

Abstract
SAMHD1 is a deoxynucleotide triphosphate (dNTP) hydrolase that plays an important role in the homeostatic balance of cellular dNTPs. Its emerging role as an effector of innate immunity is affirmed by mutations in the SAMHD1 gene that cause the severe autoimmune disease, Aicardi-Goutieres syndrome (AGS) and that are linked to cancer. Additionally, SAMHD1 functions as a restriction factor for retroviruses such as HIV. Here we review the current biochemical and biological properties of the enzyme including its structure, activity, and regulation by post-translational modifications in the context of its cellular function. We outline open questions regarding the biology of SAMHD1 whose answers will be important for understanding its function as a regulator of cell cycle progression, genomic integrity, and in autoimmunity.
Keywords: SAMHD1, dNTP metabolism, nucleotide, autoimmune disease, Aicardi-Goutieres syndrome, HIV
Deoxynucleotide Metabolism Overview
Proper regulation of intracellular deoxynucleotide triphosphates (dNTPs) is essential for normal cellular metabolism. dNTPs differ by only a single atom from ribonucleotide triphosphates (NTPs) yet are maintained at 10–1000 times lower concentration (1). A balanced supply of each of the four canonical dNTPs maintained at proper concentrations is required for accurate genomic and mitochondrial DNA synthesis and repair (2–5). As such, nucleotide metabolism is precisely regulated within cells. Organisms have evolved complex and dynamic mechanisms and checkpoints to control the synthesis and degradation of dNTPs (Fig. 1) (6, 7). The enzymes involved in these pathways are frequently regulated in a concentration dependent manner by their respective substrates and products (8–12). Synthesis of dNTPs is accomplished by way of two distinct pathways; the de novo synthesis and the salvage pathways (2, 4). During de novo synthesis, cytosolic ribonucleotide diphosphates are reduced to deoxynucleotide diphosphates (dNDPs) by the enzyme ribonucleotide reductase (RNR) (13). dNDPs are then phosphorylated by cytosolic or mitochondrial kinases. This process is dramatically upregulated during S-phase, when the cell requires large amounts of dNTPs to replicate its DNA, and intracellular dNTPs concentrations increase 5–10 fold over early G1 levels (2, 6, 7). De novo synthesis of nucleotide triphosphates is also important for supplementing intracellular nucleotide pools required for genome maintenance during other stages of the cell cycle and is carried out by a second isoform of RNR regulatory subunit at a reduced rate (14–17). The salvage pathway is constitutively active and performed in parallel in both the cytosol and mitochondrial compartments. It is essential for maintaining dNTP pool levels for nuclear and mitochondrial DNA synthesis and repair in resting cells by recycling cellular deoxynucleosides that are products of nucleic acid degradation, cellular nucleotidases, or imported from the extracellular space (6).
Figure 1. Overview of dNTP metabolism pathways and enzymes.
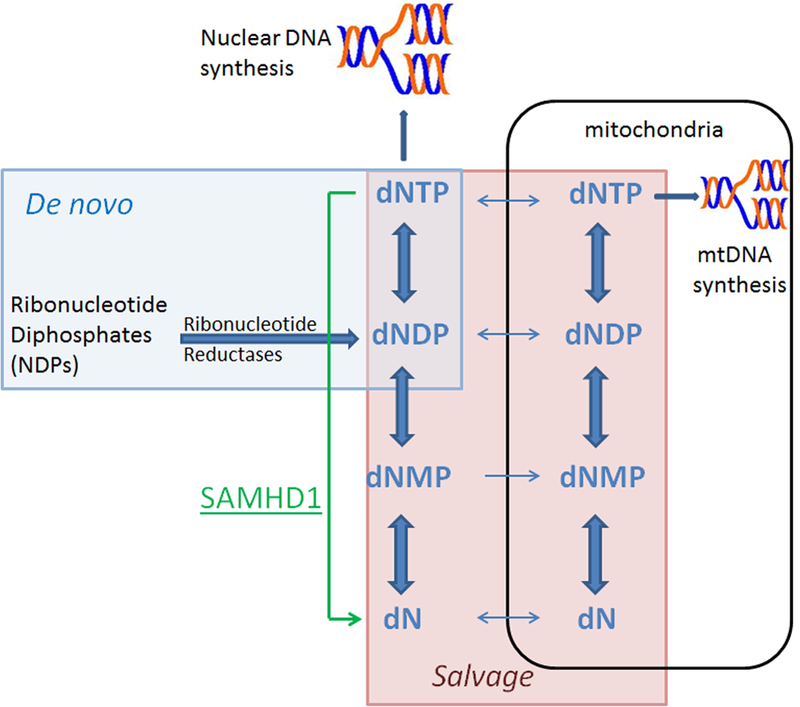
The de novo pathway of dNTP synthesis is shaded blue. The salvage pathway shaded in red consists of complementary pathways in the cytosol and mitochondria. Cellular kinases phosphorylate deoxynucleosides (dN) and deoxynucleotides (dNMP, dNDP), while 5’-deoxynucleotidases and phosphorylases catalyze the opposing reactions which degrade nucleotides to help maintain a homeostatic balance. The SAMHD1 triphosphohydrolase reaction is highlighted in green.
Opposing the de novo and salvage anabolic pathways, various enzymes function together to comprise the catabolic pathways that facilitate the degradation of dNTPs. 5’-nucleotidases, nucleoside phosphorylases, and deaminases are responsible for breaking down dNTPs into the constituent deoxynucleoside or nucleobase, at which point they can reenter the salvage pathway or be transported across the cell membrane in order to maintain the overall size and ratio of intracellular dNTPs(18). The opposing anabolic and catabolic pathways work in competing directions to maintain a dynamic equilibrium of intracellular dNTPs in a process referred to as substrate cycling (19). Isotope flow experiments of radiolabeled nucleotides have traced this phenomenon and elucidated its critical role as a regulatory mechanism for maintaining homeostatic dNTP levels (20–22). It is also important to note that cytosolic dNTP pools do not exist in isolation, but are directly related to mitochondrial dNTP pools. In fact, dNTP precursors transport between the two compartments and the nucleotide metabolism processes of one compartment can influence the other (23, 24).
Substrate cycling, DNA synthesis and repair, and nucleoside import and export form a dynamic regulatory system that maintains homeostatic dNTP concentrations within the cell. The levels are finely tuned according to the cell type and stage in the cell cycle. Each canonical dNTP is not equally abundant within the cell, however. dGTP is consistently observed to be least prevalent in eukaryotic cells (2, 4). While dNTP concentrations are asymmetric, the proper balance of the dNTP pool is crucial to cell survival and genomic integrity(25). Imbalanced dNTP pools, resulting from DNA damage, metabolic dysregulation, or mutations to the enzymes involved in maintaining dNTP equilibrium, can result in harmful consequences including: increased mutation rates and the induction of a mutator phenotype; increased DNA damage and activation of the DNA damage response; altered DNA polymerase kinetics, replication stress, and collapsed or stalled replication forks; altered replication origin spacing and usage; modified epigenetic profiles and gene expression; and restricted cell cycle progression (26–39). These molecular perturbations of genomic fidelity manifest themselves as severe pathologies including mitochondrial depletion syndromes, severe immunodeficiency disorders, induced cellular senescence, and cancer (40–47). Imbalanced dNTP pools may also impact apoptosis and inflammation, as dNTPs have been implicated in the activation of both pathways (47–50).
SAMHD1 Activation, Catalysis, and Regulation
SAMHD1 Activation and Catalysis.
Sterile Alpha Motif and Histidine-Aspartic acid domain containing protein 1 (SAMHD1) is a deoxynucleotide triphosphate (dNTP) hydrolase that catalyzes the hydrolysis of canonical dNTPs into the constituent nucleoside and inorganic tripolyphosphate. Through its dNTPase activity, SAMHD1 maintains dNTP pools within the cell at levels appropriate for DNA replication and repair but below a potentially mutagenic threshold. SAMHD1 was first discovered in 2000 as an ortholog of the mouse interferon-γ induced dendritic cell protein MG11 but the precise function of the enzyme remained unclear (51). It was not until almost 10 years after its initial discovery that SAMHD1 was implicated in nucleotide metabolism and demonstrated to exert a potent immunomodulatory effect (52). Subsequent biochemical analysis revealed the hydrolase catalytic activity of the enzyme, divalent metal ion dependence, and activation by guanosine nucleotides (53–57) (Fig. 2). Additionally, structural and functional similarities of SAMHD1 with several prokaryotic homologues have been observed (58–64). These bacterial enzymes provided an initial template for interrogating the cellular role of SAMHD1, as well as the structural mechanisms of activation and catalysis.
Figure 2. SAMHD1 is a deoxynucleotide triphosphohydrolase.
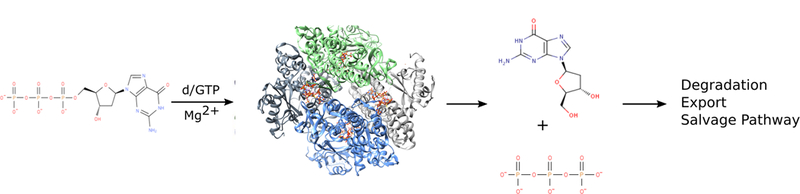
SAMHD1 hydrolyzes dNTPs in the presence of activating nucleotides and divalent metal cations into their cognate nucleoside and inorganic tripolyphosphate. Nucleosides are then degraded further, exported from the cell, or recycled through the nucleotide salvage pathway.
SAMHD1 is a 626 amino acid protein comprised of an N-terminal sterile alpha motif (SAM) and a histidine-aspartic acid containing domain (HD). While the role of the SAM domain remains an open question, SAM domains are commonly involved in protein-protein and protein-DNA/RNA interactions (65). A nuclear localization signal precedes the SAM domain and confers the nuclear occupancy observed in most studies (66, 67). The HD domain is defined by its characteristic quartet of metal coordinating histidine and aspartic acid residues within the enzyme active site. HD-domain containing proteins represent a superfamily of phosphohydrolases commonly involved in nucleic acid metabolism (68). The HD domain of SAMHD1 houses the dNTPase active site, regulatory sites, and the requisite interfaces for enzyme oligomerization. The C-terminus of SAMHD1 consists of a distinct region that is important for stabilizing the oligomeric state of the enzyme and nucleic acid interaction (69–71). X-ray crystallographic studies have illuminated the structural features and catalytic mechanism of SAMHD1 (Fig. 3). The apo-SAMHD1 protein exists in a monomer-dimer equilibrium, but tetramerizes in the presence of activating nucleotides in order to form the catalytically competent holoenzyme (57, 71, 72). Each SAMHD1 monomer contains two discrete regulatory sites (RS1 and RS2) and activating nucleotide triphosphates must sequentially bind at each site in order to induce a conformational shift that facilitates tetramerization and subsequent catalytic activation (57, 70, 71). The residues which constitute the RS1 pocket are uniquely structured such that only a guanosine triphosphate nucleotide is capable of binding with an estimated Kd between 0.1–0.4 μM (53, 55, 57, 73, 74). Given the 1000-fold excess of GTP over dGTP within cells, it is likely that RS1 is constitutively occupied by GTP as a primary activating nucleotide under physiological conditions (55, 56, 73). GTP binding at RS1 drives the oligomeric equilibrium toward the dimer as the GTP nucleotide forms contacts with both subunits. Further studies have suggested that constitutive binding of GTP by SAMHD1 can form relaxed tetramers that are primed for full assembly and high-efficiency catalysis upon binding of a second activating nucleotide at RS2 (75).
Figure 3. The sequential binding of regulatory nucleotides drives SAMHD1 tetramerization and catalytic activation.
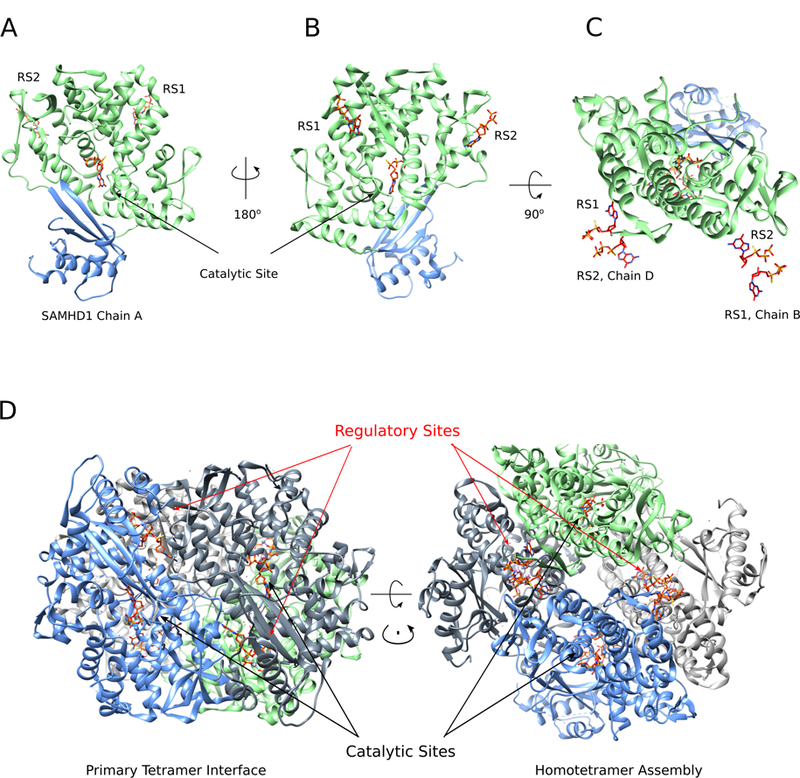
(A, B) SAMHD1 monomer depicting the HD domain major lobe (green) and C-terminal region (blue) (PDBID: 4BZC (71)). The catalytic site and regulatory site 1 (RS1) and 2 (RS2) are indicated with bound dGTP (C) SAMHD1 monomer as in A and B, with paired nucleotides from the tetrameric regulatory cleft. Each cleft contains two regulatory nucleotide binding sites from adjacent monomers that stabilize subunit interactions. (D) The catalytically active tetrameric holoenzyme of SAMHD1 reveals the primary tetramer interface (left) and the homotetrameric assembly (right).
In contrast to RS1, RS2 presents a more promiscuous binding site. RS2 can accommodate any of the four canonical dNTPs with apparent Kd values between 1–20 μM depending on the nucleobase (57, 73, 76–78). These concentrations are physiologically relevant and thus binding of a dNTP in the RS2 of SAMHD1 occurs when intracellular dNTP concentrations are elevated into the activating range (24). While RS2 is capable of binding any of the four dNTPs, there is a clear preference for purine nucleotides (57, 76). Thus, given the asymmetric nature of dNTP pools within the cell, it is likely that dATP is most frequently docked in RS2 (73). While dATP may be the primary nucleotide bound in RS2, it is possible that the specific dNTP docked in RS2 may confer differential stability and substrate specificity to the activated enzyme (77).
The binding event of a dNTP at RS2, which is preceded by docking of GTP in the guanine specific RS1 pocket, stabilizes a dimer of dimers and drives the oligomeric equilibrium towards tetramerization. Subunit assembly results in the formation of four regulatory clefts comprised of an RS1 and RS2 from adjacent monomers, as well as the residues of a third SAMHD1 subunit. The two nucleotides bound in each cleft are coordinated by a single divalent cation (commonly Mg2+) that stabilizes their phosphate tails. At each of the four occupied regulatory sites in the activated tetramer, the activating nucleotides form a network of hydrogen bonds, electrostatic, and pi-pi orbital interactions with residues from the three distinct monomers that comprise the cleft. This results in a thermodynamically stable SAMHD1 tetramer housing eight total activating nucleotides (4xd/GTP and 4xdNTP) bound in the four regulatory clefts. Tetramer assembly is further enhanced by several key structural motifs, specifically protein-protein interfaces formed by an ordering of the C-terminus (amino acids 454–599), and a long alpha helix (amino acids 352–373) that significantly contributes to the dimer-dimer interface (57, 70, 71, 74). Binding of activating nucleotides and the subsequent formation of the tetramer result in conformational changes that remodel the active site allowing substrate binding and catalysis.
It has been suggested that the catalytically active tetrameric species can persist for extended periods even after dNTP levels have diminished below the effective level for SAMHD1 activation (55, 77). This long-lived active state may be important for maintaining cellular dNTP pools at the extremely low concentrations observed in non-cycling cells. Regardless of the duration of the activity, the elegant and strictly regulated mechanism of SAMHD1 catalytic activation represents an ordered and sequential process in which dNTPs serve as both substrate and activating ligand. The finely-tuned autoregulatory mechanism enables SAMHD1 to sense small fluctuations of dNTP concentrations within the cell and respond accordingly by degrading them to physiologically appropriate levels.
Nucleobases are stabilized in the active site through a series of non-specific interactions with water molecules, which likely affords SAMHD1 substrate promiscuity (57, 70, 71, 74). In addition to the ability to hydrolyze all four canonical dNTPs and dUTP, the SAMHD1 active site is also able to accommodate base modified substrates (79). Hydrolysis of base-modified or non-canonical nucleotides may represent additional cellular functions of SAMHD1, although this aspect of its activity has not been rigorously interrogated. dNTPs are oriented for chemistry to occur through an interaction between their α-phosphate and a Mg2+ ion coordinated by the His167-His206-Asp207-Asp311 quartet characteristic of HD domains. His210, His233, and Asp218 are believed to be the residues responsible for actual catalysis, which occurs through an in-line nucleophilic attack at the α-phosphate resulting in tripolyphosphate and the cognate nucleoside as reaction products (80). Steric clashes with active site residues by the 2’-hydroxyl group found on ribonucleotides preclude efficient rNTP binding and hydrolysis.
Despite the substrates of SAMHD1 also serving as its activators, SAMHD1 displays little evidence of cooperative kinetics. The sequential activation and assembly of the SAMHD1 tetramer occur at dNTP concentrations multiple orders of magnitude below the KM. Thus, while reported KM values for SAMHD1 vary considerably from 50–350 μM depending on the study, standard Michaelis-Menten kinetic models apply under all the experimental conditions measured (55, 57, 73, 78, 81, 82). Given the steady-state conditions of an activated SAMHD1 tetramer, the turnover rate for each substrate appears to depend solely on its particular affinity for the active site and its intracellular concentration. Recent evidence however, is interjecting questions into this model. In a manner resembling ribonucleotide reductase activation, the particular dNTP bound in the RS2 may influence substrate specificity and catalytic efficiency (78). The precise nature of SAMHD1 activation and substrate specificity in vivo is an open inquiry that requires further investigation into the underlying molecular mechanism and physiological significance.
SAMHD1 Nucleic Acid Interaction.
In addition to its dNTPase activity, SAMHD1 is also a nucleic acid binding protein. SAMHD1 binds DNA and DNA:RNA duplexes, but preferentially binds single stranded nucleic acid polymers (ssNAs), with a greater affinity for ssRNA over ssDNA (83–85). The specific affinity of SAMHD1 for ssNAs may be regulated by their secondary structure (86). The phenomena of SAMHD1 ssNA interaction has also been reported in cells where multiple SAMHD1 monomers converge on sites of ssRNA or ssDNA (87). It also appears that monomeric SAMHD1 primarily interacts with nucleic acids, and that SAMHD1 monomers can form multi-subunit complexes in the presence of ssNA that are distinct from the oligomeric forms induced by dNTPs (83).
Efficient interaction with ssNAs involve residues from both the HD domain and C-terminus, with the C-terminus peptide (residues 583–626) demonstrating nucleic acid binding even in the absence of the HD and SAM domains (83, 88). Interestingly, nucleic acid binding by SAMHD1 inhibits dNTPase activity by obstructing the interfaces responsible for tetramer association. This obstruction can be relieved by increasing the concentration of activating nucleotides (88). SAMHD1 has also been reported to exhibit exonuclease activity on ssRNA and ssDNA (86, 89, 90). The proposed mechanism of catalysis for SAMHD1 exonuclease activity is a phosphorolytic cleavage of the phosphodiester bond in nucleic acid polymers (91). These findings are widely contested however, as many groups report not being able to detect SAMHD1 nuclease activity or its effect on viral restriction (53, 83, 84, 92, 93). Instead, they propose that co-purification of a contaminating nuclease is responsible for the observed exonuclease activity of SAMHD1. While the precise function of nucleic acid interaction in cells is yet to be determined, a recent report suggests that the ability of SAMHD1 to interact with DNA may play a crucial role in facilitating the repair of DNA. In this study, SAMHD1 was demonstrated to localize to sites of double stranded breaks and promote homologous recombination through its ability to form a complex with the endonuclease CtBP interacting protein (CtIP) (94). This description of a previously unidentified SAMHD1 role in DNA damage repair broadens the scope of the enzyme’s cellular functionality, and underscores the importance of further investigation of the dynamic cycle between nucleic acid bound inactive monomer-dimer and catalytically active tetramer.
SAMHD1, Cellular Regulation, and Nucleotide Metabolism
SAMHD1 expression profile and regulation.
SAMHD1 was initially termed dendritic cell derived IFN-γ induced protein (DCIP) following its identification in dendritic cells as the human ortholog of an interferon-γ induced mouse protein (51). While the precise function of DCIP (SAMHD1) remained unknown, it was subsequently determined to be upregulated in lung fibroblasts in response to TNF-⍺ or LPS induced acute injury in an IRF-1 dependent manner (95). Further studies eventually elucidated the integral function the enzyme performs in nucleotide metabolism and innate immunity within the cell, and in keeping with this critical role, SAMHD1 was revealed to be widely expressed in most human cells (96).
Although SAMHD1 is detected in almost all tissues, its expression levels vary by cell type and cell cycle phase. Quiescent primary human fibroblasts demonstrated a dramatic increase in SAMHD1 protein levels over proliferating fibroblasts, and this increase in SAMHD1 coincided with a decrease in intracellular dNTP pools (97). In contrast to fibroblasts, SAMHD1 is constitutively expressed at high levels in both cycling and non-cycling myeloid and lymphoid cells, including monocytes, macrophages, dendritic cells, and CD4+ T-cells, although its activity can be differentially regulated by post-translational modification (96, 98, 99). There is also experimental evidence to suggest that terminal differentiation of monocytes into macrophages or dendritic cells can further increase the expression levels of SAMHD1 (100). In hematopoietic cells where SAMHD1 is expressed and active, it contributes to the maintenance of dNTP levels that range from 3–250 fold lower than in those cells where it is post-translationally down-regulated, such as in activated human CD4+ T cells (101–103).
Given the cyclic and dynamic nature of nucleotide metabolism, the catalytic activity of SAMHD1 as a modulator of nucleotide pools is tightly controlled. Regulation of SAMHD1 begins at the transcriptional level, as several studies have demonstrated that the SAMHD1 promoter can be silenced by methylation (104–106). Two naturally occurring splice variants of SAMDH1 have been identified, consisting of truncated versions of the full-length protein (107). The utility of these isoforms is a subject of uncertainty however, as both were demonstrated to be metabolically unstable, catalytically inactive, and rapidly degraded in cells. In this way, human SAMHD1 diverges from its closely studied murine homologue, as mice express two catalytically competent SAMHD1 isoforms (108).
In keeping with its discovery as an interferon associated protein, SAMHD1 can be transcriptionally upregulated in some cell lines in response to cytokine (Il-12 and Il-18) or type-1 interferon (IFN1) treatment (102, 109, 110). Not all cells however, including macrophages, dendritic cells, and CD4+ T-Cells, exhibit an increase in SAMHD1 protein expression following exposure to IFN1 (96, 102, 109, 111). This discrepancy may be in part explained by the already elevated levels of SAMHD1 or by the inability of specific cell types to respond to IFN1 exposure by downregulating microRNAs that post-transcriptionally repress SAMHD1 translation (112). SAMHD1 expression can also be induced by viral challenge in an IRF3 dependent manner. Phosphorylated IRF3, a downstream transcription factor activated by intracellular pathogen pattern recognition receptors that coordinates the early immune response, was demonstrated to directly bind the SAMHD1 promoter and enhance SAMHD1 expression, thus corroborating the important role SAMHD1 plays as an effector of innate immunity (110).
Following translation of SAMHD1, little is known about its cellular half-life or subcellular interactions. Several studies however, have exposed protein interaction partners that provide clues as to the mechanism of normal SAMHD1 degradation. SAMHD1 interaction with both cyclin L2 and eukaryotic elongation faction 1A1 (eEF1A1) facilitates recruitment to the ubiquitin ligase machinery and subsequent proteosomal degradation (113, 114). A more recent study suggests that interaction with the transmembrane tetraspanin CD81 promotes proteosomal turnover of SAMHD1 (115). Interestingly, in the absence of CD81, SAMHD1 subcellular localization partially overlapped with EEA1, a marker of early endosomes. These findings reveal the initial associations and pathways in what is likely a complex network of interactions and regulatory layers that govern SAMHD1 expression and cellular half-life. Future research to clarify the precise relationships of this network will be important for understanding SAMHD1 functionality within the larger processes of nucleotide metabolism and innate immunity.
SAMHD1 regulation by post-translational modification.
While biochemical and structural studies have rigorously characterized the mechanism of SAMHD1 activation and catalysis, an emerging area of interest is the effect of post-translational modifications on SAMHD1 activity. Both human and murine SAMHD1 are phosphorylated at multiple sites (108, 116), but the most extensively studied is phosphorylation at its C-terminal T592 residue (P-T592) (98, 117). Phosphorylation of SAMHD1 at T592 occurs in a cell cycle dependent fashion by cyclin-dependent kinases 1 and 2 (CDKs) and coincides with an increase in intracellular dNTPs prior to S-phase DNA replication (93, 98–100, 118–121). Phosphorylation of the enzyme is mediated by the presence of a putative cyclin-binding motif located in the HD domain (residues 450–455), as well as a bipartite cyclinA2-CDK complex binding motif located in the C-terminus (99, 118). Further investigation of this regulatory axis reveals that SAMHD1 phosphorylation likely occurs as cells emerge from a G0/quiescent state and transition through G1 facilitated by a mitogen induced activation of the Raf/MEK/Erk kinase cascade (122). Conversely, for cells residing in a non-cycling G0/quiescent state, multiple studies report that the dephosphorylated SAMHD1 species predominates and corresponds with reduced dNTP levels (98, 99, 117).
In addition to cell cycle phase, other cellular processes modify the phosphorylation status of SAMHD1 at T592. In CD4+ lymphocytes, cytokine (IL-7 or IL-2) activation or PHA treatment, was demonstrated to increase phosphorylation of SAMHD1 at T592 (96, 119, 120). Alternatively, IFN1 treatment results in the dephosphorylation of SAMHD1 at T592 (98). IFN1 signaling impedes cell cycle progression by up-regulating the cell cycle inhibitor p21cip1/waf1 which subsequently restricts the activity of CDKs (123). Accordingly, monocyte derived dendritic cells (MDDC) matured in the presence of IFN-γ and CD40L, or M-CSF, demonstrated a decrease in phosphorylated SAMHD1 that corresponded to an increase in p21cip1/waf and a decrease in cellular dNTP pools (124, 125). DNA damage can also activate p21 and result in the concomitant loss of phosphorylated SAMHD1 in a p53 dependent manner, as described in a recent study in which macrophages were treated with topoisomerase inhibitors in order to induce a DNA damage response (126).
Given that phosphorylation of SAMHD1 occurs prior to S-phase and coincides with increased dNTP levels within cell, many have speculated that this modification is a requisite condition for altering the dNTPase capacity of the enzyme to promote DNA replication. The precise effects of phosphorylation on SAMHD1 activity are still under debate, however. Some evidence suggests phosphorylation negatively modulates SAMHD1 tetramerization and dNTPase activity (81, 99, 127), and this diminished dNTPase capacity is responsible for increased intracellular dNTP pools (93, 120, 121, 128). Other studies find limited or no effect of phosphorylation of SAMHD1 on catalytic activity(81, 108, 116, 117, 129), oligomerization equilibrium and allosteric activation(77, 129), or nucleic acid binding (83). Complicating the model further, phosphorylation may affect SAMHD1 substrate specificity (78).
Studies attempting to shed light on these discrepancies reveal that P-T592 results in altered kinetics of tetramer association and dissociation and expedited regulatory nucleotide release (77, 129). P-T592 is also less likely to form the activated tetramer at low concentration of activating nucleotides (78, 81). Structural data support these findings by identifying protein conformational shifts induced by a phosphomimetic mutant (T592E) that destabilizes the tetramer interface (127). Taken in sum, these data appear to indicate that phosphorylation at T592 is a mechanism for calibrating SAMHD1 activity through altering the thermodynamics of subunit association. The discrepant results may stem from variations in culture conditions or assay methods that lack the sensitivity to accurately capture the true effect of phosphorylation. Fine tuning SAMHD1 activity within the cell by phosphorylation, as opposed to a binary on or off state, may be important as a complementary method of regulation in order to maintain dNTP pools within the narrow window conducive to genomic integrity.
Two additional post-translational modifications have been discovered that regulate SAMHD1 catalytic activity. A recent study demonstrated that SAMHD1 catalytic activity is regulated by redox signaling (82). Redox signaling takes the form of reactive oxygen species (ROS) that are generated by cellular oxidases in response to growth factors binding and activating cellular receptors (130). These ROS act as secondary messengers that can covalently, but reversibly, interact with an enzyme to modify its tertiary structure and subsequent activity(131). SAMHD1 is catalytically inactivated when treated with the oxidizing agent H2O2 in a dose dependent but reversible manner (82). Oxidation of SAMHD1 was demonstrated to inhibit tetramerization, but this deficiency was ablated in SAMHD1 mutants containing a C522A mutation. Closer inspection of SAMDH1 crystal structures reveal that C522 comprises one member of a cysteine triad - C522, C341, and C350 - that resides adjacent to the regulatory nucleotide binding site RS2. In several available crystal structures (pdbid: 5AO4, 4RXO, 4MZ7, 3U1N)(54, 70, 74, 81), a disulfide bond exists between C341 and C350 and results in a conformational shift that may disrupt activating nucleotide binding and tetramer stability. While the experiments in the study were performed using recombinant human SAMHD1, it is interesting to note that the residues comprising the cysteine triad (C522, C341, and C350) are highly conserved among vertebrate SAMHD1 homologues. The conserved nature of these residues in varying species underscores the likely importance of redox signaling as a central regulatory mechanism for modulating SAMHD1 activity in vivo.
The redox sensitivity of SAMHD1 was recapitulated in cells during experiments in which SAMHD1 was demonstrated to be oxidized in response to proliferative signals (82). It was proposed that cells oxidize SAMHD1 in response to proliferative signaling in order to accumulate the dNTPs necessary for DNA replication. This oxidation effect is mediated by the cysteine triad, referred to as a ‘redox switch’, which can detect ROS secondary messengers and translate them into structural rearrangements that alter SAMHD1 catalytic activity. Importantly, this phenomenon is reversible and tightly controlled.
SAMHD1 was also revealed to be acetylated both in vitro and in cells on the K405 residue by the acetyltransferase Arrest Defective Protein 1 (ARD1) (132). Like the conserved cysteine residues of the redox switch, K405 is conserved in many vertebrate SAMHD1 homologues indicating acetylation at this residue may play an important role in regulating SAMHD1 activity. Acetylation of SAMHD1 peaked during the G1 phase of the cell cycle and resulted in the increased dNTPase activity of the enzyme in vitro. It was suggested, somewhat counter intuitively, that the enhanced dNTPase activity aided in G1 to S-phase transition and promoted cell cycle progression in cancer cells. In addition to identifying a novel posttranslational modification, this finding also unveils a potential method for therapeutically targeting SAMHD1 activity in cells through the use of small molecule inhibitors of acetyltransferases.
As with phosphorylation, redox regulation and acetylation of SAMHD1 represent means of control orthogonal to catalytic activation and protein expression that can be localized spatially and temporally. These post-translational modifications work as additional levers of SAMHD1 control to precisely calibrate SAMHD1 activity. The meticulously controlled dynamic gradient of SAMHD1 activity through multiple means of regulation likely performs an important role in maintaining dNTP pools within a homeostatic range at each point in the cell cycle (Fig. 4). Future research into both identified and as of yet unidentified mechanisms of SAMHD1 post-translational regulation represents a potentially fruitful line of inquiry with implications for SAMHD1 biology and broader cellular processes.
Figure 4. SAMHD1 catalytic activity is tightly controlled by regulatory nucleotides and essential for the maintenance of nucleotide homeostasis.
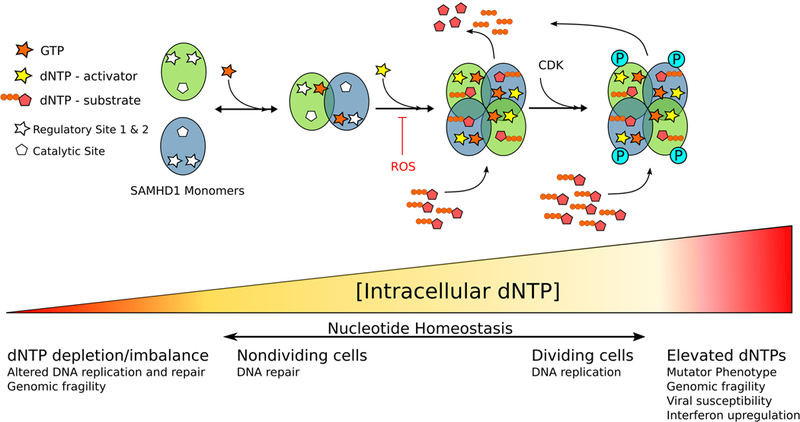
Under conditions of low dNTPs, SAMHD1 exists in a monomer-dimer equilibrium. Binding of GTP in RS1 stabilizes the dimer conformation. Elevation of intracellular dNTP concentrations above the activation threshold (1–20µM) results in dNTPs binding at RS2 and SAMHD1 tetramerization. Tetrameric SAMHD1 is able to catalyze the degradation of dNTPs and thereby prevent accumulation to levels that would be cytotoxic. Phosphorylation is proposed to destabilize tetramer stability without modifying catalytic efficiency, thereby allowing for an increase in dNTP pools necessary for DNA replication without creating mutagenic dNTP conditions.
SAMHD1, Cell Cycle Regulation, and Genomic Integrity.
Compared to its innate immune function, less attention has been given to the role of SAMHD1 in normal homeostatic cell maintenance. However, its role as a regulator of nucleotide metabolism may represent its primary biological function given the nearly ubiquitous expression of SAMHD1, the enzyme’s regulation that is synchronized with changes in dNTP pools and cell cycle stage, and the fact that proper dNTP concentrations are essential for genomic integrity and DNA replication and repair.
SAMHD1 is widely established as a central regulator of dNTP pool dynamics within cells and as such its catalytic activity can directly influence the replicative capacity of the cell (97, 133–136). Silencing or degradation of SAMHD1 results in elevated dNTP pools and altered growth kinetics as evidenced by studies that demonstrate an increased retention of cells in the G1 phase with a corresponding loss in S-phase cells (97, 133, 137). Intriguingly, the precise effect of knocking down SAMHD1 on proliferative capacity appears to depend on the specific cell type. Fibroblasts and THP-1 cells have been reported to grow either slower or faster, respectively, in the absence of SAMHD1 (97, 133). In THP-1 cells, the increased dNTP pools and enhanced proliferative capacity were accompanied by a reduction in spontaneous apoptosis (133). Conversely, up-regulation of SAMHD1 can also alter growth kinetics by contributing to a quiescent or differentiated phenotype in fibroblasts and THP-1 cells by coordinating with other nucleotide metabolizing enzymes to maintain reduced dNTP levels (97, 138). Similarly, overexpression of exogenous SAMHD1 results in reduced proliferation and increased apoptosis in HuT78 cells - a human T-cell lymphoma cell line - presumably by denying the cells the large quantities of dNTPs necessary to efficiently replicate their genomic DNA (139). These studies clearly identify SAMHD1 as a modulator of cellular growth kinetics, although additional research is required to quantify the mechanism and magnitude of the effect exerted by SAMHD1 activity.
Proper dNTP concentrations are also essential for genomic stability as imbalanced dNTP pools can hinder DNA replication and repair. By degrading dNTPs in resting or quiescent cells before they accumulate to levels that impede the efficiency or accuracy of the replication and repair machinery, SAMHD1 performs an essential function as a protector of genomic fidelity. Data from patients expressing inactive mutants of SAMHD1 underscore this point, as cells from these patients exhibit elevated dNTPs, growth arrest in G1, and hallmarks of response to DNA damage (137). These data suggest that SAMHD1 represents an important node in the cellular circuitry of tightly coordinated nucleotide synthesis and degradation that enables both proliferation and maintenance of genomic integrity. By integrating the myriad of transcriptional, translational, and post-translational regulatory mechanisms the cell can calibrate SAMHD1 activity to meet the precise metabolic needs of the specific cell cycle phase. Despite being implicated in such central processes, this aspect of SAMHD1 biology is still relatively under examined. Future work defining the role SAMHD1 plays in promoting cellular homeostasis through its effect on nucleotide pools, and the regulatory pathways that alter its activity, will likely yield important insights into the underlying mechanisms that control nucleotide metabolism and cell cycle progression, as well as the pathogenic phenotypes that result from their dysregulation.
SAMHD1 as an Effector of Innate Immunity
The discovery of SAMHD1 as an IFN1 inducible protein in dendritic cells hinted at the importance of SAMHD1 as an effector of innate immunity from the moment of its first discovery. Accordingly, virologist and immunologist have been responsible for many of the most significant advances to the understanding of SAMHD1 biology. While the following section briefly reviews these discoveries and the role of SAMHD1 as an effector of innate immunity, a recently published review exclusively explores the relationship between SAMHD1 and the innate antiviral response in greater detail (140).
SAMHD1 has garnered significant attention for its antiviral properties in non-dividing cells of hematopoietic lineage, including macrophages, dendritic cells, and resting CD4+T-cells (146, 147, 150). In these cells it is constitutively expressed and present in the dephosphorylated state, where it contributes to the maintenance of dNTP levels that are several hundred fold lower than in actively cycling CD4+ T-cells (141). Interestingly, the presence of dephosphorylated SAMHD1 in these cells coincides with the capacity of non-dividing hematopoietic cells to restrict efficient HIV-1 infection (101, 142–145). HIV-2 however, which carries the virulence accessory protein, Vpx, is able to efficiently transduce non-dividing hematopoietic cells. Incorporation of Vpx into HIV-1 virions relieved the restrictive phenotype, and suggested the presence of a specific cellular immune factor antagonized by Vpx (146–148). It was eventually revealed that SAMHD1 was the HIV cellular restriction factor counteracted by Vpx (143, 144). Through its dNTPase functionality, SAMHD1 is proposed to inhibit productive viral infection by reducing dNTP pools below the concentration required for efficient catalysis by viral DNA polymerases or reverse transcriptases (135, 149, 150). SAMHD1 thereby provides a kinetic block at the critical juncture where viral genomic content is replicated and incorporated into the host’s genome. This block is reversible upon treatment with exogenous dNTPs or knockdown of SAMHD1 (135, 143, 144, 149). The importance of SAMHD1 dNTPase activity as a barrier against viral infection was further underscored by the finding that primary monocytes deficient for SAMHD1 due to genetic mutation are highly susceptible to HIV-1 infection (145). Vpx specifically counteracts the SAMHD1 mediated depletion of cellular dNTPs to enable efficient viral infection. It accomplishes this by orchestrating the degradation of SAMHD1 (67, 143, 144, 151). Vpx facilitates the interaction of SAMHD1 with the ubiquitin ligase substrate adaptor molecule DCAF1 through the C-terminal region of SAMHD1 (152), promoting recruitment of SAMHD1 to the CRL4 ubiquitin ligase complex where it is ubiquitinylated and marked for proteosomal degradation (69, 143, 144, 153). The loss of SAMHD1 and subsequent increase in dNTPs is sufficient to permit productive viral infection for Vpx containing virions.
Intriguingly, of the two viral subtypes, HIV-2 is the less virulent. Why this is the case is not entirely understood, but it has been proposed that by using Vpx to degrade SAMHD1 and enable productive infection of macrophages and dendritic cells, which serve as the sentinel cells of the immune system, HIV-2 may in fact be alerting the host’s natural defenses to its presence. HIV-1 restriction in non-dividing immune cells may enable it to maintain an undetectable reservoir of latent virus, while avoiding activation of the host’s immune systems (154–156). Evidence supporting this model shows that SAMHD1 restriction of HIV-1 limits the innate and adaptive immune responses by preventing the activation of the cGAS/STING pathway (157). Inhibitors of SAMHD1 may therefore represent an important therapeutic tool for facilitating infection of myeloid cells and activating a robust immune response to HIV-1 infection (158).
Additional studies have broadened the scope of SAMHD1 innate immune functionality by demonstrating its antiviral restrictive capacity extends beyond HIV-1 to other retroviruses and DNA viruses such as herpes viruses (HSV-1) and hepatitis B (HBV) (85, 144, 149, 159–165). This strategy of warding off invasive pathogens through depletion of precursor metabolites is not a novel approach (150), as it finds precedence in bacteria such as Eschericia coli which express a dGTPase that reduces susceptibility to T7 bacteriophage infection (58, 166). Therapeutically targeting nucleic acid metabolizing enzymes in order to deplete intracellular dNTP pools is also an approach implemented by modern medicine in order to counter viral infection and cancer (167, 168).
More recently however, dNTP depletion has come under question as being fully sufficient to explain the entirety of SAMHD1’s antiviral restriction capacity (169). Evidence exists to suggest that tetramerization deficient SAMHD1 enzymes may still be able to exert antiviral effects (129, 170). Additionally, the antiviral capacity of SAMHD1 is strictly correlated with its phosphorylation status on its C-terminus at T592, even while debate as to the actual effect of P-T592 on SAMHD1 catalytic activity persists. As previously discussed, SAMHD1 is phosphorylated in cycling cells at T592 in a cell cycle dependent manner, but dephosphorylated in most non-dividing hematopoietic cells where it is also able to efficiently restrict HIV-1 infection (93, 98, 101, 117, 120, 142). In addition to quiescence or differentiation, viral challenge, cytokine signaling (Il-12 and Il-18), or IFN1 treatment also result in the loss of SAMHD1 phosphorylation at T592 (98, 102, 109, 110). The loss of phosphorylation at T592 coincides with the reemergence of a cellular phenotype that is refractory to HIV-1 infection and provides a clear linkage between SAMHD1 and innate immunity, while emphasizing the importance of SAMHD1 phosphorylation status as a determinant of viral restriction. While phosphorylation of T592 has been established as coinciding with the ablation of the viral restriction capacity, some data suggest that this loss of antiviral capacity is not entirely coupled to diminished SAMHD1 catalytic activity (116, 117, 125, 169). Cells expressing phosphomimetic T592D or T592E mutations support this model, as they are unable to restrict viral infection but do not exhibit elevated dNTP pools or reduced dNTPase activity (81, 116, 169). The implication of this is that SAMHD1 is able to restrict viral infection through both its dNTPase activity as well as a putative yet to be discovered mechanism, the potency of which is dependent on the phosphorylation status of the enzyme at T592. The precise mechanisms through which SAMHD1 inhibits viral infection and how post-translational modifications influence its restrictive capacity represent important research questions going forward. The answers to these questions will further reveal the role of SAMHD1 in innate immunity, as well as potential opportunities to develop therapeutic strategies for mitigating the impact of viral infections.
SAMHD1 Mutations Result in Disease
Autoimmunity.
The central role SAMHD1 performs in nucleotide metabolism and immunity increases the probability that mutations to SAMHD1 will result in disease. Many mutations to SAMHD1 have been identified and are associated with several different disease phenotypes (Fig. 5). One of these diseases is the Type I interferonopathy Aicardi-Goutieres Syndrome (AGS) – a genetically determined autoimmune disease linked to aberrant nucleic acid processing (171–173). AGS is an autosomal genetic disorder that mimics the sequela of an in utero viral infection and retains significant overlap in phenotypic presentation with that of systemic lupus erythematosus (SLE) (172, 174–176). AGS is characterized by encephalopathy, leukodystrophy, calcifications of the basal ganglia, psychomotor retardation, cerebrospinal fluid lymphocytosis, and an over-production of interferon-α. Approximately 40% of the cases result in early childhood death (177).
Figure 5. SAMHD1 gene schematic depicting location of AGS and Cancer inducing mutations.
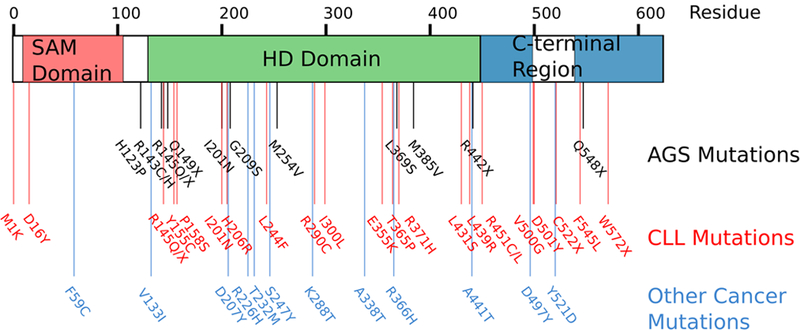
SAMHD1 consists of an N-terminal Sterile Alpha Motif domain (red), a catalytic HD domain (green), and a distinct C-terminal region. Mutations in each region have been identified in patients with autoimmune disorders and cancer (178, 200, 201, 204). AGS mutations are believed to be causative loss-of-function resulting in dysregulation of nucleotide metabolism. Questions remain as to whether SAMHD1 mutations found in cancer cells are foundational oncogenic events, secondary promoters of tumorigenesis, or symptoms of genome instability.
AGS is a genetically heterogeneous disease, with mutations to SAMHD1 accounting for approximately 13% of all documented cases (177). Its etiology can be traced to mutations in SAMHD1 and 4 other enzymes involved in nucleic acid processing, TREX1, RNase H2, ADAR1, and MDA5 (52, 177–182). Loss-of-function mutations to these enzymes may result in the cytosolic accumulation of unprocessed nucleic acids that mimic invading pathogens. Unprocessed nucleic acids can then activate endogenous nucleic acid sensing pathways and result in subsequent interferon production. The common functionality of nucleic acid metabolism which links the causative AGS enzymes has led to the hypothesis that they play a role in suppressing antigenic nucleic acid by-products. The source of the antigenic nucleic acids however is unknown, and may vary by implicated enzyme. Conjectures as to how SAMHD1 negatively regulates interferon pathways have ranged widely - from suppression of retroelement activation to control of anomalous nucleic acid species resulting from genomic instability and altered DNA replication and repair (137, 183–185). Interestingly, AGS is commonly associated with pathologies of the nervous system - a system which given the specific requirements for rapid neurogenesis in early development and neuronal longevity make it especially susceptible to replication stress and impeded DNA repair pathways (186).
The phenotypic presentation of SAMHD1 associated AGS has several novel features not observed in other AGS associated genotypes, including cerebral vasculopathy, arthropathy, and mitochondrial DNA deletions (187–190). The clinical presentation of SAMHD1 induced AGS may therefore provide clues to the mechanism through which SAMHD1 contributes to nucleotide homeostasis and negatively regulates innate immunity. Attempts to recapitulate an AGS phenotype in SAMHD1 null mice have proven perplexing however, as SAMHD1 knockout mice exhibit elevated interferon levels but do not develop autoimmune disease (161, 191). A more promising animal system for modeling SAMHD1 induced AGS might be found in zebra fish. In this alternative to mouse models, zebra fish exhibit similar interferon overexpression and cerebrovascular pathologies upon deletion of SAMHD1 to those clinically observed in AGS patients (192).
Cancer.
Ample evidence exists to connect dNTP pool dysregulation to cancer and genomic instability (40). Elevated or imbalanced dNTP pools can result in a mutator phenotype (25, 193, 194) that disrupt genomic integrity and DNA replication and repair (195–197). Conversely, dNTP pool depletion can result in genomic instability that progresses to cancer (198). Additionally, an imbalance to dNTP pools can lead to loss of genomic fidelity and impaired replication efficiency and accuracy (27–29, 199). It is therefore no surprise that recent studies have begun to implicate SAMHD1 in various cancers, including lymphocytic leukemia, lung adenocarcinoma, and colon cancer (106, 200–202) (Fig. 5). The Catalogue of Somatic Mutations in Cancer (COSMIC) has recorded 164 unique mutations to SAMHD1 found in samples obtained from various cancer tissues (203). Mutations to SAMHD1 likely result in an imbalanced dNTP pools and create the prerequisite conditions for mutagenesis and genomic instability (40, 204). It is also possible that SAMHD1 prevents mutations by sanitizing nucleotide pools via degrading base modified dNTPs before they are incorporated into DNA (205, 206). Given its role in the maintenance of genomic stability, SAMHD1 may potentially perform an additional function in cells as a tumor suppressor enzyme. It will be important to determine if mutations to SAMHD1 are foundational in that they drive tumorigenicity, or if cancer cells must down-regulate the restrictive effect of SAMHD1 in order to supply themselves with the metabolites necessary for unconstrained growth.
Lastly, SAMHD1 will likely be important in targeted health therapies, as nucleotide analogue therapeutics are a common tool used in treatment of cancer and viral infections (207, 208). Following phosphorylation by intracellular kinases, these analogues are structurally similar to the endogenous dNTP substrate, and several are substrates for SAMHD1 (209). Most notably, Ara-C, a first line therapeutic regimen against acute myelogenous leukemia (AML), is degraded by SAMHD1 in cells (210–212). This minimizes the efficacy of the treatment to such an extent that SAMHD1 expression levels were negatively correlated with Ara-C treatment success in individuals with AML. Additionally, SAMHD1 appears to reduce the efficacy of thymidine based analogs used to treat HIV, and depletion of SAMHD1 from monocyte cells affects the susceptibility of HIV-1 infections to nucleoside reverse transcriptase inhibitors (79, 213, 214). Based on these finding, the development of a robust SAMHD1 inhibitor that can potentiate nucleotide analogue therapeutic regimens should become a priority for SAMHD1 researchers. Several groups have developed high throughput assays (215, 216), but to date, no potent inhibitor of SAMHD1 has been identified.
Open Questions
Since its discovery as a participant in innate immunity and as an HIV restriction factor, SAMHD1 has been the target of significant research efforts that have extensively detailed the fundamental biochemistry and cell biology of SAMHD1. However, important questions related to SAMHD1 biology remain. In a broad context, the centrality of SAMHD1 to cellular homeostasis is becoming apparent. SAMHD1 is a vital cog in the well-oiled cellular machinery that strictly controls the anabolic and catabolic pathways that determine cellular nucleotide content. By degrading dNTPs SAMHD1 fulfills important roles as a sentinel of genomic integrity, a regulator of cell cycle progression, and an effector of innate and autoimmunity. Uncovering the details of SAMHD1 function and regulation in normal cells will provide insight into how its dysfunction leads to disease. Continued efforts to elucidate the in vivo mechanisms of regulation will be important for determining the enzyme’s operation in the intricate and dynamic pathway of nucleotide metabolism. How post-translational modifications, such as phosphorylation and oxidation, coordinate with protein activation, localization, and oligomerization to tune SAMHD1 activity and maintain dNTP homeostasis is yet to be determined. Additionally, the biological function of SAMHD1 nucleic acid interaction is an important question. Is this a potential mechanism for viral restriction, or is it primarily related to the recently discovered capacity of SAMHD1 to promote DNA end resection during homologous recombination? Given that the facilitation of DNA repair by SAMHD1 represents a novel function of the enzyme unrelated to its catalytic activity, are there additional cellular actions SAMHD1 performs that are yet to be discovered? Along the same lines, understanding the biology of the SAM domain and SAMHD1 interactions with other proteins remains an open line of inquiry. In the context of disease, the comprehensive methods of viral restriction appears to be incompletely described, as does the precise manner in which SAMHD1 acts as a negative regulator of interferon signaling. Information about the role of SAMHD1 in disease, specifically cancer and autoimmunity, is in its nascent stages, but each new discovery contributes to our knowledge and presents potential translational opportunities.
Acknowledgments:
This work has been supported through funding by NIH RO1 GM108827 (TH) and T32 GM095440 support (CHM), and the Comprehensive Cancer Center of Wake Forest University National Cancer Institute Cancer Center Support Grant P30CA012197.
Abbreviations
- AGS
Aicardi-Goutieres syndrome
- CDKs
Cyclin Dependent Kinases
- DCIP
dendritic cell derived IFN-γ induced protein
- dATP
deoxyadenosine triphosphate
- dGTP
deoxyguanosine triphosphate
- dNDP
deoxynucleotide diphosphate
- dNTP
deoxynucleotide triphosphate
- H2O2
hydrogen peroxide
- IFN1
Type-1 Interferon
- RS1
regulatory site 1
- RS2
regulatory site 2
Footnotes
Declarations of Interest: The authors declare no competing or conflicting interests.
Contributor Information
Christopher H. Mauney, Department of Biochemistry, Center for Structural Biology, Wake Forest School of Medicine, Winston Salem, NC 27157
Thomas Hollis, Department of Biochemistry, Center for Structural Biology, Wake Forest School of Medicine, Winston Salem, NC 27157.
References
- 1.Traut TW 1994. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem 140: 1–22. [DOI] [PubMed] [Google Scholar]
- 2.Reichard P 1988. Interactions between deoxyribonucleotide and DNA synthesis. Annu. Rev. Biochem 57: 349–374. [DOI] [PubMed] [Google Scholar]
- 3.Mathews CK 2014. Deoxyribonucleotides as genetic and metabolic regulators. FASEB J 28: 3832–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mathews CK 2006. DNA precursor metabolism and genomic stability. Faseb J 20: 1300–1314. [DOI] [PubMed] [Google Scholar]
- 5.Pai C, and Kearsey S. 2017. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes (Basel) 8: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rampazzo C, Miazzi C, Franzolin E, Pontarin G, Ferraro P, Frangini M, Reichard P, and Bianchi V. 2010. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat. Res. - Genet. Toxicol. Environ. Mutagen 703: 2–10. [DOI] [PubMed] [Google Scholar]
- 7.Lane AN, and Fan TW-M. 2015. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43: 2466–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofer A, Crona M, Logan DT, and Sjöberg B-M. 2012. DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol 47: 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walldén K, and Nordlund P. 2011. Structural Basis for the Allosteric Regulation and Substrate Recognition of Human Cytosolic 5′-Nucleotidase II. J. Mol. Biol 408: 684–696. [DOI] [PubMed] [Google Scholar]
- 10.Johansson K, Ramaswamy S, Ljungcrantz C, Knecht W, Piskur J, Munch-Petersen B, Eriksson S, and Eklund H. 2001. Structural basis for substrate specificities of cellular deoxyribonucleoside kinases. Nat. Struct. Biol 8: 616–620. [DOI] [PubMed] [Google Scholar]
- 11.Hunsucker SA, Mitchell BS, and Spychala J. 2005. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther 107: 1–30. [DOI] [PubMed] [Google Scholar]
- 12.Bianchi V, and Spychala J. 2003. Mammalian 5′-Nucleotidases. J. Biol. Chem 278: 46195–46198. [DOI] [PubMed] [Google Scholar]
- 13.Nordlund P, and Reichard P. 2006. Ribonucleotide Reductases. Annu. Rev. Biochem 75: 681–706. [DOI] [PubMed] [Google Scholar]
- 14.Håkansson P, Hofer A, and Thelander L. 2006. Regulation of mammalian ribonucleotide reduction and dNTP pools after DNA damage and in resting cells. J. Biol. Chem 281: 7834–41. [DOI] [PubMed] [Google Scholar]
- 15.Pontarin G, Ferraro P, Bee L, Reichard P, and Bianchi V. 2012. Mammalian ribonucleotide reductase subunit p53R2 is required for mitochondrial DNA replication and DNA repair in quiescent cells. Proc. Natl. Acad. Sci. U. S. A 109: 13302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pontarin G, Ferraro P, Rampazzo C, Kollberg G, Holme E, Reichard P, and Bianchi V. 2011. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J. Biol. Chem 286: 11132–11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pontarin G, Fijolek A, Pizzo P, Ferraro P, Rampazzo C, Pozzan T, Thelander L, Reichard PA, and Bianchi V. 2008. Ribonucleotide reduction is a cytosolic process in mammalian cells independently of DNA damage. Proc. Natl. Acad. Sci 105: 17801–17806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi J-S, and Berdis AJ. 2012. Nucleoside transporters: biological insights and therapeutic applications. Future Med. Chem 4: 1461–78. [DOI] [PubMed] [Google Scholar]
- 19.Nicander B, and Reichard P. 1985. Evidence for the involvement of substrate cycles in the regulation of deoxyribonucleoside triphosphate pools in 3T6 cells. J. Biol. Chem 260: 9216–22. [PubMed] [Google Scholar]
- 20.Bianchi V, Pontis E, and Reichard P. 1992. Dynamics of the dATP pool in cultured mammalian cells. Exp. Cell Res 199: 120–128. [DOI] [PubMed] [Google Scholar]
- 21.Spyrou G, and Reichard P. 1988. Dynamcis of the thymidine triphosphate pool during the cell cycle of synchronized 3T3 mouse fibroblasts. Mutat. Res. - Fundam. Mol. Mech. Mutagen 200: 37–43. [DOI] [PubMed] [Google Scholar]
- 22.Leanza L, Ferraro P, Reichard P, and Bianchi V. 2008. Metabolic interrelations within guanine deoxynucleotide pools for mitochondrial and nuclear DNA maintenance. J. Biol. Chem 283: 16437–16445. [DOI] [PubMed] [Google Scholar]
- 23.Rampazzo C, Ferraro P, Pontarin G, Fabris S, Reichard P, and Bianchi V. 2004. Mitochondrial Deoxyribonucleotides, Pool Sizes, Synthesis, and Regulation. J. Biol. Chem 279: 17019–17026. [DOI] [PubMed] [Google Scholar]
- 24.Gandhi VV, and Samuels DC. 2011. A review comparing deoxyribonucleoside triphosphate (dNTP) concentrations in the mitochondrial and cytoplasmic compartments of normal and transformed cells. Nucleosides. Nucleotides Nucleic Acids 30: 317–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meuth M 1989. The molecular basis of mutations induced by deoxyribonucleoside triphosphate pool imbalances in mammalian cells. Exp. Cell Res 181: 305–316. [DOI] [PubMed] [Google Scholar]
- 26.Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, and Chabes A. 2011. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res 39: 1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar D, Viberg J, Nilsson AK, and Chabes A. 2010. Highly mutagenic and severely imbalanced dNTP pools can escape detection by the S-phase checkpoint. Nucleic Acids Res 38: 3975–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buckland RJ, Watt DL, Chittoor B, Nilsson AK, Kunkel TA, and Chabes A. 2014. Increased and Imbalanced dNTP Pools Symmetrically Promote Both Leading and Lagging Strand Replication Infidelity. PLoS Genet 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watt DL, Buckland RJ, Lujan SA, Kunkel TA, and Chabes A. 2015. Genome-wide analysis of the specificity and mechanisms of replication infidelity driven by imbalanced dNTP pools. Nucleic Acids Res 44: 1669–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chabes A, and Stillman B. 2007. Constitutively high dNTP concentration inhibits cell cycle progression and the DNA damage checkpoint in yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A 104: 1183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidson MB, Katou Y, Keszthelyi A, Sing TL, Xia T, Ou J, Vaisica JA, Thevakumaran N, Marjavaara L, Myers CL, Chabes A, Shirahige K, and Brown GW. 2012. Endogenous DNA replication stress results in expansion of dNTP pools and a mutator phenotype. EMBO J 31: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gemble S, Buhagiar-Labarchède G, Onclercq-Delic R, Biard D, Lambert S, and Amor-Guéret M. 2016. A balanced pyrimidine pool is required for optimal Chk1 activation to prevent ultrafine anaphase bridge formation. J. Cell Sci 129: 3167–77. [DOI] [PubMed] [Google Scholar]
- 33.Sabouri N, Viberg J, Goyal DK, Johansson E, and Chabes A. 2008. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res 36: 5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ke PY, Kuo YY, Hu CM, and Chang ZF. 2005. Control of dTTP pool size by anaphase promoting complex/cyclosome is essential for the maintenance of genetic stability. Genes Dev 19: 1920–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hastak K, Paul RK, Agarwal MK, Thakur VS, Amin a R. M. R., Agrawal S, Sramkoski RM, Jacobberger JW, Jackson MW, Stark GR, and Agarwal ML. 2008. DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc. Natl. Acad. Sci. U. S. A 105: 6314–6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anglana M, Apiou F, Bensimon A, and Debatisse M. 2003. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell 114: 385–94. [DOI] [PubMed] [Google Scholar]
- 37.Poli J, Tsaponina O, Crabbé L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, and Pasero P. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31: 883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Papadopoulou C, Guilbaud G, Schiavone D, and Sale JE. 2015. Nucleotide Pool Depletion Induces G-Quadruplex-Dependent Perturbation of Gene Expression. Cell Rep 13: 2491–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jasencakova Z, Scharf AND, Ask K, Corpet A, Imhof A, Almouzni G, and Groth A. 2010. Replication Stress Interferes with Histone Recycling and Predeposition Marking of New Histones. Mol. Cell 37: 736–743. [DOI] [PubMed] [Google Scholar]
- 40.Mathews CK 2015. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 15: 528–539. [DOI] [PubMed] [Google Scholar]
- 41.Boss GR, and Seegmiller JE. 1982. Genetic Defects in Human Purine and Pyrimidine Metabolism. Annu. Rev. Genet 16: 297–328. [DOI] [PubMed] [Google Scholar]
- 42.El-Hattab AW, Craigen WJ, and Scaglia F. 2017. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta 1863: 1539–1555. [DOI] [PubMed] [Google Scholar]
- 43.Chabosseau P, Buhagiar-Labarchède G, Onclercq-Delic R, Lambert S, Debatisse M, Brison O, and Amor-Guéret M. 2011. Pyrimidine pool imbalance induced by BLM helicase deficiency contributes to genetic instability in Bloom syndrome. Nat. Commun 2: 368. [DOI] [PubMed] [Google Scholar]
- 44.Kimura T, Takeda S, Sagiya Y, Gotoh M, Nakamura Y, and Arakawa H. 2003. Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet 34: 440–5. [DOI] [PubMed] [Google Scholar]
- 45.Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, and Zhang R. 2013. Suppression of Nucleotide Metabolism Underlies the Establishment and Maintenance of Oncogene-Induced Senescence. Cell Rep 3: 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aird KM, and Zhang R. 2015. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett 356: 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang L, Guo R, Huang Q, and Yen Y. 2013. Chromosomal Instability Triggered by Rrm2b Loss Leads to IL-6 Secretion and Plasmacytic Neoplasms. Cell Rep 3: 1389–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliver FJ, Collins MKL, and López-Rivas A. 1996. dNTP pools imbalance as a signal to initiate apoptosis. Experientia 52: 995–1000. [DOI] [PubMed] [Google Scholar]
- 49.James SJ, Basnakian AG, and Miller BJ. 1994. In vitro folate deficiency induces deoxynucleotide pool imbalance, apoptosis, and mutagenesis in Chinese hamster ovary cells. Cancer Res 54: 5075–80. [PubMed] [Google Scholar]
- 50.Chandra D, Bratton SB, Person MD, Tian Y, Martin AG, Ayres M, Fearnhead HO, Gandhi V, and Tang DG. 2006. Intracellular Nucleotides Act as Critical Prosurvival Factors by Binding to Cytochrome C and Inhibiting Apoptosome. Cell 125: 1333–1346. [DOI] [PubMed] [Google Scholar]
- 51.Li N, Zhang W, and Cao X. 2000. Identification of human homologue of mouse IFN-γ induced protein from human dendritic cells. Immunol. Lett 74: 221–224. [DOI] [PubMed] [Google Scholar]
- 52.Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin J-PS-M, Lourenco CM, Male AM, Marques W, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, and Crow YJ. 2012. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44: 1243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Powell RD, Holland PJ, Hollis T, and Perrino FW. 2011. Aicardi-Goutieres Syndrome Gene and HIV-1 Restriction Factor SAMHD1 Is a dGTP-regulated Deoxynucleotide Triphosphohydrolase. J. Biol. Chem 286: 43596–43600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HCT, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LPS, Stoye JP, Crow YJ, Taylor IA, and Webb M. 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480: 379–382. [DOI] [PubMed] [Google Scholar]
- 55.Hansen EC, Seamon KJ, Cravens SL, and Stivers JT. 2014. GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc. Natl. Acad. Sci 111: E1843–E1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amie SM, Bambara RA, and Kim B. 2013. GTP Is the Primary Activator of the Anti-HIV Restriction Factor SAMHD1. J. Biol. Chem 288: 25001–25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ji X, Tang C, Zhao Q, Wang W, and Xiong Y. 2014. Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci 111: E4305–E4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beauchamp BB, and Richardson CC. 1988. A unique deoxyguanosine triphosphatase is responsible for the optA1 phenotype of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A 85: 2563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vorontsov II, Minasov G, Kiryukhina O, Brunzelle JS, Shuvalova L, and Anderson WF. 2011. Characterization of the deoxynucleotide triphosphate triphosphohydrolase (dNTPase) activity of the EF1143 protein from Enterococcus faecalis and crystal structure of the activator-substrate complex. J. Biol. Chem 286: 33158–33166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oganesyan V, Adams PD, Jancarik J, Kim R, and Kim SH. 2007. Structure of O67745_AQUAE, a hypothetical protein from Aquifex aeolicus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 63: 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kondo N, Kuramitsu S, and Masui R. 2004. Biochemical characterization of TT1383 from Thermus thermophilus identifies a novel dNTP triphosphohydrolase activity stimulated by dATP and dTTP. J. Biochem 136: 221–231. [DOI] [PubMed] [Google Scholar]
- 62.Kondo N, Nakagawa N, Ebihara A, Chen L, Liu ZJ, Wang BC, Yokoyama S, Kuramitsu S, and Masui R. 2007. Structure of dNTP-inducible dNTP triphosphohydrolase: Insight into broad specificity for dNTPs and triphosphohydrolase-type hydrolysis. Acta Crystallogr. Sect. D Biol. Crystallogr 63: 230–239. [DOI] [PubMed] [Google Scholar]
- 63.Mega R, Kondo N, Nakagawa N, Kuramitsu S, and Masui R. 2009. Two dNTP triphosphohydrolases from Pseudomonas aeruginosa possess diverse substrate specificities. FEBS J 276: 3211–3221. [DOI] [PubMed] [Google Scholar]
- 64.Zimmerman MD, Proudfoot M, Yakunin A, and Minor W. 2008. Structural Insight into the Mechanism of Substrate Specificity and Catalytic Activity of an HD-Domain Phosphohydrolase: The 5’-Deoxyribonucleotidase YfbR from Escherichia coli. J. Mol. Biol 378: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qiao F, and Bowie JU. 2005. The Many Faces of SAM. Sci. Signal 2005: re7–re7. [DOI] [PubMed] [Google Scholar]
- 66.Brandariz-Nuñez A, Valle-Casuso J, White TE, Laguette N, Benkirane M, Brojatsch J, and Diaz-Griffero F. 2012. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 9: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hofmann H, Logue EC, Bloch N, Daddacha W, Polsky SB, Schultz ML, Kim B, and Landau NR. 2012. The Vpx Lentiviral Accessory Protein Targets SAMHD1 for Degradation in the Nucleus. J. Virol 86: 12552–12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aravind L, and Koonin EV. 1998. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci 23: 469–472. [DOI] [PubMed] [Google Scholar]
- 69.DeLucia M, Mehrens J, Wu Y, and Ahn J. 2013. HIV-2 and SIVmac accessory virulence factor Vpx down-regulates SAMHD1 enzyme catalysis prior to proteasome-dependent degradation. J. Biol. Chem 288: 19116–19126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu C, Gao W, Zhao K, Qin X, Zhang Y, Peng X, Zhang L, Dong Y, Zhang W, Li P, Wei W, Gong Y, and Yu XF. 2013. Structural insight into dGTP-dependent activation of tetrameric SAMHD1 deoxynucleoside triphosphate triphosphohydrolase. Nat Commun 4: 2722. [DOI] [PubMed] [Google Scholar]
- 71.Ji X, Wu Y, Yan J, Mehrens J, Yang H, DeLucia M, Hao C, Gronenborn AM, Skowronski J, Ahn J, and Xiong Y. 2013. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol 20: 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan J, Kaur S, DeLucia M, Hao C, Mehrens J, Wang C, Golczak M, Palczewski K, Gronenborn AM, Ahn J, and Skowronsk S. 2013. Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J. Biol. Chem 288: 10406–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Miazzi C, Ferraro P, Pontarin G, Rampazzo C, Reichard P, and Bianchi V. 2014. Allosteric regulation of the human and mouse deoxyribonucleotide triphosphohydrolase sterile α-motif/histidine-aspartate domain-containing protein 1 (SAMHD1). J. Biol. Chem 289: 18339–18346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu C-F, Wei W, Peng X, Dong Y-H, Gong Y, and Yu X-F. 2015. The mechanism of substrate-controlled allosteric regulation of SAMHD1 activated by GTP. Acta Crystallogr. Sect. D Biol. Crystallogr 71: 516–524. [DOI] [PubMed] [Google Scholar]
- 75.Li Y, Kong J, Peng X, Hou W, Qin X, and Yu X-F. 2015. Structural Insights into the High-efficiency Catalytic Mechanism of the Sterile α-Motif/Histidine-Aspartate Domain-containing Protein. J. Biol. Chem 290: 29428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koharudin LMI, Wu Y, DeLucia M, Mehrens J, Gronenborn AM, and Ahn J. 2014. Structural Basis of Allosteric Activation of Sterile Motif and Histidine-Aspartate Domain-containing Protein 1 (SAMHD1) by Nucleoside Triphosphates. J. Biol. Chem 289: 32617–32627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Z, Bhattacharya A, Villacorta J, Diaz-Griffero F, and Ivanov DN. 2016. Allosteric activation of SAMHD1 protein by deoxynucleotide triphosphate (dNTP)-dependent tetramerization requires dNTP concentrations that are similar to dNTP concentrations observed in cycling T cells. J. Biol. Chem 291: 21407–21413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jang S, Zhou X, and Ahn J. 2016. Substrate Specificity of SAMHD1 Triphosphohydrolase Activity Is Controlled by Deoxyribonucleoside Triphosphates and Phosphorylation at Thr592. Biochemistry 55: 5635–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Amie SM, Daly MB, Noble E, Schinazi RF, Bambara RA, and Kim B. 2013. Anti-HIV host factor SAMHD1 regulates viral sensitivity to nucleoside reverse transcriptase inhibitors via modulation of cellular deoxyribonucleoside triphosphate (dNTP) levels. J. Biol. Chem 288: 20683–20691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seamon KJ, Hansen EC, Kadina AP, Kashemirov BA, McKenna CE, Bumpus NN, and Stivers JT. 2014. Small molecule inhibition of SAMHD1 dNTPase by tetramer destabilization. J. Am. Chem. Soc 136: 9822–9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arnold LH, Groom HCT, Kunzelmann S, Schwefel D, Caswell SJ, Ordonez P, Mann MC, Rueschenbaum S, Goldstone DC, Pennell S, Howell SA, Stoye JP, Webb M, Taylor IA, and Bishop KN. 2015. Phospho-dependent Regulation of SAMHD1 Oligomerisation Couples Catalysis and Restriction. PLoS Pathog 11: e1005194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mauney CH, Rogers LC, Harris RS, Daniel LW, Devarie-Baez NO, Wu H, Furdui CM, Poole LB, Perrino FW, and Hollis T. 2017. The SAMHD1 dNTP Triphosphohydrolase Is Controlled by a Redox Switch. Antioxid. Redox Signal 27: 1317–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seamon KJ, Sun Z, Shlyakhtenko LS, Lyubchenko YL, and Stivers JT. 2015. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res 43: 6486–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goncalves A, Karayel E, Rice GI, Bennett KL, Crow YJ, Superti-Furga G, and Bürckstümmer T. 2012. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutières syndrome-associated mutations. Hum. Mutat 33: 1116–1122. [DOI] [PubMed] [Google Scholar]
- 85.White TE, Brandariz-Nuñez A, Valle-Casuso JC, Amie S, Nguyen L, Kim B, Brojatsch J, and Diaz-Griffero F. 2013. Contribution of SAM and HD domains to retroviral restriction mediated by human SAMHD1. Virology 436: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, and Yakunin AF. 2013. Nuclease Activity of the Human SAMHD1 Protein Implicated in the Aicardi-Goutières Syndrome and HIV-1 Restriction. J. Biol. Chem 288: 8101–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tüngler V, Staroske W, Kind B, Dobrick M, Kretschmer S, Schmidt F, Krug C, Lorenz M, Chara O, Schwille P, and Lee-Kirsch MA. 2013. Single-stranded nucleic acids promote SAMHD1 complex formation. J. Mol. Med 91: 759–770. [DOI] [PubMed] [Google Scholar]
- 88.Seamon KJ, Bumpus NN, and Stivers JT. 2016. Single-Stranded Nucleic Acids Bind to the Tetramer Interface of SAMHD1 and Prevent Formation of the Catalytic Homotetramer. Biochemistry 55: 6087–6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choi J, Ryoo J, Oh C, Hwang S, and Ahn K. 2015. SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology 12: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ryoo J, Choi J, Oh C, Kim S, Seo M, Kim S-Y, Seo D, Kim J, White TE, Brandariz-Nuñez A, Diaz-Griffero F, Yun C-H, Hollenbaugh JA, Kim B, Baek D, and Ahn K. 2014. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med 20: 936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ryoo J, Hwang S-Y, Choi J, Oh C, and Ahn K. 2016. SAMHD1, the Aicardi-Goutières syndrome gene and retroviral restriction factor, is a phosphorolytic ribonuclease rather than a hydrolytic ribonuclease. Biochem. Biophys. Res. Commun 477: 977–981. [DOI] [PubMed] [Google Scholar]
- 92.Antonucci JM, St. Gelais C, de Silva S, Yount JS, Tang C, Ji X, Shepard C, Xiong Y, Kim B, and Wu L. 2016. SAMHD1-mediated HIV-1 restriction in cells does not involve ribonuclease activity. Nat. Med 22: 1072–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wittmann S, Behrendt R, Eissmann K, Volkmann B, Thomas D, Ebert T, Cribier A, Benkirane M, Hornung V, Bouzas NF, and Gramberg T. 2015. Phosphorylation of murine SAMHD1 regulates its antiretroviral activity. Retrovirology 12: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Daddacha W, Koyen AE, Bastien AJ, Head PE, Dhere VR, Nabeta GN, Connolly EC, Werner E, Madden MZ, Daly MB, Minten EV, Whelan DR, Schlafstein AJ, Zhang H, Anand R, Doronio C, Withers AE, Shepard C, Sundaram RK, Deng X, Dynan WS, Wang Y, Bindra RS, Cejka P, Rothenberg E, Doetsch PW, Kim B, and Yu DS. 2017. SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination. Cell Rep 20: 1921–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liao W, Bao Z, Cheng C, Mok Y-K, and Wong WSF. 2008. Dendritic cell-derived interferon-γ-induced protein mediates tumor necrosis factor-α stimulation of human lung fibroblasts. Proteomics 8: 2640–2650. [DOI] [PubMed] [Google Scholar]
- 96.Schmidt S, Schenkova K, Adam T, Erikson E, Lehmann-Koch J, Sertel S, Verhasselt B, Fackler OT, Lasitschka F, and Keppler OT. 2015. SAMHD1’s protein expression profile in humans. J. Leukoc. Biol 98: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Franzolin E, Pontarin G, Rampazzo C, Miazzi C, Ferraro P, Palumbo E, Reichard P, and Bianchi V. 2013. The deoxynucleotide triphosphohydrolase SAMHD1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci 110: 14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cribier A, Descours B, Valadão A, Laguette N, and Benkirane M. 2013. Phosphorylation of SAMHD1 by Cyclin A2/CDK1 Regulates Its Restriction Activity toward HIV-1. Cell Rep 3: 1036–1043. [DOI] [PubMed] [Google Scholar]
- 99.Yan J, Hao C, DeLucia M, Swanson S, Florens L, Washburn MP, Ahn J, and Skowronski J. 2015. CyclinA2-Cyclin-dependent Kinase Regulates SAMHD1 Protein Phosphohydrolase Domain. J. Biol. Chem 290: 13279–13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.St. Gelais C, de Silva S, Hach JC, White TE, Diaz-Griffero F, Yount JS, and Wu L. 2014. Identification of Cellular Proteins Interacting with the Retroviral Restriction Factor SAMHD1. J. Virol 88: 7689–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baldauf H-M, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, König R, Fackler OT, and Keppler OT. 2012. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med 18: 1682–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.St. Gelais C, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, and Wu L. 2012. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 9: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, and Kim B. 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem 279: 51545–51553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.De Silva S, Wang F, Hake TS, Porcu P, Wong HK, and Wu L. 2013. Downregulation of SAMHD1 Expression Correlates with Promoter DNA Methylation in Sézary Syndrome Patients. J. Invest. Dermatol 134: 562–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.De Silva S, Hoy H, Hake TS, Wong HK, Porcu P, and Wu L. 2013. Promoter methylation regulates SAMHD1 gene expression in human CD4 + T cells. J. Biol. Chem 288: 9284–9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang J, Lu F, Shen X-Y, Wu Y, and Zhao L. 2014. SAMHD1 is down regulated in lung cancer by methylation and inhibits tumor cell proliferation. Biochem. Biophys. Res. Commun 455: 229–233. [DOI] [PubMed] [Google Scholar]
- 107.Welbourn S, Miyagi E, White TE, Diaz-Griffero F, and Strebel K. 2012. Identification and characterization of naturally occurring splice variants of SAMHD1. Retrovirology 9: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bloch N, Gläsker S, Sitaram P, Hofmann H, Shepard CN, Schultz ML, Kim B, and Landau NR. 2016. A Highly Active Isoform of Lentivirus Restriction Factor SAMHD1 in Mouse. J. Biol. Chem jbc.M116.743740. [DOI] [PMC free article] [PubMed]
- 109.Pauls E, Jimenez E, Ruiz A, Permanyer M, Ballana E, Costa H, Nascimiento R, Parkhouse RM, Peña R, Riveiro-Muñoz E, a Martinez M, Clotet B, a Esté J, and Bofill M. 2013. Restriction of HIV-1 replication in primary macrophages by IL-12 and IL-18 through the upregulation of SAMHD1. J. Immunol 190: 4736–41. [DOI] [PubMed] [Google Scholar]
- 110.Yang S, Zhan Y, Zhou Y, Jiang Y, Zheng X, Yu L, Tong W, Gao F, Li L, Huang Q, Ma Z, and Tong G. 2016. Interferon regulatory factor 3 is a key regulation factor for inducing the expression of SAMHD1 in antiviral innate immunity. Sci. Rep 6: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goujon C, Schaller T, Galão RP, Amie SM, Kim B, Olivieri K, Neil SJD, and Malim MH. 2013. Evidence for IFNα-induced, SAMHD1-independent inhibitors of early HIV-1 infection. Retrovirology 10: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Riess M, Fuchs NV, Idica A, Hamdorf M, Flory E, Pedersen IM, and König R. 2017. Interferons induce expression of SAMHD1 in monocytes through down-regulation of miR-181a and miR-30a. J. Biol. Chem 292: 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kyei GB, Cheng X, Ramani R, and Ratner L. 2015. Cyclin L2 is a critical HIV dependency factor in macrophages that controls samhd1 abundance. Cell Host Microbe 17: 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Morrissey C, Schwefel D, Ennis-Adeniran V, Taylor IA, Crow YJ, and Webb M. 2015. The eukaryotic elongation factor eEF1A1 interacts with SAMHD1. Biochem. J 466: 69–76. [DOI] [PubMed] [Google Scholar]
- 115.Rocha-Perugini V, Suárez H, Álvarez S, López-Martín S, Lenzi GM, Vences-Catalán F, Levy S, Kim B, Muñoz-Fernández MA, Sánchez-Madrid F, and Yáñez-Mó M. 2017. CD81 association with SAMHD1 enhances HIV-1 reverse transcription by increasing dNTP levels. Nat. Microbiol. [DOI] [PMC free article] [PubMed]
- 116.Welbourn S, Dutta SM, Semmes OJ, and Strebel K. 2013. Restriction of Virus Infection but Not Catalytic dNTPase Activity Is Regulated by Phosphorylation of SAMHD1. J. Virol 87: 11516–11524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.White TE, Brandariz-Nuñez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, and Diaz-Griffero F. 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 13: 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.St. Gelais C, Kim SH, Ding L, Yount JS, Ivanov D, Spearman P, and Wu L. 2016. A Putative Cyclin-binding Motif in Human SAMHD1 Contributes to Protein Phosphorylation, Localization, and Stability. J. Biol. Chem 291: 26332–26342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Coiras M, Bermejo M, Descours B, Mateos E, García-Pérez J, López-Huertas M-R, Lederman MM, Benkirane M, and Alcamí J. 2016. IL-7 Induces SAMHD1 Phosphorylation in CD4+ T Lymphocytes, Improving Early Steps of HIV-1 Life Cycle. Cell Rep 14: 2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pauls E, Ruiz A, Badia R, Permanyer M, Gubern A, Riveira-Munoz E, Torres-Torronteras J, Alvarez M, Mothe B, Brander C, Crespo M, Menendez-Arias L, Clotet B, Keppler OT, Marti R, Posas F, Ballana E, and Este JA. 2014. Cell Cycle Control and HIV-1 Susceptibility Are Linked by CDK6-Dependent CDK2 Phosphorylation of SAMHD1 in Myeloid and Lymphoid Cells. J. Immunol 193: 1988–1997. [DOI] [PubMed] [Google Scholar]
- 121.Pauls E, Badia R, Torres-Torronteras J, Ruiz A, Permanyer M, Riveira-Munoz E, Clotet B, Marti R, Ballana E, and Este JA. 2014. Palbociclib, a selective inhibitor of cyclin-dependent kinase4/6, blocks HIV-1 reverse transcription through the control of sterile alpha motif and HD domain-containing protein-1 (SAMHD1) activity. Aids 28: 2213–2222. [DOI] [PubMed] [Google Scholar]
- 122.Mlcochova P, Sutherland KA, Watters SA, Bertoli C, de Bruin RA, Rehwinkel J, Neil SJ, Lenzi GM, Kim B, Khwaja A, Gage MC, Georgiou C, Chittka A, Yona S, Noursadeghi M, Towers GJ, and Gupta RK. 2017. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J 36: 604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mandal M, Bandyopadhyay D, Goepfert TM, and Kumar R. 1998. Interferon-induces expression of cyclin-dependent kinase-inhibitors p21WAF1 and p27Kip1 that prevent activation of cyclin-dependent kinase by CDK-activating kinase (CAK). Oncogene 16: 217–25. [DOI] [PubMed] [Google Scholar]
- 124.Pauls E, Ruiz A, Riveira-Muñoz E, Permanyer M, Badia R, Clotet B, Keppler OT, Ballana E, and Este JA. 2014. p21 regulates the HIV-1 restriction factor SAMHD1. Proc. Natl. Acad. Sci. U. S. A 111: E1322–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Valle-Casuso JC, Allouch A, David A, Lenzi GM, Studdard L, Barré-Sinoussi F, Müller-Trutwin M, Kim B, Pancino G, and Sáez-Cirión A. 2017. p21 Restricts HIV-1 in Monocyte-Derived Dendritic Cells through the Reduction of Deoxynucleoside Triphosphate Biosynthesis and Regulation of SAMHD1 Antiviral Activity. J. Virol 91: e01324–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mlcochova P, Caswell SJ, Taylor IA, Towers GJ, and Gupta RK. 2018. DNA damage induced by topoisomerase inhibitors activates SAMHD1 and blocks HIV-1 infection of macrophages. EMBO J 37: 50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tang C, Ji X, Wu L, and Xiong Y. 2015. Impaired dNTPase activity of SAMHD1 by phosphomimetic mutation of Thr-592. J. Biol. Chem 290: 26352–26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ruiz A, Pauls E, Badia R, Torres-Torronteras J, Riveira-Muñoz E, Clotet B, Martí R, Ballana E, and Esté JA. 2015. Cyclin D3-dependent control of the dNTP pool and HIV-1 replication in human macrophages. Cell Cycle 14: 1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bhattacharya A, Wang Z, White T, Buffone C, Nguyen LA, Shepard CN, Kim B, Demeler B, Diaz-Griffero F, Ivanov DN, Rice GI, Baldauf HM, Berger A, Descours B, Hrecka K, Laguette N, Beauchamp BB, Richardson CC, Kornberg SR, Lehman IR, Bessman MJ, Simms ES, Kornberg A, Seto D, Bhatnagar SK, Bessman MJ, Franzolin E, Fujita M, Goujon C, Kim B, Nguyen LA, Daddacha W, Hollenbaugh JA, Lahouassa H, Cribier A, Descours B, Valadao AL, Laguette N, Benkirane M, Welbourn S, Dutta SM, Semmes OJ, Strebel K, Welbourn S, Strebel K, White TE, Beloglazova N, Ryoo J, Crow YJ, Crow YJ, Seamon KJ, Sun Z, Shlyakhtenko LS, Lyubchenko YL, Stivers JT, Goncalves A, Tungler V, White TE, Ji X, Tang C, Zhao Q, Wang W, Xiong Y, Ji X, Koharudin LM, Yan J, Zhu C, Zhu CF, Nordlund P, Reichard P, Segura-Pena D, Zimanyi CM, Chen PY, Kang G, Funk MA, Drennan CL, Arnold LH, Tang C, Ji X, Wu L, Xiong Y, Yan J, Brandariz-Nunez A, Hansen EC, Seamon KJ, Cravens SL, Stivers JT, Zhao K, Amie SM, Bambara RA, Kim B, Arnold LH, Kunzelmann S, Webb MR, Taylor IA, Seamon KJ, Stivers JT, Weiss KK, Choi J, Ryoo J, Oh C, Hwang S, Ahn K, Brookes E, Cao W, Demeler B, Yee JK, Friedmann T, Burns JC, Fricke T, and Diamond TL. 2016. Effects of T592 phosphomimetic mutations on tetramer stability and dNTPase activity of SAMHD1 can not explain the retroviral restriction defect. Sci. Rep 6: 31353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Poole LB 2015. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med 80: 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Go Y-M, and Jones DP. 2013. The Redox Proteome. J. Biol. Chem 288: 26512–26520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee EJ, Seo JH, Park J-H, Vo TTL, An S, Bae S-J, Le H, Lee HS, Wee H-J, Lee D, Chung Y-H, Kim JA, Jang M-K, Ryu SH, Yu E, Jang SH, Park ZY, and Kim K-W. 2017. SAMHD1 acetylation enhances its deoxynucleotide triphosphohydrolase activity and promotes cancer cell proliferation. Oncotarget 8: 68517–68529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bonifati S, Daly MB, St. Gelais C, Kim SH, Hollenbaugh JA, Shepard C, Kennedy EM, Kim DH, Schinazi RF, Kim B, and Wu L. 2016. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 495: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Stillman B 2013. Deoxynucleoside triphosphate (dNTP) synthesis and destruction regulate the replication of both cell and virus genomes. Proc. Natl. Acad. Sci. U. S. A 110: 14120–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim B, Nguyen LA, Daddacha W, and Hollenbaugh JA. 2012. Tight Interplay among SAMHD1 Protein Level, Cellular dNTP Levels, and HIV-1 Proviral DNA Synthesis Kinetics in Human Primary Monocyte-derived Macrophages. J. Biol. Chem 287: 21570–21574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hollenbaugh JA, Tao S, Lenzi GM, Ryu S, Kim D-H, Diaz-Griffero F, Schinazi RF, and Kim B. 2014. dNTP pool modulation dynamics by SAMHD1 protein in monocyte-derived macrophages. Retrovirology 11: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kretschmer S, Wolf C, Konig N, Staroske W, Guck J, Hausler M, Luksch H, a Nguyen L, Kim B, Alexopoulou D, Dahl A, Rapp A, Cardoso MC, Shevchenko A, and Lee-Kirsch MA. 2015. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis 74: e17–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dragin L, Munir-Matloob S, Froehlich J, Morel M, Sourisce A, Lahouassa H, Bailly K, Mangeney M, Ramirez BC, and Margottin-Goguet F. 2015. Evidence that HIV-1 restriction factor SAMHD1 facilitates differentiation of myeloid THP-1 cells. Virol. J 12: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kodigepalli KM, Li M, Liu S-L, and Wu L. 2017. Exogenous expression of SAMHD1 inhibits proliferation and induces apoptosis in cutaneous T-cell lymphoma-derived HuT78 cells. Cell Cycle 16: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Antonucci JM, St. Gelais C, and Wu L. 2017. The Dynamic Interplay between HIV-1, SAMHD1, and the Innate Antiviral Response. Front. Immunol 8: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kennedy EM, Gavegnano C, Nguyen L, Slater R, Lucas A, Fromentin E, Schinazi RF, and Kim B. 2010. Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J. Biol. Chem 285: 39380–39391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, and Benkirane M. 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, and Skowronski J. 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474: 658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, Yatim A, Emiliani S, Schwartz O, and Benkirane M. 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474: 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Berger A, Sommer AFR, Zwarg J, Hamdorf M, Welzel K, Esly N, Panitz S, Reuter A, Ramos I, Jatiani A, Mulder LCF, Fernandez-Sesma A, Rutsch F, Simon V, König R, and Flory E. 2011. SAMHD1-deficient CD14+ cells from individuals with Aicardi-Goutières syndrome are highly susceptible to HIV-1 infection. PLoS Pathog 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, and Skowronski J. 2008. Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog 4: e1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pertel T, Reinhard C, and Luban J. 2011. Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology 8: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sunseri N, O’Brien M, Bhardwaj N, and Landau NR. 2011. Human immunodeficiency virus type 1 modified to package Simian immunodeficiency virus Vpx efficiently infects macrophages and dendritic cells. J Virol 85: 6263–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, and Margottin-Goguet F. 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol 13: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Amie SM, Noble E, and Kim B. 2013. Intracellular nucleotide levels and the control of retroviral infections. Virology 436: 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Berger G, Turpin J, Cordeil S, Tartour K, Nguyen XN, Mahieux R, and Cimarelli A. 2012. Functional analysis of the relationship between Vpx and the restriction factor SAMHD1. J. Biol. Chem 287: 41210–41217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schwefel D, Groom HCT, Boucherit VC, Christodoulou E, Walker PA, Stoye JP, Bishop KN, and Taylor IA. 2014. Structural basis of lentiviral subversion of a cellular protein degradation pathway. Nature 505: 234–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ahn J, Hao C, Yan J, DeLucia M, Mehrens J, Wang C, Gronenborn AM, and Skowronski J. 2012. HIV/Simian Immunodeficiency Virus (SIV) accessory virulence factor Vpx loads the host cell restriction factor SAMHD1 onto the E3 ubiquitin ligase complex CRL4 DCAF1. J. Biol. Chem 287: 12550–12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ayinde D, Bruel T, Cardinaud S, Porrot F, Prado JG, Moris A, and Schwartz O. 2015. SAMHD1 Limits HIV-1 Antigen Presentation by Monocyte-Derived Dendritic Cells. J. Virol 89: 6994–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Brégnard C, Benkirane M, and Laguette N. 2014. DNA damage repair machinery and HIV escape from innate immune sensing. Front. Microbiol 5: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Manel N, Hogstad B, Wang Y, Levy DE, Unutmaz D, and Littman DR. 2010. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature 467: 214–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Maelfait J, Bridgeman A, Benlahrech A, Cursi C, and Rehwinkel J. 2016. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep 16: 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.van Montfoort N, Olagnier D, and Hiscott J. 2014. Unmasking immune sensing of retroviruses: Interplay between innate sensors and host effectors. Cytokine Growth Factor Rev 25: 657–668. [DOI] [PubMed] [Google Scholar]
- 159.Kim ET, White TE, Brandariz-Núñez A, Diaz-Griffero F, and Weitzman MD. 2013. SAMHD1 Restricts Herpes Simplex Virus 1 in Macrophages by Limiting DNA Replication. J. Virol 87: 12949–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hollenbaugh JA, Gee P, Baker J, Daly MB, Amie SM, Tate J, Kasai N, Kanemura Y, Kim D-H, Ward BM, Koyanagi Y, and Kim B. 2013. Host factor SAMHD1 restricts DNA viruses in non-dividing myeloid cells. PLoS Pathog 9: e1003481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D, Berka U, Lienenklaus S, Peschke K, Gibbert K, Wittmann S, Lindemann D, Weiss S, Dahl A, Naumann R, Dittmer U, Kim B, Mueller W, Gramberg T, and Roers A. 2013. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep 4: 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sze A, Belgnaoui SM, Olagnier D, Lin R, Hiscott J, and van Grevenynghe J. 2013. Host Restriction Factor SAMHD1 Limits Human T Cell Leukemia Virus Type 1 Infection of Monocytes via STING-Mediated Apoptosis. Cell Host Microbe 14: 422–434. [DOI] [PubMed] [Google Scholar]
- 163.Sommer AFR, Rivière L, Qu B, Schott K, Riess M, Ni Y, Shepard C, Schnellbächer E, Finkernagel M, Himmelsbach K, Welzel K, Kettern N, Donnerhak C, Münk C, Flory E, Liese J, Kim B, Urban S, and König R. 2016. Restrictive influence of SAMHD1 on Hepatitis B Virus life cycle. Sci. Rep 6: 26616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen Z, Zhu M, Pan X, Zhu Y, Yan H, Jiang T, Shen Y, Dong X, Zheng N, Lu J, Ying S, and Shen Y. 2014. Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun 450: 1462–1468. [DOI] [PubMed] [Google Scholar]
- 165.Gramberg T, Kahle T, Bloch N, Wittmann S, Müllers E, Daddacha W, Hofmann H, Kim B, Lindemann D, and Landau NR. 2013. Restriction of diverse retroviruses by SAMHD1. Retrovirology 10: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huber HE, Beauchamp BB, and Richardson CC. 1988. Escherichia coli dGTP triphosphohydrolase is inhibited by gene 1.2 protein of bacteriophage T7. J. Biol. Chem 263: 13549–13556. [PubMed] [Google Scholar]
- 167.Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, Mavrommati I, Pai C-C, Zalmas L-P, Drobnitzky N, Dianov GL, Verrill C, Macaulay VM, Ying S, La Thangue NB, D’Angiolella V, Ryan AJ, and Humphrey TC. 2015. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell 28: 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Aye Y, Li M, Long MJC, and Weiss RS. 2015. Ribonucleotide reductase and cancer: biological mechanisms and targeted therapies. Oncogene 34: 2011–2021. [DOI] [PubMed] [Google Scholar]
- 169.Welbourn S, and Strebel K. 2016. Low dNTP levels are necessary but may not be sufficient for lentiviral restriction by SAMHD1. Virology 488: 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Brandariz-Nuñez A, Valle-Casuso JC, White TE, Nguyen L, Bhattacharya A, Wang Z, Demeler B, Amie S, Knowlton C, Kim B, Ivanov DN, and Diaz-Griffero F. 2013. Contribution of oligomerization to the anti-HIV-1 properties of SAMHD1. Retrovirology 10: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Crow YJ, and Rehwinkel J. 2009. Aicardi-Goutieres syndrome and related phenotypes: linking nucleic acid metabolism with autoimmunity. Hum. Mol. Genet 18: R130–R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Crow YJ, and Manel N. 2015. Aicardi–Goutières syndrome and the type I interferonopathies. Nat. Rev. Immunol 15: 429–440. [DOI] [PubMed] [Google Scholar]
- 173.Plander M, and Kalman B. 2016. Rare autoimmune disorders with Mendelian inheritance. Autoimmunity 49: 285–297. [DOI] [PubMed] [Google Scholar]
- 174.Goutières F, Aicardi J, Barth PG, and Lebon P. 1998. Aicardi-Goutières syndrome: an update and results of interferon-alpha studies. Ann. Neurol 44: 900–7. [DOI] [PubMed] [Google Scholar]
- 175.Ravenscroft JC, Suri M, Rice GI, Szynkiewicz M, and Crow YJ. 2011. Autosomal dominant inheritance of a heterozygous mutation in SAMHD1 causing familial chilblain lupus. Am. J. Med. Genet Part A 155: 235–237. [DOI] [PubMed] [Google Scholar]
- 176.Crow MK, Kirou KA, and Wohlgemuth J. 2003. Microarray Analysis of Interferon-regulated Genes in SLE. Autoimmunity 36: 481–490. [DOI] [PubMed] [Google Scholar]
- 177.Crow YJ, Chase DS, Schmidt JL, Szynkiewicz M, Forte GMA, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-hamid MS, Abdel-salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon-shalev S, Arellano M, Ariaudo G, Aswani V, Babul-hirji R, Baildam EM, Bahi-buisson N, Bailey KM, Barnerias C, Barth M, Battini R, Bianchi M, De TB, Beresford MW, Blair EM, Bloom M, Burlina AB, Carpanelli ML, Carvalho DR, Castro-gago M, Cavallini A, Cereda C, Chandler KE, Chitayat DA, Collins AE, Corcoles CS, V Cordeiro NJ, Crichiutti G, Dabydeen L, Dale RC, Arrigo SD, De Goede CGEL, De Laet C, De Waele LMH, Denzler I, Desguerre I, Devriendt K, Di Rocco M, Fahey MC, Gener B, Goizet C, Gowrinathan NR, Gowrishankar K, Hanrahan D, Isidor B, Khan N, King MD, Kirk EP, Kumar R, Lagae L, Lin JS, Linnankivi T, Mackay MT, Marom DR, Lourenc CM, Mckee SA, Moroni I, V Morton JE, Suri M, Tacke U, Tan TY, Naude W, Teik KW, Thomas MM, Till M, Tonduti D, Valente EM, Van Coster RN, Van Der Knaap MS, Vassallo G, Vijzelaar R, Vogt J, Wallace GB, Wassmer E, Webb HJ, Whitehouse WP, Whitney RN, Zaki MS, Zuberi SM, Livingston JH, Rozenberg F, Lebon P, Vanderver A, Orcesi S, and Rice GI. 2015. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet Part A 296–312. [DOI] [PMC free article] [PubMed]
- 178.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BCJ, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, and Crow YJ. 2009. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet 41: 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, Yasumi T, Kato H, Shirai T, Ohara O, Fujita T, and Heike T. 2014. Aicardi-Goutières Syndrome Is Caused by IFIH1 Mutations. Am. J. Hum. Genet 95: 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Déry C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martínez-Frías ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JBP, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, and Jackson AP. 2006. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet 38: 910–6. [DOI] [PubMed] [Google Scholar]
- 181.Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, Robins P, Harvey S, Hollis T, O’Hara A, Herrick AL, Bowden AP, Perrino FW, Lindahl T, Barnes DE, and Crow YJ. 2007. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet 80: 811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, Van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Michaud JL, Ponsot G, Voit T, Massey RF, Franc J, Lebon P, Bonthron DT, Jackson AP, Barnes DE, and Lindahl T. 2006. Mutations in the gene encoding the 3′−5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet 38: 917–920. [DOI] [PubMed] [Google Scholar]
- 183.Hu S, Li J, Xu F, Mei S, Le Duff Y, Yin L, Pang X, Cen S, Jin Q, Liang C, and Guo F. 2015. SAMHD1 Inhibits LINE-1 Retrotransposition by Promoting Stress Granule Formation. PLOS Genet 11: e1005367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhao K, Du J, Han X, Goodier JL, Li P, Zhou X, Wei W, Evans SL, Li L, Zhang W, Cheung LE, Wang G, Kazazian HH, and Yu X-F. 2013. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutières syndrome-related SAMHD1. Cell Rep 4: 1108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Crow MK 2010. Long interspersed nuclear elements (LINE-1): Potential triggers of systemic autoimmune disease. Autoimmunity 43: 7–16. [DOI] [PubMed] [Google Scholar]
- 186.McKinnon PJ 2013. Maintaining genome stability in the nervous system. Nat. Neurosci 16: 1523–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Xin B, Jones S, Puffenberger EG, Hinze C, Bright A, Tan H, Zhou A, Wu G, Vargus-Adams J, Agamanolis D, and Wang H. 2011. Homozygous mutation in SAMHD1 gene causes cerebral vasculopathy and early onset stroke. Proc. Natl. Acad. Sci. U. S. A 108: 5372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ramesh V, Bernardi B, Stafa A, Garone C, Franzoni E, Abinun M, Mitchell P, Mitra D, Friswell M, Nelson J, Shalev SA, Rice GI, Gornall H, Szynkiewicz M, Aymard F, Ganesan V, Prendiville J, Livingston JH, and Crow YJ. 2010. Intracerebral large artery disease in Aicardi-Goutières syndrome implicates SAMHD1 in vascular homeostasis. Dev. Med. Child Neurol 52: 725–732. [DOI] [PubMed] [Google Scholar]
- 189.Thiele H, Du Moulin M, Barczyk K, George C, Schwindt W, Nürnberg G, Frosch M, Kurlemann G, Roth J, Nürnberg P, and Rutsch F. 2010. Cerebral arterial stenoses and stroke: novel features of Aicardi-Goutières syndrome caused by the Arg164X mutation in SAMHD1 are associated with altered cytokine expression. Hum. Mutat 31: 1836–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Leshinsky-Silver E, Malinger G, Ben-Sira L, Kidron D, Cohen S, Inbar S, Bezaleli T, Levine A, Vinkler C, Lev D, and Lerman-Sagie T. 2011. A large homozygous deletion in the SAMHD1 gene causes atypical Aicardi-Goutiéres syndrome associated with mtDNA deletions. Eur. J. Hum. Genet 19: 287–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, Bieniasz PD, Towers GJ, Moita LF, Crow YJ, Bonthron DT, and Reis e Sousa C. 2013. SAMHD1-dependent retroviral control and escape in mice. EMBO J 32: 2454–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Kasher PR, Jenkinson EM, Briolat V, Gent D, Morrissey C, Zeef LAH, Rice GI, Levraud J-P, and Crow YJ. 2015. Characterization of samhd1 Morphant Zebrafish Recapitulates Features of the Human Type I Interferonopathy Aicardi-Goutières Syndrome. J. Immunol 194: 2819–2825. [DOI] [PubMed] [Google Scholar]
- 193.Kunz BA 1988. Mutagenesis and deoxyribonucleotide pool imbalance. Mutat. Res. - Fundam. Mol. Mech. Mutagen 200: 133–147. [DOI] [PubMed] [Google Scholar]
- 194.Weinberg G, Ullman B, and Martin DW. 1981. Mutator phenotypes 78: 2447–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sohl CD, Ray S, and Sweasy JB. 2015. Pools and Pols: Mechanism of a mutator phenotype. Proc. Natl. Acad. Sci 112: 201505169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Williams LN, Marjavaara L, Knowels GM, Schultz EM, Fox EJ, Chabes A, and Herr AJ. 2015. dNTP pool levels modulate mutator phenotypes of error-prone DNA polymerase ε variants. Proc. Natl. Acad. Sci. U. S. A 112: E2457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mertz TM, Sharma S, Chabes A, and V Shcherbakova P. 2015. Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity. Proc. Natl. Acad. Sci. U. S. A 112: E2467–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, and Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145: 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pajalunga D, Franzolin E, Stevanoni M, Zribi S, Passaro N, Gurtner A, Donsante S, Loffredo D, Losanno L, Bianchi V, Russo A, Rampazzo C, and Crescenzi M. 2017. A defective dNTP pool hinders DNA replication in cell cycle-reactivated terminally differentiated muscle cells. Cell Death Differ 1–11. [DOI] [PMC free article] [PubMed]
- 200.Rentoft M, Lindell K, Tran P, Chabes AL, Buckland RJ, Watt DL, Marjavaara L, Nilsson AK, Melin B, Trygg J, Johansson E, and Chabes A. 2016. Heterozygous colon cancer-associated mutations of SAMHD1 have functional significance. Proc. Natl. Acad. Sci 113: 4723–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Clifford R, Louis T, Robbe P, Ackroyd S, Burns A, Timbs AT, Wright Colopy G, Dreau H, Sigaux F, Judde JG, Rotger M, Telenti A, Lin YL, Pasero P, Maelfait J, Titsias M, Cohen DR, Henderson SJ, Ross MT, Bentley D, Hillmen P, Pettitt A, Rehwinkel J, Knight SJL, Taylor JC, Crow YJ, Benkirane M, and Schuh A. 2014. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood 123: 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Johansson P, Klein-Hitpass L, Bergmann AK, Siebert R, Scholtysik R, Przekopowitz M, Seifert M, Zenz T, Dührsen U, Küppers R, and Dürig J. 2017. SAMHD1 Is frequently involved in T-Cell prolymphocytic leukemia (T-PLL) pathogenesis. Hematol. Oncol 35: 164–164. [Google Scholar]
- 203.Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, Stefancsik R, Harsha B, Kok CY, Jia M, Jubb H, Sondka Z, Thompson S, De T, and Campbell PJ. 2017. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45: D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Kohnken R, Kodigepalli KM, and Wu L. 2015. Regulation of deoxynucleotide metabolism in cancer: novel mechanisms and therapeutic implications. Mol. Cancer 14: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mathews CK 2017. Oxidized deoxyribonucleotides, mutagenesis, and cancer. FASEB J 31: 11–13. [DOI] [PubMed] [Google Scholar]
- 206.Rudd SG, Valerie NCK, and Helleday T. 2016. Pathways controlling dNTP pools to maintain genome stability. DNA Repair (Amst) 44: 193–204. [DOI] [PubMed] [Google Scholar]
- 207.Parker WB 2009. Enzymology of Purine and Pyrimidine Antimetabolites Used in the Treatment of Cancer. Chem. Rev 109: 2880–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ewald B, Sampath D, and Plunkett W. 2008. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 27: 6522–6537. [DOI] [PubMed] [Google Scholar]
- 209.Herold N, Rudd SG, Sanjiv K, Kutzner J, Bladh J, Paulin CBJ, Helleday T, Henter J-I, and Schaller T. 2017. SAMHD1 protects cancer cells from various nucleoside-based antimetabolites. Cell Cycle 4101: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schneider C, Oellerich T, Baldauf H-M, Schwarz S-M, Thomas D, Flick R, Bohnenberger H, Kaderali L, Stegmann L, Cremer A, Martin M, Lohmeyer J, Michaelis M, Hornung V, Schliemann C, Berdel WE, Hartmann W, Wardelmann E, Comoglio F, Hansmann M-L, Yakunin AF, Geisslinger G, Ströbel P, Ferreirós N, Serve H, Keppler OT, and Cinatl J. 2016. SAMHD1 is a biomarker for cytarabine response and a therapeutic target in acute myeloid leukemia. Nat. Med. [DOI] [PubMed]
- 211.Herold N, Rudd SG, Ljungblad L, Sanjiv K, Myrberg IH, Paulin CBJ, Heshmati Y, Hagenkort A, Kutzner J, Page BDG, Calderón-Montaño JM, Loseva O, Jemth A-S, Bulli L, Axelsson H, Tesi B, Valerie NCK, Höglund A, Bladh J, Wiita E, Sundin M, Uhlin M, Rassidakis G, Heyman M, Tamm KP, Warpman-Berglund U, Walfridsson J, Lehmann S, Grandér D, Lundbäck T, Kogner P, Henter J-I, Helleday T, and Schaller T. 2017. Targeting SAMHD1 with the Vpx protein to improve cytarabine therapy for hematological malignancies. Nat. Med 23: 256–263. [DOI] [PubMed] [Google Scholar]
- 212.Herold N, Rudd SG, Sanjiv K, Kutzner J, Myrberg IH, Paulin CBJ, Olsen TK, Helleday T, Henter J-I, and Schaller T. 2017. With me or against me: Tumor suppressor and drug resistance activities of SAMHD1. Exp. Hematol 52: 32–39. [DOI] [PubMed] [Google Scholar]
- 213.Ballana E, Badia R, Terradas G, Torres-Torronteras J, Ruiz A, Pauls E, Riveira-Muñoz E, Clotet B, Martí R, and Esté JA. 2014. SAMHD1 specifically affects the antiviral potency of thymidine analog HIV reverse transcriptase inhibitors. Antimicrob. Agents Chemother 58: 4804–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Huber AD, Michailidis E, Schultz ML, Ong YT, Bloch N, Puray-Chavez MN, Leslie MD, Ji J, Lucas AD, Kirby KA, Landau NR, and Sarafianos SG. 2014. SAMHD1 Has Differential Impact on the Efficacies of HIV Nucleoside Reverse Transcriptase Inhibitors. Antimicrob. Agents Chemother 58: 4915–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Arnold LH, Kunzelmann S, Webb MR, and Taylor IA. 2015. A Continuous Enzyme-Coupled Assay for Triphosphohydrolase Activity of HIV-1 Restriction Factor SAMHD1. Antimicrob. Agents Chemother 59: 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Seamon KJ, and Stivers JT. 2015. A High-Throughput Enzyme-Coupled Assay for SAMHD1 dNTPase. J. Biomol. Screen 20: 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]