

Abstract
Purpose
60% of papillary thyroid cancers (PTC) have an oncogenic (V600E) BRAF mutation. Inhibitors of BRAF and its substrates MEK1/2 are showing clinical promise in BRAFV600E PTC. PTC progression can be decades long, which is challenging in terms of toxicity and cost. We previously found that MEK1/2 require copper (Cu) for kinase activity and can be inhibited with the well-tolerated and economical Cu chelator TM. We therefore tested TM for antineoplastic activity in BRAFV600E-positive PTC.
Experimental Design
The efficacy of TM alone and in combination with current standard-of-care lenvatinib and sorafenib or BRAF and MEK1/2 inhibitors vemurafenib and trametinib was examined in BRAFV600E-positive human PTC cell lines and a genetically engineered mouse PTC model.
Results
TM inhibited MEK1/2 kinase activity and transformed growth of PTC cells. TM was as or more potent than lenvatinib and sorafenib and enhanced the antineoplastic activity of sorafenib and vemurafenib. Activated ERK2, a substrate of MEK1/2, overcame this effect, consistent with TM deriving its antineoplastic activity by inhibiting MEK1/2. Oral TM reduced tumor burden as well as vemurafenib in a BrafV600E-positive mouse model of PTC. This effect was ascribed to a reduction of Cu in the tumors. TM reduced P-Erk1/2 in mouse PTC tumors while genetic reduction of Cu in developing tumors trended towards a survival advantage. Finally, TM as a maintenance therapy after cessation of vemurafenib reduced tumor volume in the aforementioned PTC mouse model.
Conclusions
TM inhibits BRAFV600E-driven PTC through inhibition of MEK1/2, supporting clinical evaluation of chronic TM therapy for this disease.
Introduction
The incidence of thyroid cancer is rising faster than that of any other malignancy. New cases have more than tripled over the past four decades, with the papillary thyroid cancer (PTC) histologic subtype accounting for the overwhelming majority of the rising incidence (1, 2). Tumors of all stages and sizes have increased in incidence, as has the incidence-based mortality rate, suggesting that the shift in epidemiology cannot be explained solely by over-diagnosis (3, 4). Fortunately, most PTC patients are diagnosed with early stage disease, which responds well to surgical resection with or without radioactive iodine (RAI) therapy. Indeed, the 10-year survival rate for Stage I or II disease is 90%. However, a subset of PTC cases does not respond to conventional therapy, and the 10-year survival rate for patients with regional lymph node involvement and/or distant metastases drops to 77% and 37%, respectively (5). Even in patients initially responsive to RAI, there is a 20-30% recurrence rate within 10-15 years of diagnosis (6). The therapeutic options for patients with progressive or symptomatic disease not amenable to surgical resection or RAI are limited to the tyrosine kinase inhibitors (TKIs) sorafenib or lenvatinib (7, 8). These therapies extend progression free survival by 5 to 15 months and are associated with well-documented toxicities (9, 10). As PTC is typically diagnosed in patients under 55 years of age, yet disease-specific death peaks in the 70s (11). This presents a unique, decades-long clinical challenge between balancing appropriate therapy with patient quality of life (12, 13).
In the U.S., 40% to 60% of PTC tumors have an oncogenic (V600E) mutation in the kinase BRAF (14), which is associated with a 2-fold increase in recurrent/persistent disease and increased mortality (6, 14). This mutation results in a constitutively active BRAF kinase, leading to increased phosphorylation and activation of MEK1/2 kinases, which in turn phosphorylate and activate ERK1/2 kinases, all of which constitute the MAPK pathway. Activation of the MAPK pathway in this fashion promotes expression of an array of cell survival and proliferation genes that drive tumorigenesis (14). The BRAFV600E mutation may be a driver mutation in PTC; it appears early in human PTC (15) and causes PTC when recreated in mice (16) alone or even more rapidly, when combined with the loss of PTEN(17). The MAPK pathway has been extensively drugged. There are now two BRAFV600E inhibitors (BRAFi), vemurafenib and dabrafenib, and two MEK1/2 inhibitors (MEKi) trametinib and cobimetinib, approved as combination treatments for BRAFV600E-positive late stage melanoma (18–20). Similarly, the MEKi selumetinib alone (21, 22) or in combination with vemurafenib (23) are being tested for the treatment of non-small cell lung cancer, with a host of other MAPK inhibitors (MAPKi) under clinical evaluation (24, 25). MAPKi are also being clinically evaluated for the treatment of BRAFV600E-positive PTC (26). In a phase II trial of patients with BRAFV600E-positive RAI-resistant PTC, vemurafenib treatment resulted in 15.6 and 9.8 months of progression-free survival in TKI-naïve and TKI-treated patients, with response rates of 58% and 36%, respectively (27). Similarly, dabrafenib prevented disease progression in 13 out of 14 patients with metastatic BRAFV600E-positive PTC and extended progression-free survival by 11.3 months, with only one patient progressing, and then only after 9.3 months of response (28). MEKi are also showing promise in the management of advanced PTC by restoring tumor sensitivity to RAI (29–31). Moreover, the combination of vemurafenib and trametinib was recently shown to have an overall response rate of 69% in patients with BRAFV600E-positive anaplastic thyroid cancer (ATC), with an estimated median 1-year survival rate of 80% (32).
One potential challenge to the clinical adoption of MAPKi for the treatment of BRAFV600E- positive PTC is the protracted period of time that can elapse before recurrence is identified. Even in patients initially responsive to RAI, there is a 20-30% recurrence rate within 10-15 years of diagnosis (33). It is thus conceivable that MAPKi may need to be chronically dosed. Drug toxicities may become problematic in such a prolonged treatment setting based on the experiences in other cancers. 20% of patients with BRAFV600E-positive solid tumors treated with selumetinib required dose reduction or discontinuation while 38% of BRAFV600E-positive melanoma patients treated with the same drug were unable to complete the first cycle of therapy (30, 34). Similarly, in a Phase II trial of combined dabrafenib plus trametinib in melanoma, 55% of patients experienced dose interruptions and 33% required dose reductions, with 13% treatment termination (35). Early indications suggest the same may be expected for PTC. 66% of BRAFV600E-positive PTC patients treated with vemurafenib experience Grade 3 or 4 adverse events (26). Such toxicities must be weighed against the quality of life and cost of therapy, especially in a disease like PTC for which patients can remain asymptomatic for decades. Indeed, there have been calls for the design of treatment courses that are more physically and financially appropriate for patients who require long-term management (12, 13, 36–38).
Current MEKi all target the ATP-dependent activity of these kinases (39). However, MEK1/2 require the metal copper (Cu) for kinase activity and to mediate the oncogenic signaling and tumorigenic activity of BRAFV600E. Importantly, this novel activity of MEK1/2 can be pharmacologically targeted with existing Cu chelating drugs (40–42). Oral Cu chelators have been used to lower Cu levels for the treatment of Wilson’s disease, a Cu overload condition (43, 44). These are extremely well tolerated drugs when paired with serological monitoring of Cu levels by measuring the amount of Cu bound to the plasma protein ceruloplasmin. Patients with Wilson’s disease remain on continuous Cu chelation for their entire lives (43, 44). Of these, tetrathiomolybdate (TM) exhibits higher potency in comparison to other chelators (45, 46) and functions both by binding dietary copper to prevent copper absorption and by forming a complex with free Cu and albumin in the blood (47–52). Clinical cancer trials of TM, in part based on the relationship of Cu with angiogenesis (53–57), reported that TM is well tolerated in patients with advanced malagnancies (54, 58–63). In a Phase I trial for metastatic breast cancer, patients were orally dosed three times daily with TM for upwards of 65 months(63). A Phase II trial on breast cancer patients has shown that the most common side-effects for TM treatment are Grade I and II sulfur eructation, fatigue, and neutropenia (62) – the same side effects seen in Wilson’s disease patients (45, 46, 64) – which were managed by adjusting the dose of TM (54, 58, 60–63). We therefore explored the potential of repurposing TM for long-term inhibition of the MAPK pathway in BRAFV600E-positive PTC.
Materials and methods
Cell lines
BCPAP (65) and K1 (66) cells were purchased from ATCC in 2014 and maintained in RPMI supplemented with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin. The identity of both cell lines was confirmed by DNA profiling of polymorphic short tandem repeat (STR) markers through the human cell line authentication analysis service at the Duke University DNA Analysis Facility. The resultant STR makers, assess by GenePrint 10 kit (Promega), were compared to those available for BCPAP (CVCL_0153) and K1 (CVCL_2537) cells lines through Cellosaurus (65) on February 2018. Both cell lines were also confirmed to be free of mycoplasma infection, as assessed by the Duke Cell Culture Facility using MycoAlert PLUS test (Lonza) on January 2018. Both cell lines were used within 5 passages of being thawed. BCPAP cell lines were engineered to express ERK2GOF by stable infection using established methods (67) of a retrovirus derived from the plasmid pBABEpuro-HA-ERK2GOF encoding the ERK2R67S, D321N mutant form of ERK2, termed ERKGOF (40).
Drug preparation
Trametinib, vemurafenib, sorafenib (Chemitek), lenvatinib (Selleckchem), and ammonium tetrathiomolybdate, termed TM (Sigma-Aldrich), were dissolved in 100% DMSO for in vitro experiments and 1% DMSO/1% methylcellulose for in vivo experiments.
Soft agar assay
Soft agar assays were performed as previously described (40). In brief, 1 ml of 0.3% bactoagar-RPMI solution containing 2.0×104 BCPAP cells or 1.0×104 K1 cells and the indicated concentration of drug or vehicle was plated into each well of a 6-well plate already containing a solidified 2 ml bottom layer of 0.6% bactoagar-RPMI solution also containing the relevant vehicle or drug. Cells were fed weekly with 250 μl of media containing vehicle or drug. After 3 weeks, colonies containing > 50 cells were counted in a blinded fashion. Drug concentrations were determined based on effective concentration. Specifically, we first empirically identified the lowest concentrations of each drug that inhibited colony formation (see below) to the maximum level, and termed this the EC100. Based on this, the EC0, EC12.5, EC25, EC50, EC100, and EC200 and are as follows: 0, 50, 100, 200, 400, or 800 nM for TM; 12.5, 25, 50, 100, or 200 nM for sorafenib; 12.5, 25, 50, 100, or 200 nM for lenvatinib; 12.5, 25, 50, 100, or 200 nM for vemurafenib; and 0.625, 1.25, 2.5, 5.0, or 10.0 nM for trametinib (Supplementary Table 1). Each drug was serially diluted from the highest concentration so that the total volume of DMSO added was identical in each well with the 0 nM control, namely the total volume of DMSO per well was 200 μl for TM, 50 μl for sorafenib, lenvatinib, and vemurafenib, and 25 μl for trametinib, regardless of the dilution. The concentrations of drugs in combinations experiments were combined in fixed-ratio doses in order to calculate therapeutic synergy.
Immunoblot analysis
Whole cell lysates were isolated using standard RIPA buffer containing proteases and phosphatases and quantified using the Lowery protein assay (BioRad). 50 μg of lysates from BCPAP cells treated with DMSO vehicle or increasing concentrations (25 nM, 50 nM, or 100 nM) of TM for 7 days or TM (EC6.25, 25 μM), vemurafenib (EC6.25, 6.25 μM) or both drugs at the same concentrations for 7 days were resolved by SDS-PAGE and immunoblotted with a rabbit anti-phospho(Thr 202/Tyr 204)-ERK1/2 antibody (Cell Signaling Technology, antibody # 3700 at a 1:1000 dilution), a mouse anti-ERK1/2 antibody (Cell Signaling Technology, antibody # 9101 at a 1:1000 dilution), a mouse anti-HA-Tag antibody (Cell Signaling Technology, antibody # 2367 at a 1:1000 dilution), a rabbit anti-phospho-S6 ribosomal protein (Ser235/236) antibody (Cell Signaling Technology, antibody #4858S at a 1:1000 dilution), a rabbit anti-S6 ribosomal protein (Cell Signaling Technology, antibody #2217 at a 1:1000 dilution), or a mouse anti-β-tubulin (Sigma-Aldrich, antibody # 2367 at a 1:5000 dilution) followed by a goat anti-rabbit IgG (Cell Signaling Technology, antibody # 7076) or a goat anti-mouse IgG (Cell Signaling Technology, antibody # 7074) horseradish peroxidase-conjugated secondary antibody and visualized using enhanced chemiluminescence detection (Cell Signaling Technology).
8-week treatment TBP mouse study
Mice with BrafCA (68), Ptenfl (69), and Thyro::CreERT2 (70) alleles were obtained from the Jackson Laboratory or as a kind gift from the laboratories of David Kirsch (Duke University), or Martin McMahon (University of Utah). BrafCA/CA;Ptenfl/fl and Thyro::CreERT2/+ mice were crossed to generate Thyro::CreERT2/+;BrafCA/+;Ptenfl/+(TBP) mice (17). Mice in this study were derived from a mixed BL6 and Sv129 background and littermates were used regardless of sex. At 40 days of age, TBP mice received a single 100 μl intraperitoneal injection of a 10 mg/ml tamoxifen (Sigma-Aldrich) dissolved in peanut oil to activate CreER in thyrocytes and induce tumorigenesis. 8 weeks later, cohorts of 10 mice each were randomly assigned to one of three treatment groups receiving daily oral gavage of vehicle (250 μl of 1% DMSO/1% methylcellulose), vemurafenib (50 mg/kg), or TM (80 mg/kg). The appearance, behavior, and weight of mice were monitored daily and drug holidays provided if weight dropped below 10% of the maximum weight of the animal. The drug holiday was maintained until the mouse reached its previous weight. There was only a one-day difference in the average number of treatment days between the three cohorts (Supplementary Table 2). Mice were euthanized at the end of the 8-week treatment period and their thyroids removed for analysis. Studies involving mice were conducted in accordance with protocols approved by the Duke University Institutional Animal Care and Use Committee.
Ctr1fl/fl TBP mouse study
Mice with a Ctr1fl allele (71), a kind gift of Dennis Thiele (Duke University), were used to generate BrafCA/CA;Ptenfl/fl;Ctr1fl/+ and Ctr1fl/+;Thyro::CreERT2 mice, which were crossed to generate 23 Ctr1+/+ versus 8 Ctr1fl/fl TBP littermates. Mice in this study were derived from a mixed BL6 and Sv129 background and littermates were used regardless of sex. At 40 days of age, thyroid tumorigenesis was induced by injection of tamoxifen as above, after which the appearance, behavior, and weight of these mice were monitored weekly. Mice were humanely euthanized upon reaching a maximum tumor volume (1 cm3), 15% weight loss, or moribund. Studies involving mice were conducted in accordance with protocols approved by the Duke University Institutional Animal Care and Use Committee.
24-week treatment TBP mouse study
At 40 days of age TBP mice were injected with tamoxifen as above to induce thyroid tumorigenesis. Mice in this study were derived from a mixed BL6 and Sv129 background and littermates were used regardless of sex. 8 weeks later, mice were randomly assigned into cohorts of 17 mice that were treated by daily oral gavage with vemurafenib (50 mg/kg) and either vehicle (250 μl of 1% DMSO/1% methylcellulose) or TM (80 mg/kg) for 4 weeks. Vemurafenib treatments were then terminated while vehicle or TM treatments were continued for a further 20 weeks. Mice were monitored as above and given drug holidays if required. All mice were humanely euthanized at the end of the 24-week treatment period and their tumors removed for analysis. Studies involving mice were conducted in accordance with protocols approved by the Duke University Institutional Animal Care and Use Committee.
Determining tumor volumes in thyroids
Thyroids removed from the above TBP mice at the end of the 8- or 24-week treatment period were fixed in formalin for 24 hours and paraffin embedded. Each thyroid was sectioned in its entirety on a RM2125 RTS microtome (Leica Biosystems Incorporated). Multiple serial slices from every 200 μM throughout the depth of the gland were mounted on slides. Two slides from each depth were reserved for immunohistochemical analysis (see below) while another one was hematoxylin and eosin (H&E) stained and the tumor perimeter delineated by an endocrine pathologist blinded to the genotype. One pathologist performed all tumor delineations over the span of two months and referenced control slides from a normal and 8-week post-induction thyroid from TBP mice to reduce intra-rater variability. Tumor volume was calculated based on the delineated tumor area for each slide. The tumor load, as defined by percent tumor occupying the total thyroid area, was calculated for each slide and then used to calculate the volume of thyroid occupied by tumor for each mouse.
Immunohistochemistry
The slides reserved for immunohistochemical analysis were de-paraffinized, dehydrated, and stained with a rabbit anti-phospho (Thr 202/Tyr204)-Erk1/2 (Cell Signaling Technology, antibody # 4376, 1:400 dilution) or a rabbit anti-CD31 (Abcam, antibody #28364, 1:100 dilution) by the Duke Pathology Research Histology Laboratory. A breast cancer slide and a mouse spleen slide were used as positive control for p-Erk1/2 and CD31, respectively. High-powered, stitch photographs were taken of the entire tissue on a Vanox S microscope (Olympus Corporation of the Americas). Percentage of immuno-positive area per tumor was then quantified in a blinded fashion using Image J (NIH). Both thresholding and/or optical density analyses was performed.
Statistics
Statistical analysis of tumor load, tumor volume, and metastases was performed with two-way analysis of variance (ANOVA) using Prism 6 software (GraphPad Software Incorporated). Kaplan-Meier survival curve analysis was performed using the Mantel-Cox log-rank method to compare survival curves between Ctr1+/+ versus Ctr1fl/fl TBP littermates using Prism 6 software (GraphPad Software Incorporated). Synergy between drug combinations was calculated using the previously described Bliss and Combination Indexes (72, 73). Statistical analysis of metastatic lesions, growth in soft agar, and immunohistochemical staining of P-Erk1/2 and CD31 was performed with two-way t-test using Prism 6 software (GraphPad Software Incorporated).
Results
TM reduces growth of a human BRAFV600E-positive PTC cell line through inhibition of MEK1/2
Given the rapidly evolving landscape of targeted therapies for BRAFV600E-positive cancers, we sought to evaluate the therapeutic potential of TM relative to both standard-of-care (SOC) treatment for progressive and/or symptomatic advanced iodine-resistant PTC, sorafenib and lenvatinib (36), and the promising MAPKi, vemurafenib, dabrafenib, and trametinib (24, 26–28). We elected to use a more long-term assay of soft agar growth over traditional proliferation assays. BCPAP cells, one of the very few BRAFV600E-positive human PTC cell lines available (65), were seeded in triplicate in soft agar containing either vehicle or a five-step dose escalation of each of the aforementioned five drugs. The dosages of these drugs were based on the effective concentration, as defined by the lowest concentration that inhibited colony formation to the maximum level for each drug (EC100). After three weeks of drug treatment, the number of anchorage-independent colonies was counted in a blinded fashion. This analysis revealed that TM was as or more effective than sorafenib and lenvatinib at reducing the anchorage-independent growth of BCPAP cells (Fig. 1A). The small effect seen by sorafenib and lenvatinib was not unexpected, as both drugs are TKI inhibitors while the tested cells are BRAFV600E-positive. The relative effectiveness of TM compared to vemurafenib and trametinib tracked with previous studies using BRAFV600E-positive melanoma cell lines (42), suggesting that TM targets the MAPK pathway with similar effectiveness. Similar results were observed in another BRAFV600E-positive human PTC cell line (Supplementary Fig. 1). To confirm that the effect of TM on transformed growth was through inhibition of MEK1/2 rather than other potential Cu-dependent proteins or pathways, BCPAP cells were engineered and confirmed by immunoblot to stably express ERKGOF, an activated version of this kinase (40) (Fig. 1B). The cells were then treated with TM and the levels of phosphorylation ERK1/2 (P-ERK1/2), the substrates of MEK1/2, and phosphorylated S6 (P-S6), an ERK1/2 substrate, were assessed by immunoblot analysis and transformed growth was assessed by a soft agar assay. As controls, vehicle-treated vector cells (negative control) were shown to exhibit lower P-ERK1/2 and P-S6 levels compared to vehicle-treated ERKGOF cells (positive control). In agreement with previous studies in other BRAFV600E-positive tumor and cancer cell lines (40, 42), P-ERK1/2 levels were reduced in cells treated with TM (Fig. 1B), and furthermore, TM failed to reduce the anchorage-independent growth of BCPAP cells expressing ERKGOF (Fig. 1C). Similar results were found with another Cu chelator, trientine (Supplementary Fig. 2). We conclude that TM inhibits MEK1/2 kinase activity and correspondingly retards the transformed growth of the human BRAFV600E-positive PTC cell line BCPAP.
Figure 1. TM reduces anchorage-independent growth of BCPAP cells.
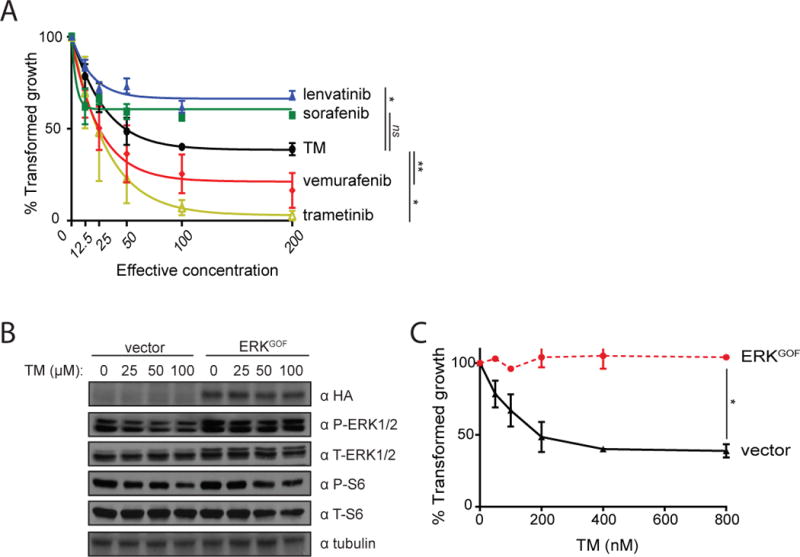
A) % transformed growth in soft agar (mean ± SEM, triplicate samples, three experiments) normalized to vehicle control of BCPAP cells treated with increasing doses (effective concentration) of lenvatinib (blue solid triangle), sorafenib (green solid square), TM (black solid circle), vemurafenib (red solid diamond), or trametinib (yellow open triangle).
B) Immunoblot detection of HA epitope-tagged ERKGOF, phosphorylated (P-) and total (T-) Erk1/2 or S6, and tubulin in BCPAP cells transduced with an expression vector encoding no transgene (vector) or ERKGOF treated with the indicated increasing concentrations of TM.
C) % transformed growth in soft agar (mean ± SEM, triplicate samples, three experiments) normalized to vehicle control of BCPAP cells stably transduced with a vector encoding no transgene (black solid triangle) or one encoding ERKGOF (red solid circle) at the indicated increasing concentrations of TM.
*p<0.05. **p<0.01. ns= not significant.
Oral TM reduces tumor load and volume in a BrafV600E-driven mouse model of aggressive PTC
To address whether TM exhibits antineoplastic activity in a more relevant in vivo setting, we turned to the genetically engineered Thyro::CreERT2/+;BrafCA/+;Ptenfl/+(TBP) mouse model of PTC. In this model, administration of tamoxifen activates CreER recombinase in the thyrocytes, leading to recombination of the BrafCA and Ptenfl alleles. This results in expression of oncogenic BrafV600E and inactivation of the tumor suppressor Pten in these cells, which leads to aggressive metastatic PTC (17). Indeed, longitudinal analysis of the thyroids from TBP mice after injection with tamoxifen revealed a stepwise progression from normal thyroid (Fig. 2A, B, and C). At two weeks post-injection, gross goiter (Fig. 2D, E, and F) was detected. At eight weeks, histologically confirmed PTC with the diagnostic papillary structures and nuclear pallor, grooves, and enlargement were present (Fig. 2G, H, and I). At 12 weeks, enlarged tumors were observed (Fig. 2J, K, and L) with half of the mice developing metastasis to the lung. The primary tumors retained their PTC characteristics (diagnostic papillary structures, and nuclear pallor, grooves, and enlargement). This lack of dedifferentiation into ATC and tumor aggression confirms the validity of TBP as a model for aggressive BrafV600E-positive PTC.
Figure 2. Histology and gross pathology of thyroid tumor development in TBP mice.
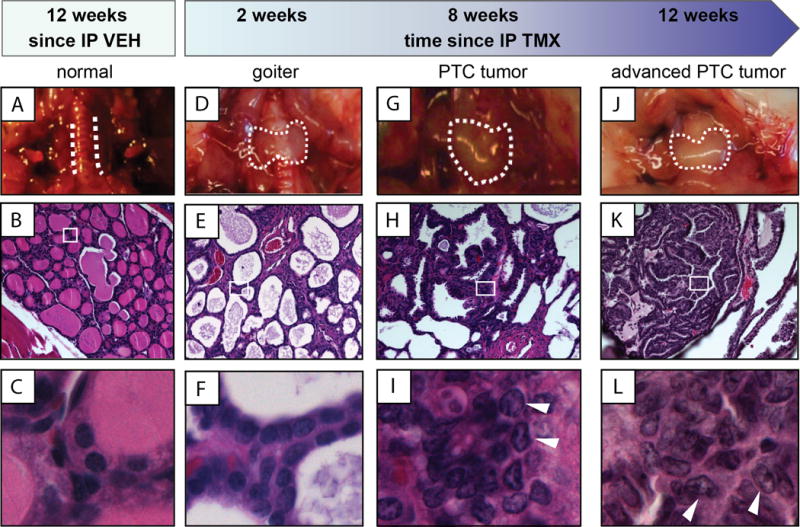
Samples from euthanized TBP mice at the indicated time points after an intraperitoneal (IP) injection of vehicle (VEH, A to C) or tamoxifen (TMX, D to L). Top: Gross pathology (dotted lines demark border of thyroid). Middle: H&E staining of thyroid section at 20× magnification. Bottom: 63× magnification of boxed region in corresponding 20× images. Arrows indicate nuclear clearing and grooves characteristic to PTC.
At 40 days of age, TBP mice were injected with tamoxifen and then randomly assigned to one of three treatment groups of ten mice each: i) a negative-control vehicle-treated cohort; ii) a positive-control vemurafenib-treated cohort (given that this drug shows clinical promise in phase II trials of RAI-resistant PTC) (26); and finally, iii) the experimental TM-treated cohort. As noted above, histologically confirmed PTC was not established until eight weeks after tamoxifen injection (Fig. 2). Thus, to ensure the presence of established disease, treatments were not initiated until eight weeks post-tamoxifen injection. As TM requires up to four weeks to reduce Cu to therapeutically low levels in both rodents and humans (47–49, 58), mice were treated for a total eight weeks in order to provide four weeks of therapeutic TM dosing. This dose of TM has previously been shown to reduce serum ceruloplasmin activity by 20% in mice (42), which compares favorably to human clinical cancer trials that reduce serum ceruloplasmin activity by up to 50% (62). To ensure the same effective treatment time, mice in the vemurafenib arm were treated for the first four weeks, then provided vehicle control for the final four weeks. After eight weeks, all mice were humanely euthanized and their thyroids were removed en-bloc, paraffin embedded, and serially sectioned every 200 m (amounting to roughly 15 sections per thyroid). Necropsy revealed a similar number of mice with grossly visible metastatic lung lesions (Supplementary Fig. 3A). The region of the thyroid occupied by tumor was then circumscribed by a pathologist who was blinded to the genotype of the samples. All mice were confirmed by the pathologist to have PTC. The tumor versus total thyroid area was determined for each section, and the total volume occupied by the tumor calculated from all sections and recorded as either the tumor volume or as a percentage of total thyroid occupied by tumor (Fig. 3A). Most thyroids contained one tumor per lobe, as demonstrated in three-dimensional reconstructions of a sample thyroid from each study cohort (Fig. 3B). As expected, vemurafenib treatment significantly reduced the average tumor load by 22% (Fig. 3C) and tumor volume by 39% (Fig. 3D) in comparison to vehicle control. Similarly, TM treatment significantly reduced the average tumor load by 34% (Fig. 3C) and tumor volume by 40% (Fig. 3D). Thus, oral TM treatments in mice with established PTC reduces tumor load as well as treatment with the clinical BRAFi vemurafenib.
Figure 3. TM treatments reduce tumor load and volume in TBP mice.
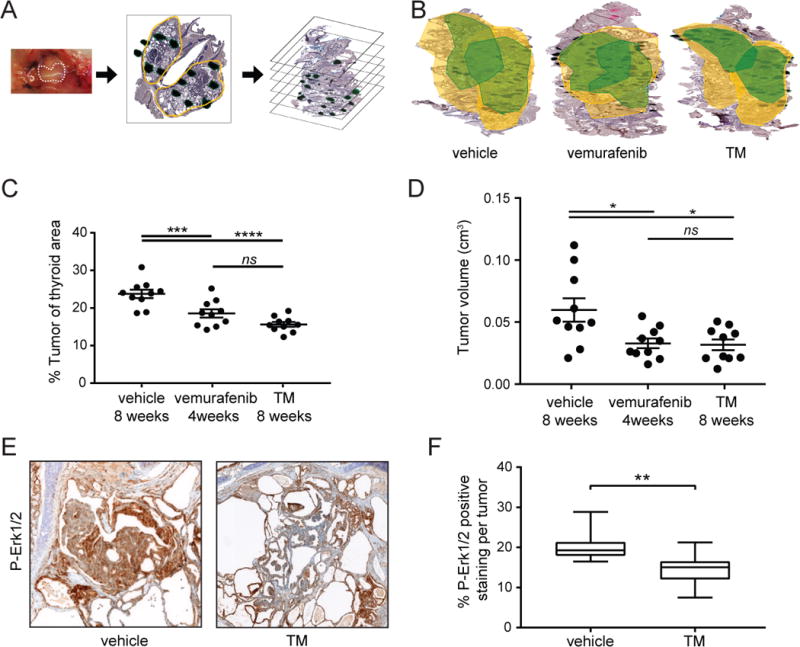
A) Diagram of tumor load and volume analysis. Left panel: Thyroids (dotted line) were removed from TBP mice euthanized 8 weeks after being treated with vehicle, vemurafenib, or TM. Middle panel: Thyroids were fixed enblock, paraffin mounted, sections taken every 200 microns, and H&E stained. Right panel: Tumor area (dark green regions) in the thyroid (yellow outline) was determined by a pathologist blinded to the mouse genotype to calculate the tumor load (% of the ratio of tumor area / thyroid area of all sections) and tumor volume (cm3).
B) A representative 3-dimensional reconstruction of a thyroid enblock from a TBP mouse euthanized 8 weeks after being treated with vehicle, vemurafenib, or TM. Yellow outlines the thyroid area while green outlines the tumor boundaries. Both lobes of the thyroid are shown.
C) % tumor load (% of thyroid occupied by tumor, each sample is a filled circle, mean ± SEM shown as bars) at the 8-week fixed endpoint in three cohorts of ten TBP mice orally treated with vehicle, vemurafenib, or TM.
D) Tumor volume (cm3, each sample is a filled circle, mean ± SEM shown as bars) at the 8-week fixed endpoint in three cohorts of 10 TBP mice orally treated with vehicle, vemurafenib, or TM.
E) Representative image of a thyroid section Hematoxylin stained (purple) and immunohistochemically stained with an anti-P-Erk1/2 antibody (brown) isolated from a TBP mouse euthanized at the 8-week fixed endpoint after being treated with vehicle (left) or TM (right).
F) Boxplot of % P-Erk1/2 positive area per tumor (18 tumors from 10 mice in each cohort) at the 8-week fixed endpoint in TBP mice treated with either vehicle or TM.
*p<0.05. **p<0.01. ***p<0.005. ****p<0.001. ns=not significant.
Oral TM inhibits Mek1/2 kinase activity in PTC tumors
To explore the mechanism of TM, we performed immunohistochemical staining for phosphorylated (P) Erk1/2. MEK1/2 kinases are well established to transmit oncogenic BRAF signaling by phosphorylating their substrates ERK1/2 (14). As such, reduction in P-Erk1/2 levels in tumors has been used to validate on-target effects of MAPKi. 36 thyroid sections from 20 TBP mice treated with TM or vehicle were stained with an anti-P-Erk1/2 antibody, after which the positive-stained area of tumors was circumscribed and expressed as a percent of the total tumor. In agreement with the effects of TM on P-Erk1/2 levels in tumors from other oncogenic BrafV600E cancer mouse models (40), there was a statistically significant reduction in percent P-Erk1/2 staining in the thyroid tumors of mice treated with TM, as assessed by threshold analysis (Fig. 3E and F), although not by optical density analysis (Supplementary Fig. 4). In agreement with previous observations that chelating Cu reduces angiogenesis (53–57), CD31 staining was also statistically reduced in the thyroid tumors of mice treated with TM compared to those treated with vehicle, as assessed by threshold analysis (Supplementary Fig. 5). Thus, TM appears to inhibit Mek1/2 kinase activity in BrafV600E-positive PTC lesions.
Crossing a Ctr1fl gene into a BrafV600E-driven mouse model of aggressive PTC trended towards an extension of lifespan
To genetically test whether the antineoplastic activity of TM was due to a reduction of Cu in PTC lesions, we compared the lifespan of TBP mice with or without a functional Ctr1 gene in their tumors. Ctr1 encodes the primary Cu-specific transporter in mammalian cells, and inactivation of this gene has been shown to reduce Cu levels in cells (71, 74), including in tumors (40, 42). A floxed version of the Ctr1 gene (71) was therefore crossed into the TBP background. TBP littermates homozygous for the wild-type (+/+, n=23) versus the floxed (fl/fl, n=8) Ctr1 alleles were treated with tamoxifen to both induce PTC and inactivate the Ctr1fl alleles when present. All mice were then regularly monitored and euthanized upon reaching disease endpoint where all mice were confirmed to have thyroid tumors. Analysis of the Kaplan-Meier survival curve revealed a trend towards an 18% increase in median survival of the Ctr1fl/fl cohort. Moreover, the near absence of mice reaching endpoint in the Ctr1fl/fl cohort until around 400 days suggests a true survival benefit until a sudden decline due to age, although admittedly all mice reached endpoint with thyroid cancer (Fig. 4). These findings support the contention that the antineoplastic activity of TM is derived from its ability to reduce Cu in PTC lesions. This in turn could lead to a survival benefit, which ultimately is the most important critical clinical outcome.
Figure 4. Loss of Ctr1 in thyrocytes of TBP mice trends towards a survival advantage.
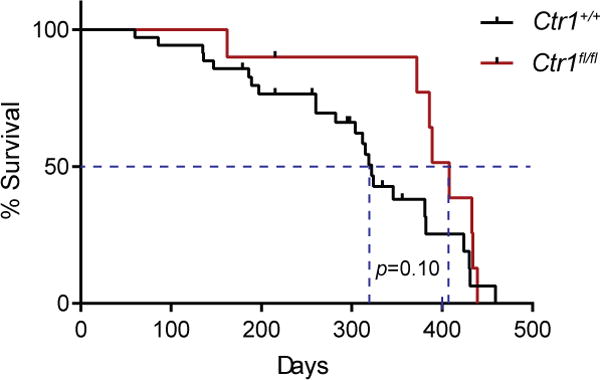
Kaplan-Meier survival curve of Ctr1+/+ (n=23, 12 on study, red line) versus Ctr1fl/fl (n=8, 2 on study, black line) TBP mice. Dotted line: median survival. Censor marks: mice still on study. p=0.10.
TM enhances the antineoplastic activity of sorafenib and vemurafenib
Given the potential of TM to therapeutically target the MAPK pathway and an increasing emphasis on multi-drug therapy, we evaluated the therapeutic potential of TM when combined with current SOC drugs, sorafenib and lenvatinib, or the clinically assessed MAPKi vemurafenib and trametinib, again using the long-term assay of growth in soft agar. BCPAP cells were seeded in triplicate in soft agar containing one of these five drugs at their EC12.5, EC25, and EC50 concentrations, or in combinations with TM, again at these three concentrations. Given the known effect of combining BRAFi and MEKi, we also tested a triple combination of TM with vemurafenib and trametinib. Vehicle-treated cells served to normalize transformed growth to 100%. After three weeks of drug treatment, the number of anchorage-independent colonies was counted in a blinded fashion, and the percent transformed growth was determined and used to calculate the Combination and BLISS Indices to assess drug synergy. TM was synergistic with vemurafenib by both indices at EC12.5 and the BLISS index at EC25 (Table 1). This agrees with the synergy observed between TM and vemurafenib in other BRAFV600E-positive cancer cell lines (42), and the clinical superiority of BRAFi and MEKi combination therapy in comparison to either drug alone (35, 75). This was attributed to a reduction in MAPK signaling, as the addition of TM at an EC6.25 to vemurafenib at an EC6.25 reduced P-ERK1/2 and P-S6 levels in BCPAP cells below those of the same cells treated with either drug alone (Supplementary Fig. 6). TM was also synergistic with sorafenib by both indices at EC12.5 and the Combination Index at EC25 with an additive effect by the BLISS Index (Table 1), which is perhaps not unexpected as sorafenib was originally designed to target BRAF paralog, CRAF (76). TM was not synergistic with lenvatinib, trametinib, or in the triple combination. In summary, TM enhances the antineoplastic activity of the SOC sorafenib and the BRAFi vemurafenib, this suggests a possible avenue to evaluate TM clinically.
Table 1.
TM is synergistic with sorafenib and vemurafenib at EC12.5 and EC25.
| TM with | Combination Index | EC12.5 | EC25 | EC50 |
|---|---|---|---|---|
| lenvatinib | BLISS | 0.68 | 0.86 | 0.71 |
| CI | 0.97 | 0.72 | 0.95 | |
| sorafenib | BLISS | 1.75 | 1.07 | 0.86 |
| CI | 0.34 | 0.52 | 1.28 | |
| vemurafenib | BLISS | 2.63 | 1.42 | 0.79 |
| CI | 0.51 | 0.88 | 1.44 | |
| trametinib | BLISS | 0.74 | 0.53 | 0.29 |
| CI | 0.88 | 2.95 | 8.27 | |
| vemurafenib trametinib | BLISS | 0.82 | 0.81 | 0.87 |
| CI | 0.72 | 1.13 | 0.57 |
Summary of BLISS and Combination Index (CI) values for anchorage independent growth of BCPAP cells at increasing fixed-ratio doses (EC12.5, 25, and 50). BLISS Index is presented in grey rows and CI in white rows. BLISS Index vales -1.2 are considered additive, while BLISS Index >1.2 are considered synergistic. CI Index vales 0.8-1 are considered additive, while CI Index <0.8 are considered synergistic. Synergistic values are bolded.
Oral TM enhances the ability of vemurafenib to reduce tumor volume in a BrafV600E-driven mouse model of aggressive PTC
As TM enhances the antineoplastic activity of vemurafenib and can be dosed for extended periods of time in late-stage cancer patients, we surmised that one clinical scenario that TM may prove valuable is long-term maintenance therapy following vemurafenib treatment. To this end, we treated mice with vemurafenib and TM for four weeks, and then continued TM treatments for a further four weeks, but saw no advantage over TM or vemurafenib alone (not shown). We therefore extended TM treatment for a total of 20 weeks. Specifically, TBP mice were injected with tamoxifen at 40 days of age as above, and eight weeks later when PTC was established to be present (e.g. Fig. 2), two cohorts of 17 mice were treated with i) vemurafenib and vehicle for four weeks, followed by 20 weeks of vehicle or ii) vemurafenib and TM for four weeks, followed by 20 weeks of TM. All mice were humanely euthanized and tumor volumes determined exactly as described above. Mice treated with vemurafenib and TM followed by maintenance therapy with TM exhibited an average tumor volume of 0.20±0.01 cm3 whereas mice treated with vemurafenib and vehicle followed by vehicle exhibited an average tumor volume of 0.40±0.04 cm3, or roughly a 50% reduction in tumor volume (Fig. 5). The number of metastatic lesions was similar between the two cohorts (Supplementary Fig. 3B). Thus, prolonged TM therapy after cessation of vemurafenib treatment reduces tumor volume in a mouse model of aggressive PTC.
Figure 5. Long-term TM treatment after cessation of vemurafenib treatment reduces tumor volume in TBP mice.
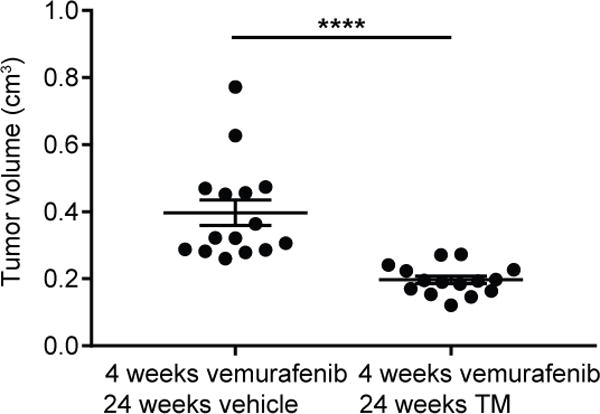
Tumor volume (cm3, each sample is a filled circle, mean ± SEM shown as bars) at the 24-week fixed endpoint in cohorts of 17 TBP mice orally treated with vemurafenib and vehicle, or vemurafenib and TM as indicated. ****p<0.001.
Discussion
The finding that 40 to 60% of PTC tumors have a BRAFV600E mutation (6) in the U.S. prompted the clinical evaluation of BRAFi and MEKi for the treatment of advanced BRAFV600E-positive PTCs (27–31), originally developed for the treatment of BRAFV600E-positive melanoma (24, 35, 75). However, melanoma and PTC behave very differently; PTC is known to be much more indolent overall, with longer associated survival observed for patients with unresectable or metastatic disease. Indeed, the average time to recurrence is 8.1 years for all stages of PTC, with 17% of deaths occurring after 20 years (33). This poses unique challenges to targeting the MAPK pathway in PTC, as cost and toxicities are amplified over prolonged treatment. Unlike most other cancers, where costs are largely incurred in the final year of life, 78% of the cost of thyroid cancer treatment accumulates over the initial and surveillance phases of the disease (12, 13). The cost of targeted therapies administered over a long period of time can be prohibitively expensive for patients and payers (12, 13, 36–38). In additional to financial toxicity, drug treatment itself often comes with non-negligible side effects. In a recent clinical trial of vemurafenib, 66% of PTC patients experienced Grade 3 and 4 adverse events (26). These toxicities are especially concerning for patients with PTC who are often asymptomatic from their disease for long periods of time. As such, quality of life considerations are of special importance in their treatment decisions (12, 13, 36–38).
Here we show that TM was as effective as the BRAFi vemurafenib in terms of inhibiting Mek1/2 kinase activity and the growth of established PTC lesion in a BRAFV600E-driven mouse model of this disease. Additionally, TM enhanced the antineoplastic activity of both the SOC sorafenib and the clinical BRAFi vemurafenib. TM is typically a well-tolerated drug. Unlike vemurafenib, which caused 66% of PTC patients to experience Grade 3 and 4 adverse events (26), the only side-effect of treatment with TM is a small likelihood of mild and transient anemia in the first week of treatment (58–63). Regular surveillance of Cu levels using the simple serum assay of ceruloplasmin activity has allowed TM to be dosed chronically in humans with few ill-effects (58–63). In fact, TM has been continually dosed thrice daily for as long as 65 months in a Phase I breast cancer trial (63). This feature makes TM particularly well suited for management of advanced PTC, especially in terms of chronic inhibition of the MAPK pathway. The synergy of TM with sorafenib in vitro suggests an immediate clinical pathway to evaluating this drug in PTC patients. Alternatively, the combination of BRAFi and MEKi is known to be clinically superior to either drug alone in the treatment of melanoma (75). Thus, TM could be added to current efforts testing BRAFi in PTC patients (26). Although combining TM with a MEKi or both a BRAFi and MEKi was not synergistic in vitro, there may still be value in adding TM to these modalities as a long-term maintenance therapy after initial treatment with these more toxic drugs. Indeed, when TM was provided as a maintenance therapy after vemurafenib treatments were terminated, tumor volume was reduced compared to control mice. As such, there are a number of potential clinical venues to explore TM for the treatment of BRAFV600E-positive PTC.
Mechanistically, we ascribe the antineoplastic effects of TM in large part to inhibition of the Cu-dependent activity of MEK1/2 in the PTC tumors themselves. This is supported by three lines of evidence. First, the ability of TM to inhibit PTC transformed growth was rescued by ERKGOF, an activated version of the MEK1/2 substrate ERK2. If TM inhibited transformation by other Cu-dependent pathways, this would not have been the case. This also highlights the specificity of this drug. Namely, like other MAPKi, BRAF mutation status predicts sensitivity to TM (40). Although TM has been evaluated across a broad spectrum of cancers with modest effects (54, 58, 60–63), it has never been matched to the BRAFV600E mutation, which will be an important inclusion criterion. Second, in TBP mice treated with TM, there was a reduction in the level of P-Erk1/2 by one criteria, a direct measure of Mek1/2 kinase activity in vivo, which was similar to what has been reported in other mouse models of oncogenic Braf-driven tumorigenesis (42). Third, crossing conditional null alleles of the primary Cu-specific transporter Ctr1 into the mouse model of PTC, ostensibly reducing Cu in the developing tumors, trended towards an extension of lifespan. This argues that the effect of TM on tumorigenesis lies in a reduction of Cu in the tumor rather than the stroma. Admittedly however, how TM specifically inhibits MEK1/2 remains to be fully elucidated. It is worth noting that a disulfiram metabolite has been shown to have copper-binding properties and is antineoplastic (77) and we also detected a reduction of CD31 staining in the thyroid tumors of TM-treated mice. These findings collectively suggest that TM inhibits the Cu-dependent activity of MEK1/2 kinases to reduce oncogenic BRAF-driven signaling.
In summary, TM represents a unique clinical opportunity in PTC. Its ability to inhibit MEK1/2 kinases while having low toxicity makes it particularly well suited for long term inhibition of the MAPK pathway in BRAFV600E-positive PTC, either in combination with current or emerging therapies and/or as a maintenance therapy.
Supplementary Material
Translational relevance.
The incidence of thyroid cancer, and in particular papillary thyroid cancer (PTC), is rising faster than that of any other malignancy. PTC often have an oncogenic (V600E) mutation in the kinase BRAF. Inhibitors against BRAF or its substrates MEK1/2 are showing promise as new therapies for this disease. However, the indolent nature of PTC may be a challenge to the clinical adaption of these inhibitors, as financial and physical toxicities may be amplified over prolonged treatment. Here we demonstrate that the well-tolerated copper chelator tetrathiomolybdate (TM) inhibits MEK1/2 and reduces transformed and tumor growth of BRAFV600E-positive PTC alone or when combined with a BRAF inhibitor. As such, TM may find utility in chronic inhibition of MEK1/2 in BRAFV600E-positive PTC.
Acknowledgments
We thank members of the Counter laboratory for helpful discussions and feedback and David Kirsch (Duke University) and Martin McMahon (University of Utah) for providing mice. This work was supported by the National Cancer Institute (R01CA177587 to CMC) and the Lymphoma Foundation (CMC).
Footnotes
Conflict of interest statement: Christopher M. Counter declares a minor conflict of interest as a founder of the company Merlon Inc.
References
- 1.Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, et al. SEER Cancer Statistics Review, 1975-2014. National Cancer Institute; Bethesda, MD: https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site, April 2017. Cited 2017 Oct 5th. [Google Scholar]
- 2.Davies L, Welch HG. Current thyroid cancer trends in the United States. JAMA Otolaryngol Head Neck Surg. 2014;140:317–22. doi: 10.1001/jamaoto.2014.1. [DOI] [PubMed] [Google Scholar]
- 3.Enewold L, Zhu K, Ron E, Marrogi AJ, Stojadinovic A, Peoples GE, et al. Rising thyroid cancer incidence in the United States by demographic and tumor characteristics, 1980-2005. Cancer Epidemiol Biomarkers Prev. 2009;18:784–91. doi: 10.1158/1055-9965.EPI-08-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974-2013. JAMA. 2017;317:1338–48. doi: 10.1001/jama.2017.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sciuto R, Romano L, Rea S, Marandino F, Sperduti I, Maini CL. Natural history and clinical outcome of differentiated thyroid carcinoma: a retrospective analysis of 1503 patients treated at a single institution. Ann Oncol. 2009;20:1728–35. doi: 10.1093/annonc/mdp050. [DOI] [PubMed] [Google Scholar]
- 6.Xing M. Prognostic utility of BRAF mutation in papillary thyroid cancer. Mol Cell Endocrinol. 2010;321:86–93. doi: 10.1016/j.mce.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harris PJ, Bible KC. Emerging therapeutics for advanced thyroid malignancies: rationale and targeted approaches. Expert Opin Investig Drugs. 2011;20:1357–75. doi: 10.1517/13543784.2011.614230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American thyroid association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American thyroid association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid. 2016;26:1–133. doi: 10.1089/thy.2015.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384:319–28. doi: 10.1016/S0140-6736(14)60421-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med. 2015;372:621–30. doi: 10.1056/NEJMoa1406470. [DOI] [PubMed] [Google Scholar]
- 11.Kitahara CM, Sosa JA. The changing incidence of thyroid cancer. Nat Rev Endocrinol. 2016;12:646–53. doi: 10.1038/nrendo.2016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lubitz CC, Sosa JA. The changing landscape of papillary thyroid cancer: Epidemiology, management, and the implications for patients. Cancer. 2016;122:3754–9. doi: 10.1002/cncr.30201. [DOI] [PubMed] [Google Scholar]
- 13.Lubitz CC, Kong CY, McMahon PM, Daniels GH, Chen Y, Economopoulos KP, et al. Annual financial impact of well-differentiated thyroid cancer care in the United States. Cancer. 2014;120:1345–52. doi: 10.1002/cncr.28562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Reviews Cancer. 2013;13:184–99. doi: 10.1038/nrc3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Biase D, Cesari V, Visani M, Casadei GP, Cremonini N, Gandolfi G, et al. High-sensitivity BRAF mutation analysis: BRAF V600E is acquired early during tumor development but is heterogeneously distributed in a subset of papillary thyroid carcinomas. J Clin Endocrinol Metab. 2014;99:E1530–8. doi: 10.1210/jc.2013-4389. [DOI] [PubMed] [Google Scholar]
- 16.Charles RP, Iezza G, Amendola E, Dankort D, McMahon M. Mutationally activated BRAF(V600E) elicits papillary thyroid cancer in the adult mouse. Cancer Res. 2011;71:3863–71. doi: 10.1158/0008-5472.CAN-10-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charles RP, Silva J, Iezza G, Phillips WA, McMahon M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol Cancer Res. 2014;12:979–86. doi: 10.1158/1541-7786.MCR-14-0158-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248–60. doi: 10.1016/S1470-2045(16)30122-X. [DOI] [PubMed] [Google Scholar]
- 19.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867–76. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 21.Casaluce F, Sgambato A, Maione P, Sacco PC, Santabarbara G, Gridelli C. Selumetinib for the treatment of non-small cell lung cancer. Expert Opin Investig Drugs. 2017;26:973–84. doi: 10.1080/13543784.2017.1351543. [DOI] [PubMed] [Google Scholar]
- 22.Stinchcombe TE, Johnson GL. MEK inhibition in non-small cell lung cancer. Lung Cancer. 2014;86:121–5. doi: 10.1016/j.lungcan.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim YH. Dual inhibition of BRAF and MEK in BRAF-mutated metastatic non-small cell lung cancer. J Thorac Dis. 2016;8:2369–71. doi: 10.21037/jtd.2016.09.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grimaldi AM, Simeone E, Festino L, Vanella V, Strudel M, Ascierto PA. MEK inhibitors in the treatment of metastatic melanoma and solid tumors. Am J Clin Dermatol. 2017;18:745–54. doi: 10.1007/s40257-017-0292-y. [DOI] [PubMed] [Google Scholar]
- 25.Volpe VO, Klufas DM, Hegde U, Grant-Kels JM. The new paradigm of systemic therapies for metastatic melanoma. J Am Acad Derm. 2017;77:356–68. doi: 10.1016/j.jaad.2017.04.1126. [DOI] [PubMed] [Google Scholar]
- 26.Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, et al. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:1272–82. doi: 10.1016/S1470-2045(16)30166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, et al. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid. 2013;23:1277–83. doi: 10.1089/thy.2013.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Falchook GS, Millward M, Hong D, Naing A, Piha-Paul S, Waguespack SG, et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid. 2015;25:71–7. doi: 10.1089/thy.2014.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368:623–32. doi: 10.1056/NEJMoa1209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leijen S, Soetekouw PM, Jeffry Evans TR, Nicolson M, Schellens JH, Learoyd M, et al. A phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;68:1619–28. doi: 10.1007/s00280-011-1732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson SM, Osborne JR, Grewal RK, Tuttle RM. Redifferentiating thyroid cancer: selumetinib-enhanced radioiodine uptake in thyroid cancer. Mol Imaging Radionucl Ther. 2017;26:80–6. doi: 10.4274/2017.26.suppl.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol. 2017 doi: 10.1200/JCO.2017.73.6785. doi: 10.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grogan RH, Kaplan SP, Cao H, Weiss RE, Degroot LJ, Simon CA, et al. A study of recurrence and death from papillary thyroid cancer with 27 years of median follow-up. Surgery. 2013;154:1436–46. doi: 10.1016/j.surg.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 34.Catalanotti F, Solit DB, Pulitzer MP, Berger MF, Scott SN, Iyriboz T, et al. Phase II trial of MEK inhibitor selumetinib (AZD6244, ARRY-142886) in patients with BRAF V600E/K-mutated melanoma. Clin Cancer Res. 2013;19:2257–64. doi: 10.1158/1078-0432.CCR-12-3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–9. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 36.Gild ML, Topliss DJ, Learoyd D, Parnis F, Tie J, Hughes B, et al. Clinical guidance for radioiodine refractory differentiated thyroid cancer. Clin Endocrinol. 2017 doi: 10.1111/cen.13508. doi: 10.1111. [DOI] [PubMed] [Google Scholar]
- 37.Ramsey S, Blough D, Kirchhoff A, Kreizenbeck K, Fedorenko C, Snell K, et al. Washington State cancer patients found to be at greater risk for bankruptcy than people without a cancer diagnosis. Health Aff. 2013;32:1143–52. doi: 10.1377/hlthaff.2012.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zafar SY. Financial toxicity of cancer care: It’s time to intervene. J Natl Cancer Inst. 2015;108 doi: 10.1093/jnci/djv370. djv370. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 40.Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature. 2014;509:492–6. doi: 10.1038/nature13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol Cell Biol. 2012;32:1284–95. doi: 10.1128/MCB.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brady DC, Crowe MS, Greenberg DN, Counter CM. Copper chelation inhibits BRAFV600E-driven melanomagenesis and counters resistance to BRAFV600E and MEK1/2 Inhibitors. Cancer Res. 2017;77:6240–52. doi: 10.1158/0008-5472.CAN-16-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- 44.Das SK, Ray K. Wilson’s disease: an update. Nat Clin Pract Neurol. 2006;2:482–93. doi: 10.1038/ncpneuro0291. [DOI] [PubMed] [Google Scholar]
- 45.Brewer GJ, Askari F, Lorincz MT, Carlson M, Schilsky M, Kluin KJ, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch Neuro. 2006;63:521–7. doi: 10.1001/archneur.63.4.521. [DOI] [PubMed] [Google Scholar]
- 46.Brewer GJ, Askari F, Dick RB, Sitterly J, Fink JK, Carlson M, et al. Treatment of Wilson’s disease with tetrathiomolybdate: V. Control of free copper by tetrathiomolybdate and a comparison with trientine. Transl Res. 2009;154:70–7. doi: 10.1016/j.trsl.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 47.Mills CF, El-Gallad TT, Bremner I. Effects of molybdate, sulfide, and tetrathiomolybdate on copper metabolism in rats. J Inorg Biochem. 1981;14:189–207. doi: 10.1016/s0162-0134(00)80000-8. [DOI] [PubMed] [Google Scholar]
- 48.Mills CF, El-Gallad TT, Bremner I, Wenham G. Copper and molybdenum absorption by rats given ammonium tetrathiomolybdate. J Inorg Biochem. 1981:163–75. doi: 10.1016/s0162-0134(00)80037-9. [DOI] [PubMed] [Google Scholar]
- 49.Bremner I, Mills CF, Young BW. Copper metabolism in rats given di- or trithiomolybdates. J Inorg Biochem. 1982;16:109–19. doi: 10.1016/s0162-0134(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 50.Gooneratne SR, Howell JM, Gawthorne JM. An investigation of the effects of intravenous administration of thiomolybdate on copper metabolism in chronic Cu-poisoned sheep. Br J Nutr. 1981;46:469–80. doi: 10.1079/bjn19810055. [DOI] [PubMed] [Google Scholar]
- 51.McQuaid A, Mason J. A comparison of the effects of Penicillamine, Trientine, and trithiomolybdate on [35S]-labeled metallothionein in vitro; implications for Wilson’s disease therapy. J Inorg Biochem. 1991;41:87–92. doi: 10.1016/0162-0134(91)80002-y. [DOI] [PubMed] [Google Scholar]
- 52.Kodama H, Fujisawa C, Bhadhprasit W. Inherited copper transport disorders: Biochemical mechanisms, diagnosis, and treatment. Curr Drug Metab. 2012;13:237–50. doi: 10.2174/138920012799320455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Juarez JC, Betancourt O, Jr, Pirie-Shepherd SR, Guan X, Price ML, Shaw DE, et al. Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1. Clin Cancer Res. 2006;12:4974–82. doi: 10.1158/1078-0432.CCR-06-0171. [DOI] [PubMed] [Google Scholar]
- 54.Brewer GJ. Copper lowering therapy with tetrathiomolybdate as an antiangiogenic strategy in cancer. Curr Cancer Drug Targets. 2005;5:195–202. doi: 10.2174/1568009053765807. [DOI] [PubMed] [Google Scholar]
- 55.Finney L, Vogt S, Fukai T, Glesne D. Copper and angiogenesis: unravelling a relationship key to cancer progression. Clin Exp Pharmacol Physiol. 2009;36:88–94. doi: 10.1111/j.1440-1681.2008.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brem SS, Zagzag D, Tsanaclis AM, Gately S, Elkouby MP, Brien SE. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am J Pathol. 1990;137:1121–42. [PMC free article] [PubMed] [Google Scholar]
- 57.Badet J, Soncin F, Guitton JD, Lamare O, Cartwright T, Barritault D. Specific binding of angiogenin to calf pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 1989;86:8427–31. doi: 10.1073/pnas.86.21.8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brewer GJ, Dick RD, Grover DK, LeClaire V, Tseng M, Wicha M, et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin Cancer Res. 2000;6:1–10. [PubMed] [Google Scholar]
- 59.Hordyjewska A, Popiolek L, Kocot J. The many “faces” of copper in medicine and treatment. Biometals. 2014;27:611–21. doi: 10.1007/s10534-014-9736-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pass HI, Brewer GJ, Dick R, Carbone M, Merajver S. A phase II trial of tetrathiomolybdate after surgery for malignant mesothelioma: final results. Ann Thorac Surg. 2008;86:383–9. doi: 10.1016/j.athoracsur.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 61.Redman BG, Esper P, Pan Q, Dunn RL, Hussain HK, Chenevert T, et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin Cancer Res. 2003;9:1666–72. [PubMed] [Google Scholar]
- 62.Chan N, Willis A, Kornhauser N, Ward MM, Lee SB, Nackos E, et al. Influencing the tumor microenvironment: A Phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin Cancer Res. 2017;23:666–76. doi: 10.1158/1078-0432.CCR-16-1326. [DOI] [PubMed] [Google Scholar]
- 63.Jain S, Cohen J, Ward MM, Kornhauser N, Chuang E, Cigler T, et al. Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse. Ann Oncology. 2013;24:1491–8. doi: 10.1093/annonc/mds654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brewer GJ, Hedera P, Kluin KJ, Carlson M, Askari F, Dick RB, et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: III. Initial therapy in a total of 55 neurologically affected patients and follow-up with zinc therapy. Arch Neurol. 2003;60:379–85. doi: 10.1001/archneur.60.3.379. [DOI] [PubMed] [Google Scholar]
- 65.Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab. 2008;93:4331–41. doi: 10.1210/jc.2008-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saiselet M, Floor S, Tarabichi M, Dom G, Hebrant A, van Staveren WC, et al. Thyroid cancer cell lines: an overview. Front Endocrinol. 2012 doi: 10.3389/fendo.2012.00133. doi: 10.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Hayer KM, Counter CM. A genetically defined normal human somatic cell system to study ras oncogenesis in vivo and in vitro. Methods Enzymol. 2006;407:637–47. doi: 10.1016/S0076-6879(05)07050-3. [DOI] [PubMed] [Google Scholar]
- 68.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, et al. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–9. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- 70.Undeutsch H, Lof C, Offermanns S, Kero J. A mouse model with tamoxifen-inducible thyrocyte-specific cre recombinase activity. Genesis. 2014;52:333–40. doi: 10.1002/dvg.22740. [DOI] [PubMed] [Google Scholar]
- 71.Kuo YM, Zhou B, Cosco D, Gitschier J. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc Natl Acad Sci USA. 2001;98:6836–41. doi: 10.1073/pnas.111057298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 73.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect. 2015;3:e00149. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee J, Prohaska JR, Thiele DJ. Essential role for mammalian copper transporter Ctr1 in copper homeostasis and embryonic development. Proc Natl Acad Sci USA. 2001;98:6842–7. doi: 10.1073/pnas.111058698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Long GV, Hauschild A, Santinami M, Atkinson V, Mandala M, Chiarion-Sileni V, et al. Adjuvant dabrafenib plus trametinib in stage III BRAF-mutated melanoma. N Engl J Med. 2017;377:1813–23. doi: 10.1056/NEJMoa1708539. [DOI] [PubMed] [Google Scholar]
- 76.Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006;407:597–612. doi: 10.1016/S0076-6879(05)07047-3. [DOI] [PubMed] [Google Scholar]
- 77.Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature. 2017;552:194–9. doi: 10.1038/nature25016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.