

Abstract
The aetiology and pathogenesis of inflammatory bowel disease (IBD) remains unclear but involves a complex interplay between genetic risk, environmental exposures, the immune system and the gut microbiota. Nearly two decades ago, the first susceptibility gene for Crohn's disease, NOD2, was identified within the IBD 1 locus. Since then, over 230 genetic risk loci have been associated with IBD and yet NOD2 remains the strongest association to date. As an intracellular innate immune sensor of bacteria, investigations into host–microbe interactions, involving both innate and adaptive immune responses, have become of particular interest in understanding the pathogenesis of IBD. Advancements in sequencing technology have lead to the groundbreaking characterization of the gut microbiota and its role in health and disease. While an altered microbiome has been described for IBD, whether it is a cause or an effect of the intestinal inflammation has yet to be determined. Moreover, the bidirectional relationship between the gut microbiota and the mucosal immune system adds to the multifaceted complexity of intestinal homeostasis. A better understanding of how host genetics, including NOD2, influence immune–microbe interactions and alter susceptibility to IBD is necessary in order to develop therapeutic and preventative treatments.
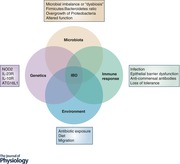
Keywords: inflammatory bowel diseases, gut microbiota, Nod2, NLRs, innate immunity, adaptive immunity, immune‐microbe interactions
Introduction
The nucleotide‐binding oligomerization domain (NOD)‐like receptors (NLRs) are a family of evolutionarily conserved, intracellular sensors that recognize a wide range of microbial‐ and damage‐associated signals during infection and inflammation. Upon activation, these innate immune receptors can lead to nuclear factor κB (NF‐κB) and mitogen‐activated protein kinase (MAPK) signalling or inflammasome cascades (Rubino et al. 2012). There are currently 22 human and 34 mouse NLR proteins that are classified based on their structure (Franchi et al. 2009). NLRs contain an N‐terminal protein‐binding domain responsible for downstream signalling, a central nucleotide‐binding domain responsible for oligomerization, and a leucine‐rich repeat domain located at the C‐terminal, responsible for ligand sensing and binding. Identification of the NLR, NOD2, as the strongest genetic risk factor for the inflammatory bowel disease (IBD), Crohn's disease, has paved the way for nearly 20 years of research into the role it plays in immunity, host–microbe interactions and, ultimately, intestinal disease. Here, we will review the genetic, immune and microbial associations with IBD, with a focus on the role for NOD2 in the pathogenesis of Crohn's disease.
Overview of IBD
Inflammatory bowel diseases encompass illnesses characterized by chronic, relapsing inflammation of the gastrointestinal tract. The highest prevalence of IBD is in western countries in Europe, Oceania and North America, with prevalence exceeding 0.3% in most (Ng et al. 2018). Interestingly, IBD incidence seems to be stabilizing in the western countries, whereas newly industrialized countries are experiencing rapid increases in IBD incidence (Kaplan, 2015). Canada continues to have one of the highest rates of IBD in the world, with 1 in every 150 Canadians (∼0.6% prevalence) living with either Crohn's disease (CD) or ulcerative colitis (UC), the two main types of IBD (Crohn's and Colitis Foundation of Canada, 2012). CD can cause inflammation at any point along the alimentary canal, from mouth to anus, whereas inflammation is localized to the colon and rectum in UC. Disease onset typically begins in late teens to early adulthood, and symptoms include severe diarrhoea, abdominal pain, bloating, fatigue and weight loss. With disease induction occurring during the most productive years and symptoms severely impacting the patient's quality of life, IBD is a huge burden to the patient, their families, society and the health care system.
While the aetiology of IBD remains unclear, it is currently hypothesized to be a multi‐hit, multi‐factorial auto‐inflammatory disease. The most widely accepted hypothesis is that a genetically susceptible person experiences an environmental trigger that leads to an inappropriate immune response against the commensal gut microbiota, resulting in chronic immune activation, inflammation, epithelial damage, bacterial translocation and further amplification of the inflammatory response (Sartor, 2006). Despite significant efforts, the environmental trigger(s) leading to this chronic inflammatory cycle remains unknown. The search for a “cure” for IBD requires understanding the fundamental principles governing the interaction between the intestinal immune system and gut microbes and in particular, mechanisms that control responses to “normal” commensal bacteria. Indeed, the challenge to the mucosal immune system is to maintain a controlled and appropriate response to the trillions of gut microbes that make up the gut microbiota. One can imagine that loss of this regulation could lead to chronic intestinal inflammation and play a key role in the pathogenesis of IBD.
Genetic risk in IBD
The genetic influence on IBD development has been well studied. Some of the first studies looking at IBD occurrence in twins identified a higher concordance in monozygotic twins than dizygotic twins, 58.3% versus 0%, respectively, for CD (Orholm et al. 2000). Moreover, a study of Swedish monozygotic twins identified a concordance of 18% in UC and 50% in CD (Halfvarson et al. 2003). This indicated that IBD development, particularly CD, is influenced by genetics. To date over 230 genetic loci have been identified that are associated with an increased risk of developing IBD, 30 of which are specific to CD (Jostins et al. 2012; Liu et al. 2015; de Lange et al. 2017). The majority of IBD susceptibility genes are linked to pathways involved in immune–microbe interactions. Pathways highlighted by these IBD‐linked genes include: microbial detection, immune activation and suppression, and fucosylation of the mucosal epithelium. Some of these genes include the interleukin (IL)‐23 receptor (IL‐23R), the autophagy‐related protein 16‐1 (ATG16L1) and the IL‐10 receptor (IL‐10R) (Jostins et al. 2012). The strongest genetic association with CD is nucleotide‐binding oligomerization domain‐containing protein 2 (NOD2) mutations, with the largest odds ratio of 3.1 (Hugot et al. 2001; Ogura et al. 2001; Jostins et al. 2012).
NOD2
NOD2 is a cytosolic pattern recognition receptor that senses muramyl dipeptide (MDP), a component of peptidoglycan found in the cell wall of Gram‐positive and ‐negative bacteria (Girardin et al. 2003). Located within the leucine rich repeat (LRR) domain responsible for microbial sensing are the three main IBD‐associated NOD2 mutations: Arg702Trp and Gly908Arg, which result in amino acid substitutions, and the 1007fs frameshift, which results in a premature stop codon and a truncated protein (Hugot et al. 2001; Ogura et al. 2001) (Fig. 1). These mutations are thought to result in “loss of function” and cause defective bacterial sensing. Up to 40% of CD patients have at least one allele mutated in NOD2, while mutations in both NOD2 alleles are found in ∼10% of CD patients (Lesage et al. 2002). Upon activation, NOD2 signalling is mediated by Rip2 kinase, which activates NF‐κB and MAPKs leading to increased immune gene expression and inflammation. These observations suggest that innate immune responses to bacteria are a key element in the pathogenesis of CD; however, the mechanism by which this occurs is still unclear (Girardin et al. 2003; Watanabe et al. 2004; Kobayashi et al. 2005; Fritz et al. 2006).
Figure 1. NOD2 gene and IBD associated mutations.
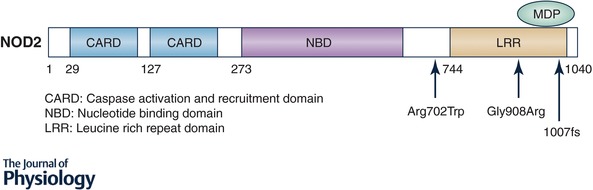
NOD2 has three functioning domains: CARD, responsible for NF‐κB activation through interactions with receptor interacting protein‐2 (RIP2); NBD, responsible for oligomerization; and the LRR domain, responsible for bacterial sensing via binding of muramyl dipeptide (MDP). The three main mutations in NOD2 are all located near or in the LRR domain and arrows indicate their locations.
NOD2 is expressed in numerous cell types, including T and B cells (Shaw et al. 2009; Petterson et al. 2011), macrophages and dendritic cells (Hedl et al. 2007; Cooney et al. 2010), plus epithelial cells, goblet cells and Paneth cells (Rosenstiel et al. 2003; Ogura et al. 2003a; Ramanan et al. 2014). Each of these cell subsets are located within the intestine and are involved in either maintaining the epithelial barrier thus limiting bacterial translocation into the tissue, or bacterial sensing and clearance. Indeed, a study showed that NOD2 is constitutively expressed within Lgr5+ stem cells within intestinal crypts, and sensing of MDP resulted in increased stem cell survival and epithelial restitution (Nigro et al. 2014). Therefore, it is clear that defects in the bacterial sensor NOD2 could impair any or all of the mechanisms available for protecting the host from bacterial‐induced inflammation in the gut. Indeed, polymorphisms in NOD2 lead to defective NF‐κB activation resulting in inefficient epithelial and macrophage clearance of invasive bacteria (Sartor, 2004). Furthermore, patients with NOD2 mutations have reduced defensin production and secretion by Paneth cells, increased T cell and humoral immune responses and a proposed loss of tolerance to the commensal gut microbiota (Kobayashi et al. 2005).
NOD2 is also involved in other cellular defence mechanisms, such as autophagy, where MDP sensing by NOD2 induces recruitment of the autophagy protein ATG16L1 to the bacterial entry site in the plasma membrane (Travassos et al. 2010). Indeed, the CD‐associated frameshift mutation of NOD2 fails to induce ATG16L1 recruitment and results in incomplete autophagasome formation. Additionally, there is evidence suggesting a role for NOD2 in antiviral immune responses; however, the exact mechanism and cells involved remain unclear (Sabbah et al. 2009; Lin et al. 2013). Together, this suggests that NOD2 mutations not only impair bacterial sensing but also influence other defence pathways that could be associated with CD.
Involvement of the immune system in IBD
Multiple components of the immune system are involved in the pathogenesis of IBD, from innate sensing of bacteria to adaptive anti‐commensal responses. The gut is home to the largest number of immune cells in the body and only a single layer of columnar epithelial cells physically separates the numerous luminal antigens from the primed mucosal immune system. The mucosal immune system exists to protect against invading pathogens, while at same time it must remain tolerant to food antigens and commensal microbes.
Structure and function of the intestine
In order to understand the role of the gut microbiota on mucosal immune development and disease, the structure of the intestine must be considered. The alimentary tract runs from mouth to anus, and while microbial colonization does exist in the mouth and stomach, the majority of the gut microbiota live in the small and large intestine (Sender et al. 2016). The small intestine can be considered as three functionally distinct sections: the proximal duodenum, responsible for enzymatic breakdown of ingested food, the medial jejunum and the distal ileum, which are responsible for nutrient absorption. Function dictates the structure of the epithelial barrier along the small intestine, from long villi and crypts in the duodenum and jejunum to increase surface area necessary for absorption, to shorter villi in the ileum (Mowat & Agace, 2014). Multipotent stem cells inhabit the base of the crypt alongside Paneth cells, and give rise to new epithelial cells to replace the constant turnover of the barrier every 4–5 days (Barker et al. 2008). As the epithelial cells migrate up the crypt and villi, they mature into one of the various cell types within the barrier: absorptive enterocytes (the most common), tuft cells, enteroendocrine cells, or mucus‐producing goblet cells. Tuft cells have come into the spotlight for their recently discovered ability to initiate type 2 immune responses in the gut, and involvement in the complex network between immune cells and the intestinal epithelium (Gerbe & Jay, 2016; Middelhoff et al. 2017). Together, these cell subsets maintain a secure barrier along the small intestine, a barrier between the microbes in the lumen and the immune cells in the mucosa. Some of the mechanisms used to protect the barrier include expression of tight junction proteins to reduce the permeability of the epithelium, release of anti‐microbial peptides including defensins from Paneth cells, and mucus production by goblet cells (Peterson & Artis, 2014). Furthermore, expression of pattern recognition receptors, such as NOD2, by epithelial cells provides another protective checkpoint. Indeed, NOD2 expression is particularly high in Paneth cells within the ileum, and NOD2 mutations have been associated with ileal inflammation in Crohn's disease patients (Hugot et al. 2001).
In contrast, the large intestine has no villi, only crypts, and its main physiological function is the reabsorption of water (Mowat & Agace, 2014). The crypts still maintain a base of regenerating stem cells (but lack Paneth cells), which populate the colonic epithelium with enterocytes, enteroendocrine cells and goblet cells. Goblet cells produce a protective mucus composed of two layers: a thin inner layer that is firmly attached to the colonic epithelium and impervious to bacterial translocation during homeostasis, and an outer layer which is significantly thicker but looser, allowing bacterial habitation (Hansson & Johansson, 2010). Mucins, such as Muc2, are the structural components of the mucus layer, which can be quantified and used as a measure of the host's response to microbes (Johansson et al. 2011). Moreover, immune mediators, such as IL‐9 and IL‐13, stimulate mucus production (Steenwinckel et al. 2009).
Immune response in IBD
The gut represents the largest and most diverse site of interaction between the host and the environment. As such, patients with IBD can experience a flare in response to numerous triggers of inflammation. As described above, the lumen is physically separated from the immune cells by highly specialized epithelial cells. IBD patients have altered intestinal permeability (i.e. leaky gut due to reduced intercellular adhesion), possibly allowing for bacterial translocation into the lamina propria (Peeters et al. 1997; Soderholm et al. 1999; Buhner et al. 2006), although it remains to be determined if this is due to the inflammation, alterations of the gut microbiota, or represents an underlying genetic defect. Recent work from our laboratory failed to show a genetic association with abnormal intestinal permeability in healthy subjects (Kevans et al. 2015).
Paneth cells, located in the base of the crypts, are secretory cells that specialize in host defence by secreting potent anti‐microbial α‐defensins. NOD2 is highly expressed in Paneth cells, suggesting that polymorphisms in NOD2 could result in defective anti‐microbial defence (Ogura et al. 2003a). Indeed, CD patients have a reduction in anti‐microbial peptide production and morphologically abnormal Paneth cells (Cadwell et al. 2008; VanDussen et al. 2014). A thick layer of mucus lines the gut epithelium acting as a chemo‐physical barrier limiting bacterial adherence and translocation (Duerkop et al. 2009). IBD is associated with a thinner mucus layer that may enhance bacterial invasion of the mucus (Pullan et al. 1994). Collectively, epithelial barrier defects are a characteristic feature in IBD patients and may represent a key feature in the breakdown of host–microbe interactions as it is the main contact point between luminal microbes and mucosal immune cells.
IBD patients have elevated levels of many pro‐inflammatory cytokines such as IL‐1β, tumour necrosis factor α (TNFα), IL‐6 and IL‐12 in serum and mucosal tissue compared to healthy controls. It has been postulated that this elevation is due primarily to the inability to control the immune response to commensal bacterial antigens. Indeed, several groups have identified increased anti‐microbial responses in IBD patients (Round & Mazmanian, 2009; Manichanh et al. 2012). Specifically, one team identified an increase in highly IgA‐coated microbes in the stool of IBD patients, indicating an increased adaptive immune response against commensals (Palm et al. 2014). Another group investigated the reactivity of serum antibodies to microbial antigens and found that CD patients have elevated serum IgG reactive to bacterial flagellin (Lodes et al. 2004). Tissue specific responses also indicate an inability to control inflammation in IBD patients. For example, the alarmin, IL‐33, is increased in inflamed tissue of IBD patients (Kobori et al. 2010). Moreover, increases in neutrophils and monocytes within the tissue result in increased reactive oxygen species and further tissue damage (Brown & Mayer, 2007). Mucosal T cells in IBD patients survive longer than normal T cells and remain within the mucosa (Neurath et al. 2001). This latter finding has resulted in a new avenue of therapeutics targeted against gut homing receptors, including the integrin α4β7 (Ghosh et al. 2003). Another effective treatment of CD is anti‐TNFα therapy, which suppresses immune activation by binding to and neutralizing TNFα produced by immune cells. Limiting the inflammatory response in this way allows for the epithelial barrier to heal thus preventing further microbial translocation into the lamina propria and continuous immune stimulation. Alternatively, some patients respond to oral antibiotic therapy, which reduces the total microbial load in the gut lumen and limits bacterial invasion into the tissue; however, the mechanism by which antibiotics mediate remission are likely due to alterations of the gut microbiota (Sartor, 2004).
Gut microbiota
The human body is truly an ecosystem, colonized by a wide variety of microbes, including Archaea, bacteria, protists, viruses and bacteriophages, and some not so “micro” organisms, such as worms and fungi. The “microbiota” collectively refers to all of these components but more specifically relates to the bacterial community both on and in the body, whereas the “microbiome” denotes the genes and genetics of this community (Hooper & Gordon, 2001). While the existence and influence of the “virome” and “mycobiome” have been established, much less is known about their role in health and disease, or the co‐evolution with gut bacteria (Cui et al. 2013; Minot et al. 2013; Lim et al. 2015). Symbiosis between the host and its resident microbiota has important consequences for human health and physiology. These interactions may have beneficial nutritional, immunological and developmental effects or pathogenic effects for the host (Penders et al. 2006). Research characterizing which bacteria are present at various anatomical locations, their abundance and function has exploded over the last decade as we have come to appreciate their essential role in both health and disease.
Quantification and assessment of the microbiota
The community of microbes varies by anatomical location, from skin to vagina, mouth to gut (Costello et al. 2009; Cho & Blaser, 2012). While many microbes are found on the skin, the highest concentration of bacteria is located within the gastrointestinal tract. Bacteria live in the mouth (109), stomach (103) and small intestine (104–108) but the majority are located in the colon, with nearly 1011 bacteria per gram of intestinal content or 3.8 × 1013 bacteria in total (Sender et al. 2016). The complexity of the community makes it difficult to ascertain exactly how many bacterial species are represented but it is estimated that 500–1000 species colonize the average human colon, the majority of which are obligate anaerobes (Claesson et al. 2009; Human Microbiome Project Consortium, 2012). Actual biodiversity and community structure, particularly how the microbes co‐habitat and interact with each other, are difficult to ascertain since many organisms cannot yet be cultured ex vivo. Some researchers are developing new ways to culture commensal bacteria in physiologically relevant conditions and in ways that attempt to address the community structure, including invention of the “robogut” and other novel culturing techniques (Petrof et al. 2013; Lau et al. 2016). Advancement of these methodologies will allow for significant advancement of microbial manipulation experiments and testing of potential therapeutics that could alter the microbiota to prevent or eliminate disease. However, until then, molecular methods for investigating the microbial population primarily involve genetic analysis via sequencing variable regions of the 16S ribosomal RNA (rRNA) genes. High throughput sequencing of selected variable regions of the 16S rRNA gene is currently the most common method for taxonomic identification and assessing community structure (Case et al. 2007; Caporaso et al. 2010). In this way, sequencing data provide insight into the presence or absence of bacterial taxa and their abundance relative to the entire population, and as such are used to discern the community structure of the microbiota at a given time. Specific measures of the community include α‐diversity, which evaluates species richness or the presence of taxa (e.g. Chao1 or Shannon diversity index), and β‐diversity, which determines the presence and abundance of taxa and how it relates to the community (e.g. Unifrac, Bray‐Curtis dissimilarity) (Human Microbiome Project Consortium, 2012). Longitudinal sampling of the same subjects allows for both inter‐ and intra‐individual comparisons of the microbial composition, providing insight into how environmental cues (e.g. diet, drugs) can influence the community over time within an individual and if the same patterns can be observed across individuals.
Microbiota in IBD
The gut microbiota in IBD has been characterized as increased abundance of Bacteroidetes and Proteobacteria, with loss of Firmicutes (Oyri et al. 2015). Moreover, IBD patients exhibit a reduction in total bacterial diversity (Manichanh et al. 2006). Loss of certain beneficial microbes in IBD patients, such as Faecalibacterium prausnitzii, has also been associated with “dysbiosis” (Sokol et al. 2008, 2009). The term ‘dysbiosis’ refers to a state of imbalance or altered composition or altered function (and not necessarily composition) of the microbiota, leading to altered host–microbe interactions. It has been proposed that a state of dysbiosis occurs when harmful microbes overtake the beneficial ones, which is particularly observed during diseased states, such as IBD, obesity, metabolic disorders and infections (Carding et al. 2015). The issue with dysbiosis in IBD is that it is nearly impossible to ascribe causation since microbial community functional or structural alterations observed during disease are likely linked to treatment regimens and/or the on‐going inflammatory state within the gut. Indeed, few studies have identified that an altered microbiome precedes disease onset or causes inflammation. One study attempted to more accurately address this issue by looking at the microbiome of newly diagnosed, treatment naive paediatric patients (Gevers et al. 2014). They found CD patients exhibited increased abundance of Enterobacteriaceae, Pasteurellaceae and Fusobacteriaceae, and decreased abundance of Erysipelotrichaceae, Bacteroidales and Clostridiales compared with unaffected controls. These differences were observed in mucosal samples (from the terminal ileum and rectum), but were not well reflected in the stool. Moreover, antibiotic treatment further exacerbated this phenotype with further loss of Erysipelotrichaceae and Clostridiales. Others have also identified a reduction in Clostridiales in IBD, particularly members of Clostridium clusters XIVa and IV, which are taxa thought to promote immune regulation (Frank et al. 2007, 2011). Another study investigated the microbial and metabolic profiles of healthy first‐degree relatives of paediatric IBD patients; the authors identified that some healthy relatives displayed microbial dysbiosis, which in some cases was also associated with a perturbed metabolome and increased fecal calprotectin indicating a possible pre‐disease, sub‐clinical state of inflammation (Jacobs et al. 2016). Clearly, there is a need for prospective studies in healthy susceptible first‐degree relatives of IBD patients, such as the Genetics, Environment, Microbial (GEM) project, to help resolve whether changes in the microbial communities precede disease onset in IBD.
In summary, IBD implicates a significant role for the gut microbiota, either in driving or perpetuating chronic relapsing inflammation (Huttenhower et al. 2014). While an altered microbiota certainly exists in patients with active IBD, whether a “dysbiotic” microbiome is a cause or an effect of IBD is yet to be determined.
Immune–microbe interactions in IBD
The mucosal immune system develops and/or matures in response to the presence of a gut microbiome (Shroff & Cebra, 1995; Duerkop et al. 2009; Tlaskalova‐Hogenova et al. 2011). From birth, the newly colonizing gut microbiota interacts with the gut epithelium and host immune system, influencing development, maturation and regulation, which, in turn, influence the development of the microbiota (Tomas et al. 2013; Francino, 2014). Indeed, IBD can be viewed as an imbalance in the bidirectional interactions between immune responses and the gut microbiome in genetically susceptible individuals; however, it is not clear if this is due to an abnormal gut microbiome or an abnormal immune response or both. A better understanding of the mechanisms involved in this bidirectional relationship is essential.
Role of microbes in host immune development
The mucosal immune system functions to distinguish between friend (non‐threatening symbiotic bacteria) and foe (pathogenic microbe). The latter requires a protective, often inflammatory response while the former, either no response or a controlled response with minimal collateral damage from inflammation. A major challenge to the mucosal immune system is ensuring an “appropriate” response to the large and diverse population of the commensal gut microbiota (Littman & Pamer, 2011).
The critical window for immune development is immediately following birth and during the first year of life (Holt & Jones, 2000; Arrieta et al. 2014; Bokulich et al. 2016; Tamburini et al. 2016). From birth, microorganisms, including bacteria, viruses and fungi, colonize humans and animals. These microorganisms stimulate the development of the local and systemic immune system, which, in turn, influences the development of the microbiota.
Innate receptors detect microbe‐associated molecular patterns (MAMPs) by germline‐encoded pattern‐recognition receptors (PRRs), e.g. Toll‐like receptors (TLRs) (Iwasaki & Medzhitov, 2004) and NOD‐like receptors (NLRs) (Fritz et al. 2006). These PRRs regulate first‐line host responses but also influence the antigen‐specific or adaptive immune responses. Tolerance developed in early life during immune and microbiota co‐development is essential for maintaining a homeostatic mucosal environment throughout life (Sartor, 2004). The concept of a mucosal “firewall” elegantly describes the multiple layers of protection involved in maintaining tolerance and limiting inflammation within the mucosa (Macpherson et al. 2009). Physically, the epithelial barrier and the mucus layer(s), along with secretory IgA and anti‐microbial peptides, limit contact of bacteria to underlying immune cells. If bacteria are able to get close to or through the epithelial layer, intestinal macrophages engulf and kill them; alternatively, dendritic cells phagocytose and transport live bacteria to mesenteric lymph nodes to initiate a local, targeted immune response (Belkaid & Hand, 2014). Thus, it is not surprising that perturbation of the gut microbiota can affect immune health. Bacteria interact with the immune system via numerous ligands, including: capsular polysaccharide (CPS), lipopolysaccharide, peptidoglycan, muramic acid, flagellin and unmethylated CpG motifs of bacterial DNA (Platt & Mowat, 2008). In response to these bacterial ligands, cytokines are produced that shape the differentiation of the adaptive immune system, including T cells. Indeed, gut immune maturation depends on colonization, as germ‐free mice possess severely depleted mucosal immune development (Shroff & Cebra, 1995; Hooper et al. 2012). Studies of germ‐free mice have highlighted the essential roles microbes play in immune, epithelial and metabolic development of the host (Backhed et al. 2004; Macpherson & Harris, 2004; Sommer & Backhed, 2013).
Using gnotobiotic mice, researchers have begun to understand the profound effects of microbial stimulation on immune development (Tlaskalova‐Hogenova et al. 2011). Specifically, studies have determined that IgA and germinal centre formation are strongly linked to gut colonization with a diverse microbiota (Macpherson et al. 2000; Fagarasan, 2006; Mora et al. 2006; Macpherson & Slack, 2007). Phenotypic differentiation of T cells is also coordinated through microbial sensing, demonstrated, for example, by development of Th17 cells in response to segmented filamentous bacteria (SFB) in mice (Ivanov et al. 2008). Moreover, microbial components, such as polysaccharide A (PSA) or cocktails of Clostridia species are potent inducers of regulatory T cells (Mazmanian et al. 2008; Round & Mazmanian, 2010; Atarashi et al. 2011, 2013). Chung et al. colonized germ‐free mice with either human‐ or mouse‐derived microbiota (Chung et al. 2012). Human versus mouse colonization results in different recipient microbial community profiles, which induces different numbers and transcriptomic profiles of mucosal T cells (Chung et al. 2012). Furthermore, gut bacteria heavily influence Treg cell development and may be of particular importance in preventing bacterial driven mouse models of colitis (Thorstenson & Khoruts, 2001). We are just beginning to appreciate the influence of microbial colonization on the modulation of mucosal B and T cell development and function. Understanding the mechanisms involved and the impact of host genetics on this process will provide opportunities for personalized treatment of IBD patients.
Animal models of IBD
The constant exposure of the intestinal tissue to gut microorganisms maintains the mucosa in a state of minimal “physiological inflammation”, which balances tolerogenic and pro‐inflammatory responses to maintain homeostasis. IBD is thought to result from an imbalance in this response to commensal microbes causing mucosal damage. Mouse models of intestinal inflammation have facilitated investigations of the role of host–microorganism interactions on the development and regulation of disease. These include pathways involved in the maintenance of intestinal epithelial barrier integrity, the promotion of protective and tolerant immune responses within the intestinal mucosa and regulation of the microbiota (Philpott et al. 2014).
Over 100 animal models of colitis exist (Strober et al. 2002; Jiminez et al. 2015), including those that are genetically driven, chemically induced or immune mediated, all of which are represented in mouse models of colitis. Genetically driven models include the interleukin‐10 (IL‐10) deficient mouse, whose inability to produce IL‐10 results in uncontrolled inflammation in the gut (Kuhn et al. 1993; Sellon et al. 1998), the SAMP1/YitFc mouse, which develops spontaneous ileitis (Pizarro et al. 2011), the TRUC mouse, whose deficiency in both T‐bet and Rag2 results in exacerbated TNFα responses and a colitogenic microbiota (Garrett et al. 2007), and the Mdr1a−/− mouse, which lacks P‐glycoprotein 170, resulting in increased gut permeability, microbial translocation and colitis development (Panwala et al. 1998). Chemically induced models involve oral or rectal administration of a compound that induces epithelial damage, allowing for microbial translocation and immune activation. These include administration of dextran sulfate sodium (DSS), piroxicam or 2,4,6‐trinitrobenzene sulfonic acid. Models that are driven primarily by immune activation include the T cell transfer model, where transfer of naive T cells into a lymphopenic recipient results in wasting disease that can be prevented by co‐transfer of regulatory T cells (Powrie et al. 1993, 1994) and anti‐CD3ε monoclonal antibody model, which induces acute small intestinal mucosal damage through T cell activation and a cytokine storm (Merger et al. 2002; Zhou et al. 2004). Infections that drive intestinal inflammation, including Citrobacter rodentium or rotavirus, are also models used to study the inflammatory response in the gut (Little & Shadduck, 1982; Higgins et al. 1999; Franco et al. 2006; Collins et al. 2014). Interestingly, many mouse models of colitis fail to develop disease under germ‐free conditions, including IL‐10−/− mice (Sellon et al. 1998), IL‐2−/− mice (Schultz et al. 1999), T cell receptor‐α deficient mice (Dianda et al. 1997) and T cell transfer colitis (Aranda et al. 1997). This indicates that the inflammatory response or perpetuation of the inflammation is driven in large part by the microbes (Kuhn et al. 1993; Simpson et al. 1998; Macpherson & Harris, 2004).
Recently, the use of gnotobiotic mice colonized with human‐derived microbiota has become a popular way to explore the role of specific human microbes on immune modulation and host–microbe interactions (Geva‐Zatorsky et al. 2017). Human microbiota‐associated mice have been used to model recurrent Clostridium difficile infections (Collins et al. 2015), asthma (Arrieta et al. 2015) and, most famously, obesity (Ridaura et al. 2013). A study investigating the effect of UC‐derived microbiota in humanized mice found expansion of Th17 cells and related gene expression, and increased sensitivity to DSS colitis compared to mice colonized with healthy donor microbiota (Natividad et al. 2015). Similar findings were observed in mice colonized with CD‐derived microbiota, where the microbes increased pro‐inflammatory immune responses but did not induce overt pathology in gnotobiotic wild‐type mice (Nagao‐Kitamoto et al. 2016). However, CD‐microbiota did induce severe colitis when used to colonize germ‐free IL‐10−/− mice. Thus, while colonization of germ‐free mice with IBD‐derived microbiota does not result in spontaneous inflammation, it increases susceptibility to induced colitis, possibly through increasing the pro‐inflammatory status of mucosal immune cells.
Although mutations in the NOD2 gene represent the strongest genetic link to Crohn's disease, Nod2‐deficient mice do not spontaneously develop colitis (Ogura et al. 2003b). Work in our laboratory has shown that Nod2−/− T cells show no overt functional defect in terms of proliferative and suppressive function and cytokine production (Zanello et al. 2013). However, there are conflicting results on whether Nod2−/− mice are more or less susceptible to various models of intestinal inflammation (Table 1). Ramanan et al. suggested that Nod2−/− mice harbouring Bacteroides vulgatus had increased piroxicam‐induced small intestinal damage (Ramanan et al. 2014). This correlates with our laboratory's recent finding that Nod2−/− mice had delayed epithelial recovery and prolonged small intestinal mucosal damage following intraperitoneal injection of anti‐CD3ε monoclonal antibody (mAb) (Zanello et al. 2016). Conversely, we were unable to identify a difference in susceptibility to T cell transfer colitis using Nod2−/− T cells (Zanello et al. 2013), and Amendola et al. identified a protective effect of Nod2 deficiency in 2,4,6‐trinitrobenzene sulfonic acid induced colitis (Amendola et al. 2014). These findings suggest that Nod2 may play a more significant role in modulating small intestinal rather than colonic inflammation. On the other hand, Natividad et al. showed that Nod1−/−;Nod2−/− (NOD‐DKO) mice have increased epithelial permeability, which leads to an exacerbated response to DSS colitis (Natividad et al. 2012). The response to infection by bacteria, viruses and parasites also varies significantly in Nod2‐deficient mice (Al Nabhani et al. 2017). Therefore, under the appropriate conditions, there could be a perpetuated inflammatory response in the gut related to the Nod2 mutation. Proper control of environmental variables, including the use of heterozygous‐derived littermate mice to equalize the microbiota and early life environmental exposures between wild‐type and Nod2‐deficient mice, will help to better elucidate the role of Nod2 in intestinal homeostasis and inflammation.
Table 1.
Susceptibility of Nod2‐deficient mice to models of intestinal inflammation
| Model of intestinal inflammation | Susceptibility in Nod2‐deficient mice | Inflammatory response | Location of inflammation | Use of littermates | Reference |
|---|---|---|---|---|---|
| Anti‐CD3ε mAb | Increased | Increased IL‐17A and myeloperoxidase (MPO) | Small intestine | Yes | (Zanello et al. 2016) |
| Piroxicam | Increased | Increased interferon gamma (IFN‐γ) | Small intestine | No | (Ramanan et al. 2014) |
| Citrobacter rodentium | Increased | Decreased IL‐17A | Caecum | No | (Geddes et al. 2011) |
| T cell transfer | No difference or reduced | None or decreased IFN‐γ | Colon | No | (Shaw et al. 2009; Zanello et al. 2013) |
| Dextran sulfate sodium (DSS) | Increased | Increased IL‐6 | Colon | No | (Natividad et al. 2012; Couturier‐Maillard et al. 2013) |
| 2,4,6‐Trinitrobenzene sulfonic acid (TNBS) | Reduced | Decreased IL‐17A | Colon | No | (Amendola et al. 2014) |
Concluding remarks
IBD is complex, multifactorial and likely no one causative agent will be identified. Instead, a combination of genetic predisposition, environmental exposures and the gut microbiota will culminate in an unfortunate perfect storm for some individuals, resulting in disease development. In order to identify new therapeutic targets and develop preventative and therapeutic strategies, a better understanding of how these factors influence each other and come together to initiate IBD pathogenesis is required. NOD2 has remained the strongest genetic risk factor associated with CD development for nearly two decades, although exactly how it is related to disease onset remains elusive. Its involvement in microbial sensing, innate and adaptive immune activation, plus its role in autophagy, the gut epithelial barrier and shaping the gut microbiota suggest that it is a versatile protein with many roles in IBD pathogenesis. To this end, further investigation into the multi‐faceted roles of NOD2 is required, at both the basic science and clinical level.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
D.J.P. is supported by the Canadian Institute for Health Research (CIHR), including a CIHR Team Grant on Chronic Inflammation (THC‐13523).
Biographies
Ashleigh Goethel obtained her PhD in immunology from the University of Toronto, where she studied the effects of antibiotic exposure on host immune–microbiota crosstalk and the development of colitis in the laboratories of Dr Ken Croitoru and Dr Dana Philpott.

Dana Philpott is a Professor in the Department of Immunology and co‐director of the Host‐Microbiome Research Network at the University of Toronto. Her research employs animal models of inflammatory bowel disease and considers how innate immunity and the microbiome shape immune homeostasis within the intestine.
Edited by: Ole Petersen & David Grundy
This review was presented at the symposium ‘Gastrointestinal Tract XVII: Current Biology of the GI Tract, Mucosa, Microbiota, and Beyond’, which took place at FASEB 2017, Steamboat Springs, Colorado, USA, 30 July–4 August 2017.
This is an Editor's Choice article from the 1 September 2018 issue.
References
- Al Nabhani Z, Dietrich G, Hugot JP & Barreau F (2017). Nod2: The intestinal gate keeper. PLoS Pathog 13, e1006177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola A, Butera A, Sanchez M, Strober W & Boirivant M (2014). Nod2 deficiency is associated with an increased mucosal immunoregulatory response to commensal microorganisms. Mucosal Immunol 7, 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda R, Sydora BC, McAllister PL, Binder SW, Yang HY, Targan SR & Kronenberg M (1997). Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J Immunol 158, 3464–3473. [PubMed] [Google Scholar]
- Arrieta MC, Stiemsma LT, Amenyogbe N, Brown EM & Finlay B (2014). The intestinal microbiome in early life: health and disease. Front Immunol 5, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist‐Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL, Subbarao P, Mandhane P, Becker A, McNagny KM, Sears MR, Kollmann T, CHILD Study Investigators , Mohn WW, Turvey SE & Finlay BB (2015). Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med 7, 307ra152. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M & Honda K (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K & Honda K (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF & Gordon JI (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101, 15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van de Wetering M & Clevers H (2008). The intestinal stem cell. Genes Dev 22, 1856–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y & Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, Lieber AD, Wu F, Perez‐Perez GI, Chen Y, Schweizer W, Zheng X, Contreras M, Dominguez‐Bello MG & Blaser MJ (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 8, 343ra382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ & Mayer L (2007). The immune response in inflammatory bowel disease. Am J Gastroenterol 102, 2058–2069. [DOI] [PubMed] [Google Scholar]
- Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, Kuechler I, Krueger S, Schmidt HH & Lochs H (2006). Genetic basis for increased intestinal permeability in families with Crohn's disease: role of CARD15 3020insC mutation? Gut 55, 342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS & Virgin HW 4th (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J & Knight R (2010). QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carding S, Verbeke K, Vipond DT, Corfe BM & Owen LJ (2015). Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 26, 26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case RJ, Boucher Y, Dahllof I, Holmstrom C, Doolittle WF & Kjelleberg S (2007). Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl Environ Microbiol 73, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I & Blaser MJ (2012). The human microbiome: at the interface of health and disease. Nat Rev Genet 13, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, Umesaki Y, Mathis D, Benoist C, Relman DA & Kasper DL (2012). Gut immune maturation depends on colonization with a host‐specific microbiota. Cell 149, 1578–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claesson MJ, O'Sullivan O, Wang Q, Nikkila J, Marchesi JR, Smidt H, de Vos WM, Ross RP & O'Toole PW (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4, e6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J, Auchtung JM, Schaefer L, Eaton KA & Britton RA (2015). Humanized microbiota mice as a model of recurrent Clostridium difficile disease. Microbiome 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB & Frankel G (2014). Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 12, 612–623. [DOI] [PubMed] [Google Scholar]
- Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D & Simmons A (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16, 90–97. [DOI] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI & Knight R (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier‐Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye‐Crespin A, Gaillot O, Schreiber S, Lemoine Y, Ryffel B, Hot D, Nunez G, Chen G, Rosenstiel P & Chamaillard M (2013). NOD2‐mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crohn's and Colitis Foundation of Canada (2012). The Impact of Inflammatory Bowel Disease in Canada. http://www.crohnsandcolitis.ca/Crohns_and_Colitis/documents/reports/ccfc-ibd-impact-report-2012.pdf.
- Cui L, Morris A & Ghedin E (2013). The human mycobiome in health and disease. Genome Med 5, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez‐Achury J, Ji SG, Heap G, Nimmo ER, Edwards C, Henderson P, Mowat C, Sanderson J, Satsangi J, Simmons A, Wilson DC, Tremelling M, Hart A, Mathew CG, Newman WG, Parkes M, Lees CW, Uhlig H, Hawkey C, Prescott NJ, Ahmad T, Mansfield JC, Anderson CA & Barrett JC (2017). Genome‐wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 49, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianda L, Hanby AM, Wright NA, Sebesteny A, Hayday AC & Owen MJ (1997). T cell receptor‐alpha beta‐deficient mice fail to develop colitis in the absence of a microbial environment. Am J Pathol 150, 91–97. [PMC free article] [PubMed] [Google Scholar]
- Duerkop BA, Vaishnava S & Hooper LV (2009). Immune responses to the microbiota at the intestinal mucosal surface. Immunity 31, 368–376. [DOI] [PubMed] [Google Scholar]
- Fagarasan S (2006). Intestinal IgA synthesis: a primitive form of adaptive immunity that regulates microbial communities in the gut. Curr Top Microbiol Immunol 308, 137–153. [DOI] [PubMed] [Google Scholar]
- Franchi L, Warner N, Viani K & Nunez G (2009). Function of Nod‐like receptors in microbial recognition and host defense. Immunol Rev 227, 106–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francino MP (2014). Early development of the gut microbiota and immune health. Pathogens 3, 769–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco MA, Angel J & Greenberg HB (2006). Immunity and correlates of protection for rotavirus vaccines. Vaccine 24, 2718–2731. [DOI] [PubMed] [Google Scholar]
- Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, Pace NR & Li E (2011). Disease phenotype and genotype are associated with shifts in intestinal‐associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 17, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N & Pace NR (2007). Molecular‐phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104, 13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz JH, Ferrero RL, Philpott DJ & Girardin SE (2006). Nod‐like proteins in immunity, inflammation and disease. Nat Immunol 7, 1250–1257. [DOI] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo‐Villarino G, Mazmanian SK, Ito S, Glickman JN & Glimcher LH (2007). Communicable ulcerative colitis induced by T‐bet deficiency in the innate immune system. Cell 131, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes K, Rubino SJ, Magalhaes JG, Streutker C, Le Bourhis L, Cho JH, Robertson SJ, Kim CJ, Kaul R, Philpott DJ & Girardin SE (2011). Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 17, 837–844. [DOI] [PubMed] [Google Scholar]
- Gerbe F & Jay P (2016). Intestinal tuft cells: epithelial sentinels linking luminal cues to the immune system. Mucosal Immunol 9, 1353–1359. [DOI] [PubMed] [Google Scholar]
- Geva‐Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz‐Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D, Benoist C & Kasper DL (2017). Mining the human gut microbiota for immunomodulatory organisms. Cell 168, 928–943.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vazquez‐Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, Gonzalez A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R & Xavier RJ (2014). The treatment‐naive microbiome in new‐onset Crohn's disease. Cell Host Microbe 15, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask‐Madsen J, Rutgeerts P, Vyhnalek P, Zadorova Z, Palmer T & Donoghue S; Natalizumab Pan‐European Study Group (2003). Natalizumab for active Crohn's disease. N Engl J Med 348, 24–32. [DOI] [PubMed] [Google Scholar]
- Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ & Sansonetti PJ (2003). Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278, 8869–8872. [DOI] [PubMed] [Google Scholar]
- Halfvarson J, Bodin L, Tysk C, Lindberg E & Jarnerot G (2003). Inflammatory bowel disease in a Swedish twin cohort: a long‐term follow‐up of concordance and clinical characteristics. Gastroenterology 124, 1767–1773. [DOI] [PubMed] [Google Scholar]
- Hansson GC & Johansson ME (2010). The inner of the two Muc2 mucin‐dependent mucus layers in colon is devoid of bacteria. Gut Microbes 1, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedl M, Li J, Cho JH & Abraham C (2007). Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci U S A 104, 19440–19445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins LM, Frankel G, Douce G, Dougan G & MacDonald TT (1999). Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun 67, 3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt PG & Jones CA (2000). The development of the immune system during pregnancy and early life. Allergy 55, 688–697. [DOI] [PubMed] [Google Scholar]
- Hooper LV & Gordon JI (2001). Commensal host‐bacterial relationships in the gut. Science 292, 1115–1118. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Littman DR & Macpherson AJ (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent‐Puig P, Gower‐Rousseau C, Macry J, Colombel JF, Sahbatou M & Thomas G (2001). Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 411, 599–603. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Kostic AD & Xavier RJ (2014). Inflammatory bowel disease as a model for translating the microbiome. Immunity 40, 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB & Littman DR (2008). Specific microbiota direct the differentiation of IL‐17‐producing T‐helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A & Medzhitov R (2004). Toll‐like receptor control of the adaptive immune responses. Nat Immunol 5, 987–995. [DOI] [PubMed] [Google Scholar]
- Jacobs JP, Goudarzi M, Singh N, Tong M, McHardy IH, Ruegger P, Asadourian M, Moon BH, Ayson A, Borneman J, McGovern DP, Fornace AJ Jr, Braun J & Dubinsky M (2016). A disease‐associated microbial and metabolomics state in relatives of pediatric inflammatory bowel disease patients. Cell Mol Gastroenterol Hepatol 2, 750–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiminez JA, Uwiera TC, Douglas Inglis G & Uwiera RR (2015). Animal models to study acute and chronic intestinal inflammation in mammals. Gut Pathog 7, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson ME, Larsson JM & Hansson GC (2011). The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host‐microbial interactions. Proc Natl Acad Sci U S A 108, Suppl. 1, 4659–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D'Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford‐Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC & Cho JH (2012). Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan GG (2015). The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol 12, 720–727. [DOI] [PubMed] [Google Scholar]
- Kevans D, Turpin W, Madsen K, Meddings J, Shestopaloff K, Xu W, Moreno‐Hagelsieb G, Griffiths A, Silverberg MS, Paterson A & Croitoru K; GEM Project (2015). Determinants of intestinal permeability in healthy first‐degree relatives of individuals with Crohn's disease. Inflamm Bowel Dis 21, 879–887. [DOI] [PubMed] [Google Scholar]
- Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G & Flavell RA (2005). Nod2‐dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734. [DOI] [PubMed] [Google Scholar]
- Kobori A, Yagi Y, Imaeda H, Ban H, Bamba S, Tsujikawa T, Saito Y, Fujiyama Y & Andoh A (2010). Interleukin‐33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol 45, 999–1007. [DOI] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K & Muller W (1993). Interleukin‐10‐deficient mice develop chronic enterocolitis. Cell 75, 263–274. [DOI] [PubMed] [Google Scholar]
- Lau JT, Whelan FJ, Herath I, Lee CH, Collins SM, Bercik P & Surette MG (2016). Capturing the diversity of the human gut microbiota through culture‐enriched molecular profiling. Genome Med 8, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Zouali H, Cezard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O'Morain C, Gassull M, Binder V, Finkel Y, Modigliani R, Gower‐Rousseau C, Macry J, Merlin F, Chamaillard M, Jannot AS, Thomas G & Hugot JP; EPWG‐IBD Group; EPIMAD Group; GETAID Group (2002). CARD15/NOD2 mutational analysis and genotype‐phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet 70, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ES, Zhou Y, Zhao G, Bauer IK, Droit L, Ndao IM, Warner BB, Tarr PI, Wang D & Holtz LR (2015). Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat Med 21, 1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin GH, Wortzman ME, Girardin SE, Philpott DJ & Watts TH (2013). T cell intrinsic NOD2 is dispensable for CD8 T cell immunity. PLoS One 8, e56014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little LM & Shadduck JA (1982). Pathogenesis of rotavirus infection in mice. Infect Immun 38, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littman DR & Pamer EG (2011). Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 10, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, Cho J, Daryani NE, Franke L, Fuyuno Y, Hart A, Juyal RC, Juyal G, Kim WH, Morris AP, Poustchi H, Newman WG, Midha V, Orchard TR, Vahedi H, Sood A, Sung JJ, Malekzadeh R, Westra HJ, Yamazaki K, Yang SK; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium , Barrett JC, Franke A, Alizadeh BZ, Parkes M, Bk T, Daly MJ, Kubo M, Anderson CA & Weersma RK (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47, 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M & Hershberg RM (2004). Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest 113, 1296–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H & Zinkernagel RM (2000). A primitive T cell‐independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ & Harris NL (2004). Interactions between commensal intestinal bacteria and the immune system. Nat Rev Immunol 4, 478–485. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ & Slack E (2007). The functional interactions of commensal bacteria with intestinal secretory IgA. Curr Opin Gastroenterol 23, 673–678. [DOI] [PubMed] [Google Scholar]
- Macpherson AJ, Slack E, Geuking MB & McCoy KD (2009). The mucosal firewalls against commensal intestinal microbes. Semin Immunopathol 31, 145–149. [DOI] [PubMed] [Google Scholar]
- Manichanh C, Borruel N, Casellas F & Guarner F (2012). The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9, 599–608. [DOI] [PubMed] [Google Scholar]
- Manichanh C, Rigottier‐Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J & Dore J (2006). Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 55, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazmanian SK, Round JL & Kasper DL (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625. [DOI] [PubMed] [Google Scholar]
- Merger M, Viney JL, Borojevic R, Steele‐Norwood D, Zhou P, Clark DA, Riddell R, Maric R, Podack ER & Croitoru K (2002). Defining the roles of perforin, Fas/FasL, and tumour necrosis factor alpha in T cell induced mucosal damage in the mouse intestine. Gut 51, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middelhoff M, Westphalen CB, Hayakawa Y, Yan KS, Gershon MD, Wang TC & Quante M (2017). Dclk1‐expressing tuft cells: critical modulators of the intestinal niche? Am J Physiol Gastrointest Liver Physiol 313, G285–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD & Bushman FD (2013). Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A 110, 12450–12455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora JR, Iwata M, Eksteen B, Song SY, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi‐Castagnoli P, Rajewsky K, Adams DH & von Andrian UH (2006). Generation of gut‐homing IgA‐secreting B cells by intestinal dendritic cells. Science 314, 1157–1160. [DOI] [PubMed] [Google Scholar]
- Mowat AM & Agace WW (2014). Regional specialization within the intestinal immune system. Nat Rev Immunol 14, 667–685. [DOI] [PubMed] [Google Scholar]
- Nagao‐Kitamoto H, Shreiner AB, Gillilland MG 3rd, Kitamoto S, Ishii C, Hirayama A, Kuffa P, El‐Zaatari M, Grasberger H, Seekatz AM, Higgins PD, Young VB, Fukuda S, Kao JY & Kamada N (2016). Functional characterization of inflammatory bowel disease‐associated gut dysbiosis in gnotobiotic mice. Cell Mol Gastroenterol Hepatol 2, 468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natividad JM, Petit V, Huang X, de Palma G, Jury J, Sanz Y, Philpott D, Garcia Rodenas CL, McCoy KD & Verdu EF (2012). Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1− / −; Nod2− / − mice. Inflamm Bowel Dis 18, 1434–1446. [DOI] [PubMed] [Google Scholar]
- Natividad JM, Pinto‐Sanchez MI, Galipeau HJ, Jury J, Jordana M, Reinisch W, Collins SM, Bercik P, Surette MG, Allen‐Vercoe E & Verdu EF (2015). Ecobiotherapy rich in Firmicutes decreases susceptibility to colitis in a humanized gnotobiotic mouse model. Inflamm Bowel Dis 21, 1883–1893. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Finotto S, Fuss I, Boirivant M, Galle PR & Strober W (2001). Regulation of T‐cell apoptosis in inflammatory bowel disease: to die or not to die, that is the mucosal question. Trends Immunol 22, 21–26. [DOI] [PubMed] [Google Scholar]
- Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, Sung JJY & Kaplan GG (2018). Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population‐based studies. Lancet 390, 2769–2778. [DOI] [PubMed] [Google Scholar]
- Nigro G, Rossi R, Commere PH, Jay P & Sansonetti PJ (2014). The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 15, 792–798. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G & Cho JH (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411, 603–606. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, Greenson JK, Keshav S & Nunez G (2003a). Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut 52, 1591–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Saab L, Chen FF, Benito A, Inohara N & Nunez G (2003b). Genetic variation and activity of mouse Nod2, a susceptibility gene for Crohn's disease. Genomics 81, 369–377. [DOI] [PubMed] [Google Scholar]
- Orholm M, Binder V, Sorensen TI, Rasmussen LP & Kyvik KO (2000). Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand J Gastroenterol 35, 1075–1081. [DOI] [PubMed] [Google Scholar]
- Oyri SF, Muzes G & Sipos F (2015). Dysbiotic gut microbiome: A key element of Crohn's disease. Comp Immunol Microbiol Infect Dis 43, 36–49. [DOI] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, Ruggiero E, Cho JH, Goodman AL & Flavell RA (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panwala CM, Jones JC & Viney JL (1998). A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol 161, 5733–5744. [PubMed] [Google Scholar]
- Peeters M, Geypens B, Claus D, Nevens H, Ghoos Y, Verbeke G, Baert F, Vermeire S, Vlietinck R & Rutgeerts P (1997). Clustering of increased small intestinal permeability in families with Crohn's disease. Gastroenterology 113, 802–807. [DOI] [PubMed] [Google Scholar]
- Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA & Stobberingh EE (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. [DOI] [PubMed] [Google Scholar]
- Peterson LW & Artis D (2014). Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14, 141–153. [DOI] [PubMed] [Google Scholar]
- Petrof EO, Gloor GB, Vanner SJ, Weese SJ, Carter D, Daigneault MC, Brown EM, Schroeter K & Allen‐Vercoe E (2013). Stool substitute transplant therapy for the eradication of Clostridium difficile infection: ‘RePOOPulating’ the gut. Microbiome 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petterson T, Jendholm J, Mansson A, Bjartell A, Riesbeck K & Cardell LO (2011). Effects of NOD‐like receptors in human B lymphocytes and crosstalk between NOD1/NOD2 and Toll‐like receptors. J Leukoc Biol 89, 177–187. [DOI] [PubMed] [Google Scholar]
- Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K & Girardin SE (2014). NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol 14, 9–23. [DOI] [PubMed] [Google Scholar]
- Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, Chieppa M, Arseneau KO, Ley K & Cominelli F (2011). SAMP1/YitFc mouse strain: a spontaneous model of Crohn's disease‐like ileitis. Inflamm Bowel Dis 17, 2566–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt AM & Mowat AM (2008). Mucosal macrophages and the regulation of immune responses in the intestine. Immunol Lett 119, 22–31. [DOI] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Caddle LB & Coffman RL (1993). Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B‐17 scid mice. Int Immunol 5, 1461–1471. [DOI] [PubMed] [Google Scholar]
- Powrie F, Leach MW, Mauze S, Menon S, Caddle LB & Coffman RL (1994). Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562. [DOI] [PubMed] [Google Scholar]
- Pullan RD, Thomas GA, Rhodes M, Newcombe RG, Williams GT, Allen A & Rhodes J (1994). Thickness of adherent mucus gel on colonic mucosa in humans and its relevance to colitis. Gut 35, 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan D, Tang MS, Bowcutt R, Loke P & Cadwell K (2014). Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus . Immunity 41, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC & Gordon JI (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D & Schreiber S (2003). TNF‐alpha and IFN‐gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 124, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Round JL & Mazmanian SK (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 9, 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL & Mazmanian SK (2010). Inducible Foxp3+ regulatory T‐cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107, 12204–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino SJ, Selvanantham T, Girardin SE & Philpott DJ (2012). Nod‐like receptors in the control of intestinal inflammation. Curr Opin Immunol 24, 398–404. [DOI] [PubMed] [Google Scholar]
- Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, Xiang Y & Bose S (2009). Activation of innate immune antiviral responses by Nod2. Nat Immunol 10, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor RB (2004). Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics. Gastroenterology 126, 1620–1633. [DOI] [PubMed] [Google Scholar]
- Sartor RB (2006). Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol 3, 390–407. [DOI] [PubMed] [Google Scholar]
- Schultz M, Tonkonogy SL, Sellon RK, Veltkamp C, Godfrey VL, Kwon J, Grenther WB, Balish E, Horak I & Sartor RB (1999). IL‐2‐deficient mice raised under germfree conditions develop delayed mild focal intestinal inflammation. Am J Physiol 276, G1461–G1472. [DOI] [PubMed] [Google Scholar]
- Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM & Sartor RB (1998). Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin‐10‐deficient mice. Infect Immun 66, 5224–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S & Milo R (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14, e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw MH, Reimer T, Sanchez‐Valdepenas C, Warner N, Kim YG, Fresno M & Nunez G (2009). T cell‐intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii . Nat Immunol 10, 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff KE & Cebra JJ (1995). Development of mucosal humoral immune responses in germ‐free (GF) mice. Adv Exp Med Biol 371A, 441–446. [DOI] [PubMed] [Google Scholar]
- Simpson SJ, de Jong YP, Comiskey M & Terhorst C (1998). T cells in mouse models of gut inflammation. Chem Immunol 71, 118–138. [DOI] [PubMed] [Google Scholar]
- Soderholm JD, Olaison G, Lindberg E, Hannestad U, Vindels A, Tysk C, Jarnerot G & Sjodahl R (1999). Different intestinal permeability patterns in relatives and spouses of patients with Crohn's disease: an inherited defect in mucosal defence? Gut 44, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez‐Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Dore J, Marteau P, Seksik P & Langella P (2008). Faecalibacterium prausnitzii is an anti‐inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 105, 16731–16736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokol H, Seksik P, Furet JP, Firmesse O, Nion‐Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P & Dore J (2009). Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis 15, 1183–1189. [DOI] [PubMed] [Google Scholar]
- Sommer F & Backhed F (2013). The gut microbiota – masters of host development and physiology. Nat Rev Microbiol 11, 227–238. [DOI] [PubMed] [Google Scholar]
- Steenwinckel V, Louahed J, Lemaire MM, Sommereyns C, Warnier G, McKenzie A, Brombacher F, Van Snick J & Renauld JC (2009). IL‐9 promotes IL‐13‐dependent Paneth cell hyperplasia and up‐regulation of innate immunity mediators in intestinal mucosa. J Immunol 182, 4737–4743. [DOI] [PubMed] [Google Scholar]
- Strober W, Fuss IJ & Blumberg RS (2002). The immunology of mucosal models of inflammation. Ann Rev Immunol 20, 495–549. [DOI] [PubMed] [Google Scholar]
- Tamburini S, Shen N, Wu HC & Clemente JC (2016). The microbiome in early life: implications for health outcomes. Nat Med 22, 713–722. [DOI] [PubMed] [Google Scholar]
- Thorstenson KM & Khoruts A (2001). Generation of anergic and potentially immunoregulatory CD25+CD4 T cells in vivo after induction of peripheral tolerance with intravenous or oral antigen. J Immunol 167, 188–195. [DOI] [PubMed] [Google Scholar]
- Tlaskalova‐Hogenova H, Stepankova R, Kozakova H, Hudcovic T, Vannucci L, Tuckova L, Rossmann P, Hrncir T, Kverka M, Zakostelska Z, Klimesova K, Pribylova J, Bartova J, Sanchez D, Fundova P, Borovska D, Srutkova D, Zidek Z, Schwarzer M, Drastich P & Funda DP (2011). The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: contribution of germ‐free and gnotobiotic animal models of human diseases. Cell Mol Immunol 8, 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas J, Wrzosek L, Bouznad N, Bouet S, Mayeur C, Noordine ML, Honvo‐Houeto E, Langella P, Thomas M & Cherbuy C (2013). Primocolonization is associated with colonic epithelial maturation during conventionalization. FASEB J 27, 645–655. [DOI] [PubMed] [Google Scholar]
- Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE & Philpott DJ (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11, 55–62. [DOI] [PubMed] [Google Scholar]
- VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, Xavier RJ, McGovern DP & Stappenbeck TS (2014). Genetic variants synthesize to produce Paneth cell phenotypes that define subtypes of Crohn's disease. Gastroenterology 146, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Kitani A, Murray PJ & Strober W (2004). NOD2 is a negative regulator of Toll‐like receptor 2‐mediated T helper type 1 responses. Nat Immunol 5, 800–808. [DOI] [PubMed] [Google Scholar]
- Zanello G, Goethel A, Forster K, Geddes K, Philpott DJ & Croitoru K (2013). Nod2 activates NF‐kB in CD4+ T cells but its expression is dispensable for T cell‐induced colitis. PLoS One 8, e82623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanello G, Goethel A, Rouquier S, Prescott D, Robertson SJ, Maisonneuve C, Streutker C, Philpott DJ & Croitoru K (2016). The cytosolic microbial receptor Nod2 regulates small intestinal crypt damage and epithelial regeneration following T cell‐induced enteropathy. J Immunol 197, 345–355. [DOI] [PubMed] [Google Scholar]
- Zhou P, Streutker C, Borojevic R, Wang Y & Croitoru K (2004). IL‐10 modulates intestinal damage and epithelial cell apoptosis in T cell‐mediated enteropathy. Am J Physiol Gastrointest Liver Physiol 287, G599–G604. [DOI] [PubMed] [Google Scholar]