


Abstract
Structural Maintenance of Chromosomes (SMC) proteins and their complex partners (ScpA and ScpB in many bacteria) are involved in chromosome compaction and segregation in all kinds of organisms. We employed single molecule tracking (SMT), tracking of chromosomal loci, and single molecule counting in Bacillus subtilis to show that in slow growing cells, ∼30 Smc dimers move throughout the chromosome in a constrained mode, while ∼60 ScpA and ScpB molecules travel together in a complex, but independently of the nucleoid. Even an Smc truncation that lacks the ATP binding head domains still scans the chromosome, highlighting the importance of coiled coil arm domains. When forming a complex, 10–15 Smc/ScpAB complexes become essentially immobile, moving slower than chromosomal loci. Contrarily, SMC-like protein RecN, which forms assemblies at DNA double strand breaks, moves faster than chromosome sites. In the absence of Smc, chromosome sites investigated were less mobile than in wild type cells, indicating that Smc contributes to chromosome dynamics. Thus, our data show that Smc/ScpAB clusters occur at several sites on the chromosome and contribute to chromosome movement.
INTRODUCTION
A fundamental question in biology is how cells can organize and segregate their DNA in ordered processes, allowing DNA compaction, unperturbed replication, transcription, translation, and the faithful production of daughter cells to occur. In eukaryotes, these processes are mediated by the action of nucleosomes, DNA topoisomerase II and the family of structural maintenance of chromosomes (SMC) proteins, a highly conserved protein family (1,2). SMCs include condensin and cohesin, which mediate chromosome compaction in prophase and sister chromosome cohesion from S phase into metaphase, as well as proteins that have prominent roles in DNA repair such as Rad50 or the SMC5/6 complex. In bacteria, chromosome segregation and replication occur in parallel, starting at the origin of replication and ending with the separation of the terminus regions. In several bacteria, replicated origins are generally found at the outer edges of the elongating chromosome, while other loci are found in between the replication origin and the more centrally located terminus, leading to a ‘longitudinal’ organization of bacterial chromosomes (3). In prokaryotes, chromosome compaction and segregation are accomplished by histone-like proteins, like HU, H-NS and Fis, by topoisomerases and by SMC proteins. Especially the molecular function of SMC proteins in both eukaryotes and prokaryotes is still unclear, yet essential in its nature. SMCs have a unique structure (2,4): the N- and C-terminal domains of Smc form a joint globular ‘head domain’, homologous to that of ATP cassettes of ABC transporters, which can bind ATP via Walker A and Walker B motifs, located in the N- or C-terminus, respectively (5). These domains are connected by long coiled-coil regions (50 nm in length), and a globular hinge domain in the middle (Figure 1A), which allows the backfolding of the coiled-coil regions, such that the globular N- and C-termini can interact with each other and formation of antiparallel coiled coils is established. SMC monomers dimerize via their hinge domains and form I- or V-shaped dimers. A conserved mode of stable DNA binding appears to be the trapping of a DNA loop or two DNA strands (from daughter chromosomes) within the closed ring structure (6–8). In analogy to ABC transporters, it was demonstrated that the head domains can engage and disengage dependent on ATP binding and hydrolysis (9,10). There is evidence that the ATPase activity of SMCs controls DNA binding at the hinge domain, based on the exposure of a DNA binding site in this domain upon ATP binding (11,12). In other studies, it was suggested that DNA binds via a positively charged surface of the head domains of SMC and MukB (13). Despite more than 20 years of SMC research, the mechanism of DNA interaction with SMC is still far from clear and there is also evidence available that an Smc dimer has multiple DNA-interacting regions (2). Importantly, both the ATPase cycle and the DNA binding activity of Smc/MukB were reported to be dependent on the accessory proteins ScpA and ScpB (termed MukE and MukF in Escherichia coli) (14–16). ScpA interacts with both Smc subunits in an asymmetric manner, and binds a dimer of ScpB, resulting to a stoichiometry within the SMC complex of Smc:ScpA:ScpB of 2:1:2 (17,18). The N-terminus of ScpA binds to the coiled-coil region directly adjacent to one head domain, while the C-terminus binds to the tip of the other head domain, thereby bridging the dimers at the head domains.
Figure 1.

(A) Domain organization of Smc and of headless Smc. Residue numbering corresponds to amino acids of Smc of Bacillus subtilis. Head-N: N-terminal part of head domain, N: Neck region, Coiled-coil-N: N-terminal coiled coil, Coiled-coil-C: C-terminal coiled coil, Head-C: C-terminal part of head domain. All proteins are tagged with the fluorescent proteins YFP or mVenus at the C-terminus. (B) The scheme indicates the organization of the complex of Smc, ScpA, and ScpB. (C) Typical image of a DAPI-stained cell of length ∼3.2 μm, (D) same as (C) after addition of chloramphenicol, (E and F) Distribution of normalized DAPI fluorescence in cells of ∼3.2 μm length in exponentially growing cells (n = 22) and in cells treated with chloramphenicol (n = 19) which results in a single compacted nucleoid in the cell middle. (G–L) The spatial organization of Smc and headless tracks reflects the organization of the nucleoid in exponentially growing B. subtilis cells, whereas ScpA tracks cover the entire cell. Upper panels show typical tracks obtained in a single cell, lower panels are histograms of localizations projected onto the long cell axis. Chloramphenicol treatment was used as a test for tracking on the nucleoid. Tracks were analyzed in cells of ∼3.2 μm length for better comparison.
Investigation of fluorescent protein fusions to bacterial SMCs using wide-field microscopy led to the prevalent view that SMC forms bipolar protein clusters, also termed ‘foci’ or ‘condensation centers’ near replication origins, dependent on the DNA-binding protein ParB (19–21). However, it has still not been demonstrated that these clusters are directly responsible for chromosome condensation and to the sites of entrapment in vivo. Only recently, single molecule microscopy studies additionally reported significant mobile fractions of Smc and the E. coli homologue MukB and its accessory proteins also outside of these clusters (22,23), which were not detectable using traditional wide-field microscopy with long exposure times due to motion blurring of these molecules. Thereby, two fractions of Smc molecules could be distinguished: static molecules mainly associated with the ‘condensation centers’ and mobile molecules that appeared to scan the chromosome. Exchange between the two fractions occurs within a range of few minutes, based on FRAP measurements (24). We wished to get further insight into the role of these fractions of Smc molecules in the model bacterium Bacillus subtilis.
Single molecules can be observed using a traditional wide-field microscope by employing laser power densities sufficiently high to detect single molecules. The center of each emission spot indicates the position of the individual molecule with 10–40 nm accuracy, dependent amongst others on the amount of photons captured from the single molecule (25). This individual molecule can then be followed in time using tracking algorithms, allowing to characterize its diffusion behavior quantitatively. An important prerequisite for tracking algorithms is to obtain a sufficiently low amount of molecules inside a cell in order to avoid spurious tracks (26). We achieved this by bleaching the majority of fluorescent molecules until three or less copies were visible in each cell at a time.
Our single molecule analysis revealed that Smc exclusively scans the chromosome, and even a truncated version of Smc lacking the head domains does so. Furthermore, we found also significant trackable fractions of diffusing ScpA and ScpB in the cell, which have not been reported before, but these molecules were not detectably enriched at the chromosome. Depletion experiments suggest that ScpA and ScpB travel together in a complex throughout the cell in vivo. By comparing chromosomal locus movement with movement of relative immobile Smc, we found that DNA moves much faster than Smc. However, in the absence of Smc, chromosome sites were less mobile than in wild type cells, pointing further to a role of Smc in the regulation of chromosome motion, either directly or indirectly. Single molecule counting shows that there is a surprisingly low number of Smc within the cell, and ScpA is in excess in the whole cell and in foci.
MATERIALS AND METHODS
Cell growth and preparation
Bacillus subtilis strains were grown in S750 defined medium (27) without casamino acids. For induction of the xylose promoter, glucose was exchanged for 0.5% fructose, and xylose was added up to 0.001%. For induction of the hyperspank promoter, the culture media were supplemented with 1 mM Isopropyl β-d-1-thiogalactopyranoside (IPTG). The antibiotics chloramphenicol (5 μg/ml), erythromycin (0.5 μg/ml), lincomycin (12.5 μg/ml) or spectinomycin (50 μg/ml) were used when necessary. Overnight cultures were grown at 21°C in S750, resulting in exponentially growing cells the next day. Chloramphenicol was added to a final concentration of 200 μg/ml to induce chromosome compaction. The agarose pads were prepared by adding these antibiotics after cooling of the agarose to 50°C to allow constant growth conditions during imaging.
Strain construction
To obtain headless-mVenus, a fragment of Smc was cloned in the vector 1193NLMV (lab stock) via isothermal assembly (28) using oligonucleotides listed in Supplementary Table S1 and transformed into competent B. subtilis PY. Expression of headless was confirmed by Western Blotting (Supplementary Figure S6). The strain SS143 was obtained by transformation of 1193NLMV in competent B. subtilis PY. All other strains were created by transformation of chromosomal DNA using standard procedures for B. subtilis.
Single molecule tracking - data acquisition
Cells were dropped onto a coverslip, and an agarose pad (1% agarose in S750 medium) was put on top to supply the bacteria with nutrients and prevent them from drying up. Cells were imaged with a high numerical aperture objective (CFI Plan Apo Lambda DM 100× Oil, NA = 1.49, Nikon) on a Nikon Eclipse Ti microscope equipped with a back-illuminated EMCCD camera (Hamamatsu ImagEM X2). An EMCCD gain of 200 was used. Illumination was achieved by focusing the excitation laser light onto the objective's back focal plane. A laser power density of 160 W cm−2 was used for single molecule tracking using 30 ms exposure time (33 Hz) for slowly moving proteins, and 310 W cm−2 for tracking of fast moving proteins with 128-135 Hz. For fast moving molecules, a shorter exposure time was used, since we observed non trackable cloud-like structures or elliptical point spread functions using 30 ms exposure time, which is expected for this time scale in a bacterial cell for relatively fast-diffusing molecules (29,30). In each movie, 1500 frames were acquired, resulting in no detectable growth defects after this illumination period (data not shown). Imaging was performed at 21°C.
Single molecule tracking and track analysis
Images were recorded using VisiView (Visitron Systems) and were subsequently analyzed by using ImageJ software Version 1.50f (W. Rasband, National Institutes of Health, Bethesda, MD, USA). For particle tracking, a published analysis software was used (31). First, the images were bandpass filtered with a lower limit of 1 au, and a higher limit of 5 au. A Threshold of 1000 au was used and a Window size of 5. Detected particles were connected by allowing a maximal jump of 5 pixel, the shortest track length was allowed to be 5 frames, and no gaps (blinking) was allowed. Then, tracks were manually checked. Single molecules were identified and trajectories of single molecules were tracked using the software MATLABtrack (31). Trajectory analysis was done with this software and also with custom software written in MATLAB. Shortly, the step length histogram of displacements was determined and fitted using the following formula (31,32):
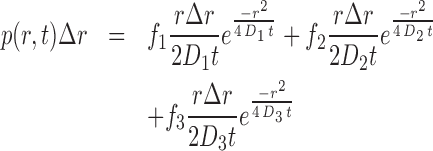 |
where D1, D2, D3 are the diffusion constants, Δr is the bin size of the histogram of one-step displacements and p is the probability of observing a molecule travelling a distance between r – Δr/2 and r + Δr/2 in a time t; f1, f2 and f3 are the fractions for the corresponding diffusing fractions, so that f1+f2+f3 = 1. For every obtained histogram of displacements, parameter estimation of D1, D2, D3 and f1, f2, f3 was done with the MATLAB function lsqnonlin. Histograms for Smc-YFP showed a clear peak at D ∼ 0.1 μm2 s−1, which we therefore assigned to be the static fraction. Therefore, this value was then used in constrained fits for the other histograms. An F-test was performed to avoid overfitting as suggested by others (33); P < 0.05 was used to justify statistical relevance. The fluorescence in the cells was bleached until only one to two diffusing molecules in the cell were detectable to avoid spuriously connected positions of single molecules (26). Importantly, for ectopically expressed proteins, the initial brightness of a cell was determined before image processing and only cells with a similar number of molecules like Smc-YFP were analyzed to avoid effects from overexpression of the protein of interest.
Determination of localization precision of single molecules and chromosomal loci
Camera counts of single molecules and diffraction-limited spots of chromosomal loci in a single frame were converted into photons by using a proper conversion factor when using an EMCCD gain of 200. The localization precision was then calculated from the amount of photons detected as was shown in (34). The standard deviation of the PSF was determined by a Gaussian fit. This gave a localization precision of 21 ± 3 nm for single molecules of Smc-YFP (100 ms exposure time), 32 ± 3 nm for single molecules imaged at 8 ms exposure time, 42 ± 12 nm for single molecules of free mVenus imaged at 4 ms exposure time, and 12 ± 2 nm for the chromosomal loci (100 ms exposure time). Chromosomal loci were imaged at 640 W cm−2. The localization precision changed during the stream acquisition with 300 frames for chromosomal loci only marginally from 12 ± 2 nm in the first frame to 14 ± 1 nm in the last frame, most likely due to photobleaching of locus-associated chromophores, essentially ruling out that faster diffusion of chromosomal loci compared to Smc-YFP (imaged at 100 ms exposure time) is caused by decreased localization precision.
Quantification of number of molecules in a cell
The integrated fluorescence of a cell in the first illuminated frame of the stream acquisition was determined, subtracted from the background and divided by the integrated intensity of a single fluorophore, identified by a single bleaching step. To determine the cell length, the cells were counterstained with SYTO 60.
Quantification of number of molecules integrated into clusters
First, clusters were identified by Gaussian fits to the average projection image of the first ten frames of the stream acquisition. The background outside the clusters was substracted. The contribution to the camera counts due to diffusive YFP molecules in the cytosol was determined as the initial intensity in the cell outside the clusters after subtracting the autofluorescence contribution measured from B. subtilis PY WT without artificially introduced fluorophores and the instrumental background (e.g. offset, contribution from fluorescence of agarose pad). Then, the integrated camera counts of the identified clusters were divided by the integrated intensity of a single fluorophore, identified by a single bleaching step, as above.
Statistical data analysis
For the diffusion coefficients and radii of confinement, statistical hypothesis testing was performed using a one-way ANOVA with Tukey's Honestly Significant Difference Procedure as post-hoc test to correct for multiple hypothesis testing. Normality was verified using a Lilliefors test on a 0.05 significance level, and equality of variances was asserted using Levene's Test which showed that equal variances could be assumed on a 0.05 significance level. ***P < 0.0005, *P < 0.05 and n.s. stands for not statistically significant. Evaluation of correlation between cell length and molecule numbers was done using the two-sided Pearson correlation coefficient. Statistical hypothesis testing and plotting was performed using MATLAB’s Statistics and Machine Learning Toolbox™ and R.
Plasmid construction and generation of mutants for protein purification
The PCR product of scpB-strep was inserted into the second MCS of pETDuet-1 (Novagen) using NdeI and XhoI to give rise to pETDuet-1(scpB-strep). This was cut with NcoI and BamHI and scpA was inserted into the first MCS of this construct resulting in pETDuet-1(scpA/B-strep). To create pCDFD(smc-strep) the antisense primer of smc was designed with a strep-tag sequence and the PCR fragment was cloned into the NcoI and XhoI cut pCDFDuet-1. For headless smc, the central region of smc lacking N- and C-terminal head domains was cloned analogously. Transformants were selected on LB-agar plates under antibiotic pressure with 100 μg/ml ampicillin or 50 μg/ml streptomycin.
Expression and sample collection
The ScpAB complex or the full SMC complex were expressed from pETDuet (ScpA/B-Strep) and pCDFDuet(SMC) in Rosetta 2 (DE3) pLysS cells and purified by strep affinity purification. The cells were incubated on a shaking platform at 37°C until OD600 0.9 was reached and were induced with 0.5 mM IPTG. After one hour cells were harvested by centrifugation and lysed by French pressing. ScpB-Strep was bound to a Streptactin column pulling down the untagged ScpA or ScpA and SMC, respectively. The complex was eluted, concentrated with a MonoQ column and rebuffered to BIAcore buffer by gel filtration (Superdex 200 30/100 GL).
Surface plasmon resonance
A 500 bp fragment of DNA containing a native parS site was amplified from chromosomal B. subtilis DNA with 5′-biotin labelled primers. The fragment was passed across one flow cell of a streptavidin coated BIAcore chip surface (sensor chip SA, GE Healthcare) at 20 μl/min in 0.5 M NaCl. The surface was washed by injection of 10 μl wash buffer (500 mM NaCl, 100 mM NaOH) to remove non-specifically adsorbed DNA. The analytes were passed across the surface of two flow cells at 20 μl/min in BIAcore buffer (50 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2 and 0.005% Tween 20). The signal of the flow cell without DNA (‘control’) was subtracted from the flow cell with the DNA coated surface (‘sample’). At the end of each interaction the surfaces were regenerated by the injection 5 μl wash buffer.
RESULTS
The mobile fraction of Smc almost exclusively moves throughout the chromosome in a constrained mode
Figure 1A and B show an overview of the domain organization of Smc and a cartoon of the arrangement of the Smc/ScpA/ScpB (short SMC) complex. In previous work, we had inferred from the pattern of Smc diffusion that the mobile fraction seems to track with the nucleoid. We used streams with 8 ms exposure time for our analysis, which also capture fast diffusing molecules and therefore give a comprehensive picture about molecular diffusive populations within a bacterial cell. We observed a bimodal distribution of diffusion coefficients, reflecting two different states of diffusion of Smc molecules: rather stationary ones and mobile ones (Supplementary Figure S1A). Fitting with a two-population model shows that the stationary fraction peaks at 0.1 ± 0.01 (SD) μm2 s−1 with 47 ± 5.5%, and the dynamic fraction at 0.53 ± 0.12 (SD) μm2 s−1 with 52 ± 4.6% (Figure 2). We therefore defined diffusion with 0.1 ± 0.01 μm2 s−1 as static. We overlaid all tracks obtained from ∼3.2 μm long cells, which generally have two segregated nucleoids under our growth conditions (Figure 1C, average fluorescence intensity shown in Figure 1E). We observed that >93% of the tracks followed the nucleoid, avoiding the space between the nucleoid lobes and the poles of the cells (Figure 1G). To confirm that Smc moves preferentially in areas of high DNA density, we treated exponentially growing cells with 200 μg/ml chloramphenicol, leading to a highly compacted single chromosome in B. subtilis (Figure 1D and F) and in other organisms (35). We observed the same effect: 95 ± 3% (three independent experiments, n = 97 cells) of cells showed a substantially condensed chromosome in the middle of the cell, and Smc continued to only move on the compacted chromosomes (Figure 1J). Additionally, we also observed 5 ± 3% anucleate cells. In these cells, no tracks of Smc were detectable (data not shown), showing that in addition to the lack of the static SMC complex in cells devoid of DNA (19) not even freely diffusive Smc is present. In conclusion, these data show that in vivo, almost all Smc molecules interact with DNA. Interestingly, Smc slows down after chloramphenicol treatment, from 0.53 ± 0.12 (SD) μm2 s−1 to 0.22 ± 0.08 μm2 s−1. As stated above, most Smc tracks were confined to the highly condensed nucleoids (Figure 1J), as observed after addition of a nucleoid stain, DAPI, directly after single molecule imaging to the cells (Supplementary Figure S2). We therefore interpret that the slowing down of Smc after chloramphenicol treatment is due to the higher density of interaction sites, resulting in slower diffusion of Smc. As a validation that movement on the nucleoid does not apply to all soluble proteins, we tracked ScpA-YFP in exponentially growing and in chloramphenicol-treated cells. ScpA-YFP tracks were found throughout the cells, and were not confined to the nucleoids (Figure 1I), which did not change after chloramphenicol treatment (Figure 1L), suggesting that mobile ScpA molecules do not substantially interact with DNA (see below for further discussion).
Figure 2.
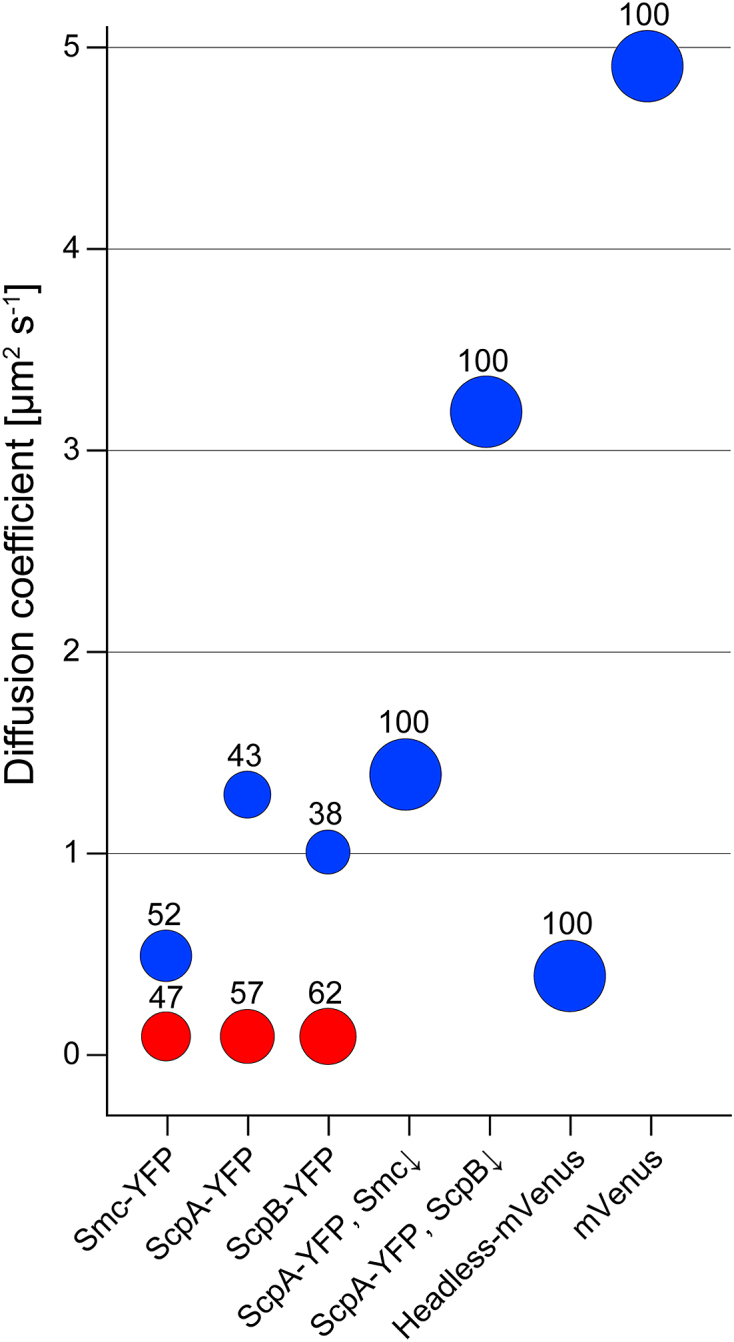
Bubble plot showing percentages of static (red) and dynamic (blue) fractions for the various constructs. Please refer to Table 1 for the detailed diffusion coefficients and fractions together with standard deviations, derived from at least three independent experiments.
Headless Smc still binds to DNA in vitro, and shows constrained movement in vivo
It has previously been shown that a ‘headless’ version of Smc comprised of residues 160–1037 (lacking the globular head domains) is still able to bind to DNA using gel shift experiments (36). In flow chamber experiments, headless was found to globally compact DNA in vitro, but with a reduced compaction rate compared to full length Smc (73% of the flow-stretched DNA length as compared to 100% for the WT) (37). We wished to obtain further quantitative data on DNA binding in vitro, and therefore used SPR experiments, with DNA attached to the surface of the chip. Full length Smc and headless Smc (containing the coiled coil up the N- and C-terminal domain, i.e. including the ScpA neck-binding region) were purified as Strep fusions via streptavidin affinity chromatography and ensuing gel filtration chromatography (Supplementary Figure S3) (23,38). Headless Smc could only be obtained up to 370 nM, which was tested for binding 500 bp DNA immobilized on an SPR chip (via biotinylation). Wild type Smc bound to the DNA in a sequence-independent fashion (the more Smc is added, the more is bound), and an 8-fold molar excess of ScpAB markedly reduced binding, ∼2-fold (Supplementary Figure S4A) (23). Addition of ATP to the reaction mixture led to a mild increase in binding avidity of Smc (Supplementary Figure S4A). Headless Smc also bound to DNA, with ∼70% efficiency relative to full length Smc (Supplementary Figure S4B). The addition of an 8-fold excess of ScpAB did not strongly reduce DNA binding of headless Smc (Supplementary Figure S4B), because of the missing ‘cap’ binding site at the head domains. These data show that the lack of head domains has a noticeable but not drastic effect on DNA binding of Smc protein.
As next step, we investigated chromosome interaction of headless in vivo, and therefore expressed a headless-mVenus fusion from a xylose inducible promoter. Headless-mVenus did not display a static fraction (Supplementary Figure S1C), nor showed foci in epifluorescence (data not shown), but the dynamic fraction still diffused slowly with 0.40 ± 0.09 μm2 s− 1 (Figure 2, Table 1), suggesting that headless can still interact with DNA in vivo. As further test, we overlaid tracks on cells of length ∼3.2 μm, showing that headless still follows the nucleoid staining (Figure 1H), in that preferential movement on two separate nucleoids is apparent. As a second test, we also treated these cells with chloramphenicol. The effect of slowing down of headless was not as drastic as for Smc, but we still observed a minor decrease of the diffusion coefficient to 0.32 ± 0.07 μm2 s−1. Importantly, headless-mVenus still tracked on the condensed DNA (Figure 1K), while ScpA-YFP showed no exclusive association with DNA (Figure 1L). Diffusion coefficients depend on the hydrodynamic radius of a particle (39), which will be altered in headless Smc, but our analysis shows that it can still interact with DNA in vivo.
Table 1.
Summary of determined diffusion coefficients
| Protein fusion/genotype |
MM (kDa) |
Diffusion coefficient [μm2 s−1 ± SD*] |
Number of tracks | Fraction (%) | Sets | Tracks per set |
|---|---|---|---|---|---|---|
| Smc-YFP | 158.8 | D1: 0.53 ± 0.12 | 1312 | 52 ± 5 | 3 | 345; 561; 406 |
| D2: 0.10 ± 0.01 | 47 ± 6 | |||||
| ScpA-YFP | 56.6 | D1: 1.32 ± 0.32 | 689 | 43 ± 10 | 3 | 213; 145; 331 |
| D2: 0.10 ± 0.01 | 57 ± 9 | |||||
| ScpB-YFP | 49.0 | D1: 1.02 ± 0.25 | 972 | 38 ± 12 | 4 | 243; 114; 312; 303 |
| D2: 0.10 ± 0.01 | 62 ± 13 | |||||
| ScpA-YFP, smc::kan, amyE::pspac-smc | 56.5 | 1.43 ± 0.35 | 858 | 100 | 3 | 321; 337; 200 |
| ScpA-YFP, scpB::kan, amyE::pspac-scpB-cfp | 56.5 | 3.22 ± 1.41 | 867 | 100 | 3 | 477; 213; 177 |
| headless-mVenus | 124.0 | 0.40 ± 0.09 | 768 | 100 | 3 | 245; 166; 357 |
| mVenus | 27.0 | 4.9 ± 0.92 | 399 | 100 | 4 | 112; 138; 98; 51 |
*Fitting to data from a set was done with a model of two populations, with a constrained fit for one population at D = 0.1 μm2 s−1. An F-test justified the fitting of two populations for these fusions, whereas it justified fitting of one population for example for mVenus. SD refers to the standard deviation from fitting of the amount of sets indicated, so at least three independent replicates (sets) were used for the determination of the mean and standard deviation of each diffusion coefficient.
ScpA and ScpB consist of a dynamic and of a static fraction, the latter depending on the presence of Smc, and move together throughout the entire cell
We wished to determine diffusion constants of non-Smc bound ScpA and ScpB molecules, to investigate their behavior in the cell. We tracked ScpA and ScpB using 8 ms exposure time, to be able to visualize and track dynamic ScpA-YFP and ScpB-YFP molecules (Supplementary Figure S5, movies S2 and S3). Both fusions were expressed from the original gene locus, as sole source of the protein, and fully supported wild type growth and chromosome compaction. For ScpA-YFP, we found 57.4 ± 9.2% static molecules, and 42.7 ± 9.5% dynamic molecules (Figure 2). ScpA-YFP had a diffusion constant of 1.32 ± 0.32 μm2 s−1 (Table 1), and thus moved much faster than dynamic Smc-YFP molecules (P = 0.002). Different from Smc, ScpA tracks could be observed within the entire cell, indicating that the movement is not primarily restrained to the chromosome (Figure 1I). Interestingly, ScpB-YFP dynamics were quite similar to those of ScpA (Supplementary Figure S1F), in that ScpB showed 61.6 ± 13% static molecules and 38.4 ± 12% dynamic molecules, and dynamic ScpB molecules also moved with a similar diffusion coefficient as ScpA, namely 1.02 ± 0.25 μm2 s−1. Thus, ScpB and ScpA showed a similar distribution with equally distributed diffusion coefficients and fractions (Figure 2).
We have previously shown that static Smc molecules depend on the presence of ScpA and of ScpB (23). We therefore wondered if in Smc depleted cells ScpA-YFP would become entirely mobile. Therefore, we expressed ScpA-YFP from its native promoter and depleted Smc that was expressed from an ectopic locus in an Smc deletion strain. After 7 hours of depletion, we observed a considerable increase in the dynamic fraction of ScpA-YFP. Most molecules moved with 1.43 ± 0.35 μm2 s−1, and no static fraction was observed, indicating that depletion was complete. Thus, the static fraction of ScpA depends on the presence of Smc, and therefore on SMC complex formation.
If ScpA and ScpB were to move together in a 1:2 complex that was previously characterized in vitro (40), a prediction would be that in the absence of ScpB, ScpA would become faster. Interestingly, this was the case (movie S5). After the depletion of ScpB, the ScpA diffusion constant increased strongly (3.22 ± 1.41 μm2 s−1), providing evidence that ScpA and ScpB move together as dynamic fraction in vivo. ScpA diffused surprisingly fast in the absence of ScpB, so as a control we wished to analyze a protein presumably diffusing even faster. Therefore, we used our setup to track mVenus in B. subtilis using 4 ms exposure time. Under our conditions, free mVenus diffused with 4.9 ± 0.92 μm2 s−1, in good agreement with the published literature for free fluorescent protein derivatives in bacteria (29,30). These data show that we are able to track very slowly and very fast diffusing molecules with our setup, with a control protein (free mVenus) resulting in similar values as reported from other groups for free GFP derivatives.
B. subtilis SMC complex compacts the chromosome from several condensation centers
Single molecule localization microscopy yields a localization precision of ∼20–25 nm, and therefore a much higher resolution than conventional light microscopy (29,41). When frames from a movie are superimposed after Gaussian centroid fitting (each spot having a size of ∼20 nm), static spots at a given position add up to a bright spot, while dynamic spots yield a cloud-like background. Using this approach (Figure 3A and B), we found several static tracks for ScpA and ScpB in each cell half, indicating that ScpA and ScpB stop at several sites on the chromosome. Detailed analysis showed that we could detect 2.1 ± 1.4 static tracks per 1 μm cell length (Figure 3C), or 6.3 static molecules per 3 μm average cell length. We then constructed histograms of the distance of static and dynamic tracks to the peak fluorescence in the first frame of the stream acquisitions (Figure 3D) showing that static tracks appeared to be closer to the peak fluorescence in the cell half. Therefore, even slow-growing B. subtilis cells that do not have multiple origin regions (1–4, and on average 2 through most of the cell cycle) contain more than two condensation centers as previously suggested based on epifluorescence microscopy (19,42), composed of the Smc–ScpAB complex. Importantly, we were not able to observe a considerable number of transitions between mobile and static events in the molecules which we classified as immobile, neither in molecules which were close to the origin region, nor in molecules further away, in agreement with the slow turnover of ScpA within the condensation centers (24).
Figure 3.

(A) Overlay of average fluorescence of ScpA-YFP from first ten frames with tracks of static molecules (diameter less than three pixels) that could be tracked later in the experiment (blue circle). The highest fluorescence intensity in the first frame in each cell half is indicated by a red circle. (B) Image of static ScpA-YFP tracks. (C) Number of static molecules per 2 μm cell length during 1500 frames (8 ms exposure time) which were >100 nm apart (n = 38 cells, 172 static tracks, mean = 4.2 ± 2.8 (SD) static molecules) (strain: ScpA-YFP). (D) Histograms of the distance of centroids of ScpA-YFP tracks to the highest fluorescence in the first frame, static molecules indicated in blue, mobile molecules in red.
DNA shows subdiffusion in B. subtilis and moves faster than static Smc molecules, while RecN moves faster than chromosome sites
Recently, it has been shown that ParB/Spo0J induction and subsequent Smc loading at origin regions leads to an extension of chromosome arm juxtapositioning away from origins with an apparent rate of 50 kb/min (43), using time-resolved Hi-C. An important open question of this discovery was if the SMC complex visibly tracks on DNA in vivo. We imaged Smc-YFP using a long exposure time (100 ms) in stream acquisition and compared the movement of static Smc-YFP to the motion of chromosomal loci, labelled at the 90° and 270° positions by using the well-established FROS (fluorescent repressor/operator) system (44). Visual inspection already revealed that static Smc was much more constrained in its movement than chromosomal loci (Figure 4B). In order to quantify our finding, we chose for our analysis plots of MSD versus lag time (Figure 4A). This method has the advantage over the distribution of displacements that one can distinguish freely diffusive, subdiffusive, and directed motions. According to our results, chromosomal loci in B. subtilis exhibited subdiffusive motion, which can be deduced from the curvature of the MSD curve towards long time lags, as was already shown for E. coli and Caulobacter crescentus (45,46). The slopes of the first points of the MSD plots yield nominal short-time diffusion constants. For the chromosomal loci at 90° and 270° we observed similar slopes and therefore similar diffusion coefficients: DLocus 270° = 0.0029 ± 5 × 10−4 (SD) μm2 s−1 and DLocus 90° = 0.0021 ± 5 × 10−4 (SD) μm2 s−1. Smc-YFP showed a much shallower slope than that of chromosome loci (Figure 4A) and therefore a lower diffusion coefficient: DSmcstatic = 0.0008 ± 1 × 10−5 μm2 s−1. This diffusion coefficient is about three times lower than that of chromosome loci, indicating highly constrained movement of Smc relative to chromosomal sites. Of note, the diffusion coefficient for static Smc determined using 100 ms acquisition is not exactly the same diffusion coefficient as determined with the 8 ms timescale above (Table 1) due to the different exposure time.
Figure 4.

Smc is immobile in the cell, while chromosomal loci show substantial movement. (A) Mean squared displacement (MSD) versus lag time. Smcstatic (red) shows about three times less movement than chromosomal loci (270° blue, 90° yellow) as can also be deduced from the linear fit to the first 3 time points of the MSD curve. The confinement is indicated by the shadowing in the plot. (B) Kymograph showing that movement of Smc-YFP is lower than that of locus 270°. (C) Smcstatic does not move significantly faster or slower if origin associated Smcstatic is compared with Smcstatic outside origins (unpaired, two-sided Student's t-test, P = 0.7305, Smcstatic origin associated: n = 12, Smcstatic outside origins: n = 19). Black dots are considered outliers (lie outside of 1.5 ITR). (D) Box plot of diffusion coefficients from seven independent experiments (with at least n = 250 tracks) for each condition. Statistical differences between static Smc-YFP and the two chromosome loci are highly significant (***), as are differences of locus 270° between wild type and smc mutant (Δsmc) cells. Differences in movement of locus 270° between wild type and ΔparB cells are small but statistically significant (*). (E) Box plot of radius of confinement, i.e. the radius reached by the molecule/locus over 2 s. Statistical evaluation analogous to panel D.
As a control for this unusually static behavior of the SMC complex, we imaged another SMC-like protein, RecN, which is involved in DNA repair (47), and stably binds to ssDNA ends (48) that occur at DNA breaks. Upon addition of Mitomycin C, a fully functional RecN-YFP fusion forms discrete foci on the chromosome, at sites of DNA breaks (49), where it is assumed to form multimers. The diffusion of RecN was in striking contrast to Smcstatic: its diffusion coefficient was ∼40-fold higher (DRecN = 0.035 ± 0.0075 μm2 s−1) than that of Smc, and ∼10-fold higher than the movement of chromosome sites (Figure 4D), indicating that Smc-like proteins can have radically different mobilities. These data show that DNA repair protein RecN displays very different diffusion constants when bound to (broken) chromosomes compared to Smc.
To investigate if the motion of SMC complexes is different near oriC or at sites further away, we determined the diffusion constants of static Smc-YFP within a radius of 150 nm around oriC versus oriC-distant Smc-YFP. Figure 4C shows no statistically significant difference in motion of oriC-proximal versus oriC-distant Smc-YFP, but overall comparable dynamics. Thus, static SMC complexes are not distinguishable in terms of mobility at different sites on the chromosome.
According to the recent loop extrusion model for condensin activity (condensin can actively extrude DNA loops (50), which is further discussed below), Smc might contribute to the motion of chromosome loci. We therefore analyzed diffusion coefficients for the locus at 270° in wild type and in smc mutant cells. Diffusion coefficients for 270° were highly reduced in the absence of Smc, while the absence of ParB increased mobility of the locus (moderately, but still statistically significantly) (Figure 4D). Likewise, mobility of origin regions on the chromosome was decreased in the absence of Smc (data not shown). ParB has been shown to generate stable DNA loops and establish stable long range interactions between the origin region and adjacent sites (51,52), which appears to lead to containment of chromosome movement. As a second method for analysis, we determined the radius of confinement of molecules or sites within 2 s. Figure 4E shows that Smc-YFP shows the highest degree of confinement (which in this case is 65 nm), while that of chromosome loci is much larger (170 nm in case of 270°). The confinement radius becomes much smaller in smc mutant cells, and slightly larger in parB mutant cells (Figure 4E), supporting that directly or indirectly, Smc contributes to chromosome mobility.
Taken together, our data reveal that SMC centers do not visibly track with the chromosome, but are somehow anchored at different sites within the cell, or may act as ‘chromosome constrainers’, which rigidly tether together chromosomal sites, as was suggested for lamin A in eukaryotic cells (53).
Smc, ScpA and ScpB are present at low, roughly equimolar concentrations in the cell
We performed fluorescence intensity measurements in single cells. Knowing the amount of fluorescence emitted by a single YFP based on one step bleaching towards the end of the stream acquisitions (Figure 5A and B), we calculated the number of molecules in a single cell from the fluorescence intensity at the beginning of the acquisition (Figure 5C and D). Of note, not all FPs are fluorescent. We estimate that only 80% of the molecules are fluorescent at a given time point (54), so the actual number of molecules in a cell must be corrected accordingly. For Smc, we found between 20 and 96 molecules per cell (Figure 5E), and 61 molecules for an average sized cell of 3 μm length (Figure 5H), corresponding to 30 dimers on average. There was no significant correlation between molecule number and cell size (Figure 5E, rho = 0.188, P = 0.166, Pearson two-sided significance, n = 57), suggesting that there is quite a high degree of noise in the expression of smc. For ScpA, we found an average of 87 molecules per cell (Figure 5F and H), so surprisingly more than for Smc, and here, we found a significant correlation between cell size and molecule number (rho = 0.574, P < 0.001; n = 60), which roughly doubles within the cell cycle. On the other hand, the number of ScpB molecules was comparable to that of Smc and had no obvious correlation to cell size (Figure 5G and H, rho = 0.169, P = 0.218, n = 55). Therefore, Smc and its two interactors are present in roughly equal amounts, and ScpA is in excess, as the ratio of Smc:ScpA:ScpB is 2:1:2 within the SMC complex as previously biochemically determined (17).
Figure 5.

(A) Fluorescence of a single molecule whose outline is indicated by a red line. The scale bar represents 2 μm. (B) Integrated fluorescence intensity of this single molecule during the stream acquisition. Please note that the acquisition of a single molecule could be clearly detected according to the bleaching step. From this bleaching step, the fluorescence of a single molecule could be measured. (C) Fluorescence of the cell shown in (A). (D) Integrated fluorescence intensity of this cell during the stream acquisition. The total fluorescence of a cell corresponds to the peak intensity minus the baseline intensity after bleaching. (E–G) Number of molecules are plotted against the cell length and a linear regression was performed. A Pearson correlation test of the correlation between cell length and cell number revealed a significant correlation for ScpA-YFP (rho = 0.574, Pearson two-sided significance, n = 60) and no significant correlation for Smc-YFP (rho = 0.188, n = 57) and ScpB (rho = 0.169, n = 55). (H) Numbers of molecules for a cell with an average length of 3 μm.
Furthermore, we quantified the amount of Smc, ScpA and ScpB integrated into foci within the cells, which would yield information on the number of molecules present within the static centers. Integrated camera counts of the clusters were divided by the integrated intensity of a single fluorophore, identified by a single bleaching step within the cell (see material and methods). Our analysis showed a surprisingly low number of proteins integrated into the clusters: for Smc, we counted that, on average, 5.2 ± 0.8 molecules were integrated into clusters, for ScpA 8.1 ± 1.7 molecules, and for ScpB 4.9 ± 0.8 molecules (Figure 6). Thus, ScpA is not only present in excess in the whole cell, but there is also an excess of ScpA present in clusters, in agreement with our determination of static molecules for ScpA from single molecule tracking.
Figure 6.

Analysis of numbers of molecules integrated in clusters. (A) Distribution of number of molecules in clusters for Smc-YFP, ScpA-YFP and ScpB-YFP. For Smc-YFP, 5.2 ± 0.8 molecules are integrated in clusters (n = 64), for ScpA-YFP 8.1 ±1.7 molecules are integrated in clusters (n = 89), and for ScpB-YFP 4.9 ± 0.8 molecules are integrated in clusters (n = 91). (B) Boxplot showing numbers of molecules integrated into clusters. There was no statistically significant difference between the numbers of molecules integrated into clusters for Smc-YFP and ScpB-YFP (P = 0.03), but ScpA-YFP showed a statistically significant higher number of molecules integrated into clusters (P < 0.005). Black dots are considered outliers (lie outside of 1.5 ITR).
DISCUSSION
Multiple SMC complexes compact the chromosome from several sites on the chromosome
More than twenty five years after the discovery of MukB, the first (bacterial) SMC protein that was discovered (55), and 16 years after the identification of the SMC complex consisting of Smc, ScpA and ScpB (MukB, MukF and MukE in E. coli) (19,56), it is still unclear how the low abundant ATPase Smc can compact an entire 4.3 Mbp chromosome in a cell. We provide quantitative data on the protein dynamics of the B. subtilis SMC complex, and of chromosome dynamics in the milliseconds range, revealing that only few Smc molecules affect chromosome dynamics by either constrained movement through the entire chromosome or formation of static complexes with ScpA and ScpB, at several sites on the chromosome arms.
Through single molecule counting in live cells, we show that 20–40 Smc dimers, 30–100 ScpA monomers and a number of ScpB molecules similar to that of Smc are present in slow growing cells (a majority of which have two origin regions). About half of Smc molecules are static and bound to ScpA and to ScpB, while the remaining Smc molecules move throughout the chromosome. Interestingly, recent in vivo crosslinking experiments have shown that about 20% of Smc molecules form a ring structure together with ScpA and ScpB, which stably encircles DNA strands (57). These data suggest that 5–10 static ScpAB bound Smc dimers are present per origin, i.e. in one cell half, in slow growing cells. Indeed, our SMT experiments reveal that many more than just one Smc/ScpAB cluster exists within each cell half, as was deduced from conventional fluorescence microscopy (19–21,42); frequently, three to five static clusters can be seen within one cell half, dependent on cell size. Fluorescence intensity determination suggests that between one and three Smc dimers are present within static SMC centers, indicating that a 4Smc/2ScpA/4ScpB complex could be the functional unit. This is an interesting parallel to the MukBEF complex, which had been suggested to act as dimer of dimers within the static clusters (22). However, given our localization precision of 21 nm, we cannot distinguish if clusters consist of multimers or of individually acting 2Smc/ScpA/2ScpB complexes. Clearly, the SMC complex does not condense the chromosome from a single site on the chromosome, or as one large multimeric cluster (40,47), but from several distinct positions.
Furthermore, our data are in agreement with and support recent data from the Rudner laboratory, who have shown that Smc/ScpAB set up several sites where chromosome arms are bridged, starting from parS sites and moving towards the terminus region (43,52). It is possible that these bridges are achieved by the two to four static SMC complexes we have observed per cell half during exponential growth.
The SMC complex is statically positioned on the chromosome
A key finding of our study is the fact that SMC complexes show smaller diffusion constants and confinement radii than chromosome loci. This finding can be interpreted by different scenarios: (a) the SMC complex binds stably to—possibly many—DNA strands and immobilizes bound DNA, or (b) the complex is anchored by unknown cellular components, and possibly DNA moves through the complex, in analogy to DNA translocases. In the first scenario, Smc could capture and release DNA loops, driven by its ATPase cycle. Stochastic Smc interactions bridging distal chromosomal loci, either within chromosome arms or between arms, could stabilize the nucleoid (58). Stabilization of long range interactions within the chromosome by SMC have been shown for Schizosaccharomyces pombe during mitosis (59).
The second scenario is supported by the fact that Smc has an intriguing structural resemblance to motor proteins like kinesin and myosin, and it has been suggested very early that Smc could possibly have the ability to actively track on DNA driven by ATP hydrolysis. Indeed, it has been shown that condensin from Saccharomyces cerevisiae is a molecular motor capable of ATP hydrolysis-dependent translocation along double-stranded DNA in vitro, using DNA curtains at a velocity of ∼60 base pairs per second (60). An argument against this model is the relative low ATPase activity of Smc (3) compared to other nucleic acid motor proteins. Recently, a model has been put forward describing the mode of action of Smc by an active loop extrusion mechanism (43), which would also imply that Smc should actively track on DNA, or that DNA tracks through an Smc ring. Our results suggest that the SMC complex does not visibly track on the nucleotides, which would imply that DNA would move through SMC complexes. A single condensin complex bound to DNA can asymmetrically reel in DNA and expel a loop in an ATP-driven, processive manner (61). Therefore, the SMC complex could anchor on DNA and extrude a loop, thereby moving along DNA, which would not be visible as movement within the cells, because the DNA is moved. We are currently not able to distinguish if low Smc mobility on DNA is based on loop extrusion or on loop constraining; or on possibly another mechanism.
We further show that in the absence of Smc, chromosome regions become more static than in wild type cells. This is in contrast to the absence of ParB, which stably binds to DNA loops and generates long range interactions between oriC regions and distant flanking regions (52); here, we observed higher mobility of chromosome regions. It should be noted that S. pombe chromosome movement is higher in the absence of condensin (59), showing that this effect in B. subtilis is not universal. Although the effect of Smc activity on chromosome movement is compatible with Smc acting as a loop extrusion motor in B. subtilis, a strong link of SMC proteins to topoisomerases and torsion-mediated DNA movement (2) could equally well explain altered mobility.
Interestingly, we found that SMC-like RecN protein showed higher mobility bound to DNA double strand breaks compared to normal motion of chromosome sites, indicating that sites of DNA breaks may become more mobile in cells actively repairing DNA damage.
The SMC complex is established from an Smc fraction moving through the chromosome and a freely diffusing ScpAB complex
At origin regions, Smc dimers start to diffuse through the chromosome (20,21), to eventually meet a freely diffusing ScpAB subcomplex and switch to a DNA-encircling mode of DNA binding (57), establishing a condensation center. ScpAB have been shown to convert Smc—that binds DNA via coiled coil opening at hinge domains (loosely-DNA bound Smc)—to stably DNA-bound Smc rings, which involves head interactions of Smc (62). We show that ∼60% of ScpA monomers (about 50 monomers per cell) are statically positioned, dependent on the presence of Smc, and therefore must be bound to Smc dimers at the SMC clusters, while 40% of ScpA are diffusing through the cell. As there are only about 15 static Smc dimers detected in our tracking analysis, there are few more static ScpAs than can be accounted for being bound to Smc. It is possible that ScpAB are transiently engaged with other proteins in the cell, as has been proposed (63). Mobile ScpA is not associated with DNA, which is also true for ScpB, both of which do not show DNA binding in vitro (14,38). These dynamics are markedly different from that of the MukBEF complex, where MukB, E, and F diffuse with the same velocity, and therefore move together through the chromosome (22). This difference suggests a different mode of setting up of condensation centers in E. coli and relatives than in B. subtilis.
Chromosome-associated movement of Smc depends on hinge and arms, while head domains are essential for cluster formation
Interestingly, our data show that headless Smc (consisting of coiled coil arms and hinge) is still able to diffuse through the chromosome similar to wild type Smc, but is unable to form condensation clusters. Indeed, in vitro, headless binds to DNA non-specifically, and only moderately less avidly compared to wild type Smc, as seen by gel shift (36) and SPR experiments (this work). Moreover, hinge/coil interactions and a correct length of the coiled coil arms are essential for DNA contacts in vivo, and for Smc functionality (12,64). These findings indicate that mobile Smc binds chromosomal DNA via coiled coil opening at hinge domains, and this loose DNA binding enables constrained movement through the chromosome. We favor the view that mobile, DNA-associated Smc is important for efficient formation of condensation centers away from oriC. While most Smc molecules will meet close to oriC, dimers that diffuse towards the terminus region (which is located in the cell center) and back towards bipolar origin regions will meet other Smc molecules away from oriC, and chromosome arm interactions are not only found close to oriC regions, but all along chromosome arms (20,42,43). Additionally, since only few Smc dimers exist per cell, a predominantly DNA-bound movement will highly increase chances for dimers to meet. The finding that headless Smc diffuses in a comparable rate as wild type Smc shows that head engagement is not needed for the loose DNA-bound state, which is in good agreement with recent biochemical data from the Gruber group (62).
Collectively, our results rule out some earlier models of formation of single large SMC clusters within a cell half (47), and show that several clusters, containing few Smc dimers bound to ScpAB, are present at wide spread positions on the chromosome. Interactions with DNA or with an unknown anchor protein render SMC complexes stationary relative to general chromosome movement, while biochemical data suggest movement of Smc relative to DNA. It will be important to further investigate if the SMC complex acts as a chromosome constrainer or a DNA loop extruder, and how ScpAB convert a DNA-diffusive Smc to a stationary DNA binder.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Benjamin Eggart and Dr. Walter Gräwe from Hamamatsu Photonics for help with the conversion of camera counts to photons, DFG Research Fellowship 2816/1-1.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
LOEWE Center for Synthetic Microbiology (SYNMIKRO); BMBF funded graduate consortium NANOKAT; Deutsche Forschungsgemeinschaft; Excellence Initiative of the German Federal and State Governments (EXC 294). Funding for open access charge: Deutsche Forschungsgemeinschaft.
Conflict of interest statement. None declared.
REFERENCES
- 1. Uhlmann F. SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016; 17:399–412. [DOI] [PubMed] [Google Scholar]
- 2. Hirano T. Condensin-based chromosome organization from bacteria to vertebrates. Cell. 2016; 164:847–857. [DOI] [PubMed] [Google Scholar]
- 3. Graumann P.L. Chromosome architecture and segregation in prokaryotic cells. J. Mol. Microbiol. Biotechnol. 2014; 24:291–300. [DOI] [PubMed] [Google Scholar]
- 4. Gligoris T., Löwe J.. Structural insights into ring formation of cohesin and related Smc complexes. Trends Cell Biol. 2016; 26:680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Löwe J., Cordell S.C., van den Ent F.. Crystal structure of the SMC head domain: an ABC ATPase with 900 residues antiparallel coiled-coil inserted. J. Mol. Biol. 2001; 306:25–35. [DOI] [PubMed] [Google Scholar]
- 6. Haering C.H., Farcas A.M., Arumugam P., Metson J., Nasmyth K.. The cohesin ring concatenates sister DNA molecules. Nature. 2008; 454:297–301. [DOI] [PubMed] [Google Scholar]
- 7. Cuylen S., Metz J., Haering C.H.. Condensin structures chromosomal DNA through topological links. Nat. Struct. Mol. Biol. 2011; 18:894–901. [DOI] [PubMed] [Google Scholar]
- 8. Gruber S., Haering C.H., Nasmyth K.. Chromosomal cohesin forms a ring. Cell. 2003; 112:765–777. [DOI] [PubMed] [Google Scholar]
- 9. Lammens A., Schele A., Hopfner K.P.. Structural biochemistry of ATP-driven dimerization and DNA-stimulated activation of SMC ATPases. Curr. Biol. 2004; 14:1778–1782. [DOI] [PubMed] [Google Scholar]
- 10. Hu B., Itoh T., Mishra A., Katoh Y., Chan K.L., Upcher W., Godlee C., Roig M.B., Shirahige K., Nasmyth K.. ATP hydrolysis is required for relocating cohesin from sites occupied by its Scc2/4 loading complex. Curr. Biol. 2011; 21:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirano M., Hirano T.. Opening closed arms: long-distance activation of SMC ATPase by hinge-DNA interactions. Mol. Cell. 2006; 21:175–186. [DOI] [PubMed] [Google Scholar]
- 12. Soh Y.M., Bürmann F., Shin H.C., Oda T., Jin K.S., Toseland C.P., Kim C., Lee H., Kim S.J., Kong M.S. et al. Molecular basis for SMC rod formation and its dissolution upon DNA binding. Mol. Cell. 2015; 57:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Woo J.S., Lim J.H., Shin H.C., Suh M.K., Ku B., Lee K.H., Joo K., Robinson H., Lee J., Park S.Y. et al. Structural studies of a bacterial condensin complex reveal ATP-dependent disruption of intersubunit interactions. Cell. 2009; 136:85–96. [DOI] [PubMed] [Google Scholar]
- 14. Hirano M., Hirano T.. Positive and negative regulation of SMC-DNA interactions by ATP and accessory proteins. EMBO J. 2004; 23:2664–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kleine Borgmann L.A., Graumann P.L.. Structural maintenance of chromosome complex in bacteria. J. Mol. Microbiol. Biotechnol. 2014; 24:384–395. [DOI] [PubMed] [Google Scholar]
- 16. Rybenkov V.V., Herrera V., Petrushenko Z.M., Zhao H.. MukBEF, a chromosomal organizer. J. Mol. Microbiol. Biotechnol. 2014; 24:371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bürmann F., Shin H.C., Basquin J., Soh Y.M., Gimenez-Oya V., Kim Y.G., Oh B.H., Gruber S.. An asymmetric SMC-kleisin bridge in prokaryotic condensin. Nat. Struct. Mol. Biol. 2013; 20:371–379. [DOI] [PubMed] [Google Scholar]
- 18. Kamada K., Miyata M., Hirano T.. Molecular basis of SMC ATPase activation: role of internal structural changes of the regulatory subcomplex ScpAB. Structure. 2013; 21:581–594. [DOI] [PubMed] [Google Scholar]
- 19. Mascarenhas J., Soppa J., Strunnikov A.V., Graumann P.L.. Cell cycle-dependent localization of two novel prokaryotic chromosome segregation and condensation proteins in Bacillus subtilis that interact with SMC protein. EMBO J. 2002; 21:3108–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gruber S., Errington J.. Recruitment of condensin to replication origin regions by ParB/SpoOJ promotes chromosome segregation in B. subtilis. Cell. 2009; 137:685–696. [DOI] [PubMed] [Google Scholar]
- 21. Sullivan N.L., Marquis K.A., Rudner D.Z.. Recruitment of SMC by ParB-parS organizes the origin region and promotes efficient chromosome segregation. Cell. 2009; 137:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Badrinarayanan A., Reyes-Lamothe R., Uphoff S., Leake M.C., Sherratt D.J.. In vivo architecture and action of bacterial structural maintenance of chromosome proteins. Science. 2012; 338:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kleine Borgmann L.A., Ries J., Ewers H., Ulbrich M.H., Graumann P.L.. The bacterial SMC complex displays two distinct modes of interaction with the chromosome. Cell Rep. 2013; 3:1483–1492. [DOI] [PubMed] [Google Scholar]
- 24. Kleine Borgmann L.A., Hummel H., Ulbrich M.H., Graumann P.L.. SMC condensation centers in Bacillus subtilis are dynamic structures. J. Bacteriol. 2013; 195:2136–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shivanandan A., Deschout H., Scarselli M., Radenovic A.. Challenges in quantitative single molecule localization microscopy. FEBS Lett. 2014; 588:3595–3602. [DOI] [PubMed] [Google Scholar]
- 26. Weimann L., Ganzinger K.A., McColl J., Irvine K.L., Davis S.J., Gay N.J., Bryant C.E., Klenerman D.. A quantitative comparison of single-dye tracking analysis tools using Monte Carlo simulations. PLoS One. 2013; 8:e64287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaacks K.J., Healy J., Losick R., Grossman A.D.. Identification and characterization of genes controlled by the sporulation-regulatory gene spo0H in Bacillus subtilis. J. Bacteriol. 1989; 171:4121–4129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gibson D.G., Young L., Chuang R.Y., Venter J.C., Hutchison C.A. 3rd, Smith H.O.. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009; 6:343–345. [DOI] [PubMed] [Google Scholar]
- 29. Bakshi S., Bratton B.P., Weisshaar J.C.. Subdiffraction-limit study of Kaede diffusion and spatial distribution in live Escherichia coli. Biophys. J. 2011; 101:2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. English B.P., Hauryliuk V., Sanamrad A., Tankov S., Dekker N.H., Elf J.. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:E365–E373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mazza D., Abernathy A., Golob N., Morisaki T., McNally J.G.. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 2012; 40:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grünwald D., Spottke B., Buschmann V., Kubitscheck U.. Intranuclear binding kinetics and mobility of single native U1 snRNP particles in living cells. Mol. Biol. Cell. 2006; 17:5017–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jaqaman K., Galbraith J., Davidson M., Galbraith C.G.. Changes in single-molecule integrin dynamics linked to local cellular behavior. Mol. Biol. Cell. 2016; 27:1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thompson R.E., Larson D.R., Webb W.W.. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002; 82:2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stracy M., Lesterlin C., Garza de Leon F., Uphoff S., Zawadzki P., Kapanidis A.N.. Live-cell superresolution microscopy reveals the organization of RNA polymerase in the bacterial nucleoid. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E4390–E4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hirano M., Hirano T.. Hinge-mediated dimerization of SMC protein is essential for its dynamic interaction with DNA. EMBO J. 2002; 21:5733–5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim H., Loparo J.J.. Multistep assembly of DNA condensation clusters by SMC. Nat. Commun. 2016; 7:10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Volkov A., Mascarenhas J., Andrei-Selmer C., Ulrich H.D., Graumann P.L.. A prokaryotic condensin/cohesin-like complex can actively compact chromosomes from a single position on the nucleoid and binds to DNA as a ring-like structure. Mol. Cell Biol. 2003; 23:5638–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dix J.A., Verkman A.S.. Crowding effects on diffusion in solutions and cells. Annu. Rev. Biophys. 2008; 37:247–263. [DOI] [PubMed] [Google Scholar]
- 40. Mascarenhas J., Volkov A.V., Rinn C., Schiener J., Guckenberger R., Graumann P.L.. Dynamic assembly, localization and proteolysis of the Bacillus subtilis SMC complex. BMC Cell Biol. 2005; 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uphoff S., Reyes-Lamothe R., Garza de Leon F., Sherratt D.J., Kapanidis A.N.. Single-molecule DNA repair in live bacteria. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:8063–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lindow J.C., Kuwano M., Moriya S., Grossman A.D.. Subcellular localization of the Bacillus subtilis structural maintenance of chromosomes (SMC) protein. Mol. Microbiol. 2002; 46:997–1009. [DOI] [PubMed] [Google Scholar]
- 43. Wang X., Brandao H.B., Le T.B., Laub M.T., Rudner D.Z.. Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science. 2017; 355:524–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Teleman A.A., Graumann P.L., Lin D.C., Grossman A.D., Losick R.. Chromosome arrangement within a bacterium. Curr. Biol. 1998; 8:1102–1109. [DOI] [PubMed] [Google Scholar]
- 45. Javer A., Long Z., Nugent E., Grisi M., Siriwatwetchakul K., Dorfman K.D., Cicuta P., Cosentino Lagomarsino M.. Short-time movement of E. coli chromosomal loci depends on coordinate and subcellular localization. Nat. Commun. 2013; 4:3003. [DOI] [PubMed] [Google Scholar]
- 46. Weber S.C., Spakowitz A.J., Theriot J.A.. Bacterial chromosomal loci move subdiffusively through a viscoelastic cytoplasm. Phys. Rev. Lett. 2010; 104:238102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Graumann P.L., Knust T.. Dynamics of the bacterial SMC complex and SMC-like proteins involved in DNA repair. Chromosome Res. 2009; 17:265–275. [DOI] [PubMed] [Google Scholar]
- 48. Sanchez H., Cardenas P.P., Yoshimura S.H., Takeyasu K., Alonso J.C.. Dynamic structures of Bacillus subtilis RecN DNA complexes. Nucleic. Acids. Res. 2008; 36:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kidane D., Graumann P.L.. Dynamic formation of RecA filaments at DNA double strand break repair centers in live cells. J. Cell Biol. 2005; 170:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L.A.. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016; 15:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Graham T.G., Wang X., Song D., Etson C.M., van Oijen A.M., Rudner D.Z., Loparo J.J.. ParB spreading requires DNA bridging. Genes Dev. 2014; 28:1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang X., Le T.B., Lajoie B.R., Dekker J., Laub M.T., Rudner D.Z.. Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev. 2015; 29:1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bronshtein I., Kepten E., Kanter I., Berezin S., Lindner M., Redwood A.B., Mai S., Gonzalo S., Foisner R., Shav-Tal Y. et al. Loss of lamin A function increases chromatin dynamics in the nuclear interior. Nat. Commun. 2015; 6:8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ulbrich M.H., Isacoff E.Y.. Subunit counting in membrane-bound proteins. Nat. Methods. 2007; 4:319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Niki H., Jaffe A., Imamura R., Ogura T., Hiraga S.. The new gene mukB codes for a 177 kd protein with coiled-coil domains involved in chromosome partitioning of E. coli. EMBO J. 1991; 10:183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Soppa J., Kobayashi K., Noirot-Gros M.F., Oesterhelt D., Ehrlich S.D., Dervyn E., Ogasawara N., Moriya S.. Discovery of two novel families of proteins that are proposed to interact with prokaryotic SMC proteins, and characterization of the Bacillus subtilis family members ScpA and ScpB. Mol. Microbiol. 2002; 45:59–71. [DOI] [PubMed] [Google Scholar]
- 57. Wilhelm L., Bürmann F., Minnen A., Shin H.C., Toseland C.P., Oh B.H., Gruber S.. SMC condensin entraps chromosomal DNA by an ATP hydrolysis dependent loading mechanism in Bacillus subtilis. eLife. 2015; 4:e06659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cheng T.M., Heeger S., Chaleil R.A., Matthews N., Stewart A., Wright J., Lim C., Bates P.A., Uhlmann F.. A simple biophysical model emulates budding yeast chromosome condensation. eLife. 2015; 4:e05565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kakui Y., Rabinowitz A., Barry D.J., Uhlmann F.. Condensin-mediated remodeling of the mitotic chromatin landscape in fission yeast. Nat. Genet. 2017; 49:1553–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Terakawa T., Bisht S., Eeftens J.M., Dekker C., Haering C.H., Greene E.C.. The condensin complex is a mechanochemical motor that translocates along DNA. Science. 2017; 358:672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ganji M., Shaltiel I.A., Bisht S., Kim E., Kalichava A., Haering C.H., Dekker C.. Real-time imaging of DNA loop extrusion by condensin. Science. 2018; 360:102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Minnen A., Bürmann F., Wilhelm L., Anchimiuk A., Diebold-Durand M.L., Gruber S.. Control of Smc coiled coil architecture by the ATPase heads facilitates targeting to chromosomal ParB/parS and release onto flanking DNA. Cell Rep. 2016; 14:2003–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dervyn E., Noirot-Gros M.F., Mervelet P., McGovern S., Ehrlich S.D., Polard P., Noirot P.. The bacterial condensin/cohesin-like protein complex acts in DNA repair and regulation of gene expression. Mol. Microbiol. 2004; 51:1629–1640. [DOI] [PubMed] [Google Scholar]
- 64. Bürmann F., Basfeld A., Vazquez Nunez R., Diebold-Durand M.L., Wilhelm L., Gruber S.. Tuned SMC arms drive chromosomal loading of prokaryotic condensin. Mol. Cell. 2017; 65:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.