

Abstract
Current mass spectrometry (MS)-based proteomics approaches are ineffective for mapping protein expression in tissue sections with high spatial resolution because of the limited overall sensitivity of conventional workflows. Here we report an integrated and automated method to advance spatially resolved proteomics by seamlessly coupling laser capture microdissection (LCM) with a recently developed nanoliter-scale sample preparation system termed nanoPOTS (Nanodroplet Processing in One pot for Trace Samples). The workflow is enabled by prepopulating nanowells with DMSO, which serves as a sacrificial capture liquid for microdissected tissues. The DMSO droplets efficiently collect laser-pressure catapulted LCM tissues as small as 20 μm in diameter with success rates >87%. We also demonstrate that tissue treatment with DMSO can significantly improve proteome coverage, likely due to its ability to dissolve lipids from tissue and enhance protein extraction efficiency. The LCM-nanoPOTS platform was able to identify 180, 695, and 1827 protein groups on average from 12-μm-thick rat brain cortex tissue sections having diameters of 50, 100, and 200 μm, respectively. We also analyzed 100-μm-diameter sections corresponding to 10–18 cells from three different regions of rat brain and comparatively quantified ∼1000 proteins, demonstrating the potential utility for high-resolution spatially resolved mapping of protein expression in tissues.
Keywords: Omics, Mass Spectrometry, Imaging, Imaging Hardware, Label-free quantification, laser capture microdissection, mapping, microfluidics, nanowell, spatially resolved proteomcis
Biological tissues are often highly heterogeneous, consisting of a variety of cell types, subpopulations, and substructures (1). Tissue cells often generate distinct microenvironments to execute biological functions, providing varied response to external stimuli, and often resulting in distinct pathology. Spatially resolved and multiplexed molecular imaging of tissue sections is of key importance for understanding biological function and pathogenesis (2, 3). The characterization of the molecular landscape in tissues often relies on targeted methods that monitor a small number of species such as immunohistochemistry (IHC) staining, fluorescence in situ hybridization (FISH) or imaging mass cytometry. These methods rely on the availability of suitable probes and require a priori knowledge of the system, thus limiting discovery. Recent advances in RNA amplification and sequencing have enabled the quantification of thousands of transcripts in tissue sections and single cells (4). To broadly measure proteins, peptides, metabolites and lipids across tissues in a label-free manner, mass spectrometry imaging techniques based on matrix-assisted laser desorption/ionization (MALDI) and other techniques have been developed (5–8). Although MS imaging can simultaneously measure hundreds of distinct molecular species, broad measurements of proteins remain a challenge because of issues related to ionization suppression and the absence of a separation dimension.
LC-MS/MS-based bottom-up proteomics has significantly advanced biomolecular science and can identify and quantify >10,000 proteins in some cases (9, 10). However, the expansion of proteomics to map protein expression in tissue sections with high spatial resolution has been hindered by overall sensitivity as each sample is typically thousands of times smaller than those used for conventional bulk measurements. The sensitivity limitation is primarily due to protein/peptide losses during sample isolation and processing, as well as ionization and transmission of ions to the mass analyzer. Substantial efforts have been devoted to improving overall sensitivity, including nanoelectrospray ionization (11) and associated nanoflow chemical separations (12, 13), high-performance ion optics (14) and high-resolution mass analyzers (e.g. Orbitrap). State-of-the-art systems are now capable of measuring hundreds of proteins from single-cell-sized samples (15), but in practice much larger samples are still required to achieve in-depth proteome coverage because of losses that occur during sample processing. Thus, the major remaining obstacles lie primarily in sampling proteins from tissue sections without losing spatial information, and efficiently processing biological samples into ready-to-analyze peptides.
To this end, microsampling techniques are being developed to extract and analyze proteins from tissue sections with high spatial resolution and proteome coverage. One in situ method developed by Caprioli and coworkers (16, 17) involves applying trypsin-impregnated hydrogels onto tissue sections of interest. Proteins are digested into peptides and transferred to the hydrogel, after which peptides are extracted from the hydrogel and analyzed by nanoLC-MS/MS. Under optimized conditions, >600 proteins were identified from a 260-μm-diameter region of rat liver (17). Another in situ protein extraction workflow was developed by Fournier and coworkers (18–21) using a liquid microjunction. A picoliter dispenser deposited protease onto a tissue section for protein digestion, after which the liquid microjunction extracted peptides (19, 20). NanoLC-MS/MS of the extracted samples resulted in identification of ∼1500 proteins from 650 μm spots (19), or >500 proteins from regions as small as 250 μm (20). Although both approaches would most likely yield greater proteome coverage by using latest-generation MS instrumentation, there are inherent limitations that are likely to preclude substantial further miniaturization. For example, minimum droplet sizes for liquid microjunction workflows are driven by droplet volume, surface tension and contact angle, requiring careful optimization to achieve smaller extraction regions. Careful placement of hydrogels over tissue regions of interest is also expected to become increasingly difficult with further miniaturization. Further, the shape of the extracted tissues would be difficult to match with small and irregular regions of interest identified in histological thin sections.
Laser-capture microdissection (LCM)1 provides an interesting alternative for microsampling because it can readily excise tissue regions as small as single cells and with any shape as guided by high-resolution optical microscopy (22). LCM has been successfully integrated into workflows for spatially resolved proteomics. For example, Wisniewski et al. (23) comparatively analyzed protein expression of LCM-purified normal and neoplastic colonic tissues, and 30 colon-cancer-specific markers were identified and verified. Clair et al. (24) comprehensively studied proteome changes of dissected alveolar tissues during lung development, and distinct proliferation-related biological processes were revealed. McDonnell and coworkers (25) applied an LCM-based proteomics workflow to study kidney substructures, and a panel of protein markers was identified. To reduce adsorptive sample losses during sample preparation and improve proteome coverage, several approaches were developed including the use of carrier species (23), immobilized enzyme reactors (24) and vial conditioning with BSA (25). Despite these efforts, large tissue samples containing > 1000 cells were still required to achieve in-depth proteome profiling.
We have recently developed an ultrasensitive proteome processing and analysis platform termed nanoPOTS (Nanodroplet Processing in One pot for Trace Samples) (26). NanoPOTS significantly increased the overall sensitivity of MS-based proteomics by reducing the sample processing volumes to the nanoliter range, thus greatly minimizing the protein and peptide losses to the reaction vessel surfaces. Quantitative profiling of >3,000 proteins was achieved for as few as 10 HeLa cells, which is a level of proteome coverage not previously attained for fewer than thousands of cells. The nanoPOTS platform was also used to analyze laser microdissected thin sections of single human pancreatic islets. However, a major challenge with the nanoPOTS platform was the inability to directly integrate LCM-based sample isolation. Rather, dissected tissues were manually transferred from a vial to the nanowells using a fine tweezer under a stereomicroscope, which required extensive expertise, limited the smallest tissue size to >100 μm, and resulted in a loss of spatial information for the islets within the original tissue. In addition, the success rates of sample transfer were only ∼50% and the transfer throughput was ∼5 min per sample. Special care was also needed to avoid protein contamination from human tissues or ambient air during the transfer process. Thus, the development of automated sample transfer directly from LCM to the nanoPOTS chip is critical for enabling large-scale proteomic studies of tissue microstructures with high spatial resolution and proteome coverage.
Here we describe the seamless integration of nanoPOTS with LCM by prepopulating the nanowells with DMSO droplets (Fig. 1A). The low-vapor-pressure DMSO serves as a highly efficient sacrificial capture solvent for dissected tissue specimens as small as single cells. Capture efficiencies of ∼90% could be achieved for both rat brain and human cancer tissue samples. Using a 12-μm-thick section of rat brain as a model tissue, we demonstrate that the LCM-nanoPOTS workflow facilitated by DMSO as capture solvent can identify and comparatively quantify ∼1000 proteins with a spatial resolution of ∼100 μm, corresponding to just 10–18 brain cells.
Fig. 1.

A, schematic diagram showing the direct integration of LCM with nanoPOTS using DMSO droplets for tissue capture. B, Image of a nanoPOTS chip with an array of 200-nL prepopulated DMSO droplets. C, Direct mounting of a nanoPOTS chip on a slide adapter for a PALM MicroBeam LCM system. D, Microdissected tissue section and E, the corresponding tissue pieces collected in nanowells with square lateral dimensions from 20 μm to 200 μm. A 12-μm-thick rat brain coronal section was used as model sample.
EXPERIMENTAL PROCEDURES
Reagents and Chemicals
Deionized water (18.2 mΩ) generated from a Barnstead Nanopure Infinity system (ThermoFisher, Waltham, MA) was used throughout. Dithiothreitol (DTT) and iodoacetamide (IAA) were from ThermoFisher, and their working solutions were freshly prepared in 50 mm ammonium bicarbonate (ABC) buffer before use. n-dodecyl-β-d-maltoside (DDM), Mayer's hematoxylin, eosin Y (alcoholic solution), Scott's Tap Water Substitute and dimethyl sulfoxide (DMSO, HPLC grade) were purchased from Sigma-Aldrich (St. Louis, MO). Trypsin (MS grade) and Lys-C (MS grade) were from Promega (Madison, WI). Other unmentioned reagents were from ThermoFisher.
Nanowell Chip Fabrication
The nanoPOTS chip consisted of three parts including a nanowell-containing substrate, a spacer and a cover plate. The nanowell substrate was fabricated with similar procedures as described previously (26). Briefly, a glass slide (25 mm × 75 mm) with pre-coated chromium and photoresist (Telic company, Valencia, CA) was used as starting material. Standard photolithography and wet etching procedures were employed to generate an array of pedestals with a diameter of 1.2 mm, a height of 10 μm, and a spacing of 4.5 mm between adjacent pedestals on the slide. The exposed surfaces surrounding the pedestals were rendered hydrophobic with 2% (v/v) heptadecafluoro-1,1,2,2-tetrahydrodecyldimethylchlorosilane in 2,2,4-trimethylpentane. After removing the chromium layer, the pedestals maintained the hydrophilicity of untreated glass and served as nanoliter-scale wells for tissue collection and proteomic sample processing. The glass spacer was laser machined (Coherent Inc., Santa Clara, CA) on a standard 1.2-mm-thick microscope slide. The machining process removed the center region of the slide, leaving an ∼5-mm-wide frame. The machined slide was glued to the nanowell substrate using a silicone adhesive and served as a spacer to limit the headspace of the nanowells after reversibly sealing to a cover plate to minimize evaporation during incubation steps while preventing contact of the droplet reactors with the cover plate. The cover plate was produced by spin coating of a thin layer of Sylgard 184 base and curing reagent (10:1, v/v) (Dow Corning) at a spin speed of 500 rpm for 10 s followed by 3000 rpm for 30 s. The cover plate was baked at 70 °C for 10 h to generate a ∼30-μm-thick polydimethylsiloxane (PDMS) layer.
Tissue Preparation
All procedures involving animals were in accordance with protocols established in the NIH/NRC Guide and Use of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee of Battelle, Pacific Northwest Division. Pregnant female Sprague-Dawley rats (gestational day 22, ∼250 g) were purchased from Charles River Laboratories Inc. (Wilmington, MA). Rats were housed in solid-bottom cages with hardwood chips under standard laboratory conditions. Rat pups post-natal day 17, were euthanized using carbon dioxide as asphyxiant. Brains were rapidly removed, quickly rinsed in phosphate buffered saline, and flash frozen in liquid nitrogen. The brains were stored at −80 °C until use. A cryostat (NX-70, ThermoFisher) was used to cut tissues to a thickness of 12 μm. The chuck and blade temperatures were set at −16 °C and −20 °C, respectively. The tissue sections were deposited on PEN membrane slides (Carl Zeiss Microscopy, Germany) and stored at −80 °C.
Before the hematoxylin and eosin (H&E) staining procedures, the tissue section was removed from the freezer or dry ice box and immediately immersed into 70% ethanol to fix proteins. The tissue was then rehydrated in deionized water for 30 s and stained in Mayer's hematoxylin solution for 1 min. Excess dye was rinsed with water and the tissue was blued in Scott's Tap Water Substitute for 15 s. Next, 70% ethanol was used to dehydrate the tissue and a 50% diluted eosin Y solution (v/v in ethanol) was applied for 1–2 s by a quick dip. The tissue sample was further dehydrated by immersion twice in 95% ethanol for 30 s, twice in 100% ethanol for 30 s, and finally in xylene for 2 min. All procedures were performed in a fume hood and the slide was blotted on absorbent paper between different solutions to minimize carryover. The processed tissue could be directly used for LCM or stored at −80 °C until use.
WHIM2 patient-derived xenograft tissue sections with a thickness of 12 μm were obtained from Dr. Sherri Davies at Washington University School of Medicine. All human tissues for these experiments were processed in compliance with NIH regulations and institutional guidelines and approved by the institutional review board at Washington University. All animal procedures were reviewed and approved by the institutional animal care and use committee at Washington University in St. Louis. The tissue sections were fixed as described above and stained with hematoxylin only.
Laser Capture Microdissection
Unless mentioned otherwise, an array of DMSO droplets with a volume of 200 nL were deposited on nanowells using a nanoliter-dispensing robotic system (Fig. 1B) (27, 28). A PALM MicroBeam laser capture microdissection system (Carl Zeiss MicroImaging, Munich, Germany) was employed. The nanowell chip was fixed on a standard microscope slide adapter (SlideCollector 48, Carl Zeiss MicroImaging) and mounted on the robotic arm of the LCM system (Fig. 1C). Both the rat brain and breast cancer tissues were cut at an energy level of 42 and catapulted into the DMSO droplet using the “CenterRoboLPC” function with an energy level of delta 15 and a focus level of delta 10. Tissue samples in the nanowell chip could be processed directly or stored at −20 °C.
NanoPOTS Proteomic Sample Processing
Before processing, DMSO droplets were evaporated to dryness by keeping the nanowell chip in a vacuum desiccator for 10 to 15 min (Fig. 1E). Reagent dispensing was performed using the robotic system as described previously (27). Briefly, 100 nL of PBS buffer containing 0.2% DDM surfactant and 5 mm DTT was added to each nanowell. The chip was incubated at 70 °C for 1 h for protein extraction and denaturation. Proteins were then alkylated by adding 50 nL of 30 mm IAA in 50 mm ABC in each reaction and then incubating for 40 min in the dark. A two-step digestion was performed at 37 °C with Lys-C and trypsin for 4 h and 8 h, respectively. Finally, the digested peptide samples were collected and stored in a fused silica capillary (4 cm long, 200 μm i.d., 360 μm o.d.). Each nanowell was washed twice with 200 nL of 0.1% formic acid aqueous buffer and the wash solution was also collected into the same capillary to maximize sample recovery. To prevent residual PEN membrane pieces from being drawn into the collection capillary, the distance between the capillary distal end and the nanowell surface was kept at 100 μm during the sample aspiration process. The capillary was sealed with Parafilm M (Sigma Aldrich) at both ends and stored at −70 °C until analyzed.
nanoLC-MS/MS
Samples in the collection capillary were desalted and concentrated on a solid phase extraction (SPE) column (75-μm-i.d. fused silica capillary packed with 3 μm diameter, 300 Å pore size C18 particles, Phenomenex, Torrance, CA) (29). Peptides were separated using a 60-cm-long, 30-μm-i.d. nanoLC column with the same C18 particles and with an integrated electrospray emitter (Self-Pack PicoFrit column, New Objective, Woburn, MA) (26, 30). A nanoUPLC pump (Dionex UltiMate NCP-3200RS, Thermo Scientific, Waltham, MI) was used to deliver mobile phase to the LC column. To obtain reproducible and smooth gradient profiles, a tee interface was used to split the LC flow rate from 300 nL/min to 50 nL/min for the 30-μm-i.d. LC column. A linear 100-min gradient starting from 8% Buffer B (0.1% formic acid in acetonitrile; Buffer A: 0.1% formic acid in water) to 22%, followed by a 15-min linear increase to 35% Buffer B. The column was washed with 90% Buffer B for 5 min and re-equilibrated with 2% Buffer B for 20 min prior to the subsequent analysis.
Peptides were ionized at the nanospray source using a potential of 2 kV. An Orbitrap Fusion Lumos Tribrid mass spectrometer (ThermoFisher) operated in data dependent mode to automatically switch between full scan MS and MS/MS acquisition with a cycle time of 2 s. The ion transfer capillary was heated to 250 °C to accelerate desolvation and the S lens was set at 30. Full-scan MS spectra (m/z 375–1600) were acquired in the Orbitrap analyzer with 120,000 resolution (m/z 200), an AGC target of 3 × 106, and a maximum ion accumulation time of 246 ms. Precursor ions with charges from +2 to +7 were isolated with an m/z window of 2 and were sequentially fragmented by high energy dissociation (HCD) with a collision energy of 30%. The AGC target was set at 1 × 105. MS/MS scan spectra were acquired in the Orbitrap with an ion accumulation time of 502 ms and a resolution of 240,000 for 50-μm-diameter tissue samples, an ion accumulation time of 246 ms and 120,000 resolution for 100-μm-diameter tissue samples, and an ion accumulation time of 118 ms and 60,000 resolution for 200-μm-diameter tissue samples.
Data Analysis
Raw data were analyzed by MaxQuant 1.5.3.30 as described previously (31). Briefly, Andromeda engine was used to search MS/MS spectra against a UniProtKB/Swiss-Prot database for Rattus norvegicus, which contained a total of 31,816 entries with 8020 reviewed entries when downloaded on Jan 4, 2018. For WHIM2 patient-derived xenograft samples, the raw files were searched against a combined UniProtKB/Swiss-Prot database containing 20,129 reviewed homo sapiens entries (Dec 29, 2016) and 16,844 Mus musculus entries (Jan 28, 2017). Trypsin was specified for protein digestion with two missed cleavages allowed for each peptide. Carbamidomethylation was set as a fixed modification, and n-terminal protein acetylation and methionine oxidation were set as variable modifications. Recalibrated MS/MS spectra were matched with a tolerance of 5 ppm on precursor mass and 20 ppm on fragment mass. The minimum peptide length was set at 6 amino acids, and maximum peptide mass was 4600 Da. A false discovery rate (FDR) of 1% was applied for both peptide and protein filtering. For the spatially resolved study of brain tissue samples, Match Between Runs (MBR) was activated to enhance identification sensitivity. The time windows for feature alignment and matching were 20 min, and 0.7 min, respectively. Label-free quantification (LFQ) was performed in each parameter group containing tissue samples of similar size.
Contamination and reverse identifications were filtered with Perseus (version 1.5.6.0) (32). The results were exported to a table and visualized with OriginPro 2017 and an online tool powered by R language (http://www.omicsolution.org/wu-kong-beta-linux/main/). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (33) partner repository with the dataset identifier PXD008844 and 10.6019/PXD008844.
Experimental Design and Statistical Rationale
Both DMSO evaluation and sensitivity evaluation experiments were repeated in triplicate to generate three biological replicates (supplemental Table S1 and S2). To reduce sample heterogeneity, all tissues were dissected from the same region. Full injection without technical replicates was employed to improve analytical sensitivity for each small tissue sample. For spatially resolved quantitative proteome mapping experiments, four biological replicates were used for each tissue region (supplemental Table S3). For relative quantification, the LFQ intensities were transformed with log2 function and then filtered to contain >70% valid values in at least one group. The missing values were imputed by normal distribution in each column with a width of 0.5 and a down shift of 2. To identify significant differences, ANOVA multiple sample testing with permutation-based FDR control was used. p value <0.01, q-value <0.05, and fold change >4 (S0 = 2) were required to obtain significant proteins.
RESULTS
DMSO as Tissue Capture Liquid
Following dissection in a conventional LCM system, tissue pieces may be collected into microtubes by gravity or catapulted into tube caps prefilled with extraction solution or an adhesive coating, depending on the instrument vendor and configuration. However, these collection approaches cannot be adapted to the nanoPOTS system because the rapid evaporation of nanoliter-scale extraction solution and the prohibitive absorptive losses of proteins onto adhesive silicone coatings (34). To address this challenge, we prepopulated nanowells in the nanoPOTS chip with DMSO droplets to serve as a sacrificial capture medium for small tissue samples (Fig. 1A). This approach has several attractive merits. First, DMSO has a very low vapor pressure (0.8 mbar at room temperature) and evaporates slowly under ambient conditions, which allows for long working times and uninterrupted sample collection. As shown in Fig. 2A, the evaporation times of 100–300 nL DMSO droplets were 194 min to 416 min, which were >50 times longer than for water droplets in our laboratory conditions. Such prolonged times are sufficient to collect up to hundreds of tissue samples in each chip. Second, DMSO can be completely removed by gentle heating or vacuum, eliminating any possible interference during subsequent sample processing and analysis steps. Third, compared with other low-vapor-pressure solvents such as dimethylformamide (DMF), DMSO has lower toxicity, and is thus widely used as a storage solvent for drug candidates and cells. Fourth, the freezing point of DMSO is 18.5 °C, which should facilitate chip and sample transfer between histology and analytical labs without the risk of sample mixing or losses during shipping. Finally, during this study we have found that DMSO significantly increases the sensitivity of protein identification in brain tissues, which we ascribe to improved protein extraction efficiency as discussed below.
Fig. 2.
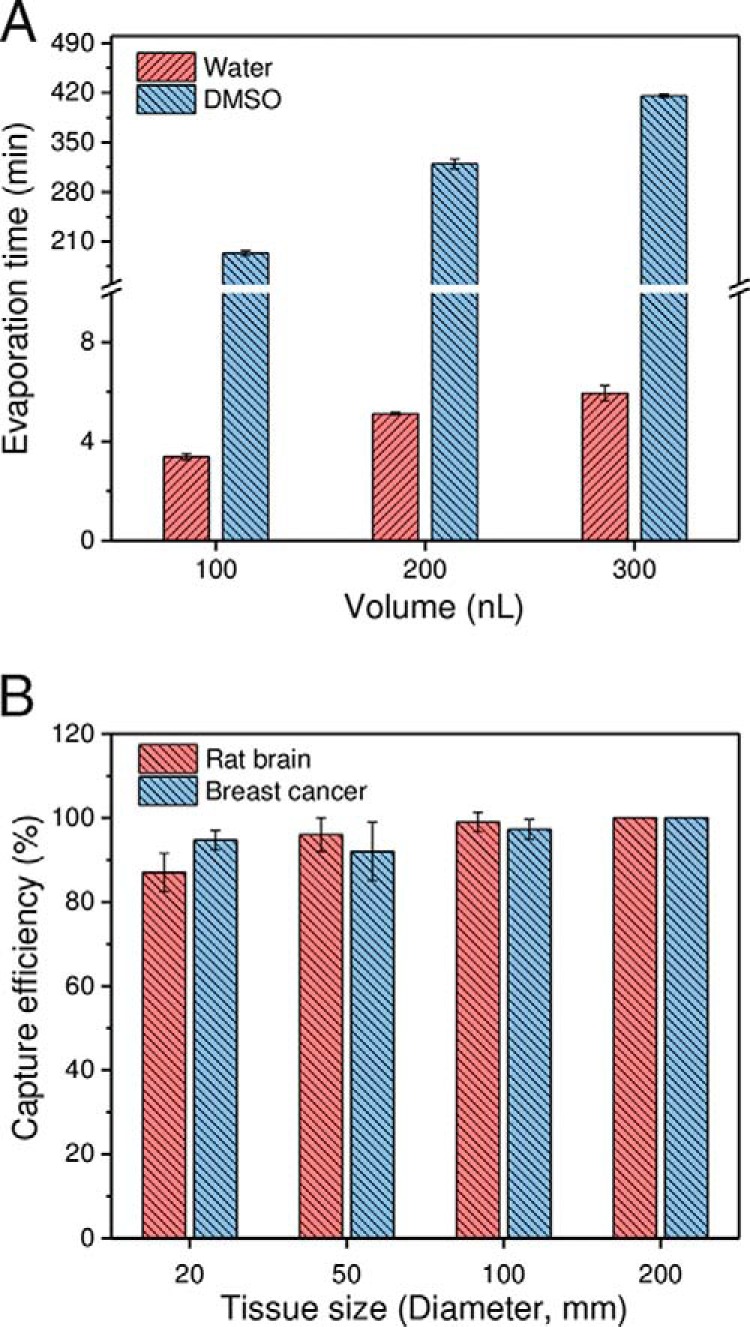
A, comparison of evaporation times for water and DMSO droplets. n = 5 for each condition. B, Evaluation of the capture efficiency of LCM tissue samples using DMSO droplets. A patient-derived xenograft and a rat brain section (12 μm thick) were used as model samples. The replicate numbers were 75, 75, 75, and 27 for tissues having lateral dimensions of 20 μm, 50 μm, 100 μm, and 200 μm, respectively. 200 nL DMSO droplets pre-deposited in nanowells with a diameter of 1.2 mm were used for tissue collection.
We first evaluated the capture efficiency with square tissues having side lengths of 20 μm, 50 μm, 100 μm, and 200 μm using both rat brain and breast cancer tissue sections with a thickness of 12-μm as model samples (35). For smaller tissue samples with square side lengths from 20 μm to 100 μm, a total of 75 cuts for each size were collected. For the 200 μm tissue samples, a total of 27 cuts were collected. The “CenterRoboLPC” function, in which the catapult laser pulse was applied at the centroid of pre-cut tissue piece, was used instead of commonly-used “RoboLPC.” We observed that the “CenterRoboLPC” function provided better control on the catapult trajectory of tissue pieces from slide to DMSO droplets. Under optimized conditions, the capture efficiencies ranged from 87% to 99% for smaller tissue samples (20 μm to 100 μm), indicating most LCM tissues can be collected (Fig. 2B). When tissue diameters were equal to or larger than 200 μm, all were successfully collected. With the increase of tissue sizes, the dissection time increased from 6 s to 15 s per tissue sample, which is still much faster than the manual transfer method (26). The high-speed dissection and high capture efficiencies, along with batch sample processing should enable many applications requiring high-throughput proteomic studies such as large-scale mapping of heterogeneous tissues. It should also be noted that tissue pieces with a diameter of 20 μm correspond to nearly single cells in most mammalian tissues, demonstrating the potential of the present approach for single-cell isolation and analysis.
Proteomic Analysis of LCM-isolated Rat Brain and WHIM2 Patient-derived Xenograft Tissues
To determine whether DMSO adversely affected tissue analysis, we analyzed rat cortex tissue samples collected with DMSO droplets and compared with those obtained using manual transfer without DMSO (supplemental Table S1). Surprisingly, we observed a 54 and 49% increase in average and total unique peptide identifications, respectively, resulting in a corresponding respective 33 and 26% increase in protein identifications, when DMSO was used for tissue collection (Fig. 3A). A Venn diagram of total protein identifications indicates that most of the proteins obtained from DMSO-free samples were included in DMSO-collected samples (Fig. 3B). This demonstrates that the use of DMSO droplets improves proteome coverage for small tissue samples without any negative effects on the proteomic analysis. A possible explanation for this is that DMSO increases overall protein extraction efficiency by disrupting the lipid content within tissues. Protein extraction from tissue samples was found to be more challenging than for cultured cells (36), especially for tissues with high lipid content such as brain. Various approaches have been developed to address this challenge by employing strong detergents (37) or organic solvents (38) in the extraction buffer. As an organic solvent, DMSO is expected to solubilize most lipids prior to protein extraction. Compared with commonly used detergent approaches, sample losses in detergent removal steps including buffer exchange (23, 37) and spin columns were avoided using our approach. These merits of DMSO have thus provided an added benefit in the workflow of spatially resolved proteomic analysis.
Fig. 3.

A–C, unique peptide (A) and protein (B) identifications for rat brain cortex tissue samples obtained by LCM followed by DMSO and DMSO-free sample collection. C, Venn diagram of total protein identifications. Tissue size: 200 μm in diameter and 12 μm in depth. D–F, Evaluation of the sensitivity of the LCM-nanoPOTS system in proteomic analysis of small rat cortex tissue samples. The relationship between tissue size and unique peptide (D) and protein (E) identifications, and (F) the overlap of total protein identifications in different sizes. G, Gene Ontology cellular component analysis of the 2098 proteins identified from 200-μm cortex tissues using the online tool DAVID (42). All peptide and protein identifications were based on MS/MS spectra with Match Between Runs disabled. Each condition was analyzed in triplicate.
We next tested the sensitivity of the LCM-nanoPOTS system with DMSO as capture solvent on proteomic analysis of small tissue samples. Circular regions from rat cortex tissue with diameters of 50 μm, 100 μm, and 200 μm were obtained as model samples. Based on hematoxylin staining of cell nuclei, the corresponding cell numbers were ∼2–6, 10–18, and 30–50 for the three different tissue diameters, respectively. Fig. 3D and 3E show the nearly linear increase of unique peptide and protein identifications with tissue size. As expected, almost all peptides and proteins identified in the smaller tissues were also identified in larger tissues (Fig. 3D), demonstrating that analytical sensitivity was the dominant factor in determining proteome coverage. The present system can identify an average of 180 ± 22, 695 ± 115, and 1827 ± 29 protein groups (n = 3) from cortex tissues with diameters of 50 μm, 100 μm, and 200 μm, respectively. With the same 4-fold increase in tissue area, we observed a 286% increase in protein identification of tissues with diameters from 50 to 100 μm, and a 163% increase from 100 to 200 μm, indicating that the overall sensitivity is a main limiting factor for proteome coverage at the low end of tissue areas. Compared with previous spatially resolved proteomic studies of brain tissues (17–20, 39–41) in which at least 0.65 mm-sized tissues were required to obtain a coverage of >1000 proteins, the LCM-nanoPOTS system provided >9 times improved spatial resolution with improved proteome coverage.
The total 2098 proteins identified from 200-μm-diameter cortex tissues were submitted for Gene Ontology cellular component (GOCC) analysis (42). As shown in Fig. 3G, we observed a high percentage (27.1%) of annotated membrane proteins, suggesting robust coverage of membrane proteins in our workflow. It is interesting to note that we observed 6.5% dendrite proteins and 5.8% axon proteins (Fig. 3G), which are vital for brain function. In brain, glutamate and GABA are two major neurotransmitters, which play excitatory and inhibitory functions, respectively. Interestingly, we identified three types of GABA receptors (GABRA1, GABRA2, GABRB2, GABRB2, and GABRG2) and a large family of glutamate receptors including GRIA1, GRIA2, GRIA3, GRM2, GRM3, GRM5, GRIN1, GRIN2a, and GRIN2b, all of which are transmembrane proteins.
We also tested the proteome coverage of WHIM2 patient-derived xenograft tissues with side lengths of 200 μm. An average of 10,345 and total 16,210 unique peptides, corresponding to an average of 2325 and total 2613 protein groups were identified in experimental triplicate. The average protein groups decreased to 1719 when 2 peptides were required for identification (supplemental Table S2). A 27% increase in protein identifications over those of brain cortex tissue samples was observed, which could be attributed to the fact that more cells/proteins were contained in the breast cancer tissues.
Spatially Resolved Quantitative Proteomic Analysis of Rat Brain Tissue
To evaluate the potential utility of the LCM-nanoPOTS system for spatially resolved quantitative proteomic tissue analysis, we performed a proof-of-concept experiment by dissecting and analyzing three different rat brain regions (cerebral cortex (CTX), corpus callosum (CC), and caudoputamen (CP)) from a 12-μm-thick coronal section (Fig. 4). Tissue samples were dissected as circular regions with a diameter of 100 μm, corresponding to an area of ∼0.008 mm2. The spatial distances (center to center) were from 116 μm to 716 μm within the same brain region, and from 424 μm to 1727 μm across different brain regions (Fig. 4A). For each region, four samples were processed and analyzed by LC-MS/MS (Fig. 4B).
Fig. 4.
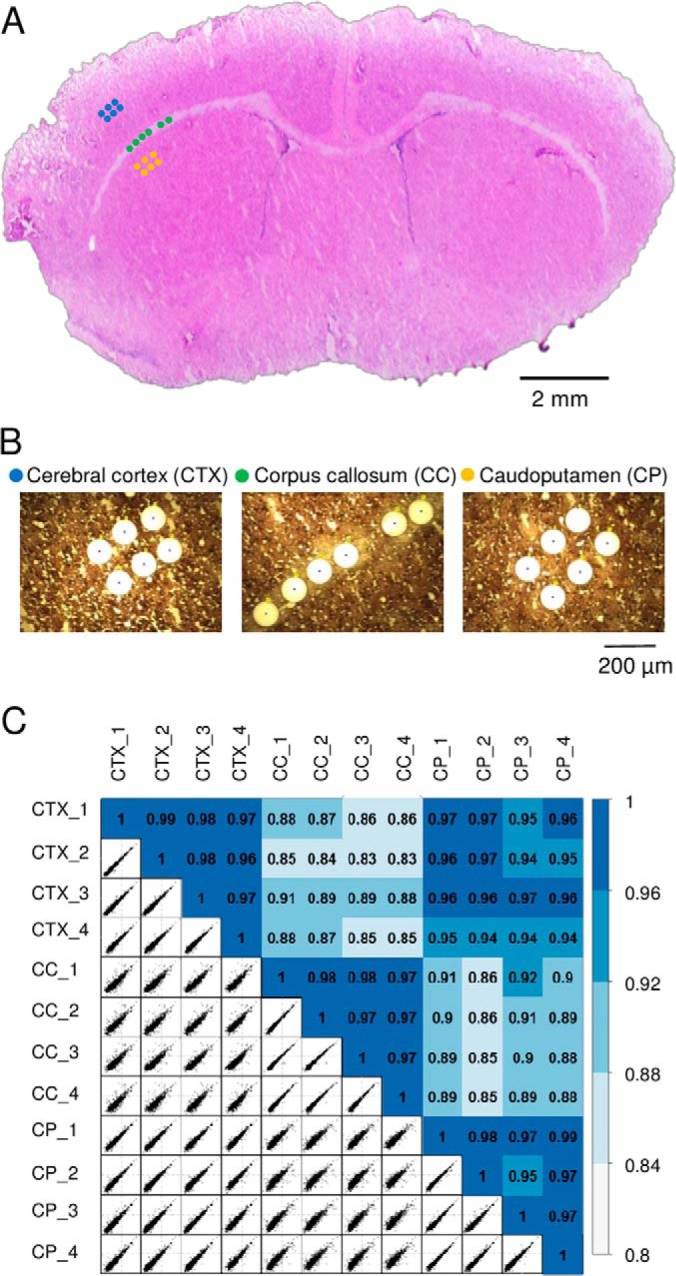
A, the 12-μm-thick rat brain coronal section used in the study. Three distinct regions including cerebral cortex (CTX), corpus callosum (CC), and caudoputamen (CP) were dissected with a spatial resolution of 100 μm in diameter. B, The corresponding microscopic images of the tissue regions after dissection. C, Pairwise correlation plots with log2-transformed LFQ intensities between 12 tissue samples from the three regions. The color codes indicate the relatively high correlations between the same tissue regions and relatively low correlations between different regions.
To increase the number of quantifiable proteins, we used the Match Between Runs (MBR) algorithm of MaxQuant (13, 31) wherein the peptides were identified based on accurate masses and LC retention times, similar to the AMT concept originally reported by our group (43, 44). A total of 2059 proteins were identified and 1484 (72.1%) were common across all the three brain regions. After stringent filtering for valid log2-transformed LFQ values, 1103 protein groups were quantifiable (supplemental Table S3). As expected, relatively high correlation coefficients from 0.95 to 0.99 were observed between biological replicates of the same brain regions (Fig. 4C). Between different tissue regions, CTX and CP displayed lower correlation coefficients from 0.94 to 0.97, whereas CC had lowest correlations (from 0.83 to 0.92) with the other two regions. Such differences are also consistent with the morphology of the brain tissue (Fig. 4A).
Next, we examined whether the quantitative proteome data obtained from the LCM-nanoPOTS system could clearly differentiate the three brain tissue regions. We used unsupervised principal component analysis (PCA) to process the LFQ intensity data from the 12 samples. As shown in Fig. 5A, the three tissue regions were clearly segregated based on component 1 and component 2, which accounted for 38.5% and 13.8%, respectively. All four biological replicates were well clustered within the corresponding tissue region without overlap with other regions, suggesting the present system can effectively distinguish differences in cell or tissue types based on their protein expression levels.
Fig. 5.
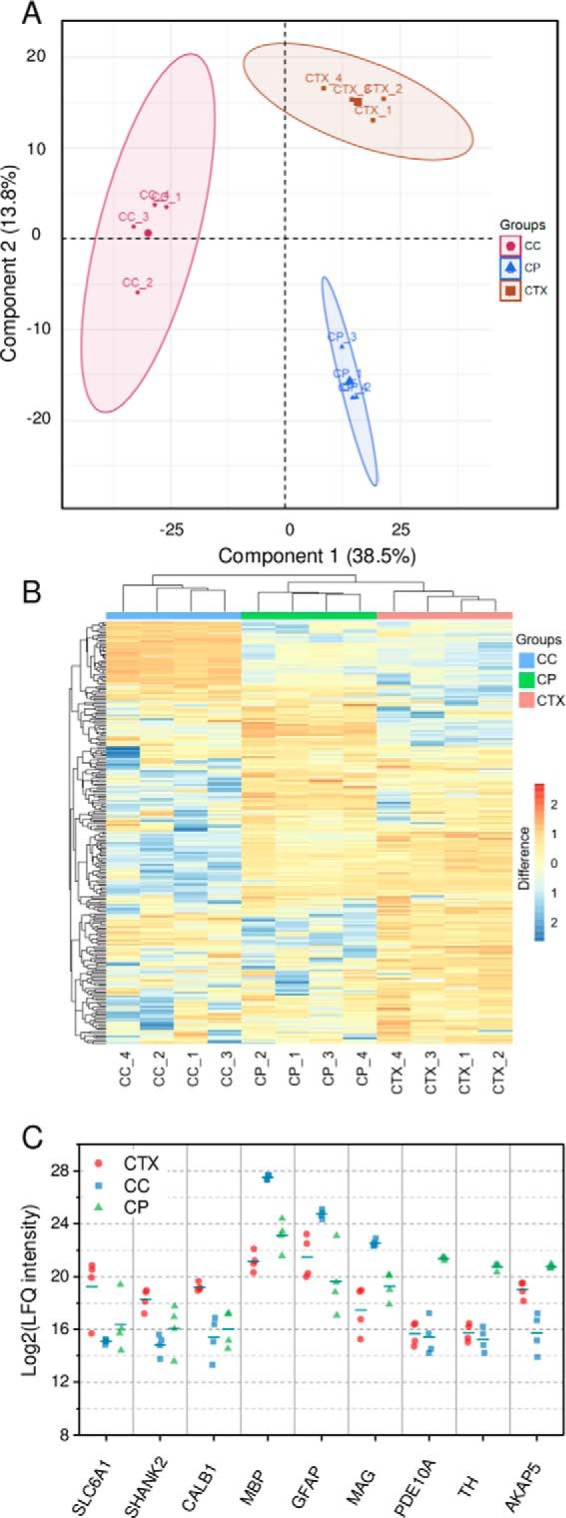
A, principle component analysis of protein expression in CTX, CC, and CP regions of rat brain section as shown in Fig. 4. B, Hierarchical clustering analysis (HCA) of the significant proteins. C, A panel of selected differentially expressed proteins among the three regions.
To identify differentially expressed proteins among the three tissue regions, we employed ANOVA test with permutation-based FDR algorithm using the Perseus data analysis platform (32). Using a fold-change of 4 and an FDR level of 0.05, 244 out of 1103 total quantifiable protein groups were revealed to have significant differences in expression among the three regions (supplemental Table S3). The most abundant proteins, such as Tuba1b, Tubb2a, Actb, Sptan1, Cltc, and Atp5b, were found to have no difference in LFQ intensity, which agrees well with previous findings (45). For the 244 significant proteins, we observed 67, 34, and 70 proteins groups enriched in CTX, CC, and CP regions, respectively, with a log2 abundance ratio >2 over the mean value for each protein across all samples. To visualize the protein expression differences, we applied unsupervised hierarchical clustering analysis (HCA) on the significant proteins with mean-centered log2 transformed abundances (Fig. 5B). Like the PCA plot, each of four replicates from the same region was clustered together. In addition, each region has a distinct cluster of proteins with higher abundances relative to other regions, indicating different biological functions for each brain region.
Fig. 5C shows a panel of selected differentially expressed proteins in each tissue region. Sodium- and chloride-dependent GABA transporter 1 (SLC6A1) was enriched in CTX region. SLC6A1 removes GABA from the synaptic cleft, which was found to have an important role in the development of neurological disorders such as Schizophrenia. SH3 and multiple ankyrin repeat domains protein 2 (SHANK2) belongs to the Shank family of synaptic proteins, which function as molecular scaffolds in the postsynaptic density. Calbindin 1 (CALB1) was observed to be highly expressed in CTX compared with the other regions. CALB1 has the function to buffer entry of calcium upon stimulation of glutamate receptors and the loss of CALB1 protein is found in patients with Huntington disease (46). In CC, several myelin-related proteins (MBP, PLP1, MAG, and MOG) were highly expressed. The myelination of CC axons is highly active during the development of mouse brain (47), which agrees well with the age of the rat model (17 days old). In another recent report, MBP was clearly shown to be highly expressed in the corpus callosum (48). In the CP region, we observed the elevated expression of two phosphodiesterases (PDE1B and PDE10A). PDE10A is well-studied dual-substrate phosphodiesterase capable of hydrolyzing cAMP and cGMP. A previous immunohistochemical study has indicated its high expression in the CP region of rat brain (49). We also observed the high expression of two phosphatase 1 regulation proteins (PPP1R1B and PPP1R9A), tyrosine 3-monooxygenase (TH), and a-kinase anchor protein 5 (AKAP5).
DISCUSSION
Spatially resolved proteome mapping of healthy or pathological tissue sections can provide important insights into physiology or pathophysiology. The ability to map the proteome (the abundances of thousands of proteins) with relatively high spatial resolution across tissue regions provides a fundamental way to understand tissue microenvironment, substructure, and cellular organization from a global proteome perspective. Moreover, this approach can easily be integrated with other spatially resolved measurement methods such as immunohistochemistry staining, FISH, spatial transcriptomics (4), and MALDI imaging (5) to provide a more complete molecular atlas of tissue sections.
The described LCM-nanoPOTS platform significantly advances spatially resolved proteomics by improving the spatial resolution and increasing the sensitivity. The use of DMSO droplets not only served to efficiently capture dissected tissue pieces as small as 20 μm in diameter (single-cell scale) into nanowells, but also significantly improved the proteome coverage. The whole workflow can be fully automated without manual transfer, and thus sample loss and protein contamination are minimized. The extension to formalin-fixed paraffin-embedded (FFPE) tissues could potentially be achieved by incorporating paraffin removing and antigen retrieval steps after sample collection. We anticipate this platform will play an important role in clinical tissue characterization at the proteome level. The technology should also be broadly applicable to other research areas such as microbial communities and plant science. With further advances of LC-MS sensitivity, we anticipate proteome mapping at single cell resolution will be achievable. Finally, the LCM-nanoPOTS platform should be readily extended to other omics studies requiring tissue isolation and nanoscale processing, such as transcriptomics, lipidomics, and metabolomics.
Compared with IHC staining and MALDI-MS imaging, the bottom-up proteomics approach is low in throughput, as each LC-MS analysis requires at least 1 h. This will limit the capacity for comprehensive mapping of whole tissue sections at high resolution. Nevertheless, the LCM-nanoPOTS platform is unique for potentially obtaining spatially resolved mechanistic insights of tissue pathology because of the depth of proteome coverage. Further improvement in throughput could be achieved by using multiple LC/MS systems (50), employing sample multiplexing based on isobaric chemical barcoding (25), or potentially by using rapid and high-resolution ion mobility separations in place of LC (51, 52). Alternatively, to mitigate this throughput limitation, well-developed staining and MALDI imaging techniques (41) can be combined with the LCM-nanoPOTS platform by guiding pathological regions and tissue substructures for in-depth proteome mapping.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (33) partner repository with the dataset identifier PXD008844 and 10.6019/PXD008844.
Supplementary Material
Footnotes
* This research was supported by the Genomic Science Program of the U.S. DOE-OBER and is a contribution of the PNNL Foundational Scientific Focus Area. This work was also supported by NIH grants R21EB020976, R33CA225248, P41GM103493, UC4DK104167, and DP3DK110844. This research was performed using EMSL, a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at PNNL.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- LCM
- laser capture microdissection
- nanoPOTS
- nanodroplet processing in one pot for trace samples
- DMSO
- dimethyl sulfoxide
- DDM
- n-dodecyl-β-D-maltoside
- GABA
- γ-aminobutyric acid
- CTX
- cerebral cortex
- CC
- corpus callosum
- CP
- caudoputamen.
REFERENCES
- 1. Satija R., Farrell J. A., Gennert D., Schier A. F., and Regev A. (2015) Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crosetto N., Bienko M., and van Oudenaarden A. (2014) Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 16, 57–66 [DOI] [PubMed] [Google Scholar]
- 3. Lein E., Borm L. E., and Linnarsson S. (2017) The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 [DOI] [PubMed] [Google Scholar]
- 4. Ståhl P. L., Salmén F., Vickovic S., Lundmark A., Navarro J. F., Magnusson J., Giacomello S., Asp M., Westholm J. O., Huss M., Mollbrink A., Linnarsson S., Codeluppi S., Borg Å Pontén F, Costea P. I., Sahlén P., Mulder J., Bergmann O., Lundeberg J., and Frisén J. (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 [DOI] [PubMed] [Google Scholar]
- 5. Van de Plas R., Yang J., Spraggins J., and Caprioli R. M. (2015) Image fusion of mass spectrometry and microscopy: a multimodality paradigm for molecular tissue mapping. Nat. Methods 12, 366–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwamborn K., and Caprioli R. M. (2010) Molecular imaging by mass spectrometry — looking beyond classical histology. Nat. Rev. Cancer 10, 639–646 [DOI] [PubMed] [Google Scholar]
- 7. Laskin J., and Lanekoff I. (2016) Ambient mass spectrometry imaging using direct liquid extraction techniques. Anal. Chem. 88, 52–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wiseman J. M., Ifa D. R., Song Q., and Cooks R. G. (2006) Tissue imaging at atmospheric pressure using Desorption Electrospray Ionization (DESI) mass spectrometry. Angew. Chemie - Int. Ed. 45, 7188–7192 [DOI] [PubMed] [Google Scholar]
- 9. Mann M., Kulak N. A., Nagaraj N., and Cox J. (2013) the coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 49, 583–590 [DOI] [PubMed] [Google Scholar]
- 10. Aebersold R., and Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 [DOI] [PubMed] [Google Scholar]
- 11. Wilm M., and Mann M. (1996) Analytical properties of the nanoelectrospray ion source. Anal. Chem. 68, 1–8 [DOI] [PubMed] [Google Scholar]
- 12. Sun L., Zhu G., Zhao Y., Yan X., Mou S., and Dovichi N. J. (2013) Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew. Chemie - Int. Ed. 52, 13661–13664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhu Y., Zhao R., Piehowski P. D., Moore R. J., Lim S., Orphan V. J., Paša-Tolić L., Qian W. J., Smith R. D., and Kelly R. T. (2018) Subnanogram proteomics: Impact of LC column selection, MS instrumentation and data analysis strategy on proteome coverage for trace samples. Int. J. Mass Spectrom. 427, 4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kelly R. T., Tolmachev A. V., Page J. S., Tang K., and Smith R. D. (2010) The ion funnel: Theory, implementations, and applications. Mass Spectrom. Rev. 29, 294–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li S., Plouffe B. D., Belov A. M., Ray S., Wang X., Murthy S. K., Karger B. L., and Ivanov A. R. (2015) An integrated platform for isolation, processing, and mass spectrometry-based proteomic profiling of rare cells in whole blood. Mol. Cell. Proteomics 14, 1672–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harris G. A., Nicklay J. J., and Caprioli R. M. (2013) Localized in situ hydrogel-mediated protein digestion and extraction technique for on-tissue analysis. Anal. Chem. 85, 2717–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rizzo D. G., Prentice B. M., Moore J. L., Norris J. L., and Caprioli R. M. (2017) Enhanced spatially resolved proteomics using on-tissue hydrogel-mediated protein digestion. Anal. Chem. 89, 2948–2955 [DOI] [PubMed] [Google Scholar]
- 18. Wisztorski M., Desmons A., Quanico J., Fatou B., Gimeno J. P., Franck J., Salzet M., and Fournier I. (2016) Spatially-resolved protein surface microsampling from tissue sections using liquid extraction surface analysis. Proteomics 16, 1622–1632 [DOI] [PubMed] [Google Scholar]
- 19. Quanico J., Franck J., Dauly C., Strupat K., Dupuy J., Day R., Salzet M., Fournier I., and Wisztorski M. (2013) Development of liquid microjunction extraction strategy for improving protein identification from tissue sections. J. Proteomics 79, 200–218 [DOI] [PubMed] [Google Scholar]
- 20. Quanico J., Franck J., Cardon T., Leblanc E., Wisztorski M., Salzet M., and Fournier I. (2017) NanoLC-MS coupling of liquid microjunction microextraction for on-tissue proteomic analysis. Biochim. Biophys. Acta -Proteins Proteomics 1865, 891–900 [DOI] [PubMed] [Google Scholar]
- 21. Wisztorski M., Quanico J., Franck J., Fatou B., Salzet M., and Fournier I. (2017) in Methods in Molecular Biology (Humana Press, New York, NY: ), pp 49–63 [DOI] [PubMed] [Google Scholar]
- 22. Cahill J. F., Kertesz V., Weiskittel T. M., Vavrek M., Freddo C., and Van Berkel G. J. (2016) Online, absolute quantitation of propranolol from spatially distinct 20- and 40-μm dissections of brain, liver, and kidney thin tissue sections by laser microdissection-liquid vortex capture-mass spectrometry. Anal. Chem. 88, 6026–6034 [DOI] [PubMed] [Google Scholar]
- 23. Wiśniewski J. R., Ostasiewicz P., and Mann M. (2011) High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J. Proteome Res. 10, 3040–3049 [DOI] [PubMed] [Google Scholar]
- 24. Clair G., Piehowski P. D., Nicola T., Kitzmiller J. A., Huang E. L., Zink E. M., Sontag R. L., Orton D. J., Moore R. J., Carson J. P., Smith R. D., Whitsett J. A., Corley R. A., Ambalavanan N., and Ansong C. (2016) Spatially-resolved proteomics: rapid quantitative analysis of laser capture microdissected alveolar tissue samples. Sci. Rep. 6, 39223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Graaf E. L., Pellegrini D., and McDonnell L. A. (2016) Set of novel automated quantitative microproteomics protocols for small sample amounts and its application to kidney tissue substructures. J. Proteome Res. 15, 4722–4730 [DOI] [PubMed] [Google Scholar]
- 26. Zhu Y., Piehowski P. D., Zhao R., Chen J., Shen Y., Moore R. J., Shukla A. K., Vladislav P. A., Campbell-Thompson M. L., Mathews C. E., Smith R. D., Qian W., and Kelly R. T. (2018) Nanodroplet processing platform for deep and quantitative proteome profiling of 10–100 mammalian cells. Nat. Commun. 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhu Y., Zhang Y. X., Cai L. F., and Fang Q. (2013) Sequential operation droplet array: An automated microfluidic platform for picoliter-scale liquid handling, analysis, and screening. Anal. Chem. 85, 6723–6731 [DOI] [PubMed] [Google Scholar]
- 28. Zhu Y., Zhang Y.-X., Liu W.-W., Ma Y., Fang Q., and Yao B. (2015) Printing 2-dimentional droplet array for single-cell reverse transcription quantitative PCR assay with a microfluidic robot. Sci. Rep. 5, 9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shen Y., Moore R. J., Zhao R., Blonder J., Auberry D. L., Masselon C., Paša-Tolić L., Hixson K. K., Auberry K. J., and Smith R. D. (2003) High-efficiency on-line solid-phase extraction coupling to 15–150-μm-i.d. Column liquid chromatography for proteomic analysis. Anal. Chem. 75, 3596–3605 [DOI] [PubMed] [Google Scholar]
- 30. Shen Y., Tolić N., Masselon C., Paša-Tolić L., Camp D. G., Hixson K. K., Zhao R., Anderson G. A., and Smith R. D. (2004) Ultrasensitive proteomics using high-efficiency on-line micro-SPE-nanoLC-nanoESI MS and MS/MS. Anal. Chem. 76, 144–154 [DOI] [PubMed] [Google Scholar]
- 31. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 32. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 33. Vizcaíno J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yu X., Xiao J., and Dang F. (2015) Surface modification of poly(dimethylsiloxane) using ionic complementary peptides to minimize nonspecific protein adsorption. Langmuir 31, 5891–5898 [DOI] [PubMed] [Google Scholar]
- 35. Li S., Shen D., Shao J., Crowder R., Liu W., Prat A., He X., Liu S., Hoog J., Lu C., Ding L., Griffith O. L., Miller C., Larson D., Fulton R. S., Harrison M., Mooney T., McMichael J. F., Luo J., Tao Y., Goncalves R., Schlosberg C., Hiken J. F., Saied L., Sanchez C., Giuntoli T., Bumb C., Cooper C., Kitchens R. T., Lin A., Phommaly C., Davies S. R., Zhang J., Kavuri M. S., McEachern D., Dong Y. Y., Ma C., Pluard T., Naughton M., Bose R., Suresh R., McDowell R., Michel L., Aft R., Gillanders W., DeSchryver K., Wilson R. K., Wang S., Mills G. B., Gonzalez-Angulo A., Edwards J. R., Maher C., Perou C. M., Mardis E. R., and Ellis M. J. (2013) Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4, 1116–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shevchenko G., Musunuri S., Wetterhall M., and Bergquist J. (2012) Comparison of extraction methods for the comprehensive analysis of mouse brain proteome using shotgun-based mass spectrometry. J. Proteome Res. 11, 2441–2451 [DOI] [PubMed] [Google Scholar]
- 37. Wiśniewski J. R., Zougman A., Nagaraj N., and Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 38. Dapic I., Uwugiaren N., Jansen P. J., and Corthals G. L. (2017) Fast and simple protocols for mass spectrometry-based proteomics of small fresh frozen uterine tissue sections. Anal. Chem. 89, 10769–10775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Donnarumma F., and Murray K. K. (2016) Laser ablation sample transfer for localized LC-MS/MS proteomic analysis of tissue. J. Mass Spectrom. 51, 261–268 [DOI] [PubMed] [Google Scholar]
- 40. Quanico J., Franck J., Wisztorski M., Salzet M., and Fournier I. (2017) Integrated mass spectrometry imaging and omics workflows on the same tissue section using grid-aided, parafilm-assisted microdissection. Biochim. Biophys. Acta - Gen. Subj. 1861, 1702–1714 [DOI] [PubMed] [Google Scholar]
- 41. Dilillo M., Pellegrini D., Ait-Belkacem R., De Graaf E. L., Caleo M., and McDonnell L. A. (2017) Mass spectrometry imaging, laser capture microdissection, and LC-MS/MS of the same tissue section. J. Proteome Res. 16, 2993–3001 [DOI] [PubMed] [Google Scholar]
- 42. Huang D. W., Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 43. Conrads T. P., Anderson G. A., Veenstra T. D., Paša-Tolić L., and Smith R. D. (2000) Utility of accurate mass tags for proteome-wide protein identification. Anal. Chem. 72, 3349–3354 [DOI] [PubMed] [Google Scholar]
- 44. Zimmer J. S. D., Monroe M. E., Qian W. J., and Smith R. D. (2006) Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom. Rev. 25, 450–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma K., Schmitt S., Bergner C. G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., Kongi K., Cantuti L., Hanisch U. K., Philips M. A., Rossner M. J., Mann M., and Simons M. (2015) Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kiyama H., Seto-Ohshima A., and Emson P. C. (1990) Calbindin D28K as a marker for the degeneration of the striatonigral pathway in Huntington's disease. Brain Res. 525, 209–214 [DOI] [PubMed] [Google Scholar]
- 47. Sturrock R. R. (1980) Myelination of the mouse corpus callosum. Neuropathol. Appl. Neurobiol. 6, 415–420 [DOI] [PubMed] [Google Scholar]
- 48. Kim K., Shin M.-S., Cho H.-S., and Kim Y.-P. (2014) Effects of endurance exercise on expressions of glial fibrillary acidic protein and myelin basic protein in developing rats with maternal infection-induced cerebral palsy. J. Exerc. Rehabil. 10, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seeger T. F., Bartlett B., Coskran T. M., Culp J. S., James L. C., Krull D. L., Lanfear J., Ryan A. M., Schmidt C. J., Strick C. A., Varghese A. H., Williams R. D., Wylie P. G., and Menniti F. S. (2003) Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 985, 113–126 [DOI] [PubMed] [Google Scholar]
- 50. Livesay E. A., Tang K., Taylor B. K., Buschbach M. A., Hopkins D. F., LaMarche B. L., Zhao R., Shen Y., Orton D. J., Moore R. J., Kelly R. T., Udseth H. R., and Smith R. D. (2008) Fully automated four-column capillary LC-MS system for maximizing throughput in proteomic analyses. Anal. Chem. 80, 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baker E. S., Burnum-Johnson K. E., Ibrahim Y. M., Orton D. J., Monroe M. E., Kelly R. T., Moore R. J., Zhang X., Théberge R., Costello C. E., and Smith R. D. (2015) Enhancing bottom-up and top-down proteomic measurements with ion mobility separations. Proteomics 15, 2766–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Deng L., Ibrahim Y. M., Hamid A. M., Garimella S. V. B., Webb I. K., Zheng X., Prost S. A., Sandoval J. A., Norheim R. V., Anderson G. A., Tolmachev A. V., Baker E. S., and Smith R. D. (2016) Ultra-high resolution ion mobility separations utilizing traveling waves in a 13 m serpentine path length structures for lossless ion manipulations module. Anal. Chem. 88, 8957–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (33) partner repository with the dataset identifier PXD008844 and 10.6019/PXD008844.