

Abstract
Domestication and selective breeding has resulted in over 1000 extant cattle breeds. Many of these breeds do not excel in important traits but are adapted to local environments. These adaptations are a valuable source of genetic material for efforts to improve commercial breeds. As a step toward this goal we identified candidate regions to be under selection in genomes of nine Russian native cattle breeds adapted to survive in harsh climates. After comparing our data to other breeds of European and Asian origins we found known and novel candidate genes that could potentially be related to domestication, economically important traits and environmental adaptations in cattle. The Russian cattle breed genomes contained regions under putative selection with genes that may be related to adaptations to harsh environments (e.g., AQP5, RAD50, and RETREG1). We found genomic signatures of selective sweeps near key genes related to economically important traits, such as the milk production (e.g., DGAT1, ABCG2), growth (e.g., XKR4), and reproduction (e.g., CSF2). Our data point to candidate genes which should be included in future studies attempting to identify genes to improve the extant breeds and facilitate generation of commercial breeds that fit better into the environments of Russia and other countries with similar climates.
Introduction
Recent advances in sequencing and genotyping technologies allow the identification of changes in genomes (‘selection signatures’) guided by positive selection in wild and domestic populations with a resolution and accuracy not previously achievable and at reasonable expense1. This has resulted in the construction of detailed selection signature maps for human populations2–4 and other species5,6. Domestic animals are of particular interest for this research because numerous traits have been affected by strong artificial selection for many generations, first during domestication and later during breed formation7. In addition, domesticated species (e.g., livestock) have been exposed to natural selection to adapt to the diverse environments into which they have been moved together with migrating human populations8 and during natural migrations prior to domestication9. Domestic cattle provide a good example of a species that has been domesticated at least twice in human history10, that has adapted to diverse environmental conditions ranging from Africa to Siberia11 and that has been under strong artificial selection to produce more than 1000 extant breeds12 exhibiting diverse levels of milk production, meat quality, feed efficiency and other economically important traits13. The main sources of modern cattle breed genetics are two Bos subspecies: B. taurus taurus and B. taurus indicus with some subsequent hybridisation events resulting in the descendant hybrid breeds being adapted to a large variety of environments14,15.
Genome studies of commercial cattle have allowed the identification of major genes and variants related to milk production (e.g., DGAT1 and ABCG2), meat quality (e.g., MSTN and TG), feed efficiency (e.g., ZIM2) and coat colour (e.g., KIT, KITLG, and MITF). Similarly, a recent study of native African cattle pointed to candidate genes for hot climate adaptation, such as HSPA4 and SOD116 and a study of Chinese cattle has confirmed selection around well-established functional candidates (e.g., MC1R, PAG1, and MYH cluster) and has also revealed novel gene candidates17. These two examples demonstrate the power and the need for detailed studies of native/local breed genomes to reveal the genetic bases of economically important traits and adaptations to specific environments. Understanding the genetic backgrounds of selected traits within local environmental contexts may be useful in the design of new breeds that will combine the high productivity of developed commercial breeds with the adaptive alleles found in native breeds18.
As a step toward this goal for Eurasian cattle, we recently described the genetic history and population structure of the major Russian native cattle breeds relative to the commercial and native breeds previously collected from around the world19. Most of the Russian breeds have ancestry that is shared with European taurine cattle breeds, while a few breeds represented by a branch of the so-called Turano-Mongolian cattle (Kalmyk, Yakut, and Buryat) share ancestry with the Asian taurines including Japanese Black (Wagyu) and Korean Hanwoo. This lineage is diverged from the European taurine cattle and shares few common haplotypes with them excluding those that have been transferred by recent admixture events in some populations19. The history of Turano-Mongolian cattle breeds traces back to early postglacial times and some researchers have hypothesised the independent domestication of the Asian taurine cattle preliminarily supported by genetic20 and paleontological evidence10.
Herein, we present the results of scans for putative signatures of selection and adaptation in the most distinct populations of the Russian cattle breeds selected based on our previous results19. The local Russian breeds are presumably adapted to an environment that is characterized by a range of conditions: e.g., rich grazing pastures and high temperatures during the short summer period, but harsh weather conditions and short daytime during the long winter. Breeds such as the Yakut, which is native to the Eastern Siberia, can overwinter in the open with temperatures as low as −50 °C21. The unique adaptations found in the Russian breeds likely also include a biotic component characterized by the resistance to indigenous pathogen infections and parasites22. Despite this wide range of ecotype adaptation, reports on signatures of selection in Russian cattle breeds have been limited to sparse genotype data (54,000 Single Nucleotide Polymorphisms (SNPs)) and to only four breeds: Yakut, Yaroslavl, Kalmyk, and Ukrainian Grey analyzed together with six breeds from Northern Europe23 and breeds from China24. The authors reported selection in the region of the ABCG2 gene related to milk production25 in the Yaroslavl breed and near virus resistance genes in the Yakut cattle which is supported by the known resistance of the breed to some viral infections22. Therefore, in this work we present analysis of signatures of selection for nine Russian native cattle breeds based on genotype data for ~139,000 SNPs combined with four additional breeds of European origin and two Asian taurine cattle breeds. We used hapFLK multipoint statistics considering the structure of haplotypes segregating within populations to reveal likely signatures of selection within three groups of related breeds and for individual breeds, de-correlated composite of multiple signals (DCMS) of selection combining H1 and H12 statistics, Tajima’s D, nucleotide diversity (Pi), and the fixation index (FST) which have previously been shown to be more efficient than each of these statistics individually for the detection of candidate selection signatures26–28. Our results demonstrate that while the Russian cattle breeds share major common signatures of selection/adaptation with world breeds, they also exhibit unique putative signatures of selection that can be related to adaptation to local environment, e.g. the cold climates.
Methods
Genotype data for nine Russian cattle breeds
GeneSeek Genomic Profiler High-Density (GGP HD150K) genotype results for nine Russian cattle breeds (Table 1) were obtained from Yurchenko et al.19. For the Russian breeds of European origin, we used 139,378 genotypes which were combined with the corresponding whole-genome sequencing (WGS)-based data originating from additional breeds of European origin (Table 1; described below). For the Russian breeds of Asian origin, we combined the GGP HD150K genotypes with 770 K Illumina BovineHD array data for the Japanese Black and with the WGS-based genotype data for the same SNPs for the Hanwoo breed from Korea resulting in 105,099 autosomal SNPs.
Table 1.
Breeds and breed groups.
| Breed | No. samples | Type | Reference | Breed group |
|---|---|---|---|---|
| Kalmyk | 23 | beef | 19 | ASIA |
| Buryat | 24 | dual purpose | 19 | ASIA |
| Yakut | 26 | dual purpose | 19 | ASIA |
| Hanwoo | 21 | beef | 30 | ASIA |
| Japanese Black | 19 | beef | 14 | ASIA |
| Kazakh Whiteheaded | 20 | beef | 19 | EUR1 |
| Kostroma | 18 | dual purpose | 19 | EUR1 |
| Jersey | 15 | dairy | 29 | EUR1 |
| Fleckvieh | 25 | dual purpose | 29 | EUR1 |
| Kholmogory | 30 | dairy | 19 | EUR2 |
| Bestuzhev | 19 | dual purpose | 19 | EUR2 |
| Yaroslavl | 18 | dairy | 19 | EUR2 |
| Black Pied | 24 | dairy | 19 | EUR2 |
| Holstein | 25 | dairy | 29 | EUR2 |
| Angus | 25 | beef | 29 | — |
| Average/Total | 22/332 | — | — | — |
SNP identification and filtering from WGS data
Sequence data for individuals from four European breeds (Holstein, Angus, Jersey, Fleckvieh) were obtained from the 1000 Bull Genomes project Run229. Sequences for 23 Hanwoo samples were downloaded from the NCBI GenBank in fastq format30. The full sequence processing protocols are described in31. Briefly, fastq reads were trimmed for adaptors and quality (>20 phred), and filtered to have <3 Ns and to pass chastity. Reads were then aligned to the UMD3.1 bovine genome assembly using bwa mem32 and PCR duplicates were removed. Variants were called using SAMtools v. 1.333 and filtered. High-quality SNPs were queried for their presence on the GGP HD150K array and genotypes from matched SNPs were extracted and merged with the GGP HD150K genotype data using PLINK34 --merge command.
Selecting animals and populations
Russian breeds with fewer than 18 samples and those breeds exhibiting recent admixture with other breeds19 were removed from the analysis. Breed groups for the hapFLK analyses were selected based on the results of principal component analysis (PCA35) of the joint dataset of 15 cattle breeds (Fig. 1). The breeds formed three clusters that demonstrated no major evidence of recent admixture between the groups. Two of the clusters (EUR1 and EUR2) included Russian and foreign breeds of European ancestry while the last cluster (ASIA) included Russian and foreign Turano-Mongolian breeds (Fig. 1; Supplementary Fig. S1; Table 1). Outlier samples from each breed were identified from the PCA plots and were removed from the analyses.
Figure 1.
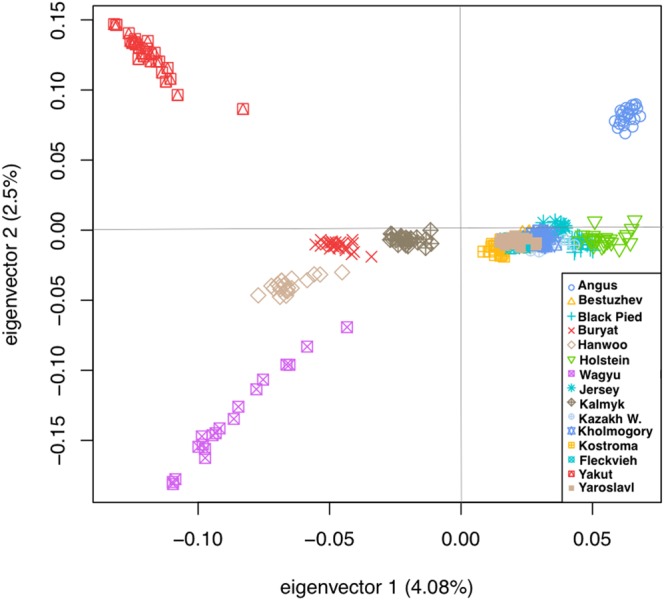
Principal component analysis of genotypes for nine native Russian breeds and additional cattle breeds of European and Asian origins used in the current study.
To eliminate sample size bias for breeds with large numbers of genotyped individuals (e.g., >100; Holstein) we removed animals with a high relatedness to other members of the same breed. We calculated indexes of relatedness within Holstein using the PLINK --genome function. Pairs of animals with unusually high values for pairwise PI_HAT statistic sum had one member removed from the analysis.
Identification of signatures of selection with hapFLK statistics
We performed a genome scan for signatures of selection within each group of breeds listed in Table 1 using a haplotype-based statistic (hapFLK)36. Because the hapFLK model assumes selection to be acting on ancestral SNP alleles we excluded rare SNPs with low minor allele frequencies (MAFs) from each of the three breed groups (MAF < 0.05). We also excluded poorly genotyped individuals (<95% of SNPs with genotypes), loci genotyped in <99% of samples, and SNPs on the sex chromosomes in PLINK using the commands: --maf 0.05, --geno 0.01, --mind 0.05, and --chr 1–29 prior to performing the genome selection scans.
The hapFLK method takes the haplotype structure of the population into account. What was important for our dataset is that this method can account for population bottlenecks and migration. Reynold’s distances and a kinship matrix were calculated by the hapFLK program v.1.436. For the hapFLK analysis, the number of haplotype clusters for each breed group was estimated with fastPhase37 and were set as –K 25, 35, 20 for the EUR1, EUR2, and ASIA breed groups, respectively. The expected maximum number of iterations was set to 30 for three groups. We used Yakut samples as outgroups to root the EUR1 and EUR2 population trees calculated by hapFLK and performed midpoint rooting for the ASIA set of breeds. Local Reynolds distances were calculated for selected regions using local_reynolds.py script and local population trees were then built with local_trees R script obtained from https://forge-dga.jouy.inra.fr/projects/hapflk/wiki/LocalTrees.
P-value calculation
For hapFLK the calculation of raw p-values was performed assuming that the selected regions represent only a small fraction of the genome38. The genome-wide distribution of hapFLK statistics could be modelled relatively well with a normal distribution except for a small fraction of outliers from potentially selected regions. Robust estimates of the mean and variance of the hapFLK statistic were obtained using the R MASS package, rlm function to eliminate influence of outlying regions following Boitard et al.39. This was done for each group (EUR1, EUR2, and ASIA). The hapFLK values were Z-transformed using these parameter estimates and p-values were calculated from the normal distribution in R. The R qvalue package was used to correct p-values for multiple testing40.
Composite measure of selection
It has recently been demonstrated, that the application of the composite measures of selection significantly improves signal to noise ratio and increases the power for the location of signals of possible selection27,28, specifically in comparison to single statistics or their simple overlaps. We combined five genome-wide statistics including the fixation index (FST41, haplotype homozygosity (H142), modified haplotype homozygosity statistics (H1242), Tajima’s D index43 and nucleotide diversity (Pi44) in the de-correlated composite of multiple signals (DCMS) framework27 which was shown to be efficient for combining p-values from the individual test statistics. DCMS combines p-values produced by several statistics for each locus into a single measure considering the correlation between the statistics. The correlation matrix was calculated genome-wide and allowed the assignment of different weights to each statistic’s p-value depending on their genome-wide correlation.
Haplotype-based statistics
Genotypes were phased separately in the two breed groups from Asia (ASIA) and Europe (EUR1 + EUR2) using SHAPEIT2 software45 with 400 conditioning states (–states 400) and effective population size parameter equal to 5000 (–effective-size 5000) as a safe provisional estimate for our diverse dataset. We used recombination rate estimates from Ma et al.46 to correct for the variability in recombination rates along chromosomes.
Both the H1 and H12 statistics42 estimate levels of haplotype-based homozygosity in windows throughout the genome, but H12 calculates homozygosity using the frequencies of the first and the second most common haplotypes which allows more efficient detection of hard and soft selective sweeps which are common in populations of wild and domestic species42,47,48. To calculate the H1 and H12 statistics we used VCF files with phased genotypes that were converted to the specific format required by the H12_H2H1.py script (https://github.com/ngarud/SelectionHapStats) from Garud et al.42. We calculated the statistics individually for each breed using overlapping windows of 14 SNPs with a step size of one SNP.
Tajima’s D statistics
Tajima’s D statistics were calculated for the same overlapping 14 SNP windows using vcftools v. 0.1.1549 and the –TajimaD function. The intervals were formed based on the output of the H1 statistic analysis for the Asian and European breeds separately and then passed to the bcftools (view) software33 along with the breed-specific gzipped VCF file before being passed to the vcftools –TajimaD function. To reduce the time of calculation all the work was carried out in parallel mode with assistance of GNU PARALLEL50.
Nucleotide diversity (Pi)
Nucleotide diversity was calculated for each breed and chromosome separately using the vcftools –site-pi option. The produced values were then smoothed using the R runmed function with the window size of 31 SNPs (k = 31, endrule = “constant”).
Fixation index (FST)
We calculated the FST index as a measure of population differentiation separately for the European (EUR1 + EUR2) and Asian breeds (ASIA). Within each of the groups, FST was calculated for each variant for each breed against the rest of the samples from the other breeds within the group using the PLINK --fst function. Negative FST values were converted in zeros and the statistics was smoothed for each chromosome using R runmed function in windows of 31 SNPs (k = 31, endrule = “constant”) to reduce noise.
De-correlated composite of multiple signals (DCMS)
To calculate the DCMS statistics for each of the breeds we combined the aforementioned statistics (H1, H12, Tajima’s D, Pi, and FST) in a single spreadsheet by merging data on the basis of the SNP name. We next used the R MINOTAUR package51 to calculate genome-wide P-values based on fractional ranks for each statistic (stat_to_pvalue MINOTAUR function) with appropriate one-tailed tests (Pi and Tajima D – left-tailed; H1, H12, and FST – right-tailed). The covariance matrix between the different statistics was estimated based on 50,000 randomly sampled SNPs using the CovNAMcd function with alpha = 0.75. The DCMS statistic was calculated using the DCMS function and the precalculated covariance matrix among the statistics. The resulting DCMS statistics for each breed were examined for normality of their distributions and then fitted to the normal distribution using the robust fitting of linear model method implemented in the rlm R function of the MASS package39. The fitted DCMS statistics were then converted in p-values using the pnorm function (lower.tail = FALSE, log.p = FALSE) and the P-values were finally converted to the corresponding q-values using the qvalue R function40.
Identification of chromosome intervals under selection and candidate genes
Bovine gene annotations from the bovine genome assembly build UMD 3.1 were downloaded from Biomart52. We then identified chromosome intervals and candidate genes predicted to have been subjected to selection. To locate the putative regions under selection we considered chromosome intervals with SNPs with adjusted p-values < 0.05 and boundaries of each interval were defined by the locations of the first flanking SNPs exhibiting adjusted p-values > 0.1. Within the selected intervals, genes were identified within 1σ value from the most significant SNP based on statistical values (DCMS or hapFLK) distribution similar to Fariello et al.38. This approach results in fewer candidate genes being reported for the “sharp” selection peaks while for the intervals with many SNPs exhibiting similar statistics values, larger numbers of genes were reported. Genes were also ranked based on their distance from the SNP with the highest statistics value in each region with larger ranks assigned to more distant genes. We visualized regions under putative selection using circlize R package53.
Results
Breed groups
The PCA analysis suggested the presence of two well-differentiated clusters of breeds in our dataset (Fig. 1) represented by the Asian Turano-Mongolian (ASIA: Yakut, Buryat, Kalmyk, Japanese Black, and Hanwoo) and European breeds matching our previous results19. In addition, in the hapFLK analysis, the European breed population was further subdivided into two sets of breeds (EUR1: Kazakh Whiteheaded, Kostroma, Jersey, Fleckvieh; and EUR2: Bestuzhev, Black Pied, Holstein, Kholmogory, Yaroslavl) matching the two major clusters of European breeds from Yurchenko et al.19 and supported by the PCA analysis (Supplementary Fig. S1). In total, 332 animals from 15 breeds (including nine Russian breeds) with a mean number of 22 individuals per breed were used in these analyses (Table 1).
The DCMS and hapFLK statistics for the Russian and foreign cattle breeds overlapped to some extent providing independent support for selected regions. However, because the hapFLK statistic detects probable signatures of selection within groups of breeds and the DCMS in our study was used to combine statistics within a breed, the hapFLK results could not be added to the DCMS framework. The hapFLK revealed additional regions under putative selection within groups of breeds missed by the DCMS while the DCMS was efficient in detecting shorter candidate regions.
Composite measure of selection
The DCMS statistic was calculated for each SNP for each breed. After fitting of the normal distribution, calculation of p-values and correction for multiple testing we obtained 29 to 90 genomic intervals under putative selection per breed (q-value < 0.05) with a total of 953 regions detected across all breeds (with some overlaps between breeds; Supplementary Table S1). The size of the genomic regions putatively under selection varied from 1 bp to 4.80 Mbp with the average size of 242.46 Kbp. The number of genes within detected regions per breed ranged from 42 to 209.
HapFLK
The total number of regions identified by the hapFLK analyses (46; Supplementary Table S1) was lower than found by the DCMS method. The largest number of hapFLK detected regions was observed in the EUR1 set (25), followed by the ASIA (12) and EUR2 (9) sets. The EUR1, EUR2, and ASIA sets all shared a common hapFLK interval (BTA5:13.2–27.00 Mbp, containing the KITLG gene), however, coordinates for sub-regions detected in the EUR1 and ASIA sets did not overlap. No other shared hapFLK regions were detected for any combination of the breed sets. Sizes of the putatively selected regions ranged from 484 Kbp to 15 Mbp with an average size of 2.8 Mbp.
Candidate genes for adaptation of the Russian cattle breeds to environmental and climate challenges
We investigated how adaptation to local environmental challenges including viral and parasite challenges and the cold climate could have shaped the genomes of the Russian native cattle breeds (Table 2). We found a region on BTA7:23.04–23.14 Mbp containing RAD50 and reported by DCMS that appeared to have been likely selected in the Russian Kholmogory, Bestuzhev, Kalmyk, Yakut, and Yaroslavl breeds as well as in the Korean Hanwoo. RAD50 is a DNA repair protein, the component of MRN complex, that plays a central role in double-strand break repair and is a key gene in antiviral protection54 suggesting its potential role in the response of the local breeds to viral challenges. The cold-resistant Kholmogory cattle had a probable signature of selection in the region on BTA5:29.68–30.17 Mbp containing the aquaporin cluster (AQP@), including the top ranked in DCMS results AQP5 gene previously reported as a cold/heat acclimation candidate due to its role in the formation of water channels55. AQP5 has been under positive selection in yaks in response to adaptation to high altitudes56 characterised by low temperatures. Seven regions under putative selection were reported by hapFLK for the cold-adapted Yakut cattle, of which four were shared with the Japanese Black and two with Hanwoo cattle (Fig. 2; Supplementary Table S2). Top-ranked genes within the two intervals that were unique to the Yakut cattle according to the hapFLK haplotype local tree analysis (p-value < 0.05) were candidates for the reaction of organisms to cold exposure. The RETREG1 is responsible for human pain insensitivity disorder caused by hereditary sensory and autonomic neuropathy (HSAN). Mutations in human RETREG1 are responsible for HSAN Type 257. HSAN Type 2 leads to inability to feel pain and temperature58. The Yakut cattle have a signature of selection in this region (p-value = 0.001) while Hanwoo possesses a suggestive signature of selection (p-value = 0.09) according to hapFLK and significant signature according to the DCMS analysis (Fig. 2; Table 2; Supplementary Fig. S2). The ribosomal large subunit protein 7 (RPL7) gene was shown to be cold-responsive in freeze tolerant frogs with higher protein levels found in the skin of cold-tolerant species compared to non-tolerant species and increased expression levels in muscles and brain under freezing conditions59. According to the hapFLK haplotype local tree analysis, this region could be under strong selection in the Yakut cattle (p-value = 0.00005) with a weak signal also observed in Hanwoo and Japanese Black (p-value = 0.05; Fig. 2; Supplementary Fig. S3). In addition, the Yakut cattle demonstrated a unique signature of putative selection in the genomic region containing the tankyrase (TNKS) gene (top ranked by the DCMS analysis in the region) that has been shown to be related to energy expenditure, feed intake and adiposity in mice60, suggesting its possible role in adaptation to variation in local environment feed availability and quality. The Yakut cattle also had a signature of selection in the region of the keramide kinase like gene (CERKL) expressed in the retina, which possesses variants responsible for retinitis pigmentosa in humans, associated with light stress response and protection of photoreceptor cells61. This putative signature of selection could be related to the adaptation of the Yakut cattle to the light regime above the Polar circle.
Table 2.
Genes in regions predicted to be under putative selection in Russian cattle breeds.
| Chr* | Region start (Mbp) | Region end (Mbp) | Most significant SNP q-value | Breed/Group | No. genes in the region | Method | Candidate gene (rank) | Function |
|---|---|---|---|---|---|---|---|---|
| 1 | 28.402 | 28.899 | 0.002273827 | Kazakh Whiteheaded | 2 | DCMS | FKBP2 (2) | Milk |
| 2 | 15.069 | 15.083 | 0.012500925 | Yakut | 1 | DCMS | CERKL (1) | Light stress |
| 2 | 73.106 | 73.393 | 0.017181621 | Yaroslavl | 1 | DCMS | GLI2 (1) | Weight, growth |
| 3 | 9.348 | 9.751 | 0.004315819 | Bestuzhev | 13 | DCMS | COPA (3) | Coat colour |
| 3 | 9.348 | 9.722 | 0.001173551 | Black Pied | 10 | DCMS | COPA (1) | Coat colour |
| 3 | 9.386 | 9.529 | 0.046081992 | Holstein | 2 | DCMS | COPA (1) | Coat colour |
| 3 | 52.040 | 52.213 | 0.001189865 | Hanwoo | 1 | DCMS | HFM1 (1) | Reproduction |
| 3 | 52.075 | 52.231 | 0.010464401 | Holstein | 1 | DCMS | HFM1 (1) | Reproduction |
| 3 | 52.148 | 52.225 | 0.009368275 | Black Pied | 1 | DCMS | HFM1 (1) | Reproduction |
| 3 | 52.153 | 52.187 | 0.018393593 | Buryat | 1 | DCMS | HFM1 (1) | Reproduction |
| 3 | 52.174 | 52.267 | 0.000324907 | Kalmyk | 1 | DCMS | HFM1 (1) | Reproduction |
| 4 | 14.188 | 15.448 | 0.040960871 | ASIA | 6 | hapFLK | RPL7 (1) | Acclimation |
| 4 | 107.007 | 107.085 | 0.043707068 | Kalmyk | 3 | DCMS | TRPV5 (1) | Milk fever |
| 5 | 13.271 | 27.026 | 5.53E-08 | EUR2 | 71 | hapFLK | KITLG (4) | Coat colour |
| 5 | 16.357 | 21.155 | 0.001789011 | Yaroslavl | 13 | DCMS | KITLG (9) | Coat colour |
| 5 | 17.154 | 20.486 | 0.000398061 | EUR1 | 8 | hapFLK | KITLG (1) | Coat colour |
| 5 | 18.335 | 19.335 | 4.47E-05 | Bestuzhev | 1 | DCMS | KITLG (1) | Coat colour |
| 5 | 18.335 | 18.966 | 0.001068985 | Kazakh Whiteheaded | 1 | DCMS | KITLG (1) | Coat colour |
| 5 | 29.684 | 30.174 | 0.011869128 | Kholmogory | 10 | DCMS | AQP5 (3) | Acclimation |
| 5 | 47.752 | 48.287 | 0.033364267 | ASIA | 1 | hapFLK | HMGA2 (1) | Growth |
| 5 | 48.017 | 48.308 | 4.53E-10 | Buryat | 1 | DCMS | HMGA2 (1) | Growth |
| 5 | 48.017 | 48.506 | 7.46E-11 | Kalmyk | 1 | DCMS | HMGA2 (1) | Growth |
| 5 | 48.045 | 48.263 | 0.001471807 | Hanwoo | 1 | DCMS | HMGA2 (1) | Growth |
| 5 | 48.094 | 48.514 | 4.68E-06 | Bestuzhev | 1 | DCMS | HMGA2 (1) | Growth |
| 5 | 48.155 | 48.280 | 0.018053667 | Japanese Black | 1 | DCMS | HMGA2 (1) | Growth |
| 5 | 48.185 | 48.270 | 0.001526729 | Kostroma | 1 | DCMS | HMGA2 (1) | Growth |
| 5 | 60.624 | 60.624 | 0.048486709 | Buryat | 1 | DCMS | HAL (1) | Milk |
| 5 | 75.685 | 75.731 | 0.037717028 | Yaroslavl | 1 | DCMS | CSF2RB (1) | Milk |
| 5 | 107.338 | 108.490 | 0.004370697 | Yakut | 18 | DCMS | KDM5A (2) | Milk |
| 6 | 37.076 | 39.850 | 0 | EUR1 | 18 | hapFLK | NCAPG (1), LCORL (2), LAP3 (6), ABCG2 (11) | Growth, Milk |
| 6 | 37.855 | 38.000 | 1.04E-06 | Kalmyk | 2 | DCMS | ABCG2 (1) | Milk |
| 6 | 37.902 | 37.948 | 0.027503049 | Fleckvieh | 1 | DCMS | ABCG2 (1) | Milk |
| 6 | 37.983 | 38.188 | 0.006537637 | Kostroma | 3 | DCMS | ABCG2 (1) | Milk |
| 6 | 38.579 | 38.656 | 0.001009406 | Kholmogory | 3 | DCMS | LAP3 (2) | Milk |
| 6 | 38.580 | 38.616 | 0.000578482 | Buryat | 3 | DCMS | LAP3 (1) | Milk |
| 6 | 38.731 | 38.924 | 7.26E-07 | Jersey | 3 | DCMS | NCAPG (1), LCORL (3) | Growth |
| 6 | 38.735 | 38.931 | 0.000382606 | Kazakh Whiteheaded | 3 | DCMS | NCAPG (1), LCORL (2) | Growth |
| 6 | 38.924 | 39.100 | 0.006598304 | Fleckvieh | 1 | DCMS | LCORL (1) | Growth |
| 6 | 38.924 | 39.023 | 0.03478244 | Yaroslavl | 1 | DCMS | LCORL (1) | Growth |
| 6 | 59.928 | 74.936 | 0 | EUR1 | 63 | hapFLK | KIT (4) | Coat colour |
| 6 | 68.059 | 72.063 | 5.67E-07 | Kazakh Whiteheaded | 26 | DCMS | KIT (4) | Coat colour |
| 6 | 69.440 | 72.476 | 1.13E-06 | Fleckvieh | 10 | DCMS | KIT (4) | Coat colour |
| 6 | 71.240 | 71.959 | 0.000166418 | Yaroslavl | 3 | DCMS | KIT (2) | Coat colour |
| 7 | 23.035 | 23.183 | 0.001289242 | Kholmogory | 1 | DCMS | RAD50 (1) | Antimicrobial protection |
| 7 | 23.078 | 23.107 | 0.046379711 | Bestuzhev | 1 | DCMS | RAD50 (1) | Antimicrobial protection |
| 7 | 23.078 | 23.261 | 3.43E-08 | Kalmyk | 3 | DCMS | IL5 (1), IRF1 (2), RAD50 (3) | Antimicrobial protection |
| 7 | 23.113 | 23.228 | 2.35E-05 | Hanwoo | 2 | DCMS | RAD50 (1), IL5 (2) | Antimicrobial protection |
| 7 | 23.136 | 23.498 | 3.66E-06 | Bestuzhev | 4 | DCMS | IRF1 (1), IL5 (3) | Antimicrobial protection |
| 7 | 23.136 | 23.216 | 0.00265416 | Yakut | 2 | DCMS | IL5 (1), RAD50 (2) | Antimicrobial protection |
| 7 | 23.144 | 23.144 | 0.037325408 | Yaroslavl | 1 | DCMS | RAD50 (1) | Antimicrobial protection |
| 7 | 23.591 | 23.695 | 0.005771811 | Holstein | 1 | DCMS | CSF2 (1) | Reproduction |
| 7 | 23.613 | 23.699 | 0.000131001 | Black Pied | 1 | DCMS | CSF2 (1) | Reproduction |
| 7 | 23.683 | 23.698 | 0.002131539 | Yaroslavl | 1 | DCMS | CSF2 (1) | Reproduction |
| 7 | 23.691 | 23.696 | 0.028601712 | Kazakh Whiteheaded | 1 | DCMS | CSF2 (1) | Reproduction |
| 7 | 54.361 | 54.735 | 0.000687902 | Yakut | 6 | DCMS | HDAC3 (5) | Brown adipose tissue |
| 7 | 93.194 | 93.360 | 2.19E-07 | Kalmyk | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 7 | 93.206 | 93.307 | 3.83E-10 | Buryat | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 7 | 93.208 | 93.276 | 0.01177964 | Fleckvieh | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 7 | 93.211 | 93.307 | 2.37E-07 | Hanwoo | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 7 | 93.214 | 93.276 | 0.010240366 | Kostroma | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 7 | 93.215 | 93.224 | 0.031503554 | Jersey | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 7 | 93.242 | 93.282 | 0.017682048 | Yakut | 1 | DCMS | ARRDC3 (1) | Adipose tissue |
| 8 | 0.264 | 0.503 | 0.04889224 | Kholmogory | 1 | DCMS | ANXA10 (1) | Reproduction |
| 8 | 79.681 | 79.759 | 0.027035565 | Yaroslavl | 1 | DCMS | NTRK2 (1) | Antimicrobial protection |
| 8 | 88.219 | 88.589 | 0.002222339 | Black Pied | 1 | DCMS | SYK (1) | Brown adipose tissue |
| 8 | 88.420 | 88.465 | 0.027629081 | Kholmogory | 1 | DCMS | SYK (1) | Brown adipose tissue |
| 12 | 44.257 | 44.839 | 0.001059257 | Bestuzhev | 1 | DCMS | KLHL1 (1) | Milk |
| 12 | 80.782 | 81.010 | 0.012757213 | Buryat | 1 | DCMS | PCCA (1) | Milk |
| 12 | 80.803 | 81.092 | 0.010062102 | Yaroslavl | 1 | DCMS | PCCA (1) | Milk |
| 12 | 80.962 | 81.085 | 0.037912516 | Japanese Black | 1 | DCMS | PCCA (1) | Milk |
| 13 | 57.572 | 57.636 | 0.021930321 | Kazakh Whiteheaded | 1 | DCMS | EDN3 (1) | Coat colour |
| 14 | 1.489 | 1.893 | 0.032866336 | Kazakh Whiteheaded | 28 | DCMS | TONSL (2), DGAT1 (20) | Milk |
| 14 | 1.651 | 1.869 | 0.001653056 | Buryat | 11 | DCMS | TONSL (1), DGAT1 (11) | Milk |
| 14 | 1.696 | 1.893 | 0.038247912 | Kholmogory | 13 | DCMS | DGAT1 (1) | Milk |
| 14 | 23.258 | 23.269 | 0.02863779 | Yaroslavl | 1 | DCMS | NPBWR1 (1) | Reproduction |
| 14 | 24.419 | 25.135 | 1.02E-08 | Black Pied | 8 | DCMS | TMEM68 (2), XKR4 (8), PLAG1 (5) | Feed intake, Growth, Weight |
| 14 | 24.421 | 24.501 | 0.00525187 | Kazakh Whiteheaded | 1 | DCMS | XKR4 (1) | Weight |
| 14 | 24.426 | 25.135 | 1.50E-06 | Kholmogory | 8 | DCMS | TMEM68 (2), PLAG1 (5), XKR4 (7) | Feed intake, Growth, Weight |
| 14 | 24.431 | 24.434 | 0.047221525 | Kalmyk | 1 | DCMS | XKR4 (1) | Weight |
| 14 | 24.512 | 24.556 | 0.032774966 | Hanwoo | 1 | DCMS | XKR4 (1) | Weight |
| 14 | 24.699 | 25.010 | 0.000703648 | Bestuzhev | 3 | DCMS | TMEM68 (2) | Feed intake |
| 14 | 24.714 | 24.909 | 1.24E-06 | Kalmyk | 3 | DCMS | TMEM68 (3) | Feed intake |
| 14 | 25.447 | 25.849 | 5.88E-06 | Kazakh Whiteheaded | 1 | DCMS | IMPAD1 (1) | Growth |
| 14 | 25.460 | 25.617 | 4.79E-10 | Holstein | 1 | DCMS | IMPAD1 (1) | Growth |
| 14 | 25.497 | 25.667 | 0.002016879 | Angus | 1 | DCMS | IMPAD1 (1) | Growth |
| 14 | 25.497 | 25.546 | 0.014138416 | Kostroma | 1 | DCMS | IMPAD1 (1) | Growth |
| 14 | 25.499 | 25.541 | 0.015989917 | Jersey | 1 | DCMS | IMPAD1 (1) | Growth |
| 14 | 25.507 | 25.530 | 0.000738867 | Buryat | 1 | DCMS | IMPAD1 (1) | Growth |
| 15 | 35.637 | 36.167 | 0.013241977 | Black Pied | 5 | DCMS | NUCB2 (1) | Feed intake, Body temperature, Growth |
| 16 | 36.496 | 36.581 | 0.045474804 | Bestuzhev | 1 | DCMS | RGS7 (1) | Domestication, Acclimation |
| 20 | 4.046 | 4.102 | 0.00256072 | Jersey | 1 | DCMS | SH3PXD2B (1) | Growth, meat |
| 20 | 4.050 | 4.084 | 0.000859753 | Bestuzhev | 1 | DCMS | SH3PXD2B (1) | Growth, meat |
| 20 | 4.067 | 4.144 | 0.003706131 | Black Pied | 1 | DCMS | SH3PXD2B (1) | Growth, meat |
| 20 | 4.067 | 4.084 | 0.019181385 | Holstein | 1 | DCMS | SH3PXD2B (1) | Growth, meat |
| 20 | 4.593 | 4.784 | 0.0029386 | Kholmogory | 4 | DCMS | CREBRF (1) | Growth |
| 20 | 4.597 | 4.730 | 0.0004249 | Bestuzhev | 3 | DCMS | CREBRF (1) | Growth |
| 20 | 4.597 | 4.791 | 0.0145958 | Jersey | 4 | DCMS | CREBRF (3) | Growth |
| 20 | 4.628 | 4.782 | 0.0002944 | Holstein | 3 | DCMS | CREBRF (1) | Growth |
| 20 | 4.660 | 4.783 | 2.94E-05 | Hanwoo | 3 | DCMS | CREBRF (1) | Growth |
| 20 | 4.673 | 4.759 | 0.0010948 | Buryat | 2 | DCMS | CREBRF (1) | Growth |
| 20 | 4.692 | 4.782 | 0.0125848 | Black Pied | 2 | DCMS | CREBRF (1) | Growth |
| 20 | 4.692 | 4.761 | 6.08E-05 | Kalmyk | 2 | DCMS | CREBRF (1) | Growth |
| 20 | 31.912 | 32.005 | 0.019009149 | Black Pied | 1 | DCMS | GHR (1) | Milk, Growth |
| 20 | 32.098 | 32.460 | 0.032750776 | Bestuzhev | 1 | DCMS | GHR (1) | Milk, Growth |
| 20 | 52.303 | 57.558 | 0.009549042 | ASIA | 8 | hapFLK | RETREG1 (1) | Acclimation |
| 20 | 56.431 | 56.876 | 0.043882656 | Yakut | 3 | DCMS | RETREG1 (2) | Acclimation |
| 20 | 56.683 | 56.890 | 0.000763043 | Hanwoo | 2 | DCMS | RETREG1 (2) | Acclimation |
| 24 | 60.758 | 62.644 | 0.000623541 | EUR2 | 15 | hapFLK | BCL2 (1) | Reproduction |
| 27 | 24.577 | 24.803 | 0.001749199 | Yakut | 1 | DCMS | TNKS (1) | Feed intake, Adipose tissue |
| 28 | 21.176 | 26.909 | 0.00551179 | EUR2 | 30 | hapFLK | SIRT1 (2) | Antimicrobial protection |
| 28 | 24.039 | 24.885 | 0.01399301 | Yaroslavl | 6 | DCMS | SIRT1 (2) | Antimicrobial protection |
| 28 | 35.332 | 35.883 | 0.008902995 | Yaroslavl | 2 | DCMS | SFTPD (2) | Antimicrobial protection |
*For a full list of selected regions and genes see Supplementary Table S1.
Figure 2.

Manhattan plots for putative signatures of selection analysis in cattle breeds of Asian origin with names of candidate genes discussed in the text. (A) hapFLK results for the combined set of five breeds (Yakut, Buryat, Kalmyk, Hanwoo, and Japanese Black), (B) DCMS results for the Buryat cattle, (C) DCMS results for Yakut cattle. Blue line indicates q-value = 0.1 and the red line q-value = 0.05. Colored peak regions highlight positions of the genes indicated on the plots.
Brown adipose tissue is an organ that provides energy to protect animals from hypothermia62. The key gene in this process is a mitochondrial uncoupling protein 1 (UCP1)62. We found several genes known to influence expression of UCP1 and that are directly involved in the regulation of adiposity in regions under putative selection in the Russian cattle breeds. The histone deacetylase 3 (HDAC3) gene, that is required to activate brown adipose tissue enhancers to ensure thermogenic aptitude63 was found in a reported region in the Yakut cattle, while the adipocyte arrestin domain-containing 3 protein (ARRDC3), that regulates the expression of UCP1 in white adipose tissue64, was found in the region to be under probable selection in four Russian breeds, Hanwoo, Jersey, and Fleckvieh. This is suggestive of its potential role in variation in economically important traits. On the other hand, only the Kholmogory and Black-Pied breeds possessed selection signals in a region that included spleen tyrosine kinase (SYK) which is involved in brown adipocyte differentiation and is known to affect the expression of UCP165. The Yaroslavl cattle expressed a strong signal of putative selection (q-value < 0.01) in the genomic region that includes SFTPD that encodes lung surfactant protein D (SP-D) which contributes to lung defense from inhaled microorganisms and was previously found to be under positive selection in human populations adapted to high altitudes66. The Black Pied cattle had been exposed to putative selection in a region including the top-ranked NUCB2 gene that encodes Nesfatin-1 which influences the regulation of body temperature and food intake67. In cattle, genetic variants in NUCB2 have been shown to be associated with growth traits in three native Chinese breeds68. A signature of likely selection in the Bestuzhev breed was detected near RGS7 that has been shown to be related to the differentiation of neurological function between dogs and wolves and, in humans, its expression in central noradrenergic neurons increases in response to chronic cold exposure69.
Morphological traits and adaptations
Of the 999 genomic intervals (953 from DCMS and 46 from hapFLK) under putative selection, 66.7% overlapped with regions previously predicted to have been under selection in cattle70 (Supplementary Table S1). Among these previously detected regions, strong signals of differentiation were obtained in the regions containing well known candidate genes related to morphology, adaptation, and domestication (e.g., KITLG, KIT, EDN3, and COPA), growth and feed intake (XKR4, TMEM68, LCORL, NCAPG, HMGA2, IMPAD1, and GLI2), reproduction (CSF2, BCL2, ANXA10, and NPBWR1), and milk traits (DGAT1, GHR, ABCG2, GLI2, LAP3, TRPV5, FKBP2, and PCCA; Fig. 3).
Figure 3.

Circos plot showing signatures of putative selection in cattle genomes. In blue are signatures of selection detected in the European breeds of non-Russian origin, in brown are signatures of selection in the Russian cattle breeds of European origin, in green are Asian breeds of non-Russian origin while in red are Russian breeds of Asian origin. Numbers correspond to cattle autosomes. Candidate genes found in putatively selected regions in at least two breeds or in both the DCMS and hapFLK analyses reported in Table 2 are placed on the outer circle. For a full list of all regions and genes see Supplementary Table S1.
Morphology and domestication
Domestication is often associated with changes in the coat colour of domesticated populations71. Consistent with previous findings in livestock and other domestic species, strong signals of putative selection in Russian breeds of European and Asian origin were detected by the hapFLK analyses in the genomic regions that include the genes KIT and KITLG, both of which contribute to coat colour in a variety of species72,73. Interestingly, in the region including KITLG a long interval under putative selection (BTA5: 13.27–27.03 Mbp (13.76 Mbp)) was reported by hapFLK in the EUR2 breed set, while in the EUR1 set the overlapping region was shorter (BTA5:17.15–20.49 (3.34 Mbp)). On the other hand, the DCMS analysis detected multiple putative signatures of selection within the 13.76 Mbp interval on BTA5 in different breeds with Bestuzhev and Kazakh Whiteheaded being the only Russian breeds that had a narrow signal near the KITLG gene, while the Black Pied and Yaroslavl had several signals of putative selection near other genes suggesting that this locus could contain multiple sequences subjected to selection (Supplementary Table S1). Consistent with this finding, in the Asian breeds a 1.60 Mbp overlapping region was revealed (BTA5: 21.97–23.57 Mbp) which did not, however, include the KITLG (Supplementary Table S1). A similar pattern was observed near the KIT gene. The hapFLK analyses identified a region exposed to putative selection in the EUR1 breed set on BTA6:59.93–74.94 Mbp (15.01 Mbp). However, the DCMS analysis detected multiple shorter intervals within this region with KIT being found in putatively selected regions in the Yaroslavl, Kazakh Whiteheaded, and Fleckvieh breeds. Two more coat colour-related genes (EDN3, COPA) were top-ranked by DCMS among the signatures of selection detected in the Kazakh Whiteheaded (EDN3), Black Pied (COPA), and Holstein (COPA) breeds. EDN3 promotes the differentiation and proliferation of melanocytes74 and has previously been found in one of 12 genomic regions associated with the UV-protective eye area pigmentation phenotype in the Fleckvieh whiteface breed75. Since our Kazakh Whiteheaded individuals did not have well-developed eye area pigmentation but all had whiteface phenotypes, we can speculate about the EDN3 be directly involved with formation of the whiteface phenotype itself. Additionally, the Black Pied, Bestuzhev, and Holstein breeds had putative signatures of selection near the coatomer protein complex, subunit alpha (COPA) gene, which is known to be related to pigment synthesis. A missense mutation within this gene is completely associated with dominant red coat phenotype in Holstein cattle76.
Growth and feed intake
A putative signature of selection has previously been detected in a region on BTA14 containing the XKR4 gene that is associated with birth weight in Nelore cattle77 as well as with feed intake and average daily gain in cattle78,79. In the Black Pied and Kholmogory the selected interval was relatively wide containing also the TMEM68 gene previously associated with feed intake79 and PLAG1 that is associated with body size, weight and reproduction in cattle80, while in the Kazakh Whiteheaded the region was smaller containing only XKR4. Kalmyk cattle may possess two separate putatively selected regions in this interval, one near XKR4 and the other near TMEM68. The LCORL-NCAPG interval on BTA6 has been demonstrated to be associated with growth traits in cattle (average daily gain, muscle development81 and carcass composition82). In our analysis, the Kazakh Whiteheaded and Jersey demonstrate relatively wide signals in this region (~190 Kb) containing both genes with NCAPG being the top-ranked by DCMS. The Yaroslavl and Fleckvieh had shorter intervals with the LCORL gene being the top-ranked. A putative signature of selection was reported by both the hapFLK in the ASIA set and DCMS in the Buryat, Kalmyk, Bestuzhev, Kostroma, Hanwoo, and Japanese Black breeds near HMGA2 (high mobility group protein A2), a transcription factor that regulates genes involved in cell differentiation and growth. This is a key gene associated with growth in cattle83, human height84, body size in dogs85, and horses86, weight in mice and carcass traits in pigs87. We identified a likely signature of selection in the region of inositol monophosphatase domain containing 1 (IMPAD1) on BTA14 reported by DCMS in the Kazakh Whiteheaded, Kostroma, and Buryat breeds. IMPAD1 plays a role in the bone-cartilage system with mutations leading to severe growth retardation in humans88. This region has also previously been found to be under selection in Canchim89 and Brahman cattle90. DCMS also detected a signature of putative selection in the region containing GLI2 (a member of the Gli gene family, which encode transcription factors) in the Yaroslavl cattle. This gene has previously been associated with bovine weight91 and growth traits in pigs92.
Reproduction
A strong signal of putative selection was detected near the colony stimulating factor 2 (CSF2) in Black Pied, Yaroslavl, and Kazakh Whiteheaded breeds with the narrowest signal (5 Kbp) observed in the Kazakh Whiteheaded. CSF2 is important for embryonic development in cattle93,94 due to its role in inhibiting apoptosis95. Another gene involved in cattle oocyte/blastocyst apoptosis (BCL296), was identified near the most significant SNP in the BTA24: 60.76–62.64 Mbp interval found for the EUR2 breed set. ANXA10 on BTA8 has previously been associated with embryonic mortality in Japanese Black through a large chromosomal deletion97 and was found in a putatively selected region in Kholmogory cattle. NPBWR1 within a likely selected region detected in the Yaroslavl breed was previously associated with fertility traits in Nelore cattle98.
Milk production traits
A putative signature of selection detected in the Kazakh Whiteheaded, Buryat, and Kholmogory breeds at BTA14:1.49–1.89 Mbp contains a major gene controlling milk fat content (DGAT199). The dairy Kholmogory cattle had the narrowest selected region (196 Kbp) with DGAT1 being the top-ranked gene by DCMS. Another major gene affecting milk yield and content, ABCG225, was found in a 2.77 Mbp region detected by hapFLK on BTA6:37.08–39.85 Mbp in the EUR1 set of breeds. However, this interval also includes other candidate genes such as LCORL and NCAPG. The DCMS approach further divided this region into smaller intervals with ABCG2 being the top-ranked gene in regions detected for the Kalmyk and Kostroma breeds. On the other hand, in the Buryat cattle, the most significant SNP in the same region was found near the LAP3 gene, also associated with milk production traits100 and identified as the most likely candidate affecting direct calving ease in the Piedmontese breed101. A gene with a pleotropic effect, GHR, affecting protein and milk yield in dairy cattle102 and growth103 was found in an interval detected by the DSMC analysis in the Black Pied and Bestuzhev breeds. The transient receptor potential cation channel subfamily V member 5 gene (TRPV5) associated with hypocalcemia in cattle and with milk fever104 was found in a 77-Kbp selected interval of BTA4 reported for the Kalmyk cattle. The Kazakh Whiteheaded breed had a reported region on BTA1 which contained FKBP2. The FKBP2 gene was previously associated with milk protein yields and percentage in Holstein GWAS studies105. In the Kazakh Whiteheaded and Buryat we identified large regions (~400 Kbp and ~200 Kbp respectively) on BTA14 which contained the gene TONSL previously identified as a putative candidate for cattle milk traits in a genotype-by-sequencing association study106. Among other genes related to milk production traits are the CSF2RB gene located in a putatively selected region in the Yaroslavl breed, associated with milk production in a large Jersey and Holstein cohort and differentially expressed in mammary gland107; KLHL1 previously shown to be associated with milk yield and lactation persistence in the Chinese Holstein breed108 and in a region predicted to be under probable selection in the Bestuzhev breed; HAL in a region predicted to be under putative selection in Buryat cattle and previously associated with the milk traits in Chinese Holstein109; KDM5A located in a region reported for the Yakut breed and identified as a key regulator of the fatty acid levels in the milk of Brown Swiss cattle110; PCCA located in a region reported for Buryat and Yaroslavl breeds and previously associated with metabolic adaptation to divergent milk production performance111.
Other candidate genes
Among the other genes in narrow putatively selected intervals found in multiple Russian breeds, we identified several that could be functional candidates for important cattle traits. Among these are the ATP-dependent DNA helicase homolog (HFM1), associated with ovarian insufficiency in humans112 and found in regions reported for the Buryat, Kalmyk, and Black Pied Russian breeds, as well as in Hanwoo and Holstein. Mutations within SH3PXD2B, located in regions predicted to be under putative selection in the Bestuzhev and Black Pied Russian breeds, are associated with skeletal abnormalities in humans113, whereas in pigs this gene has previously been associated with intramuscular fat content114 suggesting its role in growth and meat related traits. Another candidate gene found in regions predicted to be likely selected in the Buryat, Kalmyk, Black Pied, Holstein, and Hanwoo breeds is CREBRF that has been associated with obesity, weight and height in humans115–117 and identified as a key regulator of endometrial function in goats118.
We finally searched the list of putatively selected regions for genes related to disease resistance. The strongest candidates included the interferon regulatory factor 1 (IRF1) which induces inflammatory responses in macrophages119 and is known to be associated with mycobacterium susceptibility in humans119 and mice120. IRF1 was the top-ranked gene in the DCMS results in a 362 Kbp putatively selected region reported for the Bestuzhev breed and second-ranked in Kalmyk cattle. The same region also contains interleukin 5 (IL5), top-ranked for Kalmyk and Yakut cattle and known to be involved in the immune response to mycobacterium infection in humans121. Another gene previously found to be associated with tuberculosis susceptibility in wild boars122, neurotrophic tyrosine kinase receptor, type 2 (NTRK2) was top-ranked in a 77.7 Kbp region of BTA8 reported for the Yaroslavl breed. The Yaroslavl breed also had a probable signature of selection near the sirtuin 1 (SIRT1) gene, a nicotinamide adenine dinucleotide (NAD+)-dependent deacetylase expressed in monocytes/macrophages. SIRT1 is involved in the modulation of lung myeloid cells in mycobacterium-infected mice. Moreover, myeloid cell-specific Sirt1 knockout mice showed increased susceptibility to mycobacterium infection123.
Discussion
We report the first comprehensive genome-wide autosomal analysis of putative signatures of genomic selection in the genomes of nine Russian native cattle breeds utilizing comparative data originating from six additional breeds of European and Asian origins. Integration of our genotype data19 with genotype- and sequence-generated SNPs from additional breeds allowed us to differentiate between shared signatures of selection and those that might be unique to breed(s) of Russian origin. Our data suggest that while the Russian cattle breeds share a significant fraction of probably selected regions with other breeds, they also possess some unique signatures of adaptation/selection that might be related to adaptive responses to local environments. Therefore, on the one hand, our results prove the power of using native breed genomes to reveal novel candidate signatures of selection formed as adaptive responses to specific environments, while on the other hand point to selective sweeps near key genes controlling important traits suggesting that selective breeding utilizing established genetic markers could be applied to further improve some of the local Russian breeds.
As expected, the composite measurement of statistics (combining SNP and haplotype-based individual breed statistics) resulted in a larger number of intervals being reported as putatively selected compared to the haplotype-based hapFLK approach based on breed group data. However, 30% of hapFLK regions were not detected the DCMS analysis and 83% of the DCMS regions were not found in by the hapFLK analyses, suggesting that the hapFLK approach was conservative in identifying selected regions involving long haplotypes while the DCMS analysis was more sensitive in detecting shorter selected regions.
Our results demonstrate that the Yakut cattle which are adapted to survive above the Polar circle23 possess signatures of putative selection that contain the genes RETREG1 and RPL7 which may contribute to the adaptation of this breed to its harsh environment. However, suggestive signatures of selection near these genes were also found in other Turano-Mongolian breeds (Japanese Black and Hanwoo), indicating that some differentiation in these intervals could be present in the ancestral Turano-Mongolian pool of animals making these animals more suitable for future adaptation to extreme cold conditions of Northern Russia than other taurine cattle. Life above the Polar Circle also requires adaptation to specific light regime and the ability to defend against novel parasites, viral and bacterial infections. Consistent with such requirements, multiple genes related to these processes were located in genomic regions predicted to be under putative selection in the Yakut cattle. Other Russian cattle breeds, which live in less extreme environments but that are exposed to cold temperatures especially during the winter also had reported regions near genes that are related to protection against viral infections, known candidate genes for cold acclimation (e.g., the Kholmogory) and other genes that are known to change their expression levels in response to exposure to cold in other species. We observed multiple genes involved in brown adipose tissue development as being in genomic regions that are under putative selection in Russian cattle breeds, including HDAC3 and SYK. Brown adipose tissue is an organ involved in non-shivering thermogenesis, suggesting that these genes may be involved in adaptation to cold climates. However, caution is required in interpreting these results as we observed that some of these genes were also found in regions predicted to have been selected in other cattle breeds, including the European taurines. Adipose tissue is also an important component of meat, suggesting that these genes are related to meat production traits which have been artificially selected.
In addition to genes that might be related to adaptation to local environment, the genomes of the Russian cattle breeds possess signatures of putative selection in regions containing genes that are related to domestication and morphology. One region contains the KITLG gene that is known to be responsible for the roan coat colour phenotype in cattle. In our study it was predicted to be under selection in the European cattle breeds but the overlapping selected region in the Asian cattle did not include KITLG, suggesting that other genes in this region could be under selection in the Asian taurine cattle. We also failed to identify strong signal in the region containing KIT (several coat colour phenotypes) in the Asian cattle breeds, but it expressed strong signal in the European cattle. This might suggest that different mechanisms of coat colour determination exist in the two groups of breeds or that the statistical power of analysis in Asian taurines was not high enough to reveal signatures of selection in this region.
We observed a strong relationship between the beef type of the breed and known genes related to meat quality and growth located in the detected putative signatures of selection; such as XKR4 in Kazakh Whiteheaded and Kalmyk beef breeds, NCAPG, LCORL in Kazakh Whiteheaded and IMPAD1 in Kazakh Whiteheaded, Buryat, and Kostroma dual purpose breeds. Another growth-related gene, HMGA2 was located in a region predicted to be under selection in the Kalmyk beef breed and the Bestuzhev and Kostroma dual purpose breeds. On the other hand, genes related to milk production were located in regions reported for all types of breeds. For instance, the DGAT1 was reported not only for the dairy Kholmogory breed and dual purpose Buryat cattle but also for the beef Kazakh Whiteheaded. However, only the Kholmogory breed had DGAT1 as a top-ranked gene in a narrow interval, while the other two breeds had much wider regions under putative selection and DGAT1 was low in rank (11 and 20). Another major gene related to milk production, ABCG2 was located in a region predicted to be under selection in the dual-purpose Kostroma and beef Kalmyk cattle. There is some evidence, however, that Kalmyk cattle were selected for their unique high dry-matter content of the milk124.
In conclusion, results of the first scan for signatures of putative selection/adaptation in genomes of nine Russian native cattle breeds demonstrate that their genomes contain multiple intervals likely subjected to selection, some of which appear to be related to adaptation to harsh (cold) environments. This analysis demonstrates the importance and power of studying local breeds that may not be as productive as the commonly used commercial breeds world-wide, but hold a real promise for decoding mechanisms of environmental adaptation to be utilized in genetics-guided improvement of productive multinational breeds.
Electronic supplementary material
Acknowledgements
The work was supported by the Russian Science Foundation grant (RSF, 16–14–00090).
Author Contributions
D.M.L., A.Y. and N.Y. conceived the study. A.Y., H.D.D., C.J.V.J. and D.M.L. performed the analysis. R.D.S., J.F.T., H.D.D., V.S., B.L., R.P. provided samples. A.Y. and D.M.L. drafted the paper. H.D.D., J.F.T., N.Y. edited the manuscript. D.M.L. led the project. All authors approved the manuscript.
Data availability
Data available from the Dryad Digital Repository: 10.5061/dryad.fr88s7f.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-31304-w.
References
- 1.Jensen JD, Foll M, Bernatchez L. The past, present and future of genomic scans for selection. Mol Ecol. 2016;25:1–4. doi: 10.1111/mec.13493. [DOI] [PubMed] [Google Scholar]
- 2.Pickrell JK, et al. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathieson I, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan S, Hansen ME, Lo Y, Tishkoff SA. Going global by adapting local: A review of recent human adaptation. Science. 2016;354:54–59. doi: 10.1126/science.aaf5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moon S, et al. A genome-wide scan for signatures of directional selection in domesticated pigs. BMC Genomics. 2015;16:130. doi: 10.1186/s12864-015-1330-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostrander EA, Wayne RK, Freedman AH, Davis BW. Demographic history, selection and functional diversity of the canine genome. Nat Rev Genet. 2017;18:705–720. doi: 10.1038/nrg.2017.67. [DOI] [PubMed] [Google Scholar]
- 7.Wilkins AS, Wrangham RW, Fitch WT. The “domestication syndrome” in mammals: a unified explanation based on neural crest cell behavior and genetics. Genetics. 2014;197:795–808. doi: 10.1534/genetics.114.165423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Simoni Gouveia JJ, da Silva MV, Paiva SR, de Oliveira SM. Identification of selection signatures in livestock species. Genet Mol Biol. 2014;37:330–342. doi: 10.1590/S1415-47572014000300004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheu, A. et al. The genetic prehistory of domesticated cattle from their origin to the spread across Europe. Bmc Genetics16, 10.1186/s12863-015-0203-2 (2015). [DOI] [PMC free article] [PubMed]
- 10.Zhang H, et al. Morphological and genetic evidence for early Holocene cattle management in northeastern China. Nat Commun. 2013;4:2755. doi: 10.1038/ncomms3755. [DOI] [PubMed] [Google Scholar]
- 11.Barendse W. Climate Adaptation of Tropical Cattle. Annu Rev Anim Biosci. 2017;5:133–150. doi: 10.1146/annurev-animal-022516-022921. [DOI] [PubMed] [Google Scholar]
- 12.Mason, I. L. A world dictionary of livestock breed types and varieties. In Commonwealth Agricultural Bureaux (Farnham Royal, 1969).
- 13.Ibeagha-Awemu EM, Kgwatalala P, Zhao X. A critical analysis of production-associated DNA polymorphisms in the genes of cattle, goat, sheep, and pig. Mamm Genome. 2008;19:591–617. doi: 10.1007/s00335-008-9141-x. [DOI] [PubMed] [Google Scholar]
- 14.Decker JE, et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS genetics. 2014;10:e1004254. doi: 10.1371/journal.pgen.1004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Upadhyay MR, et al. Genetic origin, admixture and population history of aurochs (Bos primigenius) and primitive European cattle. Heredity (Edinb) 2017;118:169–176. doi: 10.1038/hdy.2016.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, et al. The genome landscape of indigenous African cattle. Genome biology. 2017;18:34. doi: 10.1186/s13059-017-1153-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mei, C. et al. Genetic architecture and selection of Chinese cattle revealed by whole genome resequencing. Mol Biol Evol, 10.1093/molbev/msx322 (2017). [DOI] [PubMed]
- 18.Gao Y, et al. Single Cas9 nickase induced generation of NRAMP1 knockin cattle with reduced off-target effects. Genome biology. 2017;18:13. doi: 10.1186/s13059-016-1144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yurchenko A, et al. Genome-wide genotyping uncovers genetic profiles and history of the Russian cattle breeds. Heredity. 2018;120:125–137. doi: 10.1038/s41437-017-0024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mannen H, et al. Independent mitochondrial origin and historical genetic differentiation in North Eastern Asian cattle. Mol Phylogenet Evol. 2004;32:539–544. doi: 10.1016/j.ympev.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Soini K, Ovaska U, Kantanen J. Spaces of Conservation of Local Breeds: The Case of Yakutian Cattle. Sociol Ruralis. 2012;52:170–191. doi: 10.1111/j.1467-9523.2012.00551.x. [DOI] [Google Scholar]
- 22.DAD-IS. Domestic animal diversity Information system (DAD-IS), http://www.fao.org/dad-is/ (2017).
- 23.Iso-Touru T, et al. Genetic diversity and genomic signatures of selection among cattle breeds from Siberia, eastern and northern Europe. Anim Genet. 2016;47:647–657. doi: 10.1111/age.12473. [DOI] [PubMed] [Google Scholar]
- 24.Gao Y, et al. Species composition and environmental adaptation of indigenous Chinese cattle. Sci Rep. 2017;7:16196. doi: 10.1038/s41598-017-16438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen-Zinder M, et al. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005;15:936–944. doi: 10.1101/gr.3806705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grossman SR, et al. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327:883–886. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- 27.Ma Y, et al. Properties of different selection signature statistics and a new strategy for combining them. Heredity (Edinb) 2015;115:426–436. doi: 10.1038/hdy.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lotterhos KE, et al. Composite measures of selection can improve the signal-to-noise ratio in genome scans. Methods Ecol Evol. 2017;8:717–727. doi: 10.1111/2041-210X.12774. [DOI] [Google Scholar]
- 29.Daetwyler HD, et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Genet. 2014;46:858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 30.Kim K, et al. A novel genetic variant database for Korean native cattle (Hanwoo): HanwooGDB. Genes Genom. 2015;37:15–22. doi: 10.1007/s13258-014-0224-7. [DOI] [Google Scholar]
- 31.Daetwyler HD, et al. 1000 Bull Genomes and SheepGenomesDB projects: enabling cost-effective sequence level analyses globally. Proceedings of the Australian Association for Animal Breeding and Genetics. 2017;22:201–204. [Google Scholar]
- 32.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng X, et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics. 2013;193:929–941. doi: 10.1534/genetics.112.147231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78:629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fariello, M. I. et al. Selection Signatures in Worldwide Sheep Populations. Plos One9, 10.1371/journal.pone.0103813 (2014). [DOI] [PMC free article] [PubMed]
- 39.Boitard S, Boussaha M, Capitan A, Rocha D, Servin B. Uncovering Adaptation from Sequence Data: Lessons from Genome Resequencing of Four Cattle Breeds. Genetics. 2016;203:433–450. doi: 10.1534/genetics.115.181594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population-Structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 42.Garud, N. R., Messer, P. W., Buzbas, E. O. & Petrov, D. A. Recent Selective Sweeps in North American Drosophila melanogaster Show Signatures of Soft Sweeps. PLoS genetics11, 10.1371/journal.pgen.1005004 (2015). [DOI] [PMC free article] [PubMed]
- 43.Tajima F. Statistical-Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nei M, Li WH. Mathematical-Model for Studying Genetic-Variation in Terms of Restriction Endonucleases. P Natl Acad Sci USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 46.Ma L, et al. Cattle Sex-Specific Recombination and Genetic Control from a Large Pedigree Analysis. PLoS genetics. 2015;11:e1005387. doi: 10.1371/journal.pgen.1005387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schlamp F, et al. Evaluating the performance of selection scans to detect selective sweeps in domestic dogs. Molecular Ecology. 2016;25:342–356. doi: 10.1111/mec.13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schrider DR, Kern AD. Soft Sweeps Are the Dominant Mode of Adaptation in the Human Genome. Molecular Biology and Evolution. 2017;34:1863–1877. doi: 10.1093/molbev/msx154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tange O. Gnu parallel-the command-line power tool. The USENIX Magazine. 2011;36:42–47. [Google Scholar]
- 51.Verity R, et al. minotaur: A platform for the analysis and visualization of multivariate results from genome scans with R Shiny. Mol Ecol Resour. 2017;17:33–43. doi: 10.1111/1755-0998.12579. [DOI] [PubMed] [Google Scholar]
- 52.Kasprzyk, A. BioMart: driving a paradigm change in biological data management. Database-Oxford, 10.1093/database/bar049 (2011). [DOI] [PMC free article] [PubMed]
- 53.Gu Z, Gu L, Eils R, Schlesner M, Brors B. circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 54.Roth S, et al. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1beta production. Nat Immunol. 2014;15:538–545. doi: 10.1038/ni.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wollenberg Valero KC, et al. A candidate multimodal functional genetic network for thermal adaptation. PeerJ. 2014;2:e578. doi: 10.7717/peerj.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiu Q, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44:946–949. doi: 10.1038/ng.2343. [DOI] [PubMed] [Google Scholar]
- 57.Kurth I, et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179–1181. doi: 10.1038/ng.464. [DOI] [PubMed] [Google Scholar]
- 58.Axelrod FB. & Gold-von Simson, G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis. 2007;2:39. doi: 10.1186/1750-1172-2-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu S, De Croos JN, Storey KB. Cold acclimation-induced up-regulation of the ribosomal protein L7 gene in the freeze tolerant wood frog, Rana sylvatica. Gene. 2008;424:48–55. doi: 10.1016/j.gene.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 60.Yeh TY, et al. Hypermetabolism, hyperphagia, and reduced adiposity in tankyrase-deficient mice. Diabetes. 2009;58:2476–2485. doi: 10.2337/db08-1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mandal NA, et al. Expression and localization of CERKL in the mammalian retina, its response to light-stress, and relationship with NeuroD1 gene. Exp Eye Res. 2013;106:24–33. doi: 10.1016/j.exer.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 63.Emmett MJ, et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature. 2017;546:544–548. doi: 10.1038/nature22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carroll SH, et al. Adipocyte arrestin domain-containing 3 protein (Arrdc3) regulates uncoupling protein 1 (Ucp1) expression in white adipose independently of canonical changes in beta-adrenergic receptor signaling. PLoS One. 2017;12:e0173823. doi: 10.1371/journal.pone.0173823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knoll M, et al. SYK kinase mediates brown fat differentiation and activation. Nat Commun. 2017;8:2115. doi: 10.1038/s41467-017-02162-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Valverde G, et al. A novel candidate region for genetic adaptation to high altitude in Andean populations. PLoS One. 2015;10:e0125444. doi: 10.1371/journal.pone.0125444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Konczol K, et al. Nesfatin-1 exerts long-term effect on food intake and body temperature. Int J Obes (Lond) 2012;36:1514–1521. doi: 10.1038/ijo.2012.2. [DOI] [PubMed] [Google Scholar]
- 68.Li F, et al. Novel SNPs of the bovine NUCB2 gene and their association with growth traits in three native Chinese cattle breeds. Mol Biol Rep. 2010;37:541–546. doi: 10.1007/s11033-009-9732-y. [DOI] [PubMed] [Google Scholar]
- 69.Jedema HP, et al. Chronic cold exposure increases RGS7 expression and decreases alpha(2)-autoreceptor-mediated inhibition of noradrenergic locus coeruleus neurons. Eur J Neurosci. 2008;27:2433–2443. doi: 10.1111/j.1460-9568.2008.06208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Randhawa, I. A. S., Khatkar, M. S., Thomson, P. C. & Raadsma, H. W. A Meta-Assembly of Selection Signatures in Cattle. Plos One11, 10.1371/journal.pone.0153013 (2016). [DOI] [PMC free article] [PubMed]
- 71.Wright D. The Genetic Architecture of Domestication in Animals. Bioinform Biol Insights. 2015;9:11–20. doi: 10.4137/BBI.S28902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haase B, et al. Seven novel KIT mutations in horses with white coat colour phenotypes. Anim Genet. 2009;40:623–629. doi: 10.1111/j.1365-2052.2009.01893.x. [DOI] [PubMed] [Google Scholar]
- 73.Talenti A, et al. Genomic analysis suggests KITLG is responsible for a roan pattern in two Pakistani goat breeds. J Hered. 2017 doi: 10.1093/jhered/esx093. [DOI] [PubMed] [Google Scholar]
- 74.Kaelin CB, et al. Specifying and sustaining pigmentation patterns in domestic and wild cats. Science. 2012;337:1536–1541. doi: 10.1126/science.1220893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pausch H, et al. Identification of QTL for UV-protective eye area pigmentation in cattle by progeny phenotyping and genome-wide association analysis. PLoS One. 2012;7:e36346. doi: 10.1371/journal.pone.0036346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dorshorst B, et al. Dominant Red Coat Color in Holstein Cattle Is Associated with a Missense Mutation in the Coatomer Protein Complex, Subunit Alpha (COPA) Gene. PLoS One. 2015;10:e0128969. doi: 10.1371/journal.pone.0128969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terakado, A. P. N. et al. Genome-wide association study for growth traits in Nelore cattle. Animal, 1-5, 10.1017/S1751731117003068 (2017). [DOI] [PubMed]
- 78.Bolormaa S, et al. A genome-wide association study of meat and carcass traits in Australian cattle. J Anim Sci. 2011;89:2297–2309. doi: 10.2527/jas.2010-3138. [DOI] [PubMed] [Google Scholar]
- 79.Lindholm-Perry AK, et al. A region on BTA14 that includes the positional candidate genes LYPLA1, XKR4 and TMEM68 is associated with feed intake and growth phenotypes in cattle(1) Anim Genet. 2012;43:216–219. doi: 10.1111/j.1365-2052.2011.02232.x. [DOI] [PubMed] [Google Scholar]
- 80.Utsunomiya YT, et al. A PLAG1 mutation contributed to stature recovery in modern cattle. Sci Rep. 2017;7:17140. doi: 10.1038/s41598-017-17127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu Y, Duan X, Chen S, He H, Liu X. NCAPG is differentially expressed during longissimus muscle development and is associated with growth traits in Chinese Qinchuan beef cattle. Genet Mol Biol. 2015;38:450–456. doi: 10.1590/S1415-475738420140287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nishimaki T, et al. Allelic frequencies and association with carcass traits of six genes in local subpopulations of Japanese Black cattle. Anim Sci J. 2016;87:469–476. doi: 10.1111/asj.12453. [DOI] [PubMed] [Google Scholar]
- 83.Saatchi M, Schnabel RD, Taylor JF, Garrick DJ. Large-effect pleiotropic or closely linked QTL segregate within and across ten US cattle breeds. BMC Genomics. 2014;15:442. doi: 10.1186/1471-2164-15-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weedon MN, et al. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet. 2007;39:1245–1250. doi: 10.1038/ng2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boyko AR, et al. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol. 2010;8:e1000451. doi: 10.1371/journal.pbio.1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Makvandi-Nejad S, et al. Four loci explain 83% of size variation in the horse. PLoS One. 2012;7:e39929. doi: 10.1371/journal.pone.0039929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wimmers K, et al. Associations of functional candidate genes derived from gene-expression profiles of prenatal porcine muscle tissue with meat quality and muscle deposition. Anim Genet. 2007;38:474–484. doi: 10.1111/j.1365-2052.2007.01639.x. [DOI] [PubMed] [Google Scholar]
- 88.Nizon M, et al. IMPAD1 mutations in two Catel-Manzke like patients. Am J Med Genet A. 2012;158A:2183–2187. doi: 10.1002/ajmg.a.35504. [DOI] [PubMed] [Google Scholar]
- 89.Urbinati I, et al. Selection signatures in Canchim beef cattle. J Anim Sci Biotechnol. 2016;7:29. doi: 10.1186/s40104-016-0089-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fortes MRS, et al. Finding genes for economically important traits: Brahman cattle puberty. Anim Prod Sci. 2012;52:143–150. doi: 10.1071/AN11165. [DOI] [Google Scholar]
- 91.Wang XL, et al. A novel mutation of the GLI2 gene associated with body weight in bovine (Bos taurus) Arch Tierzucht. 2009;52:334–336. [Google Scholar]
- 92.Meng QL, et al. Identification of growth trait related genes in a Yorkshire purebred pig population by genome-wide association studies. Asian Austral J Anim. 2017;30:462–469. doi: 10.5713/ajas.16.0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Loureiro B, et al. Colony-stimulating factor 2 (CSF-2) improves development and posttransfer survival of bovine embryos produced in vitro. Endocrinology. 2009;150:5046–5054. doi: 10.1210/en.2009-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Loureiro B, et al. Consequences of conceptus exposure to colony-stimulating factor 2 on survival, elongation, interferon-tau secretion, and gene expression. Reproduction. 2011;141:617–624. doi: 10.1530/REP-10-0511. [DOI] [PubMed] [Google Scholar]
- 95.Loureiro B, Oliveira LJ, Favoreto MG, Hansen PJ. Colony-stimulating factor 2 inhibits induction of apoptosis in the bovine preimplantation embryo. Am J Reprod Immunol. 2011;65:578–588. doi: 10.1111/j.1600-0897.2010.00953.x. [DOI] [PubMed] [Google Scholar]
- 96.Boruszewska D, Sinderewicz E, Kowalczyk-Zieba I, Grycmacher K, Woclawek-Potocka I. The effect of lysophosphatidic acid during in vitro maturation of bovine cumulus-oocyte complexes: cumulus expansion, glucose metabolism and expression of genes involved in the ovulatory cascade, oocyte and blastocyst competence. Reprod Biol Endocrinol. 2015;13:44. doi: 10.1186/s12958-015-0044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sasaki S, Ibi T, Akiyama T, Fukushima M, Sugimoto Y. Loss of maternal ANNEXIN A10 via a 34-kb deleted-type copy number variation is associated with embryonic mortality in Japanese Black cattle. BMC Genomics. 2016;17:968. doi: 10.1186/s12864-016-3312-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mota RR, et al. Genome-wide association study and annotating candidate gene networks affecting age at first calving in Nellore cattle. J Anim Breed Genet. 2017;134:484–492. doi: 10.1111/jbg.12299. [DOI] [PubMed] [Google Scholar]
- 99.Grisart B, et al. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc Natl Acad Sci USA. 2004;101:2398–2403. doi: 10.1073/pnas.0308518100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zheng X, et al. Single nucleotide polymorphisms, haplotypes and combined genotypes of LAP3 gene in bovine and their association with milk production traits. Mol Biol Rep. 2011;38:4053–4061. doi: 10.1007/s11033-010-0524-1. [DOI] [PubMed] [Google Scholar]
- 101.Bongiorni S, Mancini G, Chillemi G, Pariset L, Valentini A. Identification of a short region on chromosome 6 affecting direct calving ease in Piedmontese cattle breed. PLoS One. 2012;7:e50137. doi: 10.1371/journal.pone.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Xiang R, MacLeod IM, Bolormaa S, Goddard ME. Genome-wide comparative analyses of correlated and uncorrelated phenotypes identify major pleiotropic variants in dairy cattle. Sci Rep. 2017;7:9248. doi: 10.1038/s41598-017-09788-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pereira AGT, et al. Pleiotropic Genes Affecting Carcass Traits in Bos indicus (Nellore) Cattle Are Modulators of Growth. PLoS One. 2016;11:e0158165. doi: 10.1371/journal.pone.0158165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martin-Tereso J, Verstegen MW. A novel model to explain dietary factors affecting hypocalcaemia in dairy cattle. Nutr Res Rev. 2011;24:228–243. doi: 10.1017/S0954422411000126. [DOI] [PubMed] [Google Scholar]
- 105.Cole, J. B. et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary US Holstein cows. Bmc Genomics12, 10.1186/1471-2164-12-408 (2011). [DOI] [PMC free article] [PubMed]
- 106.Ibeagha-Awemu, E. M., Peters, S. O., Akwanji, K. A., Imumorin, I. G. & Zhao, X. High density genome wide genotyping-by-sequencing and association identifies common and low frequency SNPs, and novel candidate genes influencing cow milk traits. Sci Rep-Uk6, 10.1038/srep31109 (2016). [DOI] [PMC free article] [PubMed]
- 107.Raven LA, et al. Targeted imputation of sequence variants and gene expression profiling identifies twelve candidate genes associated with lactation volume, composition and calving interval in dairy cattle. Mammalian Genome. 2016;27:81–97. doi: 10.1007/s00335-015-9613-8. [DOI] [PubMed] [Google Scholar]
- 108.Yue SJ, et al. A genome-wide association study suggests new candidate genes for milk production traits in Chinese Holstein cattle. Animal Genetics. 2017;48:677–681. doi: 10.1111/age.12593. [DOI] [PubMed] [Google Scholar]
- 109.Wang, H. F. et al. Associations between variants of the HAL gene and milk production traits in Chinese Holstein cows. Bmc Genetics15, 10.1186/s12863-014-0125-4 (2014). [DOI] [PMC free article] [PubMed]
- 110.Pegolo, S. et al. SNP co-association and network analyses identify E2F3, KDM5A and BACH2 as key regulators of the bovine milk fatty acid profile. Sci Rep-Uk7, 10.1038/s41598-017-17434-7 (2017). [DOI] [PMC free article] [PubMed]
- 111.Weikard R, Goldammer T, Brunner RM, Kuehn C. Tissue-specific mRNA expression patterns reveal a coordinated metabolic response associated with genetic selection for milk production in cows. Physiol Genomics. 2012;44:728–739. doi: 10.1152/physiolgenomics.00007.2012. [DOI] [PubMed] [Google Scholar]
- 112.Qin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update. 2015;21:787–808. doi: 10.1093/humupd/dmv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zrhidri A, et al. Identification of two novel SH3PXD2B gene mutations in Frank-Ter Haar syndrome by exome sequencing: Case report and review of the literature. Gene. 2017;628:190–193. doi: 10.1016/j.gene.2017.07.011. [DOI] [PubMed] [Google Scholar]
- 114.Wang Y, et al. Dynamic transcriptome and DNA methylome analyses on longissimus dorsi to identify genes underlying intramuscular fat content in pigs. BMC Genomics. 2017;18:780. doi: 10.1186/s12864-017-4201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Berry SD, et al. Widespread prevalence of a CREBRF variant amongst Maori and Pacific children is associated with weight and height in early childhood. Int J Obes (Lond) 2017 doi: 10.1038/ijo.2017.230. [DOI] [PubMed] [Google Scholar]
- 116.Minster RL, et al. A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat Genet. 2016;48:1049–1054. doi: 10.1038/ng.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Naka I, et al. A missense variant, rs373863828-A (p.Arg457Gln), of CREBRF and body mass index in Oceanic populations. J Hum Genet. 2017;62:847–849. doi: 10.1038/jhg.2017.44. [DOI] [PubMed] [Google Scholar]
- 118.Yang D, et al. CREB3 Regulatory Factor -mTOR-autophagy regulates goat endometrial function during early pregnancy. Biol Reprod. 2018 doi: 10.1093/biolre/ioy044. [DOI] [PubMed] [Google Scholar]
- 119.Roy S, et al. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J Immunol. 2015;194:6035–6044. doi: 10.4049/jimmunol.1402521. [DOI] [PubMed] [Google Scholar]
- 120.Yamada H, Mizuno S, Sugawara I. Interferon regulatory factor 1 in mycobacterial infection. Microbiol Immunol. 2002;46:751–760. doi: 10.1111/j.1348-0421.2002.tb02760.x. [DOI] [PubMed] [Google Scholar]
- 121.Oliver BG, et al. Interferon-gamma and IL-5 production correlate directly in HIV patients co-infected with mycobacterium tuberculosis with or without immune restoration disease. AIDS Res Hum Retroviruses. 2010;26:1287–1289. doi: 10.1089/aid.2010.0004. [DOI] [PubMed] [Google Scholar]
- 122.Queiros J, Alves PC, Vicente J, Gortazar C, de la Fuente J. Genome-wide associations identify novel candidate loci associated with genetic susceptibility to tuberculosis in wild boar. Sci Rep. 2018;8:1980. doi: 10.1038/s41598-018-20158-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cheng, C. Y. et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci Immunol2, 10.1126/sciimmunol.aaj1789 (2017). [DOI] [PMC free article] [PubMed]
- 124.Dmitriev, N. G. & Ernst, L. K. Animal genetics resources of the USSR. Food and Agriculture Organization of the United Nations, Rome, Italy (1989).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available from the Dryad Digital Repository: 10.5061/dryad.fr88s7f.