J M, Mahan M J, Michetti P, Neutra M R, Mekalanos J J. Infect Immun. 1995;63:437–441. doi: 10.1128/iai.63.2.437-441.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Steiner T S, Nataro J P, Poteet-Smith C E, Smith J A, Guerrant R L. J Clin Invest. 2000;105:1769–1777. doi: 10.1172/JCI8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hardt W D, Chen L M, Schuebel K E, Bustelo X R, Galan J E. Cell. 1998;93:815–826. doi: 10.1016/s0092-8674(00)81442-7. [DOI] [PubMed] [Google Scholar]
- 23.Komoriya K, Shibano N, Higano T, Azuma N, Yamaguchi S, Aizawa S I. Mol Microbiol. 1999;34:767–779. doi: 10.1046/j.1365-2958.1999.01639.x. [DOI] [PubMed] [Google Scholar]
- 24.Gewirtz A T, Simon P O, Schmitt C K, Taylor L J, Hagedorn C H, O'Brien A D, Neish A S, Madara J L. J Clin Invest. 2001;107:99–109. doi: 10.1172/JCI10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winstanley C, Morgan J A. Microbiology. 1997;143:3071–3084. doi: 10.1099/00221287-143-10-3071. [DOI] [PubMed] [Google Scholar]
- 26.Ciacci-Woolwine F, Blomfield I C, Richardson S H, Mizel S B. Infect Immun. 1998;66:1127–1134. doi: 10.1128/iai.66.3.1127-1134.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eaves-Pyles T, Murthy K, Liaudet L, Virag L, Ross G, Soriano F G, Szabo C, Salzman A L. J Immunol. 2000;166:1248–1260. doi: 10.4049/jimmunol.166.2.1248. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi F, Smith K D, Ozinsky A, Hawn T R, Yi E C, Goodlett D R, Eng J K, Akira S, Underhill D M, Aderem A. Nature (London) 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 29.Krutzik S R, Sieling P A, Modlin R L. Curr Opin Immunol. 2001;13:104–108. doi: 10.1016/s0952-7915(00)00189-8. [DOI] [PubMed] [Google Scholar]
- 30.Cario E, Podolsky D K. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedlund M, Frendeus B, Wachtler C, Hang L, Fischer H, Svanborg C. Mol Microbiol. 2001;39:542–552. doi: 10.1046/j.1365-2958.2001.02205.x. [DOI] [PubMed] [Google Scholar]
- 32.Schultsz C, Van Den Berg F M, Ten Kate F W, Tytgat G N, Dankert J. Gastroenterology. 1999;117:1089–1097. doi: 10.1016/s0016-5085(99)70393-8. [DOI] [PubMed] [Google Scholar]
- 33.Neish A S, Gewirtz A T, Zeng H, Young A N, Hobert M E, Karmali V, Rao A S, Madara J L. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 34.Garside P, Mowat A M, Khoruts A. Gut. 1999;44:137–142. doi: 10.1136/gut.44.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao F, Rabin R L, Smith C S, Sharma G, Nutman T B, Farber J M. J Immunol. 1999;162:186–194. [PubMed] [Google Scholar]
- 36.Hopkins S A, Niedergang F, Corthesy-Theulaz I E, Kraehenbuhl J P. Cell Microbiol. 2000;2:59–68. doi: 10.1046/j.1462-5822.2000.00035.x. [DOI] [PubMed] [Google Scholar]
- 37.Niedergang F, Sirard J C, Blanc C T, Kraehenbuhl J P. Proc Natl Acad Sci USA. 2000;97:14650–14655. doi: 10.1073/pnas.97.26.14650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pron B, Boumaila C, Jaubert F, Berche P, Milon G, Geissmann F, Gaillard J L. Cell Microbiol. 2001;3:331–340. doi: 10.1046/j.1462-5822.2001.00120.x. [DOI] [PubMed] [Google Scholar]
- 39.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl J P, Ricciardi-Castagnoli P. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}