

Abstract
AMPK activated protein kinase (AMPK), a master regulator of energy homeostasis, is activated in response to an energy shortage imposed by physical activity and caloric restriction. We here report on the identification of PAN-AMPK activator O304, which — in diet-induced obese mice — increased glucose uptake in skeletal muscle, reduced β cell stress, and promoted β cell rest. Accordingly, O304 reduced fasting plasma glucose levels and homeostasis model assessment of insulin resistance (HOMA-IR) in a proof-of-concept phase IIa clinical trial in type 2 diabetes (T2D) patients on Metformin. T2D is associated with devastating micro- and macrovascular complications, and O304 improved peripheral microvascular perfusion and reduced blood pressure both in animals and T2D patients. Moreover, like exercise, O304 activated AMPK in the heart, increased cardiac glucose uptake, reduced cardiac glycogen levels, and improved left ventricular stroke volume in mice, but it did not increase heart weight in mice or rats. Thus, O304 exhibits a great potential as a novel drug to treat T2D and associated cardiovascular complications.
Keywords: Metabolism
Keywords: Cardiovascular disease, Diabetes
AMPK activator O304 mitigates diabetes and fatty liver in DIO mice and improved glucose homeostasis, increased vascular microvascular perfusion, and reduced blood pressure in T2D patients.
Introduction
The increase in type 2 diabetes (T2D) is strongly linked to the global increase in obesity, caused by physical inactivity and a Western diet, as well as an aging population. During development of T2D, insulin resistance and reduced glucose uptake in skeletal muscle result in an initial combined hyperglycemia and hyperinsulinemia, which are key metabolic abnormalities associated with the development of T2D (1–3) as well as key risk factors for cardiovascular diseases (4, 5). In the context of persistent insulin resistance, pancreatic β cells progressively fail to match the increasing demand for insulin, which leads to the development of severe hyperglycemia and diabetes (6). Despite the introduction of novel, additional glucose-lowering agents, many people with T2D fail to reach their glycemic targets, and the low success rates cause significant morbidity and mortality (7, 8). The poor outcome may, at least in part, be due to the recently described heterogeneity of T2D patients and the apparent shortcomings in using glucose as the sole diagnosis criteria (9).
Exercise and diet/caloric restriction are the first recommendations offered to newly diagnosed T2D patients (10–13), but compliance is a major problem. In particular, exercise appears more effective than insulin treatment to promote glucose uptake in insulin-resistant skeletal muscle of T2D patients — an effect that, at least in part, is mediated by activation of AMP-activated protein kinase (AMPK), which is activated in response to an energy shortage imposed by physical activity and caloric restriction (10, 14). In line with this notion, the recently described allosteric AMPK activators PF-793 and MK-8722 increase glucose uptake in skeletal muscle in an insulin-independent manner (15, 16).
Increased peripheral glucose disposal will relieve the demand on β cells to produce and secrete insulin, thus indirectly ameliorating β cell stress (7, 8, 17) and, by increasing glucose uptake in skeletal muscle AMPK, may indirectly preserve/restore β cell function. There is little evidence, however, that any of the currently used glucose-lowering agents acts directly on β cells to reduce β cell stress and impose β cell rest, and the quest for such agents is ongoing (8, 18). Notably, AMPK appears to ensure both normal β cell glucose sensing and glucose-stimulated insulin secretion (GSIS), and reduced AMPK activity has been associated with β cell dysfunction in diet-induced obese (DIO) mice (19–23). Moreover, AMPK stimulates macroautophagy, herein referred to as autophagy, which not only is important for β cell function and survival (24, 25), but which — by clearing aggregating proteins (26, 27) — also may prevent accumulation of toxic islet amyloid polypeptide (IAPP) aggregates/amyloid in β cells (28–30). Thus, pharmacological activation of AMPK may, in addition to increasing glucose uptake in skeletal muscle, have the additional therapeutic potential to preserve/restore β cell function by promoting β cell rest.
T2D is associated with devastating microvascular complications due to reduced capillary function and perfusion, which also reduce the supply of glucose and insulin to the muscle interstitium (4, 31), but no currently used drug to treat T2D appears to directly increase microvascular blood flow (32). Notably, exercise stimulates peripheral blood flow, improves cardiac function, reduces blood pressure, and increases endurance capacity in man (33), and AMPK activation in endothelial and smooth muscle cells increases vasodilation and microvascular perfusion in muscle and reduces blood pressure (34). Moreover, exercise can activate AMPK in the heart, increase cardiac glucose uptake, reduce glycogen levels, and improve left ventricular (LV) stroke volume (35, 36). Therefore, AMPK activation may mitigate microvascular complications and reduce macrovascular events in T2D.
As part of the energy-sensing mechanism of AMPK, ADP and AMP, in competition with ATP, increases AMPK activity by suppressing the dephosphorylation of p-T172 AMPKα (pAMPK) of the AMPKα, -β, and -γ hetereotrimer, which is a key mechanism to increase AMPK activity (37). In addition, AMP, but not ADP, binds to sites in the γ subunits to allosterically activate AMPK (37). Recently, compounds PF-793 and MK-8722 that interact with the allosteric drug and metabolite (AdAM) site located between α- and β subunits and allosterically activated both β1- and β2-containing AMPK trimers were described (15, 16). However, PF-793 showed a very limited chronic effect on blood glucose levels (15), and MK-8722 promoted cardiac hypertrophy and glycogen accumulation in preclinical species (16), making clinical development a challenge.
We here describe the identification of a PAN-AMPK activator, denoted O304, which increased AMPK activity by suppressing the dephosphorylation of pAMPK. In DIO mice, O304 increased glucose uptake in skeletal muscle, reduced β cell stress, and promoted β cell rest. Consistently, in a 28-day proof-of-concept phase IIa clinical trial in T2D patients treated with Metformin, O304 reduced fasting plasma glucose (FPG) and homeostasis model assessment of insulin resistance (HOMA-IR), and it was well tolerated. Moreover, O304 improved peripheral microvascular perfusion and reduced blood pressure both in animals and T2D patients. O304 also activated AMPK in the heart, increased cardiac glucose uptake, reduced cardiac glycogen levels, and improved LV stroke volume in mice, but it did not increase heart weight in mice or rats. Thus, the major metabolic and vascular effects in animals translated to T2D patients.
Results
O304 suppresses dephosphorylation of pAMPK in vitro and acts as a PAN-AMPK activator in cells.
We used a cellular screen to identify O304 (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.99114DS1) that, instead of allosterically activating AMPK, increased pAMPK levels without reducing cellular ATP. Consistently, in vitro O304 suppressed protein phosphatase 2C–mediated (PP2C-mediated) dephosphorylation of p-T172 of human recombinant AMPKα, -β, and -γ trimers (Figure 1A) without inhibiting the activity of PP2C (Supplemental Figure 2A). O304 also protected pAMPK from dephosphorylation in the presence of excess ATP (Figure 1B) and acted in an additive manner with ADP (Supplemental Figures 2, B and C), but it did not allosterically activate AMPK (Supplemental Figure 2D). Thus, O304 mimicked the effects of ADP, but not of AMP, on AMPK activity (37).
Figure 1. O304 increases p-T172 AMPK in vitro and increases p-T172 AMPK and ATP in cells.
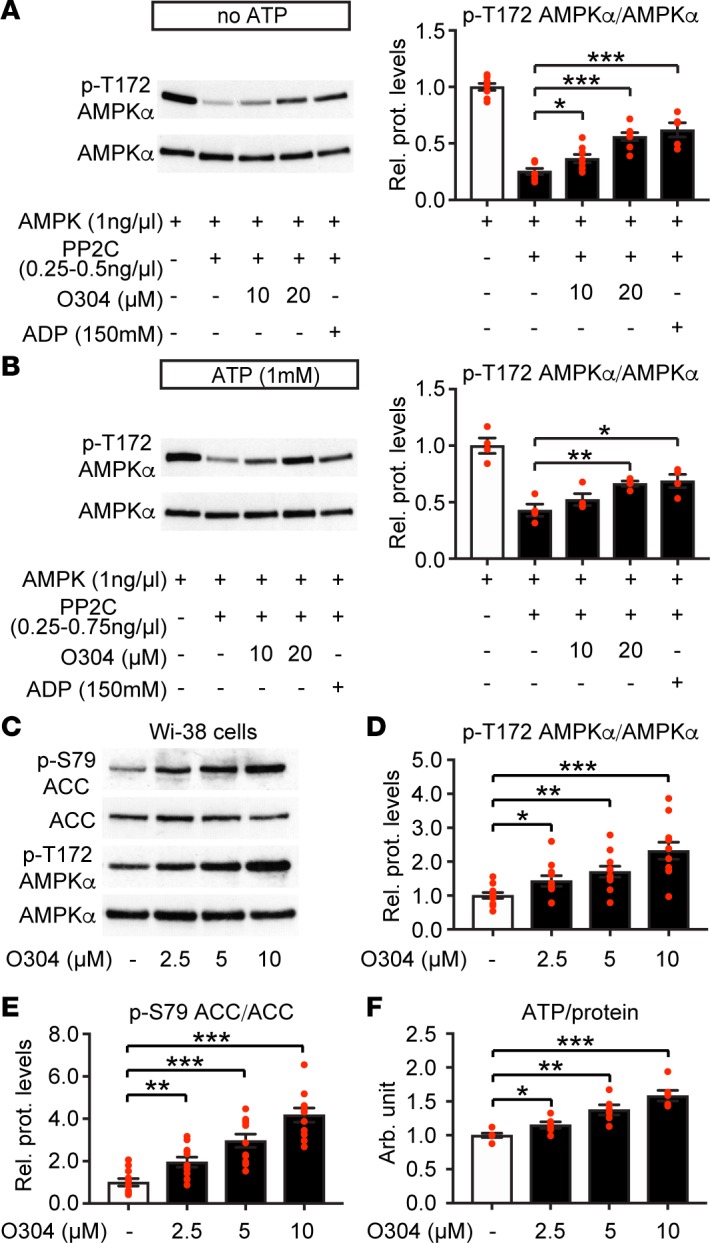
(A and B) Representative immunoblot analysis and quantification of O304 dose-dependent suppression of PP2C-mediated dephosphorylation of p-T172 AMPK in absence (A) (n = 8 per condition) and presence (B) (n = 4 per condition) of 1.0 mM ATP. (C–E) Representative immunoblot analysis (C) and quantification of O304 dose-dependent increase of p-T172 AMPK (D) and p-S79 ACC (E) phosphorylation (n = 11 per condition) in Wi-38 human lung fibroblast cells. (F) Dose-dependent increase in ATP/protein levels in O304-treated Wi-38 human lung fibroblast cells (n = 6 per condition). Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
In nontransformed human Wi-38 lung fibroblast cells, O304 increased the levels of pAMPK, the downstream target p-S79 ACC (pACC), and the ATP/protein ratio in a dose-dependent manner (Figure 1, C–F). Notably, O304 increased pAMPK in many different cell types containing a variety of different AMPK heterotrimers, which expressed either the β1 or β2 subunit, including cells implicated in T2D, such as human skeletal myotubes and hepatocytes that preferentially express the β2 subunit (Supplemental Figure 2E). Thus, O304 acts as a PAN-AMPK activator in cells. The mechanism of action of O304 requires that cells express the major upstream kinase LKB1. Consistently, in HeLa cells, which are phenotypical LKB1 null (38), O304 failed to increase the very low basal levels of pAMPK and pACC, whereas as a control, the Ca2+ ionophore ionomycin, which activates AMPK via calcium/calmodulin-dependent protein kinase kinase (CaMKK), readily activated AMPK in these cells (Supplemental Figure 2F). Thus, O304 will only further increase AMPK activity in physiologically relevant cells with intrinsic AMPK activity.
O304 prevents insulin resistance and dysglycemia in DIO mice.
In rodents, O304 is orally available with a long plasma half-life (Supplemental Figure 3A) but does not cross the blood-brain barrier (Supplemental Figure 3B). To address whether O304 alone or, as in the clinical setting, in combination with Metformin could mitigate dysglycemia and insulin resistance in vivo, mice were fed a high-fat diet (HFD), denoted DIO mice, and treated by oral gavage with vehicle, O304, Metformin, or O304+Metformin (100 mg/kg/day) each for 8 weeks (w) (Figure 2A). With this regimen, O304 and O304+Metformin, but not Metformin, averted the HFD-provoked increase in fasted glucose and plasma insulin levels (Figures 2, B and C). Consequently, compared with vehicle, O304 and O304+Metformin–treated DIO mice did not develop insulin resistance as assessed by HOMA-IR calculations (Figure 2D). Moreover, in line with the potent prevention of hyperglycemia, hyperinsulinemia, and insulin resistance, O304 and O304+Metformin, but not Metformin, significantly increased pAMPK (Figure 2E), reduced Txnip mRNA levels, and increased Glut1 mRNA levels (Figure 2F) in calf muscle of DIO mice, which is consistent with both insulin-dependent and insulin-independent effects (39–41). In summary, O304 increased pAMPK in calf muscle and potently protected against hyperglycemia, hyperinsulinemia, and insulin resistance in DIO mice; Metformin showed no significant effect, whereas O304+Metformin appeared most effective and significantly reduced HOMA-IR compared with O304 alone.
Figure 2. O304 prevents dysglycemia and insulin resistance in diet-induced obese mice.

(A) Timeline in weeks for B6 mice fed a high-fat diet (HFD) and oral gavaged with vehicle or O304 ± Metformin. (B and C) Fasted glucose (B) and fasted insulin (C) levels in B6 mice on HFD treated with vehicle (n = 10), O304 (n = 10), Metformin (n = 10), and O304+Metformin (n = 10) for 6w. (D) HOMA-IR calculations from B and C. (E) Representative immunoblot analysis and quantification of p-T172 AMPK levels in calf muscle of B6 mice on HFD treated with vehicle (n = 10), O304 (n = 10), Metformin (n = 10), and O304+Metformin (n = 10) for 8w. (F) Relative mRNA levels of Txnip and Glut1 in calf muscle of B6 mice on HFD treated with vehicle (n = 10), O304 (n = 10), Metformin (n = 9), and O304+Metformin (n = 10) for 8w. (G and H) Fasted glucose (G) and insulin (H) levels in B6 mice fed either a regular diet (RD) (n = 40) or a HFD for 7w (=Start; n = 10 + 10). The HFD-fed mice were then continued on HFD and oral gavaged with vehicle (n = 10) or O304+Metformin (n = 10) for an additional 4w. (I) HOMA-IR calculations from G and H. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
In patients with T2D, O304 would be used in combination with Metformin to reduce established hyperglycemia. To mimic these conditions, mice were fed HFD for 7w, which resulted in hyperglycemia and insulin resistance as compared with mice fed a regular diet (RD) (Figures 2, G–I), and were then treated with vehicle or O304+Metformin while continued on HFD for 4w. Whereas a reduction in fasted insulin levels and HOMA-IR was evident after 1w of treatment with O304+Metformin, fasted blood glucose levels were significantly reduced first after 2w of treatment with O304+Metformin as compared with vehicle (Figure 2, G–I). Prolonged treatment reduced blood glucose further and, after 4w of treatment, fasted glucose levels were reduced to those of mice fed RD (Figures 2G). Thus, the metabolic effects of O304+Metformin (i.e., reduction of hyperglycemia) largely resemble the effects of exercise and/or caloric restriction on hyperglycemia observed in man (10, 12).
O304 prevents and reverts diabetes in hIAPPtg DIO mice.
DIO mice become hyperglycemic but not overtly diabetic, and we therefore next explored the effect of O304 in a mouse model mimicking human T2D (i.e., HFD-induced insulin resistance/dysglycemia combined with β cell dysfunction). To this end, we used mice expressing the amyloidogenic human IAPP (hIAPP) gene under control of the rat insulin 2 promoter, denoted hIAPPtg mice, which were fed a HFD diet for 6w (Figure 3A). In hIAPPtg DIO mice, as compared with vehicle, O304 gavaged at 100 mg/kg/day averted the increase in 6h fasted blood glucose and plasma insulin levels (Figure 3, B and C). I.p. (Figure 3D) and oral (Figure 3E) glucose-tolerance tests (GTTs) confirmed that O304 prevented the development of glucose intolerance and compensatory hyperinsulinemia, indicating a relative normalization of insulin hypersecretion that mirrored that of 10w-old hIAPPtg mice on RD (Figure 3F). Additionally, HOMA-IR and the Matsuda index model of whole-body insulin sensitivity showed that O304 suppressed the development of systemic insulin resistance in hIAPPtg DIO mice (Figure 3, G and H).
Figure 3. O304 prevents diabetes in hIAPPtg diet-induced obese mice.

(A) Timeline in weeks for hIAPPtg mice fed a high-fat diet (HFD) and oral gavaged with vehicle or O304. (B and C) Fasted blood glucose (B) and insulin (C) levels in hIAPPtg mice on HFD treated with vehicle (n = 25) and O304 (n = 27) for 6w. (D and E) Blood glucose, plasma insulin profiles, and AUC, during i.p. glucose tolerance test (IPGTT) (D) and oral glucose tolerance test (OGTT) (E) in hIAPPtg mice on HFD treated with vehicle (IPGTT, n = 13; OGTT, n = 7) and O304 (IPGTT, n = 16; OGTT, n = 7) for 6w. (F) Plasma insulin profiles and AUC during IPGTT of 16w-old, hIAPPtg mice on HFD treated with O304 for 6w (from D, n = 16) compared with that of 10w-old hIAPPtg mice on regular diet (RD) (n = 7). (G) HOMA-IR calculations from glucose and insulin levels from B and C. (H) Matsuda index calculations from IPGTT (D) and OGTT (E) in vehicle and O304-treated hIAPPtg mice. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01 (Student’s t test).
To test whether O304 could revert established diabetes and obesity in obese, diabetic hIAPPtg mice, and to avoid potential confounding effects of oral gavage on HFD-induced obesity, we formulated HFD with O304, denoted O304-HFD. To test the dose response effect of O304 on glucose homeostasis, we next fed CBA mice HFD or O304-HFD with 0.4, 0.8, and 2 mg/g of O304 for 7w. In CBA mice, O304-HFD dose-dependently increased pAMPK in calf muscle (Figure 4A) and potently prevented dysglycemia, hyperinsulinemia, and insulin resistance (Figures 4, B–D). To test whether O304 could revert established diabetes, hIAPPtg mice were fed HFD for 9w and then switched to O304-HFD (2 mg/g) for 7w (Figure 4E). At 6w after the switch, O304-HFD had reverted established hyperglycemia, hyperinsulinemia, and insulin resistance — and, thus, diabetes (Figure 4, F–H). Moreover, O304 induced body weight and body fat loss, despite increased food intake (Supplemental Figure 4, A–C).
Figure 4. O304 dose-dependently averts dysglycemia in diet-induced obese mice and reverts diabetes in hIAPPtg diet-induced obese mice.

(A) Representative immunoblot analysis and quantification of p-T172 AMPK levels in calf muscle of CBA mice on high-fat diet (HFD) (n = 10) and O304-HFD with 0.4 (n = 5), 0.8 (n = 10), and 2 mg/g (n = 10) O304 for 7w. (B–D) Fasted blood glucose (B) and fasted insulin (C) levels, as well as HOMA-IR (D; from B and C), in CBA mice on HFD (n = 10) and O304-HFD with 0.4 (n = 5), 0.8 (n = 10), and 2 mg/g (n = 10) O304 for 6w. (E) Timeline in weeks for hIAPPtg mice fed HFD for 9w and then either continued on HFD or switched to O304-HFD (2 mg/g in F–H; 0.8 mg/g in I–M) for an additional 7w. (F–H) Fasted blood glucose (F) and insulin (G) levels, as well as HOMA-IR (H; from F and G), in hIAPPtg mice at start, at 9w, and 15w on HFD (n = 10), and in hIAPPtg mice at start, at 9w on HFD, and at 9w HFD+6w O304-HFD (2 mg/g) (n = 12). (I and J) Body weight (I) and body fat (J) change in hIAPPtg mice on HFD for 15w (n = 12) or HFD for 9w + 6w O304-HFD (0.8 mg/g) (n = 7). (K–M) Fasted blood glucose (K) and insulin (L) levels, and HOMA-IR (M; from K and L) at start, 9w, and 15w in hIAPPtg mice on HFD for 15w (n = 12) and in hIAPPtg mice at start, at 9w on HFD, and at 9w + 6w O304-HFD (0.8 mg/g) (n = 7). Data are presented as mean ± SEM, **P < 0.01, **P < 0.01, ***P < 0.001 (Student’s t test [A–D, I, and J]; paired 2-tailed t test.
The potent effect of O304 on established diabetes and obesity in hIAPPtg mice on HFD formulated with 2 mg/g O304 leaves open the possibility that the beneficial metabolic effects (Figure 4, F–H) observed in these mice are secondary to the effects on weight and body fat (Supplemental Figures 4, A and B). To address this issue, we therefore performed diet switch experiments on hIAPPtg mice using a lower O304-HFD concentration, where mice were fed HFD for 9w and then either continued on HFD or switched to O304-HFD (0.8 mg/g) for 7w. With this regimen, the switch to O304-HFD (0.8 mg/g) for 7w did not provoke body weight or body fat loss (Figure 4, I and J). Nonetheless, at 6w after the switch to O304-HFD (0.8 mg/g), glucose and insulin levels — as well as HOMA-IR — were significantly reduced (Figure 4, K–M), showing that, under these conditions, the beneficial metabolic effects of O304 are independent of any effect on weight and body fat loss. Together, these results show that O304 potently averts insulin resistance, hyperinsulinemia, hyperglycemia, and overt diabetes in a T2D mouse model of obesity-induced diabetes.
O304 increases glucose uptake in skeletal myotubes ex vivo and in skeletal muscle in vivo.
In skeletal muscle, AMPK activation has been implicated both in increasing insulin-independent glucose uptake and in reducing insulin resistance (2, 34, 42–44). Accordingly, in skeletal muscle myotubes, O304 increased 2-Deoxy-D-glucose (2-DG) uptake in a dose- and AMPK-dependent manner in the absence of insulin (Figures 5, A–D). Moreover, using PET analysis of tail vein injection of the radiolabeled glucose analog [18F]-Fluorodeoxyglucose ([18F]-FDG) a significant increase of [18F]-FDG uptake was observed in calf and thigh muscle of mice fed O304-HFD (2 mg/g) for 2w compared with mice fed HFD (Figure 5E), demonstrating that O304 promotes glucose uptake in skeletal muscle in vivo. Taken together, these findings provide evidence that the positive effects of O304 on glucose homeostasis is, at least in part, mediated by O304 stimulation of glucose uptake in skeletal muscle.
Figure 5. O304 increases glucose uptake in skeletal muscle.

(A) 2-deoxy-D-glucose (2-DG) uptake in rat skeletal L6 myotubes treated with O304 as indicated (vehicle, n = 8; 2.5 μM O304, n = 6; 5.0 μM O304, n = 6; and 10.0 μM O304, n = 3). (B–D) Representative immunoblot analysis (B), quantification of AMPK expression (C) (n = 6), and 2-DG glucose uptake (D) in siRNA transfected rat skeletal L6 myotubes treated with O304 as indicated (n = 8 for each condition). (E) [18F]-Fluorodeoxyglucose ([18F]-FDG) levels in calf and thigh muscle of CBA mice on high-fat diet (HFD) (n = 8) or O304-HFD (2 mg/g) (n = 6) for 2w. Data are presented as mean ± SEM, *P < 0.05, ***P < 0.001 (Student’s t test).
O304 reduces β cell stress and promotes β cell rest.
In T2D, toxic IAPP aggregates/amyloid is associated with β cell stress and β cell deterioration (45). In hIAPPtg HFD mice switched from HFD to O304-HFD (2 mg/g) for 7w, the amount of islet amyloid formed was significantly reduced compared with that in mice continued on HFD for 7w (Figures 6, A and B). The reduced amount of amyloid observed in O304-HFD–fed hIAPPtg mice may, however, be secondary to the amelioration of hyperglycemia and insulin resistance. Nonetheless, O304 directly increased pAMPKα in rat insulinoma INS-1 cells, isolated primary mouse WT and hIAPPtg islets, and human islets (Figure 6C). To explore a potential direct effect of O304 on islet cells, we therefore next provoked amyloid formation by culturing isolated primary hIAPPtg islets at high glucose (22 mM) levels. O304 potently, in a dose-dependent manner, attenuated amyloid formation in hIAPPtg islets cultured at 22 mM glucose (Figures 6, D and E). Basal autophagy has been shown to protect β cells from hIAPP oligomer toxicity (28–30, 45, 46) and AMPK activation promotes autophagy. Consistently, O304 enhanced autophagic flux in the β cell line INS-1E (Supplemental Figure 5A) and in the presence of the autophagy inhibitor 3-MA; the preventive effect of O304 on amyloid formation at 22 mM glucose was significantly attenuated (Figures 6, F and G). AMPK activation has, however, also been shown to improve function and survival of metabolically stressed β cells through preservation of ER function (47), and O304 largely prevented an increased expression of unfolded protein response genes (i.e., indicative of ER stress; ref. 48) in primary mouse islets cultured at 22 mM glucose (Supplemental Figure 5B). Thus, O304 averts β cell amyloid formation in an obesity-induced T2D mouse model, as well as in isolated mouse islets cultured ex vivo at high glucose levels. Taken together, our findings suggest that O304 counteracts metabolically induced β cell stress and amyloid formation in vivo both by reducing hyperglycemia and systemic insulin resistance and by enhancing autophagy and/or ER function in β cells, although the exact mechanisms require further analyses.
Figure 6. O304 reduces amyloid formation in hIAPPtg diet-induced obese mice and improves arginine-induced insulin secretion in diet-induced obese mice.

Representative images (A) and quantification (B) of Thio-S+ amyloid deposits in hIAPPtg mice on high-fat diet (HFD) for 16w (n = 9) and in mice on HFD for 9w and then switched to O304-HFD (2 mg/g) for an additional 7w (n = 9). (C) Representative immunoblot analysis and quantification of O304 stimulation of p-T172 AMPK in INS-1 insulinoma cells (vehicle, 2.5 μM and 5 μM O304, n = 9; 10 μM O304, n = 6, per condition, respectively), mouse primary islets (n = 6 per condition), hIAPPtg mouse primary islets (n = 6 per condition), and human islets (n = 8 per condition). (D and E) Representative images (D) and quantification (E) of Thio-S+ amyloid deposits in hIAPPtg islets ex vivo cultured for 96 hours in 11 mM glucose (n = 36 islets), 22 mM glucose (n = 45 islets), and 22 mM glucose with 2.5 μM (n = 42 islets), 5.0 μM (n = 41 islets), and 10 μM O304 (n = 36 islets) as indicated (n = 3 experiments for each ). (F and G) Representative images (F) and quantification (G) of Thio-S+ amyloid deposits in hIAPPtg islets ex vivo cultured for 96 hours in 11 mM glucose (n = 43 islets), 11 mM glucose with 5.0 μM 3-MA (n = 59 islets), 22 mM glucose (n = 54 islets), 22 mM glucose with 5.0 μM O304 (35 islets), and 22 mM glucose with 5.0 μM O304 and 5.0 μM 3-MA (n = 44 islets) as indicated (n = 3 experiments for each). (H) Plasma insulin profiles and AUC following i.p. injection of arginine (1 g/kg) in CBA mice fed a HFD (n = 10) and O304-HFD (0.8 mg/g) (n = 10) for 11w. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
The apparent ability of O304 to reduce β cell stress, likely both indirectly and directly, raises the question of whether O304 also promotes β cell rest that, in turn, would preserve long-term β cell function. Arginine stimulation of insulin secretion assesses first-phase insulin release (i.e., the ready releasable pool of granules) and provides an estimate of functional β cell reserve (49, 50). To assess the effect of O304 on β cell function, we therefore next analyzed arginine stimulation of insulin secretion. Arginine stimulation of insulin secretion was increased 2-fold in mice that had been fed O304-HFD (0.8 mg/g) for 11w compared with that of mice fed HFD (Figure 6H), providing further evidence that O304 mitigates β cell stress and promotes β cell rest, which in turn preserves/restores β cell function.
O304 reduces obesity at thermoneutral conditions and increases energy expenditure.
To further explore the effect of O304 on obesity, we performed crossover experiments. Mice fed HFD for 14 days rapidly gained weight, whereas those fed O304-HFD (2 mg/g) gained almost no weight, although they consumed more food than mice fed HFD during day 1–14 (Figures 7, A and B). When HFD and O304-HFD were switched between these 2 groups of mice, mice that were switched from HFD to O304-HFD at day 15 rapidly started to lose weight, again with a relative increase in food intake; reciprocally, mice that switched from O304-HFD to HFD gained weight while reducing the relative food intake (Figures 7, A and B). We next tested whether O304 induced weight loss at thermoneutrality, and mice transferred from housing temperature to 30°C from day 49 onwards while continued on O304-HFD still averted weight gain, whereas mice fed HFD continued to gain weight (Figure 7A). Moreover, when mice housed at 30°C switched diet from HFD to O304-HFD at day 57, they rapidly started to lose weight; reciprocally, mice that switched from O304-HFD to HFD started to rapidly gain weight (Figure 7A). Under these conditions, only a small (0.2°C) nonsignificant increase in core temperature was observed (37.7°C ±0.12°C in HFD-, n = 5, and 37.9°C ± 0.08°C in O304-HFD-treated mice, n = 5). Thus, O304 also reduces obesity at thermoneutrality.
Figure 7. O304 reverts established obesity at thermo-neutral conditions.

(A and B) Body weight change over time (A) and food intake (B) in CBA mice switched between high-fat diet (HFD) (n = 5) and O304-HFD (2 mg/g) (n = 5) at housing and thermo-neutral conditions as indicated. (C–E) Oxygen consumption (VO2) (C), respiratory exchange ratio (RER) (D), and energy expenditure (EE) rates (E) in CBA mice on HFD (n = 8) and O304-HFD (0.8 mg/g) (n = 8) for 11w. (F) Representative immunoblot analysis and quantification of ATGL and of p-S406 ATGL in inguinal white adipose tissue (iWAT) of CBA mice on HFD (n = 10) and O304-HFD with 0.4 (n = 5), 0.8 (n = 10), and 2 mg/g (n = 10) O304 for 7w. (G) Relative mRNA levels of Atgl, Cpt1b, Ppargc1a, and Cox8b in iWAT of 19w-old CBA mice fed HFD (n = 10) and O304-HFD and 2 mg/g O304 (n = 10) for 7w. (H) Relative mRNA levels of Cd36, Fas, Scd1, Acc1, and Cpt1b in brown adipose tissue (BAT) of 19w-old CBA mice fed HFD (n = 10) and O304-HFD and 2 mg/g O304 (n = 10) for 7w. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
To directly address whether O304 averts obesity by increasing energy expenditure (EE), we measured oxygen consumption (VO2), respiratory exchange ratio (RER), and EE for 3 days in mice that had been fed HFD or O304-HFD (0.8 mg/g) for 11w. VO2 was significantly increased during both light and dark periods in mice on O304-HFD compared with mice on HFD (Figure 7C). RER was significantly decreased at day 2 during the light period and throughout the 3-day measurements during the dark period, providing evidence that mice fed O304-HFD switched their main energy source from carbohydrates to fatty acids (FAs) (Figure 7D). As expected, EE was significantly increased during both light and dark periods (Figure 7E). Taken together, these data strongly suggest that O304 suppresses weight gain by enhancing energy metabolism.
O304 increases ATGL activity and expression of genes associated with FA oxidation in WAT and BAT.
In agreement with reduced body fat, O304-HFD–fed (2 mg/g) mice had markedly lower weights of inguinal white adipose tissue (iWAT) and epididymal WAT (eWAT) fat pads than HFD-fed mice (Supplemental Figure 6A). To reduce WAT depots, lipolysis needs to be enhanced. Desnutrin/Atgl, which encodes the rate-limiting enzyme catalyzing basal triglyceride (TG) hydrolysis is a direct target of AMPK, and phosphorylation of S406 by AMPK increases ATGL activity (51), which should increase lipolysis. Accordingly, O304-HFD increased both p-S406 ATGL levels and Atgl mRNA levels in iWAT (Figure 7, F and G). Moreover, Cpt1b, which increases mitochondrial FA uptake, and Cox8b, which would increase mitochondrial activity/FA oxidation (52), were also increased in iWAT of mice fed O304-HFD (2 mg/g) compared with mice fed HFD (Figure 7G). O304 slightly, in a dose-dependent manner, reduced UCP1 expression in brown adipose tissue (BAT) as well as the low-level UCP1 expression in iWAT, arguing against ectopic expression of UCP1 in WAT as a mechanism for the antiobesity effect of O304 (Supplemental Figure 6B). Together, these data provide evidence that O304, at least in part, averts obesity by increasing lipolysis and FA oxidation in WAT.
Activation of AMPK in BAT increases FA uptake, metabolic activity, and EE (53, 54). Consistently, compared with HFD fed mice, weights of BAT pads were reduced in mice fed O304-HFD (2 mg/g) (Supplemental Figure 6A), indicative of increased BAT metabolic activity. Notably, O304-HFD (2 mg/g) significantly increased the expression of Cd36, indicating enhanced FA uptake, as well as that of Cpt1b and potently reduced the expression of genes encoding FA synthase (Fas), Stearoyl-CoA desaturase 1 (Scd1), and Acc1 in BAT of DIO mice, which — in combination — should reduce de novo lipogenesis (DNL) and increase mitochondrial FA uptake/oxidation in BAT (Figure 7H). Moreover, recent results provide evidence that heat can be produced in brown fat without intracellular lipolysis and that BAT can take up and burn FAs derived from lipolysis in WAT pads (55, 56). Taken together, these findings leave open the possibility that an increase in both WAT and BAT activity in combination promote an increase in EE and reduced fat/body weight in O304-treated DIO mice.
Increased lipolytic flux from WAT to the liver may cause fatty liver (17). However, O304 dose-dependently suppressed lipid synthesis in human primary hepatocytes (Supplemental Figure 6C). O304 also reduced, by ~45%, hepatic DNL (Supplemental Figure 6D); dose-dependently increased Cpt1b and decreased Acc2, Fas, and Scd1 mRNA (Supplemental Figure 6E) levels in livers of DIO mice; and prevented and reduced hepatic steatosis in DIO mice (Supplemental Figures 6, F and G).
O304 increases cardiac pAMPK levels, increases stroke volume, and reduces cardiac glycogen but does not induce cardiac hypertrophy.
Exercise activates AMPK in the heart, increases glucose uptake, and reduces cardiac glycogen levels (35, 36). Compared with mice fed HFD, mice fed O304-HFD at 0.8 or 2 mg/g for 7w showed significantly increased pAMPK levels in the heart (Supplemental Figure 7A), and heart glycogen content was reduced in a dose-dependent manner (Figure 8A). In a separate experiment, a significant increase of [18F]-FDG uptake was observed in the hearts of mice fed O304-HFD (2 mg/g) for 2w compared with mice fed HFD (Figure 8B). Thus, the cardiac effects of O304 resemble the cardiac effects of exercise (35, 36). Compared with HFD, O304-HFD at 0.8 or 2 mg/g for 7w did not cause an increase in heart weight/tibia length (Figure 8C). Moreover, rats fed RD and gavaged for 6 months with O304 at 100, 300, and 600 mg/kg/day did not show increased heart/brain weight compared with vehicle (Supplemental Figure 7B). Therefore, O304-mediated AMPK activation in heart did not cause cardiac hypertrophy.
Figure 8. O304 reduces heart glycogen and improves stroke volume in diet-induced obese mice but does not cause cardiac hypertrophy.

(A) Heart glycogen content in CBA mice fed high-fat diet (HFD) (n = 10) and O304-HFD with 0.8 (n = 10), and 2 mg/g (n = 10) O304 for 7w. (B) [18F]-Fluorodeoxyglucose ([18F]-FDG) levels in heart of CBA mice fed HFD (n = 8) or O304-HFD (2 mg/g) (n = 6) for 2w. (C) Heart weight in CBA mice fed HFD (n = 10) and O304-HFD with 0.8 (n = 10) and 2 mg/g (n = 10) O304 for 7w. (D–F) End-diastolic volume (EDV) (D), end-systolic volume (ESV) (E), and stroke volume (SV) (F) in 16w-old CBA mice fed regular diet (RD) (n = 9) and in 18w-old CBA mice fed a HFD (n = 9) and O304-HFD with 0.8 mg/g O304 (n = 10) or 2 mg/g (n = 10) O304 for 6w. (G) Heart rate (HR) in 16w-old CBA mice fed RD (n = 9), and 18w-old CBA mice fed HFD (n = 9) and O304-HFD with 0.8 (n = 10) mg/g or 2 mg/g (n = 10) O304 for 6w. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
Exercise improves cardiac function by increasing stroke volume (33). We therefore examined the effects of O304 on LV function by echocardiography in mice fed RD, HFD, or O304-HFD at 0.8 mg/g or 2 mg/kg. Compared with RD, HFD caused a significant reduction in both end-diastolic volume and end-systolic volume and a small, but nonsignificant, decrease in stroke volume (Figures 8, D–F). O304-HFD normalized end-diastolic volume and dose-dependently improved, but did not fully restore, end-systolic volume (Figures 8, D and E). Importantly, O304-HFD 0.8 mg/g and 2 mg/g induced a significant increase (~20%) in stroke volume compared with both RD and HFD (Figure 8F). Notably, under these anesthetized conditions, HFD caused a significant increase in heart rate as compared with RD, whereas O304-HFD at 0.8 mg/g normalized and O304-HFD at 2 mg/g reduced heart rate (Figure 8G). Thus, O304 normalized the HFD-induced decrease in end-diastolic volume and induced a significant increase in stroke volume, indicating that O304 mimics the beneficial effects of exercise on LV function.
O304 improves microvascular function and endurance capacity in mice and reduces blood pressure in dogs.
Reduced microvascular function and peripheral blood flow cause severe complications in T2D (34). AMPK activation in endothelial and smooth muscle cells promotes vasodilation (31, 34), and AMPK activator 5-aminoimidazole-4-carboxyamide-1-β-D-ribofuranoside (AICAR) increases microvascular perfusion in muscle (57, 58). Thus, to elucidate a potential effect of O304 on peripheral blood flow, we used laser doppler imaging to monitor blood perfusion in left hind paws of DIO mice gavaged with vehicle or with O304 (40 mg/kg/day) for 8w. Under these conditions and without affecting body weight (mice on vehicle increased from 24.5–34 g and mice on O304 from 25.6–36.5 g), O304 — compared with vehicle — significantly increased microvascular blood flow in hind legs (Figures 9, A and B). In support of the notion that O304 increases microvascular blood flow, which would increase dissipation of heat, skin-surface temperature was increased in O304-treated Zucker rats (Supplemental Figure 8, A and B).
Figure 9. O304 improves microvascular blood flow and endurance in mice.

(A and B) Representative laser Doppler image (A) and quantification (B) of peripheral blood fusion in left hind paw in vehicle- (n = 10) and O304-treated (n = 10) B6CBAF1/J (F1) mice on 8w high-fat diet (HFD). (C and D) Endurance test (C) and lactate levels (D) after endurance test in vehicle- (n = 14) and O304-treated (n = 14) aged, lean B6 mice after 30 days of O304 treatment. (E and F) Systolic (E) and diastolic (F) blood pressure in dogs single dosed with vehicle or O304 at indicated concentrations. Data are presented as mean ± SEM, *P < 0.05, #P < 0.05, **P < 0.01, ##P < 0.01, ***P < 0.001, ###P < 0.001 (Student’s t test). In E and F, * refers to vehicle versus 540 mg/kg O304 and # refers to vehicle versus 180 mg/kg O304.
Enhanced cardiovascular function is associated with improved endurance in humans and animals (59, 60). To test whether O304 could improve endurance, we monitored running distance to exhaustion, and to avoid the confounding effect of varying degrees of obesity, we used weight-matched, 14-month-old mice fed RD and gavaged with vehicle or with O304 (20 mg/kg/day) for 30 days. The treadmill exercise reduced body weights to a similar extent in mice on vehicle (from 33.4 to 31.4 g) and in mice on O304 (from 34.1 to 32.2 g). Compared with vehicle, O304 significantly improved endurance capacity monitored as running distance to exhaustion (Figure 9C), while significantly reducing the increase in blood lactate levels (Figure 9D), indicating increased oxidative metabolism. Thus, consistent with the observed beneficial cardiovascular effects in DIO mice, O304 improves endurance capacity in lean sedentary aged mice fed RD.
The AMPK activator AICAR has been shown to acutely lower blood pressure and relax isolated resistance arteries of hypertensive rats (61). Thus, as part of the investigative new drug toxicological package, a telemetric study in conscious dog after single doses of O304 was conducted, and under these conditions, O304 acutely reduced blood pressure (Figures 9, E and F). Thus, O304 improves cardiac stroke volume, increases microvascular perfusion, and reduces blood pressure.
O304 improves glucose homeostasis in T2D patients on Metformin.
Based on the beneficial metabolic and cardiovascular effects of O304 in preclinical species, O304 was selected for clinical development, and toxicological studies in rat and dog and a phase I safety clinical trial was successfully concluded. Thus, an exploratory 28-day proof-of-concept phase IIa clinical trial, denoted TELLUS, of O304 in 65 T2D patients stably on Metformin was performed. Apart from safety, FPG, insulin, and blood pressure were monitored, and microvascular perfusion in calf muscle was examined by MRI.
T2D patients needed to perform and pass MRI examinations before start of treatment to be included in the TELLUS study; therefore, HbA1c ≥6.5% and ≤9.0% at screening, and not FPG at day 1, was used as inclusion criteria. Thus, a post hoc analysis was conducted of patients with FPG range >7 to <13.3 mmol/l, (>126 to <240 mg/dl) at day 1 (Supplemental Figure 9A), where 13.3 mmol/l (240 mg/dl) represents uncontrolled hyperglycemia. The mean absolute reduction in FPG at day 28 compared with day 1 was –0.10 mM in the placebo group and –0.60 mM in the O304 group (Figures 10, A and B, and Supplemental Figure 9A). In the Wilcoxon’s rank sum test there was a statistically significant absolute (P = 0.010) and relative (P = 0.018) reduction in FPG in the O304 group compared with the placebo group, with P = 0.049 for absolute change in the mix model ANOVA 2-way test and P = 0.037 for relative change in the mix model ANOVA 1-way test. In the Wilcoxon test within the O304 group, but not the placebo group, there was significant absolute (P = 0.0002) (Figure 10A) and relative (P = 0.0003) reduction in FPG at day 28 compared with day 1. In DIO mice, a significant reduction in fasting blood glucose is observed after 2w of treatment with O304+Metformin, and efficacy increases with duration of treatment (Figure 2G). Thus, any effect of O304 on FPG in T2D patients would likely take at least 2w to observe. Consistently, the significant reduction in FPG within in the O304 group occurred between day 21 and day 28 (Figure 10B), which is in accordance with the corresponding 14-day timeframe in DIO mice (Figure 2G). Moreover, due to the long plasma t1/2 of O304, the plasma steady-state concentration is not reached until day 14 in T2D patients (Supplemental Figure 9B). Notably, in the Wilcoxon test within the O304 group, but not the placebo group, a statistically significant both absolute (P = 0.0097) and relative (P = 0.017) reduction in HOMA-IR were observed at day 28 compared with day 1 (Figure 10C). Thus, O304 improved glucose homeostasis in T2D patients on Metformin.
Figure 10. O304 reduces fasting plasma glucose and blood pressure and increases microvascular perfusion in type 2 diabetes (T2D) patients on Metformin.

(A–C) Fasting plasma blood glucose (FPG) (A and B) and HOMA-IR (C) at day 1 and day 28 in placebo- (n = 24) and O304-treated (n = 25) T2D patients on Metformin with the FPG range >7 to <13.3 mmol/l (>126 to <240 mg/dl) at day 1. (D) Hyperemic microvascular perfusion assessed by dynamic T2*-quantification monitored by MRI at screening (MRI1) and at day 27–29 (MRI2) in calf muscle of the T2D patients. The O304 group and the placebo group were split in half based on the time-to-peak (TTP) at baseline, where short TTP (placebo A [n = 14], O304 A [n = 14]) and long TTP (placebo B [n = 13], O304 B [n = 14]) represent a relative higher and lower rate of hyperemic perfusion, respectively. A significant shortening of TTP (P = 0.043) and increase in Δ-T2* (P = 0.034) was observed in subjects with relative lower rate of perfusion at baseline (long TTP) in the O304 group (i.e., comparing O304 B MRI1 with O304 B MRI2) but not in subjects with short TTP, and there was no difference in subjects with either short or long TTP at baseline in the placebo group. (E) Absolute and relative change in systolic and diastolic blood pressure from day 1–28 in T2D patients on Metformin treated with placebo (n = 27) or O304 (n = 30). Data are presented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 (Signed Wilcoxon’s rank sum test).
O304 increases peripheral microvascular perfusion in calf muscle of T2D patients on Metformin.
Since T2D is associated with severe microvascular complications (4) and O304 increased peripheral microvascular perfusion in mice, hyperemic microvascular perfusion was monitored in the TELLUS study by MRI and dynamic T2*-quantification (the time constant for transversal relaxation caused by local magnetic-field inhomogeneities) at screening and at days 27–29 in calf muscle of the T2D patients. The obtained time graphs of T2* values were analyzed on an individual basis, and a set of parameters were extracted via automated curve fitting. As expected, when compared with the literature (62), the peripheral circulation status of the patients in the TELLUS study was at large not depressed at baseline, and a strong intervention effect signal could not be expected. Nevertheless, at day 28 compared with baseline in the 2-way ANOVA test, there was a statistically significant increase in Δ-T2* (P = 0.026) in the O304 group compared with the placebo group, defined as the difference between the minimum ischemic value and the peak hyperemic value, indicating increased hyperemic perfusion. Moreover, in the Wilcoxon test, there was a significant relative increase in the T2* gradient (P = 0.012), defined as the rate of increase of hyperemic perfusion in the O304 group but not in the placebo group at day 28 compared with baseline. However, in subjects with comparably reduced peripheral circulation, peaks were poorly defined, making it difficult to correctly identify the peak properties following reactive hyperemia. Thus, in a post hoc analysis, curve fitting was instead performed at the group level, since image noise and signal drift was averaged over many subjects, and significance testing was performed by means of a permutation test, a nonparametric resampling technique (63). Under these conditions, compared with the placebo group, there was a significant increase in both Δ-T2* (P = 0.037) and T2* gradient (P = 0.024) at day 28 compared with baseline in the O304 group (Table 1).
Table 1. O304 increases microvascular perfusion in calf muscle of T2D patients on Metformin as assessed by changes in T2*-gradient and Δ-T2* at day 28 compared with baseline.

Finally, to elucidate whether it was subjects with relatively lower perfusion at baseline that responded to treatment, the O304 group and the placebo group were split in half based on the time-to-peak (TTP) at baseline, where short TTP and long TTP represent a relative higher and lower rate of hyperemic perfusion, respectively. MRI at baseline (MRI1) compared with MRI at end of treatment (MRI2) was then investigated with permutation analysis. From this stratified analysis, a significant shortening of TTP (P = 0.043) and increase in Δ-T2* (P = 0.034) was observed in subjects with a relative lower rate of perfusion at baseline (long TTP) in the O304 group, but not in subjects with short TTP, and there was no difference in subjects with either short or long TTP at baseline in the placebo group (Figure 10D). Thus, O304 preferentially increases hyperemic microvascular perfusion in calf muscle of T2D patients with a relative lower rate of perfusion at baseline.
O304 reduces blood pressure in T2D patients on Metformin.
Microcirculation regulates peripheral vascular resistance, which — in combination with cardiac output — determines arterial blood pressure (64). AICAR acutely reduced blood pressure in spontaneously hypertensive rats (61), and O304 acutely reduced blood pressure in dogs (Figure 9, E and F). Consistently, a mean absolute reduction in systolic (–5.8 mmHg) and in diastolic (–3.8 mmHg) blood pressure was observed at day 28 compared with day 1 in the O304 group, whereas small increases of +1.2 mmHg and +0.9 mmHg, respectively, were observed in the placebo group. In the Wilcoxon test, within the O304 group but not the placebo group, there was a statistically significant absolute reduction in both systolic (P = 0.030) and diastolic (P = 0.009) blood pressure and relative reduction in systolic (P = 0.036) and diastolic (P = 0.014) blood pressure (Figure 10E). In the 1-way ANOVA test, there was a statistically significant relative reduction in systolic blood pressure (P = 0.047) and diastolic blood pressure (P = 0.044) in the O304 group, compared with the placebo group. No significant change in mean heart rate was observed in either group at day 28 compared with baseline: placebo, –0.48; O304, –1.6 bpm. Thus, O304 reduces systolic and diastolic blood pressure in people with T2D. Hence, the effects of O304 on FPG, microvascular perfusion, and blood pressure translate from animals to T2D patients.
Discussion
With time, T2D patients often fail to reach their glycemic targets despite the use of multiple glucose-lowering agents, leading to cardiovascular diseases with significant increased morbidity and mortality (8). Potentially novel treatments that can (a) increase glucose disposal, (b) relieve β cell stress and impose β cell rest to preserve long-term β cell function, and (c) reduce cardiovascular complications in T2D patients are, thus, highly desirable. AMPK is activated by exercise that enhances cardiovascular function and endurance (33, 35, 36), and pharmacological AMPK activation increases glucose uptake in skeletal muscle (15, 16, 44) and may preserve β cell function (65). However, no direct AMPK activator is currently in clinical use. By monitoring clinically relevant parameters in DIO and aged mice, and in T2D patients in a 28-day proof-of-concept phase IIa clinical trial (TELLUS), we here report on the clinical utility of PAN-AMPK activator O304.
We show that O304, like AMPK activators PF-793 and MK-8722 (15, 16), increased glucose uptake in skeletal myotubes ex vivo in an AMPK-dependent but insulin-independent manner. In DIO mice, O304 increased pAMPK levels and stimulated glucose uptake in skeletal muscle and potently reduced hyperglycemia, hyperinsulinemia, and insulin resistance. O304 also prevented amyloid formation in hIAPPtg mice fed a HFD and in ex vivo cultured hIAPPtg islet cells. Moreover, O304 averted increased UPR gene expression in mouse islets ex vivo cultured at high glucose levels, indicating preservation of ER homeostasis. O304 also improved the insulin secretory response to arginine in DIO mice in vivo, indicative of β cell rest. Thus, O304 increased glucose disposal, which indirectly reduced the demand on β cells for insulin secretion, and also directly reduced β cell stress, including amyloid formation, under glucotoxic conditions. In the clinic, O304 should be used in combination with Metformin, and in DIO mice, O304+Metformin significantly reduced fasted blood glucose levels. Consistently, O304 reduced FPG and HOMA-IR in T2D patients on Metformin. Moreover, the reduction in FPG in T2D patients treated with O304 occurred between day 21 and day 28, indicating that increased duration of treatment of T2D patients with O304 will further reduce FPG.
O304 increased VO2 and EE, and at the highest dose tested, O304 rapidly and potently both prevented and reduced obesity in DIO mice, whereas — at a lower dose that showed no effect on obesity — O304 still reduced established dysglycemia. The reduction in obesity at higher doses of O304 is likely, however, to further enhance the antiglycemic effect of O304. O304 appears to increase EE, thus averting obesity, by increasing BAT metabolic activity rather than via browning of WAT. Notably, recent results provide evidence that heat can be produced in brown fat without intracellular lipolysis and that BAT can take up and burn FAs derived from lipolysis in WAT pads (55, 56). Moreover, selective inactivation of AMPK adipocytes demonstrate a role for AMPK in ensuring mitophagy, and thus mitochondrial integrity and function, in BAT (66). Together, these findings leave open the possibility that an increase in both WAT and BAT activity, in combination, promote an increase in EE and reduced fat/body weight in O304-treated DIO mice. Whether O304 also stimulates additional pathways that increase UCP1-independent uncoupling in WAT or increase basal physical activity will require additional analyses.
Strict glycemic control reduces microvascular complications; however, no currently used antiglycemic agent directly increases microvascular perfusion (32). Exercise, as well as AMPK activation by AICAR and the indirect AMPK activator R118, increases microvascular perfusion in muscle (57, 58), and O304 increased microvascular perfusion in hind legs of DIO mice and in calf muscle of T2D patients in the TELLUS study. Therefore, by improving glycemic control and by directly increasing microvascular perfusion, O304 may reduce microvascular complications associated with T2D. Peripheral arterial disease (PAD) is common among the elderly (67) and a common initial manifestation of cardiovascular disease in T2D patients (68). Moreover, the hyperemic response in calf muscle, monitored by MRI T2* analysis, is highly correlated with disease severity in PAD patients (62). Thus, by increasing microvascular perfusion in calf muscle, O304 may show beneficial effects on PAD both in patients with or without T2D.
LV diastolic dysfunction (i.e., decreased relaxation/filling and reduced stroke volume) are frequently observed in T2D, and T2D is a risk factor for heart failure (69). Like exercise, O304 increased cardiac pAMPK levels, glucose uptake, and stroke volume and reduced cardiac glycogen content, but it neither induced cardiac hypertrophy in DIO mice nor increased heart/brain weight in rats treated with high daily doses of O304 for 6 months. Moreover, in the TELLUS study, O304 did not cause any ECG abnormalities or any effect on LV mass. Thus, O304-mediated AMPK activation in heart mimics the effects of exercise (59, 60) and not the adverse cardiac effects of PRKAG2 mutations (34). In line with these effects, O304 further increased pAMPK levels only in cells that express the upstream kinase LKB1 and irrespective of expression of AMPK β1 or -β2 subunits. Whether O304 protects against the dephosphorylation of pAMPK by the same mechanism as ADP (i.e., by interacting with sites in the γ subunits) or binds to a distinct site in the AMPK trimer will the subject of future studies but is beyond the immediate scope of the current study. Nevertheless, O304 may be beneficial in mitigating T2D-associated cardiovascular diseases, including heart failure. Further clinical studies are, however, required to elucidate these possibilities.
MK-8722, which interacts with the AdAM site to allosterically activate AMPK, caused reversible cardiac hypertrophy and glycogen accumulation in several preclinical species, indicating an imbalance in synthesis and breakdown of glycogen (16). MK-8722 did not, however, cause the ECG changes associated with PRKAG2 mutations, and although no increase in stroke volume was observed in rats, the effects were rather reminiscent of cardiac hypertrophy observed in elite athletes (16, 33). Moreover, MK-8722 allosterically activated AMPK even in the absence of p-T172 AMPK and showed a ~10 fold preference for β1- over β2-containing AMPK trimers (16), indicating a nonphysiological (over) activation of cardiac AMPK by MK-8722.
Limitations of the study.
The beneficial metabolic, cardiac, and vascular effects of O304 in preclinical species require different doses and different times of exposure of O304, and the aim of TELLUS as a short (28 days), small (~60 patients), exploratory, single-dose phase IIa study was to explore whether all or some of the beneficial preclinical effects of O304 translated to T2D patients. Although the study groups were generally well balanced with regard to age and sex, there was a numerical imbalance in weight; the placebo group’s mean weight being approximately 8 kg higher (93 kg, BMI 31) than the group treated with O304 (85 kg, BMI 28). In addition, due to MRI analyses, HbA1c at screening, not FPG at baseline, was used as an inclusion criterion. Subsequently, a wide range of FPG values, both <7 to >13.3 mmol/l, were observed at baseline. In patients with FPG range >7 to <13.3 mmol/l (>126 to <240 mg/dl), there was an imbalance in FPG and HOMA-IR at day 1, with higher FPG but lower HOMA-IR/insulin resistance in the O304 group compared with the placebo group. Compared with other antiglycemic drugs currently used in the clinic (70), the reduction in FPG in the O304 group at day 28 compared with day 1 FPG was relatively modest. However, since a main preclinical effect of O304 on glucose homeostasis is reduction of obesity-induced insulin resistance in mice, the relative low weight/BMI and low HOMA-IR but high FPG at day 1 of T2D patients treated with O304 have likely impacted the study results.
The increase in glucose uptake in skeletal muscle and reduction in insulin resistance by O304 in DIO mice may, at least in part, require rewiring of gene expression in target tissues. In line with this idea, it takes ~2w for O304 to start to significantly reduce FPG in DIO mice. Accordingly, the reduction in FPG in T2D patients starts to occur after day 21 and is significant at day 28, and the reduction in HOMA-IR also indicates that reduced insulin resistance is part of the mechanism. Moreover, due to the long plasma t1/2 of O304, the mean plasma steady-state concentration of O304 is not reached until day 14 in the T2D patients. Thus, collectively, these results also indicate that 28 days of treatment may be too short to observe the full effect of O304 on glucose homeostasis in T2D patients and that increased duration of treatment with O304 will further reduce FPG. Moreover, O304 was administered as a suspension in the phase IIa study, and work to develop a more efficacious and suitable formulation for use in future clinical trials has been initiated.
The T2D patients in the phase IIa study were treated with Metformin, which appears to act primarily in the liver to reduce gluconeogenesis but which also seems to increase glucose uptake in the intestine (71). Indirect activation of AMPK by Metformin may contribute to these effects, raising the question of whether the efficacy of O304 may be limited if Metformin-treated patients already have some degree of AMPK activation. Such potential blunting of O304’s effect by Metformin appears, however, less plausible, since O304+Metformin was more potent than either compound alone in DIO mice and since — due to the requirement for active transport — Metformin is unlikely to have any major effect on glucose uptake and/or insulin resistance in skeletal muscle.
In summary, by increasing AMPK activity, O304 mimics the beneficial metabolic and cardiovascular effects of long-term exercise and caloric restriction, and our findings provide evidence that O304 holds great potential as treatment for T2D and associated cardiovascular complications, especially for the recently described subgroup of T2D patients with severe insulin resistance who are more likely to develop end-stage renal disease and coronary events (9). Thus, our results support the further development of O304 for treatment of T2D and associated cardiovascular complications, including PAD.
Methods
Animals.
The different cohorts of mice and tests performed are presented in Supplemental Methods.
Clinical study design.
An exploratory proof-of-concept randomized, parallel-group, double-blinded, placebo-controlled phase IIa 28-day study (TELLUS) of the first-in-class AMPK activator O304 (1,000 mg/day) was conducted in 65 T2D patients on Metformin for ≥3 months, aiming at further exploring safety of O304 and the effect of O304 on FPG at a single-dose level. The predefined inclusion and exclusion criteria and further details of the clinical study are described in the Supplemental Methods.
O304.
The small hetercyclic compound O304, developed by Betagenon AB, is detailed in Supplemental Methods.
Glucose tolerance and insulin secretion tests.
GTT and GSIS were performed on 6h fasted and sedated Hypnorm (Veta Pharma)/Midazolam (Hamlenmice) following i.p. or oral administration of glucose (2 g/kg body weight; Gibco) (see also Supplemental Methods).
Cell culture and isolation and in vitro culture of islets.
For detailed information on islets, cell lines, culture, and assay conditions, see Supplemental Methods and Supplemental Table 1.
Western blot analysis.
All Western blot expression data was normalized toward AMPKα, β-actin, GAPDH, or the respective nonphosphorylated counterpart. For detailed information and antibodies used, see Supplemental Methods and Supplemental Table 2.
Quantitative PCR.
For detailed information and primers used for quantitative PCR, see Supplemental Methods and Supplemental Table 3.
Quantification analyses.
Quantification of Western blot experiments was performed using Image Lab (Bio-Rad, version 4.1, build 16) and ImageJ software (version 1.45s). Amyloid content quantification was performed using ImageJ software (version 1.49m).
Statistics.
All the statistical analyses of in vitro and in vivo data were performed by 2-tailed Student’s t tests. We considered a value of P < 0.05 to be statistically significant. Patient data analyses were performed by the mix model ANOVA test (throughout, 2-way ANOVA test was used for absolute changes and 1-way ANOVA test for percentage changes) and the nonparametric Wilcoxon Rank Sum test.
Study approval.
Animal experiments were approved by the Animal Review Board at the Court of Appeal of Northern Norrland in Umeå (approval numbers A40-12, A75-14, A17-12, A74-12, A29-16, and A11-17) and conducted in accordance with Guidelines for the Care and Use of Laboratory animals. The TELLUS trial is registered with ClinicalTrials.gov (NCT00508287) and ClinicalTrials.gov (NCT01167881). The study protocol was approved by the Regional Ethics Commitee in Uppsala, Sweden (project O304-2016-02).
Author contributions
PS and E. Lindahl contributed to the design and performance of experiments, and to the writing and editing of the manuscript. UD, JS, FB, E. Lidh, EK, JW, ME, EB, IL, IB, KL, and BE contributed to the performance of experiments and editing of the manuscript. TE and HE designed and directed the research, secured funding, analyzed and interpreted the data, and wrote the manuscript. HE is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Supplementary Material
Acknowledgments
We thank members of the laboratory of author HE for insightful discussions and Jan Axelsson (Department of Radiation Sciences, Umeå University) for assistance with PET imaging data analyses, and we acknowledge the facilities and technical assistance of Umeå Center for Comparative Biology (UCCB), Umeå University. The research leading to these results has received funding from the Knut and Alice Wallenberg foundation (KAW 2015.0278) to HE and (KAW 2012.0060, KAW 2015.0295) to HE and TE, as well as from Diabetes Wellness Network Sweden (757_2015PG) to HE. ME was supported by grants from the European Commission’s Seventh Framework Programme (HEALTH 2013-2.4.2-1/602936; project CarTarDis). Betagenon AB and TE have received funding from EU’s research and innovation framework program Horizon 2020 (EU project 754268, AMPK-DIAB). We also acknowledge support from the Strategic Research Program in Diabetes at Umeå University.
Version 1. 06/21/2018
Electronic publication
Funding Statement
KAW 2015.0278; 2012.0060; 2015.0295
757_2015PG
HEALTH 2013-2.4.2-1/602936; project CarTarDis
project 754268-AMPK-DIAB
Footnotes
Conflict of interest: HE is a cofounder, shareholder, and consultant of the unlisted biotech company Betagenon AB. TE is employed by, a cofounder, shareholder, and consultant of the unlisted biotech company Betagenon AB. EB, IL, IB, KL, and BE are all employed by, and hold shares in, the unlisted biotech company Betagenon AB. JW is a consultant and shareholder in Betagenon AB, and UD also holds shares in Betagenon AB.
Reference information: JCI Insight. 2018;3(12):e99114. https://doi.org/10.1172/jci.insight.99114.
Contributor Information
Pär Steneberg, Email: par.steneberg@umu.se.
Emma Lindahl, Email: emma.lindahl@umu.se.
Ulf Dahl, Email: ulf.dahl@umu.se.
Emmelie Lidh, Email: emmelie.lidh@umu.se.
Jurate Straseviciene, Email: jurate.straseviciene@umu.se.
Elisabet Kjellkvist, Email: elisabet.kjellkvist@umu.se.
Eva Berggren, Email: eva.berggren@betagenon.com.
Ingela Lundberg, Email: ingela.berglund-dahl@betagenon.com.
Ingela Bergqvist, Email: ingela.bergqvist@betagenon.com.
Madelene Ericsson, Email: madelene.ericsson@umu.se.
Björn Eriksson, Email: bjorn.eriksson@betagenon.com.
Kajsa Linde, Email: kajsa.linde@betagenon.com.
Jacob Westman, Email: jacob.westman@medchemcon.com.
Thomas Edlund, Email: thomas.edlund@umu.se.
Helena Edlund, Email: helena.edlund@umu.se.
References
- 1.DeFronzo RA, Tripathy D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care. 2009;32 Suppl 2:S157–S163. doi: 10.2337/dc09-S302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jaspers RT, et al. Exercise, fasting, and mimetics: toward beneficial combinations? FASEB J. 2017;31(1):14–28. doi: 10.1096/fj.201600652R. [DOI] [PubMed] [Google Scholar]
- 3.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeFronzo RA, Abdul-Ghani M. Assessment and treatment of cardiovascular risk in prediabetes: impaired glucose tolerance and impaired fasting glucose. Am J Cardiol. 2011;108(3 Suppl):3B–24B. doi: 10.1016/j.amjcard.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Levelt E, et al. Cardiac energetics, oxygenation, and perfusion during increased workload in patients with type 2 diabetes mellitus. Eur Heart J. 2016;37(46):3461–3469. doi: 10.1093/eurheartj/ehv442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795. doi: 10.2337/db09-9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Raalte DH, Verchere CB. Improving glycaemic control in type 2 diabetes: Stimulate insulin secretion or provide beta-cell rest? Diabetes Obes Metab. 2017;19(9):1205–1213. doi: 10.1111/dom.12935. [DOI] [PubMed] [Google Scholar]
- 9.Ahlqvist E, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361–369. doi: 10.1016/S2213-8587(18)30051-2. [DOI] [PubMed] [Google Scholar]
- 10.Stanford KI, Goodyear LJ. Exercise and type 2 diabetes: molecular mechanisms regulating glucose uptake in skeletal muscle. Adv Physiol Educ. 2014;38(4):308–314. doi: 10.1152/advan.00080.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bloem CJ, Chang AM. Short-term exercise improves beta-cell function and insulin resistance in older people with impaired glucose tolerance. J Clin Endocrinol Metab. 2008;93(2):387–392. doi: 10.1210/jc.2007-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim EL, Hollingsworth KG, Aribisala BS, Chen MJ, Mathers JC, Taylor R. Reversal of type 2 diabetes: normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia. 2011;54(10):2506–2514. doi: 10.1007/s00125-011-2204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomes RM, et al. Moderate exercise restores pancreatic beta-cell function and autonomic nervous system activity in obese rats induced by high-fat diet. Cell Physiol Biochem. 2013;32(2):310–321. doi: 10.1159/000354439. [DOI] [PubMed] [Google Scholar]
- 14.Musi N, Goodyear LJ. AMP-activated protein kinase and muscle glucose uptake. Acta Physiol Scand. 2003;178(4):337–345. doi: 10.1046/j.1365-201X.2003.01168.x. [DOI] [PubMed] [Google Scholar]
- 15.Cokorinos EC, et al. Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab. 2017;25(5):1147–1159.e10. doi: 10.1016/j.cmet.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Myers RW, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. 2017;357(6350):507–511. doi: 10.1126/science.aah5582. [DOI] [PubMed] [Google Scholar]
- 17.Samuel VT, Shulman GI. The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest. 2016;126(1):12–22. doi: 10.1172/JCI77812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailey CJ, Tahrani AA, Barnett AH. Future glucose-lowering drugs for type 2 diabetes. Lancet Diabetes Endocrinol. 2016;4(4):350–359. doi: 10.1016/S2213-8587(15)00462-3. [DOI] [PubMed] [Google Scholar]
- 19.Beall C, et al. Loss of AMP-activated protein kinase alpha2 subunit in mouse beta-cells impairs glucose-stimulated insulin secretion and inhibits their sensitivity to hypoglycaemia. Biochem J. 2010;429(2):323–333. doi: 10.1042/BJ20100231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beall C, Watterson KR, McCrimmon RJ, Ashford ML. AMPK modulates glucose-sensing in insulin-secreting cells by altered phosphotransfer to KATP channels. J Bioenerg Biomembr. 2013;45(3):229–241. doi: 10.1007/s10863-013-9509-9. [DOI] [PubMed] [Google Scholar]
- 21.Lim A, et al. Glucose deprivation regulates KATP channel trafficking via AMP-activated protein kinase in pancreatic beta-cells. Diabetes. 2009;58(12):2813–2819. doi: 10.2337/db09-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun G, et al. Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia. 2010;53(5):924–936. doi: 10.1007/s00125-010-1692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pepin É, et al. Pancreatic β-Cell Dysfunction in Diet-Induced Obese Mice: Roles of AMP-Kinase, Protein Kinase Cε, Mitochondrial and Cholesterol Metabolism, and Alterations in Gene Expression. PLoS One. 2016;11(4):e0153017. doi: 10.1371/journal.pone.0153017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ebato C, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008;8(4):325–332. doi: 10.1016/j.cmet.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Jung HS, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008;8(4):318–324. doi: 10.1016/j.cmet.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 26.Knaevelsrud H, Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 2010;584(12):2635–2645. doi: 10.1016/j.febslet.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 27.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shigihara N, et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest. 2014;124(8):3634–3644. doi: 10.1172/JCI69866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J, et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J Clin Invest. 2014;124(8):3311–3324. doi: 10.1172/JCI69625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivera JF, Costes S, Gurlo T, Glabe CG, Butler PC. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J Clin Invest. 2014;124(8):3489–3500. doi: 10.1172/JCI71981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirwany NA, Zou MH. AMPK in cardiovascular health and disease. Acta Pharmacol Sin. 2010;31(9):1075–1084. doi: 10.1038/aps.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smits MM, et al. GLP-1-Based Therapies Have No Microvascular Effects in Type 2 Diabetes Mellitus: An Acute and 12-Week Randomized, Double-Blind, Placebo-Controlled Trial. Arterioscler Thromb Vasc Biol. 2016;36(10):2125–2132. doi: 10.1161/ATVBAHA.116.307930. [DOI] [PubMed] [Google Scholar]
- 33.Scharhag J, Schneider G, Urhausen A, Rochette V, Kramann B, Kindermann W. Athlete’s heart: right and left ventricular mass and function in male endurance athletes and untrained individuals determined by magnetic resonance imaging. J Am Coll Cardiol. 2002;40(10):1856–1863. doi: 10.1016/S0735-1097(02)02478-6. [DOI] [PubMed] [Google Scholar]
- 34.Salt IP, Hardie DG. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway With Key Roles in the Cardiovascular System. Circ Res. 2017;120(11):1825–1841. doi: 10.1161/CIRCRESAHA.117.309633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldfarb AH, Bruno JF, Buckenmeyer PJ. Intensity and duration effects of exercise on heart cAMP, phosphorylase, and glycogen. J Appl Physiol. 1986;60(4):1268–1273. doi: 10.1152/jappl.1986.60.4.1268. [DOI] [PubMed] [Google Scholar]
- 36.Coven DL, et al. Physiological role of AMP-activated protein kinase in the heart: graded activation during exercise. Am J Physiol Endocrinol Metab. 2003;285(3):E629–E636. doi: 10.1152/ajpendo.00171.2003. [DOI] [PubMed] [Google Scholar]
- 37.Hardie DG, Carling D, Gamblin SJ. AMP-activated protein kinase: also regulated by ADP? Trends Biochem Sci. 2011;36(9):470–477. doi: 10.1016/j.tibs.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Chen H, Wang X, Zhao W, Chen JJ. Expression and transcriptional profiling of the LKB1 tumor suppressor in cervical cancer cells. Gynecol Oncol. 2014;134(2):372–378. doi: 10.1016/j.ygyno.2014.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parikh H, et al. TXNIP regulates peripheral glucose metabolism in humans. PLoS Med. 2007;4(5):e158. doi: 10.1371/journal.pmed.0040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson ML, et al. Mechanism by Which Caloric Restriction Improves Insulin Sensitivity in Sedentary Obese Adults. Diabetes. 2016;65(1):74–84. doi: 10.2337/db15-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu N, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49(6):1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narkar VA, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134(3):405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Neill HM. AMPK and Exercise: Glucose Uptake and Insulin Sensitivity. Diabetes Metab J. 2013;37(1):1–21. doi: 10.4093/dmj.2013.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marcinko K, et al. The AMPK activator R419 improves exercise capacity and skeletal muscle insulin sensitivity in obese mice. Mol Metab. 2015;4(9):643–651. doi: 10.1016/j.molmet.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91(3):795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 46.Rivera JF, et al. Human-IAPP disrupts the autophagy/lysosomal pathway in pancreatic β-cells: protective role of p62-positive cytoplasmic inclusions. Cell Death Differ. 2011;18(3):415–426. doi: 10.1038/cdd.2010.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wikstrom JD, et al. AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol Endocrinol. 2013;27(10):1706–1723. doi: 10.1210/me.2013-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen X, Zhang K, Kaufman RJ. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat. 2004;28(1-2):79–92. doi: 10.1016/j.jchemneu.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 49.Sjostrand M, et al. Assessment of beta-cell function in young patients with type 2 diabetes: arginine-stimulated insulin secretion may reflect beta-cell reserve. J Intern Med. 2014;275(1):39–48. doi: 10.1111/joim.12116. [DOI] [PubMed] [Google Scholar]
- 50.Robertson RP, et al. Arginine is preferred to glucagon for stimulation testing of β-cell function. Am J Physiol Endocrinol Metab. 2014;307(8):E720–E727. doi: 10.1152/ajpendo.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim SJ, Tang T, Abbott M, Viscarra JA, Wang Y, Sul HS. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol Cell Biol. 2016;36(14):1961–1976. doi: 10.1128/MCB.00244-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geerling JJ, et al. Metformin lowers plasma triglycerides by promoting VLDL-triglyceride clearance by brown adipose tissue in mice. Diabetes. 2014;63(3):880–891. doi: 10.2337/db13-0194. [DOI] [PubMed] [Google Scholar]
- 54.van Dam AD, Kooijman S, Schilperoort M, Rensen PC, Boon MR. Regulation of brown fat by AMP-activated protein kinase. Trends Mol Med. 2015;21(9):571–579. doi: 10.1016/j.molmed.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 55.Shin H, et al. Lipolysis in Brown Adipocytes Is Not Essential for Cold-Induced Thermogenesis in Mice. Cell Metab. 2017;26(5):764–777.e5. doi: 10.1016/j.cmet.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schreiber R, et al. Cold-Induced Thermogenesis Depends on ATGL-Mediated Lipolysis in Cardiac Muscle, but Not Brown Adipose Tissue. Cell Metab. 2017;26(5):753–763.e7. doi: 10.1016/j.cmet.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bradley EA, et al. Activation of AMP-activated protein kinase by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside in the muscle microcirculation increases nitric oxide synthesis and microvascular perfusion. Arterioscler Thromb Vasc Biol. 2010;30(6):1137–1142. doi: 10.1161/ATVBAHA.110.204404. [DOI] [PubMed] [Google Scholar]
- 58.Baltgalvis KA, et al. Exercise performance and peripheral vascular insufficiency improve with AMPK activation in high-fat diet-fed mice. Am J Physiol Heart Circ Physiol. 2014;306(8):H1128–H1145. doi: 10.1152/ajpheart.00839.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ericsson M, et al. Exercise training before cardiac-specific Serca2 disruption attenuates the decline in cardiac function in mice. J Appl Physiol. 2010;109(6):1749–1755. doi: 10.1152/japplphysiol.00282.2010. [DOI] [PubMed] [Google Scholar]
- 60.Kemi OJ, Loennechen JP, Wisløff U, Ellingsen Ø. Intensity-controlled treadmill running in mice: cardiac and skeletal muscle hypertrophy. J Appl Physiol. 2002;93(4):1301–1309. doi: 10.1152/japplphysiol.00231.2002. [DOI] [PubMed] [Google Scholar]
- 61.Ford RJ, Teschke SR, Reid EB, Durham KK, Kroetsch JT, Rush JW. AMP-activated protein kinase activator AICAR acutely lowers blood pressure and relaxes isolated resistance arteries of hypertensive rats. J Hypertens. 2012;30(4):725–733. doi: 10.1097/HJH.0b013e32835050ca. [DOI] [PubMed] [Google Scholar]
- 62.Englund EK, et al. Multiparametric assessment of vascular function in peripheral artery disease: dynamic measurement of skeletal muscle perfusion, blood-oxygen-level dependent signal, and venous oxygen saturation. Circ Cardiovasc Imaging. 2015;8(4):e002673. doi: 10.1161/CIRCIMAGING.114.002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Good P. Permutation Test: A practical guide to resampling methods for testing hypotheses. New York, NY: Springer-Verlag New York; 2013. [Google Scholar]
- 64.Gutterman DD, et al. The Human Microcirculation: Regulation of Flow and Beyond. Circ Res. 2016;118(1):157–172. doi: 10.1161/CIRCRESAHA.115.305364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. mTORC1 Signaling: A Double-Edged Sword in Diabetic β Cells. Cell Metab. 2018;27(2):314–331. doi: 10.1016/j.cmet.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 66.Mottillo EP, et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016;24(1):118–129. doi: 10.1016/j.cmet.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hamburg NM, Creager MA. Pathophysiology of Intermittent Claudication in Peripheral Artery Disease. Circ J. 2017;81(3):281–289. doi: 10.1253/circj.CJ-16-1286. [DOI] [PubMed] [Google Scholar]
- 68.Shah AD, et al. Type 2 diabetes and incidence of cardiovascular diseases: a cohort study in 1·9 million people. Lancet Diabetes Endocrinol. 2015;3(2):105–113. doi: 10.1016/S2213-8587(14)70219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12(3):144–153. doi: 10.1038/nrendo.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bianchi C, Daniele G, Dardano A, Miccoli R, Del Prato S. Early Combination Therapy with Oral Glucose-Lowering Agents in Type 2 Diabetes. Drugs. 2017;77(3):247–264. doi: 10.1007/s40265-017-0694-4. [DOI] [PubMed] [Google Scholar]
- 71.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.