

Summary
Protection from infectious disease relies on two distinct strategies: antimicrobial “resistance” directly inhibits pathogen growth, whereas infection “tolerance” protects from the negative impact of infection on host health. A single immune-mediator can differentially contribute to these strategies in distinct contexts, confounding our understanding of protection to different pathogens. For example, the NADPH-dependent phagocyte oxidase complex (Phox) produces antimicrobial superoxide and protects from tuberculosis (TB) in humans. However, Phox-deficient mice display no sustained resistance defects to M. tuberculosis, suggesting a more complicated role for NADPH phagocyte oxidase complex than strictly controlling bacterial growth. We examined the mechanisms by which Phox contributes to protection from TB and found that mice lacking the Cybb subunit of Phox suffered from a specific defect in tolerance, which was due to unregulated Caspase 1 activation, interleukin 1β (IL-1β) production, and neutrophil influx into the lung. These studies imply that a defect in tolerance alone is sufficient to compromise immunity to Mtb and highlight a central role for Phox and Caspase 1 in regulating TB disease progression.
Introduction
Protective defense to infectious disease involves functionally overlapping responses that can be divided into two fundamentally different categories (1, 2). Infection “resistance” refers to functions that directly target the infecting pathogen to prevent its growth and dissemination. Resistance pathways act by a variety mechanisms including disrupting the bacterial niche, serving as metabolic poisons, and sequestering critical nutrients (3–5). In addition, the extent of disease is also influenced by “tolerance” strategies that enhance host survival but do not directly impact pathogen growth (6–8). Tolerance pathways control a broad range of functions that protect the infected tissues from the direct cytotoxic properties of the pathogen, the inflammation-mediated immunopathology, and enhance the overall health of the host in the face of an ongoing infection. While it is well appreciated that both resistance and tolerance mechanisms are required to limit disease, the relative importance of these pathways vary for different infections (1). Furthermore, since individual immune effectors can promote both tolerance and resistance, the specific role for each mediator can change in different contexts (1, 6, 9–11). In the context of chronic infections, where resistance mechanisms are insufficient and the pathogen persists in the tissue, tolerance is likely to play a particularly important role (11).
Like many other chronic infections, the outcome of an encounter with Mycobacterium tuberculosis (Mtb) varies dramatically between individuals (12). Only 5–10% of those that are infected with this pathogen progress to active tuberculosis (TB), and disease progression is influenced by a wide-variety of genetic and environmental factors that modulate either tolerance or resistance (13–15). For example, observations from humans and mice indicate that several specific changes in T cell function may contribute to a failure of resistance and disease progression due to loss of antimicrobial resistance (16–18). In addition, studies in animal models indicate that a failure of host tolerance, which is necessary to preserve lung function or granuloma structure, influences the extent of disease (19, 20). While these studies suggest that variation in overall tolerance may be an important determinant TB risk, the specific tolerance mechanisms that influence disease progression in natural populations remain ill defined. Furthermore, since most immune mediators are pleiotropic and affect both tolerance and resistance, it remains unclear if a specific failure of tolerance alone can promote TB disease progression.
During many bacterial infections the production of reactive oxygen species (ROS) by the NADPH phagocyte oxidase (Phox) is involved in protecting the host from disease (21). Phox is a multi-protein complex, including the subunits Cybb (gp91) and Ncf1 (p47) that assemble in activated immune cells to produce superoxide radicals by transferring electrons from NADPH to molecular oxygen (22). Humans with deleterious mutations in the Phox complex develop a clinical syndrome known as chronic granulomatous disease (CGD). Leukocytes from patients with CGD are unable to kill a number of bacterial pathogens, such as Staphylococcus aureus and Serratia marcescens, and this defect is associated with the susceptibility to infection with these organisms (23). Because ROS contributes to the microbicidal activity of phagocytes, previous studies in Mtb-infected mice focused on the role of Phox in antimicrobial resistance. Several such studies found that mice deficient in Phox show no long-term differences in Mtb bacterial levels compared to wild type animals (24–26) . Cooper et al observed a transient increase in bacterial numbers between 15 and 30 days after infection of Ncf1−/− mice, but this difference was not sustained (24). Two subsequent studies found that no difference in Mtb burden could be attributed to Cybb deficiency (25, 26). The lack of an obvious antimicrobial role for Phox during Mtb infection is likely due to the expression of mycobacterial ROS defenses. These defenses include the catalase/peroxidase, KatG, which detoxifies ROS directly, and the MRC complex that detoxifies a variety of radicals (26–29). Together these bacterial defense mechanisms may allow Mtb to withstand the oxidative killing mechanisms (30). In contrast to the apparent dispensability of Phox in short-term mouse infections with Mtb, human mutations in the Cybb gene are strongly associated with susceptibility to mycobacterial diseases, including TB (31–35). Mutations that specifically reduce Cybb activity in macrophages produce a similar clinical presentation, highlighting the importance of the macrophage-derived ROS in protection (31). Taken together, while Phox is clearly important for controlling mycobacterial diseases including TB, the absence of long-term resistance defects in Mtb-infected mice suggest that the phagocyte oxidase may play a more complex role than simply limiting bacterial replication.
Here we examined the mechanisms of Phox-mediated protection in the context of Mtb infection. We found that loss of the Phox subunit Cybb does not alter the growth or survival of Mtb during infection. Instead, Cybb−/− animals suffered from a hyper-inflammatory disease caused by increased activation of the NLRP3-dependent Caspase-1 inflammasome and IL-1-dependent neutrophil accumulation in the lung. Thus, the protective effect of Phox can be solely attributed to increased tolerance to Mtb infection instead of a direct antimicrobial effect. These studies provide a mechanism to explain the association between Phox expression and TB disease in natural populations, and implicate control of Caspase-1 activation as an important regulator of infection tolerance.
Materials and Methods
Mice
C57BL/6J (Stock # 000664), Cybb−/− (B6.129S-Cybbtm1Din/J stock # 002365), Nos2−/− (B6.129P2-Nos2tm1Lau/j, stock # 002609), B6.SJL-Ptprca Pepcb carrying the pan leukocyte marker CD45.1 (stock # 002014) were purchased from the Jackson Laboratory. Mice were housed under specific pathogen-free conditions and in accordance with the University of Massachusetts Medical School, IACUC guidelines. All animals used for experiments were 6–12 weeks except mixed chimeras that were infected at 16 weeks following 8 weeks of reconstitution.
Mouse Infection
Wild type M. tuberculosis strain H37Rv was used for all studies unless indicated. This strain was confirmed to be PDIM-positive. Prior to infection bacteria were cultured in 7H9 medium containing 10% oleic albumin dextrose catalase growth supplement (OADC) enrichment (Becton Dickinson) and 0.05% Tween-80. H37Rv expressing sfYFP has been previously described and the episomal plasmid was maintained with selection in Hygromycin B (50ug/ml) added to the media (10). For low and high dose aerosol infections, bacteria were resuspended in phosphate-buffered saline containing Tween-80 (PBS-T). Prior to infection, bacteria were sonicated then delivered via the respiratory route using an aerosol generation device (Glas-Col). Infections of mice with the streptomycin dependent strain of Mtb (18b) have been previously described (36). In short, mice were infected via intra-tracheal infection and treated daily with 2mg of streptomycin daily for two weeks. For anti-IL1R treatment mice were injected with 200ug of anti-IL1R antibody or Isotype control (Bio-xcell) every other day starting at day 14. Both male and female mice were used throughout the study and no significant differences in phenotypes were observed between sexes.
Immunohistochemistry
Lungs from indicated mice were inflated with 10% buffered formalin and fixed for at least 24 hours then embedded in paraffin. Five-Micrometer—thick sections were stained with hematoxylin and eosin (H&E). All staining was done by the Diabetes and Endocrinology Research Center Morphology Core at the University of Massachusetts Medical School.
Flow Cytometry
Lung tissue was harvested in DMEM containing FBS and placed in C-tubes (Miltenyi). Collagenase type IV/DNase I was added and tissues were dissociated for 10 seconds on a GentleMACS system (Miltenyi). Tissues were incubated for 30 minutes at 37°C with oscillations and then dissociated for an additional 30 seconds on a GentleMACS. Lung homogenates were passaged through a 70-micron filter or saved for subsequent analysis. Cell suspensions were washed in DMEM, passed through a 40-micron filter and aliquoted into 96 well plates for flow cytometry staining. Non-specific antibody binding was first blocked using Fc-Block. Cells were then stained with anti-Ly-6G Pacific Blue, anti-CD4 Pacific Blue, anti-CD11b PE, anti-CD11c APC, anti-Ly-6C APC-Cy7, anti-CD45.2 PercP Cy5.5, anti-CD3 FITC, anti-CD8 APC-Cy7, anti-B220 PE-Cy7 (Biolegend). Live cells were identified using fixable live dead aqua (Life Technologies). For infections with fluorescent H37Rv, lung tissue was prepared as above but no antibodies were used in the FITC channel. All of these experiments contained a non-fluorescent H37Rv infection control to identify infected cells. Cells were stained for 30 minutes at room temperature and fixed in 1% Paraformaldehyde for 60 minutes. All flow cytometry was run on a MACSQuant Analyzer 10 (Miltenyi) and was analyzed using FlowJo_V9 (Tree Star).
Macrophage and Dendritic Cell Generation
To generate bone marrow derived macrophages (BMDMs), marrow was isolated from femurs and tibia of age and sex matched mice. Cells were then incubated in DMEM (Sigma) containing 10% fetal bovine serum (FBS) and 20% L929 supernatant. Three days later media was exchanged with fresh media and seven days post-isolation cells were lifted with PBS-EDTA and seeded in DMEM containing 10% FBS for experiments.
To generate bone marrow derived dendritic cells (BMDCs), marrow was isolated from femurs and tibia of age- and sex-matched mice. Cell were then incubated in iMDM media (GIBCO) containing 10% FBS, L-Glutamine, 2μM 2-mercaptoethanol and 10% B16-GM-CSF supernatant (37). BMDCs were then purified on day six using Miltenyi LS columns first using negative selection for F480 followed by CD11c positive selection. Cells were then plated and infected the following day.
Macrophage and Dendritic Cell Infection
Mtb or Mycobacterium bovis-BCG were cultured in 7H9 medium containing 10% oleic albumin dextrose catalase growth supplement (OADC) enrichment (Becton Dickinson) and 0.05% Tween 80. Before infection cultures were washed in PBS-T, resuspended in DMEM containing 10%FBS and passed through a 5-micron filter to ensure single cells. Multiplicity of infection (MOI) was determined by optical density (OD) with an OD of 1 being equivalent to 3×108 bacteria per milliliter. Bacteria were added to macrophages for 4 hours then cells were washed with PBS and fresh media was added. At the indicated time points supernatants were harvested for cytokine analysis and the cells were processed for further analysis. Cell death was assessed using Cell-Titer-Glo luminescent cell viability assay (Promega) following manufacturer’s instructions. For inhibitor treatments cells were treated with the indicated concentrations of IFNγ (Peprotech), MCC950 (Adipogen) or VX-765 (Invivogen) or vehicle control overnight prior to infection and maintained in the media throughout the experiment.
Mixed Bone Marrow Chimera generation and cell sorting
Mixed bone marrow chimera experiments were done essentially as previously described (10). Wild type CD45.1+ mice were lethally irradiated with two doses of 600 rads. The following day, bone marrow from CD45.1+ wild-type mice and CD45.2+ knockout mice (wild type or Cybb−/−) was isolated, red blood cells were lysed using Tris-buffered ammonium chloride (ACT), and the remaining cells were quantified using a haemocytometer. CD45.1+ and CD45.2+ cells were then mixed equally at a 1:1 ratio and 107 cells from this mixture were injected intravenously into lethally irradiated hosts that were placed on sulfatrim for three weeks. 8 weeks later mice were then infected by low-dose aerosol with M. tuberculosis H37Rv. Four weeks following infection, the lungs of chimera mice were processed for flow cytometry. An aliquot of this suspension was saved for flow cytometry analysis of the lung population and overall bacterial levels. The remaining cells were split equally and stained with either anti-CD45.1 APC or anti-CD45.2 PE. Stained populations were then incubated with either anti-APC or anti-PE magnetic beads (Miltenyi) following the manufacturer’s instructions and sorted using LS-columns (Miltenyi). Purified cells were divided equally and then plated for M. tuberculosis on 7H10 agar or counted and stained for analysis of cellular purity. Cells from the input homogenate, flow through and the positive sort fractions were stained with for purity. Samples with >90% purity were used for subsequent analysis. At 21 days after plating, colonies were enumerated and the Mtb levels per sorted cells were determined.
qRT-PCR
Cells were lysed in Trizol-LS (Thermofisher), RNA was purified using Direct-zol RNA isolation kits (Zymogen) and quantified on nanodrop. RNA was diluted to 5ng/μl and 25 ng total RNA was used for each reaction. Ct values for each sample were determined in technical duplicates for β-Actin and IL-1β using one-step RT-PCR Kit (Qiagen) on a Viia7 Real-time PCR system (Life Technologies). ΔΔct values were then determined for each sample.
Immunoblotting, immunoblotting quantification and Cytokine quantification
Murine cytokine concentrations in culture supernatants and cell-free lung homogenates were quantified using commercial enzyme-linked immunosorbent assay (ELISA) kits (R&D). All samples were normalized for total protein content. Caspase1 activation in macrophage lysates was determined by western blotting with Caspase1 antibody purchased from Adipogen. Immunoblots were quantified using ImageJ software.
Results
Cybb−/− mice are susceptible to TB disease, but maintain control of bacterial replication.
In order to examine the role of Phox in mediating protection against Mtb, we compared disease progression and the immune responses in wild type and Cybb−/− C57BL/6 mice after infection via aerosol with 50–100 bacteria. We found no significant difference in the survival or bacterial levels in the lung between groups of mice up to 3 months following infection, confirming that Cybb is not required for surviving the early stages of Mtb infection (Figure 1A and S1). However, after 100 days of infection, Cybb−/− infected mice lost an average of 10% of their body weight while wild type animals gained weight (Figure 1B). Histopathological inspection of the lungs indicated a difference in disease between these groups, with Cybb−/− lungs containing larger and less organized lesions than wild type (Figure 1C). These data suggested that wild type and Cybb−/− animals might tolerate Mtb infection differently even while harboring identical levels of bacteria.
Figure 1. Anti-inflammatory activity of Cybb protects mice from TB disease.
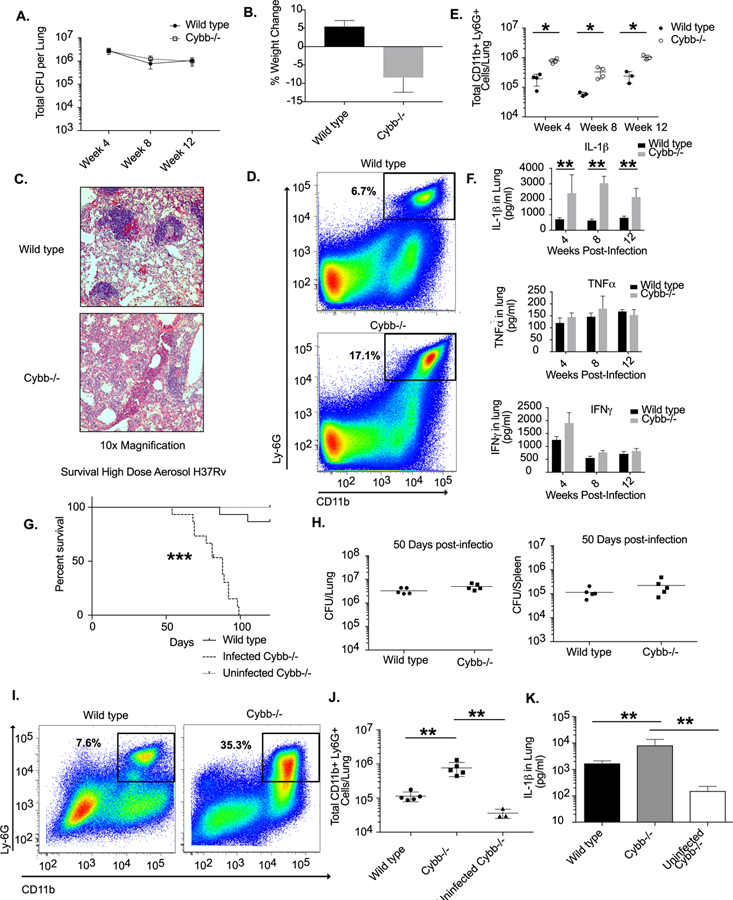
A. Following low dose aerosol infection (Day 20 of ~50–100 colony forming units, cfu) total bacterial burden (expressed in cfu, mean +/− s.d.) was determined in the lungs of wild type or Cybb−/− mice at the indicated time points with 4–5 mice per group. B. Percentage weight loss (mean change +/− s.d) from Day 30 to Day 100 was determined for wild type and Cybb−/− mice. Representative of two experiments with 3–5 mice per group. C. Immunohistochemical staining for Haemotoxylin and Eosin is shown for representative lung sections from wild type and Cybb−/− mice at 48 weeks post-infection at 20x magnification. D. Representative flow cytometry plot showing increased Ly56G+ CD11b+ neutrophil recruitment to the lungs of Cybb−/− mice 4 weeks following infection (gated on live/singlets/CD45+). E. Quantification of neutrophil recruitment to the lungs at the indicated time points following infection for wild type and Cybb−/− mice is shown as absolute number of Ly66G+ CD11b+ cells per lung (mean +/− s.d). * p-value <.05 by unpaired two-tailed t-test. Representative of 4 experiments with 3–5 mice per group. F. Lung homogenates from wild type or Cybb−/− mice infected for the indicated time were probed for the cytokines IL-71β, IFNγ, and TNFα by ELISA (mean +/− s.d). Results shown in A-D are representative of 3 independent experiments with 3–5 mice per group. ** p-value <.01 by unpaired two-tailed t-test. G. Survival of infected wild type and Cybb−/− and uninfected Cybb−/− mice was determined following high dose infection. Data are representative of two independent experiments with 814–15 mice per group. *** p-value <.001 Mantel-Cox text. H. Fifty days following high dose aerosol infection (Day 90 of 5000–7500 CFU) total bacterial burden (expressed in cfu, mean +/− s.d.) was determined in the lungs and spleen of wild type or Cybb−/− mice. Data are representative of two experiments 4–5 mice per group. I. Representative flow cytometry plot showing increased Ly106G+ CD11b+ neutrophil recruitment to the lungs of Cybb−/− mice 50 days following infection (gated on live/singlets/CD45+). J. Quantification of the absolute number of neutrophils recruited to the lungs 1150 days following high dose infection for wild type and Cybb−/− mice is shown (mean +/− s.d). ** p-value <.01 by one-way ANOVA with tukey correction. Representative of two experiments with 5 mice per group. K. Lung homogenates from uninfected Cybb−/− mice and wild type or Cybb−/− mice infected for 1250 days following high dose aerosol were probed for IL-1β by ELISA (mean +/− s.d). ** p-value <.01 by unpaired two-tailed t-test. Results shown in G-J are representative of 2 independent experiments with 5 mice per group. ** p-value <.01 by unpaired two-tailed t-test.
In order to dissect the mechanisms controlling tolerance to Mtb disease in Cybb−/− mice, we profiled the infected lungs of animals during infection by flow cytometry. We found no significant differences in the numbers of dendritic cells, macrophages, B cells, as well as total and activated T cells between wild type and Cybb−/− mice (Figure S1). In contrast, we observed an early and sustained increase of Ly6G+ CD11b+ neutrophils in the infected lungs of Cybb−/− mice (Figure 1D and 1E). A 3–5-fold increase in the total number neutrophils was observed as early as 4 weeks following infection and was maintained throughout the 12-week study.
The cytokine IL-1β promotes neutrophil-mediated disease during Mtb infection of other susceptible mouse strains (10, 36). Similarly, when we assayed cytokine levels in lung homogenates, we noted a dramatic and specific increase in IL-1β concentration in Cybb−/− animals compared to wild type at all time points (Figure 1F). In contrast, no significant differences were noted for IFNγ or TNFα at any time point between groups (Figure 1F). Thus, while the adaptive immune response to Mtb appeared to be intact, Cybb−/− animals produced excess IL-1β and the concentration of this cytokine correlated with neutrophil infiltration into the lung.
Previous studies have shown that following low dose aerosol infection mice deficient in Phox (both Cybb−/− or Ncf1−/−) survive for at least 60 days (24, 25). However, longer infection is likely necessary to determine whether the enhanced disease we noted in Cybb−/− mice would result in a survival defect. These long-term survival experiments following low dose aerosol proved difficult, since uninfected Cybb−/− mice develop arthritis as they age (38). To avoid this confounder, we quantified the survival of mice in a shorter-term study using a high dose aerosol infection. When mice were infected with ~5000 CFU per animal, Cybb−/− mice succumbed to disease significantly more rapidly than wild type animals. Cybb−/− mice had a median survival time of 88 days while only two out of fifteen wild type mice succumbed during the 120-day study (Figure 1G). In order to distinguish survival effects not related to Mtb infection, a cohort of uninfected age-matched Cybb−/− mice were maintained for the duration of the experiment. None of these animals required euthanasia over the 120 days and no animals included in this experiment developed arthritis.
During this high-dose study, we also examined a cohort of mice 50 days following infection and found identical levels of bacteria in the lungs and spleen between wild type and Cybb−/− groups (Figure 1H). Consistent with our earlier findings, Cybb−/− mice showed a significant increase in neutrophils and IL-1β in the lung (Figure 1I-K). We also found minimal levels of IL-1β and neutrophils in uninfected Cybb−/− lungs indicating that these phenotypes are dependent on Mtb infection. Therefore, the loss of Cybb leads to more severe Mtb disease that is associated with increased IL-1β levels and neutrophil recruitment, even though the number of viable Mtb in the lung did not appear to be affected.
Cybb controls tolerance to Mtb infection.
Our initial results suggested that Cybb protects mice by promoting tolerance to Mtb infection rather than directly controlling bacterial replication. However, while viable bacterial numbers were similar in wild type and Cybb−/− mice, we could not rule out that the course of disease was altered by subtle changes in the dynamics of bacterial growth and death. To more rigorously address this question, we employed two additional animal models that allowed us to differentiate tolerance and direct antimicrobial resistance in vivo.
To more formally exclude the possibility that Cybb alters the intracellular growth of Mtb during infection, we used a previously optimized mixed bone marrow chimera approach (10). These experiments normalize potential inflammatory differences between wild type and Cybb−/− mice allowing us to specifically quantify differences in bacterial control (Figure 2A). Irradiated wild type mice were reconstituted with a 1:1 mixture of CD45.1+ wild type and CD45.2+ Cybb−/− or wild type cells. Five weeks following infection, both CD45.1+ and CD45.2+ cells were sorted from the lungs and the levels of Mtb in each genotype was determined by plating and the purity of populations was determined by flow cytometry (Figure 2B and 2C and Figure S2). We found that the relative abundance of wild type and Cybb−/− cells was similarly maintained throughout infection in both the myeloid and lymphoid compartments, indicating that Cybb does not alter cellular recruitment or survival in a cell-autonomous manner. When Mtb was enumerated in sorted cells, we found identical levels of H37Rv in wild type CD45.1+ and Cybb−/− CD45.2+ populations from the same mouse, similar to the results from mice where both populations were reconstituted with congenically mismatched wild type cells. In contrast, when chimeric mice were infected with a ROS-sensitive ∆katG mutant of Mtb, we found higher levels of bacteria in Cybb−/− cells compared to wild type cells from the same mouse. These data show that the assay is able to detect the cell-autonomous antimicrobial activity of ROS against a KatG-deficient Mtb strain, but Cybb-dependent ROS did not restrict the intracellular replication of wild type Mtb.
Figure 2. The primary protective role of Cybb is anti-inflammatory.
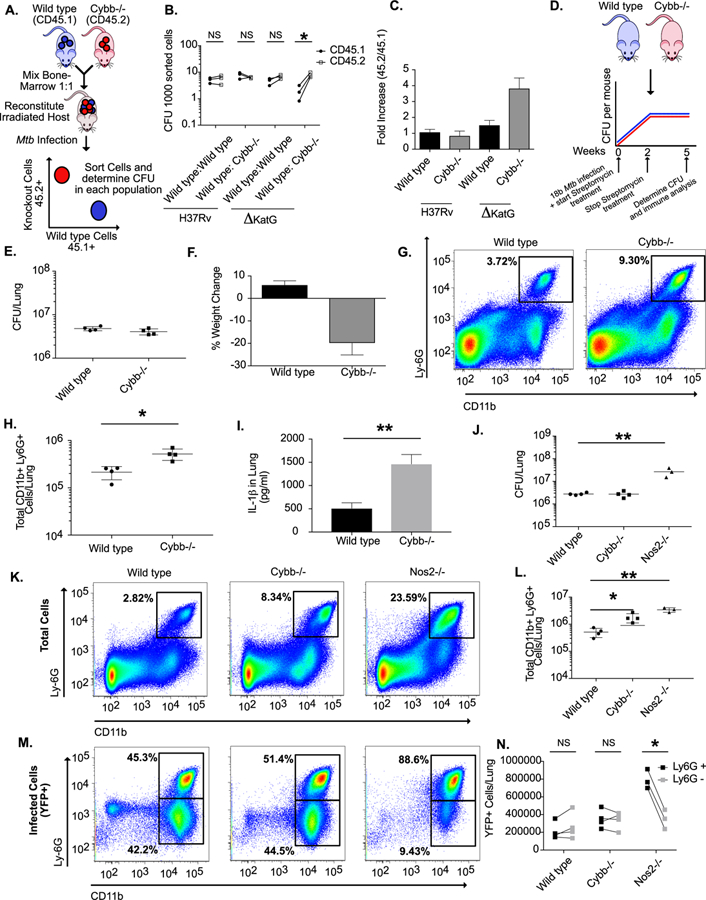
A. Schematic for the generation of mixed bone marrow chimeras. Mixed bone marrow chimeras were infected by low dose aerosol with either H37Rv or ΔKatG mutant. Five weeks following infection CFU levels were determined in purified hematopoietic cells of indicated genotypes. B. Shown are the normalized CFU per sorted cells in each population from each mouse. * p<.05 by unpaired two-tailed t-test. C. The fold-increase of bacterial levels in CD45.2+ cells (experimental) compared to CD45.1+ cells (wild type control) (mean +/− s.d.). The results in B and C are representative of three independent experiments with 3–4 mice per group. D. Schematic for streptomycin-dependent infection. Wild type and Cybb−/− mice were infected intratracheally with Mtb strain 18b and treated for two weeks daily with streptomycin. Mice were then removed from streptomycin for three weeks halting active growth of the bacteria. E. Five weeks after infection the total levels of viable Mtb in the lungs was determined by CFU plating on streptomycin (mean +/− s.d.). F. Percentage weight loss (mean change +/− s.d) from Day 0 to Day 35 was determined for wild type and Cybb−/− mice. G. Representative flow cytometry plot showing increased Ly6G+ CD11b+ neutrophil recruitment to the lungs of Cybb−/− mice 5 weeks following infection (gated on live/singlets/CD45+). H. Quantification of neutrophil recruitment to the lungs at the indicated time points following infection for wild type and Cybb−/− mice is shown as absolute number of Ly6G+ CD11b+ cells per lung (mean +/− s.d). * p-value <.05 by unpaired two-tailed t-test. Data in E-H are representative of 4 independent experiments with 4–5 mice per group. I. Lung homogenates from wild type or Cybb−/− mice infected with 18b were probed for IL-1β by ELISA (mean +/− s.d). ** p-value <.01 by unpaired two-tailed t-test. J. Following low dose aerosol infection with sfYFP H37Rv (Day 0 of ~50–100 colony forming units, cfu) total bacterial burden (expressed in cfu, mean +/− s.d.) was determined in the lungs of wild type, Cybb−/− or Nos2−/− mice 4 weeks post-infection. ** p-value <.01 by one-way ANOVA with tukey correction. K. Shown are representative flow cytometry plots from each genotype of total Ly6G+ CD11b+ cells in the infected lungs. L. Quantification of total neutrophil recruitment to the lungs of the indicated genotypes four weeks following infection (mean +/− s.d.). ** p-value <.01 * p-value <.05 by one-way ANOVA with tukey correction. M. Shown are representative flow cytometry plots from each genotype of infected (YFP+ Ly6G+ CD11b+) cells in the lung. N. Enumeration of infected (YFP+) neutrophils (Ly6G+ CD11b+) or monocytes/macrophages (Ly6G- CD11b+) in the indicated genotypes. * p-value <.05 by unpaired two-tailed t-test. Data in J-N are representative of three independent experiments with 3–5 mice per group.
To specifically determine if the loss of Cybb decreased tolerance to a given burden of bacteria, wild type and Cybb−/− mice were infected with a streptomycin dependent strain of Mtb that allows exogenous control of bacterial replication during infection. Streptomycin is provided for the first two weeks of infection, allowing the pathogen to reach the burden observed in a wild type Mtb infection. Upon streptomycin withdrawal, the pathogen is unable to replicate but remains viable and able to drive inflammatory responses (Figure 2D) (10, 36, 39). Five weeks after infection, Cybb−/− mice lost more weight than wild type animals while harboring identical levels of non-replicating bacteria (Figure 2E and2F). Lungs from Cybb−/− mice contained significantly more neutrophils and higher levels of IL-1β compared to wild type animals (Figure 2G-2I). Thus, even when the need for antimicrobial resistance is obviated by artificially inhibiting bacterial replication, Cybb−/− animals continued to exhibit a hyper-inflammatory disease.
The granulocytic inflammation observed in Cybb−/− mice was reminiscent of several other susceptible mouse strains. However, the neutrophil recruitment in other models is generally associated with a concomitant increase in bacterial growth (40–42) and a transition of the intracellular Mtb burden from macrophages to granulocytes (10). We hypothesized that Cybb−/− mice may be able to retain control of Mtb replication because the bacteria remain in macrophages. To test this hypothesis, we used a YFP-expressing Mtb strain to compare the distribution of cells harboring bacteria in wild type and Cybb−/− mice with Nos2−/− animals in which Mtb replicates to high numbers in association with infiltrating granulocytes (10). Four weeks following infection we found that lungs from both Cybb−/− and Nos2−/− mice contain higher levels of IL-1β and neutrophils than wild type animals, although the loss of Nos2−/− produced a much more severe phenotype than Cybb−/− (Figure 2J-2L and Figure S2). However, the cells harboring Mtb in these two susceptible mouse strains differed. In wild type and Cybb−/− mice, YFP-Mtb was evenly distributed between CD11b+/Ly6G+ neutrophils and the CD11b+/Ly6G- population that consists of macrophages and dendritic cells (43). This proportion was dramatically altered in Nos2−/− mice, where close to 90% of bacteria were found in the neutrophil compartment (Figure 2M and 2N). Thus, unlike other susceptible mouse models, the loss of Cybb does not alter bacterial replication or the distribution of Mtb in different myeloid subsets. Instead, this gene plays a specific role in controlling IL-1β activation, neutrophil recruitment to the infected lung, and disease progression. As a result, we conclude that Cybb specifically promotes tolerance to Mtb infection.
Enhanced IL-1β activation by Cybb−/− macrophages and dendritic cells is due to deregulated Caspase-1 inflammasome activation
To investigate the mechanism underlying increased IL-1β production in Cybb−/− mice, we quantified the release of mature cytokine from bone-marrow derived macrophages (BMDMs) and bone-marrow derived dendritic cells (BMDCs). Compared to wild type, we found that Cybb−/− BMDMs and BMDCs produced 4–5 fold more IL-1β after 24 hours of Mtb infection (Figure 3A and 3B). Under these conditions, wild type and Cybb−/− cells remained equally viable and produced equivalent amounts of TNF (Figure 3C-F), suggesting that the effect of Cybb on IL-1β activation was specific to this cytokine.
Figure 3. Cybb controls Caspase1 activation in macrophages and dendritic cells during Mtb infection.
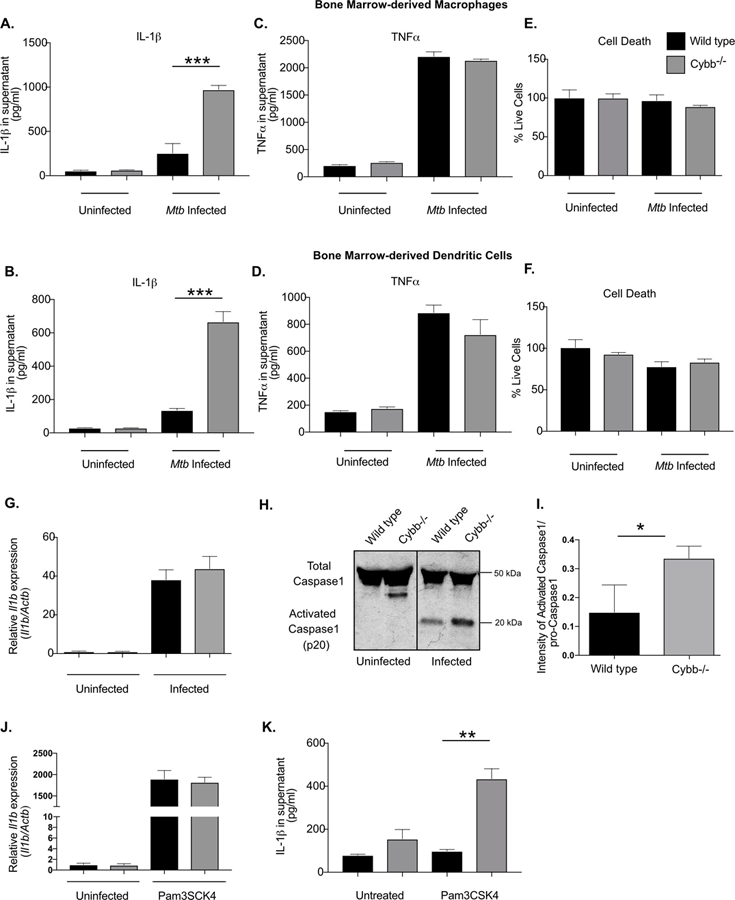
Bone marrow-derived macrophages (BMDMs) or Bone marrow-derived dendritic cells (BMDCs) from wild type or Cybb−/− mice were left untreated or infected with Mtb for 4 hours then washed with fresh media. 18 hours later supernatants were harvested and the levels of A. and B. IL-1β and C. and D. TNFα were quantified in the supernatants by ELISA. Shown is the mean of 4 biological replicates normalized to a standard curve +/− s.d. *** p<.001 by unpaired two-tailed t-test. E. and F. Viability of remaining cells was determined by quantifying ATP via luminescence and compared to cells at 4 hours post-infection (mean % viability +/− s.d.). Data in A, C and E are representative of five independent experiments with at least 3 biological replicates per experiment. Data in B, D and F are representative of three independent experiments with 4 biological replicates per experiment. G. Relative RNA expression of Il1b (compared to β-Actin) was determined from wild type and Cybb−/− BMDMs left untreated or infected for 24 hours with Mtb (mean +/− s.d.) by qRT-PCR. Data are representative of two independent experiments with 3–4 biological replicates per group. H. Immunoblot analysis was used to assess the activation of Caspase 1 from Wild type and Cybb−/− BMDMs infected for 24 hours with Mtb. Data are representative of 3 independent experiments with at least 3 biological replicates analyzed per experiment. I. Immunoblots were quantified by comparing the intensity of activated p20-Caspase 1 to total pro-Caspase1 bands. Quantification was done on three biological replicates. * p<.05 by unpaired two-tailed t-test. J. Relative RNA expression of Il1b (compared to Actb) was determined from wild type and Cybb−/− BMDMs left untreated or treated with Pam3CSK4 for 24 hours (mean +/− s.d.) by qRT-PCR. Data are representative of two independent experiments with 3–4 biological replicates per group. K. Wild type and Cybb−/− BMDMs were left untreated or treated with PAM3CSK4 for 12 hours, supernatants were harvested and the levels of IL-1β were quantified by ELISA (mean +/− s.d.). ** p<.01 by unpaired two-tailed t-test. Data are representative of three independent experiments with 4 biological replicates per experiment.
The release of mature IL-1β requires two distinct signals (44). The first signal induces the expression of Il1b mRNA and subsequent translation of pro-IL-1β, and a second signal activates Caspase 1, which is necessary for the processing and secretion of mature IL-1β. To understand what step of IL1β production was altered in Cybb−/− cells, we quantified these two signals. The expression of Il1b mRNA in uninfected and infected BMDMs was unchanged between wild type and Cybb−/− BMDMs (Figure 3G). In contrast, under the same conditions, the processing of Caspase 1 to its active form was increased in infected Cybb−/− BMDMs compared to wild type cells (Figure 3H and 3I).
These observations suggested that caspase-1 activity is increased in Cybb−/− cells, which could allow mature IL-1β secretion in the absence of an inflammasome activator. To test this hypothesis, we stimulated cells with the TLR2 agonist, PAM3CSK4, to induce pro-IL-1β expression. PAM3CSK4 stimulation induced Il1b mRNA to similar levels between wild type and Cybb−/− BMDMs, albeit over 100 times higher than infection with Mtb (Figure 3J). In wild type cells, this induction of Il1b expression produced little mature IL-1β secretion, consistent with the need for subsequent inflammasome activation. In contrast, induction of Il1b expression led to robust secretion of mature IL-1β from Cybb−/− BMDM, consistent with unregulated inflammasome activity in these cells (Figure 3K). Together these data show that loss of Cybb leads to hyper-activation of Caspase1 and increased release of IL-1β during Mtb infection of both BMDMs and BMDCs.
Loss of tolerance is reversed in Cybb−/− macrophages and mice by blocking the production or activity of IL-1β
The NLRP3 inflammasome consists of NLRP3, ASC, and Caspase 1. While this complex is generally responsible for IL-1β processing in Mtb infected macrophages (45, 46), it remained unclear whether the enhanced IL-1β secretion from Cybb−/− cells also relied on these components. To identify the responsible complex, we blocked the activation of the NLRP3 inflammasome in several distinct ways. The NLRP3 inflammasome is specifically inhibited by IFNγ stimulation, via the nitric oxide-dependent nitrosylation of the NLRP3 protein (36). Pretreatment of wild type and Cybb−/− BMDMs with varying concentrations of IFNγ, inhibited the secretion of mature IL-1β from both wild type and Cybb−/− BMDMs compared to untreated cells. While this result indicated an important role for NLRP3, IFNγ pretreatment did not completely suppress IL-1β secretion and there remained significant differences in the IL-1β release between Cybb−/− and wild type cells at all concentrations of the cytokine (Figure 4A).
Figure 4. Hyper-inflammation in Cybb−/− is reversed by inflammasome and IL1 inhibition.
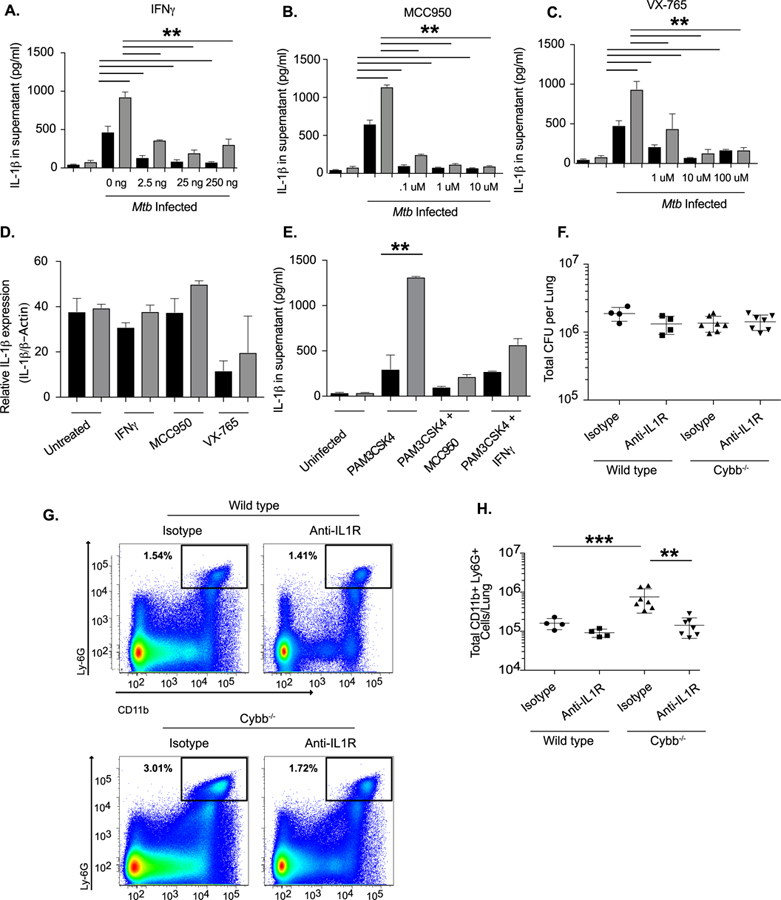
A. Wild type (Black Bars) and Cybb−/− (Grey Bars) BMDMs were left untreated or treated with the indicated concentrations of IFNγ for 12 hours. Cells were then infected with Mtb for 4 hours then washed with fresh media. 18 hours later supernatants were harvested and levels of IL-1β from each condition were quantified by ELISA (mean +/− s.d.). ** p-value <.01 * p-value <.05 by one-way ANOVA with tukey correction. Data are representative of three independent experiments with at least 3 biological replicates per experiment. B. Wild type and Cybb−/− BMDMs were left untreated or treated with the indicated concentrations of MCC950 for 2 hours. Cells were then infected with Mtb for 4 hours then washed with fresh media with inhibitor. 18 hours later supernatants were harvested and levels of IL-1β from each condition were quantified by ELISA (mean +/− s.d.). ** p-value <.01 by one-way ANOVA with tukey correction. Data are representative of three independent experiments with at least 3 biological replicates per experiment. C. Wild type and Cybb−/− BMDMs were left untreated or treated with the indicated concentrations of VX-765 for 2 hours. Cells were then infected with Mtb for 4 hours then washed with fresh media with inhibitor. 18 hours later supernatants were harvested and levels of IL-1β from each condition were quantified by ELISA (mean +/− s.d.). ** p-value <.01 by one-way ANOVA with tukey correction. Data are representative of three independent experiments with at least 3 biological replicates per experiment. D. Relative RNA expression of IL-1β (compared to b-Actin) was determined from wild type and Cybb−/− BMDMs left infected for 24 hours with Mtb in the presence or absence of the indicated inhibitors (mean +/− s.d.) by qRT-PCR. Data are representative of two experiments with 4 biological replicates per group. E. Wild type and Cybb−/− BMDMs were left untreated or treated 25ng/ml IFNγ or 1μM MCC950 overnight. The following day cells were treated with PAM3CSK4 for 12 hours, supernatants were harvested and the levels of IL-1β were quantified by ELISA (mean +/− s.d.). ** p<.01 by unpaired two-tailed t-test. Data are representative of two independent experiments with 4 biological replicates per experiment. F. Wild type and Cybb−/− mice were infected intratracheally with Mtb strain 18b and treated for two weeks daily with streptomycin then were injected every other day for two weeks with 200ug of either isotype control antibody or anti-IL1R antibody. The total levels of viable Mtb in the lungs was determined by CFU plating on streptomycin (mean +/− s.d.) with 4–7 mice per group. G. Representative flow cytometry plot showing Ly6G+ CD11b+ neutrophil recruitment to the lungs of Cybb−/− mice during control and IL1R blockade conditions (gated on live/singlets/CD45+). H. Quantification of neutrophil recruitment to the lungs at the indicated time points following infection for wild type and Cybb−/− mice during control and IL1R blockade conditions is shown as an absolute number of Ly6G+ CD11b+ cells per lung (mean +/− s.d). *** p-value <.001 ** p-value <.01 by one-way ANOVA with tukey correction. Data in F-G are representative of two independent experiments with 4–7 mice per group.
To more directly assess the role of NLRP3 and Caspase-1 in IL-1β maturation in Cybb−/− cells, we employed specific small molecule inhibitors. Treatment of Mtb-infected BMDMs with either the NLRP3 inhibitor, MCC950 (45), or the Caspase-1 inhibitor, VX-765 (47) caused a dramatic reduction in IL-1β in both wild type and Cybb−/− BMDMs compared to untreated cells (Figure 4B and 4C). This ten-fold decrease in IL-1β secretion could not be attributed to inhibition of pro-IL-1β levels, as none of these inhibitors affected Il1b mRNA by more than two-fold. Similarly, the spontaneous IL-1β secretion observed upon PAM3CSK4 stimulation was also inhibited by MCC950 and IFNγ (Figure 4E). In each case, inflammasome inhibition reduced IL-1β secretion to the same level in both wild type and Cybb−/− cells, indicating that the NLRP3 inflammasome was responsible for the enhanced processing and secretion of this cytokine in Cybb−/− BMDM.
Based on these studies, we hypothesized that the tolerance defect observed in the intact mouse was due to inflammasome-dependent IL-1 signaling. The contributions of IL-1β during Mtb infection are complex (48, 49). Production of this cytokine is important for antimicrobial immunity, but persistent IL-1 signaling can promote pathology. In order to focus on the role of over-production of IL-1β on tolerance, we inhibited IL-1 signaling in mice infected with non-replicating streptomycin-dependent Mtb to normalize the bacterial burden. Two weeks after infection, wild type and Cybb−/− mice were treated with either an isotype control antibody or an anti-IL1R antibody to block the effect of increased IL-1β activity. As expected, Mtb levels were similar in all mice, but more neutrophils accumulated in the lungs Cybb−/− animals (Figure 4F-H). While anti-IL-1R treatment had no effect in wild type animals, inhibition of IL-1 signaling reduced neutrophil infiltration in Cybb−/− mice to the level observed in wild type animals. Taken together, our data show that Cybb−/− contributes to protective immunity to Mtb not by controlling bacterial replication, but instead by preventing an IL-1-dependent inflammatory response that increases neutrophil recruitment to the lung and exacerbates disease progression.
Discussion
The role of the Phox complex in protection from TB has presented a paradox (34). Based on the well-described antimicrobial properties of Phox-derived ROS, previous studies have focused on examining the function of Phox components in controlling Mtb replication in mice (24, 25, 50). The lack of a long-term differences in bacterial levels observed in these studies suggested that Phox may not play a strong antimicrobial role during Mtb infection. Our dissection of disease progression in Cybb-deficient mice shows that Phox plays no discernable role in antimicrobial resistance to Mtb over long-term infections. However, we uncovered a previously unknown role for this complex in promoting tolerance to Mtb infection and inhibiting TB disease.
While we were able to clearly delineate the role of Phox during Mtb infection, the role(s) played by this complex in any given infection is likely to vary. Phox-deficient mice are unable to control the growth of several bacterial pathogens that are known to cause serious infections in CGD patients, including non-tuberculous mycobacteria (50–53). In the context of these infections, the antimicrobial functions of Phox may predominate. In fact, several studies suggest an important role for Phox in controlling replication of pathogenic mycobacteria other than Mtb. High-dose intravenous or intratracheal infection with M. avium, M. marinum, and M. bovis BCG all produce higher bacterial burdens and more severe disease in Phox-deficient mice than wild type animals (50, 53, 54). While these observations indicate that Phox can play an antimicrobial role in these infections, the specific role of this complex in promoting tolerance could not be discerned due to the difference in bacterial numbers in wild type and mutant animals.
The differential dependency on Phox for antimicrobial resistance to Mtb versus these other mycobacterial species likely reflects a differential susceptibility to ROS-mediated killing. Several studies support this model by demonstrating that mechanisms by which neutrophils kill these different organisms are distinct. Neutrophil-mediated killing of Mtb is independent of Phox derived ROS, as neutrophils from CGD-patients show no defect in Mtb killing and inhibitors of ROS do not alter this activity (55, 56). In contrast, M. marinum is killed by neutrophils in a ROS-dependent manner (57). Similarly, neutrophil depletion promotes the growth of BCG in the lung but has little effect in the context of Mtb (10, 58). This effect can be explained based on the ability of the ESX-1 secretion system, which is absent from BCG, to inhibit neutrophil killing (59). Based on these differences in susceptibility to neutrophil killing, we speculate that Phox-derived ROS may contribute to both tolerance and resistance to mycobacteria, such as M. avium or M. bovis BCG, but this remains to be rigorously tested. For a pathogen such as Mtb that is resistant to ROS-mediated toxicity and persists in the tissue to promote continual inflammatory damage, the tolerance-promoting activity of Phox predominates.
During Mtb infection, we found that the ROS produced by Phox are critical to control the activation of the NLRP3 inflammasome. In contrast, mitochondrial ROS are well known to activate inflammatory cascades (60), suggesting that the context by which ROS are produced influences the inflammatory outcome of activated cells. Despite this complexity, Phox-deficient mice and CGD patients suffer from hyper-inflammatory diseases including arthritis, colitis, and prolonged inflammatory reactions to microbial products, indicating that the dominant immunoregulatory role for Phox-derived ROS is anti-inflammatory (61–63). While the noninfectious granulomatous lesions of CGD patients are typified by mononuclear infiltrate, the histopathology of infectious foci are more variable. Pulmonary infections generally produce either pneumonia or abscess, which might resemble the neutrophil-associated disease that we observe in mice (64).
CGD patients are more likely to develop disseminated BCG infections following vaccination. While it is likely M. bovis is differentially susceptible to ROS killing as discussed above it is also possible enhanced inflammasome activation contributes to this disease. While inflammasome activation in wild type animals requires the ESX-1 secretion system that is deleted in BCG, we observed robust stimulation of inflammasome activation in Phox deficient cells treated with TLR stimulation alone. These results suggest that activation of Caspase1 may occur in Phox-deficient cells even upon infection with ESX-1 deficient pathogens.
Several non-mutually exclusive mechanisms could explain the anti-inflammatory effect of Phox-derived ROS. For example, ROS has been proposed to inhibit the production of inflammatory mediators by inhibiting autophagy (65). In the absence of ROS, the reduced levels of autophagy may drive enhanced inflammasome activation. Another mechanism was described in superoxide dismutase1 (Sod1) deficient cells, where the accumulation of ROS inhibits Caspase1 activation through glutathionation of reactive cysteines (66). It is possible that the loss of ROS leads to loss of glutathionation and subsequently results in the hyper-activation of Caspase1. This latter mechanism is reminiscent of the process by which nitric oxide (NO), inhibits inflammasome activation via S-nitrosylation of NLRP3 (36). The intersection of these two important anti-inflammatory pathways at the NLRP3 inflammasome indicates that this complex may be a critical point of integration where inflammatory cascades are controlled during chronic infections.
Our findings are consistent with a growing body of literature suggesting that inflammasome-derived IL-1 promotes TB disease progression. For example, genetic polymorphisms that increase the expression of IL1β or the production of IL-1 dependent pro-inflammatory lipid mediators are associated with TB disease progression (10, 67). Similarly, transcriptional signatures of inflammasome activation have been observed in severe forms of TB disease, such as meningitis (68) and TB-associated immune reconstitution syndrome (69). Together with our work, these finding imply that a failure in tolerance alone can compromise protective immunity to Mtb, even in the context of fully functional antimicrobial resistance responses, and that Caspase-1 represents a critical point at which tolerance is regulated.
Supplementary Material
Acknowledgements
We are thankful to members of the Sassetti, Behar and Fitzgerald lab for helpful discussions. This work was funded by the Arnold and Mabel Beckman Postdoctoral Fellowship (AO), Charles King Trust Postdoctoral Fellowship (CMSmith), and NIH Grant AI132130 (CMSassetti)
Footnotes
Author Contributions
AO and CMSassetti conceived of and designed the experiments. AO, CMSmith, and MK performed the experiments and analyzed the data. AO and CMSasssetti wrote the initial manuscript. All authors edited the manuscript.
References
- 1.Medzhitov R, Schneider DS, and Soares MP. 2012. Disease tolerance as a defense strategy. Science 335: 936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schneider DS, and Ayres JS. 2008. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol 8: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olive AJ, and Sassetti CM. 2016. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol 14: 221–234. [DOI] [PubMed] [Google Scholar]
- 4.Pilla-Moffett D, Barber MF, Taylor GA, and Coers J. 2016. Interferon-Inducible GTPases in Host Resistance, Inflammation and Disease. J Mol Biol 428: 3495–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hood MI, and Skaar EP. 2012. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10: 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ayres JS, and Schneider DS. 2008. A signaling protease required for melanization in Drosophila affects resistance and tolerance of infections. PLoS Biol 6: 2764–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, Larsen R, Rebelo S, Schauble S, Del Barrio L, Mithieux G, Rajas F, Lindig S, Bauer M, and Soares MP. 2017. Metabolic Adaptation Establishes Disease Tolerance to Sepsis. Cell 169: 1263–1275 e1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamieson AM, Pasman L, Yu S, Gamradt P, Homer RJ, Decker T, and Medzhitov R. 2013. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 340: 1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeney V, Ramos S, Bergman ML, Bechmann I, Tischer J, Ferreira A, Oliveira-Marques V, Janse CJ, Rebelo S, Cardoso S, and Soares MP. 2014. Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep 8: 126–136. [DOI] [PubMed] [Google Scholar]
- 10.Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, Palace SG, Goguen JD, Baker RE, Nambi S, Mishra R, Booty MG, Baer CE, Shaffer SA, Dartois V, McCormick BA, Chen X, and Sassetti CM. 2017. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol 2: 17072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meunier I, Kaufmann E, Downey J, and Divangahi M. 2017. Unravelling the networks dictating host resistance versus tolerance during pulmonary infections. Cell Tissue Res 367: 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cadena AM, Fortune SM, and Flynn JL. 2017. Heterogeneity in tuberculosis. Nat Rev Immunol [DOI] [PMC free article] [PubMed]
- 13.Chen RY, Dodd LE, Lee M, Paripati P, Hammoud DA, Mountz JM, Jeon D, Zia N, Zahiri H, Coleman MT, Carroll MW, Lee JD, Jeong YJ, Herscovitch P, Lahouar S, Tartakovsky M, Rosenthal A, Somaiyya S, Lee S, Goldfeder LC, Cai Y, Via LE, Park SK, Cho SN, and Barry CE 3rd. 2014. PET/CT imaging correlates with treatment outcome in patients with multidrug-resistant tuberculosis. Sci Transl Med 6: 265ra166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TT, Vary JC, Hawn TR, Dunstan SJ, Farrar JJ, Thwaites GE, King MC, Serhan CN, and Ramakrishnan L. 2012. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 148: 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez B, Aguilar D, Orozco H, Burger M, Espitia C, Ritacco V, Barrera L, Kremer K, Hernandez-Pando R, Huygen K, and van Soolingen D. 2003. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol 133: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redford PS, Murray PJ, and O’Garra A. 2011. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol 4: 261–270. [DOI] [PubMed] [Google Scholar]
- 17.Larson RP, Shafiani S, and Urdahl KB. 2013. Foxp3(+) regulatory T cells in tuberculosis. Adv Exp Med Biol 783: 165–180. [DOI] [PubMed] [Google Scholar]
- 18.Jayaraman P, Jacques MK, Zhu C, Steblenko KM, Stowell BL, Madi A, Anderson AC, Kuchroo VK, and Behar SM. 2016. TIM3 Mediates T Cell Exhaustion during Mycobacterium tuberculosis Infection. PLoS Pathog 12: e1005490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desvignes L, Weidinger C, Shaw P, Vaeth M, Ribierre T, Liu M, Fergus T, Kozhaya L, McVoy L, Unutmaz D, Ernst JD, and Feske S. 2015. STIM1 controls T cell-mediated immune regulation and inflammation in chronic infection. J Clin Invest 125: 2347–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pasipanodya JG, McNabb SJ, Hilsenrath P, Bae S, Lykens K, Vecino E, Munguia G, Miller TL, Drewyer G, and Weis SE. 2010. Pulmonary impairment after tuberculosis and its contribution to TB burden. BMC Public Health 10: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segal AW 2005. How neutrophils kill microbes. Annu Rev Immunol 23: 197–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panday A, Sahoo MK, Osorio D, and Batra S. 2015. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell Mol Immunol 12: 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston RB Jr., and Baehner RL. 1970. Improvement of leukocyte bactericidal activity in chronic granulomatous disease. Blood 35: 350–355. [PubMed] [Google Scholar]
- 24.Cooper AM, Segal BH, Frank AA, Holland SM, and Orme IM. 2000. Transient loss of resistance to pulmonary tuberculosis in p47(phox−/−) mice. Infect Immun 68: 1231–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung YJ, LaCourse R, Ryan L, and North RJ. 2002. Virulent but not avirulent Mycobacterium tuberculosis can evade the growth inhibitory action of a T helper 1-dependent, nitric oxide Synthase 2-independent defense in mice. J Exp Med 196: 991–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng VH, Cox JS, Sousa AO, MacMicking JD, and McKinney JD. 2004. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol 52: 1291–1302. [DOI] [PubMed] [Google Scholar]
- 27.Colangeli R, Haq A, Arcus VL, Summers E, Magliozzo RS, McBride A, Mitra AK, Radjainia M, Khajo A, Jacobs WR Jr., Salgame P, and Alland D. 2009. The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc Natl Acad Sci U S A 106: 4414–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, Nguyen HP, Shaffer SA, and Sassetti CM. 2015. The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe 17: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, Grigsby SJ, Mittal E, Park HS, Jones V, Hsu FF, Jackson M, Sassetti CM, and Philips JA. 2017. Mycobacterium tuberculosis is protected from NADPH oxidase and LC3-associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci U S A 114: E8711–E8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nathan C, and Shiloh MU. 2000. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A 97: 8841–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, and Casanova JL. 2011. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 12: 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan TA, Kalsoom K, Iqbal A, Asif H, Rahman H, Farooq SO, Naveed H, Nasir U, Amin MU, Hussain M, Tipu HN, and Florea A. 2016. A novel missense mutation in the NADPH binding domain of CYBB abolishes the NADPH oxidase activity in a male patient with increased susceptibility to infections. Microb Pathog 100: 163–169. [DOI] [PubMed] [Google Scholar]
- 33.Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, Mak PH, Fok SF, Yang X, and Lau YL. 2008. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J 27: 224–230. [DOI] [PubMed] [Google Scholar]
- 34.Deffert C, Cachat J, and Krause KH. 2014. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol 16: 1168–1178. [DOI] [PubMed] [Google Scholar]
- 35.Lau YL, Chan GC, Ha SY, Hui YF, and Yuen KY. 1998. The role of phagocytic respiratory burst in host defense against Mycobacterium tuberculosis. Clin Infect Dis 26: 226–227. [DOI] [PubMed] [Google Scholar]
- 36.Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, and Sassetti CM. 2013. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat Immunol 14: 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, and Kagan JC. 2016. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352: 1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee K, Won HY, Bae MA, Hong JH, and Hwang ES. 2011. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci U S A 108: 9548–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honore N, Marchal G, and Cole ST. 1995. Novel mutation in 16S rRNA associated with streptomycin dependence in Mycobacterium tuberculosis. Antimicrob Agents Chemother 39: 769–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, and Stallings CL. 2015. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 528: 565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kramnik I, Dietrich WF, Demant P, and Bloom BR. 2000. Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 97: 8560–8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nandi B, and Behar SM. 2011. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J Exp Med 208: 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, and Ernst JD. 2007. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179: 2509–2519. [DOI] [PubMed] [Google Scholar]
- 44.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, and Vance RE. 2013. Recognition of bacteria by inflammasomes. Annu Rev Immunol 31: 73–106. [DOI] [PubMed] [Google Scholar]
- 45.Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Nunez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, and O’Neill LA. 2015. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: 248–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorhoi A, Nouailles G, Jorg S, Hagens K, Heinemann E, Pradl L, Oberbeck-Muller D, Duque-Correa MA, Reece ST, Ruland J, Brosch R, Tschopp J, Gross O, and Kaufmann SH. 2012. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol 42: 374–384. [DOI] [PubMed] [Google Scholar]
- 47.Stack JH, Beaumont K, Larsen PD, Straley KS, Henkel GW, Randle JC, and Hoffman HM. 2005. IL-converting enzyme/caspase-1 inhibitor VX-765 blocks the hypersensitive response to an inflammatory stimulus in monocytes from familial cold autoinflammatory syndrome patients. J Immunol 175: 2630–2634. [DOI] [PubMed] [Google Scholar]
- 48.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, and Sher A. 2010. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184: 3326–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, and Behar SM. 2014. In search of a new paradigm for protective immunity to TB. Nat Rev Microbiol 12: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deffert C, Schappi MG, Pache JC, Cachat J, Vesin D, Bisig R, Ma Mulone X, Kelkka T, Holmdahl R, Garcia I, Olleros ML, and Krause KH. 2014. Bacillus calmette-guerin infection in NADPH oxidase deficiency: defective mycobacterial sequestration and granuloma formation. PLoS Pathog 10: e1004325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jackson SH, Gallin JI, and Holland SM. 1995. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 182: 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dinauer MC, Deck MB, and Unanue ER. 1997. Mice lacking reduced nicotinamide adenine dinucleotide phosphate oxidase activity show increased susceptibility to early infection with Listeria monocytogenes. J Immunol 158: 5581–5583. [PubMed] [Google Scholar]
- 53.Fujita M, Harada E, Matsumoto T, Mizuta Y, Ikegame S, Ouchi H, Inoshima I, Yoshida S, Watanabe K, and Nakanishi Y. 2010. Impaired host defence against Mycobacterium avium in mice with chronic granulomatous disease. Clin Exp Immunol 160: 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chao WC, Yen CL, Hsieh CY, Huang YF, Tseng YL, Nigrovic PA, and Shieh CC. 2017. Mycobacterial infection induces higher interleukin-1beta and dysregulated lung inflammation in mice with defective leukocyte NADPH oxidase. PLoS One 12: e0189453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones GS, Amirault HJ, and Andersen BR. 1990. Killing of Mycobacterium tuberculosis by neutrophils: a nonoxidative process. J Infect Dis 162: 700–704. [DOI] [PubMed] [Google Scholar]
- 56.Kisich KO, Higgins M, Diamond G, and Heifets L. 2002. Tumor necrosis factor alpha stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect Immun 70: 4591–4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang CT, Cambier CJ, Davis JM, Hall CJ, Crosier PS, and Ramakrishnan L. 2012. Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe 12: 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fulton SA, Reba SM, Martin TD, and Boom WH. 2002. Neutrophil-mediated mycobacteriocidal immunity in the lung during Mycobacterium bovis BCG infection in C57BL/6 mice. Infect Immun 70: 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Francis RJ, Butler RE, and Stewart GR. 2014. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis 5: e1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weinberg SE, Sena LA, and Chandel NS. 2015. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42: 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, and Blackwell TS. 2010. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One 5: e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgenstern DE, Gifford MA, Li LL, Doerschuk CM, and Dinauer MC. 1997. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J Exp Med 185: 207–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schappi M, Deffert C, Fiette L, Gavazzi G, Herrmann F, Belli D, and Krause KH. 2008. Branched fungal beta-glucan causes hyperinflammation and necrosis in phagocyte NADPH oxidase-deficient mice. J Pathol 214: 434–444. [DOI] [PubMed] [Google Scholar]
- 64.van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, Espanol T, Fischer A, Kurenko-Deptuch M, Mouy R, Petropoulou T, Roesler J, Seger R, Stasia MJ, Valerius NH, Weening RS, Wolach B, Roos D, and Kuijpers TW. 2009. Chronic granulomatous disease: the European experience. PLoS One 4: e5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, Begun J, Plantinga TS, Joosten LA, van der Meer JW, Chamilos G, Netea MG, Xavier RJ, Dinarello CA, Romani L, and van de Veerdonk FL. 2014. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A 111: 3526–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Meissner F, Molawi K, and Zychlinsky A. 2008. Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol 9: 866–872. [DOI] [PubMed] [Google Scholar]
- 67.Zhang G, Zhou B, Li S, Yue J, Yang H, Wen Y, Zhan S, Wang W, Liao M, Zhang M, Zeng G, Feng CG, Sassetti CM, and Chen X. 2014. Allele-specific induction of IL-1beta expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog 10: e1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Marais S, Lai RPJ, Wilkinson KA, Meintjes G, O’Garra A, and Wilkinson RJ. 2017. Inflammasome Activation Underlying Central Nervous System Deterioration in HIV-Associated Tuberculosis. J Infect Dis 215: 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tan HY, Yong YK, Shankar EM, Paukovics G, Ellegard R, Larsson M, Kamarulzaman A, French MA, and Crowe SM. 2016. Aberrant Inflammasome Activation Characterizes Tuberculosis-Associated Immune Reconstitution Inflammatory Syndrome. J Immunol 196: 4052–4063.. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.