

Abstract
In desmoplastic melanoma, tumor cells and tumor-associated fibroblasts are the major dominators playing a critical role in the fibrosis morphology as well as the immunosuppressive tumor microenvironment (TME), compromising the efficacy of therapeutic options. To overcome this therapeutic hurdle, we developed an innovative chemo-immuno strategy based on targeted delivery of mitoxantrone (MIT) and celastrol (CEL), two potent medicines screened and selected with the best anti-cancer and anti-fibrosis potentials. Importantly, CEL worked in synergy with MIT to induce immunogenic tumor cell death. Here, we show that when effectively co-delivered to the tumor site at their optimal ratio by a TME-responsive nanocarrier, the 5:1 combination of MIT and CEL significantly triggered immunogenic tumor apoptosis and recovered tumor antigen recognition, thus eliciting overall anti-tumor immunity. Furthermore, the strong synergy benefitted the host in reduced drug exposure and side-effects. Collectively, the nanocarrier-mediated chemo-immuno therapy successfully remodeled fibrotic and immunosuppressive TME, arrested cancer progression, and further inhibited tumor metastasis to major organs. The affected tumors remained dormant long after dosing stopped, resulting in a prolonged progression-free survival and sustained immune surveillance of the host bearing desmoplastic melanoma.
Keywords: desmoplastic melanoma, chemo-immuno therapy, mitoxantrone, celastrol, immunogenic cell death, tumor microenvironment, tumor dormancy
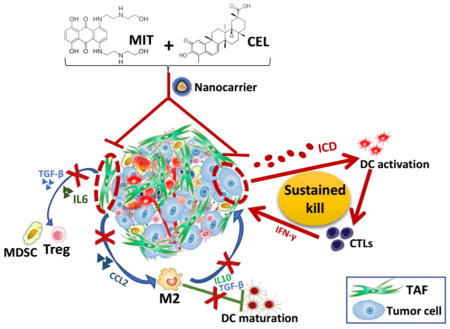
Graphical Abstract
Figure legend depicting nanocarrier-mediated chemo-immuno therapy by co-delivering MIT and CEL. MIT, mitoxantrone; CEL, celastrol; TAF, tumor associated fibroblast; ICD, immunogenic cell death; DC, dendritic cell; CTL, cytotoxic T lymphocyte; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cell; M2, M2 macrophage.
Melanoma, the most lethal skin cancer, has an incremental incidence, few durable therapies and a low survival rate of less than 10 %.1 Conventional melanoma treatment, whether radiotherapy or chemotherapy, presented a rather short therapeutic window and a high incidence of recurrence/metastasis.2 The major drawbacks of such therapies are that the tumor-specific memory response is insufficiently evoked and that the lack of specificity results in side effects to the whole-body immunity.3 Notably, the tumor microenvironment (TME) has been poorly studied. TME comprises not only tumor cells but also immune and interstitial cells. Thus, an effective therapy should be based upon a specific cancer type and a thorough understanding of its TME.4, 5
In desmoplastic melanoma (DM), a rare histological variant of melanoma, the malignant tumor cells are surrounded by rich fibrous tissues.6 This highly fibrotic property led to distinct clinical behavior when compared with other melanoma subtypes, thus hindering treatment efficacy.7 The interstitial cells, especially tumor-associated fibroblasts (TAFs) construct an extracellular matrix-rich structure and cytokine crosstalk, thus facilitating aggressive and highly metastatic tumor growth. Moreover, the fibrosis raises delivery barriers for effective therapies.8, 9, 10 In this work, we grafted murine models of DM mimicking clinical settings,11, 12 and further confirmed tumor cells and TAFs as major dominators within TME. Such domination resulted in the recruitment of immune cells - especially myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs) and tumor-associated macrophages - that collectively form the suppressive immune microenvironment.13 Consequentially, the highly immunosuppressive TME supports “tumor immunoediting”, thus inducing the tumor progression and furthering the drug-resistancy.14, 15
The field of onco-immunology recently validates that conventional cancer therapies may achieve a sustained patients’ outcome by arising innate and adaptive immunity against tumor growth.16, 17 For such purposes, specialized chemo-drugs which empowering immunogenic cell death (ICD) and immune-stimulatory side effects have been employed.18, 19 Such “Chemo-immuno therapy” offers alternative therapeutic options for conventional drugs. The common mechanism of inducing ICD by reported chemo-drugs (mitoxantrone, doxorubicin, oxaliplatin, bortezomib) involves the induction of endoplasmic reticulum (ER) stress,20 but their potency requires enhancement. Thus, in addition to targeted depletion of the dominators (tumor cell and TAFs) within the TME and more importantly, to effectively trigger ICD in DM, we screened drugs that could work synergistically. Our hypothesis was that the synergy will significantly reduce the effective drug dose if the drugs could be delivered together at an optimal dose ratio. We further hypothesized that improved ICD could induce a long lasting anti-tumor immunity which would elicit prolonged progression free survival of the host. To achieve the highest anti-cancer efficacy, we used a nanoparticle (NP) delivery system that specifically targets and triggers drug-release in order to deliver sufficient drugs with high concentrations and with the synergistically optimal combination ratio at the tumor site. This chemo-immuno therapy strategy holds promise to prime robust innate and adaptive immune responses, arrest cancer progression and induce tumor dormancy. (see Graphical Table of Contents).
RESULTS AND DISCUSSION
The design of chemo-immuno therapy
DM presents a highly fibrotic TME in comparison to non-desmoplastic subtypes. Figure 1A depicts a typical collagen-rich morphology (predominantly produced by TAFs) in DM, compared with relatively non-desmoplastic subtype, in both human patients and preclinical mouse models. Importantly, the metastatic rate of clinical primary melanoma significantly increased from 44.56 % (non-desmoplastic) to 50.45 % (desmoplastic subtypes). Among which, lung and distant lymph nodes are the major identified metastasis loci (analyzed from The Cancer Genome Atlas, data not shown).
Figure 1. The design of chemo-immuno therapy.

(A) Masson’s trichrome staining illustrating desmoplastic type of melanoma with collagen-rich TME (blue-stained collagen highlighted with black arrows), compared with relatively non-desmoplastic samples from patients and mouse tumor specimens. Scale bar indicates 300 μm. (B) Flow cytometry analysis showing the major cell populations within TME. (Tumor cells: MART1+; TAFs: FAP+). (C) A total of 25 candidate drugs were screened by MTT assay. MIT and CEL were selected among lowest IC50. n = 5. (D) IC50 and CI of MIT, CEL, and optimized ratios of MIT+CEL on both BPD6 cells (desmoplastic melanoma cells) and 3T3-T cells (TGF-β activated fibroblast, mimicking TAFs in vivo). 24 h incubation. n = 5–8. (E) ICD induced by MIT and MIT+CEL (combination ratio of 5:1) on tumor cells. For MIT: 16 μM, for MIT+CEL: 4.5 μM of MIT and 0.9 μM of CEL. Fluorescence imaging detecting ICD markers: CRT and HMGB1. Cell nuclei were stained with Hoechst 33432. Scale bar indicates 10 μm. n = 3.
In study of desmoplastic TME, we performed a whole tumor profiling assay (Figure 1B, using flow cytometry) on the murine tumor model, where the presence of ~23 % tumor cells and ~17 % TAFs were found. To target depletion of these two dominators of tumor mass, as well as inducing tumor ICD, we designed a cancer-specific “chemo-immuno therapy”. A total of 25 candidate drugs were screened by MTT assay in vitro. Among these were 4 well-reported ICD-inducing chemo-drugs and 21 active compounds from traditional Chinese medicine extracts (for full drug list, see Materials section). As illustrated in Figure 1C, among 25 candidates, mitoxantrone (MIT) and celastrol (CEL) were the most two effective drugs to treat both the tumor cell line (BPD6) and a model TAF cell line (TGF-β activated NIH 3T3 cells, referred to as the “3T3-T”).
MIT, clinically used in treating malignant melanoma,21, 22 can trigger ICD in various cancer types.23 CEL, a pentacyclic triterpene extracted from Tripterygium wilfordii, which is reportedly an immune-stimulatory and anti-cancer agent,24, 25 has also been identified as an effective chaperone inhibitor thus would theoretically facilitate ICD.26 In this work, CEL further elicited potent anti-tumor and anti-fibrosis potentials in the murine DM model (Figure 1D). Active compounds from extracts have been reported to increase efficacy of chemo-drugs as well as to decrease toxicity in various anti-cancer studies.27, 28 Thus, we explored the possible synergy between these two drugs.
As shown in Figure 1D, various combinations of these two drugs were examined for their half maximal inhibitory concentration (IC50) and the combination index (CI). In both tumor and TAF cell lines, significantly lower IC50 of MIT was achieved after combining MIT with CEL, indicating that CEL increased the sensitivity of cells to MIT. For example, IC50 of MIT decreased from 16.0 to 4.5 μM in BPD6 cells and from 30.8 to 1.5 μM in 3T3-T cells, respectively, when tested in a drug ratio of 5:1 (marked in red). IC50 values for CEL were similarly decreased in both cell lines. We have also examined the synergistic effect29 among 7 combination ratios of MIT to CEL (1:10, 1:5, 1:2, 1:1, 10:1, 5:1, and 2:1). Among these, 5:1 and 10:1 showed a strong synergy. The strongest CIs were 0.2 in TAF cell line and 0.6 in tumor cell line. Thus, CEL enables enhanced cytotoxicity of MIT in a best combination ratio of 5:1 (MIT: CEL=5:1, referred to as MIT+CEL 5:1). Such a high therapeutic efficacy achieved at low-doses would allow the reduction of side-effects and satisfies the prerequisite of ICD-inducing chemo-drugs which should be used in a low-dose range.
The application of CEL in anti-cancer therapies has elucidated its role in calcium-mediated activation of ER stress.30 When calcium is released from ER and mitochondria, un/mis-folded proteins accumulate within the ER. The impaired activities of chaperones (e.g., Hsp90) and disabled processing of proteins further promote proteasome function, thus initiating cell apoptosis and release of tumor antigens. Thus, CEL as an effective Hsp90 inhibitor, has demonstrated its great potential in facilitating ICD. Upon stress and apoptosis, calreticulin (CRT) translocated from ER lumen to the surface of cancer cells, with increased release of high mobility group box 1 (HMGB1).18, 31 With CRT exposure and HMGB1 release as standard markers for drug-induced tumor cell immunogenicity,32 we confirmed that both MIT itself and MIT+CEL 5:1 effectively induced ICD in tumor cell line (Figure 1E), indicating the potential for synergistic chemo-immuno therapy.
The TME-responsive NP delivery platform
Despite the advantages and great promise of combination drugs, obstacles aligned between the ideal optimal synergistic ratio and the ratio at the tumor sites. Conventional drugs distributed all over the body, with only a minor portion insufficiently delivered to the tumor sites. Thus, we applied a TME-responsive NP platform in purpose to enhance precisely-targeted and fast-released delivery of drugs, and subsequently to induce ICD onsite.
To construct such a NP delivery platform as aforementioned, the APS (AEAA-Polymer-Disulfide bond) NPs were synthesized by Michael addition polymerization (Figure 2A), based on the synthesis scheme of disulfide linkages in (β-amino ester) copolymers that was previously reported.33 In addition, we synthesized and added aminoethylanisamide (AEAA) ligand to the copolymers for targeting purposes. AEAA is a high-affinity (Kd = 9 nM) ligand for sigma receptors which are over-expressed on both melanoma and TAFs.34 The signal peaks in Figure 2A (1H NMR spectrum) represent functional groups such as PEG-AEAA and tertiary amine, indicating a successful synthesis of the APS copolymer. The analysis of characteristic peaks is described in Materials section.
Figure 2. The TME-responsive NP delivery platform.

(A) Synthesis scheme and 1H-NMR spectrum result of copolymer. (B) Graphical structure and composition of APS NP. (C) Measurement of NP size and TEM image. Scale bar indicates 500 nm. (D) The in vitro MIT release from NPs in changing pH and glutathione (GSH) conditions. n = 4. (E) NP hemolysis assay. n =4.
Based on the optimized combination ratio as described above, MIT and CEL were co-loaded (5:1, molar ratio) into APS NPs with a solvent evaporation procedure. Its illustrative feature (Figure 2B), TEM morphology image and NP size distribution (Figure 2C) are shown alongside. Essential characterizations of drug-loaded NP and NP itself, including nanoparticle size, zeta potential, polydispersity index, drug loading efficacy and drug encapsulation efficiency are listed in Figure S1A. Importantly, the zeta potential was reversed from a negative charge (pH 7.4) to a positive charge (pH 6.5) due to tertiary amines protonated in the copolymer. In blood (pH ~7.4), the negative charge of APS NPs can be leveraged to reduce rapid NP clearance in the circulation without interacting with blood components which are mostly also negatively-charged. Due to acidic microenvironment (pH ~6.5) in solid tumors, APS NPs triggered a charge-reversal process thus attracting their combination with negatively-charged cell membranes. In turn, this charge-reversal effect enables NPs to be internalized at the TME and minimizes their distribution among normal tissues.35, 36 These features offered excellent possibility for inducing local ICD at the tumor sites.
An effective drug delivery system allows rapid drug release inside the cancer cell.37, 38 To promptly achieve the optimal drug ratio, as well as improve anti-cancer efficacy, TME-specific glutathione (GSH) property has been employed. The concentration of GSH in the cytosol of cancer cells (~2–10 mM) was significantly higher than that of extracellular matrix and blood (~2–20 μM).39, 40, 41 Furthermore, GSH level in cancer cells are over 4-fold higher than normal cells.42 To confirm such TME-responsive property, NPs were tested in changing pH and GSH conditions. As shown in Figure S1B, the particle sizes dramatically increased in the presence of 10 mM GSH compared with no GSH condition, possibly due to disulfide-bond breakage in a reductive manner.43 A pH change from 7.4 to 6.5 further facilitated such change. Such results were also confirmed by the gel permeation chromatography assay (GPC) measuring the molecular weight distribution of copolymer in respond to pH and GSH changes (Figure S1C, for assay details see Materials and methods section). As shown in the GPC assay, the nano-carrier (copolymer) with lower PDI helped in maintaining its stability and self-assembly properties, in similar to reports from other copolymer systems.44, 45, 46, 47, 48 Importantly, the nano-carrier was unstable in either acidic or reductive conditions. Both are favorable properties for anti-cancer drug delivery.49
As a result, MIT (Figure 2D) and CEL (Figure S2A) were being triggered to release from NPs. In the condition of pH 7.4, MIT and CEL loaded NPs released less than 20 % in 24 h. However, at pH 6.5 (simulating the acidic tumor extracellular microenvironment), the drug release slightly increased to ~40 % due to ionization of tertiary amines. The slightly acidic environment led to NP leakage but did not collapse the NPs. Moreover, in the addition of 10 mM GSH (which simulated the reductive environment inside the cancer cells), the release increased to ~60 % due to the rupture of disulfide linkage, indicating that the core of NPs had collapsed. A combination effect was observed in pH 6.5 and 10 mM GSH condition, where more than 80 % of drugs released. Such a high releasing property would help achieving the optimal ratio for drug-induced ICD at the tumor sites. Similarly, previous studies using polymeric micelles or liposomes have also achieved a high MIT encapsulation efficacy, and also drug loading efficacy with reasonable stabilities and drug release behaviors.50, 51, 52, 53
To mimic the blood physiological conditions, APS NPs were added to 10 % fetal bovine serum (FBS), PBS, blood serum, blood plasma, and whole blood in vitro for 72 h. No significant change of size was found, suggesting the good stability of NP (Figure S2B). To further evaluate the biocompatibility, a hemolysis assay was employed investigating the interaction between NPs and red blood cells (RBCs). As shown in Figures 2E and Figure S2C, 1 % Triton X-100 was used as a positive control which had roughly a 100 % hemolysis rate. At pH 7.4, four concentrations of NPs (0.1, 0.5, 1, and 2 mg/mL) were observed with less than 10 % hemolysis. However, the hemolysis rate increased at pH 6.5 and 5.0; especially in maximum conditions (1 or 2 mg/mL of NP at pH 5.0), the hemolysis rate was close to 100 %, indicating strong interactions between NPs and RBCs. These were similar to report from Kelsch’s group on polymeric micelles’ stability.44 The anti-blood dilution property can also be evaluated by the critical micelles concentration (CMC) assay. As shown in Figure S2D, the nano-carrier itself has shown a rather low CMC of ~7.5 × 10−4 mg/mL (~7.76 × 10−8 M), indicating reasonable anti-dilution stability within bloodstream. Such property would further facilitate circulation within body and improve targeting efficacy. Importantly, blank NP itself showed no significant cytotoxicity in tumor cells, TAFs, normal fibroblasts, nor immune cell such as macrophages (Figure S2E). To conclude, the NP delivery platform responded in accordance to TME-mimicking factors and showed a high biocompatibility profile.
Nanocarrier-mediated chemo-immuno therapy significantly improved anti-tumor response and remodeled suppressive TME
To achieve sufficient efficacy, we first examined the delivery of MIT and CEL in TME-responsive NP among DM tumor-bearing mice. Through IVIS imaging, NPs were detected mainly distributed in the tumor 24 h after i.v. injection (Figure 3A), compared with other major organs. Although liver took up NPs, it was significantly lower than the tumors on the per gram weight basis (p < 0.01) (Figure 3B). With the help of sigma receptor-targeting, we found that most of the NPs were taken up by tumor cells and TAFs within the TME (measured by flow cytometry, data not shown).
Figure 3. Effective therapy significantly improved anti-tumor response and remodeled suppressive TME.

(A) NP distribution in tumor-bearing mice. Mice were i.v. injected with Cy5-loaded (3 μg/kg) NPs and measured by IVIS imaging 24 h post-injection. n = 3; (B) Region-of-interest intensities of fluorescence signals among tumor and organs. n = 3; (C) Pharmaco-distribution of MIT and CEL within tumor were measured by LC/MS, n = 5; (D) Tumor inhibition study and tumor weight comparison. Arrows indicate days of drug injection. Dosage: for M+C group: ~2 mg/kg of CEL per dose; for M+C NP group: ~160 μg/kg of CEL per dose. Tumors were surgically removed from the hosts at endpoint of study, weighted and compared between groups. n = 10–12; (E) Cell apoptosis were measured by TUNEL staining and collagen morphology changes were measured by Masson’s trichrome staining, scale bar indicates 300 μm. n = 3. (F) Flow cytometry analysis of immune functioning cells within TME. n = 3. *: p < 0.05, **: p < 0. 01, ***: p < 0. 001.
Furthermore, pharmaco-distribution profiles of MIT and CEL were characterized after i.v. administration of drug-loaded NPs and free drug suspension, respectively. As shown in Figure 3C, the injected NPs offered targeted delivery of MIT and CEL to the tumor for a significantly increased amount compared with injected free drugs (measured by LC/MS). Within the tumor, the area-under-curve values of nano-delivered drugs were significantly higher than those of the free drugs (2.9-fold increase in MIT and 3.8-fold increase in CEL). Importantly, the amounts of drugs in the tumor resembled the designed optimal ratio in vitro at least in the early time points (at 4 h, ratio was 6.1:1; 8 h was 5.0:1; 12 h was 4.0:1, 24 h was 3.1:1). In comparison, the free drugs failed to retain such ratio (7.1:1; 13.9:1; 9.9:1; 8.9:1, respective to 4 measured time points). To add on, the plasma concentration-time of drugs were also compared. As shown in Figure S3A, drug-loaded NPs can circulate for a longer time in the blood than free drugs, with 8-fold (for MIT) and 12-fold (for CEL) increase in half-life. These results indicated the benefit of using a controlled-release nanocarrier system.
Through NP encapsulation and delivery, the cytotoxicity of drugs was ~6-fold enhanced in comparison to free drugs in combination (measured by IC50, Figure S3B). Furthermore, on animal model bearing DM tumors (Figure 3D), the mice treated with MIT and CEL loaded NPs (denoted as the M+C NP group) presented a significant tumor-killing effect, with only ~1/13 dosage compared with free drugs administrated in combination (the M+C group). As shown in Figure 3D, the delivery of low-dose drugs largely inhibited tumor growth. Moreover, tumor mass was significantly restrained with lower tumor weight. Such therapeutic strategy also achieved the best anti-cancer effect in comparison to single drug-loaded NP groups (Figure S3C).
Desmoplastic type of tumors is well-known for its immunoediting ability and resistance to immunotherapy. The significant tumor inhibition by delivered drug-combination, also referred to as “the elimination phase” of cancer immunoediting, is mainly resulted from the following factors:
Effective apoptosis induced by MIT and CEL (shown in vivo by TUNEL assay in Figure 3E, and also in vitro by quantitative cell apoptotic assay and cell cycle analysis in Figure S4). In the cell apoptosis assay, M+C NP caused a dramatic increase in the total apoptosis rate (> 60 %) in both tumor cells and TAFs (Figure S4A and C). Enhanced apoptosis could be the result of the G1 phase shift to the G2/M phase in the cell cycle. As shown in Figure S4B and D, M+C NP group also had a higher percentage of G2/M phase cells, suggesting a synergistically blockage of cell mitosis compared with all controls. These data indicated an increased level of cell apoptosis, possibly due to ICD.
Changes of TME morphology that facilitated further delivery of therapeutics. As shown in Figure 3E, collagen deposition and fibrosis were abundant in tumor sections of the untreated group. By contrast, the in vivo tumor model depicted significantly decreased collagen density and increased NP penetration under the treatment of M+C NP (Figure S3D). Collagens were predominantly produced by TAFs, which were the target of both MIT and CEL.
The immune-stimulatory effects that counteracted immune-suppression induced by TME dominators. As shown in Figure 3F and Figure S5, the effective elimination of tumor cells and TAFs dampened the secretion of immune-suppressive TGF-β, IL6, CCL2 and IL-10 network. Thus, suppressive immune cells (such as MDSC, Treg, and tumor associated macrophages) were significantly reduced. Antigen presenting cells within TME, mostly dendritic cells (DCs) were normally inhibited in antigen-recognition functions and were tolerogenic to immune-stimulators under an immune-suppressive environment.54 After treatment, DC functions were largely recovered with CD103+ DC significantly increased, suggesting that the local effective ICD would help release tumor-associated antigens and facilitate DC maturation with cross-priming ability to CD8+ cytotoxic T lymphocytes (CTLs). As a result, IFN-γ was increasingly released to stimulate tumor-specific immunity, thus inhibiting the tumor growth. It is known that perforin and granzymes released by CTLs enable non-apoptotic pathways of cell death, thus offering an effective treatment of cancer by modulating the immune system.55, 56
To examine the observed therapeutic efficacy in other models of desmoplastic melanoma, we established a second DM model using D4M cells (BRAFV600E, syngeneic with C57BL/6 mice).57 This cell line was not PTEN−/− in comparison to BPD6 cell line. As shown in Figure S6A, similar to the BPD6 tumor model, these tumors also presented a desmoplastic morphology, but comparably less in the amount of collagen structure (less TAFs within TME), this explains the compromised efficacy of our therapy in targeting both tumor cells and TAFs. Nevertheless, the NP group achieved a significantly stronger tumor inhibition (p<0.01) in comparison to that of free drugs. Following the same treatment protocols, the M+C NP group mitigated the desmoplastic structure compared with the PBS group, resulting in superior tumor growth inhibition as compared with all other controls (Figure S6B).
Enhancement of long-term immune surveillance, host survival, and anti-metastasis efficacy of therapy
Clinically, a strong cancer immunoediting implies that the “elimination phase” can be hardly completed, thus resulted in a balance between surviving tumor cells and the modified immune system. Such balance may last for years for patients. In our animal model, as the tumor inhibition study continued, we also noticed a long-term sustained tumor inhibition effect in the M+C NP group (Figure 4A). In particular, tumor volumes were restrained around 200 mm3 for over two weeks after the last dose of treatment, with a significant prolongation of progression-free host survival (Figure 4B). A typical “tumor dormancy” phenomenon suggested the existence of endogenous immune surveillance.
Figure 4. Enhancement of long-term immune surveillance, host survival, and memory immunity.

(A) Long term tumor inhibition study. Dosing schedule was consistent for all in vivo studies. Arrows indicate days of drug injection. M+C group: 2 mg/kg of CEL per dose. M+C NP group: 160 μg/kg of CEL per dose. n = 10–12; (B) Long term survival between different treatments. n = 6–8; (C) In M+C NP group, figures depict tumor tissue’s collagen staining at different days after tumor cell inoculation. Scale bar indicates 300 μm. n = 3; (D) Tumor tissue immunostaining analysis of memory immune cells within TME at endpoint day of survival study. n = 3; (E) In M+N NP group, tumor-bearing mice were treated with 3 daily injections of anti-CD8 or anti-CD4 antibody or Isotype control (300 μg/mice i.p. started from 10 days after the last dose of therapy, arrows) to deplete either CD8+ or CD4+ T cells in vivo. n = 3–4. ***: p < 0. 001.
Tumor dormancy can be categorized into two types: cell cycle arrested at G0-1 phase at the cell level, or mostly on a population level represents a balance between tumor cell proliferation and death. Herein, the tumors were restrained as a result of several possible key factors:
A turnover of TME morphology. As depicted in Figure 4C, the density of fibrosis correlatively increased along with tumor growth (from I to II). The TME was heavily desmoplastic at the start of therapy (depicted in II, ~200 mm3 in tumor volume at day 13 after tumor cell inoculation), and then subjected to de-fibrosis treatment by targeted co-delivery of MIT and CEL, aiming at the depletion or deactivation of TAFs (depicted in III, M+C NP group). During over two weeks of host survival observation, the morphological structure of the tumor was largely reframed. At the endpoint of study, an inner necrosis-rich pattern was found in the core area of the residual tumors, along with only a minor level of fibrosis (depicted in IV).
Increased levels of immune-surveilling cells. Local CTLs and natural killer cells are crucial components, but more importantly tumor-specific memory T cells were significantly recruited (Figure 4D). The increased memory CD4+ and CD8+ T cells inside the tumor suggested the effectiveness of ICD in vivo. Reasonably, residual tumor cells may express low but persistent levels of tumor antigens to be recognized and cleared by the immune system. To address such action mechanism, we further depleted CD4+ or CD8+ T cells in mice bearing the dormant tumor. As shown in Figure 4E, the sustained tumor restrain effect was abolished by the treatment with either anti-CD8 or anti-CD4 antibody, whereas it was not affected when an isotype-matched IgG control was used. The result suggested that a strong immune-surveillance might be the main reason for such dormancy.
High expression of CD69 (Figure 5A). CD69 is a T cell activation marker. Recent studies in metastatic melanoma patients have reported that the expression of CD69 positively correlates with survival and negatively corelates with metastasis.58 We found CD69 highly expressed in the remaining cells within the TME, which would help contributing in metastasis inhibition (in both lung and liver) compared with the untreated hosts (Figure 5B).
A high percentage of remaining dormant tumor cells showing the characteristics of cancer stem cells (CSCs) (Figure 5C). Compare with untreated group (PBS control), in M+C NP group, the majority of tumor mass with strong immunogenicity were under immune surveillance and subjected to elimination, but the CSCs are poorly immunogenic with chemo-resistance potential, thus avoiding immune-surveillance and clearance.59 When remaining tumors are examined by flow cytometry, a high percentage of remaining tumors in such group was CSCs. CSCs are not favorable prognostic factor but rather a remaining question that ask for alternative immunotherapeutic options. Nevertheless, CSCs are reported with a slow growth rate and are unable to grow into overt tumor mass.60 Any immunogenic daughter cells from CSCs would possibly be eliminated by immune-surveilling cells within TME, thus present restrained tumor volume.
Figure 5. Long-term tumor dormancy and anti-metastasis efficacy of therapy.

(A) Tumor flow cytometry analysis of CD69+ leukocytes within TME at endpoint day of survival study. n = 3; (B) Tumor metastasis in liver and lung, observed at endpoint day of survival study between PBS and M+C NP group, measured by H&E staining. Scale bar indicates 300 μm; (C) Flow cytometry analysis of CD44+CD133+ tumor cells within TME at endpoint day of survival study. n = 3. *: p < 0.05, ***: p < 0. 001.
Interestingly, we found that two out of ten mice with remaining dormant tumors eventually had their tumors grown back at a late stage (Figure 4B), indicating equilibrium was disturbed in favor of tumor-escape. This suggested the possibilities underlying genetic or epigenetic changes which further allowed tumor progression. Main factors to be investigated in such probabilities can be summarized as follows: a) Due to genomic instability, dormant tumor cells may express undiscovered tumor antigens with specific mutations. Such probability may serve as effective target for the design of neoantigen vaccines; b) The remaining dormant cells may overexpress immune checkpoint ligands, such as PD-L1, gained by gradual increase in resistance. Thus, the checkpoint inhibitors and kinase inhibitors might be applied in combination with targeted therapy; c) At a late stage, dormant tumors would induce MDSC or Treg proliferation as well as their active suppression of immunity; d) A depletion of T cells or reduction in IFN-γ or IL12 would also trigger immune-escape mechanisms; e) An over-activation of angiogenesis would facilitate tumor progression and even metastasis.
Safety evaluation for chemo-immuno therapy
Safety is an essential aspect for the development of both effective and translational therapy. Thus, biosafety-related toxicological pathology analyses were performed. As shown in Figure 6A and B, the serum biochemical parameter analysis and the whole blood cell counts were remained within the normal ranges, this suggested that the treatment strategy led to undetectable systemic anemia nor inflammation. Liver and kidney function markers were remained normal. No severe weight loss in hosts were found (Figure 6C). Moreover, the H&E staining results also indicated no significant morphological damage among major organs, including kidney, lung, spleen, liver, and lung (Figure 6D). Lack of toxicity of the combination chemo-immuno therapy was certainly the direct result of low drug doses required.
Figure 6. Toxicity evaluation of therapies.

(A) and (B) are whole cell counts and serum biochemical marker analysis of tumor bearing mice. Fresh whole blood and serum were collected at endpoint of study. All indicators were among normal biological range. (C) Mice body weight changes under tumor inhibition study. (D) H&E morphology under different therapies. Major organs were collected at endpoint of study and sectioned, stained for H&E analysis. Scale bar indicates 300 μm. n = 5. n.s.: p > 0.05.
CONCLUSIONS
In summary, the success described here stems from the strong synergy between MIT and CEL in inducing ICD. The combination also targets TAFs to reduce the desmoplasia of the tumor. The chemo-immuno therapy significantly remodeled immune-suppressive TME, as well as triggered a robust immune memory response. Since only low doses of both drugs were used, the treatment was without any toxicity to the host. Low dose drug-synergy is also clinically beneficial, especially for patients who suffering from chemo-toxicities, but the optimization of such synergy needs further investigation. In comparison to personalized vaccines, no matter preventive or therapeutic, 61, 62 our strategy offers a broader application to patient subjects in a way of priming immune system against cancer.
MATERIALS AND METHODS
Materials
Mitoxantrone, Doxorubicin, Oxaliplatin, and Bortezomib were obtained from Sigma-Aldrich (MO, USA). Active compounds with over 99% purity, including Dihydroartemisinin, Glycyrrhizin, Curcumin, Tetramethylpyrazine, Resveratrol, Epigallocatechin, Celastrol, Salvianolic acid B, Salvianolic acid A, Scutellarin, Oleanolic acid, Dihydromyricetin, Osthole, Berberine, Cryptotanshinone, Baicalein, Sinomenine, Lupeol, Wogonin, Cepharanthine, and Adiponectin were obtained from Chengdu Mansite Biotech Co. Ltd (Sichuan, China). NH2-PEG-OH (MW 2k, CAS No. 32130-27-1) was obtained from Biochempeg Scientific Inc. (MA, USA). Synthetic materials as previous reported were obtained from Sigma-Aldrich MO, USA).
Cell lines and animals
Desmoplastic melanoma models were built according to previously published protocol.11 In detail, murine melanoma cell lines BPD6 (BRAFV600E, PTEN−/−) and D4M (BRAFV600E, syngeneic with C57BL/6) was kindly provided by Brent Hanks (Duke Cancer Institute) and cultivated in RPMI-1640 Medium added with 10 % FBS and 1 % Penicillin/Streptomycin (PS) (Invitrogen, Carlsbad, CA) at 37 °C and 5 % CO2. NIH-3T3 cell line (which were activated with 10 ng/mL TGF-β mimicking TAFs in vitro) were cultivated in DMEM Medium also with FBS and PS. Six-week-old female C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). All animal regulations and procedures were accepted by Institutional Animal Care and Use Committee of University of North Carolina at Chapel Hill.
Antibodies
InVivoMAb anti-mouse CD8α (clone 53–6.7) and anti-mouse CD4 (clone GK1.5) were purchased from BioXcell (West Lebanon, NH). Primary and secondary antibodies used for immunostainings (IF) and flow cytometry (flow cytr) were listed in Table S1.
ICD determination
ICD dosing was determined by in vitro CRT exposure and HMGB1 release.63 Briefly, BPD6 cells treated with MIT alone or MIT+CEL (5:1 molar ratio), then harvested, PBS washed and fixed in 0.25 % PFA in PBS for 5 min. Cells were washed twice in PBS, and a primary antibody, diluted in cold blocking buffer, was added for 30 min. After three washes in cold PBS, cells were incubated for 30 min with the appropriate FITC-conjugated secondary antibody diluted in a cold blocking buffer. Cells were fixed with 4 % PFA for 20 min and then nuclei were stained with Hoechst 33432 (ThermoFisher Scientific), followed by Confocal imaging. For intracellular HMGB1 staining, cells were washed with PBS, fixed with 0.4 % PFA for 20 min, permeabilized with 0.1 % Triton X-100 for 10 min, rinsed three times with PBS, and nonspecific binding sites were blocked with 10 % goat serum in PBS for 30 min. A primary antibody for HMGB1 was added for 1 h. Subsequently, cells were washed three times with PBS and incubated for 30 min with a secondary antibody for fluorescence imaging.
Synergistic effects of MIT and CEL at various combination ratios
Synergy of MIT in combination with CEL were tested using MTT assays. BPD6 and 3T3-T cells were seeded onto 96-well plates (5×103 cells per well). After overnight incubation, cells in each well were incubated with various concentrations of MIT, CEL, or combinations of MIT and CEL at a molar ratio of 1:10, 1:5, 1:2, 1:1, 2:1, 5:1, or 10:1 (MIT+CEL). After 24 h incubation, cell cytotoxicity was assessed by MTT assay. The synergy of MIT and CEL combination treatment was evaluated by calculating the combination index (CI) based on the level of synergism (median-effect analysis).
Where (Dose)1 and (Dose)2 are the concentrations of drug 1 and drug 2, respectively, in the combination that inhibits x % of cells; (Dose x)1 and (Dose x)2 are the concentrations of drug 1 and drug 2, respectively, as a single drug that inhibits x % of cells. The classificatory of synergy are: additive (CI = 1), synergistic (CI < 1), or antagonistic (CI > 1). IC50 value was calculated using GraphPad Prism software.
Synthesis and characterization of the drug-loaded AEAA-modified NP (APS NP)
The APS copolymers were obtained by previous reported method.33 Briefly, AEAA ligand (71.3 % yield) and AEAA-PEG-2,2-bis(acryloyloxymethyl)propionate (AEAA-PEG-BAP) (91.1 % yield) were first synthesized. Then, all the synthetic materials were dissolved in anhydrous DMSO for Michael-type step-polymerization. The reaction was gently stirred in oil bath (60 °C) for 60 h under nitrogen protection. To obtain the final products of APS NP, the mixture was dropped into excessively cold ethyl ether and dialyzed in a dialysis tube in PBS (p H 7.4) with a molecular weight of MW 5000, followed by freeze-drying (78.4 % yield).
The APS copolymer was confirmed by 1H NMR (at 500 MHz, Bruker, USA) with deuterated chloroform (CDCl3) as the solvent. As shown in Figure 2A, the signals at 3.66 ppm are the characteristic peaks for –OCH2CH2– in PEG-AEAA; the four peaks at 5.31, 3.15, 3.10, and 2.77 ppm correspond to the -NH-CH2-CH2-NH- moieties; the peak at 3.58 ppm is characteristic for the -O-CH3 end group in PEG-AEAA. The signals at 2.69, 1.32, and 1.51 ppm indicate a C–C single bond in 1,6-hexanediol. The signals at 4.01 and 2.48 ppm were generated by -O-CH2- and O-CO-CH2- moieties in the 1,6-hexanediol diacrylate, respectively. The signal at 2.57 ppm was generated by the C–C single bond and the tertiary amine in the piperazine ring.
The molecular weight and polydispersity index of the copolymer in respond to changing pH (pH 6.5, 16h) and GSH (10 mM GSH, 16 h) conditions were measured by a gel permeation chromatography (GPC) equipped with a Waters e2695 HPLC system, a laser light-scattering detector, and a refractive index detector (Malvern Instruments, Worcestershire, UK).
Dimethylformamide (DMF) with 0.1 % v/v Lithium bromide was used as the mobile phase. The block copolymer can be synthesized with controlled molecular weight and PDI. To achieve this, the final polymer was dropped into excessive cold ethyl ether (twice) and dialyzed against DMSO in a dialysis tube with a molecular weight cut off of 5,000 Daltons (for 12 h), thus to remove unreacted oligomers and copolymers with low molecular weight in a controlled manner. The final copolymers with different block compositions were obtained by dialysis against PBS (pH 7.4), followed by freeze-drying.
APS NP were prepared using the solvent evaporation method. In brief, 10 mg of the APS copolymer and MIT+CEL combination (5:1) were dissolved in 1 mL acetonitrile/methanol (1: 1) and gently added to 10 mL PBS (pH 7.4). The residual methanol and acetonitrile were water-bathed (45 °C) for 15 min. The solution was centrifuged at 4,000 rmp (10 min) and filtered by 0.18 μm filter membranes to remove the unloaded MIT and CEL. The Non-drug loaded blank APS NP were prepared by the same process but without loading any cargo. To evaluate the redox-sensitivity and pH-triggered charge reversal, the zeta potential and nanoparticle size were measured DLS (Malvern, United Kingdom) under different conditions: the normal physiological conditions (pH 7.4), the acidic extracellular tumor microenvironmental (pH 6.5), and the intracellular reductive environment (10 mM GSH). The stability of drug-loaded NPs was recorded for 72 h. Drug loading efficiency and encapsulation efficiency of MIT and CEL in NP were quantified using HPLC (Shimadzu LC-20AT, Japan). The morphological examination was detected under JEOL 100CX II TEM (JEOL, Japan). The particle size changes of the APS NP (1 mg/mL) under reductive (10 mM GSH) and/or acidic environments (pH 6.5 or 5.0) were investigated using DLS measurements.
Controlled MIT and CEL release from APS NP in vitro
The release kinetics of MIT and CEL from the NP was investigated in four different mediums using the dialysis tube method. The medium consisted of either 1) pH 7.4, 0.01 M PBS buffer, 2) pH 7.4, 0.01 M PBS buffer with 10 mM GSH, 3) pH 6.5, 0.01 M PBS buffer, or 4) pH 6.5, 0.01 M PBS buffer with 10 mM GSH. Tween 80 (0.5 % w/w) were added to all medium, then 1.0 mL of the MIT+CEL-loaded NPs solutions (0.1 mg/mL) were transferred into dialysis bags (MW 3500) with 30 mL of such medium. The release of drugs in different mediums were measured using HPLC.
Critical micelles concentration (CMC) assay
The CMC of NPs was investigated by the standard pyrene method using a fluorescence probe. Briefly, the acetone solution of pyrene (6.0 × 10−5 M) was added into the test tube and evaporated completely in a 45 °C water bath. Then, different concentrations of NPs were added to each test tube and mixed with a pyrene residue to achieve a final pyrene concentration of 6.0 × 10−7 M. After ultrasonic mixing for 1 h and subsequently equilibrating overnight in the dark (room temperature), a series of mixture solutions were determined (excitation spectra: 300–400 nm, emission: 390 nm). The intensity ratios (I373/I384) were analyzed and plotted against the logarithm of the concentration. The CMC value was the point of intersection of two straight lines.
Biocompatibility assay
The hemolytic activity was measured at pH 7.4, pH 6.5, and pH 5.0 with red blood cells (RBCs). Mice RBCs were extracted from plasma by 2,000 rpm centrifugation for 15 min and two washes. Triton X-100 was used as a positive control. The suspensions of RBCs were added to copolymer samples (0.1 to 2 mg/mL) or Triton X-100, and then compared between groups.
Drug distribution assays
To investigate the distribution of drug-loaded NP in vivo, Cy5-loaded NP (3 μg/kg) was prepared by the same method as previously mentioned.33 Mice were injected with Cy5-loaded APS NP and sacrificed after 24 h. Tumors and major organs were collected accordingly and subject to IVIS® Kinetics Optical System (Perkin Elmer, CA) for imaging and quantifications.
The intra-tumoral cellular uptake of NPs was evaluated using flow cytometer. Briefly, tumor tissues were dissociated with 1 mg/mL collagenase and 200 μg/mL DNAase (Invitrogen) in DMED/2 % FBS for 40 min to generate a single-cell suspension. Major cell populations within TME, such as tumor cells (MART1+), TAFs (αSMA+), macrophages (F4/80+), MDSCs (CD11b+Gr1+), Tregs (CD4+Foxp3+), and DCs (CD11c+) were stained for quantification. The ratios of Cy5-loaded NP distributed in these different cell populations were calculated accordingly.
LC/MS instrument (Shimadzu LCMS-2020, Japan) was also utilized to quantitatively analyze the accumulation of drugs (delivered by APS NP or delivered i.v. as free drug) in the tumor site at predetermined times (4, 8, 12, 24 h) and to study the pharmacokinetics profile. The separation of analyses was by using Thermo Scientific C18 column (100 mm × 4.6 mm, 2.6 μm) with flow rate of 0.2 mL/min and 35 °C column temperature.
Assays on tumor growth, metastasis and host survival
On day 0, mice were inoculated subcutaneously on lower flank with 1×106 BPD6 cells. Tumor growth were measured by digital caliper where volume = 0.5 × length × width × height. Mice were randomized into treatment groups (n = 15–20) when tumor volume reach ~200 mm3. Groups were named as follows: PBS treated control (the PBS group), APS NP with no drug loaded (the blank NP group), mitoxantrone+celastrol (5:1) administrated i.v. (2 mg/kg of celastrol per dose) (the M+C group), mitoxantrone+celastrol (5:1) formulated into APS NP and administrated i.v. (160 μg/kg of CEL per dose) (the M+C NP group), and as single controls, mitoxantrone loaded APS NP (the M NP group), as well as celastrol loaded APS NP (the C NP group). Treatment groups were administrated on day 13, 15, 17, 19 after tumor inoculation. Mice weight and health conditions were monitored every 2 days. Once tumors reached ~20 mm in one dimension, mice were humanely sacrificed. Long-term host survival was monitored and calculated by Kaplan-Meier curves in GraphPad Prism software. At the endpoint of survival monitor, metastasis study was performed as major organs were collected, fixed in 4% PFA, and H&E stained for pathology observation.
Cell cycle assay
Briefly, BPD6 and 3T3-T cells were seeded in 6-well plate (1.5 × 105 cells per well) and incubated for 48 h. Then, cells were treated with either blank APS NPs (100 mg/mL), MIT, CEL, MIT and CEL in combination (the MIT+CEL group), or MIT+CEL-loaded NPs (the MIT+CEL NP group) for 48 h. Cells were then collected, fixed, stained with PI staining solution, and then measured by flow cytometer (BD Biosciences, USA). For each sample, 10,000 events were recorded and compared between groups.
Apoptosis assay
Cell apoptosis assay was performed by Annexin V-FITC/PI double staining. Briefly, cells were seeded in 6-well plate (1.5 × 105 cells per well) and incubated for 24 h. Then, cells were treated with either blank APS NPs (100 mg/mL), MIT, CEL, MIT+CEL, or MIT+CEL NPs for 48 h. Cells were stained with Annexin V-FITC assay kit (Biovision, USA) and then measured by flow cytometer (BD Biosciences, USA). For each sample, 10,000 events were collected.
Flow cytometry assay
The flow cytometry assay mainly characterized the change of immune cells within TME, as previously reported.64 In brief, mice were humanely sacrificed and the whole tumors were collected and incubated at 37 °C for 40–50 min, with the addition of collagenase A and DNAase. After three rounds of PBS washes, single cell suspensions were harvested in MACs buffer (1× PBS+2 mM EDTA+0.5 % BSA, filter sterile), then subjected to fluorescein-conjugated staining. For intracellular staining, penetration buffer (BD, Franklin Lakes, NJ) must be applied before adding antibodies. All stained cells were subject to flow cytometer.
Immunofluorescence staining and Masson trichrome staining
Staining was performed on paraffin-embedded sections from tumor tissues. Briefly, all tissues for paraffin-embedding were resected, rinsed in PBS, and placed in 4 % PFA for over 48 h at 4 °C. Immunofluorescence staining was performed by deparaffinization, antigen retrieval, permeabilization, and blocking in 1 % bovine serum albumin. All antibodies conjugated with fluorophores were added to tissue slides for at least 12 h at 4 °C. Then, nuclei were counterstained with Prolong® Diamond Antifade Mountant with DAPI (ThermoFisher Scientific). Stained slides were imaged with Zeiss 880 Confocal microscopy (Germany). Five randomly microscopic fields were selected and quantified by Image J software. The Masson Trichrome assay was performed to detect collagen among tumor tissue. Tumor slides were stained using a Masson Trichrome Kit by the UNC Tissue Procurement Core.
Quantitative real-time PCR (RT-PCR) assay
Total RNA was extracted from the whole tumor using RNeasy® Microarray Tissue Mini Kit (Qiagen, Hilden, Germany). We then reverse-transcribed cDNA with iScript™ cDNA Synthesis Kit and amplify cDNA with iScript™ Reverse Transcription Supermix for RT-PCR (Bio-Rad, Hercules, CA). RT-PCR primers are listed in Table S2 with specific catalog number attached and are all mouse specific. RT-PCR reactions were run in 7500 Real-Time PCR System and subject to analysis with 7500 Software, compared to and normalized by GAPDH endogenous control.
TUNEL assay
According to DeadEnd Fluorometric TUNEL System (Promega, Madison, WI) instructions, tumor tissue slides were stained and subject to fluorescence microscopy imaging. Fragmented DNA of apoptotic cells (FITC-positive) indicate TUNEL-positive nuclei. Slides were mounted, and nuclei were stained with Prolong® Diamond Antifade Mountant with DAPI (ThermoFisher Scientific), followed by imaging under Confocal microscopy.
Safety of treatments
All mice under different treatments were humanely sacrificed at endpoint of tumor inhibition study, where whole blood and serum of hosts were harvested and subject to test by UNC histology facility. Creatinine, blood urea nitrogen (BUN), serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) indicate renal and liver functions. Red blood cells (RBC), white blood cell (WBC), platelets (PLT), hemoglobin (HGB) and hematocrits (HCT) indicate myelosuppression level. Major organs were collected, H&E stained and compared. Throughout the tumor inhibition study, mice body weights were monitored and recorded every other day starting from the treatment.
Patient Tumor Samples
Masson’s trichrome staining sections from paraffin-embedded biopsies of melanoma patients were obtained from Department of Pathology, Xinhua Hospital, China according to an approved patient sample management protocol. Informed consent was obtained from patients before evaluation.
Statistical analysis
One-way ANOVA and a two tailed Student’s t-test were performed in Prism 5.0 Software. Data were compare with PBS control group and between groups. Data averages from each group were present as mean ± SD. *: p < 0.05, **: p < 0. 01, ***: p < 0. 001.
Supplementary Material
Acknowledgments
Funding
The work was supported by NIH Grant CA198999.
We appreciate Dr. Wenbin Guan from Department of Pathology, Xinhua Hospital, China for providing the human melanoma samples. We appreciate Dr. K.H. Lee and his group, especially Menghan Zhang, for their help in the use of LC/MS and HPLC. We appreciate the Nanomedicines Characterization Core Facility at UNC and Dr. Alexander Kabanov for their help in the use of GPC tracer.
Footnotes
Author contributions
Q.L., F.C., D.W. and L.H. (Leaf Huang) designed the project and drafted the manuscript; Q.L., F.C., L.H. (Lin Hou), L.S., X.Z. performed experimental designs and analysis; Q.L. prepared the graphical abstract. All authors have proofread and agreed on manuscript submission.
Competing interests
The authors declare that they have no competing interests.
Supporting Information available:
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org. The supporting information file contains: Figure S1 to Figure S6, the antibody list (Table S1) and the primer list for real-time PCR (Table S2).
References
- 1.Gloster HM, Jr, Brodland DG. The Epidemiology of Skin Cancer. Dermatol Surg. 1996;22:217–226. doi: 10.1111/j.1524-4725.1996.tb00312.x. [DOI] [PubMed] [Google Scholar]
- 2.Azijli K, Stelloo E, Peters GJ, AJVDE New Developments in the Treatment of Metastatic Melanoma: Immune Checkpoint Inhibitors and Targeted Therapies. Anticancer Res. 2014;34:1493–1505. [PubMed] [Google Scholar]
- 3.Dudley DJ. The Immune System in Health and Disease. Baillieres Clin Obstet Gynaecol. 1992;6:393–416. doi: 10.1016/s0950-3552(05)80003-3. [DOI] [PubMed] [Google Scholar]
- 4.Liu Q, Das M, Liu Y, Huang L. Targeted Drug Delivery to Melanoma. Adv Drug Delivery Rev. 2017 doi: 10.1016/j.addr.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Ye Y, Wang C, Zhang X, Hu Q, Zhang Y, Liu Q, Wen D, Milligan J, Bellotti A, Huang L, Dotti G, Gu ZA. Melanin-Mediated Cancer Immunotherapy Patch. Sci Immunol. 2017;2 doi: 10.1126/sciimmunol.aan5692. [DOI] [PubMed] [Google Scholar]
- 6.Chen LL, Jaimes N, Barker CA, Busam KJ, Marghoob AA. Desmoplastic Melanoma: A Review. J Am Acad Dermatol. 2013;68:825–833. doi: 10.1016/j.jaad.2012.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patients with Desmoplastic Melanoma May Respond to Pd-1 Blockade. Cancer Discov. 2018 doi: 10.1158/2159-8290.CD-RW2018-012. [DOI] [PubMed] [Google Scholar]
- 8.Miao L, Guo S, Lin CM, Liu Q, Huang L. Nanoformulations for Combination or Cascade Anticancer Therapy. Adv Drug Delivery Rev. 2017 doi: 10.1016/j.addr.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miao L, Liu Q, Lin CM, Luo C, Wang Y, Liu L, Yin W, Hu S, Kim WY, Huang L. Targeting Tumor-Associated Fibroblasts for Therapeutic Delivery in Desmoplastic Tumors. Cancer Res. 2017;77:719–731. doi: 10.1158/0008-5472.CAN-16-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F, Huang L. Nano-Delivery of Fraxinellone Remodels Tumor Microenvironment and Facilitates Therapeutic Vaccination in Desmoplastic Melanoma. Theranostics. 2018;8(14):3781–3796. doi: 10.7150/thno.24821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q, Zhu H, Tiruthani K, Shen L, Chen F, Gao K, Zhang X, Hou L, Wang D, Liu R, Huang L. Nanoparticle-Mediated Trapping of Wnt Family Member 5a in Tumor Microenvironments Enhances Immunotherapy for B-Raf Proto-Oncogene Mutant Melanoma. ACS Nano. 2018 doi: 10.1021/acsnano.7b07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madhunapantula SV, Robertson GP. The Pten-Akt3 Signaling Cascade as a Therapeutic Target in Melanoma. Pigm Cell Melanoma Res. 2009;22:400–419. doi: 10.1111/j.1755-148X.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joyce JA, Fearon DT. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 14.Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, Lv ZQ, Gao CY, Wang BL, Zhang YM, Huang RP. Tumor-Induced Perturbations of Cytokines and Immune Cell Networks. Biochim Biophys Acta. 2014;1845:182–201. doi: 10.1016/j.bbcan.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Miao L, Li J, Liu Q, Feng R, Das M, Lin CM, Goodwin TJ, Dorosheva O, Liu R, Huang L. Transient and Local Expression of Chemokine and Immune Checkpoint Traps to Treat Pancreatic Cancer. ACS Nano. 2017;11:8690–8706. doi: 10.1021/acsnano.7b01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuai R, Sun X, Yuan W, Xu Y, Schwendeman A, Moon JJ. Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy. Bioconjug Chem. 2018 doi: 10.1021/acs.bioconjchem.7b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu G, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang H, Ma Y, Zhang F, Tian R, Ni Q, Cheng S, Wang Z, Lu N, Yung BC, Wang Z, Lang L, Fu X, Jin A, Weiss ID, Vishwasrao H, et al. Albumin/Vaccine Nanocomplexes That Assemble in Vivo for Combination Cancer Immunotherapy. Nat Commun. 2017;8:1954. doi: 10.1038/s41467-017-02191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gebremeskel S, Johnston B. Concepts and Mechanisms Underlying Chemotherapy Induced Immunogenic Cell Death: Impact on Clinical Studies and Considerations for Combined Therapies. Oncotarget. 2015;6:41600–41619. doi: 10.18632/oncotarget.6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, Dorosheva O, Liu T, Liu R, Huang L. Synergistic and Low Adverse Effect Cancer Immunotherapy by Immunogenic Chemotherapy and Locally Expressed Pd-L1 Trap. Nat Commun. 2018;9:2237. doi: 10.1038/s41467-018-04605-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kepp O, Menger L, Vacchelli E, Locher C, Adjemian S, Yamazaki T, Martins I, Sukkurwala AQ, Michaud M, Senovilla L, Galluzzi L, Kroemer G, Zitvogel L. Crosstalk between Er Stress and Immunogenic Cell Death. Cytokine Growth Factor Rev. 2013;24:311–318. doi: 10.1016/j.cytogfr.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Presant CA, Gams R, Bartolucci A. Mitoxantrone in Malignant Melanoma. Cancer Treat Rep. 1984;68:903–905. [PubMed] [Google Scholar]
- 22.Arseneau JC, Schoenfeld DA, Borden EC. A Phase Ii Study of Dihydroxyanthracenedione (Dhad, Mitoxantrone, Nsc 301739) in Advanced Malignant Melanoma. Invest New Drugs. 1986;4:53–56. doi: 10.1007/BF00172017. [DOI] [PubMed] [Google Scholar]
- 23.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic Cell Death and Damps in Cancer Therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 24.Kannaiyan R, Shanmugam MK, Sethi G. Molecular Targets of Celastrol Derived from Thunder of God Vine: Potential Role in the Treatment of Inflammatory Disorders and Cancer. Cancer Lett. 2011;303:9–20. doi: 10.1016/j.canlet.2010.10.025. [DOI] [PubMed] [Google Scholar]
- 25.Guo L, Luo S, Du Z, Zhou M, Li P, Fu Y, Sun X, Huang Y, Zhang Z. Targeted Delivery of Celastrol to Mesangial Cells Is Effective against Mesangioproliferative Glomerulonephritis. Nat Commun. 2017;8:878. doi: 10.1038/s41467-017-00834-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. A Novel Hsp90 Inhibitor to Disrupt Hsp90/Cdc37 Complex against Pancreatic Cancer Cells. Mol Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 27.Hu K, Miao L, Goodwin TJ, Li J, Liu Q, Huang L. Quercetin Remodels the Tumor Microenvironment to Improve the Permeation, Retention, and Antitumor Effects of Nanoparticles. ACS Nano. 2017;11:4916–4925. doi: 10.1021/acsnano.7b01522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zou L, Chen F, Bao J, Wang S, Wang L, Chen M, He C, Wang Y. Preparation, Characterization, and Anticancer Efficacy of Evodiamine-Loaded Plga Nanoparticles. Drug Delivery. 2016;23:908–916. doi: 10.3109/10717544.2014.920936. [DOI] [PubMed] [Google Scholar]
- 29.Zhou X, Seto SW, Chang D, Kiat H, Razmovski-Naumovski V, Chan K, Bensoussan A. Synergistic Effects of Chinese Herbal Medicine: A Comprehensive Review of Methodology and Current Research. Front Pharmacol. 2016;7:201. doi: 10.3389/fphar.2016.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng L, Zhang D, Fan C, Ma C, Yang W, Meng Y, Wu W, Guan S, Jiang B, Yang M, Liu X, Guo D. Er Stress-Mediated Apoptosis Induced by Celastrol in Cancer Cells and Important Role of Glycogen Synthase Kinase-3beta in the Signal Network. Cell Death Dis. 2013;4:e715. doi: 10.1038/cddis.2013.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, Bracci L, Breckpot K, Brough D, Buque A, Castro MG, Cirone M, Colombo MI, Cremer I, Demaria S, Dini L, et al. Consensus Guidelines for the Detection of Immunogenic Cell Death. Oncoimmunology. 2014;3:e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue S, Setoyama Y, Odaka A. Doxorubicin Treatment Induces Tumor Cell Death Followed by Immunomodulation in a Murine Neuroblastoma Model. Exp Ther Med. 2014;7:703–708. doi: 10.3892/etm.2014.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen F, Zhang J, Wang L, Wang Y, Chen M. Tumor Ph(E)-Triggered Charge-Reversal and Redox-Responsive Nanoparticles for Docetaxel Delivery in Hepatocellular Carcinoma Treatment. Nanoscale. 2015;7:15763–15779. doi: 10.1039/c5nr04612b. [DOI] [PubMed] [Google Scholar]
- 34.Goodwin TJ, Huang L. On the Article “Findings Questioning the Involvement of Sigma-1 Receptor in the Uptake of Anisamide-Decorated Particles” [J. Control. Release 224 (2016) 229–238]: Letter to the Editor 1 (September 14, 2016) J Control Release. 2016;243:382–385. doi: 10.1016/j.jconrel.2016.11.022. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Chen R, Chen F, Chen M, Wang Y. Nucleolin Targeting As1411 Aptamer Modified Ph-Sensitive Micelles: A Dual-Functional Strategy for Paclitaxel Delivery. J Control Release. 2015;213:e137–138. doi: 10.1016/j.jconrel.2015.05.232. [DOI] [PubMed] [Google Scholar]
- 36.Hu Y, Gong X, Zhang J, Chen F, Fu C, Li P, Zou L, Zhao G. Activated Charge-Reversal Polymeric Nano-System: The Promising Strategy in Drug Delivery for Cancer Therapy. Polymers. 2016;8:99. doi: 10.3390/polym8040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu N, Gao J, Liu Q, Wang J, Shen Y. Enzyme-Responsive Charge-Reversal Polymer-Mediated Effective Gene Therapy for Intraperitoneal Tumors. Biomacromolecules. 2018 doi: 10.1021/acs.biomac.8b00440. [DOI] [PubMed] [Google Scholar]
- 38.Hou L, Zheng Y, Wang Y, Hu Y, Shi J, Liu Q, Zhang H, Zhang Z. Self-Regulated Carboxyphenylboronic Acid-Modified Mesoporous Silica Nanoparticles with “Touch Switch” Releasing Property for Insulin Delivery. ACS Appl Mater Interfaces. 2018 doi: 10.1021/acsami.8b06998. [DOI] [PubMed] [Google Scholar]
- 39.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione Metabolism and Its Implications for Health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 40.Meng F, Zhong Z, Feijen J. Stimuli-Responsive Polymersomes for Programmed Drug Delivery. Biomacromolecules. 2009;10:197–209. doi: 10.1021/bm801127d. [DOI] [PubMed] [Google Scholar]
- 41.Chen F, Zhang J, He Y, Fang X, Wang Y, Chen M. Glycyrrhetinic Acid-Decorated and Reduction-Sensitive Micelles to Enhance the Bioavailability and Anti-Hepatocellular Carcinoma Efficacy of Tanshinone Iia. Biomater Sci. 2016;4:167–182. doi: 10.1039/c5bm00224a. [DOI] [PubMed] [Google Scholar]
- 42.Kuppusamy P, Li H, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, Krishna MC, Mitchell JB. Noninvasive Imaging of Tumor Redox Status and Its Modification by Tissue Glutathione Levels. Cancer Res. 2002;62:307–312. [PubMed] [Google Scholar]
- 43.Chen FQ, Zhang JM, Fang XF, Yu H, Liu YL, Li H, Wang YT, Chen MW. Reversal of Paclitaxel Resistance in Human Ovarian Cancer Cells with Redox-Responsive Micelles Consisting of Alpha-Tocopheryl Succinate-Based Polyphosphoester Copolymers. Acta Pharmacol Sin. 2017;38:859–873. doi: 10.1038/aps.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelsch A, Tomcin S, Rausch K, Barz M, Mailander V, Schmidt M, Landfester K, Zentel R. Hpma Copolymers as Surfactants in the Preparation of Biocompatible Nanoparticles for Biomedical Application. Biomacromolecules. 2012;13:4179–4187. doi: 10.1021/bm301453g. [DOI] [PubMed] [Google Scholar]
- 45.Zeng X, Tao W, Mei L, Huang L, Tan C, Feng SS. Cholic Acid-Functionalized Nanoparticles of Star-Shaped Plga-Vitamin E Tpgs Copolymer for Docetaxel Delivery to Cervical Cancer. Biomaterials. 2013;34:6058–6067. doi: 10.1016/j.biomaterials.2013.04.052. [DOI] [PubMed] [Google Scholar]
- 46.Hemmelmann M, Knoth C, Schmitt U, Allmeroth M, Moderegger D, Barz M, Koynov K, Hiemke C, Rosch F, Zentel R. Hpma Based Amphiphilic Copolymers Mediate Central Nervous Effects of Domperidone. Macromol Rapid Commun. 2011;32:712–717. doi: 10.1002/marc.201000810. [DOI] [PubMed] [Google Scholar]
- 47.Scheibe P, Barz M, Hemmelmann M, Zentel R. Langmuir-Blodgett Films of Biocompatible Poly(Hpma)-Block-Poly(Lauryl Methacrylate) and Poly(Hpma)-Random-Poly(Lauryl Methacrylate): Influence of Polymer Structure on Membrane Formation and Stability. Langmuir. 2010;26:5661–5669. doi: 10.1021/la903725k. [DOI] [PubMed] [Google Scholar]
- 48.Talelli M, Barz M, Rijcken CJ, Kiessling F, Hennink WE, Lammers T. Core-Crosslinked Polymeric Micelles: Principles, Preparation, Biomedical Applications and Clinical Translation. Nano Today. 2015;10:93–117. doi: 10.1016/j.nantod.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meng F, Cheng R, Deng C, Zhong Z. Intracellular Drug Release Nanosystems. Mater Today. 2012;15:436–442. [Google Scholar]
- 50.Zhang L, Ren Y, Wang Y, He Y, Feng W, Song C. Pharmacokinetics, Distribution and Anti-Tumor Efficacy of Liposomal Mitoxantrone Modified with a Luteinizing Hormone-Releasing Hormone Receptor-Specific Peptide. Int J Nanomed. 2018;13:1097–1105. doi: 10.2147/IJN.S150512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guissi NEI, Li H, Xu Y, Semcheddine F, Chen M, Su Z, Ping Q. Mitoxantrone-and Folate-Tpgs2k Conjugate Hybrid Micellar Aggregates to Circumvent Toxicity and Enhance Efficiency for Breast Cancer Therapy. Mol Pharmaceutics. 2017;14:1082–1094. doi: 10.1021/acs.molpharmaceut.6b01009. [DOI] [PubMed] [Google Scholar]
- 52.Tzur-Balter A, Rubinski A, Segal E. Designing Porous Silicon-Based Microparticles as Carriers for Controlled Delivery of Mitoxantrone Dihydrochloride. J Mater Res. 2013;28:231–239. [Google Scholar]
- 53.Tao X, Jin S, Wu D, Ling K, Yuan L, Lin P, Xie Y, Yang X. Effects of Particle Hydrophobicity, Surface Charge, Media Ph Value and Complexation with Human Serum Albumin on Drug Release Behavior of Mitoxantrone-Loaded Pullulan Nanoparticles. Nanomaterials (Basel) 2015;6 doi: 10.3390/nano6010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J Immunol. 2015;194:2985–2991. doi: 10.4049/jimmunol.1403134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ricci MS, Zong WX. Chemotherapeutic Approaches for Targeting Cell Death Pathways. Oncologist. 2006;11:342–357. doi: 10.1634/theoncologist.11-4-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martinez-Lostao L, Anel A, Pardo J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin Cancer Res. 2015;21:5047–5056. doi: 10.1158/1078-0432.CCR-15-0685. [DOI] [PubMed] [Google Scholar]
- 57.Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, Brinckerhoff CE. Multiple Murine Braf(V600e) Melanoma Cell Lines with Sensitivity to Plx4032. Pigm Cell Melanoma Res. 2014;27:495–501. doi: 10.1111/pcmr.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hillen F, Baeten CIM, van de Winkel A, Creytens D, van der Schaft DWJ, Winnepenninckx V, Griffioen AW. Leukocyte Infiltration and Tumor Cell Plasticity Are Parameters of Aggressiveness in Primary Cutaneous Melanoma. Cancer Immunol Immunother. 2008;57:97–106. doi: 10.1007/s00262-007-0353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The Immune Contexture in Human Tumours: Impact on Clinical Outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 60.Bruttel VS, Wischhusen J. Cancer Stem Cell Immunology: Key to Understanding Tumorigenesis and Tumor Immune Escape? Front Immunol. 2014;5:360. doi: 10.3389/fimmu.2014.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Q, Zhu H, Liu Y, Musetti S, Huang L. Braf Peptide Vaccine Facilitates Therapy of Murine Braf-Mutant Melanoma. Cancer Immunol Immunother. 2017 doi: 10.1007/s00262-017-2079-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J, Huang L. Combination Immunotherapy of Muc1 Mrna Nano-Vaccine and Ctla-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Mol Ther. 2018;26:45–55. doi: 10.1016/j.ymthe.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bianchi ME. Killing Cancer Cells, Twice with One Shot. Cell Death Differ. 2014;21:1–2. doi: 10.1038/cdd.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Young YK, Bolt AM, Ahn R, Mann KK. Analyzing the Tumor Microenvironment by Flow Cytometry. Methods Mol Biol. 2016;1458:95–110. doi: 10.1007/978-1-4939-3801-8_8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.