

Abstract
The tumor-cell microenvironment is recognized as a dynamic place where critical cell interactions occur and play an important role in altering tumorigenesis. While many studies have investigated the effects of cellular cross-talk within distinct tumor microenvironments, these interactions have yet to be fully examined in bone. It is well-established that many common cancers metastasize to bone, resulting in the development of tumor-induced bone disease (TIBD), a multi-facetted illness that is driven by complex cell interactions within the bone marrow. Our group has previously published that myeloid progenitor cells expand in the presence of tumors in bone, aligning with the notion that myeloid cells can act as tumor promotors. Several groups, including ours, have established that transforming growth factor β (TGF-β), an abundant growth factor in bone, can regulate both TIBD and myeloid expansion. TGF-β inhibitors have been shown to increase bone volume, decrease bone destruction, and reduce but not eliminate tumor. Therefore, we hypothesize that inhibiting TGF-β will reduce myeloid expansion leading to a reduction of tumor burden in bone and osteoclast-mediated bone loss, causing to an overall reduction in TIBD. To address this hypothesis, two different mouse models of breast cancer bone colonization were pre-treated with the TGF-β neutralizing antibody, 1D11, prior to tumor inoculation (athymic: MDA-MB-231, BALB/c: 4T1) and continuously treated until sacrifice. Additionally, a genetically modified mouse model with a myeloid specific deletion of transforming growth factor beta receptor II (TGF-βRII) (TGF-βRIIMyeKO) was utilized in our studies. Systemic inhibition of TGF-β lead to fewer osteolytic lesions, and reduced tumor burden in bone as expected from previous studies. Additionally, early TGF-β inhibition affected expansion of distinct myeloid populations and shifted the cytokine profile of pro-tumorigenic factors in bone, 4T1 tumor cells, and bone-marrow derived macrophages. Similar observations were seen in tumor-bearing TGF-βRIIMyeKO mice, where these mice contained fewer bone lesions and significantly less tumor burden in bone, suggesting that TGF-β inhibition regulates myeloid expansion leading to a significant reduction in TIBD.
Keywords: TGF-β, Myeloid cells, Breast cancer, Tumor-induced bone disease, Metastasis
1. Introduction
Breast cancer is the second leading cause of death among women (1), and up to 80% of metastatic breast cancer patients will develop bone metastases (2). Once established in bone, tumors can induce bone destruction, increase fracture, and hypercalcemia, all factors associated with high morbidity and mortality rates (2). While current therapies, including bisphosphonates and denosumab, targeting osteoclast-mediated bone destruction have successfully improved patient morbidity and time to skeletal-related events, they have limited effect on tumor growth and patient survival (2). Thus, therapies that reduce tumor growth and extend survival are needed to help improve patient outcomes.
Once tumors have established in bone they secrete factors (e.g. parathyroid hormone-related protein (PTHrP)) that stimulate osteoclast-mediated bone resorption. Previous work has established transforming growth factor beta (TGF-β) as a regulator of PTHrP expression, and our lab and others have shown that when you inhibit TGF-β systemically or within tumor cells, tumor burden and tumor-induced bone disease (TIBD) is reduced (3-6). While this has been a promising target, tumors have not been completely eliminated when treated with TGF-β inhibitors, especially in models where tumors were allowed to establish first (5, 7, 8). This observation is supported by computational modeling for the treatment of prostate cancer metastases that suggested a minimal effect on tumor growth when using TGF-β inhibitors after the establishment of tumors in bone (9). Several studies have proposed that in the context of TIBD, adjuvant TGF-β inhibition may be advantageous over treatment of active disease (5, 7, 9). However, the mechanisms of how TGF-β inhibition reduces TIBD, remains unclear.
Work from our group and others has established the CD11b+ Gr-1+ myeloid cells as contributors to TIBD through their expression of TGF-β (10-12). Outside of the bone, myeloid cells have been implicated in the expression and secretion of TGF-β and regulation of tumorigenesis (13, 14). Since myeloid cells abundantly express and secrete TGF-β, investigators have focused on targeting TGF-β receptor II (TGF-βRII), which when removed in myeloid cells reduces primary tumor growth, lung metastases and osteolytic lesions as compared to control mice (15-17). Past publications have focused on the role of myeloid cells in the primary tumor microenvironment (15, 18-21) and it is only recently that investigators have started to explore the role of myeloid cells in the context of TIBD (11, 12, 22, 23).
Since previous work has provided evidence of interactions between tumors and the bone microenvironment that can drive TIBD(11, 12, 22, 23), we hypothesized that pre-treatment with TGF-β inhibitors would reduce myeloid expansion, tumor burden in bone, and osteoclast-mediated bone destruction. In this study, we show that early pharmacological or genetic TGF-β inhibition remodels the bone microenvironment by altering myeloid populations and pro-tumorigenic factors leading to a decrease in osteolytic lesions and tumor burden in bone.
2. Materials and Methods
2.1 Cell lines and reagents
Bone metastatic clones of the human breast cancer cell line MDA-MB-231 and mouse mammary carcinoma cell line 4T1 were used in these experiments as previously described by our laboratory (12, 24). A bone metastatic clone of 4T1 cells was stably transduced with GFP. Detailed methods can be found in supplementary methods. MDA-MB-231 cells-GFP were maintained in DMEM (Corning, 10-013-CV) supplemented with 10% fetal bovine serum (FBS, Peak Serum PS-500A) and 1% penicillin/streptomycin (Corning, 30-002-CI). 4T1-GFP cells were maintained in RPMI 1640 (Corning, 10-041-CV) supplemented with 10% FBS and 1% penicillin/streptomycin. The MMTV-PyMT cells were generously provided by Harold Moses (Vanderbilt University) and derived as described previously (25, 26). Prior to experimentation, the MMTV-PyMT cells were injected into the tibia of wild-type 6-week-old female FVB mice. These cells were harvested and expanded once mice developed bone lesions as observed though x-ray analysis. This bone-derived MMTV-PyMT variant cell line was maintained in DMEM/F12 (Corning, 10-092-CV) with 5% adult bovine serum (ABS, HyClone Laboratories SH30075) and 1% penicillin/streptomycin. Cells were cultured in a 37°C atmosphere with 5% CO2. The TGF-β neutralizing antibody (1D11) and its isotype control (13C4) were generously provided by SANOFI. TGF-β was purchased from R&D systems (240-B).
2.2 Animal Studies
All procedures were performed with the approval of the Vanderbilt University Institutional Animal Care and Use Committee and in accordance with Federal guidelines. For bone metastasis experiments, n=12 mice per group, and tumorigenicity (intratibial) experiments, n=8 mice per group, were calculated by a power analysis using a probability of type I error ± 0.05; probability of type II error ± 0.20 based on previous data obtained in our laboratory. Female 4-5-week-old athymic nude and BALB/c mice were purchased from Envigo. Mice were inoculated with 10mg/kg of a TGF-β neutralizing antibody (1D11) or the isotype control antibody (13C4) by intraperitoneal injection seven days prior to tumor inoculation and were treated continuously 3 times per week until sacrifice. After 7 days of treatment, 6-7-week-old athymic nude mice were injected with MDA-MB-231-GFP cells and BALB/c mice were injected with 4T1-GFP cells by intracardiac injection. Mice were imaged weekly by x-ray and sacrificed at three separate time points post tumor inoculation (athymic: 10, 14, 32 days, BALB/c: 7, 10, 14 days). Female mice were specific pathogen free and housed 4-5 per cage, and female athymic nude mice were housed in sterile cages, also 4-5 per cage. Mice were fed an irradiated laboratory rodent diet (Lab Diet, 5LOD). TGF-βRIIMyeWT (WT) and TGF-βRIIMyeKO mice were generously provided by Harold Moses (Vanderbilt University). To generate these mice, LysM-Cre mice were crossed with TGFβRII floxed mice which were established and maintained as described (27-29). These mice are on a pure FVB background. Female 5-6-week-old FVB TGF-βRIIMyeWT (WT) and TGF-βRIIMyeKO mice (6-8 per group) were injected with MMTV-PyMT tumor cells by intratibial injection. Mice were imaged weekly by x-ray and sacrificed at 10, 14, 28 days post tumor inoculation.
2.3 Intracardiac injections
MDA-MB-231-GFP cells and 4T1-GFP cells were trypsinized, washed and resuspended in ice-cold PBS at a final concentration of 1 × 106 cells/mL. Female, 6-7 week old mice (12 per group) were anesthetized and inoculated as previously described (30). Each mouse received 1 × 105 cells in a 100 μL volume or PBS as a control.
2.4 Intratibial Injections
The bone-derived MMTV-PyMT cell line was trypsinized, washed and resuspended in ice-cold PBS at a final concentration of 1 × 105 cells/10 μl. Female 5-6 week old FVB TGF-βRIIMyeWT (WT) and TGF-βRIIMyeKO mice (6-8 per group) were injected with tumor cells as previously described (30). Each mouse received 1 × 105 cells/10 μl or PBS as a control.
2.5 Western Blot
Briefly, spleens from naïve BALB/c treated with 1D11 or 13C4 were homogenized in Complete Lysis-M (Roche) as per manufacturer’s instructions. Detailed methods can be found in supplementary methods.
2.6 Bone marrow derived macrophages (BMDM)
Bone marrow was harvested from 6 week and younger female BALB/mice. Bone marrow was flushed from the tibia and femur of the mice and resuspended in PBS. Cell were incubated in red blood cell lysis buffer for 5 mins on ice. After two PBS washes, cells were resuspended in RPMI 1640 media (Corning, 10-041-CV) supplemented with 10% FBS, 1% penicillin/streptomycin, and 20 ng/ml of recombinant macrophage colony stimulating factor (M-CSF, Sigma M6518) for 7 days on 150mm dishes (Corning, 353025) (31). On the 7th day, BMDM were plated in 6-well plates at a concentration of 8×105 cells/well in low serum RPMI (RPMI 1640 media supplemented with 1% FBS, 1% penicillin/streptomycin) and used for subsequent experiments.
2.7 In vitro experiments
For in vitro macrophage expression experiments, 8×105 BMDM/well were kept in a naïve state or stimulated with mouse IL-4 (40ng/ml, Sigma-Aldrich SRP3211), IFN-γ + LPS (each at 50ng/ml, Sigma-Aldrich SRP3058), tumor-conditioned media (TCM) collected from 4T1 cells, for 24 hrs. After 24 hrs of stimulation, BMDM derived from BALB/c mice were treated with 1D11 or 13C4 (20μg/ml) +/− TGF-β (5ng/ml, R&D systems 240-B) for 24 hours. 4T1 cells were plated at 2×105 cells/well and treated with 1D11 or 13C4 (20μg/ml) +/− TGF-β (5ng/ml, R&D systems 240-B) for 24 hours. Supernatants were then collected and cells were harvested and used for subsequent experiments.
2.8 Protein cytokine and chemokine array
BALB/c femurs were flash frozen at the time of sacrifice for all stages of TIBD (early, middle and late disease) and stored at −80°C. BMDM and 4T1 cell supernatants were obtained as previously described above and stored at −20°C. Detailed methods can be found in supplementary methods.
2.9 Histomorphometry
Mouse hind limbs were excised at death; soft tissues were removed from the tibias and femurs; and tibias were fixed for 48 h in 10% neutral-buffered formalin as previously described (12). Detailed methods can be found in supplementary methods.
2.10 Quantitative PCR (qPCR)
Cells were harvested by direct lysis using RNeasy Mini Kit (Qiagen), per manufactor’s instructions. Detailed methods can be found in supplementary methods.
2.11 Flow Cytometry
Bone marrow was flushed from the femurs of BALB/c and WT/TGF-βRIIMyeKO and resuspended in PBS (12). Cells were stained with the following antibodies purchased from Miltenyi Biotec (with the exception of F4/80 pacific blue (AbD Serotec) and propidium iodide (Life Technologies); anti-GR-1 PE, CD11b APC, Ly6C PE-Vio770, Ly6G VioBlue, F4/80 PE. Flow Cytometry experiments were performed on a BD LSRII instrument. Analysis was performed using Flowjo Software v.10.1 (Tree Star, Inc).
2.12 X-ray Imagining and Analysis
Mice were sedated using isoflurane vaporizer (2.5% Isoflurane: 2-3 L/min O2) and x-ray images were taken at 35 kVp for 8 seconds using a digital radiography system (Faxitron LX-60). Mice were monitored by x-ray imaging and three stages of disease were established: early, middle and late disease. Images were saved and lesion numbers were determined using MetaMorph Microscopy Automation and Image Analysis Software (Metamorph, Molecuar Devices, Inc.). Lesion number was calculated as total number of tibial lesions per mouse.
2.13 Micro-computed X-ray Tomography (μCT)
μCT analysis was performed in the Vanderbilt Institute of Small Animal Imaging. The long axis of each specimen was aligned with the scanning axis. One hundred slices from the proximal tibia were scanned at a 12-μm resolution (μCT Scanco Medical, Switerland). The region of interest was trabeculae within the proximal metaphysis of the tibia below the growth plate. Images were acquired using 55 kV, 114 μA, 300-ms integration and 500 projections per 180° rotation (12). Images were analyzed using the Scanco Medical Imaging software to determine the bone volume/total volume (BV/TV), trabecular number and thickness, and tissue mineral density.
2.14 Osteoclast Resorption Assays
Bone marrow was harvested from the hindlimbs of 5-6-week-old BALB/c, WT and TGF-βRIIMyeKO mice and plated in MEMα (Corning, 10-022-CV) supplemented with 10% FBS and 1% penicillin/streptomycin for 2 hours. Detailed methods can be found in supplementary methods.
2.15 Statistics and Replicates
All data are presented as means ± the standard error mean (SEM). All in vitro experiments were done in triplicate with a minimum of three independent replicates. For experiments comparing two groups, a standard two-tailed unpaired student’s t test was used unless otherwise stated in the figure legend. For experiments comparing more than 2 groups, one-way ANOVA was used with a Tukey’s post-hoc test unless specifically stated in the figure legend. For all tests, a p value less than .05 was considered significant. All statistical analyses were done using GraphPad Prism 7.01 (GraphPad Software, La Jolla, CA). n.s, not significant.
3. Results
3.1 Inhibition of TGF-β alters expansion of different myeloid subpopulations throughout the progression of tumor-induced bone disease (TIBD)
5-6-week-old female athymic nude and BALB/c mice were pre-treated with either the TGF-β neutralizing antibody, 1D11, or its isotype control, 13C4, beginning seven days prior to tumor inoculation by intracardiac injection (athymic: MDA-MB-231-GFP, BALB/c: 4T1-GFP) and continuously treated until sacrifice (Figure 1A). To determine the direct role for TGF-β in myeloid cells, we injected bone-derived MMTV-PyMT cells directly into the tibia of genetically modified mice that lacked the transforming growth factor beta receptor II (TGF-βRII) specifically in myeloid cells (TGF-βRIIMyeKO) and its wild-type control (TGF-βRIIMyeWT, called from here on WT) (Figure 1B). The mice were monitored by x-ray imaging and three stages of disease were established based on the appearance of bone lesions: early, middle and late disease. Specifically, early disease was defined as no evidence of bone loss, middle disease as minor to moderate bone loss, and late disease as extensive bone loss (Figure 1C–1E).
Figure 1. Mouse models utilized for TGF-β inhibition studies.
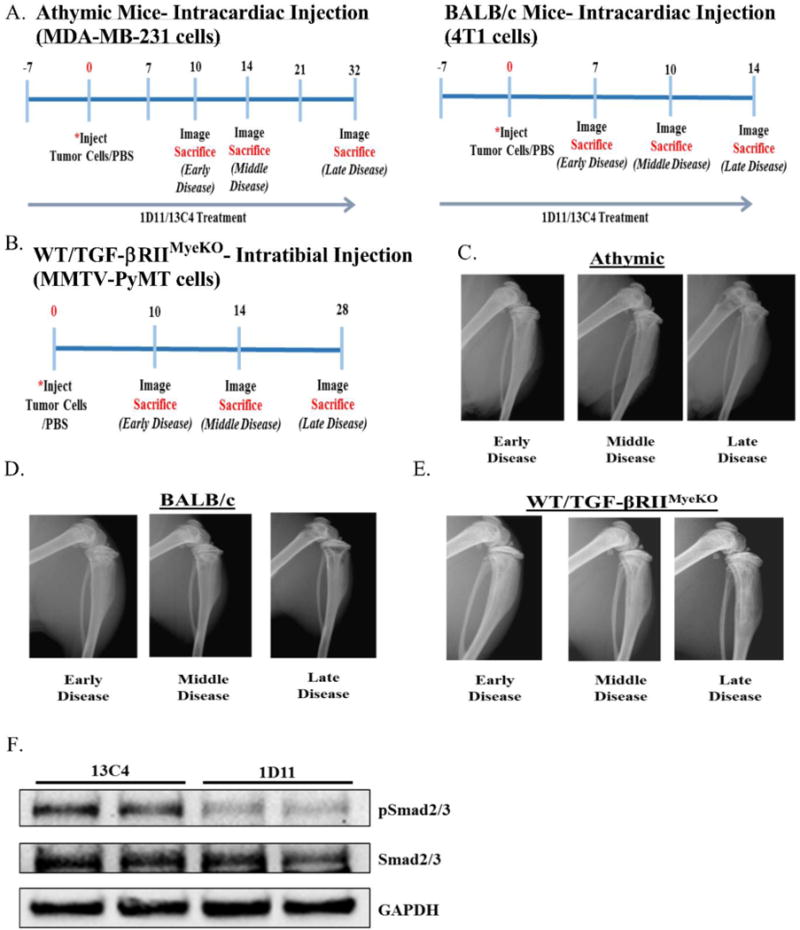
A. Experimental outline for 1D11 experiments, athymic (left), BALB/c (right). B. Experimental outline for WT/TGF-βRIIMyeKO mice. C. Representative athymic x-ray images of early, middle and late disease. D. Representative BALB/c x-ray images of early, middle and late disease. E. Representative x-ray images of early, middle and late disease for WT/TGF-βRIIMyeKO mice. F. Western blot analysis from naïve BALB/c mice treated with 13C4 or 1D11. Spleen lysate from a mouse treated with 1D11 demonstrates a decrease in p-Smad 2/3 levels.
In order to determine how TGF-β inhibition alters myeloid cell expansion in bone, flow cytometry analysis was performed at each stage of TIBD, defined by the degree of bone loss, on bone marrow from both tumor bearing and non-tumor bearing BALB/c mice treated with 1D11 or 13C4 or the WT/TGF-βRIIMyeKO mice. Gating strategy for all flow cytometry analysis can be found in Supplementary figure 2A. When TGF-β was systemically inhibited with 1D11 before tumor inoculation, monocytic immature cells (CD11b+ CD11c− Ly6G− Ly6C+) significantly decreased by 6% and the granulocytic immature cells (CD11b+ CD11c− Ly6C− Ly6G+) significantly increased by 14% (Figure 2A-2B). As TIBD progressed, specifically in this model, there were no significant expansion changes in either monocytic or granulocytic immature myeloid cells while there was a significant decrease (4%) in mature myeloid cells (CD11b+ F4/80+, Figure 2C) at middle disease.
Figure 2. Early TGF-β inhibition alters immature myeloid cell expansion at early disease and mature myeloid cells at middle disease in tumor-bearing BALB/c.
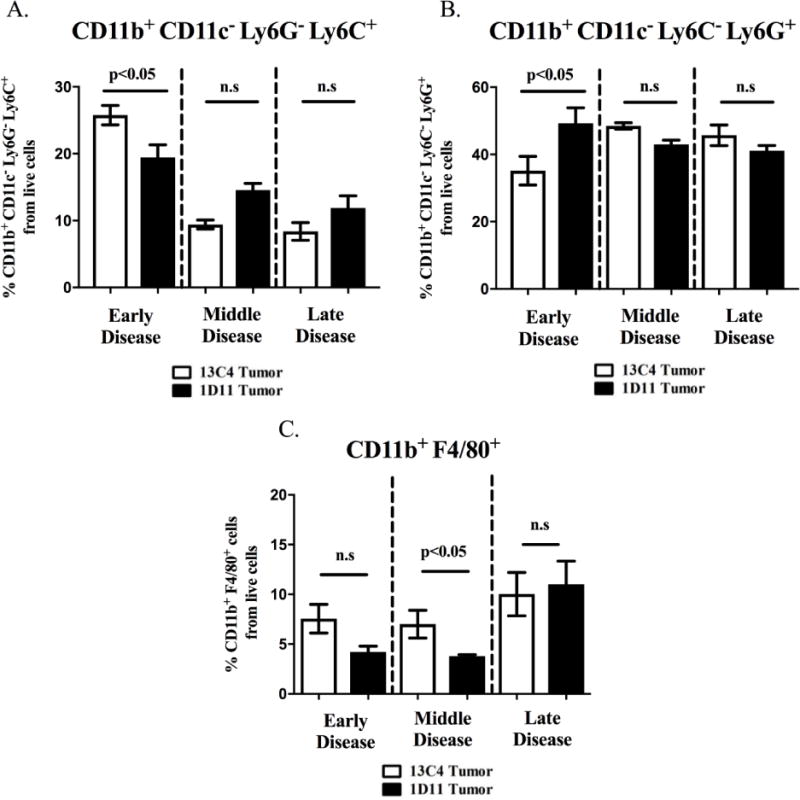
Bone marrow was harvested and prepared for flow cytometry as described in the methods section. A. Tumor-bearing BALB/c mice treated with 1D11 have a cell percentage decrease in the monocytic immature myeloid cells (CD11b+ CD11c− Ly6G− Ly6C+) at early disease. B. Granulocytic immature myeloid cells (CD11b+ CD11c− Ly6C− Ly6G+) from tumor-bearing BALB/c mice treated with 1D11 have an increase in their expansion at early disease. C. Mature myeloid cells (CD11b+ F4/80+) decrease at middle disease when TGF-β is inhibited. (5-6 mice per group).
In regards to the genetically modified model, there were no significant expansion changes in either monocytic or granulocytic immature myeloid cell (CD11b+ CD11c− Ly6G− Ly6C+, CD11b+ CD11c− Ly6C− Ly6G+) expansion between tumor-bearing WT and TGF-βRIIMyeKO at any stage of TIBD (Figure 3A-3B). In contrast, mature myeloid cells (CD11b+ F4/80+) increased by 4% in tumor-bearing TGF-βRIIMyeKO mice as TIBD progressed to the middle disease state (Figure 3C). Considering that there were no significant changes in myeloid cell expansion at late disease for either model of TGF-β inhibition (systemic and genetically modified, Figure 2A-2C, Figure 3A-3C), these studies suggest that TGF-β signaling may play an important role in regulating myeloid cell expansion in the earlier stages of disease, but not when extensive bone loss is present in our models of TIBD.
Figure 3. Early TGF-β inhibition alters mature myeloid expression at middle disease for tumor-bearing TGF-βRIIMyeKO mice.
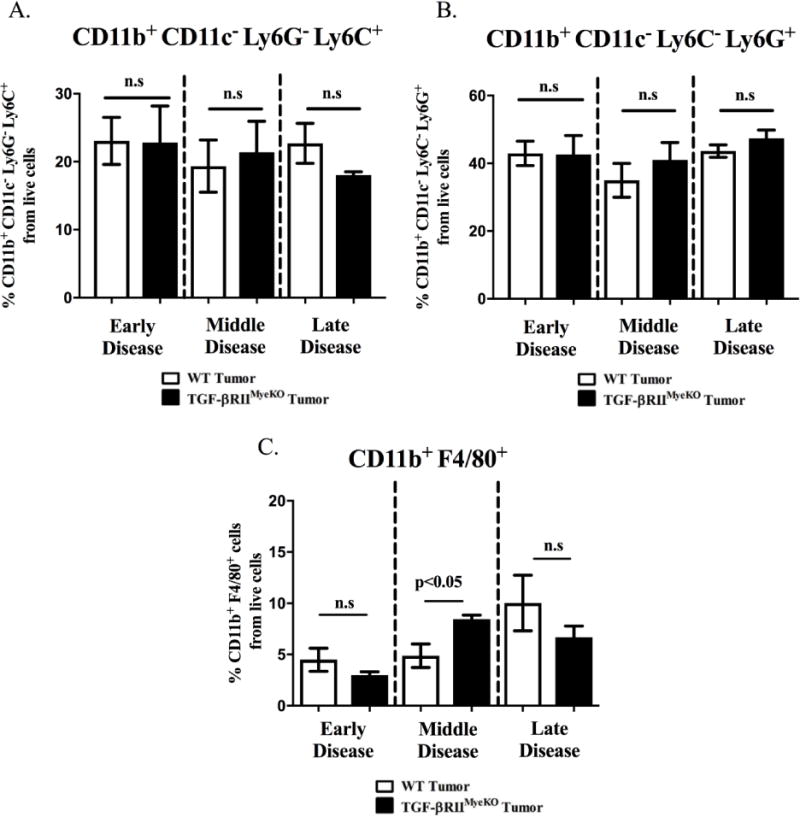
Bone marrow was harvested and prepared for flow cytometry as described in the methods section. A. No significant expansion differences in monocytic immature myeloid cells (CD11b+ CD11c− Ly6G− Ly6C+)) between tumor-bearing WT and TGF-βRIIMyeKO mice. B. No significant expansion differences in granulocytic immature myeloid cells (CD11b+ CD11c− Ly6C− Ly6G+) between tumor-bearing WT and TGF-βRIIMyeKO mice. C. Mature myeloid cells (CD11b+ F4/80+) from tumor-bearing TGF-βRIIMyeKO mice increase in cell percentage at middle disease. (5-6 mice per group).
3.2 TGF-β inhibition modulates expression and secretion of factors associated with tumor progression and immune suppression
Since changes in myeloid expansion were observed when TGF-β signaling was inhibited and TGF-β itself can stimulate the production of pro-tumorigenic factors that can expand myeloid cells, we investigated whether systemic TGF-β inhibition alters pro-tumorigenic factors in whole bone from PBS and tumor-bearing BALB/c 13C4/1D11 treated mice at all stages of TIBD (i.e. early, middle and late disease). Femurs were subjected to a cytokine and chemokine analysis using a multiplex assay system and results are outlined in Supplementary table 1A–1I. Analyses indicated that significant protein changes in femurs only occurred at late disease (Supplementary table 1C), thus, emphasis was only placed on significant differences observed between cytokine and chemokines at late disease. Compared to control femurs, two factors significantly increased in femurs from tumor-bearing mice; granulocyte colony-stimulating factor (G-CSF) and leukemia inhibitory factor (LIF); but only G-CSF significantly decreased with TGF-β inhibition (Figure 4A). Unexpectedly, the pro-tumorigenic factor, vascular endothelial growth factor (VEGF) was significantly increased in femurs from 1D11 treated tumor-bearing mice (Figure 4C). This data demonstrates that pre-treatment with a TGF-β inhibitor can both reduce and increase pro-tumorigenic proteins in bone at the late disease state, suggesting multiple mechanisms are at play.
Figure 4. TGF-β inhibition modulates secretion of factors associated with tumor promotion.
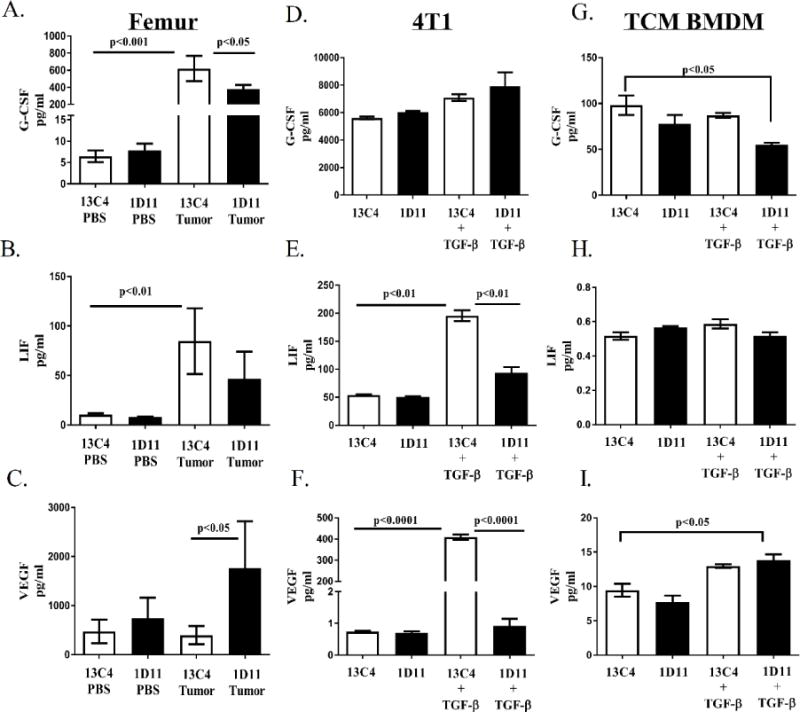
BALB/c femurs and cell supernatants were collected and prepared for the protein cytokine/chemokine array as described in the methods section. A. G-CSF protein levels increase in femur homogenates from tumor-bearing mice and levels decrease when tumor-bearing mice were treated with 1D11 at late disease. B. LIF protein levels increase in femur homogenates from tumor-bearing mice at late disease. C. VEGF protein levels increase in femur homogenates from 1D11 treated tumor-bearing mice at late disease. D. G-CSF proteins levels do not change when TGF-β is inhibited in 4T1 cells. E. LIF protein levels increase with TGF-β stimulation and decrease when 4T1 cells are treated with 1D11. F. VEGF protein levels increase with TGF-β stimulation and decrease when 4T1 cells are treated with 1D11. G. G-CSF levels decrease when TGF-β is inhibited in TCM stimulated BMDM. H. Treatment with 1D11 does not change LIF protein levels in TCM stimulated BMDM. I. VEGF protein levels increase with TGF-β stimulation in TCM stimulated BMDM. Protein cytokine/chemokine array data was analyzed using a two-way ANOVA with a Tukey’s post-hoc test (3-4 femurs per group, 3 independent in vitro cell supernatants per group).
Since TGF-β inhibition affects the cytokine and chemokine profile within bone and TGFβ-responsive tumor and myeloid cell populations reside in the tumor-bearing femurs that were analyzed, these two cell subsets were further evaluated in vitro. To further investigate the cytokine and chemokine profile of tumor and myeloid cells in response to TGF-β inhibition, 4T1 tumor cells and differentially polarized bone marrow derived macrophages (BMDM) were stimulated and simultaneously inhibited for TGF-β for 24 hours, followed by a chemokine/cytokine analysis performed on all cell supernatants (Supplementary table 1D–1I). TGF-β stimulation increases protein levels of LIF and VEGF, while inhibition of TGF-β with 1D11 reduces these cytokines significantly in 4T1 tumor cells (Figure 4E, 4F). Likewise, G-CSF levels were reduced while VEGF protein levels were increased in tumor-conditioned media (TCM) stimulated BMDM under TGF-β inhibitory conditions (Figure 4G, 4I). Thus, this in vitro data supports the notion that the changes in G-CSF and VEGF observed in bone may be myeloid-dependent.
Since arginase has been implicated in promoting immune suppression in cancer (14, 16, 32, 33), we investigated the effect of TGF-β inhibition on arginase expression in differentially polarized BMDMs. Arginase, a marker of immune suppression, was decreased significantly in IL-4 stimulated macrophages (Supplementary figure 3B), and while TCM stimulated macrophages responded similarly, their decrease in arginase expression was not statistically significant (Supplementary figure 3C). Interestingly, all BMDM independent of their stimulation profile had a statistically significant increase in arginase expression after TGF-β stimulation (Supplementary figure 3A–3D). While publications have addressed the link between arginase expression and TGF-β stimulation (32, 33), our findings suggest that TGF-β may promote immune suppression in bone, a microenvironment that contains an abundant amount of TGF-β when tumor is present (34).
3.3 TGF-β inhibition decreases osteoclast function
Published work by our group and others has demonstrated that myeloid cells found in the bone marrow, such as macrophages and CD11b+ Gr-1+ cells, have the potential to differentiate into osteoclasts (10-12, 35). Therefore, in order to explore if pre-treatment with a TGF-β inhibitor would alter osteoclast number and activity, histological analyses were performed on the tibias of tumor-bearing mice that were treated with 1D11. These analyses indicated that although the number of TRAP+ osteoclasts were decreased in both tumor-bearing athymic nude and BALB/c mice treated with 1D11, only athymic nude mice had a statistically significant decrease when TGF-β was inhibited at late stage disease (Figure 5A, 5B). When osteoclast activity was examined in vitro, treatment with 1D11 decreased both the percent of resorption area and the number of resorption pits that were made by TRAP+ osteoclasts derived from BALB/c bone marrow (Figure 5C, 5D). These data suggest that pre-treatment with TGF-β inhibitors can reduce osteoclast number and function.
Figure 5. TGF-β inhibition decreases osteoclast activity in mouse models of TIBD.
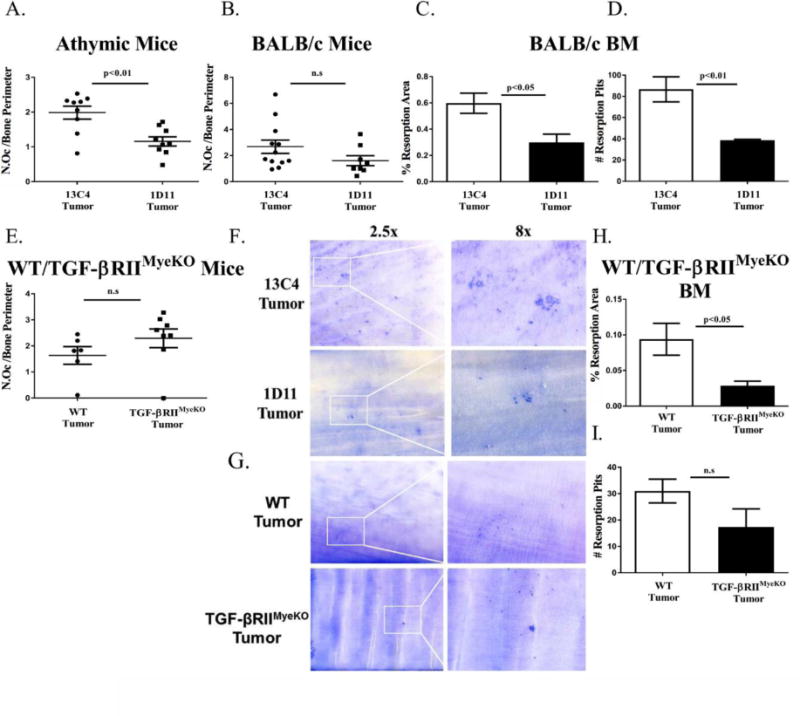
Tibial sections were stained with tartrate resistance acid phosphatase (TRAP) for osteoclast analyses and in vitro osteoclast resorption assays were performed as described in the methods section. A&B. Histological osteoclast analyses for tumor-bearing athymic and BALB/c mice at late disease. At late disease only tumor-bearing athymic mice that were treated with 1D11 have a significant reduction in their osteoclast number per bone perimeter (11-12 tibias per group). C&D. Quantification of osteoclast resorption area and resorption pits for systemic TGF-β inhibition experiments. Treatment with 1D11 significantly decreases osteoclast resorption area and resorption pits. E. At late disease, there is no significant difference in osteoclast number per bone perimeter between tumor-bearing WT and TGF-βRIIMyeKO mice (6-8 tibias per group). F. Representative images of dentin discs containing osteoclast resorption area and resorption pits for systemic TGF-β inhibition experiments. G. Representative images of dentin discs containing osteoclast resorption area and resorption pits for WT and TGF-βRIIMyeKO experiments. H&I. Quantification of osteoclast resorption area and resorption pits for WT and TGF-βRIIMyeKO experiments. TRAP+ osteoclasts derived from TGF-βRIIMyeKO bone marrow create a significantly smaller resorption area as compared to their WT control. BM, bone marrow.
Histological analyses of the WT/TGF-βRIIMyeKO tibiae indicated that there was no change in TRAP+ osteoclasts between tumor-bearing TGF-βRIIMyeKO and WT mice (Figure 5E). Despite no significant changes in osteoclast number, TGF-βRIIMyeKO TRAP+ osteoclasts have a significant decrease in percent resorption area in vitro (Figure 5H, 5I). Together this suggests that TGF-βRIIMyeKO specific osteoclasts may be impaired in their ability to resorb dentin, indicating a decrease in osteoclast function. While osteoclast number varies between the two models of TGF-β inhibition (systemic and genetic) through histological analysis, osteoclast activity is significantly reduced in both tumor-bearing 1D11 treated mice and TGF-βRIIMyeKO mice implying a reduction in overall TIBD.
3.4 Early TGF-β inhibition reduces osteolytic lesions and tumor burden in bone
Since TGF-β inhibition has an effect on the bone microenvironment once tumors are established, we hypothesized that TGF-β inhibition prior to tumor inoculation would decrease TIBD. By x-ray imaging, we identified that tumor-bearing athymic nude and BALB/c mice that were treated with 1D11 had reduced osteolytic lesions as compared to their isotype control, 13C4, at late disease (Figure 6A, 6C). While it has been previously published that 1D11 can increase overall bone volume in a naïve mouse model (36) and our studies recapitulate those results (Supplementary figure 5A–5C), we demonstrate that treatment with 1D11 increases overall bone volume in both mouse models of early systemic TGF-β inhibition at all stages of TIBD (Figure 6B and 6D). Additionally, mice treated with 1D11 (athymic and BALB/c mice) have an overall decrease in tumor burden (as determined by histomorphometry) compared to 13C4 treated mice at late stage disease (Figure 7A, 7B).
Figure 6. TGF-β inhibition reduces osteolytic lesions and improves overall bone volume.
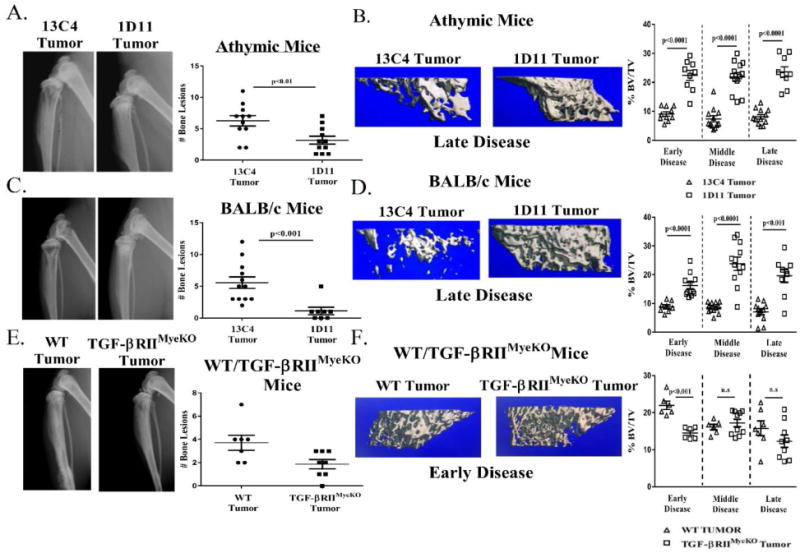
X-ray analysis of tumor-bearing mice at late stage disease prior to sacrifice. Representative images are from late stage disease. Formalin fixed tibias were scanned and analyzed by μCT as described in the methods section. Percent bone volume normalized to total volume (BV/TV) for each treatment group. A. Athymic mouse model: X-ray images, 13C4 treated (left), 1D11 treated (right) and quantification of bone lesions. Tumor-bearing athymic mice treated with 1D11 have less osteolytic lesions than isotype control. B. Left: three-dimensional (3D) rendered μCT images from two tumor-bearing athymic mice at late stage disease, 13C4 treated (left), 1D11 treated (right). Right: percent BV/TV over the course of tumor-induced bone disease (TIBD) in the athymic mouse model. Treatment with 1D11 increases overall bone volume over the course of TIBD. C. BALB/c mouse model: X-ray images, 13C4 treated (left), 1D11 treated (right) and quantification of bone lesions. Tumor-bearing BALB/c mice treated with 1D11 have less osteolytic lesions than isotype control. D. Left: 3D rendered μCT images from two tumor-bearing BALB/c mice at late stage disease, 13C4 treated (left), 1D11 treated (right). Right: percent BV/TV over the course of tumor-induced bone disease in the BALB/c mouse model. Treatment with 1D11 increases overall bone volume over the course of TIBD. E. WT/TGF-βRIIMyeKO mice mouse model: X-ray images, WT tumor (left), TGF-βRIIMyeKO tumor (right) and quantification of bone erosions. Tumor-bearing TGF-βRIIMyeKO have a reduced incidence of bone lesions as compared to tumor-bearing WT mice. F. Left: 3D rendered μCT images from two tumor-bearinF-βRIIMyeKO (right). Right: percent BV/TV over the course of tumor-induced bone disease in the WT/TGF-βRIIMyeKO mice mouse model. Tumor-bearing TGF-βRIIMyeKO mice only have a significant decrease in their overall bone volume at early disease. (9-13 tibias per group).
Figure 7. TGF-β inhibition reduces tumor burden at late stage disease in all three mouse models of TIBD.
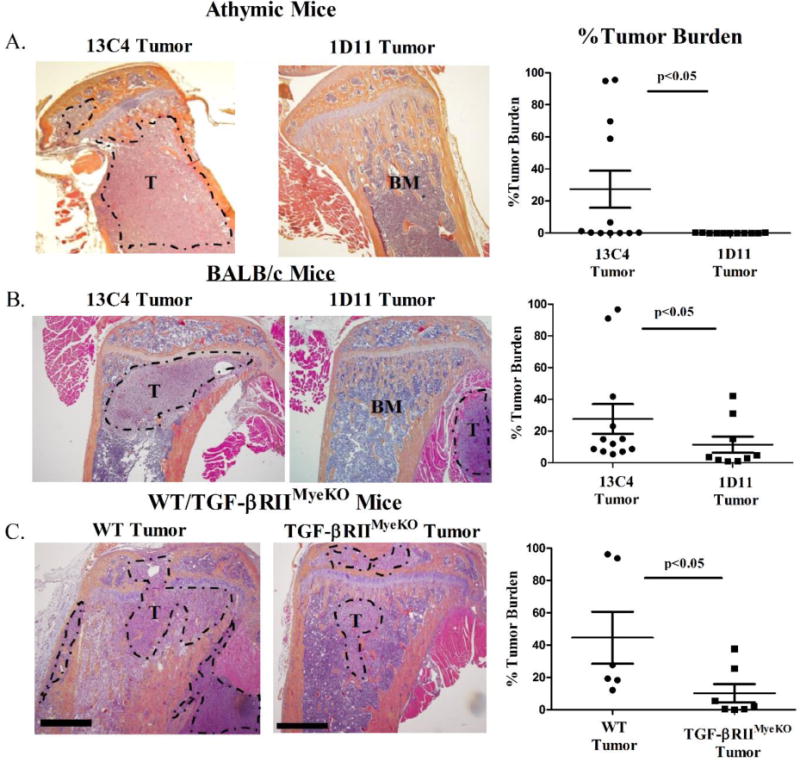
Tibal sections were stained with hematoxylin & eosin (H+E) for tumor burden analysis at late stage disease as described in the methods section. A&B. 4× magnification H+E sections from two tumor-bearing mice (athymic and BALB/c) at late stage disease, 13C4 treated (left), 1D11 treated (right) and quantification of percent tumor burden between treatment groups at late stage disease. Tumor-bearing mice (athymic and BALB/c) treated with 1D11 have a reduction in tumor burden as compared to isotype control. C. 4× magnification H+E sections from two tumor-bearing WT and TGF-βRIIMyeKO mice at late stage disease, WT (left), TGF-βRIIMyeKO (right) and quantification of percent tumor burden between mice at late stage disease. Tumor-bearing TGF-βRIIMyeKO mice have a reduction in tumor burden as compared to isotype control. T, Tumor. BM, Bone Marrow. Scale, 50 μm.
In the genetic TGF-β inhibition mouse model, tumor-bearing TGF-βRIIMyeKO mice had a significant decrease in the number of bone lesions at late disease (Figure 6E) while significant changes in overall bone volume between WT and TGF-βRIIMyeKO mice were only seen at early disease (Figure 6F). μCT analysis demonstrated that tumor-bearing TGF-βRIIMyeKO mice had a decrease in overall bone volume (BV/TV), trabecular spacing and trabecular number as compared to WT mice at early disease (Supplementary table 2). While significant changes in these bone parameters were only seen during early disease between tumor-bearing WT and TGF-βRIIMyeKO mice, changes in overall bone volume did not decrease as TIBD progressed in tumor-bearing TGF-βRIIMyeKO mice (Figure 6F). This may imply that loss of functional TGF-β signaling specifically in myeloid cells may stabilize bone loss in the presence of tumor. Analyses at late stage disease revealed that TGF-βRIIMyeKO mice had lower tumor burden than WT tumor-bearing mice (Figure 7C). Overall, this data suggests that early TGF-β inhibition prior to tumor inoculation can prevent TIBD by reducing osteolytic lesions and decreasing tumor burden in bone.
4. Discussion
This study illustrates that TGF-β inhibition alters myeloid expansion that leads to a reduction in tumor burden and bone destruction, and an increase in bone volume. Specifically, we show that early TGF-β inhibition alters the tumor-bone microenvironment by affecting expansion of myeloid cells that are at different stages of differentiation, modulating pro-tumorigenic factors and decreasing osteoclast function. Since systemic treatment with TGF-β inhibitors may alter other cell types, we utilized a transgenic mouse model that lacked TGF-βRII specifically in myeloid cells (TGF-βRIIMyeKO). In this model, mature myeloid cells expand in the presence of tumor but have less bone lesions, contain deficient osteoclasts and have a significant reduction in tumor burden in bone. Together these data strengthen the notion of the complexity of the tumor-bone microenvironment and that early TGF-β inhibition is a promising potential therapeutic target for reducing TIBD.
While it is well known that myeloid cells, specifically CD11b+ Gr-1+ cells, expand in the presence of a tumor, and that this expansion contributes to the production of pro-tumorigenic and immune suppressive factors that support tumor progression and metastasis (12, 18, 26, 37), the regulation and function of these cells in TIBD is still unclear. Since TGF-β has been shown to play an important role in myeloid differentiation, chemotaxis and polarization (13, 14, 32), we reasoned that inhibition of TGF-β signaling would decrease myeloid expansion in bone in the context of TIBD. Our data reveals that systemic TGF-β inhibition with 1D11 can both increase and decrease immature myeloid cells (decrease: CD11b+ CD11c−Ly6G− Ly6C+, increase: CD11b+ CD11c−Ly6C− Ly6G+) at early disease and reduce expansion of mature myeloid cells (CD11b+ F4/80+) at the middle disease state (Figure 1A-1C). However, loss of TGF-βRII in myeloid cells does not alter the expansion profile in either monocytic or granulocytic immature myeloid cells (CD11b+ CD11c−Ly6G− Ly6C+, CD11b+ CD11c−Ly6C− Ly6G+, Figure 3A-3B) at any stages of TIBD. On the other hand, CD11b+ F4/80+ myeloid cells from tumor-bearing TGF-βRIIMyeKO are increased as TIBD progresses to the middle disease state, where minor to moderate bone loss was detected by x-ray analysis (Figure 3C). Since the strategies to neutralize TGF-β within our mouse models were different, one model was injected with a TGF-β neutralizing antibody that would affect all TGF-β responsive cells while the other model contained a genetic deletion of TGF-βRII in myeloid cells; it is not surprising that distinct myeloid expansion profiles are observed in our studies. While it is possible that the changes in expansion are due to myeloid effects on other cell types, our findings imply that TGF-β signaling plays a role in regulating the expansion of immature and mature myeloid cells when tumors in bone are first developing. However, once these tumors become established in bone and begin promoting symptoms of TIBD (i.e. bone loss and overwhelming tumor burden), myeloid expansion may no longer be controlled by TGF-β signaling but by other mechanisms that regulate myeloid expansion (38).
Recent studies have focused on investigating the cross-talk between tumors and the immune system and how it contributes to immune suppression and evasion. Immune evasion is currently considered an emerging hallmark of cancer, and it involves the ability for tumors to evade immunological destruction by T and B cells, natural killer cells, and macrophages (39).
Specifically in breast cancer, TGF-β has been shown to promote pro-tumorigenic and immunosuppressive factors (25, 26), therefore we expected a significant reduction in these factors when TGF-β was systemically inhibited in our BALB/c model of breast cancer metastasis to bone. Surprisingly, our femur analyses revealed that systemic TGF-β inhibition can both increase and decrease factors that contribute to tumor progression at late disease. For example, the pro-tumorigenic factor G-CSF is significantly reduced in femurs from tumor-bearing mice that were treated with the TGF-β inhibitor 1D11 (Figure 4A). G-CSF has been shown to contribute to chemoresistance, migration and expansion of myeloid cells, and is correlated with a reduced overall survival in patients (40-43). Evidence from pre-clinical studies indicate that G-CSF can act directly on myeloid cells promoting their mobilization from the bone marrow to the peripheral circulation (44). Reduction of G-CSF by 1D11 could imply that bone marrow residing myeloid cells are prevented from entering circulation and migrating to tertiary organs to set up the pre-metastatic niche, allowing for additional metastases to form. Nonetheless, systemic TGF-β inhibition also increases VEGF and CCL4, factors known to support tumor progression in bone (Figure 4C, supplementary table 1C) (45, 46). Since significant cytokine changes in bone were only observed at late disease, it suggests that these changes occur when severe symptoms of TIBD are present, including but not limited to tumor burden.
Considering that both myeloid cells and 4T1 tumor cells are TGF-β-responsive cells that reside in the tumor-bearing femurs that were analyzed, we wanted to investigate whether the changes observed in bone when TGF-β was inhibited were tumor or myeloid-dependent. Thus, these two distinct cell populations were evaluated for changes in their cytokine and chemokine profiles in vitro.
When TGF-β was inhibited, 4T1 tumor cells significantly decreased both VEGF and LIF protein levels (Figure 4E, 4F), while TCM stimulated BMDM reduced G-CSF but increased VEGF levels significantly (Figure 4G, 4I). This implies that both tumor cells and BMDM have contrasting responses to TGF-β inhibition, and that both of these cell types contribute differently to the changes in pro-tumorigenic factors we observe in bone. For example, the decrease in bone G-CSF when TGF-β is inhibited suggests that this decrease is mediated through a myeloid-dependent mechanism because a similar trend is seen in TCM-stimulated BMDM (Figure 4A, Figure 4G). The same rational can be applied to the changes in VEGF in bone, the increase in bone VEGF is also observed in TCM-stimulated BMDM, suggesting a myeloid-dependent mechanism. It is also reasonable that other cells of the bone microenvironment, such as osteoblasts, are contributing to the changes in factors we observe in bone. For example, osteoblasts produce an abundant amount of VEGF (45) and LIF (47, 48), and while the role of LIF in tumorigenesis is complex (49, 50), angiogenesis and osteogenesis has been established as a coupled process in normal bone remodeling (51). Thus, the increased levels of VEGF in bone and TCM-stimulated BMDM in response to TGF-β inhibition may reflect the increase in bone volume seen with 1D11 treatment. Therefore, the increase in VEGF may be beneficial and this benefit may outweigh the consequences of an increased blood supply to the tumor.
Since our in vitro analyses only included 4T1 tumor cells and BMDM cells, we acknowledge that we may have missed important in vivo interactions and changes in other cell types due to TGF-β inhibition. Despite these limitations and the increase in pro-tumorigenic factors with TGF-β inhibition, there is an overall reduction in TIBD in our mouse model of breast cancer metastasis to bone. It is currently unknown the concentration of factors that are needed to promote a pro-tumorigenic environment. We reason that perhaps it is a certain ratio of cytokines and chemokines that are needed to tip the scale between tumor promoting and cytotoxicity and that a combination of the right factors can determine which direction the scale tips.
TGF-β inhibitors are currently in clinical trials for the treatment of gliomas, metastatic breast cancer, non-small cell lung cancer, melanoma, mesothelioma and renal cell carcinoma (52-54). While many of these clinical trials are still ongoing, evidence from preclinical studies indicate that TGF-β inhibitors, like the human monoclonal antibody fresolimumab, would be most effective if used in combination with chemotherapy and/or radiation (55-58). A preclinical study using an orthotropic model of breast cancer demonstrated that 1D11 enhanced the effects of chemotherapy by normalizing the tumor stroma (57). Similar observations are seen in our studies where pre-treatment with 1D11 remodels the bone-tumor microenvironment by regulating the expansion of the myeloid population, the ratio of pro-tumorigenic factors and decreasing the function of osteoclasts leading to an overall reduction in TIBD. While the combination of a TGF-β inhibitor and a chemotherapeutic are not yet the standard of care for metastatic breast cancer patients, it is worth exploring in preclinical setting in the context of TIBD. In conclusion, our work supports the notion that early TGF-β inhibition is a potential therapeutic approach for reducing TIBD.
Supplementary Material
Highlights.
Early TGF-β inhibition reduces tumor burden in bone and bone destruction.
TGF-β inhibition alters myeloid populations in early Tumor-induced bone disease.
TGF-β inhibitions alters osteoclast activity in multiple pre-clinical models.
TGF-β inhibition changes the cytokine profile in bone, tumor, and myeloid cells.
Acknowledgments
We would like to acknowledge the following people for their generosity and support: Dr. Linda Sealy, Dr. Scott Guelcher, Dr. Rachelle Johnson, Dr. Harold Moses, Dr. Philip Owens, Dr. Ushashi Dadwal, Dr. Shellese Cannonier, Anna Chytil, Ryan Murray, David Florian, Vera Mayhew and SANOFI for the 1D11.
Grant Support:
We recognize the following financial support: VA Merit Award 1I01BX001957 (J.A.S), NIH grant R01 CA163499 (J.A.S), DOD funding W81XWH-15-1-0622 (J.A.S), core funding 1S10RR027027631 (μCT). Initiative to Maximize Student Diversity Award 5R25GM062459 (D.B, K.A.K), Cellular, Biochemical and Molecular Sciences Training Program 5T32GM008554 (D.B), Microenvironmental Influences in Cancer Training Program 2T32CA009592 (K.A.K), NIAID 1 F31 AI133926 (N.E.P), NIAID grant K08 AI113107 (J.E.C) and internal funding from the Vanderbilt-Ingram Cancer Center. Flow Cytometry experiments were performed in the VMC Flow Cytometry Shared Resource. The VMC Flow Cytometry Shared Resource is supported by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Digestive Disease Research Center (DK058404). J.E.C is also supported by a Burroughs Wellcome Fund Career Award for Medical Scientists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures:
All authors have no conflicts of interest to disclose.
Authors’ roles:
Study design: D.B, A.R.M, K.AK, J.A.S. Study conduct, data analysis and collection: D.B, A.R.M, K.AK, N.E.P, J.R.J., J.E.C. Data interpretation: D.B, J.A.S, A.R.M, K.A.K, N.E.P. Drafting and revising manuscript: D.B, A.R.M, J.A.S, K.A.K, J.E.C, J.R.J, N.E.P. Approval of final version of manuscript: D.B and J.A.S.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: A Cancer Journal for Clinicians. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treatment Reviews. 2001;27(3):165–76. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- 3.Biswas S, Nyman JS, Alvarez J, Chakrabarti A, Ayres A, Sterling J, et al. Anti-Transforming Growth Factor ß Antibody Treatment Rescues Bone Loss and Prevents Breast Cancer Metastasis to Bone. PLoS ONE. 2011;6(11):e27090. doi: 10.1371/journal.pone.0027090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. The Journal of Clinical Investigation. 1999;103(2):197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, et al. TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer research. 2011;71(1):175–84. doi: 10.1158/0008-5472.CAN-10-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nyman JS, Merkel AR, Uppuganti S, Nayak B, Rowland B, Makowski AJ, et al. Combined treatment with a transforming growth factor beta inhibitor (1D11) and bortezomib improves bone architecture in a mouse model of myeloma-induced bone disease. Bone. 2016;91:81–91. doi: 10.1016/j.bone.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Juárez P, Mohammad KS, Yin JJ, Fournier PGJ, McKenna RC, Davis HW, et al. Halofuginone Inhibits the Establishment and Progression of Melanoma Bone Metastases. Cancer research. 2012;72(23):6247–56. doi: 10.1158/0008-5472.CAN-12-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korpal M, Yan J, Lu X, Xu S, Lerit DA, Kang Y. Imaging transforming growth factor-[beta] signaling dynamics and therapeutic response in breast cancer bone metastasis. Nat Med. 2009;15(8):960–6. doi: 10.1038/nm.1943. [DOI] [PubMed] [Google Scholar]
- 9.Cook LM, Araujo A, Pow-Sang JM, Budzevich MM, Basanta D, Lynch CC. Predictive computational modeling to define effective treatment strategies for bone metastatic prostate cancer. 2016;6:29384. doi: 10.1038/srep29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X, et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer research. 2013;73(2):672–82. doi: 10.1158/0008-5472.CAN-12-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhuang J, Zhang J, Lwin ST, Edwards JR, Edwards CM, Mundy GR, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS ONE. 2012;7(11):e48871. doi: 10.1371/journal.pone.0048871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danilin S, Merkel AR, Johnson JR, Johnson RW, Edwards JR, Sterling JA. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology. 2012;1(9):1484–94. doi: 10.4161/onci.21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer research. 2006;66(2):605–12. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 14.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature reviews Immunology. 2012;12(4):253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Novitskiy SV, Pickup MW, Chytil A, Polosukhina D, Owens P, Moses HL. Deletion of TGF-beta signaling in myeloid cells enhances their anti-tumorigenic properties. Journal of leukocyte biology. 2012;92(3):641–51. doi: 10.1189/jlb.1211639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang Y, Gara SK, Achyut BR, Li Z, Yan HH, Day CP, et al. TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer discovery. 2013;3(8):936–51. doi: 10.1158/2159-8290.CD-12-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng X, Vander Ark A, Lee P, Hostetter G, Bhowmick NA, Matrisian LM, et al. Myeloid-specific TGF-beta signaling in bone promotes basic-FGF and breast cancer bone metastasis. Oncogene. 2016;35(18):2370–8. doi: 10.1038/onc.2015.297. [DOI] [PubMed] [Google Scholar]
- 18.Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–21. doi: 10.1016/j.ccr.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 19.Pollard JW. Macrophages define the invasive microenvironment in breast cancer. Journal of leukocyte biology. 2008;84(3):623–30. doi: 10.1189/jlb.1107762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer research. 2004;64(19):7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 21.Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-β Signaling in Generation of CD39<sup>+</sup>CD73<sup>+</sup> Myeloid Cells in Tumors. The Journal of Immunology. 2014;193(6):3155–64. doi: 10.4049/jimmunol.1400578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SI, Lee C, Sadler WD, Koh AJ, Jones J, Seo JW, et al. Parathyroid hormone-related protein drives a CD11b+Gr1+ cell-mediated positive feedback loop to support prostate cancer growth. Cancer research. 2013;73(22):6574–83. doi: 10.1158/0008-5472.CAN-12-4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D’Amico L, Mahajan S, Capietto A-H, Yang Z, Zamani A, Ricci B, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. The Journal of experimental medicine. 2016;213(5):827–40. doi: 10.1084/jem.20150950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson RW, Nguyen MP, Padalecki SS, Grubbs BG, Merkel AR, Oyajobi BO, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer research. 2011;71(3):822–31. doi: 10.1158/0008-5472.CAN-10-2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bierie B, Chung CH, Parker JS, Stover DG, Cheng N, Chytil A, et al. Abrogation of TGF-β signaling enhances chemokine production and correlates with prognosis in human breast cancer. The Journal of Clinical Investigation. 119(6):1571–82. doi: 10.1172/JCI37480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGFβ Signaling in Mammary Carcinomas Recruits Gr-1+CD11b+ Myeloid Cells that Promote Metastasis. Cancer Cell. 2008;13(1):23–35. doi: 10.1016/j.ccr.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chytil A, Magnuson MA, Wright CVE, Moses HL. Conditional inactivation of the TGF-β type II receptor using Cre:Lox. genesis. 2002;32(2):73–5. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 28.Yang J, Hawkins OE, Barham W, Gilchuk P, Boothby M, Ayers GD, et al. Myeloid IKKβ Promotes Antitumor Immunity by Modulating CCL11 and the Innate Immune Response. Cancer research. 2014;74(24):7274–84. doi: 10.1158/0008-5472.CAN-14-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar A-R, et al. Effect of Conditional Knockout of the Type II TGF-β Receptor Gene in Mammary Epithelia on Mammary Gland Development and Polyomavirus Middle T Antigen Induced Tumor Formation and Metastasis. Cancer research. 2005;65(6):2296–302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 30.Campbell JP, Merkel AR, Masood-Campbell SK, Elefteriou F, Sterling JA. Models of Bone Metastasis. 2012;(67):e4260. doi: 10.3791/4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villagómez D, Barry DP, et al. EGFR regulates macrophage activation and function in bacterial infection. The Journal of Clinical Investigation. 126(9):3296–312. doi: 10.1172/JCI83585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC immunology. 2012;13:31. doi: 10.1186/1471-2172-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boutard V, Havouis R, Fouqueray B, Philippe C, Moulinoux JP, Baud L. Transforming growth factor-beta stimulates arginase activity in macrophages. Implications for the regulation of macrophage cytotoxicity. The Journal of Immunology. 1995;155(4):2077–84. [PubMed] [Google Scholar]
- 34.Sterling JA, Edwards JR, Martin TJ, Mundy GR. Advances in the biology of bone metastasis: How the skeleton affects tumor behavior. Bone. 2011;48(1):6–15. doi: 10.1016/j.bone.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 35.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(18):7260–4. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards JR, Nyman JS, Lwin ST, Moore MM, Esparza J, O’Quinn EC, et al. Inhibition of TGF-beta signaling by 1D11 antibody treatment increases bone mass and quality in vivo. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2010;25(11):2419–26. doi: 10.1002/jbmr.139. [DOI] [PubMed] [Google Scholar]
- 37.Buenrostro D, Mulcrone PL, Owens P, Sterling JA. The Bone Microenvironment: a Fertile Soil for Tumor Growth. Current osteoporosis reports. 2016;14(4):151–8. doi: 10.1007/s11914-016-0315-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature reviews Immunology. 2009;9(3):162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Kawano M, Mabuchi S, Matsumoto Y, Sasano T, Takahashi R, Kuroda H, et al. The significance of G-CSF expression and myeloid-derived suppressor cells in the chemoresistance of uterine cervical cancer. Scientific reports. 2015;5:18217. doi: 10.1038/srep18217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(16):6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hollmen M, Karaman S, Schwager S, Lisibach A, Christiansen AJ, Maksimow M, et al. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology. 2016;5(3):e1115177. doi: 10.1080/2162402X.2015.1115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delitto D, Black BS, Sorenson HL, Knowlton AE, Thomas RM, Sarosi GA, et al. The inflammatory milieu within the pancreatic cancer microenvironment correlates with clinicopathologic parameters, chemoresistance and survival. BMC cancer. 2015;15:783. doi: 10.1186/s12885-015-1820-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim HK, De La Luz Sierra M, Williams CK, Gulino AV, Tosato G. G-CSF down-regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood. 2006;108(3):812–20. doi: 10.1182/blood-2005-10-4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulcrone PL, Campbell JP, Clement-Demange L, Anbinder AL, Merkel AR, Brekken RA, et al. Skeletal Colonization by Breast Cancer Cells Is Stimulated by an Osteoblast and beta2AR-Dependent Neo-Angiogenic Switch. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2017;32(7):1442–54. doi: 10.1002/jbmr.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sasaki S, Baba T, Nishimura T, Hayakawa Y, Hashimoto S-I, Gotoh N, et al. Essential roles of the interaction between cancer cell-derived chemokine, CCL4, and intra-bone CCR5-expressing fibroblasts in breast cancer bone metastasis. Cancer Letters. 2016;378(1):23–32. doi: 10.1016/j.canlet.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Allan EH, Hilton DJ, Brown MA, Evely RS, Yumita S, Metcalf D, et al. Osteoblasts display receptors for and responses to leukemia-inhibitory factor. Journal of Cellular Physiology. 1990;145(1):110–9. doi: 10.1002/jcp.1041450116. [DOI] [PubMed] [Google Scholar]
- 48.Grimaud E, Blanchard F, Charrier C, Gouin F, Redini F, Heymann D. Leukaemia Inhibitory Factor (Lif) Is Expressed in Hypertrophic Chondrocytes and Vascular Sprouts during Osteogenesis. Cytokine. 2002;20(5):224–30. doi: 10.1006/cyto.2002.2002. [DOI] [PubMed] [Google Scholar]
- 49.Li X, Yang Q, Yu H, Wu L, Zhao Y, Zhang C, et al. LIF promotes tumorigenesis and metastasis of breast cancer through the AKT-mTOR pathway. Oncotarget. 2014;5(3):788–801. doi: 10.18632/oncotarget.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Douglas AM, Goss GA, Sutherland RL, Hilton DJ, Berndt MC, Nicola NA, et al. Expression and function of members of the cytokine receptor superfamily on breast cancer cells. Oncogene. 1997;14(6):661–9. doi: 10.1038/sj.onc.1200882. [DOI] [PubMed] [Google Scholar]
- 51.Maes C. Role and regulation of vascularization processes in endochondral bones. Calcified tissue international. 2013;92(4):307–23. doi: 10.1007/s00223-012-9689-z. [DOI] [PubMed] [Google Scholar]
- 52.Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I Study of GC1008 (Fresolimumab): A Human Anti-Transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLOS ONE. 2014;9(3):e90353. doi: 10.1371/journal.pone.0090353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.den Hollander MW, Bensch F, Glaudemans AW, Oude Munnink TH, Enting RH, den Dunnen WF, et al. TGF-beta Antibody Uptake in Recurrent High-Grade Glioma Imaged with 89Zr-Fresolimumab PET. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2015;56(9):1310–4. doi: 10.2967/jnumed.115.154401. [DOI] [PubMed] [Google Scholar]
- 54.de Gramont A, Faivre S, Raymond E. Novel TGF-beta inhibitors ready for prime time in oncoimmunology. Oncoimmunology. 2017;6(1):e1257453. doi: 10.1080/2162402X.2016.1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen X, Yang Y, Zhou Q, Weiss JM, Howard OZ, McPherson JM, et al. Effective chemoimmunotherapy with anti-TGFbeta antibody and cyclophosphamide in a mouse model of breast cancer. PLoS ONE. 2014;9(1):e85398. doi: 10.1371/journal.pone.0085398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y, et al. TGF-beta-Neutralizing Antibody 1D11 Enhances Cytarabine-Induced Apoptosis in AML Cells in the Bone Marrow Microenvironment. PLoS ONE. 2013;8(6):e62785. doi: 10.1371/journal.pone.0062785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proceedings of the National Academy of Sciences. 2012;109(41):16618–23. doi: 10.1073/pnas.1117610109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. TGFβ1 Inhibition Increases the Radiosensitivity of Breast Cancer Cells <em>In Vitro</em> and Promotes Tumor Control by Radiation <em>In Vivo</em>. Clinical Cancer research. 2011;17(21):6754–65. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.