

Abstract
Polycomb group (PcG) proteins regulate the expression of target genes by modulating histone modifications and are representative epigenetic regulators that maintain the stemness of embryonic and hematopoietic stem cells. Histone methyltransferases enhancer of zeste homolog 1 and 2 (EZH1/2), which are subunits of polycomb repressive complexes (PRC), are recurrently mutated or highly expressed in many hematological malignancies. EZH2 has a dual function in tumorigenesis as an oncogene and tumor suppressor gene, and targeting PRC2, in particular EZH1/2, for anticancer therapy has been extensively developed in the clinical setting. Here, we review the oncogenic function of EZH1/2 and introduce new therapeutic drugs targeting these enzymes.
Keywords: enhancer of zeste homolog 1 and 2, epigenetics, histone methyltransferase, polycomb repressive complexes, transcriptional repression
1. INTRODUCTION
Epigenetic regulation by DNA methylation, histone modifications, and non‐coding RNAs modulates gene expression without affecting DNA base sequences. Because aberrant DNA methylation or histone modifications are present in many malignant tumors, epigenetic dysregulation is considered a cause of tumor progression. Polycomb group (PcG) proteins regulate the expression of target genes by modulating histone modifications and are representative epigenetic regulators that maintain the “stemness” of embryonic and hematopoietic stem cells (HSC). Dysregulation of PcG proteins associated with mutations or gene overexpression is positively correlated with tumor progression in many hematological malignancies. In this review, we summarize the current knowledge of PcG proteins, focusing on the function of the histone methyltransferases enhancer of zeste homolog 1 and 2 (EZH1/2), which are subunits of polycomb repressive complexes (PRC), in hematological malignancies. In addition, we introduce novel therapeutic drugs for targeting these enzymes.
2. FUNCTION OF PRC
The basic structural unit of chromatin consists of DNA wrapped around histone proteins. Histone proteins have an N‐terminal region termed the histone tail, which undergoes various chemical modifications including acetylation, methylation, phosphorylation, and ubiquitination. These chemical modifications contribute to the regulation of target gene expression by modifying the spatial structure of chromatin. PcG proteins play an important role in maintaining the transcriptional repression of target genes.
PcG genes were first identified for their role in regulating the expression of homeotic genes, which control the body plan of embryos along the longitudinal axis and segmentation in Drosophila.1 PcG proteins form PRC in the nucleus. PRC are classified into PRC1 and PRC2 according to their biological characteristics. The core subunits of the mammalian PRC2 complex include EZH1/2, SUZ12, RbAp46/48, and EED. EZH1/2, which are histone methyltransferases, trimethylate histone H3 at lysine 27 (H3K27).2, 3 SUZ12 and EED activate methyltransferases and recruit PRC2 to the nucleosome, respectively.4, 5 The histone modification H3K27me3 represses target genes and mediates the recruitment of PRC1 to the nucleosome by serving as a docking site for the PRC1 component CBX (Figure 1A). PRC1 functions in transcriptional repression by catalyzing the monoubiquitination of histone H2A at lysine 119 (H2AK119). H2A ubiquitination blocks RNA polymerase II‐mediated transcriptional elongation.6 In addition, PRC1 induces chromatin condensation, which also contributes to the transcriptional repression of target genes. PRC1 complexes have 4 subunits, including Ring1A/B, CBX, PCGF, and PHC. Ring1A/B has ubiquitin ligase activity and ubiquitinates H2AK119.7 CBX is a chromodomain protein that recognizes H3K27me3 and recruits PRC1 to the nucleosome.8 PCGF (MEL18, BMI1) is a cofactor of Ring1A/B, and promotes its ubiquitin ligase activity.9 A non‐canonical PRC1 complex composed of RYBP and KDM2B, but not CBX, was recently identified and shown to target genes and ubiquitinate H2AK119 independently of PRC2 (Figure 1B).10, 11, 12 H2AK119ub1 recruits PRC2 to target genes and induces H3K27 trimethylation.13, 14 Canonical and non‐canonical PRC1 complexes have similar activity in the ubiquitination of H2A in a PRC2‐dependent or ‐independent method, resulting in the maintenance of gene silencing.
Figure 1.
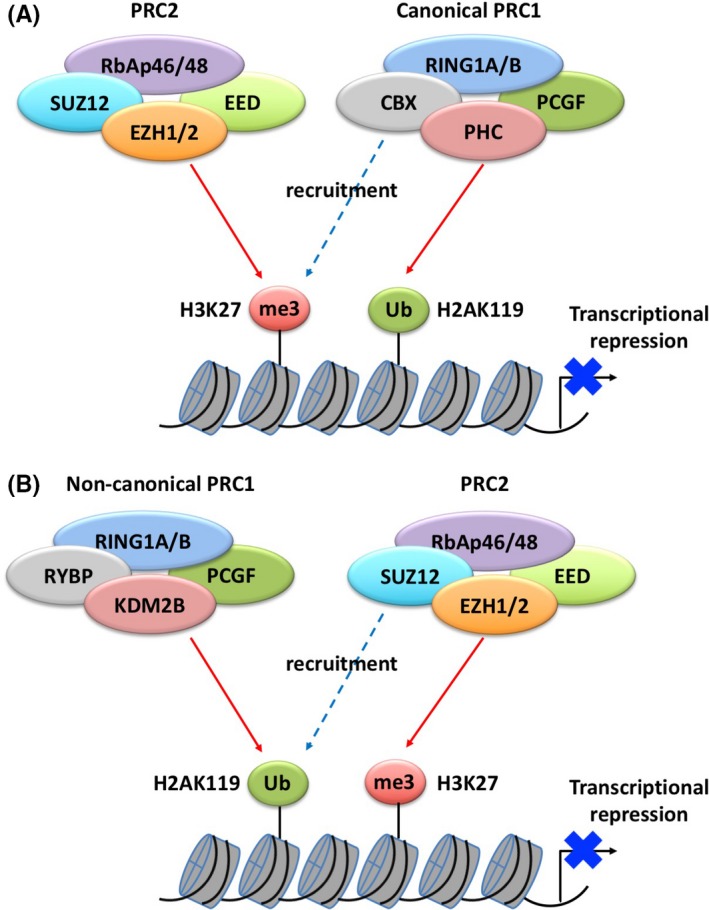
Epigenetic regulation by polycomb group (PcG) complexes. Canonical and non‐canonical PRC1 ubiquitinate H2AK119 with or independently of PRC2, respectively, resulting in transcriptional repression. A, EZH1/2 trimethylates H3K27, and the recruitment of canonical PRC1 to target sites is mediated by the recognition of H3K27me3 by CBX, followed by PRC1 ubiquitination of H2AK119. B, Non‐canonical PRC1 ubiquitinates H2AK119 independently of PRC2. H2AK119ub1 then recruits PRC2 to target genes and induces the trimethylation of H3K27. EZH1/2, enhancer of zeste homolog 1 and 2; PRC, polycomb repressive complexes
3. ROLES OF EZH1/2 IN HSC
Polycomb complexes maintain the stemness of embryonic stem (ES) cells and HSC by repressing transcription through histone modifications. In ES cells, bivalent domains are formed at promoter sites of target genes through active and repressive histone modifications catalyzed by PRC1/2 and trithorax group (TrxG) complexes, respectively.15 These domains control gene expression associated with differentiation and cell cycle signaling, resulting in the regulation of ES cell differentiation. HSC have similar systems for regulating differentiation.16
Analysis of genetically modified mice clarified the role of PcG proteins in HSC. Overexpression of Ezh2 preserves the reconstitution capacity of HSC, whereas normal HSC are rapidly exhausted after serial transplantation.17 Ezh2 deficiency impairs expansion of HSC and progenitor cells in the fetal liver, resulting in lethality at early stages of mouse development.18 These findings indicate that Ezh2‐mediated stabilization of chromatin structure is important for the self‐renewal of HSC. Furthermore, inactivation of Ezh2 in fetal liver endothelium results in embryonic lethality with severe anemia despite normal emergence of functional HSC and overexpression of MMP‐9 which cell‐extrinsically depleted the membrane‐bound form of Kit ligand.19 These results indicate that modulation of epigenetic regulators in niche components can exert a marked cell‐extrinsic impact on hematopoiesis. However, Ezh2 knock‐in mice develop myeloproliferative disease, suggesting that stem cell‐specific Ezh2 plays an oncogenic role in myeloid disorders.20
Regarding the homolog Ezh1, analysis of Ezh1‐deficient mice shows the importance of Ezh1 in bone marrow (BM) HSC.21 Ezh1 deficiency in the BM strongly induces a senescence response, leading to impairment of HSC. Deletion of Cdkn2a on the Ezh1 null background rescues HSC proliferation ability, suggesting that Ezh1 maintains adult BM HSC by repressing Cdkn2a. Ezh2 conditional knock‐out mice show dysregulation of T‐ and B‐cell development in the adult BM, whereas the function of HSC is not affected.22, 23, 24 Although H3K27me3 levels are markedly reduced in fetal liver cells of Ezh2‐deficient mice, they are mostly preserved in the adult BM, indicating that loss of Ezh2 is complemented by the homolog Ezh1 in adult BM cells. These results suggest that Ezh1 compensates for the loss of Ezh2 and that the 2 enzymes function together to maintain hematopoiesis.
4. ROLE OF EZH2 IN MALIGNANT TUMORS
Aberrant epigenetic status is associated with the malignant transformation of normal cells in addition to gene mutations and gene abnormalities. Mutations and abnormal expression of PcG group genes are reported in various types of cancer such as melanoma, lymphoma, prostate cancer, ovarian cancer, and synovial sarcoma, suggesting that dysregulation of the PRC1/2 complexes is involved in carcinogenesis.25, 26
Association of EZH2, a methyltransferase of H3K27, with carcinogenesis has been studied in PcG genes.27 High expression of EZH2 is associated with tumor aggressiveness in several cancers (Table 1).28 In hematological malignancies, EZH2 is overexpressed in AML, multiple myeloma (MM), and B‐ and T‐cell lymphomas. High expression of EZH2 in MM is associated with poor patient outcomes and high‐risk disease features, and pharmacological inhibition of EZH2 has anticancer effects in MM cell lines.33, 34 In contrast, monoallelic gain‐of‐function mutations in tyrosine residue 641 of the SET domain in EZH2 are reported in diffuse large B‐cell lymphoma (DLBCL) and follicular lymphoma (FL).35 Mutations in alanine residue 677 are also observed in DLBCL.36 These gain‐of‐function mutations result in a higher efficiency of mono‐ to di‐ and di‐ to tri‐methylation than that of the wild‐type enzyme.37 Furthermore, EZH2 gain‐of‐function mutations contribute to the widespread redistribution of H3K27me3, inducing not only persistent transcriptional repression but also increased transcription at many loci.53 These results suggest that activation of EZH2 contributes to malignant transformation and that EZH2 plays an oncogenic role in many malignant tumors.
Table 1.
Aberrant expression of EZH2 in cancers
| Types of cancer | EZH2 status | References |
|---|---|---|
| AML | Overexpression | 29 |
| B‐NHL, ATL | Overexpression | 30, 31, 32 |
| MM | Overexpression | 33, 34 |
| FL, DLBCL | Gain‐of‐function mutation (Tyr641, Ala677) | 35, 36, 37 |
| T‐ALL, ETP‐ALL | Loss‐of‐function mutation | 38, 39 |
| MDS, MDS/MPN, MF | Loss‐of‐function mutation | 40, 41 |
| Melanoma | Overexpression | 42, 43 |
| Prostate | Overexpression | 44, 45 |
| Ovarian | Overexpression | 46, 47 |
| Lung | Overexpression | 48, 49, 50 |
| Synovial sarcoma | Overexpression | 51, 52 |
AML, acute myeloid leukemia; ATL, adult T‐cell leukemia/lymphoma; B‐NHL, B‐cell non‐Hodgkin lymphomas; DLBCL, diffuse large B‐cell lymphoma; ETP‐ALL, early T‐cell precursor acute lymphoblastic leukemia; EZH2, enhancer of zeste homolog 2; FL, follicular lymphoma; MDS, myelodysplastic syndrome; MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; MF, myelofibrosis; MM, multiple myeloma; T‐ALL, T‐cell acute lymphoblastic leukemia.
EZH2 loss‐of‐function mutations or deletions are also detected in various hematological malignancies including T‐cell acute lymphoblastic leukemia, myelodysplastic syndrome (MDS), myelodysplastic syndrome/myeloproliferative neoplasm (MDS/MPN), and myelofibrosis.38, 40, 41, 54, 55 Loss‐of‐function mutations are associated with poor outcomes in these tumors,40, 41, 54, 56 suggesting that dysfunction of PRC2 promotes tumor progression. Taken together, these findings support the dual function of EZH2 as oncogene and tumor suppressor gene.
5. ROLE OF EZH1 IN HEMATOLOGICAL MALIGNANCIES AND TARGETING THERAPY AGAINST EZH1/2
Enhancer of zeste homolog 2 plays an oncogenic role, especially in lymphoma and AML. A transgenic mouse model with a combination of Myc and EZH2 Y641F showed accelerated lymphoma development.57 EZH2 deletion inhibits tumor progression concomitant with the induction of differentiation programs in mixed lineage leukemia (MLL)‐AF9 fusion AML mice, suggesting that EZH2 is important for leukemogenesis.58, 59 These studies identify EZH2 as a potential therapeutic target, and several EZH2 inhibitors, which are highly selective for the methyltransferase activity of EZH2, have been developed to target the oncogenic function of EZH2 (Table 2).
Table 2.
Overview of preclinical and clinical studies with selective EZH1/2 or EZH2 inhibitors in hematological malignancies
| Agent | Target | Types of cancer | Status | Clinical study (NCT#) | Preclinical reference(s) |
|---|---|---|---|---|---|
| El1 | EZH2 | DLBCL | Preclinical | 60 | |
| GSK2816126 | EZH2 | Non‐Hodgkin lymphoma, MM | Phase 1 | NCT02082977 | 61, 62 |
| EPZ‐6438 | EZH2 |
B‐cell lymphomas DLBCL, FL |
Phase 1 Phase 2 |
NCT01897571 NCT01897571 |
63, 64 |
| CPI‐1205 | EZH2 | B‐cell lymphomas | Phase 1 | NCT02395601 | 65, 66 |
| UNC1999 | EZH1/2 | DLBCL, AML, MM | Preclinical | 33, 67, 68, 69 | |
| DS‐3201b | EZH1/2 |
Non‐Hodgkin lymphoma AML, ALL |
Phase 1 Phase 1 |
NCT02732275 NCT03110354 |
70, 71 |
| SAH‐EZH2 peptide | EZH2‐EED complex | AML | Preclinical | 72 |
ALL, acute lymphoblastic leukemia; DLBCL, diffuse large B‐cell lymphoma; EZH1/2, enhancer of zeste homolog 1 and 2; FL, follicular lymphoma; MM, multiple myeloma; NCT, National Clinical Trial.
EZH2 inhibitors show a strong growth inhibitory effect in EZH2‐mutated lymphoma cell lines and xenograft models.61, 63, 73 Preclinical and clinical trials evaluating EZH2 inhibition are currently underway mainly in lymphoma. However, inactivation of EZH2 alone is not sufficient to impair MLL‐rearranged AML, and complete disruption of PRC2 is required.58 UNC1999, an EZH1/2 dual inhibitor, impairs the proliferation of MLL‐rearranged leukemia cells in vitro and in vivo.67 This indicates that leukemia stem cells (LSC), which are responsible for drug resistance and relapse of AML, are dependent not only on EZH2, as remaining EZH1 activity is sufficient for the self‐renewal activity of LSC, despite the fact that EZH1 only partially compensates for the loss of EZH2.74, 75
Work from our group showed that quiescent LSC express the highest levels of Ezh1/2, and dual inactivation of Ezh1/2 eradicates quiescent LSC to cure AML in experiments comparing Ezh1/2 dKO with Ezh2 sKO mice.70 Furthermore, quiescent LSC are associated with PRC2‐mediated suppression of Cyclin D, and dual inactivation of Ezh1/2 induces cell cycle progression and differentiation in quiescent LSC (Figure 2). A novel EZH1/2 dual inhibitor, OR‐S1 is an orally bioavailable small molecule compound that inhibits the histone methyltransferase activity of both EZH1 and EZH2 strongly and selectively.71 OR‐S1 reduces the number of LSC, suppresses leukemia progression, and prolongs survival without serious side‐effects.70, 71 This drug, which induces differentiation and eradicates quiescent LSC, shows a synergistic effect on LSC with conventional chemotherapy agents, similar to the effect of all‐trans‐retinoic acid in acute promyelocytic leukemia. Thus, dual inhibition of EZH1/2 with an EZH1/2 dual inhibitor is effective for disrupting PRC2. Indeed, OR‐S1 suppresses H3K27me3 more potently in cells than UNC1999 and other EZH2 inhibitors.71 This drug is more effective than other selective EZH2 inhibitors in hematological cell lines including AML and acute lymphoblastic leukemia (ALL) cell lines harboring fusion genes, DLBCL cell lines with EZH2 gain‐of‐function mutations, and peripheral T‐cell lymphoma and MM cell lines.71 Clinical trials in patients with relapsed or refractory non‐Hodgkin lymphoma (NHL) and those with AML and ALL have started (NCT02732275, NCT03110354). Preliminary results of a phase 1 trial show that DS‐3201b (a derivative of OR‐S1) has early clinical activity and indicates the potential of this drug as a novel therapeutic option for patients with B‐cell and T‐cell lymphoma.76 Further investigation in AML, ALL, and NHL is currently being pursued.
Figure 2.
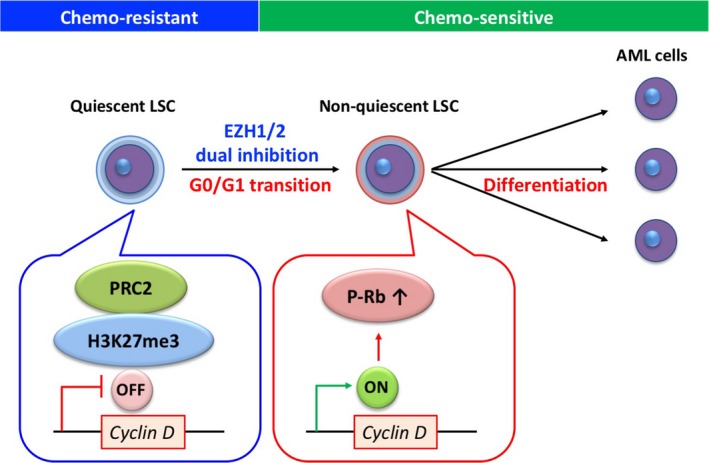
Schematic illustration of the proposed model by which dual inhibition of enhancer of zeste homolog 1 and 2 (EZH1/2) eradicates AML leukemia stem cells (LSC). Quiescent LSC show PRC2‐mediated suppression of Cyclin D. Both genetic deletion of EZH1/2 and a novel EZH1/2 inhibitor induce cell cycle progression of quiescent LSC and differentiation to non‐quiescent LSC, resulting in eradication of quiescent LSC. These conditioned AML cells show a synergistic effect with conventional chemotherapy agents. PRC, polycomb repressive complexes
6. DRUG RESISTANCE AND COMBINATION THERAPIES
Cancer cells can escape the effect of drugs, and drug resistance has been reported for EZH2 inhibitors and other drugs.26 In an EZH2‐mutated lymphoma cell line model, the acquisition of resistance was related to secondary mutations (Y111L and Y661D) in both wild‐type and gain‐of‐function Y641N EZH2 alleles.77 These resistant cells maintained a high level of H3K27me3 in the presence of EZH2 inhibitors. In addition, loss of EZH2 and subsequent reduction of H3K27me3 may be a novel pathway of acquired resistance in AML against drugs such as tyrosine kinase inhibitors because of derepression of HOX genes.78 In contrast, PRC2 loss decreases the levels of H3K27me3 and contributes to H3K27 acetylation (ac), resulting in Ras signaling amplification in malignant peripheral nerve sheath tumors.79, 80 Combination treatment with a MEK inhibitor and BRD4 inhibitor impairs tumor progression by disrupting Ras signaling and BRD4‐H3K27ac interaction, respectively.81 These results may help the identification of combination therapies with EZH2 inhibitors. Potential approaches for combination studies have been proposed in various malignant tumors. In preclinical models of EZH2 mutant germinal center NHL cells, EPZ‐6438 in combination with a glucocorticoid receptor agonist showed a dramatic synergistic cell‐killing effect. This suggests that the combination of EPZ‐6438 and CHOP, which is the current standard of care for DLBCL, enhances the inhibition of proliferation in germinal center NHL.82 In addition, bortezomib combined with UNC1999 remarkably inhibits the growth of myeloma cells in vitro and in vivo because proteasome inhibitors also repress EZH2 transcription by suppressing the RB‐E2F pathway.68 AMP‐activated protein kinase (AMPK)‐mediated phosphorylation of EZH2 at T311 inhibits PRC2 oncogenic function by disrupting the interaction between EZH2 and SUZ12.83 This indicates that AMPK agonists could be a promising sensitizer for EZH2‐targeting drugs in anticancer treatment. Taken together, these studies provide evidence of resistance mechanisms and potential approaches for combination therapy, and further investigation is warranted.
7. CONCLUSIONS AND FUTURE PERSPECTIVES
Polycomb group proteins play an important role in the maintenance of normal HSC. They have a dual function as oncogene and tumor suppressor gene in the process of tumorigenesis. Recent advances in epigenetic research have contributed to the design of new polycomb‐targeted drugs. However, despite the identification of molecular mechanisms underlying the anticancer effects of EZH1/2 and EZH2 inhibitors, many of the complex functions of PcG proteins remain to be clarified. It will be important to uncover the mechanism of resistance against polycomb‐targeted drugs, and to identify useful biomarkers for evaluating the effect of treatment and to stratify patients. Ongoing clinical trials will show whether targeting EZH1/2 is an effective treatment against various diseases, and these results will be of value for the development of epigenetic therapies.
CONFLICT OF INTEREST
IK received research funding from Daiichi Sankyo. MN has no conflicts of interest to declare.
ACKNOWLEDGMENTS
This work was supported in part by Acceleration Transformative Research for Medical Innovation from the Japan Agency for Medical Research and Development and by the National Cancer Center Research and Development Fund (I.K.).
Nakagawa M, Kitabayashi I. Oncogenic roles of enhancer of zeste homolog 1/2 in hematological malignancies. Cancer Sci. 2018;109:2342–2348. 10.1111/cas.13655
REFERENCES
- 1. Lewis EB. A gene complex controlling segementation in Drosophila. Nature. 1978;276:565‐570. [DOI] [PubMed] [Google Scholar]
- 2. Cao R, Wang L, Wang H, et al. Role of histone H3 Lysine 27 methylation in X inactivation. Science. 2002;298:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 3. Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185‐196. [DOI] [PubMed] [Google Scholar]
- 4. Margueron R, Justin N, Ohno K, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461:762‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED‐EZH2 complex. Mol Cell. 2004;15:57‐67. [DOI] [PubMed] [Google Scholar]
- 6. Stock JK, Giadrossi S, Casanova M, et al. Ring1‐mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat Cell Biol. 2007;9:1428‐1435. [DOI] [PubMed] [Google Scholar]
- 7. Wang H, Wang L, Erdjument‐Bromage H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873‐878. [DOI] [PubMed] [Google Scholar]
- 8. Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl‐lysine marks in histone H3 by polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870‐1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Buchwald G, Van Der Stoop P, Weichenrieder O, Perrakis A, Van Lohuizen M, Sixma TK. Structure and E3‐ligase activity of the Ring‐Ring complex of Polycomb proteins Bmi1 and Ring1b. EMBO J. 2006;25:2465‐2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Morey L, Aloia L, Cozzuto L, Benitah SA, Di Croce L. RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells. Cell Rep. 2013;3:60‐69. [DOI] [PubMed] [Google Scholar]
- 11. Gao Z, Zhang J, Bonasio R, et al. PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol Cell. 2012;45:344‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tavares L, Dimitrova E, Oxley D, et al. RYBP‐PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell. 2012;148:664‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blackledge NP, Farcas AM, Kondo T, et al. Variant PRC1 complex‐dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014;157:1445‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalb R, Latwiel S, Baymaz HI, et al. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol. 2014;21:569‐571. [DOI] [PubMed] [Google Scholar]
- 15. Ku M, Koche RP, Rheinbay E, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cui K, Zang C, Roh TY, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kamminga LM, Bystrykh LV, De Boer A, et al. The Polycomb group gene. Blood. 2006;107:2170‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mochizuki‐kashio M, Mishima Y, Miyagi S, et al. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood. 2011;118:6553‐6561. [DOI] [PubMed] [Google Scholar]
- 19. Neo WH, Booth CAG, Azzoni E, et al. Cell‐extrinsic hematopoietic impact of Ezh2 inactivation in fetal liver endothelial cells. Blood. 2018;131:2223‐2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herrera‐Merchan A, Arranz L, Ligos JM, De Molina A, Dominguez O, Gonzalez S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun. 2012;3:611‐623. [DOI] [PubMed] [Google Scholar]
- 21. Hidalgo I, Herrera‐Merchan A, Ligos JM, et al. Ezh1 is required for hematopoietic stem cell maintenance and prevents senescence‐like cell cycle arrest. Cell Stem Cell. 2012;11:649‐662. [DOI] [PubMed] [Google Scholar]
- 22. O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb‐group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330‐4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mochizuki‐Kashio M, Aoyama K, Sashida G, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous malignancies in an Ezh1‐dependent manner. Blood. 2015;126:1172‐1183. [DOI] [PubMed] [Google Scholar]
- 24. Su IH, Basavaraj A, Krutchinsky AN, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4:124‐131. [DOI] [PubMed] [Google Scholar]
- 25. Sauvageau M, Sauvageau G. Polycomb group proteins: multi‐faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim KH, Roberts CWM. Targeting EZH2 in cancer. Nat Med. 2016;22:128‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kondo Y, Shen L, Cheng AS, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40:741‐750. [DOI] [PubMed] [Google Scholar]
- 28. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu F, Li X, Wu L, et al. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol. 2011;90:643‐653. [DOI] [PubMed] [Google Scholar]
- 30. Van Kemenade FJ, Raaphorst FM, Blokzijl T, et al. Coexpression of BMI‐1 and EZH2 polycomb‐group proteins is associated with cycling cells and degree of malignancy in B‐cell non‐Hodgkin lymphoma. Blood. 2001;97:3896‐3901. [DOI] [PubMed] [Google Scholar]
- 31. Visser HPJ, Gunster MJ, Kluin‐Nelemans HC, et al. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112:950‐958. [DOI] [PubMed] [Google Scholar]
- 32. Sasaki D, Imaizumi Y, Hasegawa H, et al. Overexpression of enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T‐cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica. 2011;96:712‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Agarwal P, Alzrigat M, Párraga AA, et al. Genome‐wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget. 2016;7:6809‐6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pawlyn C, Bright MD, Buros AF, et al. Overexpression of EZH2 in multiple myeloma is associated with poor prognosis and dysregulation of cell cycle control. Blood Cancer J. 2017;7:e549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B‐cell lymphomas of germinal‐center origin. Nat Genet. 2010;42:181‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McCabe MT, Graves AP, Ganji G, et al. Mutation of A677 in histone methyltransferase EZH2 in human B‐cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci USA. 2012;109:2989‐2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild‐type plus mutant EZH2 drive tumor‐associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B‐cell lymphomas. Proc Natl Acad Sci USA. 2010;107:20980‐20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ntziachristos P, Tsirigos A, Van Vlierberghe P, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18:296‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T‐cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722‐726. [DOI] [PubMed] [Google Scholar]
- 41. Nikoloski G, Langemeijer SMC, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665‐667. [DOI] [PubMed] [Google Scholar]
- 42. Bachmann IM, Halvorsen OJ, Collett K, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268‐273. [DOI] [PubMed] [Google Scholar]
- 43. Zingg D, Debbache J, Schaefer SM, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:1‐17. [DOI] [PubMed] [Google Scholar]
- 44. Zhou M, Barrette TR, Kumar‐Sinha C, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624‐629. [DOI] [PubMed] [Google Scholar]
- 45. Xu K, Wu ZJ, Groner AC, et al. EZH2 oncogenic activity in castration‐resistant prostate cancer cells is polycomb‐independent. Science. 2012;338:1465‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu C, Han HD, Mangala LS, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010;18:185‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li H, Cai Q, Godwin AK, Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res. 2010;8:1610‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Behrens C, Solis LM, Lin H, et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non‐small cell lung carcinoma. Clin Cancer Res. 2013;19:6556‐6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Zhao H, Lv L, Bao L, Wang X, Han S. Prognostic significance of EZH2 expression in non‐small cell lung cancer: a meta‐analysis. Sci Rep. 2016;6:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang H, Qi J, Reyes JM, et al. Oncogenic deregulation of EZH2 as an opportunity for targeted therapy in lung cancer. Cancer Discov. 2016;6:1007‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Changchien YC, Tatrai P, Papp G, et al. Poorly differentiated synovial sarcoma is associated with high expression of enhancer of zeste homologue 2 (EZH2). J Transl Med. 2012;10:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen JK, Cote GM, Gao Y, et al. Targeting EZH2‐mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci Rep. 2016;6:25239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Souroullas GP, Jeck WR, Parker JS, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med. 2016;22:632‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guglielmelli P, Biamonte F, Score J, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011;118:5227‐5234. [DOI] [PubMed] [Google Scholar]
- 55. Iwama A. Polycomb repressive complexes in hematological malignancies. Blood. 2017;130:23‐29. [DOI] [PubMed] [Google Scholar]
- 56. Bejar R, Stevenson K, Abdel‐Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496‐2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Berg T, Thoene S, Yap D, et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb‐group gene EZH2 in lymphomagenesis. Blood. 2014;123:3914‐3924. [DOI] [PubMed] [Google Scholar]
- 58. Neff T, Sinha AU, Kluk MJ, et al. Polycomb repressive complex 2 is required for MLL‐AF9 leukemia. Proc Natl Acad Sci USA. 2012;109:5028‐5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107‐1117. [DOI] [PubMed] [Google Scholar]
- 60. Qi W, Chan H, Teng L, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109:21360‐21365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McCabe MT, Ott HM, Ganji G, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2‐activating mutations. Nature. 2012;492:108‐112. [DOI] [PubMed] [Google Scholar]
- 62. Zeng D, Liu M, Pan J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell‐like myeloma cells. Oncotarget. 2017;8:3396‐3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Knutson SK, Kawano S, Minoshima Y, et al. Selective inhibition of EZH2 by EPZ‐6438 leads to potent antitumor activity in EZH2‐mutant non‐hodgkin lymphoma. Mol Cancer Ther. 2014;13:842‐854. [DOI] [PubMed] [Google Scholar]
- 64. Hernando H, Gelato KA, Lesche R, et al. EZH2 inhibition blocks multiple myeloma cell growth through upregulation of epithelial tumor suppressor genes. Mol Cancer Ther. 2016;15:287‐298. [DOI] [PubMed] [Google Scholar]
- 65. Moros A, Rodríguez V, Saborit‐Villarroya I, et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib‐resistant mantle cell lymphoma. Leukemia. 2014;28:2049‐2059. [DOI] [PubMed] [Google Scholar]
- 66. Bradley WD, Arora S, Busby J, et al. EZH2 inhibitor efficacy in non‐hodgkin's lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21:1463‐1475. [DOI] [PubMed] [Google Scholar]
- 67. Xu B, On DM, Ma A, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL‐ rearranged leukemia. Blood. 2015;125:346‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rizq O, Mimura N, Oshima M, et al. Dual inhibition of EZH2 and EZH1 sensitizes PRC2‐dependent tumors to proteasome inhibition. Clin Cancer Res. 2017;23:4817‐4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Konze KD, Ma A, Li F, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324‐1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fujita S, Honma D, Adachi N, et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia. 2018;32:855‐864. [DOI] [PubMed] [Google Scholar]
- 71. Honma D, Kanno O, Watanabe J, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108:2069‐2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim W, Bird GH, Neff T, et al. Targeted disruption of the EZH2‐EED complex inhibits EZH2‐dependent cancer. Nat Chem Biol. 2013;9:643‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Campbell JE, Kuntz KW, Knutson SK, et al. EPZ011989, A potent, orally‐available EZH2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6:491‐495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shen X, Liu Y, Hsu YJ, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Margueron R, Li G, Sarma K, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32:503‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Maruyama D, Tobinai K, Makita S, et al. First‐in‐human study of the EZH1/2 dual inhibitor DS‐3201b in patients with relapsed or refractory non‐hodgkin lymphomas — preliminary results. Blood. 2017;130(suppl 1):4070. LP‐4070. [Google Scholar]
- 77. Gibaja V, Shen F, Harari J, et al. Development of secondary mutations in wild‐type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Göllner S, Oellerich T, Agrawal‐Singh S, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med. 2017;23:69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Baude A, Lindroth AM, Plass C. PRC2 loss amplifies Ras signaling in cancer. Nat Genet. 2014;46:1154‐1155. [DOI] [PubMed] [Google Scholar]
- 80. De Raedt T, Walton Z, Yecies JL, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras‐driven tumors. Cancer Cell. 2011;20:400‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras‐driven transcription and confers sensitivity to BRD4‐based therapies. Nature. 2014;514:247‐251. [DOI] [PubMed] [Google Scholar]
- 82. Knutson SK, Warholic NM, Johnston LD, et al. Synergistic anti‐tumor activity of EZH2 inhibitors and glucocorticoid receptor agonists in models of germinal center non‐Hodgkin lymphomas. PLoS ONE. 2014;9:1‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wan L, Xu K, Wei Y. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol Cell. 2018;69:279‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]