

Here, Bruzzone et al. explore the relative contributions of the transcriptional coactivators Mediator and two histone acetyltransferase (HAT) complexes, NuA4 and SAGA, to RNA polymerase II association at specific genes and gene classes by rapid nuclear depletion of key complex subunits. They show that the NuA4 HAT Esa1 differentially affects certain groups of genes, whereas the SAGA HAT Gcn5 has a weaker but more uniform effect, and their findings suggest that at least three distinct combinations of coactivator deployment are used to generate moderate or high transcription levels.
Keywords: transcription, histone acetylation, Esa1, Gcn5, Mediator, Tra1
Abstract
The transcriptional coactivators Mediator and two histone acetyltransferase (HAT) complexes, NuA4 and SAGA, play global roles in transcriptional activation. Here we explore the relative contributions of these factors to RNA polymerase II association at specific genes and gene classes by rapid nuclear depletion of key complex subunits. We show that the NuA4 HAT Esa1 differentially affects certain groups of genes, whereas the SAGA HAT Gcn5 has a weaker but more uniform effect. Relative dependence on Esa1 and Tra1, a shared component of NuA4 and SAGA, distinguishes two large groups of coregulated growth-promoting genes. In contrast, we show that the activity of Mediator is particularly important at a separate, small set of highly transcribed TATA-box-containing genes. Our analysis indicates that at least three distinct combinations of coactivator deployment are used to generate moderate or high transcription levels and suggests that each may be associated with distinct forms of regulation.
Nucleosomes are generally considered impediments to various DNA-related processes. One mechanism used by cells to counteract this repressive property of nucleosomes is post-translational modification of histone tails, such as histone acetylation, which has long been recognized to influence transcription (Kouzarides 2007). In higher eukaryotic cells, conditions that lead to massive perturbations of gene expression, such as developmental cues or cancer, are accompanied by modifications in the histone acetylation pattern on both enhancers and promoters (Gong et al. 2016; Kinnaird et al. 2016; Podobinska et al. 2017). Similarly, in yeast, variation in growth conditions and environmental stress induce changes in gene expression concomitant with alterations in the histone acetylation state at gene promoters (Kuang et al. 2014; Weiner et al. 2015).
Histone acetylation states result from the antagonistic action of two classes of enzymes: histone acetyltransferases (HATs; or lysine acetyltransferases [KATs]) and histone deacetylases (HDACs). In Saccharomyces cerevisiae, there are two main HATs: Esa1 and Gcn5. Esa1 (TIP60/KAT5 in humans) is the only essential HAT in yeast and is part of the NuA4 complex (Allard et al. 1999), whereas Gcn5 is a nonessential HAT that belongs to the SAGA complex (Grant et al. 1997). While Esa1 is mainly responsible for acetylation of histones H4 and H2A (Boudreault et al. 2003; Xu et al. 2016), Gcn5 acetylates almost exclusively histone H3 (Suka et al. 2001). Early studies measuring steady-state mRNA levels in esa1 temperature-sensitive mutant strains have hinted at a specific role at ribosomal protein genes (RPGs) (Reid et al. 2000; Rohde and Cardenas 2003) or pointed to more global effects on transcription (Durant and Pugh 2007).
On the other hand, Gcn5 has been reported to play an important role in transcription of only ∼10% of yeast genes, the so-called “SAGA-dominated genes” (Huisinga and Pugh 2004). These genes are primarily stress-induced and contain a consensus TATA-box sequence in their promoters, in contrast to the “TFIID-dominated genes,” which lack a well-defined TATA box (Huisinga and Pugh 2004; Bhaumik 2011). Nevertheless, even in these early studies, double-mutant analysis indicated that most genes use both TFIID and SAGA. This view has been reinforced by recent studies using more direct measures of transcription that implicate both Gcn5 and SAGA in the activation of nearly all yeast genes (Bonnet et al. 2014; Baptista et al. 2017), consistent with the observation that SAGA, similar to NuA4, binds to the upstream activating sequences (UASs) of most yeast genes (Ohtsuki et al. 2010; Bonnet et al. 2014; Kuang et al. 2014; Baptista et al. 2017).
Both SAGA and NuA4 are large complexes organized in at least two main modules: a HAT module that contains the catalytic activity (Gcn5 and Esa1, respectively) and a recruitment module responsible for binding of the complex to at least some promoters (Chittuluru et al. 2011; Lee et al. 2011). Interestingly, the recruitment modules of SAGA and NuA4 contain a single common subunit: the essential protein Tra1 (TRRAP in mammalian cells) (Brown et al. 2001). However, both Gcn5 and Esa1 can bind promoters in the absence of the recruitment module in two different complexes: the ADA complex for Gcn5 and the Piccolo–NuA4 (picNuA4) complex for Esa1 (Grant et al. 1997; Boudreault et al. 2003).
Mediator is an additional highly conserved large multisubunit coactivator complex proposed to play a genome-wide role in stabilizing the preinitiation complex (PIC) by mediating interactions between general transcription factors (GTFs) and the RNA polymerase II (RNAPII) holoenzyme (Allen and Taatjes 2015; Soutourina 2018). Conditional inactivation or degradation of essential Mediator subunits (Esnault et al. 2008; Ansari et al. 2009; Plaschka et al. 2015; Warfield et al. 2017) or weakening of a Mediator interaction with the Rpb3 subunit of RNAPII (Soutourina et al. 2011) has widespread effects on transcription in growing cells. In addition, several studies reported a role for Mediator together with SAGA in activation of inducible genes (Bhoite et al. 2001; Bryant and Ptashne 2003; Govind et al. 2005; Leroy et al. 2006). The possibility that promoter histone acetylation influences Mediator binding has not been addressed, although in vitro studies suggest that Mediator binding could be partially dependent on histone acetylation (Zhu et al. 2011; Liu and Myers 2012).
Here we investigated the importance of histone acetylation for transcription in optimal growth conditions and for regulation following oxidative stress in the budding yeast S. cerevisiae. By carrying out RNAPII ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) analysis in cells in which either Esa1 or Gcn5 was depleted from the nucleus using the anchor-away technique (Haruki et al. 2008), we revealed that Esa1 and Gcn5 have a positive but nonessential role in transcription at individual genes. We also investigated the role of Mediator by rapid depletion of Med17, an essential subunit of the head domain. We found that Med17 anchoring leads to a partial loss of function that reveals a crucial role for Mediator—or at least its head domain—at a small set of ∼200 genes whose promoters are characterized by a strong TATA-binding protein (TBP) ChIP signal, weak binding of TBP-associated factors (TAFs; forming TFIID), and little or no sensitivity to Esa1 depletion. Taking advantage of the chromatin endogenous cleavage (ChEC) technique (Schmid et al. 2004) combined with high-throughput sequencing (ChEC-seq) (Zentner et al. 2015), we found that Mediator binding genome-wide is enhanced by histone acetylation. Finally, we showed that two classes of growth-promoting “housekeeping” genes—the ribosome biogenesis (RiBi) genes and RPGs—display different sensitivities to variations in histone acetylation, with the RiBi genes being predominantly dependent on Esa1 and the RPGs being equally dependent on both Esa1 and Gcn5.
Results
HATs contribute differently, but neither is strictly required for transcription in growing cells
In order to investigate the role of the HATs Esa1 and Gcn5 in transcription, we first rapidly depleted the proteins from the nucleus in exponentially growing cells using the anchor-away technique (Haruki et al. 2008) by treating the cells with rapamycin for 60 min and then measured both histone acetylation and RNAPII binding genome-wide by ChIP-seq (Fig. 1A). As a rough gauge of nuclear depletion efficiency, we measured cell growth of the anchor-away strains in solid medium containing rapamycin (Supplemental Fig. S1A). As expected, rapamycin prevented colony formation of the Esa1-FRB strain and caused a slow-growth phenotype of the Gcn5-FRB strain similar to that of a GCN5 deletion. A more direct measure indicates that Esa1 depletion was indeed efficient, since by ChIP we observed a massive genome-wide decrease in histone H4 acetylation (H4ac), with 5038 out of 5043 genes showing a decrease in promoter histone H4ac of >1.5-fold and an average decrease of 8.4-fold (Fig. 1B, left panel). Furthermore, the effectiveness of Esa1 depletion was confirmed by the strong decrease in the recruitment of Epl1 (another subunit of NuA4), as measured by ChIP-qPCR (ChIP combined with quantitative PCR) (Supplemental Fig. S1B). At the same time, depletion of Esa1 resulted in only minor changes in nucleosome occupancy, as measured by histone H3 ChIP-seq (Supplemental Fig. S1C,D, left panel), or in nucleosome positioning, as measured by micrococcal nuclease (MNase) digestion followed by high-throughput sequencing (MNase-seq) (Supplemental Fig. S1E). Although Esa1 acts directly on H4 and H2A, we also observed a significant although weaker decrease in H3ac upon Esa1 depletion (Supplemental Fig. S1C,D, right panel). We note here that our determination of changes in histone acetylation, histone occupancy, and RNAPII binding all relied on normalization to a “spiked-in” control (Schizosaccharomyces pombe chromatin), which others have shown to be crucial to precisely quantify genome-wide changes in ChIP-seq experiments (Bonhoure et al. 2014; Chen et al. 2015; Hu et al. 2015). Finally, RNAPII ChIP-seq was performed using an antibody that specifically recognizes the Ser5 phosphorlyated residue on the Rpb1 C-terminal domain (CTD), a modification of RNAPII associated with active transcription (Harlen and Churchman 2017).
Figure 1.

(A) Genome browser tracks showing a region of chromosome XII for H4ac and RNAPII ChIP-seq in Esa1 nuclear-depleted and nondepleted cells and H3K9ac and RNAPII ChIP-seq in Gcn5 nuclear-depleted and nondepleted cells. Gene annotations are shown below the tracks. (B) Scatter plots showing H4ac (left panel) and RNAPII (right panel) ChIP-seq in Esa1 nuclear-depleted cells (Esa1−; Y-axis) versus nondepleted cells (Esa1+; X-axis). Each dot represents a gene (5038 genes in total); the color of the dots represents the density of the points (from more dense [red] to less dense [blue]). The signal for H4ac was normalized to the H3 signal. The average signal for H4ac and H3 ChIP was quantified in a window of 500 base pairs (bp) centered on the transcription start site (TSS). For RNAPII, the average signal was quantified from the TSS to the transcription termination site (TTS). Blue and red lines on the plots represent the threshold of 1.5-fold change. The number next to the blue and red arrows indicates the number of genes above the threshold. (Blue arrow) Negatively affected genes; (red arrow) positively affected genes. The scale for both the X-axis and the Y-axis is log10. (C) Scatter plot comparing the change in H4ac (normalized to H3; X-axis) and the change in RNAPII (Y-axis), calculated as log2 ratio between the signal in Esa1 nuclear-depleted cells and nondepleted cells (Esa1−/Esa1+). Pearson correlation coefficient and P-value are shown. (D) Scatter plots showing H3K9ac (left panel) and RNAPII (right panel) ChIP-seq in Gcn5 nuclear-depleted cells versus nondepleted cells, calculated and plotted as in B. (E) Box plots showing RNAPII change in Esa1 (left) and Gcn5 (right) nuclear-depleted cells (calculated as log2 ratio of nuclear-depleted vs. nondepleted cells) for 4682 genes categorized in TFIID-dominated and SAGA-dominated according to Huisinga and Pugh (2004).
In contrast to the greater than eightfold average decrease in H4ac at promoters observed following Esa1 depletion, we observed a more modest decrease in RNAPII occupancy, with only slightly more than half of all genes (2736) down-regulated by >1.5-fold (Fig. 1B, right panel). Importantly, though, there is a significant correlation (R = 0.64; P-value < 0.001, Mann-Whitney rank sum test) between the effect on H4ac and the effect on RNAPII levels induced by Esa1 nuclear depletion (Fig. 1C), suggesting that Esa1's effect on transcription is dependent on its HAT activity.
In examining the genes most strongly affected by Esa1 depletion in terms of loss of H4ac and RNAPII binding, we noticed that many belong to the large suite of RiBi genes (Supplemental Table S2). Indeed, the 232 annotated RiBi genes (Jorgensen et al. 2004) displayed a highly significant correlation between the loss of H4ac and RNAPII binding (Supplemental Fig. S1F, left panel). In contrast, the RPGs, although showing a strong but heterogeneous decrease in H4ac, displayed only a modest decrease in RNAPII levels that was not as strongly correlated with the loss of H4ac (Supplemental Fig. S1F, right panel). This latter finding suggests that RPGs, unlike the RiBi genes, might achieve full expression through a more complex pattern of histone modifications.
Gcn5 nuclear depletion also resulted in a very significant genome-wide decrease in histone acetylation at promoter regions (in this case, measuring the K9 position of histone H3), consistent with previous findings in a gcn5Δ strain (Bonnet et al. 2014). Nearly all genes (4968 out of 5043) were affected at least 1.5-fold, with an average decrease of threefold genome-wide (Fig. 1D, left panel; Supplemental Fig. S1G). Consistent with effective Gcn5 depletion, binding of the SAGA subunit Ada2 was also strongly reduced (Supplemental Fig. S1H). As was the case for Esa1, Gcn5 depletion does not affect either H3 occupancy or nucleosome positioning (Supplemental Fig. S1G,I [left panel], J). Furthermore, Gcn5 acetylation is highly specific for histone H3, since only very few genes showed a >1.5-fold change in H4ac levels in Gcn5-depleted cells (Supplemental Fig. S1I, right panel). This observation indicates that the recruitment and activity of Esa1 operates independently of Gcn5-mediated H3K9 acetylation (H3K9ac).
In contrast to Esa1, we found that Gcn5 depletion results in a relatively modest decrease of RNAPII binding, albeit at a large number of genes, consistent with its global effect on H3K9ac (Bonnet et al. 2014) and a recent report that measured transcription in gcn5Δ cells (Baptista et al. 2017). Thus, although we observed a genome-wide decrease in RNAPII binding upon Gcn5 nuclear depletion, only 525 genes displayed a >1.5-fold decrease (Fig. 1D, right panel; Supplemental Table S3). Nevertheless, the correlation between the changes in histone acetylation and RNAPII levels in Gcn5-depleted cells is highly significant (R = 0.48; P-value < 0.001 in a Mann-Whitney rank sum test) (Supplemental Fig. S1K), as was the case for Esa1 depletion. Interestingly, we observed that while Esa1's primary targets are mainly TATA-less TFIID-dominated genes (Huisinga and Pugh 2004), Gcn5-affected genes are equally distributed between previously defined TFIID- and SAGA-dominated genes (Fig. 1E; Supplemental Fig. S1L), clearly indicating that Gcn5 does not exclusively function at the so-called SAGA-dominated genes even though these genes are indeed overrepresented among the group of 525 most strongly Gcn5-dependent (P = 8.1 × 10−11 for SAGA-dominated vs. P = 1 for TFIID-dominated genes) (Supplemental Fig. S1M).
Esa1 and Gcn5 enhance the transcriptional response to oxidative stress
To investigate possible roles for Esa1- or Gcn5-mediated histone acetylation in gene regulation, we decided to analyze the effect of Esa1 and Gcn5 depletion on the transcriptional response to oxidative stress induced by diamide. Diamide treatment has been shown to result in a rapid and dramatic reorganization of the yeast transcriptome, including activation of many general stress response genes and pervasive down-regulation of many growth-promoting genes (Gasch et al. 2000; Weiner et al. 2012, 2015).
To this end, we depleted Esa1 and Gcn5 by anchor-away in exponentially growing cells (60 min of rapamycin treatment) and then treated the cells with diamide before harvesting them (5, 20, and 60 min after diamide addition) for RNAPII ChIP-seq analysis (Fig. 2A). As expected, diamide treatment induced rapid changes in transcription, with many genes up-regulated (1210) or down-regulated (746) by >1.5-fold only 5 min following treatment (Supplemental Fig. S2A, left panel). The maximum response to diamide was observed after 20 min (1348 genes up-regulated and 984 genes down-regulated), whereas, after 60 min, the number of affected genes was reduced (Supplemental Fig. S2A, middle and right panels). Importantly, the RNAPII changes provoked by diamide were nearly identical (R = 0.98 at 20 min) in the two different anchor-away strains not treated with rapamycin, suggesting that the FRB-tagged proteins themselves do not significantly affect the response (Supplemental Fig. S2B).
Figure 2.

(A) Experimental outline. Yeast cells were treated with 1 µg/mL rapamycin to induce Esa1 or Gcn5 nuclear depletion. After 60 min of rapamycin (or vehicle control) treatment, 1.5 mM diamide was added to the cultures, and cells were harvested for cross-linking and RNAPII ChIP-seq at the indicated time points. (B) Plot showing average RNAPII signal (log2) normalized to time 0 (time 0 = 0) separately for Esa1 nuclear-depleted (Esa1−) and nondepleted (Esa1+) cells for the diamide-up-regulated genes (calculated as up-regulated genes at 20 min following diamide addition in the Esa1-FRB-tagged strain untreated with rapamycin, as in Supplemental Fig. S2A) in Esa1 nuclear-depleted (Esa1−; dashed line) and nondepleted (Esa1+; continuous line) cells. (C) Heat map representing the result of a k-means clustering analysis on the change in RNAPII occupancy (average signal from the TSS to the TTS) on the 1346 diamide-up-regulated genes, calculated as log2 ratio of the RNAPII signal in Esa1 nuclear-depleted (Esa1−) versus nondepleted (Esa1+) cells. (D) Plots showing average RNAPII signal for the gene groups identified in C and plotted as in B. (E) Bar plots showing the percentage of genes defined as SAGA-dominated (light brown) and TFIID-dominated (dark brown) as reported in Huisinga and Pugh (2004) for each of the five groups identified in C. (F) Plots showing average RNAPII signal (log2) for the diamide-down-regulated genes (calculated as down-regulated genes at 20 min following diamide addition in the Esa1-FRB-tagged strain not treated with rapamycin), plotted as in B. (G) Plots showing average RNAPII signal (log2) for the diamide-up-regulated genes (calculated as up-regulated genes at 20 min following diamide addition in the Gcn5-FRB-tagged strain not treated with rapamycin) in Gcn5 nuclear-depleted (Gcn5−; dashed line) and nondepleted (Gcn5+; continuous line) cells, plotted as in B. (H) Same as in G for the diamide-down-regulated genes (calculated as down-regulated genes at 20 min following diamide addition in the Gcn5-FRB-tagged strain not treated with rapamycin).
To quantify the effect of Esa1 and Gcn5 depletion on the diamide stress response, we considered only those genes whose expression level changed by >1.5-fold after 20 min of diamide treatment (both up-regulated and down-regulated). Furthermore, since both Esa1 and Gcn5 depletion led to reduced transcription of many genes prior to diamide addition (Fig. 1), we analyzed the kinetics of the diamide response by normalizing each time point to the t = 0 value. The diamide response in Esa1-depleted cells was overall quite similar to that observed in wild-type cells, with many genes being up-regulated and down-regulated, although to a different extent than in wild-type cells (Fig. 2B). Because this analysis groups together >1000 induced genes, we performed a k-means clustering analysis on the RNAPII occupancy kinetics following diamide treatment. This revealed five groups of genes that differ in their magnitude and kinetics of response to Esa1 depletion as well as their gene ontology (GO) term enrichments (Fig. 2C; Supplemental Table S4). Although some sets of genes (clusters IV and V, enriched in genes implicated in organic substrate catabolic processes and protein complex assembly, respectively) showed a stronger dependence on Esa1 for their up-regulation, we were unable to identify any subset of genes that was not up-regulated in the absence of Esa1 (Fig. 2D). Notably, clusters IV and V are enriched in TFIID-dominated genes, consistent with the observation (Fig. 1E, left panel) that Esa1 is more important for transcription regulation of these genes as opposed to SAGA-dominated genes (Fig. 2E). As expected, diamide treatment lead to down-regulation of many genes, including growth-promoting genes such as RPGs and RiBi genes. Genes down-regulated by diamide stress were slightly more affected in Esa1-depleted cells, with small differences observed between different subsets of growth-promoting genes (Fig. 2F; Supplemental Fig. S2C,D; Supplemental Table S5).
Compared with Esa1, Gcn5 nuclear depletion had a minor effect on gene up-regulation in response to diamide, as the kinetics and magnitude of gene activation in the absence of Gcn5 were almost identical to wild type (Fig. 2G). Contrary to Esa1 depletion, the effect of Gcn5 depletion was strongest on repressed genes, whose down-regulation following Gcn5 depletion was apparently dampened (Fig. 2H). We caution, however, that we cannot rule out a role for Gcn5 in transcriptional elongation at these genes that masks the full extent of the initiation decrease at these genes. While k-means clustering identified subsets of genes with different sensitivities to nuclear depletion of Gcn5 for both diamide-up-regulated (Supplemental Fig. S2E) and diamide-down-regulated (Supplemental Fig. S2G) genes, the differences in kinetics and the magnitude of the effects were relatively subtle (Supplemental Fig. S2F,H; Supplemental Tables S6,S7).
Altogether, these results show that gene activation can occur under conditions of reduced histone acetylation, suggesting that histone acetylation has a positive but likely not essential role in gene transcription. Furthermore, analyzing the effects of Gcn5 nuclear depletion during diamide response revealed that Gcn5's role in transcription is not always positive and suggests that it might vary depending on specific promoter features and/or environmental conditions.
Role of the Nua4–SAGA shared subunit Tra1 and evidence for HAT redundancy in transcription
Given the relatively modest effect of either Esa1 or Gcn5 depletion on RNAPII association, we speculated that these two HATs might have a partially redundant role in transcription. Testing this hypothesis by simultaneous anchoring of both proteins proved to be problematic, since a strain in which the two proteins were FRB-tagged displayed a strong growth defect even in the absence of rapamycin. We thus chose to deplete the only shared subunit of SAGA and NuA4 complexes: the essential protein Tra1 (Supplemental Fig. S3A). To verify that Tra1 nuclear depletion impairs both SAGA and NuA4 function, we measured H3K9ac and H4ac levels. As expected, Tra1 depletion led to a significant decrease in both H3K9ac and H4ac (3459 and 5038 genes affected >1.5-fold, respectively) with essentially no effect on H3 occupancy (Fig. 3A, left and middle panels; Supplemental Fig. S3B,C). Although these effects are qualitatively similar to the sum of those observed for Gcn5 and Esa1 anchoring, their magnitude is reduced (Supplemental Fig. S3D,E). This might be explained by two nonexclusive possibilities: (1) a decreased efficiency of Tra1 anchoring compared with that of either HAT or (2) delivery of HAT activity in the absence of Tra1 targeting in the case of Gcn5 through the ADA subcomplex (Grant et al. 1997) and for Esa1 through picNuA4 (see below; Boudreault et al. 2003). Importantly, ChIP-qPCR of Esa1 and Gcn5 in Tra1 nuclear-depleted cells revealed a clear decrease in Esa1 binding to two RPG promoters, while the effect on Gcn5 binding was much more modest (Supplemental Fig. S3F), suggesting that Gcn5 can still associate at least to some promoters in a Tra1-independent fashion.
Figure 3.
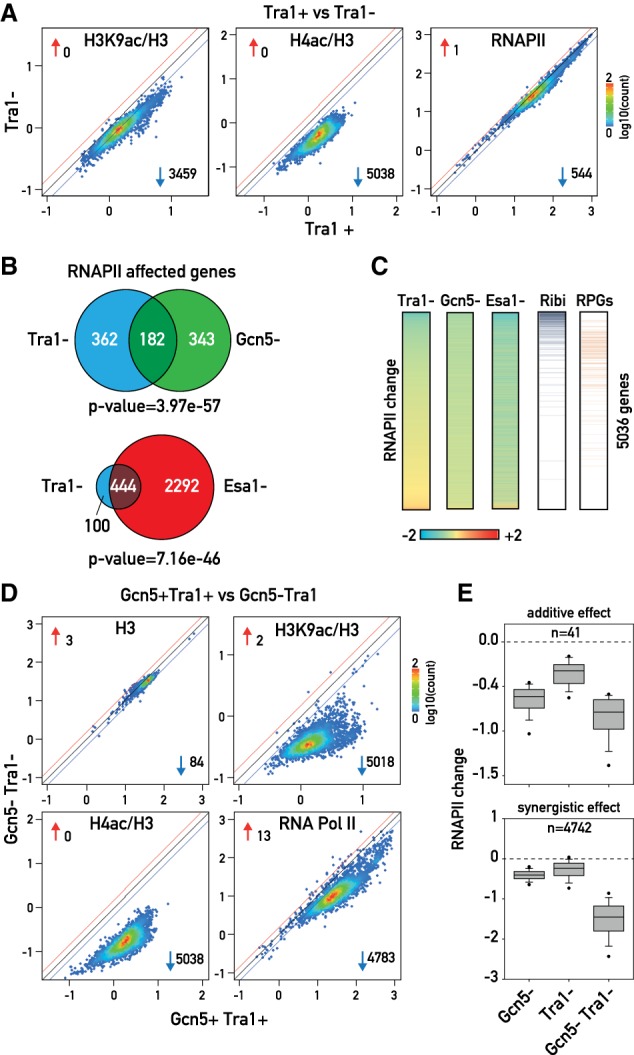
(A) Scatter plots showing H3K9ac (left), H4ac (middle), and RNAPII (right) ChIP-seq in Tra1 nuclear-depleted versus nondepleted cells, calculated and plotted as in Figure 1B. (B) Venn diagram depicting the overlap between genes affected at least 1.5-fold in RNAPII occupancy by Tra1 nuclear depletion and Gcn5 nuclear depletion (top) and by Tra1 nuclear depletion and Esa1 nuclear depletion (bottom). (C) Heat maps showing 5037 genes (rows) sorted according to RNAPII change in Tra1 nuclear-depleted versus nondepleted cells. (Blue) RiBi genes; (orange) RPGs. (D) Scatter plots showing H3 (top left), H3K9ac (top right), H4ac (bottom left), and RNAPII (bottom right) ChIP-seq in Tra1 and Gcn5 nuclear-depleted cells, calculated and plotted as in Figure 1B. (E) Box plots showing RNAPII change in Gcn5, Tra1, and Gcn5 and Tra1 nuclear-depleted cells (calculated as log2 ratio of nuclear-depleted vs. nondepleted cells) for 4783 genes, divided according to the effect of Gcn5 and Tra1 double depletion in “additive effect” (41 genes; top panel) and “synergistic effect” (4742 genes; bottom panel).
Consistent with the more modest decrease in histone acetylation caused by Tra1 nuclear depletion, we observed a concomitant decrease in RNAPII recruitment at fewer genes than would be expected from a simple superposition of Esa1 and Gcn5 depletion effects (544 decreased >1.5-fold) (Fig. 3A, right panel). Thus, although the overlap between Tra1-affected genes and those affected by either Gcn5 or Esa1 depletion is highly significant, many HAT target genes were not identified as Tra1 targets at the >1.5-fold cutoff (Fig. 3B). Nevertheless, the decrease in RNAPII binding seen in Tra1-depleted cells correlated well with that observed in either Esa1-depleted (R = 0.48) or Gcn5-depleted (R = 0.53) cells (Supplemental Fig. S3G), as expected.
Considering those genes whose RNAPII binding was affected by Tra1 depletion above the 1.5-fold threshold, we found that 82% of them were also Esa1 targets, whereas only 33% were similarly affected by Gcn5 depletion (Fig. 3B). Thus, the genes whose RNAPII recruitment is most affected by Tra1 nuclear depletion are the genes that are most sensitive to a decrease in Esa1-driven H4ac (Fig. 3C) either because Tra1 depletion more efficiently removes NuA4 compared with SAGA or because the ADA subcomplex promoter binding is more prevalent than that of Piccolo-NuA4. GO analysis of these genes revealed a significant enrichment for RiBi genes, genes involved in RNA metabolism and processing, and, last, RPGs (Supplemental Table S8).
Our finding that Tra1 depletion appeared to more fully capture the effect of Esa1 depletion compared with that of Gcn5 suggests that a significant amount of Gcn5 may have remained nuclear under these conditions, actively acetylating promoter nucleosomes in the context of either the full SAGA complex or the ADA subcomplex. We thus performed a Tra1–Gcn5 double-depletion experiment to better address the issue of HAT redundancy. This resulted in a similarly massive loss of H4ac compared with Esa1 depletion and a markedly more robust decrease in H3K9ac than that observed for Tra1 depletion alone (4.4-fold average decrease compared with three- and 1.8-fold for Gcn5 or Tra1 depletion alone, respectively) (Fig. 3D). What was particularly striking was the transcriptional effect of this double depletion (Fig. 3D,E), where we observed a >1.5-fold decrease at 4783 genes and an average fold decrease of 3.1-fold. Significantly, the double-depletion effect was more than additive at the vast majority of genes (4742 of 4783) (Fig. 3E), indicative of a synergistic effect of H4ac and H3K9ac on transcription genome-wide.
Requirement for the Mediator head module is most pronounced at a set of genes with high TBP binding
We next turned our attention to Mediator and performed anchor-away nuclear depletion of an essential subunit of the head module, Med17, whose loss is expected to destabilize the entire head module (Supplemental Fig. S4A; Takagi et al. 2006). Genome-wide RNAPII ChIP-seq following 1 h of Med17 depletion revealed a widespread drop in RNAPII binding but with only 315 genes exceeding a >1.5-fold decrease (Fig. 4A; Supplemental Fig. S4B). In line with this relatively mild effect, we found that Med17 depletion had only subtle effects on apparent promoter nucleosome occupancy and position, as determined by MNase mapping (Supplemental Fig. S4C). These results were surprising given previous reports showing that essentially all RNAPII transcription is strongly reduced in a med17-ts mutant at the nonpermissive temperature (Holstege et al. 1998; Paul et al. 2015; Plaschka et al. 2015). Although Med8 binding (as measured by Med8-MNase ChEC-seq) was strongly reduced in the Med17 anchor-away strain following rapamycin addition, indicating that efficient nuclear depletion of at least the head module of Mediator had been achieved (Supplemental Fig. S4D,E), we cannot rule out the possibility that small amounts of Mediator remain in the nucleus and are sufficient to drive normal levels of transcription at most genes. Consistent with this idea, Mediator appears not to be limiting for most transcription (Grunberg et al. 2016). In any event, we conclude that Med17 anchoring leads to a partial loss-of-function (hypomorphic) phenotype that provides a unique opportunity to identify those genes whose expression is most strongly dependent on Mediator (see the Discussion).
Figure 4.

(A) Scatter plots showing RNAPII ChIP-seq in Med17 nuclear-depleted cells, calculated and plotted as in Figure 1B. (B) Box plots showing RNAPII change in Med17 nuclear-depleted cells for 3992 genes, calculated and plotted as in Figure 1E and categorized according to the presence or absence of the TATA box and the promoter binding of TAF1 as reported in Rhee and Pugh (2012). TATA+ TAF1− group, n = 390; TATA+ TAF1+ group, n = 307; TATA− TAF1− group, n = 503; TATA− TAF1+ group, n = 2790. (C) Heat maps showing TBP occupancy (Kubik et al. 2018) (right) and Med8 ChEC signal (left) of 5036 yeast genes aligned to the TSS and sorted according to the change in RNAPII occupancy in Med17 nuclear-depleted cells. (D) Genome browser tracks showing a region of chromosome II for Med8 ChEC-seq in Esa1 and Gcn5 nuclear-depleted and nondepleted cells. (E) Scatter plots showing Med8 occupancy in Esa1 (left) or Gcn5 (right) nuclear-depleted cells (Y-axis) and nondepleted cells (X-axis) for the 2000 genes having the strongest Med8 ChEC signal in their UASs.
In accord with previous findings (Ansari et al. 2009), we observed that Med17 depletion modestly reduced RNAPII association at both RPGs and RiBi genes. In contrast, the most strongly affected genes were primarily members of the SAGA-dominated gene group (Supplemental Fig. S4F; Huisinga and Pugh 2004) that are characterized by a conserved TATA box and weak Taf1 binding (Fig. 4B; Rhee and Pugh 2012). These genes tend to be highly transcribed and are associated with metabolic functions, as revealed by GO analysis (Supplemental Table S9). Interestingly, these genes displayed the highest levels of both TBP (ChIP-seq) and Med8 (ChEC-seq) binding in genome-wide analyses (Fig. 4C).
Low histone acetylation impairs Mediator binding to UASs but not transcription
Since previous studies have pointed to general functions of Mediator and the major HATs Gcn5 and Esa1 in global transcription, we further explored possible functional relationships between these two classes of coactivators. To determine whether the recruitment of Mediator to UASs is dependent on histone acetylation, we used ChEC-seq to measure Med8 binding in cells depleted of either Esa1 or Gcn5 (Fig. 4D). For comparison of control and anchor-away conditions, we considered the 2000 UAS regions with the strongest Med8 ChEC-seq signal in order to avoid artifacts due to background variability at UASs with very low signal. Depletion of Esa1 led to an overall genome-wide decrease in Med8 binding to UASs (Fig. 4D [top panel], E [left panel]), suggesting that Esa1-mediated histone acetylation generally promotes Mediator association with the genome. Interestingly, there was no correlation between the decrease in Med8 binding to the UAS and the decrease in RNAPII binding on the gene body (Pearson R = 0.096) (Supplemental Fig. S4G, left panel), consistent with previous findings suggesting a broad uncoupling between Mediator UAS occupancy and transcription level (Grunberg et al. 2016). A similar global decrease, although to a lesser magnitude, was observed with Gcn5 anchor-away (Fig. 4E, right panel). As for Esa1 depletion, there was no correlation between the reduction of Med8 at UASs and RNAPII binding (Pearson R = 0.028) (Supplemental Fig. S4G, right panel). Our results show that Mediator recruitment is generally stimulated by histone acetylation.
Coactivator usage defines five specific promoter classes
To obtain a more comprehensive genome-wide picture of the genes regulated by the coactivators that we examined (Esa1, Gcn5, Tra1, and Med17), k-means clustering analysis of the effect of their depletion on RNAPII binding was performed. Strikingly, this analysis revealed five distinct clusters of genes (Fig. 5A; Supplemental Fig. S5A,B) whose unique features are discussed in more detail below. One feature that distinguishes the clusters, based on which we ordered them, is expression level. Clusters 1 and 2, which comprise more than half of all yeast genes, display the lowest average expression. Clusters 3 and 4 show moderate to high expression, whereas cluster 5 contains only 246 predominantly highly expressed genes (Fig. 5B; Supplemental Fig. S5C).
Figure 5.

(A) Heat map representing the result of a k-means clustering analysis on the change in RNAPII occupancy measured in Esa1, Gcn5, Tra1, and Med17 nuclear-depleted cells. (B) Box plots showing the transcription rate measured by nascent elongating transcript sequencing (NET-seq) (Churchman and Weissman 2011) for the genes in the five clusters described in A. (C,D) Bar plots showing the number of Ribi genes (C) and RPGs (D) in each of the five clusters. (E) Box plot showing TBP promoter occupancy for the genes in the five clusters (Kubik et al. 2018). (F) Bar plots showing the percentage of genes having a well-defined TATA box in their promoter (green) and significant TAF1 binding measured by ChIP (purple) as reported in Rhee and Pugh (2012) for each of the five clusters.
Cluster 1 genes (709 in total) are characterized by a relative lack of response to depletion of Esa1, Tra1, or Med17. However, like all genes, cluster 1 genes are modestly but uniformly affected by Gcn5 depletion. GO analysis showed that this group is enriched in genes involved in stress-related processes (Supplemental Table S10), consistent with promoter enrichment for the DNA-binding motifs of two stress-related TFs: Msn2 and Msn4 (P-value 2.2 × 10−12). This may explain their low transcription levels in standard growth conditions (measured by nascent elongating transcript sequencing [NET-seq] [Churchman and Weissman 2011] or RNAPII ChIP-seq [Fig. 5B; Supplemental Fig. S5C]). Cluster 2 genes (2407 in total) represent almost half of the RNAPII transcriptome and differ from cluster 1 genes by a much stronger dependence on Esa1 and a—probably related—slight increase in Tra1 dependence. Cluster 3 genes (712 in total) are the genes most strongly affected by both Esa1 and Tra1 depletion. As pointed out above, this cluster is highly enriched in genes involved in ribosome biogenesis (190 out of 232 RiBi genes are in cluster 3; P-value 1.25 × 10−83) (Fig. 5C) and RNA-related processes (P-value 1.72 × 10−93) (Supplemental Table S10). RiBi gene promoters are characterized by the presence of two well-conserved motifs named PAC and RRPE (Hughes et al. 2000; Wade et al. 2001). Not surprisingly, cluster 3 gene promoters are enriched for both motifs (Supplemental Fig. S5D). Cluster 4 genes (962) are characterized by a very similar response to depletion of all four coactivators. These genes are predominantly classified as TFIID-dominated (Supplemental Fig. S5E) and are moderately to highly transcribed (Fig. 5B; Supplemental Fig. S5C). The cluster is enriched in the so-called “growth-promoting” genes, specifically those involved in “cytoplasmic translation” (Supplemental Table S10), and RPGs are part of this cluster (132 out of 137 RPGs) (Fig. 5D). Finally, cluster 5 is comprised of a small set of 246 genes that are strongly affected by Med17 depletion. Notably, like all genes, this subset of genes responds modestly to Gcn5 depletion but not to either Esa1 or Tra1 depletion. Interestingly, expression of cluster 5 genes, like that of those in cluster 1, shows little dependence on Esa1 despite the fact that, in both cases, H4ac is decreased when Esa1 is depleted (Supplemental Fig. S5F,G). Cluster 5 genes are highly transcribed (Fig. 5B; Supplemental Fig. S5C) and are characterized by strong promoter TBP binding and a well-defined TATA box (Fig. 5E,F). Importantly, this cluster of genes is classified as being Taf1-depleted (Fig. 5F; Huisinga and Pugh 2004; Rhee and Pugh 2012). This suggests that when Mediator is essential for transcription, HAT activity at the promoter—particularly that of Esa1—plays little or no role in expression. Notably, genes in cluster 5 displayed the highest average Med8 enrichment at the UASs (Supplemental Fig. S5H), consistent with previous findings (Grunberg et al. 2016). The UASs of genes associated with clusters 1 and 4 displayed moderate to average Med8 signal, while this signal was notably lower at cluster 2 and 3 genes (Supplemental Fig. S5H).
Overall, this analysis highlights some important features of the yeast RNAPII transcriptome: (1) Gcn5 is important for transcription of virtually all genes, (2) Esa1 controls transcription of the majority of genes but to a more variable extent than does Gcn5, and (3) Mediator is particularly critical for transcription of a well-defined and small group of genes whose expression is largely independent of Esa1.
Discussion
The SAGA HAT Gcn5 plays a genome-wide role in transcription
Contrary to expectation when we began this work, we found that Gcn5 nuclear depletion leads to decreased RNAPII association at most yeast genes, albeit to a relatively modest extent, with only ∼500 genes showing a reduction >30%. Our findings are consistent with a recent report (Baptista et al. 2017) in which comparative dynamic transcriptome analysis (Sun et al. 2012) was used to measure transcription in a gcn5Δ strain and suggest that Gcn5 has a more general but modest role in transcription than previously thought. It is worth noting that Baptista et al. (2017) observed stronger effects in other SAGA deletion mutants (spt7Δ and spt20Δ) and upon anchor-away of Spt7. This difference might stem from the fact that these genetic perturbations lead to a complete disassembly of the SAGA complex (Lee et al. 2011), whereas Gcn5 anchor-away does not (Baptista et al. 2017), thus permitting promoter targeting, deubiquitinylation, and TBP-interacting functions of SAGA to operate. We found that although Gcn5 depletion resulted in a comparable decrease of RNAPII association at TFIID-dominated and SAGA-dominated genes, those genes most strongly affected (i.e., >1.5-fold decreased) were in fact biased toward the SAGA-dominated class, consistent with the early findings of Pugh and colleagues (Basehoar et al. 2004; Huisinga and Pugh 2004).
Interestingly, we noted an apparently repressive effect of Gcn5 at genes that are down-regulated by diamide treatment (Fig. 2H). At present, we cannot rule out the possibility that this reflects a positive role of Gcn5 in transcriptional elongation that masks a decrease in initiation rate rather than a genuine role for Gcn5 in repression of these genes upon stress. Indeed, Gcn5 has been implicated in RNAPII elongation (Govind et al. 2007). On the other hand, Gcn5 has also been shown to down-regulate transcription following glucose induction of RPGs through acetylation of a nonhistone target, the Ifh1 activator (Downey et al. 2013). Additional work will be required to decipher the precise role of Gcn5 during stress, which could be multifaceted.
Even though we observed a quantitatively greater effect of Gcn5 depletion on H3K9ac compared with depletion of Tra1, it is unclear whether this is due to more efficient depletion of Gcn5, action of Gcn5 in the absence of Tra1, or both. Future quantitative binding analysis of Gcn5 and unique ADA complex subunits following Tra1 depletion may resolve this issue. We note that Tra1, which is not essential in S. pombe, regulates only a subset of SAGA-dominated genes in this yeast (Helmlinger et al. 2011). It is thus tempting to speculate that, in budding yeast, Gcn5 may act at some genes in the absence of Tra1 targeting, presumably through the ADA complex.
Promoter type specificity of Esa1 action
Consistent with its requirement for viability, we found that Esa1 depletion has a considerably stronger effect on RNAPII association than does Gcn5 at a large number of genes. Furthermore, and unlike Gcn5, Esa1 has a relatively variable effect, with two groups of genes (clusters 1 and 5) displaying essentially little or no Esa1 dependence and three groups (clusters 2–4) displaying moderate to strong dependence. This distinction is correlated with a relatively high TATA-box frequency and low Taf1 binding in clusters 1 and 5 and the opposite in clusters 2–4. Taken together, these findings suggest that NuA4 action is associated with a TFIID mode of TBP recruitment and PIC assembly. This observation is consistent with the fact that the TFIID-associated proteins Bdf1 and Bdf2 (and the Taf1 subunit in humans) (Jacobson et al. 2000) contain two bromodomains, known to bind specific acetylated lysines on histone H4 (Matangkasombut et al. 2000). Conversely, at promoters where TFIID and Esa1 are less important, transcription levels are either low (cluster 1) or high and are dependent on the Mediator head module (cluster 5). In summary, our findings suggest the existence of two promoter types associated with low expression (NuA4-dependent and NuA4-independent) and three distinct strategies leading to moderate or high expression levels (Fig. 6).
Figure 6.
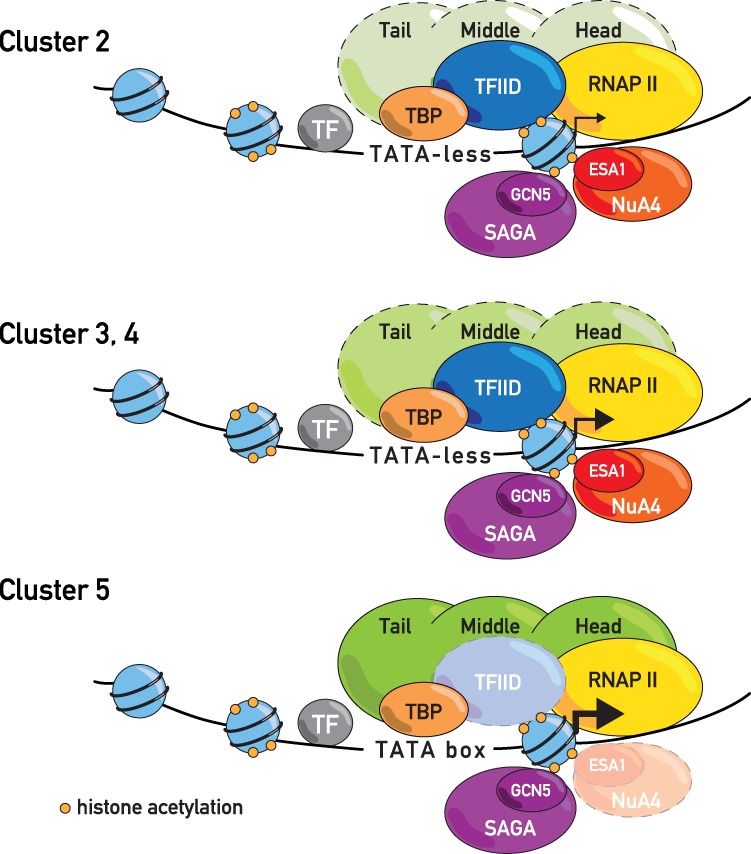
Schematic representation of three promoter configurations associated with moderate and high transcription. The intensity of the colors (Mediator, NuA4, and Esa1) reflects the importance of the coactivator in transcription. The size of the arrow next to the +1 nucleosome indicates the transcription level. The sizes and positions of the coactivators in the cartoon are illustrative. For simplicity, TFIID binding downstream from the TSS is not depicted.
What might distinguish the different NuA4-responsive clusters (2–4)? We note that cluster 2 is relatively more dependent on Esa1 than Tra1, suggesting that these genes may engage Esa1 in the absence of Tra1 (presumably through picNuA4) more frequently than genes in clusters 3 and 4. This may reflect more prevalent recruitment of NuA4 to promoters of genes in clusters 3 and 4 through specific TF interactions with Tra1, which is generally considered to be a NuA4 (or SAGA) recruitment module. Alternatively, or in addition, cluster 2 promoters may display features absent from cluster 3 and 4 promoters that favor the recruitment and/or action of picNuA4. To further evaluate these models and clarify the role of Esa1 in transcription, it may be useful to develop quantitative measures of Esa1 and picNuA4 promoter binding in both wild-type and Tra1-depleted cells.
Evidence for widespread HAT redundancy in transcription
Given the genome-wide role of Gcn5 reported here and elsewhere (Bonnet et al. 2014; Baptista et al. 2017) and the requirement of the NuA4 HAT Esa1 for viability, we were surprised to find that the significant reduction (average threefold) in histone acetylation provoked by anchoring of either acetylase had a quantitatively smaller effect on transcription genome-wide in growing cells either under steady-state conditions or during a transcriptional response to stress. Interestingly, Gcn5–Tra1 double depletion, which resulted in a stronger reduction in H3K9ac than was achieved by either single depletion, led to a more pronounced down-regulation of the vast majority of genes that was greater than an additive effect of the two single depletions. This finding indicates that transcription responds in a nonlinear fashion to a severe loss of histone acetylation. We suggest two nonexclusive explanations for this observation, the first of which is related to the long-standing in vitro finding that acetylation disfavors chromatin compaction and may thus promote a more open conformation that increases PIC assembly or subsequent steps in transcription initiation (Garcia-Ramirez et al. 1995; Tse et al. 1998; Shogren-Knaak et al. 2006). Alternatively, a minimum level of acetylation might be required to promote binding through acetyl lysine-interacting bromodomains that are common to several coactivators or coactivator complexes (Dhalluin et al. 1999).
Our data provide evidence for another general function of HATs that may not directly affect transcription. Using ChEC-seq, we show that acetylation of histones H4 by Esa1 and, to a lesser extent, H3 by Gcn5 enhances for Mediator recruitment to UASs in vivo. The function of widespread UAS binding of Mediator remains unclear, but previous studies (Grunberg et al. 2016; Soutourina 2018) strongly suggest that Mediator occupancy and expression levels are broadly uncoupled, with the exception of highly transcribed genes.
A small set of TATA-box-containing genes is strongly dependent on Mediator
Med17 is one of 10 essential subunits of Mediator. Transcriptomic analysis of a med17-ts strain revealed a strong genome-wide impairment of transcription at a nonpermissive temperature (Holstege et al. 1998; Paul et al. 2015; Plaschka et al. 2015), highlighting the importance of the Mediator head module for transcription of most genes. While measurements of additional med17-ts alleles yielded more heterogeneous results (Eyboulet et al. 2015), recent auxin-induced degradation of the critical Mediator subunit Med14 revealed that a functional Mediator complex is required for transcription of all genes (Warfield et al. 2017). Thus, given that Med17 is essential for viability, our finding that its nuclear depletion strongly reduced RNAPII binding at only a small set of highly transcribed TATA-box-containing genes with high levels of TBP binding was unexpected. However, data from a recent study (Petrenko et al. 2017) that also reported RNAPII ChIP-seq measurements following nuclear depletion of Med17 correlate well with our data (see Supplemental Fig. S6A,B). In agreement with our findings, Petrenko et al. (2017) show that SAGA-dominated genes are most strongly affected by Med17 anchoring. Furthermore, analysis of data for TFIIB binding in Med17-depleted cells (Jeronimo and Robert 2014) revealed a strong correlation with our RNAPII results (Supplemental Fig. S6A,B). Finally, depletion of another essential Mediator head subunit, Med22 (Srb6), leads to a transcriptional phenotype very similar to that of Med17 depletion (Supplemental Fig. S6C; Petrenko et al. 2017). Taken together, our data strongly suggest that Med17 depletion generates a Mediator hypomorph phenotype (partial loss of function) that allowed us to uncover those genes most strongly dependent on Mediator activity.
Variable deployment of coactivators at distinct gene groups drives PIC assembly and transcription
Our work provides striking evidence that despite their widespread roles in transcriptional activation, coactivators may exhibit different functions at specific gene groups, determining their expression levels under steady-state growth and their regulatory responses to stress.
For example, RPGs and RiBi genes, prominent members of two major gene clusters, differ in their requirement for NuA4 activity. Even though Esa1 is required for expression of >80% of genes, genes in cluster 3 are significantly more dependent on NuA4 compared with genes in cluster 4 despite comparable expression levels, TBP and Taf1 occupancy, and TATA-box content. This finding suggests the existence of an important and specific function for NuA4 at cluster 3 genes. We speculate that as yet unidentified cluster-specific UAS and promoter features other than simply the quality of the TATA sequence element may determine these distinct dependencies, which might be related to the acceleration of additional rate-limiting steps at these genes.
Another example of coactivator specificity is the strong sensitivity for Mediator that we observed for a small cluster of highly expressed TATA-box-containing genes. Interestingly, this group of genes is largely unaffected by Esa1 depletion and is highly overlapping with a group of genes recently shown to have a relatively low TFIID dependence (cluster 1 in Warfield et al. 2017). We also found significant overlap of the weakly TFIID-dependent genes identified by Warfield et al. (2017) with our cluster 1 genes, which are also essentially unaffected by Esa1 depletion (Supplemental Table S11). Thus, at promoters where TFIID and Esa1 are less important, transcription levels are either low (cluster 1) or high and strongly dependent on Mediator activity (cluster 5). In summary, our findings suggest the existence of two promoter types associated with low expression (one NuA4-independent [cluster 1] and one NuA4-dependent [cluster 2]) and three moderately to highly expressed promoter types that show distinct requirements for coactivator functions (clusters 3–5) (Fig. 6).
Recent data suggest that TFIID and SAGA have important functions for transcription initiation at all genes (Baptista et al. 2017; Warfield et al. 2017). Our findings extend this picture to other transcriptional coactivators (NuA4 and Mediator) and provide evidence for distinct roles of these coactivators at different gene groups (Fig. 6). We suggest two general mechanistic interpretations of these findings. One hypothesis is that at genes strongly Mediator-dependent but unresponsive to NuA4 (cluster 5), TBP deposition and PIC formation are preferentially facilitated by SAGA, which may require Mediator for its stable recruitment (Govind et al. 2005; Qiu et al. 2005). The particularly strong requirement for Mediator at these genes may reflect the fact that SAGA, unlike TFIID (Louder et al. 2016), does not independently bind promoter DNA. In contrast, we propose that TBP recruitment and PIC assembly at all other genes most often occur through a NuA4- and TFIID-facilitated pathway that operates independently of SAGA. An alternative scenario is that SAGA, Mediator, and NuA4-TFIID directly facilitate every PIC formation event at all promoters. In both interpretations, we imagine that a combination of gene-specific TFs, chromatin remodelers, histone mark “readers,” and other factors—probably determined by yet unknown cluster-specific UAS and/or promoter elements—determines which coactivator combinations most strongly influence the rate of PIC formation at the different promoter types that we identified. It is worth pointing out in this regard that mechanisms underlying the activation of the significant fraction of yeast genes whose expression is linked to growth rate (so-called “housekeeping” genes) are still poorly understood. A better understanding of the factors and promoter DNA signals that operate at these genes may be an important step in deciphering the rules governing coactivator usage that we describe here.
Materials and methods
Yeast strains
Yeast strains used in this study are listed in Supplemental Table S1.
Growth assay
Tenfold serial dilutions of log-phase growing cells (OD600 = 0.3) were spotted on plates containing complete medium with or without 1 µg/mL rapamycin. Plates were imaged after 24 and 48 h of growth at 30°C.
Anchor-away, diamide treatment, and cross-link
Anchor-away experiments were performed as described in Haruki et al. (2008). Log-phase growing cells (OD600 = 0.3–0.4) were treated with 1 µg/mL rapamyicin (resuspended in 90% ethanol, 10% Tween-20) for 60 min before collection unless indicated otherwise. For Gcn5 and Tra1 double anchor-way, cells were treated with rapamycin for 120 min. For diamide stress experiments, cells were treated with 1.5 mM diamide (from 1 M stock in TE buffer), collected at the indicated time points, and then cross-linked.
ChIP-qPCR and ChIP-seq
After cross-linking, ChIP-qPCR and ChIP-seq were performed as described in Ribaud et al. (2012) with the following modifications for ChIP-seq. A defined amount (5% of total chromatin) of cross-linked and sonicated chromatin from S. pombe was added to S. cerevisiae chromatin prior to the immunoprecipitation step. Following steps in the ChIP protocol were performed as described in Ribaud et al. (2012). ChIP was performed using the following antibodies: for histone H3 ChIP, Abcam ab1791; for histone H3K9ac, Millipore 07-352; for histone H4ac, Millipore 06-866; for RNAPII, Abcam ab5131; for Flag, Sigma F3165; and for Myc, a homemade antibody (clone 9E10). DNA libraries where prepared using TruSeq Illumina kit according to the manufacturer's instructions and sequenced in single-end mode on the Illumina HiSeq 2500 platform at the Institute of Genetics and Genomics of Geneva (iGE3; http://www.ige3.unige.ch/genomics-platform.php). For ChIP-qPCR, primer sequences are available on request.
ChIP-seq: mapping, spike control normalization, and data analysis
FASTQ files were mapped to the sacCer3 and the 972-h pombe genome assemblies using HTS Station (http://htsstation.epfl.ch) (David et al. 2014), and reads that mapped to both genomes were discarded. For each sample, reads mapped uniquely to the sacCer3 genome were first normalized to the total number of mapped reads in sacCer3 and then to the total number of uniquely mapped reads in 972-h pombe in the same sample as described in Hu et al. (2015). For RNAPII, the signal was quantified for each gene between the transcription start site (TSS) and transcription termination site (TTS). For H3, H3K9ac, and H4ac, the signal was quantified in a window of 500 base pairs (bp) centered on the TSS. A list of TSSs and TTSs for each gene was obtained from van Bakel et al. (2013). To compare depleted versus nondepleted cells, we divided the signal from the +rapamycin sample by the one from the −rapamycin sample and log2 transformed this value. All data from publicly available databases were mapped using HTS Station (http://htsstation.epfl.ch) (David et al. 2014).
Motif search
Motif search was performed using the DREME algorithm (Bailey 2011). For each cluster, promoter sequences (400-bp-long sequences upstream of the TSS) were used as input, and analogous sequences randomly selected among the other clusters were used as controls. Comparison of the identified motifs with binding sites for known DNA-binding proteins was performed with the TOM TOM algorithm (Gupta et al. 2007). The FIMO algorithm (Gupta et al. 2007) was used to determine the distribution of the PAC and RRPE motifs (Hughes et al. 2000; Wade et al. 2001) in the promoters (defined as the 400-bp-long sequence upstream of the TSS).
Heat maps and plots
The heat maps in Figure 4C and the scatter plots in Figure 1C and Supplemental Figures S1I; S2B; S3, D–F; S4G; and S6B were generated using EaSeq (http://easeq.net) (Lerdrup et al. 2016). In all of the box plots, the box shows the 25th–75th percentile, whiskers show the 10th–90th percentile, and dots show the 5th and 95th percentiles. R-values for all correlation measurements are the Pearson's correlation coefficients. Statistical significance of difference between groups was evaluated using the Mann-Whitney rank sum test.
MNase-seq
MNase digestion, DNA isolation, and library preparation were performed as described in Kubik et al. (2015). Libraries were sequenced in paired-end mode on the Illumina HiSeq 2500 platform at the iGE3 genomics platform of the University of Geneva. Mapping of paired-end reads and data analysis were performed as described in Kubik et al. (2015).
ChEC-seq
Med8 was tagged with MNase using pGZ109, and ChEC was performed as described previously (Zentner et al. 2015). As we determined previously that the Med8 ChEC-seq signal is stable over time (Grunberg et al. 2016), we used time points between 1 and 5 min for the experiments described here. Libraries were constructed as described previously (Zentner et al. 2015) and sequenced for 25 cycles in paired-end mode on the Illumina HiSeq 2500 platform at the Fred Hutchinson Cancer Research Center Genomics Shared Resource. Pairs of FASTQ files were mapped to the sacCer3 genome assembly using Novoalign (Novocraft), generating SAM files that were then converted to tag directories with HOMER (http://homer.ucsd.edu) (Heinz et al. 2010) for further analysis. ChEC-seq data were visualized as reads per million (RPM)-normalized single-base resolution bedgraph tracks. For quantification of Mediator signal, we determined the RPM-normalized tags falling within bases −500 to −100 relative to the TSS of each gene for which RNAPII ChIP-seq was measured. This window was chosen to be inclusive of the UASs and exclusive of the core promoters for most yeast genes. For further analyses, the top 2000 genes in terms of Mediator UAS signal were used to avoid artifacts due to low signal. To compare control and anchor-away conditions, we divided the +rapamycin UAS counts by the −rapamycin UAS counts and log2 transformed this value.
Accession numbers
All deep sequencing data sets have been submitted to the NCBI Gene Expression Omnibus under accession code GSE109235.
Supplementary Material
Acknowledgments
We thank members of the Shore laboratory for useful discussion; Steven Hahn for comments on the manuscript; Uli Laemmli for help with the anchor-away strains and method; Mylène Docquier and the Institute of Genetics and Genomics of Geneva (iGE3; http://www.ige3.unige.ch/genomics-platform.php) for high-throughput DNA sequencing; Nicolas Roggli for expert artwork; Jacques Rougemont, Jean-Luc Falcone, and Srinivas Ramachandran for discussions on spike-in controls; and Thomas Schalch for the use of his local Galaxy server. M.J.B. was supported in part by an iGE3 Ph.D. student fellowship, and S.G. was supported by National Institutes of Health grant GM075114. D.S. acknowledges support from the Swiss National Fund and the Republic and Canton of Geneva.
Author contributions: M.J.B. and D.S. conceived the study. M.J.B., S.G., S.K., G.E.Z., and D.S. designed the experiments. M.J.B. and S.K. performed the experiments. S.G. performed the ChEC-seq experiments. M.J.B, S.K., S.G., and G.E.Z. carried out the bioinformatics analysis. M.J.B., S.G., and D.S. wrote the manuscript with input from all of the authors.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.312173.118.
References
- Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Cote J. 1999. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J 18: 5108–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ. 2015. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16: 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari SA, He Q, Morse RH. 2009. Mediator complex association with constitutively transcribed genes in yeast. Proc Natl Acad Sci 106: 16734–16739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL. 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27: 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista T, Grunberg S, Minoungou N, Koster MJE, Timmers HTM, Hahn S, Devys D, Tora L. 2017. SAGA is a general cofactor for RNA polymerase II transcription. Mol Cell 68: 130–143.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF. 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709. [DOI] [PubMed] [Google Scholar]
- Bhaumik SR. 2011. Distinct regulatory mechanisms of eukaryotic transcriptional activation by SAGA and TFIID. Biochim Biophys Acta 1809: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoite LT, Yu Y, Stillman DJ. 2001. The Swi5 activator recruits the Mediator complex to the HO promoter without RNA polymerase II. Genes Dev 15: 2457–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoure N, Bounova G, Bernasconi D, Praz V, Lammers F, Canella D, Willis IM, Herr W, Hernandez N, Delorenzi M, et al. 2014. Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res 24: 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Wang CY, Baptista T, Vincent SD, Hsiao WC, Stierle M, Kao CF, Tora L, Devys D. 2014. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev 28: 1999–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreault AA, Cronier D, Selleck W, Lacoste N, Utley RT, Allard S, Savard J, Lane WS, Tan S, Cote J. 2003. Yeast enhancer of polycomb defines global Esa1-dependent acetylation of chromatin. Genes Dev 17: 1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Howe L, Sousa K, Alley SC, Carrozza MJ, Tan S, Workman JL. 2001. Recruitment of HAT complexes by direct activator interactions with the ATM-related Tra1 subunit. Science 292: 2333–2337. [DOI] [PubMed] [Google Scholar]
- Bryant GO, Ptashne M. 2003. Independent recruitment in vivo by Gal4 of two complexes required for transcription. Mol Cell 11: 1301–1309. [DOI] [PubMed] [Google Scholar]
- Chen K, Hu Z, Xia Z, Zhao D, Li W, Tyler JK. 2015. The overlooked fact: fundamental need for spike-in control for virtually all genome-wide analyses. Mol Cell Biol 36: 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittuluru JR, Chaban Y, Monnet-Saksouk J, Carrozza MJ, Sapountzi V, Selleck W, Huang J, Utley RT, Cramet M, Allard S, et al. 2011. Structure and nucleosome interaction of the yeast NuA4 and Piccolo–NuA4 histone acetyltransferase complexes. Nat Struct Mol Biol 18: 1196–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. 2011. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David FP, Delafontaine J, Carat S, Ross FJ, Lefebvre G, Jarosz Y, Sinclair L, Noordermeer D, Rougemont J, Leleu M. 2014. HTSstation: a Web application and open-access libraries for high-throughput sequencing data analysis. PLoS One 9: e85879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. 1999. Structure and ligand of a histone acetyltransferase bromodomain. Nature 399: 491–496. [DOI] [PubMed] [Google Scholar]
- Downey M, Knight B, Vashisht AA, Seller CA, Wohlschlegel JA, Shore D, Toczyski DP. 2013. Gcn5 and sirtuins regulate acetylation of the ribosomal protein transcription factor Ifh1. Curr Biol 23: 1638–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durant M, Pugh BF. 2007. NuA4-directed chromatin transactions throughout the Saccharomyces cerevisiae genome. Mol Cell Biol 27: 5327–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esnault C, Ghavi-Helm Y, Brun S, Soutourina J, Van Berkum N, Boschiero C, Holstege F, Werner M. 2008. Mediator-dependent recruitment of TFIIH modules in preinitiation complex. Mol Cell 31: 337–346. [DOI] [PubMed] [Google Scholar]
- Eyboulet F, Wydau-Dematteis S, Eychenne T, Alibert O, Neil H, Boschiero C, Nevers MC, Volland H, Cornu D, Redeker V, et al. 2015. Mediator independently orchestrates multiple steps of preinitiation complex assembly in vivo. Nucleic Acids Res 43: 9214–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ramirez M, Rocchini C, Ausio J. 1995. Modulation of chromatin folding by histone acetylation. J Biol Chem 270: 17923–17928. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong F, Chiu LY, Miller KM. 2016. Acetylation reader proteins: linking acetylation signaling to genome maintenance and cancer. PLoS Genet 12: e1006272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind CK, Yoon S, Qiu H, Govind S, Hinnebusch AG. 2005. Simultaneous recruitment of coactivators by Gcn4p stimulates multiple steps of transcription in vivo. Mol Cell Biol 25: 5626–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govind CK, Zhang F, Qiu H, Hofmeyer K, Hinnebusch AG. 2007. Gcn5 promotes acetylation, eviction, and methylation of nucleosomes in transcribed coding regions. Mol Cell 25: 31–42. [DOI] [PubMed] [Google Scholar]
- Grant PA, Duggan L, Cote J, Roberts SM, Brownell JE, Candau R, Ohba R, Owen-Hughes T, Allis CD, Winston F, et al. 1997. Yeast Gcn5 functions in two multisubunit complexes to acetylate nucleosomal histones: characterization of an Ada complex and the SAGA (Spt/Ada) complex. Genes Dev 11: 1640–1650. [DOI] [PubMed] [Google Scholar]
- Grunberg S, Henikoff S, Hahn S, Zentner GE. 2016. Mediator binding to UASs is broadly uncoupled from transcription and cooperative with TFIID recruitment to promoters. EMBO J 35: 2435–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. 2007. Quantifying similarity between motifs. Genome Biol 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, Churchman LS. 2017. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat Rev Mol Cell Biol 18: 263–273. [DOI] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, Laemmli UK. 2008. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol Cell 31: 925–932. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmlinger D, Marguerat S, Villen J, Swaney DL, Gygi SP, Bahler J, Winston F. 2011. Tra1 has specific regulatory roles, rather than global functions, within the SAGA co-activator complex. EMBO J 30: 2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 95: 717–728. [DOI] [PubMed] [Google Scholar]
- Hu B, Petela N, Kurze A, Chan KL, Chapard C, Nasmyth K. 2015. Biological chromodynamics: a general method for measuring protein occupancy across the genome by calibrating ChIP-seq. Nucleic Acids Res 43: e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol 296: 1205–1214. [DOI] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF. 2004. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell 13: 573–585. [DOI] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, Tjian R. 2000. Structure and function of a human TAFII250 double bromodomain module. Science 288: 1422–1425. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Robert F. 2014. Kin28 regulates the transient association of Mediator with core promoters. Nat Struct Mol Biol 21: 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. 2004. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev 18: 2491–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnaird A, Zhao S, Wellen KE, Michelakis ED. 2016. Metabolic control of epigenetics in cancer. Nat Rev Cancer 16: 694–707. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- Kuang Z, Cai L, Zhang X, Ji H, Tu BP, Boeke JD. 2014. High-temporal-resolution view of transcription and chromatin states across distinct metabolic states in budding yeast. Nat Struct Mol Biol 21: 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone JL, Rougemont J, Shore D. 2015. Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol Cell 60: 422–434. [DOI] [PubMed] [Google Scholar]
- Kubik S, O'Duibhir E, de Jonge WJ, Mattarocci S, Albert B, Falcone JL, Bruzzone MJ, Holstege FCP, Shore D. 2018. Sequence-directed action of RSC remodeler and general regulatory factors modulates +1 nucleosome position to facilitate transcription. Mol Cell 71: 89–102.e5. [DOI] [PubMed] [Google Scholar]
- Lee KK, Sardiu ME, Swanson SK, Gilmore JM, Torok M, Grant PA, Florens L, Workman JL, Washburn MP. 2011. Combinatorial depletion analysis to assemble the network architecture of the SAGA and ADA chromatin remodeling complexes. Mol Syst Biol 7: 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerdrup M, Johansen JV, Agrawal-Singh S, Hansen K. 2016. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat Struct Mol Biol 23: 349–357. [DOI] [PubMed] [Google Scholar]
- Leroy C, Cormier L, Kuras L. 2006. Independent recruitment of mediator and SAGA by the activator Met4. Mol Cell Biol 26: 3149–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Myers LC. 2012. Med5(Nut1) and Med17(Srb4) are direct targets of mediator histone H4 tail interactions. PLoS One 7: e38416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louder RK, He Y, Lopez-Blanco JR, Fang J, Chacon P, Nogales E. 2016. Structure of promoter-bound TFIID and model of human pre-initiation complex assembly. Nature 531: 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. 2000. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev 14: 951–962. [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki K, Kasahara K, Shirahige K, Kokubo T. 2010. Genome-wide localization analysis of a complete set of Tafs reveals a specific effect of the taf1 mutation on Taf2 occupancy and provides indirect evidence for different TFIID conformations at different promoters. Nucleic Acids Res 38: 1805–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul E, Zhu ZI, Landsman D, Morse RH. 2015. Genome-wide association of mediator and RNA polymerase II in wild-type and mediator mutant yeast. Mol Cell Biol 35: 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko N, Jin Y, Wong KH, Struhl K. 2017. Evidence that Mediator is essential for Pol II transcription, but is not a required component of the preinitiation complex in vivo. Elife 6: e28447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaschka C, Lariviere L, Wenzeck L, Seizl M, Hemann M, Tegunov D, Petrotchenko EV, Borchers CH, Baumeister W, Herzog F, et al. 2015. Architecture of the RNA polymerase II–Mediator core initiation complex. Nature 518: 376–380. [DOI] [PubMed] [Google Scholar]
- Podobinska M, Szablowska-Gadomska I, Augustyniak J, Sandvig I, Sandvig A, Buzanska L. 2017. Epigenetic modulation of stem cells in neurodevelopment: the role of methylation and acetylation. Front Cell Neurosci 11: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Hu C, Zhang F, Hwang GJ, Swanson MJ, Boonchird C, Hinnebusch AG. 2005. Interdependent recruitment of SAGA and Srb mediator by transcriptional activator Gcn4p. Mol Cell Biol 25: 3461–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JL, Iyer VR, Brown PO, Struhl K. 2000. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol Cell 6: 1297–1307. [DOI] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF. 2012. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 483: 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribaud V, Ribeyre C, Damay P, Shore D. 2012. DNA-end capping by the budding yeast transcription factor and subtelomeric binding protein Tbf1. EMBO J 31: 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde JR, Cardenas ME. 2003. The tor pathway regulates gene expression by linking nutrient sensing to histone acetylation. Mol Cell Biol 23: 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Durussel T, Laemmli UK. 2004. ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 16: 147–157. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847. [DOI] [PubMed] [Google Scholar]
- Soutourina J. 2018. Transcription regulation by the Mediator complex. Nat Rev Mol Cell Biol 19: 262–274. [DOI] [PubMed] [Google Scholar]
- Soutourina J, Wydau S, Ambroise Y, Boschiero C, Werner M. 2011. Direct interaction of RNA polymerase II and mediator required for transcription in vivo. Science 331: 1451–1454. [DOI] [PubMed] [Google Scholar]
- Suka N, Suka Y, Carmen AA, Wu J, Grunstein M. 2001. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol Cell 8: 473–479. [DOI] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Lariviere L, Maier KC, Seizl M, Tresch A, Cramer P. 2012. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22: 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Calero G, Komori H, Brown JA, Ehrensberger AH, Hudmon A, Asturias F, Kornberg RD. 2006. Head module control of mediator interactions. Mol Cell 23: 355–364. [DOI] [PubMed] [Google Scholar]
- Tse C, Sera T, Wolffe AP, Hansen JC. 1998. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol Cell Biol 18: 4629–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bakel H, Tsui K, Gebbia M, Mnaimneh S, Hughes TR, Nislow C. 2013. A compendium of nucleosome and transcript profiles reveals determinants of chromatin architecture and transcription. PLoS Genet 9: e1003479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade C, Shea KA, Jensen RV, McAlear MA. 2001. EBP2 is a member of the yeast RRB regulon, a transcriptionally coregulated set of genes that are required for ribosome and rRNA biosynthesis. Mol Cell Biol 21: 8638–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield L, Ramachandran S, Baptista T, Devys D, Tora L, Hahn S. 2017. Transcription of nearly all yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Mol Cell 68: 118–129 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Chen HV, Liu CL, Rahat A, Klien A, Soares L, Gudipati M, Pfeffner J, Regev A, Buratowski S, et al. 2012. Systematic dissection of roles for chromatin regulators in a yeast stress response. PLoS Biol 10: e1001369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A, Hsieh TH, Appleboim A, Chen HV, Rahat A, Amit I, Rando OJ, Friedman N. 2015. High-resolution chromatin dynamics during a yeast stress response. Mol Cell 58: 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Li C, Chen Z, Jiang S, Fan S, Wang J, Dai J, Zhu P, Chen Z. 2016. The NuA4 core complex acetylates nucleosomal histone H4 through a double recognition mechanism. Mol Cell 63: 965–975. [DOI] [PubMed] [Google Scholar]
- Zentner GE, Kasinathan S, Xin B, Rohs R, Henikoff S. 2015. ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat Commun 6: 8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhang Y, Bjornsdottir G, Liu Z, Quan A, Costanzo M, Davila Lopez M, Westholm JO, Ronne H, Boone C, et al. 2011. Histone modifications influence mediator interactions with chromatin. Nucleic Acids Res 39: 8342–8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.