

Abstract
Carboxylesterase 1 (CES1) metabolizes methylphenidate and other drugs. CES1 gene variation only partially explains pharmacokinetic (PK) variability. Biomarkers predicting the PKs of drugs metabolized by CES1 are needed. We identified lipids in plasma from 44 healthy subjects that correlated with CES1 activity as determined by PK parameters of methylphenidate including a ceramide (q value = 0.001) and a phosphatidylcholine (q value = 0.005). Carriers of the CES1 143E allele had decreased methylphenidate metabolism and altered concentration of this phosphatidylcholine (q value = 0.040) and several high polyunsaturated fatty acid lipids (PUFAs). The half‐maximal inhibitory concentration (IC50) values of chenodeoxycholate and taurocholate were 13.55 and 19.51 μM, respectively, consistent with a physiological significance. In silico analysis suggested that bile acid inhibition of CES1 involved both binding to the active and superficial sites of the enzyme. We initiated identification of metabolites predicting PKs of drugs metabolized by CES1 and suggest lipids to regulate or be regulated by this enzyme.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ The activity of CES1, the enzyme responsible for degrading MPH, is highly variable. The underlying contributors to that variability are unclear.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study shows that the PKs of MPH may be used to identify metabolic pathways, which regulate CES1 activity or are regulated by CES1.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The present study identified CES1 as a potential regulator of the concentration of EPA, an anti‐inflammatory fatty acid, in lipids. It also identified bile acids that were correlated with the PK of MPH and subsequently shown to inhibit CES1 activity in vitro. Finally, the study has linked MPH PK and plasma lipid concentrations to CES1 genotype and apparent enzyme activity.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ Metabolomics is a tool, which may be able to identify metabolic pathways contributing to variability in drug concentration.
Methylphenidate (MPH) is a central nervous system stimulant used in the treatment of attention deficit hyperactivity disorder.1 Its medical use began in 1960; the drug became increasingly prescribed by the late 1980s and early 1990s when the diagnosis of attention deficit hyperactivity disorder itself became more widely accepted and prevalent.2 Up to 30% of patients treated with MPH either do not achieve the desired improvement in symptom severity or are intolerant to the treatment.3
The individual variation in the response to MPH is poorly understood but factors affecting the activity of CES1 may be important in determining individual variation in plasma concentration and clinical outcome of the drug.4
The pharmacokinetic (PK) properties of a drug are determined by the processes of its absorption, distribution, metabolism, and excretion. Among these, the metabolism is particularly interesting because the activity of several drug metabolizing enzymes are significantly influenced by variation in the genes coding them.5, 6, 7 This opens up for the use of genotyping of drug metabolizing enzymes to predict PKs and potentially provide information about decrease in efficacy or susceptibility to adverse reactions.6, 7
Carboxylesterase 1 (CES1) is a hydrolase, and is the principal enzyme in the metabolism of commonly prescribed drugs, such as MPH,8 clopidogrel,9 and the angiotensin‐converting enzyme inhibitor prodrugs.10, 11 This enzyme has also been implicated in the metabolism of endogenous compounds, such as esters of cholesterol and triglycerides.12 Besides serving as potential substrates for CES1, endogenous molecules with a cholesterol‐like structure have been implicated in the regulation of the activity of the enzyme by allosteric binding to its superficial ligand binding site, the so‐called Z‐site.13, 14, 15
Some individuals carry a hybrid of carboxylesterase 1 pseudogene 1 (CES1P1) and a segment of CES1. This hybrid has been designated CES1A2, whereas CES1 frequently is referred to as CES1A1.16, 17 Collectively, CES1A1 and CES1A2 are often referred to as CES1A. CES1A2 is composed of the promoter and exon 1 of CES1P1 and a duplicated segment of CES1 containing exons 2–14. Because the most common promoter in CES1A2, is markedly weaker than that of CES1A1, presence of CES1A2 is usually not associated with significantly increased amounts of CES1 mRNA.16 However, there is a promoter haplotype of CES1A2 with a CES1A1‐derived segment containing two overlapping Sp1 sites that exhibits significantly higher transcriptional activity as compared with CES1A2 haplotypes without these sites, the high‐activity promoter.18 Several single nucleotide polymorphisms (SNPs) of CES1 have been reported, including rs71647871, also designated G143E or p.Gly143Glu as it leads to a change of glycine to glutamic acid at position 143, a variation that has been associated with a significantly decreased rate of drug metabolism.19, 20 Another variant, CES1A1c, is a haplotype defined by a series of SNPs, preferentially located in exon 1, that has been associated with reduction in CES1 mRNA expression but without marked impact on drug metabolizing activity.21, 22
Because the frequencies of variants of CES1 with a functional impact are low (Ferrero‐Milliani, unpublished) it is likely that variation in this gene explains only a relatively small proportion of the variation in the PKs of MPH. Hence, other types of biomarkers may be required for prediction of the kinetics of drugs metabolized by CES1 on the individual level.
Using metabolomics approaches, totally new insights have been gained about the mode of action of a variety of drugs and the mechanisms implicated in variation in response to treatment with key classes of therapies, including antidepressants,23 statins,24 and antihypertensives.25
Accordingly, metabolomics is a tool, which may prove helpful in the identification of biomarkers for prediction of drug response.
Several enzymes implicated in drug metabolism also have endogenous substrates.26, 27 Knowledge about endogenous substrates and small molecules of endogenous origin modulating the activity of such an enzyme may provide clues to the identification of biomarkers in the blood that correlate with its activity in the liver.28
The aim of the present study was to identify predose endogenous metabolites that correlate with the PKs of MPH and variation in the gene encoding CES1, the principal enzyme in the metabolism of this drug, besides gaining new insights into the physiological role of this enzyme.
MATERIALS AND METHODS
Study design, sampling, genetic analyses, and pharmacokinetic analyses
Data from a previous study in which we investigated the influence of variation in the gene encoding CES1 on the PK variables of MPH in 44 healthy volunteers,29 were made available to the present study. In brief, the 44 volunteers, consisting of 19 men and 25 women aged 20–29 years and with a body mass index ranging from 18–28, had been selected from a total of 200 recruited subjects on the basis of CES1 genotyping and classified into six CES1 genotype groups, which included the CES1 wildtype genotype and five genotypes of CES1 with presumed or documented effect on drug metabolism. These six groups were designated 1–6 and consisted of group 1: wild type with 2 copies of CES1 and without nonsynonymous SNPs, n = 16; group 2: carriers of four CES1 copies including two CES1A2 copies, n = 5; group 3: carriers of the 143E allele (rs71647871), n = 6; group 4: carriers of three CES1 copies including one CES1A2 copy with overlapping Sp1 sites conferring increased transcriptional activity, n = 2; group 5: carriers of the CES1A1c variant, n = 4; and group 6: carriers of three copies of CES1 including one CES1A2 copy, which carries the common and low‐activity promoter, n = 10. One of 17 subjects with the wild‐type genotype caught a cold with fever and did not complete the study according to the protocol. Hence, this group consisted of 16 subjects. The characteristics of the study subjects in the six genotype groups are listed in Table S1. The study had been designed as a prospective, open labeled, single armed trial in which the subjects from all genotype groups received a single dose of 10 mg MPH (Ritalin; Novartis, Basel, Switzerland) after a standardized breakfast. Serial blood samples for analysis of plasma concentrations of MPH (d‐MPH and l‐MPH) and its metabolites, which are d‐ritalinic and l‐ritalinic acid (d‐RA and l‐RA), were collected 0.5, 1, 1.5, 2, 2.5, 3,4, 6, 8, 10, 24, and 33 hours after dose administration. Noncompartmental methods were used to determine area under the curve (AUC)0‐inf, area under the concentration‐time curve from time 0 to infinity; Cmax, the maximum post‐dose concentration; Tmax, time at which Cmax occurs; and t½, terminal elimination half‐life of d‐MPH, l‐MPH, d‐RA, and l‐RA. We examined whether these PK variables differed between CES1 genotypes using an unpaired Wilcoxon Test with correction by the Benjamini & Hochberg method to control for false discovery rates based on significance set to q < 0.2.30 The PK variables of the six genotype groups are listed in Table S2.
Samples for metabolomics profiling
Blood samples for metabolomics profiling had been collected immediately before the standardized breakfast using 4‐mL tubes containing EDTA (Becton Dickinson). After centrifugation of the tubes, plasma was harvested and immediately frozen by submergence in a mixture of ethanol and dry ice. The plasma samples were stored at −80° until analysis.
Metabolomics profiling: liquid chromatography and mass spectroscopy targeted metabolomics analyses
Targeted metabolomics analyses, summarized below, were performed using standard operating procedures based on previously published methods. More detailed method descriptions and target lists are given in the Supplementary Material.
Biogenic amine profiling
Internal standards were added to 5 μl plasma, reduced with tris(2‐carboxyethyl)phosphine, extracted via a protein precipitation step, and derivatized using AccQ‐Tag (Waters Chromatography, Etten‐Leur, The Netherlands), after which the samples were analyzed using an AccQ‐Tag Ultra column coupled to a Xevo‐TQS ultra‐performance liquid chromatography‐tandem mass spectrometry system (Waters Chromatography).31
Positive and negative lipid profiling
We took 10‐μL and 20‐μL plasma samples that were spiked with calibration and internal standards and extracted using isopropyl alcohol (for the positive lipid platforms) or methanol (for the negative lipid platforms). Samples were analyzed using an ACQUITY UPLC system with an HSS T3 column coupled to a 6530 Accurate Mass QToF (Agilent Technologies, Santa Clara, CA).32
Bile acid profiling
Bile acid extraction was performed by adding methanol containing internal standards to 50 μL plasma. Samples were analyzed using an ACQUITY UPLC system with an HSS T3 column coupled to a 6530 Accurate Mass QToF (Agilent Technologies).
Following liquid chromatography‐mass spectrometry analysis, data was preprocessed by performing peak integration, background correction, and determination of the relative ratios between metabolites and their corresponding internal standards. The metabolite units were expressed as the peak area ratios of the target analyte to the respective internal standard. An in‐house written tool was applied using the QC samples to compensate for shifts in the sensitivity of the mass spectrometer throughout the batches. Both internal standard correction and QC correction were applied to the dataset before reporting results. Quality assurance of metabolite measurements was performed using the QC relative standard deviation (RSDqc). For amines, reported compounds had an RSDqc <15%, for positive and negative lipids RSDqc <20%, and for bile acids RSDqc <30%.
Processing of metabolomics data and statistical analysis
Data processing and analysis was performed in the open‐source statistical software, R version 3.2.2.33 Initial testing determined much of the data was skewed from normal. Hence, log transformation of the data was used for all subsequent analyses. Significant correlation between outcomes and metabolites was tested by linear regression.
Correlation coefficients (r) were calculated using the Pearson's product‐moment correlation test as used in the cor.test function. Comparisons of genotype variants to wild‐type genotype were tested using an unpaired Wilcoxon Test. A retrospective power analysis was conducted on the dataset in Stage et al.29 2017 showing that a minimum of 6 subjects per group, more precisely 5.06 subjects, was required to detect a difference between groups. Therefore, genotype comparisons were restricted to only those groups that had at least >5 subjects, namely groups 1 (wild type) and groups 3 and 6. Multiple testing correction for each set of comparisons was performed using the Benjamini & Hochberg correction method to control for false discovery rates with significance set to q < 0.1.30 With the significance set at q < 0.1, the results should include at least 90% true positives.
In vitro inhibition study
The ester substrate, p‐nitrophenyl acetate, the bile acids sodium glycocholate hydrate, sodium taurochenodeoxycholate, sodium taurocholate, lithocholic acid, deoxycholic acid, sodium glycochenodeoxycholate, chenodeoxycholate, and cholic acid were purchased from Sigma‐Aldrich (St. Louis, MO). Diltiazem hydrochloride, an inhibitor of CES1, served as a positive control and was purchased from Napp Pharmaceuticals Research (Cambridge, UK). Recombinant human CES1 (CES1b/CES1A1), the major isoform in the human liver, was from BD Gentest (Woburn, MA), and the p‐nitrophenol was purchased from Fluka (Buchs, Switzerland). Other chemicals were of liquid chromatography‐mass spectrometry grade and also commercially available. Glycocholic acid was only available as a hydrate with an unspecified number of water molecules. For calculation of the molecular mass of this compound, we assumed presence of two water molecules in it.
Inhibition of CES1 by the bile acids was investigated by incubation with the recombinant enzyme in the presence of p‐nitrophenyl acetate. The final concentration of this substrate and the enzyme was 100 µM and 10 µg/mL, respectively. The bile acids were incubated at six concentration levels ranging from 0.33–1200 µM. Reactions containing enzyme but without bile acids were also prepared. A reaction mixture with substrate but without enzyme was included as a negative control. Dimethyl sulfoxide in a final concentration of 2% v/v was used for dissolution of the bile acids. This dimethyl sulfoxide concentration was previously found to have only negligible effect on CES1 activity.11 All incubations were performed in 96‐well pureGrade BRAND plates (BRAND, Wertheim, Germany) in 100 mM phosphate buffer at 37°C in a final volume of 200 µL. The concentration of the hydrolytic product of the substrate, that is, p‐nitrophenol, was determined by measurement of its absorbance at 405 nm after 3 minutes using a Sunrise microplate reader (Tecan, Grödig, Austria). The absorbance readings were corrected for spontaneous hydrolysis by subtracting the absorbance of the negative control. The sampling time was found to be within the linear range of the reaction. All reactions were performed in triplicate. The half‐maximal inhibitory concentration (IC50) constants were determined using nonlinear regression in Prism, version 6.07 (GraphPad Software, San Diego, CA).
In silico analysis
A docking study was carried out to decipher the preferred CES1 binding site of the seven bile acids for which we determined IC50 values in the in vitro analysis, namely, glycocholate, taurochenodeoxycholate, taurocholate, deoxycholic acid, glycochenodeoxycholate, chenodeoxycholic, and cholic acid. For this purpose, the coordinates of human CES1 in complex with taurocholate in the catalytic site and in the Z‐site (Protein Data Bank code 2DR0) were obtained from the Research Collaborator for Structural Bioinformatics Protein Data Bank. The structure preparation and docking calculations were performed using CLC Drug Discovery Workbench (CLC Drug Discovery Workbench version 2.0. CLC Bio‐Qiagen, Aarhus, Denmark). Water molecules and cofactors were removed from the complex and the monomeric form of the protein was protonated at pH 7.2. The binding sites were defined with a 15 Å radius around taurocholate, both in the catalytic site and the Z‐site. To assess whether the docking program was able to reproduce crystallographic binding modes, taurocholate was docked back in the two binding sites. Subsequently, the seven bile acids were docked into the defined binding sites assuming the protein to be completely rigid in all docking studies to reduce computational costs. A total of 100 conformations were computed for each compound and the first ranked conformation of each of the compounds, that is, the conformations with the lowest docking scores and the most stable interactions inside a cavity, were considered for further analysis. Analysis of binding was performed using the software Molecular Operating Environment, 2015.10 (Chemical Computing Group, Montreal, QC, Canada).
RESULTS
Lipid metabolites were measured either in positive or negative mode. In positive mode, 153 lipid metabolites were measured (Supplementary Material). They belonged to the following lipid classes: ceramide (Cer), cholesterol esters (CEs), diacylglycerol (DG), lysophosphatidylcholine (LPC), phosphatidylcholine (PC), phosphatidylethanolamine (PE), sphingomyelin (SM), and triglycerides (TG). In the negative mode, 61 lipids were measured belonging to: fatty acids (FAs), LPC, lysophosphatidylethanolamine (LPE), SN1, and SN2 classes. A measure of 13 individual bile acids (primary and secondary) and a panel of 32 amines were also measured.
Predose metabolomics data were correlated with the PK profile of MPH and its metabolite RA (Table 1). Because the study subjects were of similar age (20–29 years) and body mass index (18–28 kg/m2) evaluation of these covariates indicated that they did not possess enough variance to justify adding them to the model (data not shown).
Table 1.
Significant correlations between lipids or bile acids and outcomes by testing full lipid panels and after classification of lipids
| Lipid | Outcome | Correlation (r) | P value | Q valuea |
|---|---|---|---|---|
| Positive lipids – all | ||||
| PC(38:5) | AUC d‐Ratio | −0.61912 | 0.000012 | 0.018930 |
| PC(38:5) | AUC d‐MPH | −0.60946 | 0.000018 | 0.028101 |
| Cer(d18:1/24:1) | Half‐life l‐RA | 0.59135 | 0.000037 | 0.057003 |
| PC(38:5) | Cmax d‐MPH | −0.56358 | 0.000102 | 0.155770 |
| Positive lipids ‐ by class | ||||
| Cer(d18:1/24:1) | Half‐life l‐RA | 0.59135 | 0.000037 | 0.001490 |
| PC(38:5) | AUC d‐Ratio | −0.61912 | 0.000012 | 0.004825 |
| PC(38:5) | AUC d‐MPH | −0.60946 | 0.000018 | 0.007163 |
| PC(38:5) | Cmax d‐MPH | −0.56358 | 0.000102 | 0.039706 |
| CE(20:5) | AUC d‐MPH | −0.45900 | 0.002232 | 0.089283 |
| PC(36:3) | AUC l‐RA | 0.53377 | 0.000272 | 0.106196 |
| DG(36:3) | AUC d‐RA | 0.40624 | 0.007597 | 0.151942 |
| Negative lipids ‐ by class | ||||
| SN2‐LPC(20:5) | Cmax d‐MPH | −0.53120 | 0.000421 | 0.050481 |
| SN1‐LPC(22:5‐w6) | AUC d‐RA | 0.52534 | 0.000500 | 0.094993 |
| High PUFA triglycerides | ||||
| Half‐life d‐MPH | −0.30436 | 0.050028 | 0.400225 | |
| Tmax d‐MPH | −0.28994 | 0.062524 | 0.500195 | |
| AUC d‐MPH | −0.25876 | 0.097997 | 0.783973 | |
| Bile acids | ||||
| Primary bile acids | Half‐life l‐RA | 0.369441 | 0.024425 | 0.195402 |
| Primary conjugated bile acids | Half‐life l‐RA | 0.428832 | 0.008090 | 0.064723 |
AUC, area under the curve; Cmax, peak plasma concentration; MPH, methylphenidate; RA, ritalinic acid; PUFA, polyunsaturated fatty acid lipid; Tmax, time of maximum plasma concentration.
The figures in italics represent p‐value < 0.1 and q‐value > 0.1.
Q values for the positive and negative lipids are by classs and are for guidance for future research rather than indicating significance.
When testing all positive lipids as a set, baseline measurements of two lipids showed significance with measured outcomes (Table 1). PC(38:5) was negatively correlated with both AUC of d‐MPH and the Cmax of d‐MPH, and Cer(d18:1/24:1) was positively correlated with the half‐life of the metabolite l‐RA.
Separating positive lipids by class before testing results revealed three additional suggestive findings, namely a negative correlation between CE(20:5) and AUC of d‐MPH, and positive correlations between DG(36:3) and AUC of d‐RA and PC(36:3) and AUC of l‐RA.
Testing all negative lipids as a set did not reveal any significant correlations between baseline measurement and outcome (Table 1). Again, separating the negative lipids by class identified two additional lipids having suggestive correlations between baseline measurements and various outcomes. Specifically, SNI‐LPC(22:5‐w6) was positively correlated with the AUCs of d‐RA and l‐RA. SN2‐LPC(20:5) was negatively correlated with Cmax of d‐MPH.
Although no significance was found after correction for multiple testing of the correlations between the PK outcomes and the baseline measurements of triglycerides with highly unsaturated fatty acids, low P values were observed for the half‐life, Tmax, and AUC of d‐MPH (P < 0.10; q < 1.00). These results may be suggestive of a need for future study.
No significant correlations were found with triglycerides having lower numbers of double bonds in the fatty acids, or using a measurement of TG/PC ratio.
When testing predose measurements of bile acids individually for correlation with outcomes (Table 1), although some P values were low, there was no significance after multiple‐testing correction. However, when categorized based on primary, secondary, and conjugated status, as detailed in Supplementary Material, there were significant correlations with half‐life of l‐RA in both primary and primary‐conjugated bile acids. Other categorized measures were not significant, nor were there any correlations with other bile acid categories.
Testing predose measurements of the 32 amines for correlation with outcomes showed no significant results.
On testing correlations between CES1 genotype and PK, significance was mostly seen in the comparison between the normal genotype (group 1) and the group with the rs71647871 143E allele (n = 6) with a higher AUC of d‐MPH, half‐life of d‐MPH, and Cmax of d‐MPH in the latter of these two groups (Table 2). Detailed analysis of the genotype/PK comparisons shown in Table 2 have been previously reported using a slightly different statistical approach, which involved application of the Kruskal‐Wallis test.29
Table 2.
Significant results from testing between genotypes for outcomes of pharmacokinetics
| Outcome | Direction | P value | Q value |
|---|---|---|---|
| Group 1 vs. group 3 (143E allele) | |||
| AUC d‐MPH | Group 3 higher | 0.000454 | 0.003630 |
| Half‐life d‐MPH | Group 3 higher | 0.000982 | 0.007860 |
| Cmax d‐MPH | Group 3 higher | 0.007112 | 0.056892 |
AUC, area under the curve; Cmax, peak plasma concentration; MPH, methylphenidate.
Due to the large impact of the 143E allele of rs71647871 on MPH metabolism, testing of correlation between genotype groups and levels of predose metabolites was restricted to comparison of carriers of 143E allele with the control group (Table 2 , Figure S1). For positive lipids, the only significant comparison found with genotype was for PC(38:5), which was present at a higher concentration at baseline in group 1 (median log value: 1.23) than group 3 (median log value: 0.96; Table 3 , Figure S2). Group 3 had decreased concentrations of metabolites with high polyunsaturated fatty acid lipids (PUFAs), specifically TG(56:6) and TG(56:5), when compared to group 1. There were no other significant correlations.
Table 3.
Significant results from testing between genotypes for outcomes of lipids
| Lipid | Direction | P value | Q value |
|---|---|---|---|
| Group 1 vs group 3 positive lipids | |||
| PC(38:5) | Group 1 higher | 0.007988 | 0.039939 |
| Group 1 vs. group 3 high PUFA triglycerides | |||
| TG(56:6) | Group 1 higher | 0.006085 | 0.091271 |
| TG(56:5) | Group 1 higher | 0.007988 | 0.119818 |
PUFA, polyunsaturated fatty acid lipid.
The IC50 values differed significantly between the bile acids (Figure 1). We found that chenodeoxycholic acid and taurocholic acid inhibited with IC50 of 13.55 and 19.51 μM, respectively, which is in a range similar to diltiazem, a known inhibitor of CES1.
Figure 1.
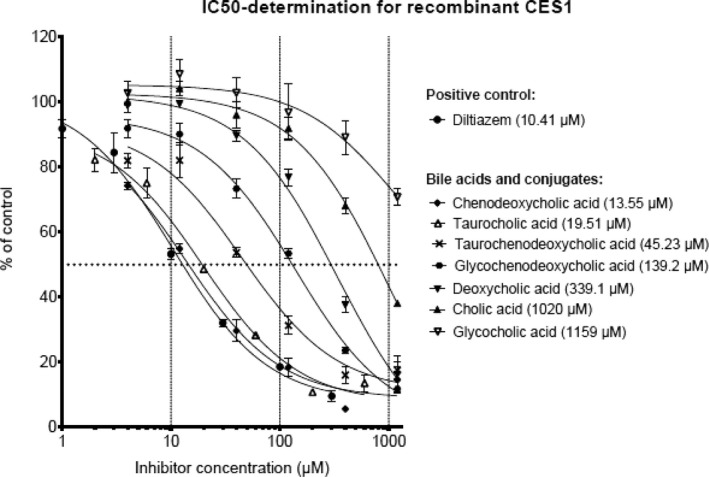
Dose response curves for compounds inhibiting carboxylesterase 1 (CES1) activity. Half‐maximal inhibitory concentrations (IC50) are shown in brackets.
Taurochenodeoxycholic acid had an IC50 value, which was more than fourfold higher; whereas glycochenodeoxycholic acid had an IC50 ∼10‐fold higher than diltiazem. We were not able to determine the IC50 value of lithocholic acid because it could not be dissolved in aqueous buffer.
Docking analysis predicted a preference of all bile acids for the catalytic site over that of the Z‐site of the enzyme, except for deoxycholic acid (Table 4). Taurocholic acid, taurochenodeoxycholic acid, and glycochenodeoxycholic acid were predicted to bind to the active site with the highest affinity. Chenodeoxycholate, taurocholate, and taurochenodeoxycholic acid, the strongest inhibitors of CES1 in our in vitro analysis, were found to interact more strongly with the Z‐site of the enzyme than the four other bile acids in our docking analysis (Table 4). There was a weak correlation between docking scores of the bile acids for the catalytic pocket of CES1 and the IC50 values (r = 0.44). The correlation between the docking scores of the bile acids for the Z‐site and the IC50 values was strong (r = 0.86). The docking analysis also revealed that taurocholate and other compounds with a sulfate group are stabilized by an ionic interaction with Lys92 at the entrance of the active site, thus potentially compromising access of substrate to this site (Figure 2).
Table 4.
Docking scores of bile acids for the active site and Z‐site of carboxylesterase 1
| Compound | Docking score active site | Docking score Z‐site |
|---|---|---|
| Chenodeoxycholic acid | −53.00 | −51.50 |
| Taurocholic acid | −67.10 | −54.41 |
| Taurochenodeoxycholic acid | −63.20 | −51.00 |
| Glycochenodeoxycholic acid | −68.00 | −48.80 |
| Deoxycholic acid | −41.50 | −47.20 |
| Cholic acid | −52.00 | −46.70 |
| Glycocholic acid | −53.20 | −43.60 |
Figure 2.
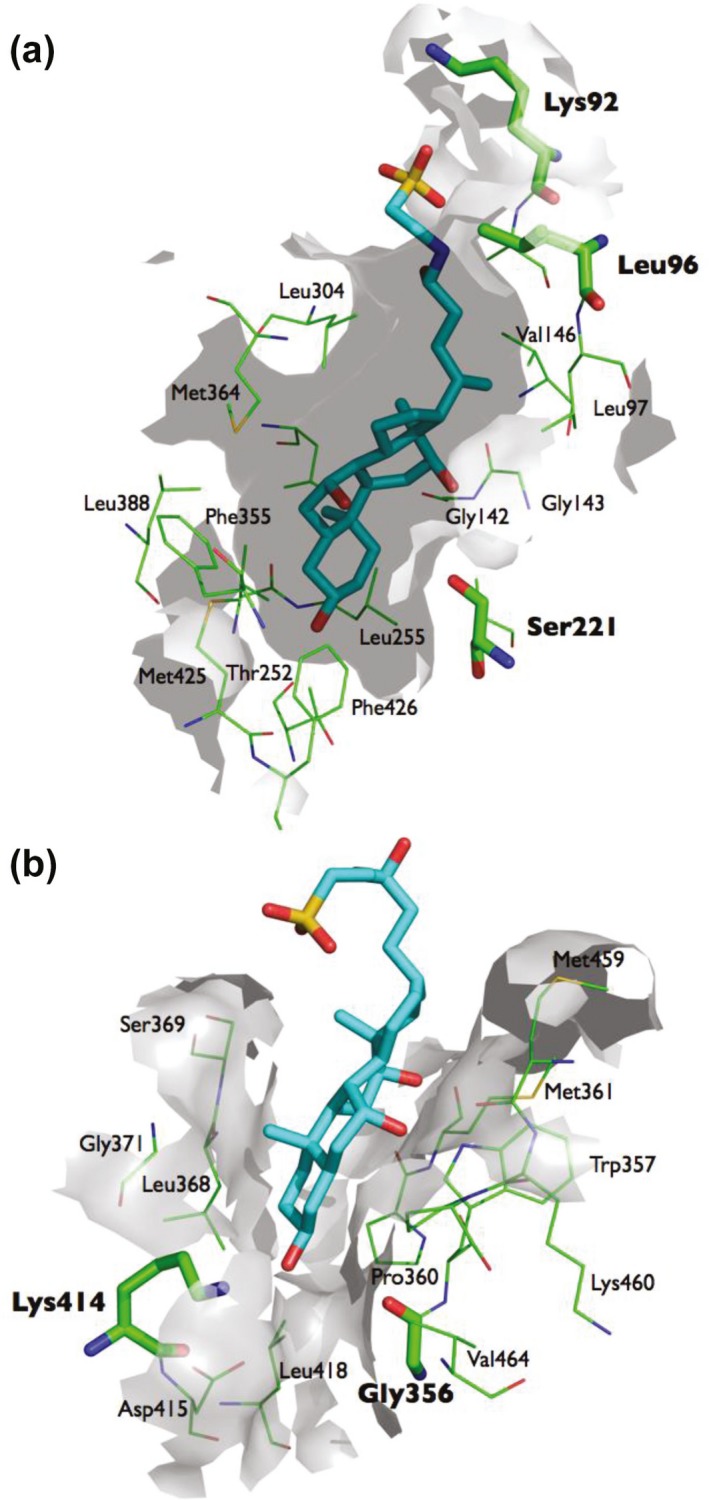
Docking of taurocholate into the catalytic site (a) and the Z‐site (b). The binding of taurocholate in the catalytic pocket of carboxylesterase 1 is stabilized by an ionic interaction with Lys92 at the entrance of this site. Binding of taurocholate to the Z‐site appears to be stabilized by Lys414.
The carboxyl group containing bile acids, such as chenodeoxycholate, are located with their carboxyl group close to the catalytic triad amino acid Ser221, which could hinder nucleophilic substrate attack by this amino acid. The binding of taurocholate and chenodeoxycholate to the Z‐site seem to be stabilized by Gly356 and Lys414 (Figure 3).
Figure 3.
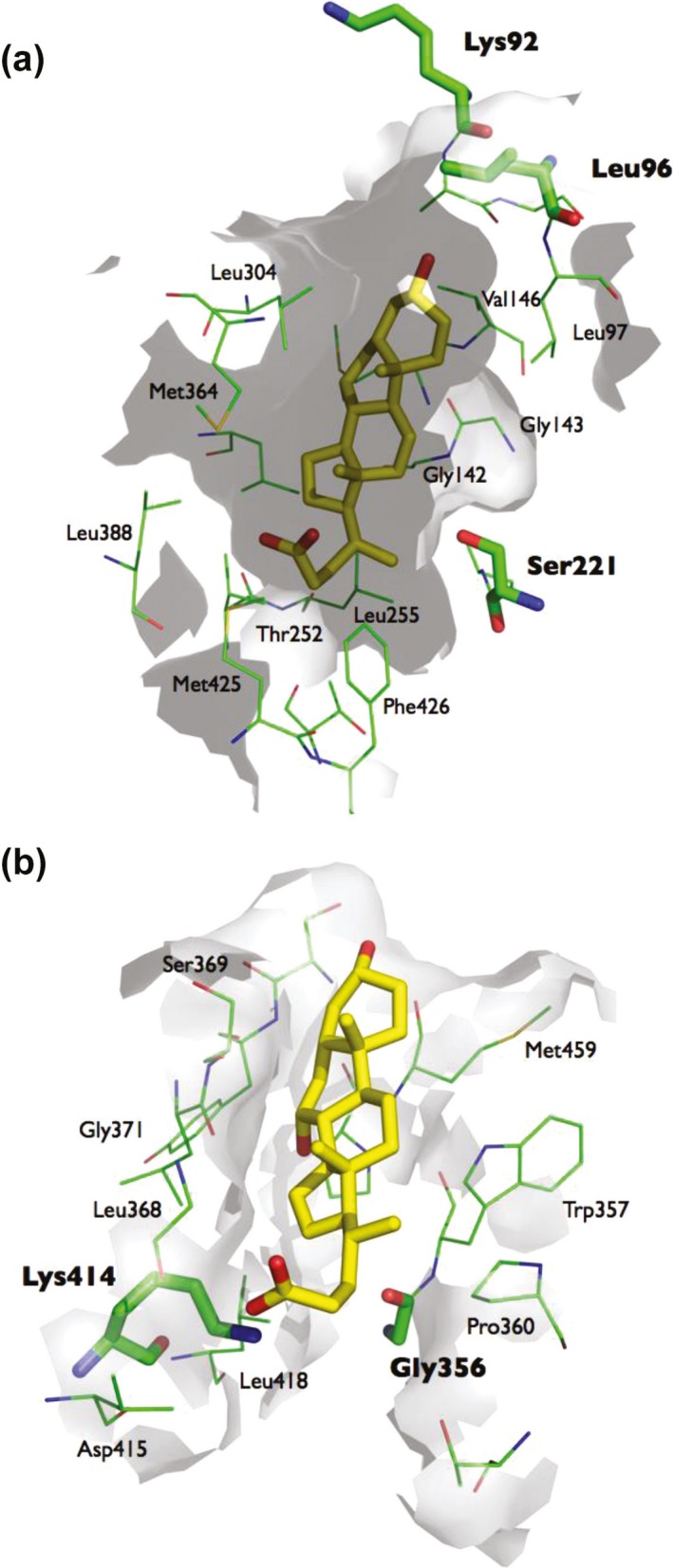
Docking of chenodeoxycholate into the catalytic site (a) and the Z‐site (b). Chenodeoxycholate is located deep in the catalytic pocket Ser221 close to the catalytic triad amino acid Ser221. The binding of chenodeoxycholate to the Z‐site appears to be stabilized by Lys414.
DISCUSSION
This study is the first to correlate small endogenous molecules in the blood with hepatic activity of CES1 as determined by the PKs of a drug, MPH. Its main findings were the identification of lipids that not only correlated with the PK of MPH and RA but also with CES1 genotype and suggested a role of CES1 in lipid metabolism. Involvement of specific bile acids in the regulation of the activity of CES1 was supported by the finding of an ability of two bile acids to inhibit CES1 in vitro, which was supplemented with in silico observations to determine the mechanism underlying the inhibition.
The metabolites that correlated with PK variables of d‐MPH included lipid metabolites containing eicosapentaenoic acid (EPA) 20:5 and docosapentaenoic acid (DPA) 22:5, FAs commonly found in fish oil. The EPA and DPA in FA, LPC, CE, and PC containing metabolites were found to be correlated with the Cmax and AUC of d‐MPH. Both EPA and DPA are long‐chain PUFAs that are important in production of eicosanoids, signaling molecules, which regulate inflammation and metabolism.34
Lipids containing EPA were negatively correlated with d‐MPH concentration, thus decreased EPA would be associated with increased MPH level and, hence decreased CES1 activity. This supports the hypothesis that CES1 activity determines the quantity of PUFAs in triglycerides and regulates release of lipoproteins.35 Lower CES1 activity would result in decreased PUFAs in hepatic lipids and the release of those lipids modulating the composition and release of lipoproteins from the liver.35, 36 The significant lipid metabolites with 20:5 as the FA moiety identified in this study indicate that CES1 preferentially regulates the quantity of EPA in lipids. Alternatively, higher levels of omega 6 PUFA, such as DPA, which, in our study, had a positive correlation with RA AUC, may inhibit CES1 activity14 providing a feedback regulation to control lipoprotein production. This is in line with previous in vitro findings that fatty acids inhibit CES1, particularly unsaturated fatty acids.14
The finding of lower levels of PUFA in PC and TG in the genotype group with the 143E allele, which was associated with significantly decreased CES1 activity, should be interpreted with caution due to the small sample size. However, it connects CES1 activity with physiological functions and allows for improved prediction of this enzyme activity based on the combination of genetics and metabolomics information.
We found that levels of bile acids marginally correlated with l‐RA and d‐MPH PK. Bile acids possess a variety of physiological functions37 and have been linked with response to drugs, such as simvastatin.24 Moreover, cholesterol‐like molecules, including the bile acids cholate and taurocholate, have been shown to bind to CES1, which is thought to shift the trimer‐hexamer equilibrium of the enzyme toward the trimer, the catalytically active form.13 Hence, we hypothesize that the correlations of bile acids with l‐RA and d‐MPH PK in the present study reflect involvement of bile acids in the regulation of CES1 activity. However, we cannot exclude that these correlations have occurred as a result of a bias due to the small sample size of our cohort.
Additionally, the correlations may have resulted from indirect regulation of lipid metabolism by farnesoid X receptor.
Using in vitro analysis, we revealed large differences between bile acids in the ability to inhibit CES1. Two bile acids had IC50 values in a range sufficiently low to inhibit CES in vivo,38 thus arguing in favor of a role of bile acids in the regulation of CES1 activity.
Our in vitro analysis of the bile acids were supplemented with an in silico study of these molecules to provide insights into their molecular interactions with CES1 and resultant effect on the enzyme. It suggested that bile acids are capable of binding to the catalytic pocket as well as the Z‐site of CES1 but have larger affinity to the former of these two sites. Using X‐ray crystallography, a previous study observed binding of taurocholate to both the Z‐site and the active site of CES1, which is consistent with our findings. Inhibitors that both bind to the active site and the Z‐site of CES1 are most likely mixed‐type inhibitors, which may include bile acids.9 The docking scores of the ligands in the catalytic pockets and the Z‐site may not necessarily be comparable with respect to impact on enzyme activity. The strong correlation between the docking scores for the Z‐site and the IC50 values suggests that the docking values for this site directly translates into enzyme activity.
Several factors determine the level of FA and bile acids, including diet and body weight. Hence, it is possible that these and similar factors have an impact on the activity of CES1 and the metabolism of several of drugs.39
The lack of correlation between the level of amines and PK of MPH is noteworthy as amines have not been associated with CES1. This is opposed to lipids that correlated with the PK of MPH in the present study and previously have been identified as substrates and allosteric regulators of CES1.13 Hence, our findings are in accordance with the existing knowledge about substrates and endogenous regulators of CES1 activity.
In summary, the findings of the present study that lipid and bile acid metabolites were correlated with apparent activity and genotype of CES1 are suggestive of a role of CES1 in lipid metabolism and a physiological role of the enzyme. Our findings also open up for prediction of the PK of drugs metabolized by CES1 on the individual level and for improved therapy with these drugs.
Source of Funding
This research was supported by grant 10–092792/DSF from the Danish Council for Strategic Research, Programme Commission on Individuals, Disease and Society.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
E.H.S., R.B., O.T., G.S.N., R.T., G.J., and A.A.M. wrote the manuscript. R.K.D., H.B.R., and T.H. designed the research. A.C.H., C.S., K.P.D., H.B.R., O.T., G.S.N., R.T., K.L., G.J., and T.H. performed the research. E.H.S., R.B., C.S., K.P.D., O.T., G.S.N., R.T., K.L., G.J., and A.A.M. analyzed the data.
Supporting information
Supplementary Material: Metabolite analysis methods
Supplementary Table S1. Subject characteristics
Supplementary Table S2. Pharmacokinetic parameters in CES1 genotype groups
Supplementary Figure S1: Violin plots representing the distribution of vlaues for the significant outcomes for group 1 subjects and group 3 subjects as shown in Table 2.
Supplementary Figure S2: Violin plots representing the distribution of values for the significant lipids for group 1 subjects and group 3 subjects as shown in Table 3.
Supplementary Figure S3: Diagram of the flow for the statistical tests.
Supplementary Figure S4: Scatter plots for the significant metabolites with (FDR<0.1) in Table 1.
Acknowledgments
The project INDICES (INDIvidualised drug therapy based on pharmacogenomics: focus on CES1) aims at developing strategies for individualized treatment with methylphenidate and angiotensin‐converting enzyme inhibitors.
Contributor Information
Rima Kaddurah‐Daouk, Email: rima.kaddurahdaouk@duke.edu.
Thomas Hankemeier, Email: hankemeier@lacdr.leidenuniv.nl.
Henrik B. Rasmussen, Email: Henrik.Berg.Rasmussen@regionh.dk
References
- 1. Kendall, T. , Taylor, E. , Perez, A. , Taylor C. & Guideline Development Group . Diagnosis and management of attention‐deficit/hyperactivity disorder in children, young people, and adults: summary of NICE guidance. BMJ 337, a1239 (2008). [DOI] [PubMed] [Google Scholar]
- 2. Safer, D.J. & Krager, J.M. A survey of medication treatment for hyperactive/inattentive students. JAMA 260, 2256–2258 (1988). [PubMed] [Google Scholar]
- 3. Mohammadi, M.R. & Akhondzadeh, S. Pharmacotherapy of attention‐deficit/hyperactivity disorder: nonstimulant medication approaches. Expert Rev. Neurother. 7, 195–201 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Frölich, J. , Banaschewski, T. , Döpfner, M. & Görtz‐Dorten, A. An evaluation of the pharmacokinetics of methylphenidate for the treatment of attention‐deficit/hyperactivity disorder. Expert Opin. Drug Metab. Toxicol. 10, 1169–1183 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Gaedigk, A. , Sangkuhl, K. , Whirl‐Carrillo, M. , Klein, T. & Leeder, J.S. Prediction of CYP2D6 phenotype from genotype across world populations. Genet. Med. 19, 69–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ingelman‐Sundberg, M. , Sim, S.C. , Gomez, A. & Rodriguez‐Antona, C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol. Ther. 116, 496–526 (2007). [DOI] [PubMed] [Google Scholar]
- 7. Wu, A.H. Drug metabolizing enzyme activities versus genetic variances for drug of clinical pharmacogenomic relevance. Clin. Proteomics 8, 12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun, Z. et al Methylphenidate is stereoselectively hydrolyzed by human carboxylesterase CES1A1. J. Pharmacol. Exp. Ther. 310, 469–476 (2004). [DOI] [PubMed] [Google Scholar]
- 9. Tang, M. et al Antiplatelet agents aspirin and clopidogrel are hydrolyzed by distinct carboxylesterases, and clopidogrel is transesterificated in the presence of ethyl alcohol. J. Pharmacol. Exp. Ther. 319, 1467–1476 (2006). [DOI] [PubMed] [Google Scholar]
- 10. Takai, S. et al Hydrolytic profile for ester‐ or amide‐linkage by carboxylesterases pl 5.3 and 4.5 from human liver. Biol. Pharm. Bull. 20, 869–873 (1997). [DOI] [PubMed] [Google Scholar]
- 11. Thomsen, R. , Rasmussen, H.B. , Linnet K. & INDICES Consortium . In vitro drug metabolism by human carboxylesterase 1: focus on angiotensin‐converting enzyme inhibitors. Drug Metab. Dispos. 42, 126–133 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Ross, M.K. , Streit, T.M. & Herring, K.L. Carboxylesterases: dual roles in lipid and pesticide metabolism. J. Pestic. Sci. 35, 257–264 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bencharit, S. et al Multisite promiscuity in the processing of endogenous substrates by human carboxylesterase 1. J. Mol. Biol. 363, 201–214 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crow, J.A. , Herring, K.L. , Xie, S. , Borazjani, A. , Potter, P.M. & Ross, M.K. Inhibition of carboxylesterase activity of THP1 monocytes/macrophages and recombinant human carboxylesterase 1 by oxysterols and fatty acids. Biochim. Biophys. Acta 1801, 31–41 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fleming, C.D. et al Structural insights into drug processing by human carboxylesterase 1: tamoxifen, mevastatin, and inhibition by benzil. J. Mol. Biol. 352, 165–177 (2005). [DOI] [PubMed] [Google Scholar]
- 16. Fukami, T. et al Structure and characterization of human carboxylesterase 1A1, 1A2, and 1A3 genes. Pharmacogenet. Genomics 18, 911–920 (2008). [DOI] [PubMed] [Google Scholar]
- 17. Sai, K. et al Association of carboxylesterase 1A genotypes with irinotecan pharmacokinetics in Japanese cancer patients. Br. J. Clin. Pharmacol. 70, 222–233 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoshimura, M. et al Functional polymorphisms in carboxylesterase1A2 (CES1A2) gene involves specific protein 1 (Sp1) binding sites. Biochem. Biophys. Res. Commun. 369, 939–942 (2008). [DOI] [PubMed] [Google Scholar]
- 19. Zhu, H.J. et al Two CES1 gene mutations lead to dysfunctional carboxylesterase 1 activity in man: clinical significance and molecular basis. Am. J. Hum. Genet. 82, 1241–1248 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shi, J. et al Association of oseltamivir activation with gender and carboxylesterase 1 genetic polymorphisms. Basic Clin. Pharmacol. Toxicol. 119, 555–561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sanford, J.C. et al Regulatory effects of genomic translocations at the human carboxylesterase‐1 (CES1) gene locus. Pharmacogenet. Genomics 26, 197–207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang, X. et al CES1 genetic variation affects the activation of angiotensin‐converting enzyme inhibitors. Pharmacogenomics J. 16, 220–230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abo, R. et al Merging pharmacometabolomics with pharmacogenomics using “1000 Genomes” single‐nucleotide polymorphism imputation: selective serotonin reuptake inhibitor response pharmacogenomics. Pharmacogenet. Genomics 22, 247–253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaddurah‐Daouk, R. et al Lipidomic analysis of variation in response to simvastatin in the Cholesterol and Pharmacogenetics Study. Metabolomics 6, 191–201 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wikoff, W.R. et al Pharmacometabolomics reveals racial differences in response to atenolol treatment. PLoS One 8, e57639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El‐Sherbeni, A.A. & El‐Kadi, A.O. The role of epoxide hydrolases in health and disease. Arch. Toxicol. 88, 2013–2032 (2014). [DOI] [PubMed] [Google Scholar]
- 27. Ozdemir, V. , Gunes, A. , Dahl, M.L. , Scordo, M.G. , Williams‐Jones, B. & Someya, T. Could endogenous substrates of drug‐metabolizing enzymes influence constitutive physiology and drug target responsiveness? Pharmacogenomics 7, 1199–1210 (2006). [DOI] [PubMed] [Google Scholar]
- 28. Rasmussen, H.B. et al Individualization of treatments with drugs metabolized by CES1: combining genetics and metabolomics. Pharmacogenomics 16, 649–665 (2015). [DOI] [PubMed] [Google Scholar]
- 29. Stage, C. et al The impact of CES1 genotypes on the pharmacokinetics of methylphenidate in healthy Danish subjects. Br. J. Clin. Pharmacol. 83, 1506–1514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995). [Google Scholar]
- 31. Noga, M.J. et al Metabolomics of cerebrospinal fluid reveals changes in the central nervous system metabolism in a rat model of multiple sclerosis. Metabolomics 8, 253–263 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hu, C. et al RPLC‐ion‐trap‐FTMS method for lipid profiling of plasma: method validation and application to p53 mutant mouse model. J. Proteome Res. 7, 4982–4991 (2008). [DOI] [PubMed] [Google Scholar]
- 33. R Core Team . R: A language and environment for statistical computing. <http://www.R-project.org/> (2014).
- 34. Wiktorowska‐Owczarek, A. , Berezińska, M. & Nowak, J.Z. PUFAs: structures, metabolism and functions. Adv. Clin. Exp. Med. 24, 931–941 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Quiroga, A.D. et al Deficiency of carboxylesterase 1/esterase‐x results in obesity, hepatic steatosis, and hyperlipidemia. Hepatology 56, 2188–2198 (2012). [DOI] [PubMed] [Google Scholar]
- 36. Quiroga, A.D. , Lian, J. & Lehner, R. Carboxylesterase1/esterase‐x regulates chylomicron production in mice. PLoS One 7, e49515 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wahlström, A. , Sayin, S.I. , Marschall, H.U. & Bäckhed, F. Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 (2016). [DOI] [PubMed] [Google Scholar]
- 38. Angelin, B. , Björkhem, I. , Einarsson, K. & Ewerth, S. Hepatic uptake of bile acids in man. Fasting and postprandial concentrations of individual bile acids in portal venous and systemic blood serum. J. Clin. Invest. 70, 724–731 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang, D. , Pearce, R.E. , Wang, X. , Gaedigk, R. , Wan, Y.J. & Yan, B. Human carboxylesterases HCE1 and HCE2: ontogenic expression, inter‐individual variability and differential hydrolysis of oseltamivir, aspirin, deltamethrin and permethrin. Biochem. Pharmacol. 77, 238–247 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material: Metabolite analysis methods
Supplementary Table S1. Subject characteristics
Supplementary Table S2. Pharmacokinetic parameters in CES1 genotype groups
Supplementary Figure S1: Violin plots representing the distribution of vlaues for the significant outcomes for group 1 subjects and group 3 subjects as shown in Table 2.
Supplementary Figure S2: Violin plots representing the distribution of values for the significant lipids for group 1 subjects and group 3 subjects as shown in Table 3.
Supplementary Figure S3: Diagram of the flow for the statistical tests.
Supplementary Figure S4: Scatter plots for the significant metabolites with (FDR<0.1) in Table 1.