

Thackray et al. show authentic propagation of mammalian prions in Drosophila melanogaster rendered transgenic solely for PrP. The molecular and cellular co-factors necessary for strain-specific prion propagation are not therefore unique to mammals, suggesting that Drosophila may also be a suitable host for studying prion transmission.
Keywords: PrPSc, prion, infectivity, Drosophila, strains
Abstract
Mammalian prions propagate by template-directed misfolding and aggregation of normal cellular prion related protein PrPC as it converts into disease-associated conformers collectively referred to as PrPSc. Mammalian species may be permissive for prion disease because these hosts have co-evolved specific co-factors that assist PrPC conformational change and prion propagation. We have tested this hypothesis by examining whether faithful prion propagation occurs in the normally PrPC-null invertebrate host Drosophila melanogaster. Ovine PrP transgenic Drosophila exposed at the larval stage to ovine scrapie showed a progressive accumulation of transmissible prions in adult flies. Strikingly, the biological properties of distinct ovine prion strains were maintained during their propagation in Drosophila. Our observations show that the co-factors necessary for strain-specific prion propagation are not unique to mammalian species. Our studies establish Drosophila as a novel host for the study of transmissible mammalian prions.
Introduction
Mammalian prions cause fatal neurodegenerative diseases such as Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle and scrapie disease of sheep (Prusiner, 2004). These conditions are transmissible between individuals of the same or different species and as a consequence, animal prion diseases pose a realistic threat to human health through their zoonotic potential (Bruce et al., 1997). Prions lack a conventional nucleic acid-based genome. The ‘protein only’ concept that relates to prion diseases predicts that infectious prion particles consist of PrPSc in the form of aggregates of misfolded conformers of the normal host protein PrPC (Prusiner, 1982). Prion propagation occurs by template-directed nucleation whereby a prion seed induces conformational change within PrPC and promotes the incorporation of nascent misfolded prion protein into growing PrPSc assemblies, which subsequently undergo fragmentation (Collinge, 2016).
PrPC is a cell-surface glycoprotein attached to the membrane by a glycosylphosphatidyl inositol (GPI) anchor (Stahl et al., 1987). It is now well established that PrPC expression is obligatory for prion replication and prion-induced neurotoxicity (Bueler et al., 1993; Mallucci et al., 2003). A remarkable feature of mammalian prions is the existence of different strains of the transmissible agent that directly influence host range and the clinico-pathological features of prion disease in the affected host (Prusiner, 2004). According to the ‘protein only’ concept (Prusiner, 1982), prion strain-specific properties are dictated by the conformational arrangement of PrPSc (Bessen and Marsh, 1994; Telling et al., 1996; Safar et al., 1998). Changes in the replication environment of prions can induce mutational change in their strain properties (Collinge and Clarke, 2007), an event that may occur during inter-species prion transmission where PrP primary structures differ (Kimberlin et al., 1987; Bartz et al., 2000).
Several fundamental features of mammalian prion biology remain unresolved despite considerable efforts aimed at their elucidation (Collinge, 2016). These include lack of a full description of the molecular and cellular machinery necessary to generate infectious neurotoxic prions (Supattapone, 2014). The possibility exists that mammalian hosts, in which PrPC is ubiquitously expressed and well conserved, are permissive for prion formation because they have co-evolved specific co-factors that assist in prion protein misfolding and propagation. Here we have tested this hypothesis by examining whether authentic prion propagation occurs in Drosophila melanogaster, an invertebrate species that is phylogenetically separated from mammals by millions of years of divergent evolution. Drosophila and other members of the arthropod phylum do not show the presence of an orthologous PrP gene (PRNP) in their genome and therefore do not normally express PrPC (Rivera-Milla et al., 2006).
In this study, we tested the capacity of mammalian prions to faithfully propagate in a strain-specific manner in Drosophila genetically engineered to express PrPC (Thackray et al., 2012b, 2014). Exposure of larval stage PrP transgenic Drosophila to mammalian prions resulted in a progressive accumulation of bona fide prions in adult flies. Strikingly, the biological properties of the original prion strain used as inoculum were maintained during their passage in the insect host. These observations show for the first time that the molecular and cellular co-factors necessary for strain-specific prion propagation are conserved between phylogenetically diverse species and are not unique to mammalian hosts. Our studies provide a fundamental advance in the understanding of mammalian prion biology and also establish Drosophila as a novel methodology for the study of transmissible mammalian prions.
Materials and methods
Fly stocks
The UAS-PrP fly lines w; M{VRQ-PrP(GPI), 3xP3-RFP.attP}ZH-51D and w; M{ARQ-PrP(GPI), 3xP3-RFP.attP}ZH-51D transgenic for ovine V136R154Q171 (VRQ) or A136R154Q171 (ARQ) PrP, respectively, expressed with an N-terminal leader peptide and C-terminal GPI signal sequence, were generated by PhiC31 site-specific transformation by BestGene as previously described (Thackray et al., 2012c). The following fly lines were obtained from the Department of Genetics, University of Cambridge, UK:
Cre-mediated removal of the red fluorescent protein (RFP) gene from the VRQ and 51D fly genome was performed by conventional fly crosses (Thackray et al., 2014). PrP transgenic Drosophila were crossed with either the Elav-GAL4 or Actin-5C-GAL4 driver fly lines to derive transgenic flies that expressed PrP pan neuronally or ubiquitously, respectively. 51D Drosophila crossed with either driver fly line were used as control flies where appropriate. All fly lines were raised on standard cornmeal media at 25°C and maintained at low to medium density, and pre-mated before experimental use.
Prion inoculation of Drosophila
Primary transmission of sheep scrapie: sheep-to-fly
Drosophila at the larval stage of development were exposed to brain homogenate of cerebral cortex tissue from a confirmed PG127 (alternatively referred to as DAW or G338) scrapie-positive sheep (SE1848/0005) (Thackray et al., 2008). New Zealand-derived VRQ/VRQ scrapie-free brain tissue was used as control material. Two hundred and fifty microlitres of a 1% (w/v) sheep brain homogenate prepared in phosphate-buffered saline (PBS) pH 7.4, were added to the top of the cornmeal that contained third instar Drosophila larvae in 3-inch plastic vials. Following eclosion (i.e. hatching) flies were transferred to fresh non-treated vials.
Secondary transmission of sheep scrapie: fly-to-fly
Drosophila head homogenates were prepared from 30-day-old flies that had been exposed at the larval stage to scrapie-positive or scrapie-negative sheep brain material. Two hundred and fifty microlitres of a 10−1 (v/v) dilution of the original fly brain homogenate were added to the top of the cornmeal that contained third instar Drosophila larvae in 3-inch plastic vials. Flies were transferred to fresh, non-treated vials following eclosion.
Transmission of defined ovine prion strains in PrP transgenic Drosophila
Drosophila at the larval stage of development were exposed to 1% (w/v) mouse brain homogenate infected with a defined ovine prion strain or prion-free mouse brain homogenate as control. The different mouse-adapted prion strains used here were isolated by serial passage of sheep scrapie isolates in ovine PrP transgenic mice as previously described (Thackray et al., 2011, 2012a).
Preparation of Drosophila head homogenate
Whole flies in an Eppendorf tube were frozen in liquid nitrogen for 10 min and then vortexed for 2 min to cause decapitation. Individual fly heads were isolated and placed in clean Eppendorf tubes using a fine paint brush. PBS pH 7.4 was added to give 1 µl/head and homogenates were prepared by manual grinding of the fly heads with sterilized plastic pestles. For western blot analysis, fly head homogenate was mixed with an equal volume of 20% scrapie-free sheep brain homogenate prior to extraction and proteinase K (PK) digestion as previously described (Lacroux et al., 2012) using monoclonal antibody Sha31 (Feraudet et al., 2005).
Protein misfolding cyclic amplification
Protein misfolding cyclic amplification (PMCA) was carried out as previously described (Lacroux et al., 2012). The substrate consisted of 10% (w/v) ovine VRQ PrP transgenic mouse (tg338) brain homogenate in PBS pH 7.4, 0.1% Triton™ X-100 and 150 mM NaCl buffer. Five microlitres of fly head homogenate were mixed with 45 µl of substrate in 0.2 ml thin-wall PCR tubes. Sealed tubes were then placed in the horn of a Misonix 4000 sonicator for one round of 96 cycles. Each cycle consisted of a 10-s sonication step (70% of power) followed by a 14 min and 50 s incubation step. Twenty microlitres of each reaction mix were subsequently treated with PK (4 µg of PK per mg of protein) at 37°C for 2 h and the reaction stopped by adding Pefabloc® (4 mM final concentration). PK-resistant PrP was detected by western blot as previously described (Lacroux et al., 2012) using monoclonal antibody Sha31 (Feraudet et al., 2005).
Mouse prion bioassay
Mouse prion bioassays were carried out in tg338 mice, which are transgenic for ovine VRQ PrP and are highly efficient for the detection of sheep scrapie infectivity (Le Dur et al., 2005). Mice (n = 6 per inocula) were injected intracerebrally with 20 µl of diluted fly head homogenate (to give approximately two fly-head equivalents per mouse) and monitored daily until the occurrence of clinical signs of mouse prion disease. Inoculated mice were euthanized when they started to show locomotor disorders and any impairment in their capacity to feed, or at a predefined end-point for the assay (either >250 days or in some cases >670 days post-inoculation) (Andreoletti et al., 2011). Brain tissue (cerebral cortex) was collected from euthanized mice and frozen for PrPSc analysis by western blot (TeSeE, Bio-Rad) or paraffin-embedded tissue (PET) blot analysis (Andreoletti et al., 2011).
Negative geotaxis climbing assay
The locomotor ability of flies was assessed in a negative geotaxis climbing assay initiated with 45 (3 × n = 15) age-matched, pre-mated female flies in each treatment group. Drosophila were placed in adapted plastic 25 ml pipettes that were used as vertical climbing columns and allowed to acclimatize for 30 min prior to assessment of their locomotor ability. Flies were tapped to the bottom of the pipette (using the same number and intensity of taps on each occasion) and then allowed to climb for 45 s. At the end of the climbing period the number of flies above the 25 ml mark, the number below the 2 ml mark and the number in-between the 2 ml and 25 ml mark was recorded. This procedure was performed three times at each time point. The performance index (PI) was calculated for each group of 15 flies (average of three trials) using the formula:
PI = 0.5 × (ntotal + ntop − nbottom) / ntotal where ntotal is the total number of flies, ntop is the total number of flies at the top, and nbottom is the total number of flies at the bottom. A PI value of 1 is recorded if all flies climb to the top of the tube whereas the value is 0 if no flies climb the tube past the 2 ml mark. The mean PI ± SD (standard deviation) at individual time points for each treatment group was plotted as a regression line.
Statistical analysis
Statistical analysis of the negative geotaxis climbing assay data was performed by the paired Student t-test, using Prism (GraphPad Software Inc, San Diego, USA).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material.
Results
Mammalian prions replicate in PrP transgenic Drosophila
Transgenic Drosophila that express membrane-bound VRQ ovine PrP (VRQ Drosophila fly line) and control non-transgenic flies (51D fly line) were exposed at the larval stage to brain material prepared from either PG127 scrapie-infected or healthy prion-free sheep (Thackray et al., 2012c, 2014). After hatching, Drosophila were transferred to prion-free culture tubes. At various time points (≤40 days) during their adult lifespan, groups of Drosophila were euthanized, decapitated and homogenate prepared from the isolated fly heads. These homogenates were used to seed in vitro PMCA reactions in order to reveal the presence of prion seeding activity as shown by the data in Fig. 1. PMCA mimics prion replication in vivo, but in an accelerated form, allowing amplification of minute quantities of PrPSc and prion infectivity (Saborio et al., 2001). In PMCA reactions, a PrPC-containing substrate (in this study brain homogenate prepared from ovine PrP transgenic mice) is combined with the seed (in this study Drosophila head homogenate) that may contain minute amounts of PrPSc. Following repeated cycles of incubation and sonication, the amount of PrPSc seed increases to levels at which it can be detected by conventional biochemical means, such as western blot detection of PK-resistant PrPSc as was used here.
Figure 1.

Prion seeding activity in scrapie-exposed PrP transgenic Drosophila. Elav × VRQ(GPI) PrP transgenic (VRQ) and Elav × 51D (51D) Drosophila were exposed at the larval stage to PG127 scrapie-infected or prion-free control sheep brain material. At various times after hatching, head homogenate was prepared from harvested flies and used as seed in PMCA reactions. (A) End-point titration of PMCA prion seeding activity in head homogenate from control or PG127-exposed Drosophila. (B) Western blot detection of PK-resistant PrP27–30 in PMCA reaction products seeded with control or PG127-exposed Drosophila head homogenate. Molecular mass markers in kDa shown on the left.
No prion seeding activity was detected in PMCA reactions with seed prepared from mock-infected VRQ Drosophila head homogenate, or from scrapie-exposed or mock-infected 51D control flies (Fig. 1A). Similarly, no prion seeding activity was detected in head homogenates prepared from 5- or 10-day-old scrapie-exposed VRQ Drosophila. Strikingly, prion seeding activity was detected in head homogenate prepared from scrapie-infected VRQ Drosophila aged ≥20 days. An end-point titration of prion seeding activity, carried out using serial 10-fold dilutions of original fly head homogenate, indicated a progressive increase of prion seeding titre in the heads of prion-exposed VRQ Drosophila from 20 to 40 days of age (Fig. 1B). The seeding activity in 40-day-old prion-exposed VRQ Drosophila was 10−4 lower than that seen in a terminal disease-PG127 sheep scrapie inoculum (Thackray et al., 2016).
We determined if the presence of the prion seeding activity in scrapie-inoculated VRQ Drosophila was associated with the presence of PK-resistant PrPSc, a pathognomonic marker of prion disease in mammalian hosts (Prusiner, 2004). Head homogenate from 40-day-old Drosophila was PK-digested and analysed directly by SDS-PAGE and western blot probed with an anti-PrP monoclonal antibody. The data in Fig. 2 show that no PK-resistant PrPSc signal was detected in Drosophila exposed to scrapie-free sheep brain material (Fig. 2A and B), or from scrapie-treated 51D flies (Fig. 2C). Notably, a PK-resistant PrP signal was detected in head homogenate from scrapie-exposed VRQ Drosophila (Fig. 2D). The molecular profile of the PK-resistant PrP in VRQ Drosophila was clearly different to that present in the original PG127 inoculum, an expected feature considering differences in N-linked glycosylation that exist between Drosophila and mammalian hosts (Choi et al., 2010; Thackray et al., 2012c). The intensity of the PK-resistant PrP signal in the VRQ Drosophila head homogenate was similar to that obtained in a 10−4 dilution of a terminal disease-PG127 sheep scrapie inoculum. This result was consistent with the data obtained in the PMCA endpoint titration where 40-day-old VRQ Drosophila displayed a 10−4 lower seeding activity than a terminal disease-PG127 scrapie inoculum.
Figure 2.

PK-resistant PrP in scrapie-exposed PrP transgenic Drosophila. Elav × VRQ(GPI) PrP transgenic (VRQ) and Elav × 51D (51D) Drosophila were exposed at the larval stage to PG127 scrapie-infected or prion-free control sheep brain material. At 30 days post-hatching, head homogenate was prepared from harvested flies, treated with PK and analysed by western blot to detect PK-resistant PrP. (A and B) Drosophila exposed to control brain homogenate. (C and D) Drosophila exposed to PG127 scrapie-infected sheep brain homogenate. (A and C) Elav × 51D Drosophila. (B and D) Elav × VRQ(GPI) PrP transgenic Drosophila. A titration of original PG127 scrapie-infected sheep brain homogenate is included on each blot for comparative purposes. Molecular mass markers in kDa shown in the centre.
Fly-to-mouse prion transmission
To unequivocally demonstrate that authentic prions replicated in scrapie-exposed VRQ Drosophila we performed fly-to-mouse transmission. Head homogenate from flies used as a seed in the PMCA experiment shown above was inoculated into ovine VRQ PrP transgenic (tg338) mice. The transmission data are shown in Table 1. No evidence of clinical prion disease or abnormal PrP accumulation were observed in mice inoculated with head homogenates prepared from VRQ Drosophila and 51D flies exposed to control sheep brain homogenate. Similarly, no prion infectivity was detected in tg338 mice inoculated with head homogenates prepared from 5- or 10-day-old PG127-exposed VRQ Drosophila. An incomplete attack rate for clinical prion disease transmission was observed in tg338 mice inoculated with head homogenate from 20-day-old scrapie-infected VRQ Drosophila. Mice inoculated with head homogenates prepared from 30- and 40-day-old scrapie-exposed VRQ Drosophila developed a 100% attack rate for clinical prion disease transmission. The brains of tg338 mice inoculated with fly-head homogenate were probed for disease-associated PrP as shown in Fig. 3. Prion disease in mice that showed clinical signs of the condition was confirmed by western blot detection of PK-resistant PrPSc (Fig. 3A). PET blot analysis of the brains of clinically affected mice showed the typical distribution of PrPSc in PG127 scrapie-inoculated tg338 mice (Fig. 3B). No clinical signs of mouse prion disease or abnormal PrP accumulation were observed in tg338 mice inoculated with scrapie-exposed 51D fly head homogenate. This result excludes the suggestion that carry-over of the original sheep inoculum was an explanation for prion infectivity detection in scrapie-exposed VRQ Drosophila.
Table 1.
Prion infectivity accumulates in scrapie-exposed PrP transgenic Drosophila
| Fly line | Inoculum | Age of Drosophila inoculated into tg338 mice | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 days | 10 days | 20 days | 30 days | 40 days | |||||||
| Attack Rate | IP | Attack Rate | IP | Attack Rate | IP | Attack Rate | IP | Attack Rate | IP | ||
| 51D | Control | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 |
| PG127 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | |
| VRQ | Control | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 | 0/6 | >250 |
| PG127 | 0/6 | >250 | 0/6 | >250 | 4/6 | 103 ± 11 | 6/6 | 89 ± 3 | 6/6 | 89 ± 2 | |
Elav × VRQ(GPI) PrP transgenic (VRQ) and Elav × 51D (51D) Drosophila were exposed at the larval stage to PG127 scrapie-infected or prion-free control sheep brain material. At various times after hatching, head homogenate was prepared from harvested flies and intracerebrally inoculated into ovine PrP transgenic (tg338) mice. Inoculated mice were euthanized when they showed clinical signs of prion infection or after 250 days for those that did not develop clinical disease. Mice were considered positive for prion disease when PK-resistant PrP27–30 was detected in brain tissue by western blot. The attack rate (number of prion positive mice/total number of mice inoculated) is reported for each treatment group. The incubation period (IP) for inoculated mice, which represents the average time from inoculation to euthanasia for each inoculated group of animals, is reported in days ± SD.
Figure 3.
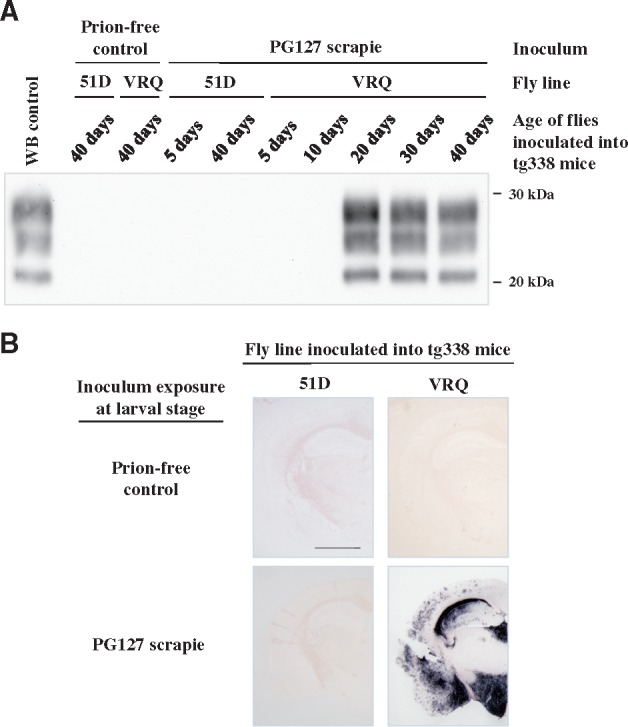
Detection of prion infectivity in scrapie-exposed PrP transgenic Drosophila. Elav × VRQ(GPI) PrP transgenic (VRQ) and Elav × 51D (51D) Drosophila were exposed at the larval stage to PG127 scrapie-infected or prion-free control sheep brain material. At various times after hatching, head homogenate was prepared from harvested flies and inoculated into tg338 mice. Inoculated mice were euthanized when they showed clinical signs of prion infection or after 250 days for those that did not develop clinical disease. Mice were considered positive for prion disease when PK-resistant PrP27–30 was detected in brain tissue by western blot. (A) Western blot detection of PK-resistant PrP27–30 in the brains of tg338 mice with clinical prion disease. Molecular mass markers in kDa shown on the right. (B) PET blot analysis of tg338 mouse brains from animals inoculated with 40-day-old Elav × VRQ(GPI) PrP transgenic (VRQ) or Elav × 51D (51D) Drosophila exposed to PG127 scrapie-infected or prion-free control sheep brain material. Scale bar = 150 µm.
To further increase the robustness of this observation, the fly-to-mouse transmission experiment was repeated twice using samples obtained in two independent experiments (using the same fly lines and the same sheep scrapie prion strain). The observed incubation times and attack rates in these repeat experiments were consistent to those recorded in the original experiment, as shown in the Supplementary material, S1. Collectively these data unequivocally demonstrate that ovine prion infectivity progressively accumulated in scrapie-exposed ovine VRQ PrP transgenic Drosophila.
Fly-to-fly prion propagation
We next investigated whether the prion infectivity that accumulates in VRQ Drosophila at first passage could be serially propagated in the same fly line. Accordingly, head homogenate from 30-day-old adult Drosophila (first passage flies) was used to inoculate fresh VRQ Drosophila (second passage flies) at the larval stage. Second passage VRQ Drosophila were allowed to hatch and groups of flies were euthanized at 5, 30 and 40 days of age when head homogenate was prepared for PMCA analysis. The data in the Supplementary material (S2) show, as expected, no PMCA prion seeding activity was detected in VRQ Drosophila that were exposed to head homogenate from first passage mock-infected VRQ Drosophila, or first passage scrapie- or mock infected 51D flies. However, prion seeding activity was detected in second passage VRQ Drosophila exposed to 30- and 40-day-old, but not 5-day-old, head homogenate from first passage scrapie-exposed VRQ Drosophila. Endpoint titration of the PMCA-positive second passage VRQ Drosophila samples indicated an increase in prion seeding activity titre in these flies between 30 and 40 days of age.
Second passage VRQ Drosophila head homogenate was inoculated into tg338 mice to assess prion infectivity in these samples. Mice that received 30-day-old PMCA-positive second passage VRQ Drosophila head homogenate showed 100% attack rate for clinical signs of mouse prion disease and an incubation period of 89 ± 4 days. The brains of inoculated mice were examined for the presence of disease-associated PrP as shown by the data in the Supplementary material (S3). PK-resistant PrPSc was evidenced by western blot of the brains of clinically affected mice and PET blot analysis of the brains of clinically affected mice revealed the typical distribution of PrPSc in PG127 scrapie-inoculated tg338 mice. No clinical signs or abnormal PrP accumulation was observed in tg338 mice inoculated with 5-day-old second passage VRQ Drosophila head homogenate or from VRQ Drosophila exposed to first passage 51D control flies. These results demonstrate that mammalian prions propagated in VRQ Drosophila can be serially transmitted in flies.
Prion-induced toxic phenotype in PrP transgenic Drosophila
Since prion-induced toxicity occurs concomitantly with prion replication in mammalian hosts (Bueler et al., 1993; Mallucci et al., 2003) we next investigated whether the propagation of prions in VRQ Drosophila induced a toxic phenotype in these flies. To do so, we performed a negative geotaxis climbing assay (Thackray et al., 2012c) using adult Drosophila previously exposed at the larval stage to ovine scrapie prions. The data in the Supplementary material (S4, together with the accompanying statistical analysis described in the Supplementary material for this figure) show that there was no difference in the climbing ability between 51D flies exposed to PG127 scrapie or control prion-free sheep brain homogenate. Strikingly, VRQ Drosophila developed a toxic phenotype after exposure to PG127 prions, evidenced by an accelerated decrease in locomotor ability compared to control treated flies that became progressively more severe with age. Collectively, these data show that the prion-induced toxic fly phenotype was associated with prion propagation in adult PrP transgenic Drosophila as evidenced by the data in Table 1 and Fig. 3.
Prion strain properties are maintained in PrP transgenic Drosophila
A comparison of the incubation period and PK-resistant PrPSc molecular profile observed in tg338 mice inoculated with either a sample of the original PG127 sheep scrapie inoculum, or this material passaged in VRQ Drosophila, suggested that mammalian prion strain properties were unaltered following propagation in PrP transgenic Drosophila. To confirm this was the case we propagated three distinct ovine prion strains in PrP transgenic Drosophila prior to their re-isolation in tg338 mice. The panel of ovine prion strains used here have been previously characterized by bioassay in tg338 mice and are differentiated by unique incubation times and on the basis of their PK-resistant PrPSc western blot molecular profile (Thackray et al., 2011, 2012a). We exposed ovine PrP transgenic Drosophila at the larval stage to each ovine scrapie prion strain. VRQ Drosophila were exposed to ovine prions isolated in VRQ PrP transgenic mice and ARQ Drosophila were exposed to ovine prions isolated in ARQ PrP transgenic mice. Collectively, the ovine PrP transgenic Drosophila were susceptible to all three different prion strains shown by the development of a progressive toxic phenotype in the form of an accelerated decline in performance in a negative geotaxis climbing assay (Supplementary material, S4).
An aliquot of each original prion strain isolated in PrP transgenic mice and samples of these prions propagated in PrP transgenic Drosophila were transmitted to tg338 mice (for either one or two passages). The attack rate and incubation period observed in tg338 mice inoculated with the original prion strains were similar to those obtained following inoculation with the equivalent prion strain propagated in PrP transgenic Drosophila as shown in Table 2. The brains of clinically prion-diseased tg338 mice were examined for disease-associated PrP and neuropathology as shown in Fig. 4. Irrespective of the number of passages, the molecular profile of PK-resistant PrPSc observed in the brains of tg338 mice inoculated with the original ovine prions and those propagated in PrP transgenic Drosophila were indistinguishable (Fig. 4A). In addition, the vacuolar lesion profile in the brains of tg338 mice inoculated with the original ovine prion strain and with the equivalent prion strain propagated in PrP transgenic Drosophila were virtually identical (Fig. 4B). Collectively, these data demonstrate that the biological properties of distinct ovine prion strains were maintained after their propagation in ovine PrP transgenic Drosophila.
Table 2.
Scrapie strains retain transmission properties after passage in PrP transgenic Drosophila
| Scrapie strain | Original strain isolated in tg338 mice | Drosophila-passaged strain isolated in tg338 mice | ||||
|---|---|---|---|---|---|---|
| First passage | Second passage | |||||
| Attack rate | IP | Attack rate | IP | Attack rate | IP | |
| PG127 | 6/6 | 61 ± 2 | 6/6 | 87 ± 4 | 6/6 | 59 ± 1 |
| Pa59 | 6/6 | 143 ± 3 | 6/6 | 187 ± 4 | 6/6 | 142 ± 1 |
| Apl338 | 6/6 | 631 ± 83 | 3/5 | 642 ± 57 | NA | NA |
The ovine classical scrapie prion strains PG127 and Apl338 (isolated in ovine VRQ tg338 mice) and Pa59 (isolated in ovine ARQ tg59 mice) were transmitted to tg338 ovine PrP transgenic mice. The ovine classical scrapie prion strains PG127, Pa59 or Apl338 were transmitted to tg338 ovine PrP transgenic mice. In parallel, Actin × VRQ(GPI) PrP transgenic (VRQ) Drosophila were exposed to the PG127 and Apl338, prion strains, and Elav × ARQ(GPI) PrP transgenic (ARQ) Drosophila were exposed to the Pa59 prion strain, at the larval stage. Head homogenates were prepared from adult flies aged 30 days and serially transmitted in tg338 mice (two iterative passages) by intracerebral inoculation. Mice were euthanized when they showed clinical signs of prion infection and after 250 days for those that did not develop clinical disease. Mice were considered positive for prion disease when PK-resistant PrP27–30 was detected in brain tissue by western blot. The attack rate (number of prion positive mice/total number of mice inoculated) is reported for each treatment group. The incubation period (IP) for inoculated mice, which represents the mean time from inoculation to euthanasia for each inoculated group of animals, is reported in days ± SD. NA = data not available, still ongoing.
Figure 4.

Authentic prion replication in PrP transgenic Drosophila. Actin × VRQ(GPI) PrP transgenic (VRQ) Drosophila were exposed to the PG127 and Apl338, prion strains, and Elav × ARQ(GPI) PrP transgenic (ARQ) Drosophila were exposed to the Pa59 prion strain, at the larval stage. Actin × 51D and Elav × 51D (both referred to as 51D in appropriate graph) Drosophila were used as control flies where appropriate. Head homogenates were prepared from adult flies aged 30 days and inoculated into tg338 mice. Inoculated mice were euthanized when they showed clinical signs of prion infection or after 250 days for those that did not develop clinical disease. Mice were considered positive for prion disease when PK-resistant PrP27–30 was detected in brain tissue by western blot. Mouse brains were analysed for the presence of PK-resistant PrP and subjected to neuropathological assessment by PET blot and lesion profile analysis. (A) Western blot detection of PK-resistant PrP27–30 in the brains of tg338 mice during the isolation of PG127, Pa59 or Apl338, or prion strains (original inoculum), or these prion strains after passage in Drosophila (Drosophila passaged). Molecular mass markers in kDa are shown on the left. (B) PET blot and lesion profile analysis of tg338 mouse brains after exposure to the original mouse-derived PG127, Pa59 or Apl338, or prion strains (original inoculum), or these prion strains after passage in Drosophila (Drosophila passaged). Scale bar = 150 µm.
Discussion
In our study presented here we have demonstrated that scrapie-exposed Drosophila transgenic for ovine PrP expressed pan neuronally displayed a progressive accumulation of prion seeding activity and showed the presence of PK-resistant PrP. Crucially, we also demonstrated a progressive accumulation of prion infectivity in these Drosophila by bioassay in tg338 mice. These features unequivocally demonstrate for the first time the replication of bona fide mammalian prions in a PrP transgenic invertebrate host.
The central role of PrP in prion replication has been verified by infectivity studies in mammalian hosts, principally mice, with either gene knock-out or transgenic over-expression of endogenous prion protein expression (Brandner and Jaunmuktane, 2017). These extensive studies have collectively established that mammalian prions do not replicate in hosts that fail to express PrPC. However, several lines of evidence have suggested that expression of PrPC alone is not sufficient to confer susceptibility to prion propagation (Raeber et al., 1999; Enari et al., 2001; Courageot et al., 2008). This has led to the suggestion that specific molecular co-factors and/or cellular machinery are crucial for prion propagation. The nature and function of co-factors in prion propagation remains undefined (Ma, 2012), although their role is highlighted by in vitro reconstitution experiments. To date, RNA or lipid has been shown to be required for the generation of prions with high infectious titre (Deleault et al., 2003; Wang et al., 2010; Deleault et al., 2012). However, it is possible that other molecular components participate in the process of prion replication and the definitive list of these co-factors and their mode of action remain to be established (Castilla et al., 2008). Insects are invertebrate hosts that are phylogenetically separated in evolutionary terms from mammals by millions of years. We have demonstrated here that Drosophila, a normally PrP-null insect host, rendered transgenic solely for mammalian PrP, are permissive for the faithful replication of a panel of three mammalian prion strains. This provides a cogent argument that the nature of the molecular and cellular factors that facilitate prion replication do not result from a co-evolutionary selection process between the infectious agent and their natural hosts.
An intriguing feature in mammalian prion biology is the existence of prion strains. These are identified as different prion isolates from individuals of the same species that produce distinct disease phenotypes upon serial passage in isogenic hosts (Bruce and Dickinson, 1987). Glycosylation is an important factor in the determination and maintenance of conformation, function and interactions of glycoproteins (O’Connor and Imperiali, 1996). Mammalian PrP expressed in the natural host has two N-linked carbohydrate moieties attached to asparagine residues located in the C-terminal domain of the protein (Endo et al., 1989). Transmission experiments carried out in mice transgenic for PrP devoid of either one or both N-linked carbohydrate moieties, as a result of changes to the prion protein primary structure, indicated an alteration in the biological properties of some of the prion strains tested (Cancellotti et al., 2013). These results supported the view that N-linked glycosylation of PrPC could participate in the transfer and/or maintenance of the prion strain-specific information. Mammalian N-linked glycoproteins, including those attached to PrPC expressed in the natural host, typically comprise complex glycans that consist of N-acetylglucosamine (GlcNAc), mannose, galactose, and terminal sialic acid residues (Endo et al., 1989). In contrast, the majority of neurons in the Drosophila brain synthesize N-linked glycans with core structures similar or identical to those produced by all eukaryotes but fail to acquire complex carbohydrate structures (März et al., 1995). Our study here demonstrates that the biological properties of distinct ovine prion strains were maintained after propagation in PrP transgenic Drosophila. Therefore, differences in N-linked glycosylation moieties between mammalian and insect species had no apparent impact on the biological properties of the different prion strains that we propagated in PrP transgenic Drosophila.
We have shown that VRQ Drosophila exposed to scrapie prions at the larval stage show an accelerated decline in locomotor ability at adulthood (Thackray et al., 2012b, 2014, 2016). The severity of this prion-induced toxic fly phenotype increased as the flies aged and correlates with the accumulation of prion seeding activity and infectivity in PrP transgenic Drosophila. In mammalian species clinical signs in prion affected individuals arise as a consequence of neurodegenerative processes initiated by the conversion of PrPC to PrPSc. Only cells that express PrPC are susceptible to prion-induced toxicity and formation of the toxic moiety occurs concomitantly with prion replication (Bueler et al., 1993; Mallucci et al., 2003). Despite significant advances (Moreno et al., 2012), the mechanisms responsible for neurodegeneration in prion disease and the role of PrPC and PrPSc remain incompletely defined. Unravelling events that are directly triggered by prion protein conversion from those arising as a consequence of the perturbation of the CNS micro-environment is extremely challenging. PrP was expressed pan neuronally in the VRQ PrP transgenic fly line used to assess prion seeding activity and prion infectivity in the scrapie-infected Drosophila we have used here. We have previously shown that a prion-induced toxic phenotype occurs in Drosophila transgenic for pan neuronal expression of PrP (Thackray et al., 2014, 2016). Consequently, we consider that the prion-induced decline of the locomotor activity observed in PrP transgenic Drosophila reflects a neurotoxic phenotype that arises as a consequence of prion replication. This supports the view that PrP transgenic Drosophila can serve as a tractable system to probe for genetic modifiers of prion-induced neurodegeneration. Drosophila are increasingly used as a model system in the study of mammalian neurodegenerative disease including the pathobiology of Alzheimer’s disease, Parkinson’s disease and tauopathies (Lu and Vogel, 2009). This arises because the brains of Drosophila and mammalian species are composed of similar components (i.e. neurons and neuronal circuitry), and the nature of ion channels, neurotransmitters and synaptic proteins are highly conserved between mammals and the fly (Lessing and Bonini, 2009). Our use of PrP transgenic Drosophila to recapitulate mammalian prion infection provides an unprecedented opportunity to unravel the mechanisms of prion-induced neurotoxicity in a new animal system. Drosophila have already proved to be an important animal model to analyse the genetics of mammalian neurodegeneration. For example, resultant behavioural defects in Drosophila that arise through the deletion of a fly homologue of a human neurodegenerative gene can be compensated for by a functional human homologous gene (Luo et al., 1992). In addition, mutagenesis screens in Drosophila have led to the isolation of flies that show late-onset progressive degeneration of the adult nervous system that resembles human diseases (Min and Benzer, 1999).
Prion bioassays, which are generally carried out in conventional or transgenic mice, or primates or small ruminants, continue to provide a central role in the study of mammalian prion diseases. However, because of ethical and economic reasons, such as the cost of maintaining the vertebrate species during long bioassays, there is increasing pressure to limit their use and adopt alternative approaches to assess mammalian prion infectivity. Over the last decade, in vivo prion biology studies have been supplemented by in vitro techniques such as PMCA (Saa et al., 2005) and quaking-induced conversion (QuIC) (Atarashi et al., 2007). Both of these methodologies have been used for the detection of prion seeding activity, a biochemically and biologically relevant surrogate marker of prion infectivity. In this context, PMCA and QuIC provide rapid and sensitive quantitative detection of prion-infected samples. However, in vitro amplification methods remain refractory to many questions and experimental needs that require to be addressed in prion research. These methods are also obviously not adapted for investigating prion-induced neurodegeneration. Drosophila have several important positive experimental advantages as an animal model to study neurodegenerative disease. Large numbers of flies can be produced in a short time, which correlated with their relatively short life span and simple genetics, allow rapid, statistically robust data collection. The ability of Drosophila to faithfully propagate mammalian prion strains has multiple potential applications. In relation to prion diagnostics, our Drosophila-based prion bioassay has already been shown to be capable of the sensitive detection of prion infectivity in brain tissue (Thackray et al., 2012b, 2016) and blood (Thackray et al., 2016). PrP transgenic Drosophila could also help to address the need for new animal models that allow a relevant, rapid, robust and reasonably high throughput screening of therapeutic compounds against prion disease.
In our studies presented here we have demonstrated that core features of transmissible mammalian prion disease can be recapitulated in PrP transgenic Drosophila. This opens a new era for the investigation of prion-induced neurotoxicity and prion replication mechanisms in a genetically well-defined tractable animal system. This new invertebrate model of mammalian prion disease will provide the opportunity to exploit the power of genetics in the fly to identify potential genetic modifiers of prion-induced neurotoxicity. Such genetic modifiers may serve as candidate diagnostic markers or therapeutic targets of human prion disease and prion-like diseases. In addition, the ease of transgenesis in Drosophila will allow the development of fly lines that express different species forms of PrP, such as bovine, cervid and human PrP to bioassay prion infectivity from these different mammalian hosts. For appropriate biosecurity, these future studies will require the BSE, chronic wasting disease (CWD) or CJD-inoculated Drosophila to be maintained at containment level 3 for prion research, an enhanced level of safety above the containment level 2 used for the sheep scrapie studies described here. In this manner, PrP transgenic Drosophila can be used to begin to address important questions on the pathogenic potential of known and possible zoonotic prions.
Funding
This work was supported in part by funds from the NC3Rs Project (Grant NC/K000462/1).
Supplementary Material
Glossary
Abbreviations
- ARQ
A136R154Q171 ovine PrP
- GPI
glycosylphosphatidyl inositol
- PMCA
protein misfolding cyclic amplification
- PrPC
cellular prion related protein
- PrPSc
disease-associated conformers of prion related protein
- VRQ
V136R154Q171 ovine PrP
References
- Andreoletti O, Orge L, Benestad SL, Beringue V, Litaise C, Simon S et al. . Atypical/Nor98 scrapie infectivity in sheep peripheral tissues. PLoS Pathog 2011; 7: e1001285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA et al. . Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 2007; 4: 645–50. [DOI] [PubMed] [Google Scholar]
- Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol 2000; 74: 5542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol 1994; 68: 7859–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner S, Jaunmuktane Z. Prion disease: experimental models and reality. Acta Neuropathol 2017; 133: 197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce ME, Dickinson AG. Biological evidence that scrapie agent has an independent genome. J Gen Virol 1987; 68 (Pt 1): 79–89. [DOI] [PubMed] [Google Scholar]
- Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A et al. . Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997; 389: 498–501. [DOI] [PubMed] [Google Scholar]
- Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M et al. . Mice devoid of PrP are resistant to scrapie. Cell 1993; 73: 1339–47. [DOI] [PubMed] [Google Scholar]
- Cancellotti E, Mahal SP, Somerville R, Diack A, Brown D, Piccardo P et al. . Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 2013; 32: 756–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla J, Morales R, Saa P, Barria M, Gambetti P, Soto C. Cell-free propagation of prion strains. EMBO J 2008; 27: 2557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Jeon YC, Lee DW, Oh JM, Lee HP, Jeong BH et al. . A Drosophila model of GSS syndrome suggests defects in active zones are responsible for pathogenesis of GSS syndrome. Hum Mol Genet 2010; 19: 4474–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016; 539: 217–26. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science 2007; 318: 930–6. [DOI] [PubMed] [Google Scholar]
- Courageot MP, Daude N, Nonno R, Paquet S, Di Bari MA, Le Dur A et al. . A cell line infectible by prion strains from different species. J Gen Virol 2008; 89: 341–7. [DOI] [PubMed] [Google Scholar]
- Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature 2003; 425: 717–20. [DOI] [PubMed] [Google Scholar]
- Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J et al. . Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci USA 2012; 109: E1938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci USA 2001; 98: 9295–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Groth D, Prusiner SB, Kobata A. Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 1989; 28: 8380–8. [DOI] [PubMed] [Google Scholar]
- Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C et al. . Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 2005; 280: 11247–58. [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Cole S, Walker CA. Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J Gen Virol 1987; 68 (Pt 7): 1875–81. [DOI] [PubMed] [Google Scholar]
- Lacroux C, Vilette D, Fernandez-Borges N, Litaise C, Lugan S, Morel N et al. . Prionemia and leukocyte-platelet-associated infectivity in sheep transmissible spongiform encephalopathy models. J Virol 2012; 86: 2056–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Dur A, Beringue V, Andreoletti O, Reine F, Lai TL, Baron T et al. . A newly identified type of scrapie agent can naturally infect sheep with resistant PrP genotypes. Proc Natl Acad Sci USA 2005; 102: 16031–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessing D, Bonini NM. Maintaining the brain: insight into human neurodegeneration from Drosophila melanogaster mutants. Nat Rev Genet 2009; 10: 359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Vogel H. Drosophila models of neurodegenerative diseases. Annu Rev Pathol 2009; 4: 315–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron 1992; 9: 595–605. [DOI] [PubMed] [Google Scholar]
- Ma J. The role of cofactors in prion propagation and infectivity. PLoS Pathog 2012; 8: e1002589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003; 302: 871–4. [DOI] [PubMed] [Google Scholar]
- März L, Altmann F, Staudacher E, Kubelka V. Protein glycosylation in insects. In: Montreuil J, Schachter H, Vliegenthart JFG, editors. Glycoproteins. Amsterdam, The Netherlands: Elsevier; 1995. p. 543–63. [Google Scholar]
- Min KT, Benzer S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science 1999; 284: 1985–8. [DOI] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG et al. . Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 2012; 485: 507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor SE, Imperiali B. Modulation of protein structure and function by asparagine-linked glycosylation. Chem Biol 1996; 3: 803–12. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–44. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prion biology and diseases, 2nd edn. New York, NY: Cold Spring Harbor Laboratory Press; 2004. [Google Scholar]
- Raeber AJ, Sailer A, Hegyi I, Klein MA, Rulicke T, Fischer M et al. . Ectopic expression of prion protein (PrP) in T lymphocytes or hepatocytes of PrP knockout mice is insufficient to sustain prion replication. Proc Natl Acad Sci USA 1999; 96: 3987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Milla E, Oidtmann B, Panagiotidis CH, Baier M, Sklaviadis T, Hoffmann R et al. . Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J 2006; 20: 317–19. [DOI] [PubMed] [Google Scholar]
- Saa P, Castilla J, Soto C. Cyclic amplification of protein misfolding and aggregation. Methods Mol Biol 2005; 299: 53–65. [DOI] [PubMed] [Google Scholar]
- Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411: 810–13. [DOI] [PubMed] [Google Scholar]
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M et al. . Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 1998; 4: 1157–65. [DOI] [PubMed] [Google Scholar]
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987; 51: 229–40. [DOI] [PubMed] [Google Scholar]
- Supattapone S. Synthesis of high titer infectious prions with cofactor molecules. J Biol Chem 2014; 289: 19850–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R et al. . Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 1996; 274: 2079–82. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Andreoletti O, Bujdoso R. Bioassay of prion-infected blood plasma in PrP transgenic Drosophila. Biochem J 2016; 473: 4399–412. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Di Y, Zhang C, Wolf H, Pradl L, Vorberg I et al. . Prion-induced and spontaneous formation of transmissible toxicity in PrP transgenic Drosophila. Biochem J 2014; 463: 31–40. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Hopkins L, Lockey R, Spiropoulos J, Bujdoso R. Emergence of multiple prion strains from single isolates of ovine scrapie. J Gen Virol 2011; 92: 1482–91. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Hopkins L, Lockey R, Spiropoulos J, Bujdoso R. Propagation of ovine prions from “poor” transmitter scrapie isolates in ovine PrP transgenic mice. Exp Mol Pathol 2012a; 92: 167–74. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Hopkins L, Spiropoulos J, Bujdoso R. Molecular and transmission characteristics of primary-passaged ovine scrapie isolates in conventional and ovine PrP transgenic mice. J Virol 2008; 82: 11197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray AM, Muhammad F, Zhang C, Denyer M, Spiropoulos J, Crowther DC et al. . Prion-induced toxicity in PrP transgenic Drosophila. Exp Mol Pathol 2012b; 92: 194–201. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Muhammad F, Zhang C, Di Y, Jahn TR, Landgraf M et al. . Ovine PrP transgenic Drosophila show reduced locomotor activity and decreased survival. Biochem J 2012c; 444: 487–95. [DOI] [PubMed] [Google Scholar]
- Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010; 327: 1132–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary material.