

Abstract
para‐Hydrogen‐induced polarization (PHIP) is a method to rapidly generate hyperpolarized compounds, enhancing the signal of nuclear magnetic resonance (NMR) experiments by several thousand‐fold. The hyperpolarization of metabolites and their use as contrast agents in vivo is an emerging diagnostic technique. High degrees of polarization and extended polarization lifetime are necessary requirements for the detection of metabolites in vivo. Here, we present pulsed NMR methods for obtaining hyperpolarized magnetization in two metabolites. We demonstrate that the hydrogenation with para‐hydrogen of perdeuterated vinyl acetate allows us to create hyperpolarized ethyl acetate with close to 60 % 1H two‐spin order. With nearly 100 % efficiency, this order can either be transferred to 1H in‐phase magnetization or 13C magnetization of the carbonyl function. Close to 60 % polarization is experimentally verified for both nuclei. Cleavage of the ethyl acetate precursor in a 20 s reaction yields ethanol with approximately 27 % 1H polarization and acetate with around 20 % 13C polarization. This development will open new opportunities to generate metabolic contrast agents in less than one minute.
Keywords: hyperpolarization, metabolites, NMR spectroscopy, para-hydrogen induced polarization, pulsed NMR
The phenomenon of nuclear magnetic resonance (NMR) comes with an inherent low sensitivity. To overcome such a limitation, hyperpolarization techniques have been developed that allow for signal enhancements over four orders of magnitude.1, 2, 3, 4, 5, 6 Hyperpolarization techniques include dynamic nuclear polarization (DNP),1, 2, 3, 4, 5, 6, 7, 8, 9 spin exchange optical pumping (SEOP),10, 11, 12, 13 and para‐hydrogen‐based methods such as signal amplification by reversible exchange (SABRE)14, 15, 16, 17, 18, 19, 20, 21, 22, 23 and para‐hydrogen‐induced polarization (PHIP).24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41 Notably, the former technique (SABRE) represents a non‐hydrogenative form of PHIP. One emerging application of hyperpolarized NMR is the direct observation of metabolism in vitro and in vivo.1, 2, 3, 4, 5 PHIP applications in medical settings have been limited, as the technique requires unsaturated bonds in contrast agent precursors, which can undergo a hydrogenation reaction.40 To extend the range of polarizable substrates, approaches with side arms that can be attached to potential metabolites of interest have been developed. In early experiments, these side arms mainly served the purpose of preventing keto–enol tautomerism to hyperpolarize ethanol starting from vinyl acetate.29, 30 Acetate can be seen as a protection group that is cleaved after hydrogenation of vinyl acetate into ethyl acetate to reveal hyperpolarized ethanol. Reineri et al. subsequently introduced the PHIP‐SAH approach (PHIP by side arm hydrogenation), which allows the transfer of proton polarization from a hydrogenated sidearm to a 13C nucleus of a metabolite.31 Cleavage of the precursor molecule leads to hyperpolarized metabolites. In the case of lactate, it was demonstrated that, after the polarization procedure, lactate can enzymatically be converted into pyruvate.32 So far, the most successful approach of hyperpolarizing metabolites with para‐hydrogen relies on a field cycling technique, for which 13C polarization values of about 5 % have been experimentally demonstrated in highly concentrated solutions of acetate and pyruvate.31, 32, 33, 34, 35, 36 The theoretical polarization limits of the field cycling technique have been investigated and simulations for an ideal case have shown that more than 30 % 13C polarization can be achieved in the precursor molecule (before cleavage), whereas 50 % was suggested experimentally.33, 34, 35
In this Communication, we demonstrate a pulsed NMR PHIP approach that surpasses the current theoretical limits in metabolite polarization for field cycling. We have investigated perdeuterated vinyl acetate (with natural abundant 13C) as a precursor that can be hydrogenated, thus hyperpolarized, and upon cleavage yields acetate and ethanol. By using precursors with 13C at natural abundance, we experimentally verify close to 60 % 1H and 13C polarization in the ethyl acetate product after hydrogenation. Furthermore, we show that after cleavage, hyperpolarized 1–13C‐acetate‐d3 is obtained with 19.3±0.2 % 13C polarization and ethanol‐d3 with 26.7±1.6 % 1H polarization.
The procedure for generating the hyperpolarized metabolites is schematically depicted in Figure 1. Hydrogenation of vinyl acetate‐d6 leads to ethyl acetate‐d6, in which the chemical shift difference between the two added protons is δ=2.88 ppm (864 Hz at B 0=7 T) and the homonuclear J coupling is J HA,HB=7.1 Hz. The 13C nucleus of interest for the hyperpolarization of acetate can be found at the 1‐position (carbonyl‐13C) of the corresponding moiety. The three‐spin system of two protons and one 13C is characterized by the two proton‐carbon couplings J C,HB=3.2 Hz and J C,HA <0.4 Hz. All J‐coupling parameters are depicted in Figure 1. Following incoherent hydrogenation with para‐enriched gas of the vinyl bond in high magnetic field, the initial spin density operator is assumed to be proportional to the two‐spin longitudinal order operator ρ 0∝I 1z I 2z. For 1H polarization experiments (left path of Figure 1), sequence 1 converts the two‐spin longitudinal order of protons after the addition of para‐hydrogen, ρ 0=I 1z I 2z, into in‐phase magnetization of one of the two protons. After the first 90y pulse, the first block exploits the large chemical shift difference to transfer two‐spin transverse order I 1x I 2x into I 1x I 2y with negligible effect from the J‐coupling evolution. The second 90−x pulse brings the spin density operator into I 1x I 2z, which is then refocused into I 1y/2 by homonuclear J evolution during the second block. For 13C polarization experiments (right path of Figure 1), once that para‐hydrogen is added and ethyl acetate has formed, the couplings and chemical shift difference are ideal for utilizing the recently introduced ESOTHERIC (efficient spin order transfer to heteronuclei via relayed inept chains) sequence to transfer ρ 0=I 1z I 2z/2 into heteronuclear in‐phase magnetization S 3y/4, with a theoretical efficiency close to 95 %.39
Figure 1.
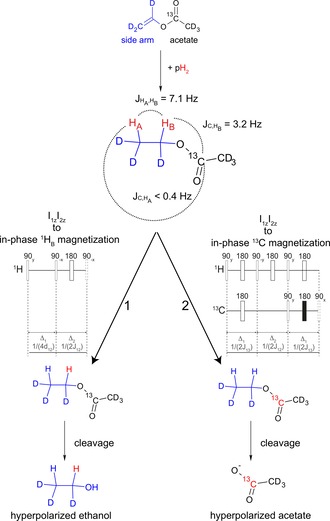
Schematic of the procedure leading to the hyperpolarization of ethanol and acetate. Both sequences result in in‐phase y magnetization and the following experimental timings are used in sequence 1: Δ1=0.29 ms and Δ2=70.4 ms. All pulses are applied on resonance on the HB proton. In sequence 2, we have used the following timings: Δ1=156.3 ms and Δ2=70.4 ms. The dotted 90° pulses at the end of both sequences are utilized before the cleavage to flip the magnetization into the z direction. After the cleavage, the polarization is detected following another 90° pulse.
Samples were prepared from [D4]MeOH solutions, containing 0.9 mm of vinyl acetate, 2 mm of commercially available rhodium catalyst, [1,4‐Bis(diphenylphosphino)butane](1,5‐cyclooctadiene) rhodium(I) tetrafluoroborate, and 9 mm toluene‐13Cα enriched (>99 %) as internal reference for determining 13C polarization levels. The precursor concentration was chosen so to avoid radiation damping effects after hyperpolarization and to perform quantitative experiments. For all the polarization experiments, the signal enhancements were determined in the assumption that all the precursor has reacted into ethyl acetate during the hydrogenation. The full procedure of verifying sample stability as well as calculating polarization levels is discussed in detail in the Supporting Information. To carry out the experiments, we used hydrogen gas in which the ortho and para spin isomers were equilibrated at 36 K, corresponding to a 92 % para‐H2 fraction.
PASADENA‐type experiments were performed, in which the para‐enriched hydrogen gas was bubbled for 10 s to a solution of perdeuterated vinyl acetate in [D4]MeOH at 320 K in the presence of a homogeneous rhodium catalyst in high field (here: B 0=7 T). After hydrogenation in high field, a 45° pulse is typically used to detect a PASADENA spectrum (Figure 2 a) that displays only half of the nascent 1H polarization, as previously discussed.39 Here, sequence 1 was utilized to convert in full the 1H longitudinal two‐spin order into in‐phase magnetization on the methylene proton of ethyl acetate‐d6, Figure 2 b. Comparison with the thermally polarized proton spectrum of the same sample (Figure 2 c) gives an average observable polarization P(1H)=54.9±3.2 %. Application of the ESOTHERIC sequence directly after hydrogenation leads to the 13C spectrum of hyperpolarized carbonyl carbon in ethyl acetate presented in Figure 2 d. Considering that the observed 13C signal originates from natural abundant 13C, which is 1.08 % of all the carbon nuclei of the sample, we estimate a polarization P(13C)=59.1±1.4 %. For comparison, the thermal 13C–{1H} spectrum of the internal 13Cα‐labelled toluene (13C>99 %) standard is shown in Figure 2 e.
Figure 2.
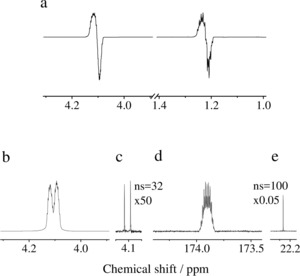
a) 1H spectrum of hyperpolarized protons in ethyl acetate‐d6 detected with a 45° pulse at B 0=7 T. b) 1H spectrum of the hyperpolarized (P=56.9 %, highest achieved polarization) methylene protons of ethyl acetate‐d6 in 0.6 mL solution after pulse sequence 1. c) Corresponding 2H‐decoupled 1H spectrum after thermal equilibration. d) 13C spectrum of the hyperpolarized (P=60.5 %, highest achieved polarization) carbonyl carbon of ethyl acetate‐d6 (at natural abundance) in 0.6 mL solution after utilizing sequence 2. e) 1H‐decoupled 13C spectrum of toluene‐α‐13C (enrichment >99 %) used as internal reference after thermal equilibration. Note: The chemical shift scale is the same in both spectra for (d) and (e). Owing to hardware limitations, we could not decouple deuterium in the same experiment as the polarization transfer.
In a series of subsequent experiments, we investigated the metabolite cleavage from the sidearm. As preparation for the cleavage, we utilized an additional 90° pulse at the end of both sequences to flip the magnetization into the z direction. To a solution of 0.4 mL of hyperpolarized ethyl acetate, 0.05 mL 1 m NaOD was added and mixed with the sample for 20 s inside the 7 T magnet by bubbling nitrogen gas. Acquisition of the 1H or 13C spectrum following a 90° pulse showed the cleaved ethanol‐d4 with P(1H)=26.7±1.6 % (Figure 3 a) and acetate with P(13C)=19.3±0.2 % (Figure 3 c). An increase in utilized NaOD will result in a fully cleaved product as shown recently.31 We have utilized a reduced amount of base to demonstrate the change in chemical shift between the cleaved and uncleaved sample in the same experiment. The reduction in polarization can be accounted for by two factors. Firstly, the longitudinal relaxation of the polarization during 20 s gives a factor of 0.6 for 13C nuclei (T 1=41 s) in the uncleaved acetate (in [D4]MeOH) and 0.74 for 1H (T 1=69 s) nuclei of ethanol‐d4 (in [D4]MeOH and NaOD/D2O). An additional factor of about 0.5 comes from the fact that only part of the sample is excited and detected by the B1 coil. This volume is approximately 0.2 mL, where the polarization transfer occurs, whereas the sample volume is 0.45 mL. After application of the pulse sequence, the pressure is released and NaOD is added. The sample is mixed with nitrogen bubbling to ensure the ester bond cleavage that leads to a diluted signal from hyperpolarized sample, which is the value that we report.
Figure 3.
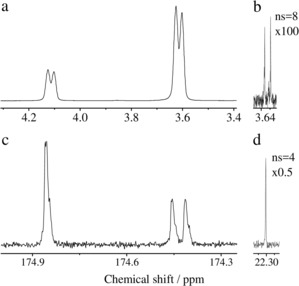
NMR spectra of hyperpolarized a) ethanol‐d3 [P(1H)=29.3 %, highest achieved polarization] and c) acetate‐d3 [P(13C)=19.4 %, highest achieved polarization] after hydrogenation with para‐enriched H2 gas of vinyl acetate‐d6 with 13C at natural abundance, polarization transfer and ester bond cleavage with 50 μL of NaOD (1 m) added to the reaction solution (0.4 mL). In the 1H spectrum (a), non‐cleaved ethyl acetate appears at approximately 4.1 ppm and the cleaved ethanol appears at 3.6 ppm. b) Corresponding thermal 2H‐decoupled 1H spectrum after the reaction. In the 13C spectrum (c), the doublet at approximately 174.4 ppm corresponds to the 13C carbonyl in ethyl acetate (J C,HA=3.2 Hz) and the singlet peak corresponds to acetate‐d3 at approximately 174.9 ppm (no heteronuclear 1H‐13C coupling is present). d) Corresponding thermally polarized 1H‐decoupled 13C spectrum of toluene‐α‐13C (enrichment >99 %) used as internal reference after the cleavage reaction.
Although we obtained well above 50 % polarization for protons and 13C in ethyl acetate‐d6, it is conceivable that higher polarization values can be achieved in pulsed PHIP experiments. In a publication from Anwar et al., it was shown that close to unity nascent 1H polarization is experimentally possible.41 A necessary step towards this goal would require the use of 100 % para‐enriched hydrogen gas as opposed to the 92 % used in the experiments presented above. According to the theory, for a given para‐hydrogen fraction f, the amount of singlet order available for successive polarization transfers is ps=(1–4f)/3.38 The formula implies a usable 89 % initial two‐spin order in our experiments, and indicates that the achievable polarization can be improved by a factor of 1.1. As the proton longitudinal two‐spin order relaxation time of the product ethyl acetate‐d6 is T 1 (I1zI 2z)≈100 s (in [D4]MeOH) (see the Supporting Information), at most 5 % polarization loss to relaxation is expected during the bubbling time of para‐hydrogen (10 s) before the polarization experiments. Such loss could be decreased by reducing the reaction time from 10 s to 1–3 s. We speculate that the remaining factor of 1.5 in polarization loss may be attributed to para–ortho conversion of hydrogen at the catalytic sites. In recently published experiments, it was shown that after a few seconds of reaction time, more than 50 % para‐hydrogen can be converted back into its ortho state, leading to a significant polarization loss.34 In our experiments, a para–ortho conversion of about 20 % (i.e. from 92 to 73 % total para enrichment) during the hydrogenation reaction would reduce the theoretical achievable polarization to 64 %, which is close to the observed experimental value. This point will be part of our future investigations, requiring the construction of an optimized reaction setup to achieve shorter reaction times and potentially mitigate para–ortho conversion. Other future technological challenges, that we are currently addressing, concern the increase of the final concentration of hyperpolarized metabolites, the speeding up of the cleavage procedure, and the separation of the catalyst to finally obtain an injectable hyperpolarized metabolite in water.
In conclusion, we have shown that high levels of 1H and 13C polarization of over 50 % in the metabolite precursor ethyl acetate‐d6 are achievable with para‐hydrogen within seconds. Cleavage of the precursor yields hyperpolarized ethanol‐d3 P(1H)=26.7±1.6 % and acetate‐d3 with P(13C)=19.3±0.2 %. These values correspond to signal enhancements of 12 000 for 1H and 34 000 for 13C at 310 K and 7 T. The reported polarization values of metabolite precursors are about one order of magnitude higher than previously achieved, partially owing to the adoption of effective pulsed methods for polarization transfer. We anticipate the future application of 1H hyperpolarized ethanol as a contrast agent for metabolic studies, owing to the relatively long proton relaxation time (T 1≈70 s at 7 T). The consistent achievement of polarization levels >20 % in metabolites fundamentally proves that the proposed pulsed PHIP‐SAH approach can generate hyperpolarized metabolites above the threshold requested for in vivo experiments.40
Experimental Section
Further experimental details can be found in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge generous funding from the Max‐Planck‐Society and would like to thank Prof. Christian Griesinger for helpful discussion and access to his facilities.
S. Korchak, S. Mamone, S. Glöggler, ChemistryOpen 2018, 7, 672.
References
- 1. Golman K., in ′t Zandt R., Thaning M., Proc. Natl. Acad. Sci. USA 2006, 103, 11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Day S. E., Kettunen M. I., Gallagher F. A., Hu D. E., Lerche M., Wolber J., Golman K., Ardenkjaer-Larsen J. H., Brindle K. M. Nat. Med. 2007, 13, 1382. [DOI] [PubMed] [Google Scholar]
- 3. Gallagher F. A., Kettunen M. I., Day S. E., Hu D. E., Ardenkjaer-Larsen J. H., in ′t Zandt R., Jensen P. R., Karlsson M., Golman K., Lerche M. H., Brindle K. M., Nature 2008, 453, 940. [DOI] [PubMed] [Google Scholar]
- 4. Nelson S. J., Kurhanewicz J., Vigneron D. B., Larson P. E. Z., Harzstark A. L., Ferrone M., van Criekinge M., Chang J. W., Bok R., Park I., Reed G., Carvajal L., Small E. J., Munster P., Weinberg V. K., Ardenkjaer-Larsen J. H., Chen A. P., Hurd R. E., Odegardstuen L.-I., Robb F. J., Tropp J., Murray J. A., Sci. Transl. Med. 2013, 198, 198ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kurhanewicz J., Vigneron D. B., Brindle K., Chekmenev E. Y., Comment A., Cunningham C. H., DeBerardinis R. J., Green G. G., Leach M. O., Rajan S. S., Rizi R. R., Ross B. D., Warren W. S., Malloy C. R., Neoplasia 2011, 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ardenkjaer-Larsen J. H., Fridlund B., Gram A., Hansson G., Hansson L., Lerche M. H., Servin R., Thanning M., Golman K., Proc. Natl. Acad. Sci. USA 2003, 100, 10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dzien P., Kettunen M. I., Marco-Ruis I., Serrao E. M., Rodrigues T. B., Larkin T. J., Timm K. N., Brindle K. M., Magn. Reson. Med. 2015, 73, 1733. [DOI] [PubMed] [Google Scholar]
- 8. Lesage A., Lelli M., Gajan D., Caporini M. A., Vitzthum V., Miévilee P., Alauzun J., Roussey A., Thieuleux C., Mehdi A., Bodenhausen G., Coeperet C., J. Am. Chem. Soc. 2010, 132, 15459. [DOI] [PubMed] [Google Scholar]
- 9. Thankamony A. S. L., Wittmann J. J., Kaushik M., Corzilius B., Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 120. [DOI] [PubMed] [Google Scholar]
- 10. Walker T. G., Happer W., Rev. Mod. Phys. 1997, 69, 629. [Google Scholar]
- 11. Appelt S., Baranga A. B.-A., Erickson C. J., Romalis M. V., Young A. R., Happer W., Phys. Rev. A 1998, 58, 1412. [Google Scholar]
- 12. Möller H. E., Chen X. J., Saam B., Hagspiel K. D., Johnson G. A., Altes T. A., de Lange E. E., Kauczor H.-U., Magn. Reson. Med. 2002, 47, 1029. [DOI] [PubMed] [Google Scholar]
- 13. Schröder L., Lowery T. J., Hilty C., Wemmer D. E., Pines A., Science 2006, 314, 446. [DOI] [PubMed] [Google Scholar]
- 14. Adams R. W., Aguilar J. A., Atkinson K. D., Cowley M. J., Elliott P. I. P., Duckett S. B., Green G. G. R., Khazal I. G., Lopez-Serrano J., Williamson D. C., Science 2009, 323, 1708. [DOI] [PubMed] [Google Scholar]
- 15. Rayner P. J., Burns M. J., Olaru A. M., Norcott P., Fekete M., Green G. G. R., Highton L. A. R., Mewis R. E., Duckett S. B., Proc. Natl. Acad. Sci. USA 2017, 114, E3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barskiy D. A., Kovtunov K. V., Koptyug I. V., He P., Groome K. A., Best Q. A., Shi F., Goodson B. M., Shchepin R. V., Coffey A. M., Waddell K. W., Chekmenev E. Y., J. Am. Chem. Soc. 2014, 136, 3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suefke M., Lehmkuhl S., Liebisch S. A., Blümich B., Appelt S., Nat. Phys. 2017, 13, 568. [Google Scholar]
- 18. Eshuis N., Hermkens N., van Weerdenburg B. J. A., Feiters M. C., Rutjes F. P. J., Wijmenga S. S., Tessari M., J. Am. Chem. Soc. 2014, 136, 2695. [DOI] [PubMed] [Google Scholar]
- 19. Theis T., Truong M. L., Coffey A. M., Shchepin R. V., Waddell K. W., Shi F., Goodson B. M., Warren W. S., Chekmenev E. Y., J. Am. Chem. Soc. 2015, 137, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spannring P., Reile I., Emondts M., Schleker P. P. M., Hermkens N. K. J., van der Zwaluw N. G. J., van Weerdenburg B. J. A., Tinnemans P., Tessari M., Blümich B., Rutjes F. P. J. T., Feiters M. C., Chem. Eur. J. 2016, 22, 9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Truong M. L., Shi F., He P., Yuan B., Plunkett K. N., Coffey A. M., Shchepin R. V., Barskiy D. A., Kovtunov K. V., Koptyug I. V., Waddell K. W., Goodson B. M., Chekmenev E. Y., J. Phys. Chem. B 2014, 118, 13882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iali W., Olaru A. M., Green G. G. R., Duckett S. B., Chem. Eur. J. 2017, 23, 10491. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Colell J. F. P., Emondts M., Logan A. W. J., Shen K., Bae J., Shchepin R. V., G. X. Ortiz, Jr. , Spannring P., Wange Q., Malcolmson S. J., Chekmenev E. Y., Feiters M. C., Rutjes F. P. J. T., Blümich B., Theis T., Warrem W. S., J. Am. Chem. Soc. 2017, 139, 7761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bowers C. R., Weitekamp D. P., Phys. Rev. Lett. 1986, 57, 2645. [DOI] [PubMed] [Google Scholar]
- 25. Schmidt A. B., Berner S., Schimpf W., Müller C., Lickert T., Schwaderlapp N., Knecht S., Skinner J. G., Dost A., Rovedo P., Hennig J., von Elverfeldt D., Hövener J.-B., Nat. Commun. 2017, 8, 14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bhattacharya P., Chekmenev E. Y., Perman W. H., Harris K. C., Lin A. P., Norton V. A., Tan C. T., Ross B. D., Weitekamp D. P., J. Magn. Reson. 2007, 186, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bhattacharya P., Chekmenev E. Y., Reynolds W. F., Wagner S., Zacharias N., Chan H. R., Bünger R., Ross B. D., NMR Biomed. 2011, 24, 1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goldman M., Johannesson H., Axelsson O., Karlsson M., C. R. Chim. 2006, 9, 357. [Google Scholar]
- 29. Trantzschel T., Bernarding J., Plaumann M., Lego D., Gutmann T., Ratajczyk T., Dillenberger S., Buntkowsky G., Bargon J., Bommerich U., Phys. Chem. Chem. Phys. 2012, 14, 5601. [DOI] [PubMed] [Google Scholar]
- 30. Salnikov O. G., Kovtunov K. V., Koptyug I. V., Sci. Rep. 2015, 5, 13930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reineri F., Boi T., Aime S., Nat. Commun. 2015, 6, 5858. [DOI] [PubMed] [Google Scholar]
- 32. Cavallari E., Carrera C., Aime S., Reineri F., Chem. Eur. J. 2017, 23, 1200–1204. [DOI] [PubMed] [Google Scholar]
- 33. Cavallari E., Carrera C., Boi T., Aime S., Reineri F., J. Phys. Chem. B 2015, 119, 10035. [DOI] [PubMed] [Google Scholar]
- 34. Cavallari E, Carrera C., Aime S., Reineri F., J. Magn. Reson. 2018, 289, 12. [DOI] [PubMed] [Google Scholar]
- 35. Shchepin R. V., Barskiy D. A., Coffey A. M., Esteve I. V. Manzanera, Chekmenev E. Y., Angew. Chem. Int. Ed. 2016, 55, 6071; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6175. [Google Scholar]
- 36. Kovtunov K. V., Barskiy D. A., Shchepin R. V., Salnikov O. G., Prosvirin I. P., Bukhtiyarov A. V., Kovtunova L. M., Bukhtiyarov V. I., Koptyug I. V., Chekmenev E. Y., Chem. Eur. J. 2016, 22, 16446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haake M., Natterer J., Bargon J., J. Am. Chem. Soc. 1996, 118, 8688. [Google Scholar]
- 38. Natterer J., Bargon J., Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293. [Google Scholar]
- 39. Korchak S., Yang S., Mamone S., Glöggler S., ChemistryOpen 2018, 7, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hövener J.-B., Pravdivtsev A. N., Kidd B., Bowers C. R., Glöggler S., Kovtunov K. V., Plaumann M., Katz-Brull R., Buckenmaier K., Jerschow A., Reineri F., Theis T., Shchepin R. V., Wagner S., Bhattacharya P., Zacharias N. M., Chekmenev E. Y., Angew. Chem. Intl. Ed. 2018, 10.1002/anie.201711842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Anwar M. S., Blazina D., Carteret H. A., Duckett S. B., Halstead T. K., Jones J. A., Kozak C. M., Taylor R. J. K., Phys. Rev. Lett. 2004, 93, 040501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary