

Abstract
Adoptive cell therapy with chimeric antigen receptor (CAR)-redirected T cells induced spectacular regressions of leukemia and lymphoma, however, failed so far in the treatment of solid tumors. A cause is thought to be T cell repression through TGF-β, which is massively accumulating in the tumor tissue. Here, we show that T cells with a CD28-ζ CAR, but not with a 4-1BB-ζ CAR, resist TGF-β-mediated repression. Mechanistically, LCK activation and consequently IL-2 release and autocrine IL-2 receptor signaling mediated TGF-β resistance; deleting the LCK-binding motif in the CD28 CAR abolished both IL-2 secretion and TGF-β resistance, while IL-2 add-back restored TGF-β resistance. Other γ-cytokines like IL-7 and IL-15 could replace IL-2 in this context. This is demonstrated by engineering IL-2 deficient CD28ΔLCK-ζ CAR T cells with a hybrid IL-7 receptor to provide IL-2R β chain signaling upon IL-7 binding. Such modified T cells showed improved CAR T cell activity against TGF-β+ tumors. Data draw the concept that an autocrine loop resulting in IL-2R signaling can make CAR T cells more potent in staying active against TGF-β+ solid tumors.
Keywords: CAR, chimeric antigen receptor, T cell, immune suppression, TGF-β, τ, ν, μ, o, ρ
Graphical Abstract
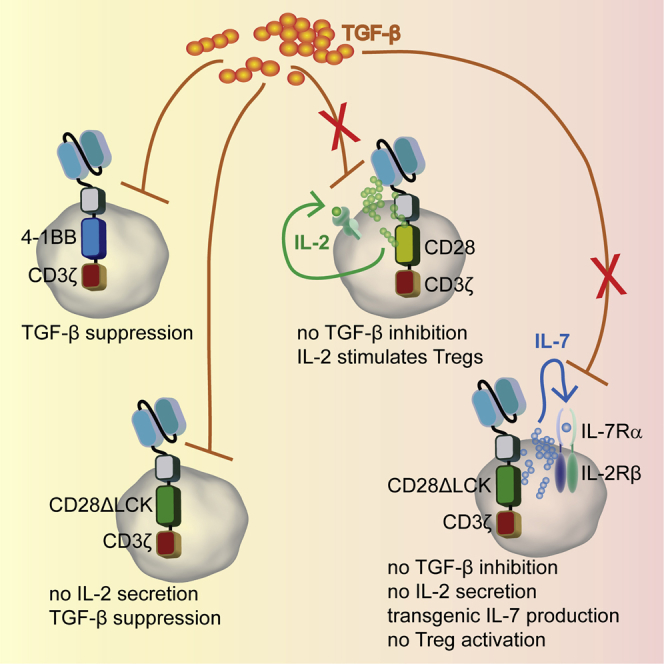
T cells with CD28 CAR, but not 4-1BB CAR, resist TGF-β suppression by IL-2 signaling, which can be replaced by IL-7. T cells, which are deficient in CAR-mediated IL-2 release and autocrine stimulated by transgenic IL-7, do not activate Treg cells, resist TGF-β suppression, and show superior tumor eradication.
Introduction
So far, adoptive cell therapy with chimeric antigen receptor (CAR)-modified T cells largely failed in the treatment of solid cancer while successful in the treatment of refractory leukemia and lymphoma.1 The reason for this is thought to be a hostile tumor environment that represses the immune cell response through multiple mechanisms such as suppressive cells and cell-bound and soluble factors, including repressive cytokines like transforming growth factor-β1 (TGF-β). The main sources of TGF-β in the tumor environment are cancer and stroma cells, both of which repress T cell amplification and finally prevent a lasting anti-tumor response.2 Tremendous efforts are currently made to shield CAR T cells against immune suppression; for instance, a transgenic dominant-negative (dn) form of the TGF-β receptor confers some resistance in a preclinical melanoma model3 and a model of Epstein-Barr virus (EBV)-positive Hodgkin’s lymphoma;4 the same dnTGF-β receptor was used to protect T cells in a recent trial.5 Resistance against suppression can also be provided by a hybrid receptor which binds interleukin-4 (IL-4) by the ectodomain and transmits an activating γ-cytokine signal through the IL-7 receptor α chain6 or the IL-2 and IL-15 receptor β chain.7
We previously showed that CAR T cell repression through TGF-β can be overcome by CD28 co-stimulation provided by the CD28-ζ CAR; in contrast, “first-generation” CD3ζ CAR T cells are suppressed by TGF-β.8 In order to define how CD28-ζ CAR T cells counteract TGF-β repression, we dissected the CD28 endodomain and identified LCK activation and subsequent IL-2 release and autocrine IL-2 receptor signaling crucial in counteracting TGF-β repression. By replacing IL-2 through other γ-cytokines and using a hybrid IL-2 receptor, we revealed that an engineered autocrine loop with IL-2 receptor signaling capacities can improve the anti-tumor efficacy of CAR T cells in a TGF-β+ environment independently of IL-2.
Results
CD28-ζ CAR T Cells, but Not 4-1BB-ζ CAR T Cells, Are Resistant to TGF-β-Mediated Suppression
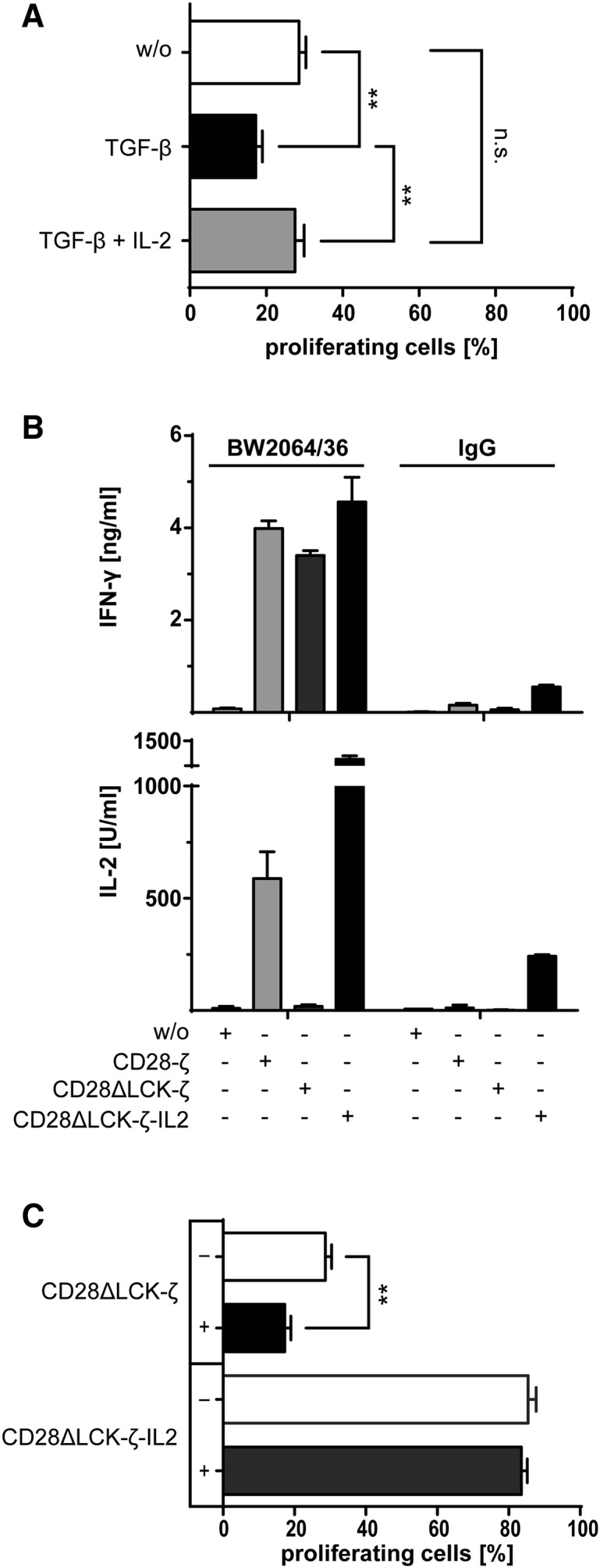
We recorded the impact of TGF-β on the CAR-triggered T cell response in a thoroughly controlled system. Human T cells were engineered with carcinoembryonic antigen (CEA)-specific CARs containing the CD3ζ (ζ), CD28-ζ, or 4-1BB-ζ endodomains, respectively (Figure S1), and activated through CAR signaling upon engaging immobilized cognate antigen, i.e., the BW2064/36 antibody, which is an anti-idiotypic antibody against the anti-CEA single-chain fragment of variable region (scFv) of the CAR. In the presence of TGF-β, T cell proliferation was suppressed when activated by the ζ CAR or the 4-1BB-ζ CAR. However, CD28-ζ CAR T cells resisted TGF-β-mediated suppression displaying similar proliferation rates as without TGF-β (Figure 1A). In the presence of TGF-β, interferon (IFN)-γ and IL-2 release by CAR T cells was decreased after antigen-based activation (Figure 1B), while the cytolytic activity toward CEA+ LS174T target cells was not substantially altered (Figure 1C). Overall, CD28-ζ CAR T cells resisted TGF-β suppression, whereas the amplification of T cells with the ζ and the 4-1BB-ζ CAR was suppressed.
Figure 1.

The CD28-ζ CAR, but Not the 4-1BB-ζ CAR, Overcomes the TGF-β-Mediated Repression in T Cell Amplification
(A) Human peripheral blood T cells were engineered with the anti-CEA CAR with ζ, 4-1BB-ζ or CD28-ζ signaling domain, respectively, and labeled with CFSE. CAR T cells (2.5 × 104 T cells per well) were incubated for 4 days on 96-well plates coated with the anti-idiotypic mAb BW2064/36 (1.5 μg/mL coating concentration), which served as surrogate antigen and is directed against the anti-CEA scFv binding domain of the CAR. Cells were incubated in the presence or absence of TGF-β (10 ng/mL). CAR T cell amplification was recorded by flow cytometric recording of CFSE dilution, and CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody, which binds to the extracellular IgG1 spacer of the CAR. (B) CAR T cells with different signaling domains were incubated on plates (2.5 × 104 CAR T cells per well) coated with the anti-idiotypic mAb BW2064/36 or mouse IgG1 as isotype control (1.5 μg/mL each). T cells without CAR (w/o) served as control. The cells were incubated in the presence or absence of TGF-β (10 ng/mL). IFN-γ and IL-2 secreted into the culture supernatants were recorded by ELISA. (C) T cells with or without CAR (0.125 − 4 × 104 CAR T cells per well) were incubated with CEA+ LS174T cells (2 × 104 cells per well) in the presence or absence of TGF-β (10 ng/mL). The specific cytotoxicity after 2 days was determined by the XTT-based viability assay. Data represent the mean of triplicates ± SD. The assays were repeated three times; a representative assay is shown. Statistical analyses were performed using a two-tailed Student’s t test (*p < 0.05).
LCK Activation and Released IL-2 Mediate Resistance of CD28-ζ CAR T Cells toward TGF-β
In order to identify the CD28-ζ CAR signal responsible for TGF-β resistance, we modified the LCK- or PI3K-activating motif or both in the CD28 endodomain to ablate CAR-induced LCK and PI3K signaling, respectively9 (Figure 2A). CARs with the respective CD28 mutants were as equally expressed by T cells as the wild-type CAR (Figure S1). Upon antigen engagement, CD28-ζ CAR T cells displayed increased LCK phosphorylation compared with T cells without CAR; CD28ΔLCK-ζ CAR T cells showed abrogated LCK phosphorylation (Figure 2B). LCK is required for IL-2 release by T cells;10 CD28ΔLCK-ζ CAR T cells were accordingly deficient in IL-2 release upon antigen-based CAR stimulation, while the IFN-γ release was not altered (Figure 2C). Of note, CD28ΔPI3K-ζ CAR T cells with a mutated phosphatidylinositol 3-kinase (PI3K)-binding domain secreted both IL-2 and IFN-γ at similar levels as unmodified CD28-ζ CAR T cells upon antigen engagement.
Figure 2.

Resistance of CD28-ζ CAR T Cells to TGF-β Repression Requires LCK, but Not PI3K Activation
(A) The wild-type and mutated CD28 signaling domain with the PI3K and LCK binding sides. Proline at the P187 and P190 positions were substituted by alanine (A) to destroy the LCK binding side, and tyrosine Y170 was replaced by phenylalanine F170 to eliminate the PI3K binding side. (B) Mutation of the LCK binding side in the CD28 signaling moiety results in less phosphorylated LCK (pLCK) upon CAR signaling. T cells were engineered with the CD28-ζ or the CD28ΔLCK-ζ CAR; T cells without CAR (w/o) served as control. CAR T cells (106 per well) were incubated for 5 min on 96-well plates coated with the anti-idiotypic mAb BW2064/36 (1.5 μg/mL) as surrogate antigen, fixed, permeabilized, and stained with the PE-conjugated anti-phospho-LCK antibody. The cells were analyzed by flow cytometry. The number of pLCK+ cells (%) is shown. (C) Deletion of the LCK binding side abrogates the CAR-induced secretion of IL-2, but not of IFN-γ. T cells with or without (w/o) CAR were incubated on plates (2.5 × 104 CAR T cells per well) coated with anti-idiotypic mAb BW2064/36 or mouse IgG1 (1.5 μg/mL each) as an isotype control. IFN-γ and IL-2 in culture supernatants were recorded by ELISA. (D) T cells were engineered with the respective CAR, labeled with CFSE and incubated (2.5 × 104 T cells per well) with or without TGF-β (10 ng/mL) for 4 days on 96-well plates coated with anti-idiotypic mAb BW2064/36 (1.5 μg/mL) as surrogate antigen. T cell amplification was monitored by flow cytometric recording of CSFE dilution, and CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. Data were transformed to demonstrate the loss or gain in CAR T cell amplification in the presence of TGF-β (number of proliferating cells without TGF-β − number of proliferating cells in the presence of TGF-β+ proliferating cells without TGF-β) × 100. (E) T cells with or without CAR (0.125 − 4 × 104 CAR T cells per well) were incubated with CEA+ LS174T cells (2 × 104 cells per well) in the presence or absence of TGF-β (10 ng/mL). The specific cytotoxicity after 2 days was determined by the XTT-based viability assay. (C–E) Data represent the mean of triplicates ± SD. The assays were repeated three times; a representative assay is shown. Statistical analyses were performed using a two-tailed Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001).
To address whether resistance to TGF-β-mediated suppression requires CD28-mediated LCK and/or PI3K activation, T cells with the respective CARs were stimulated through their cognate antigen in the presence or absence of TGF-β. As summarized in Figure 2D, CAR T cell amplification was suppressed when stimulated by the CD28ΔLCK-ζ CAR with abrogated LCK phosphorylation, whereas T cells with the CD28ΔPI3K-ζ CAR amplified as the CD28-ζ CAR T cells. In addition, the cytolytic activity of CD28ΔLCK-ζ CAR T cells was suppressed in the presence of TGF-β, while the activity of CD28-ζ CAR T cells was not altered (Figure 2E). We concluded that CD28-mediated LCK activation is required for resisting TGF-β repression of CAR T cells. Of note, the CD28ΔLCK-ζ CAR-induced T cell amplification in the absence of TGF-β was much lower than that of T cells containing the unmodified CD28-ζ CAR and at a level of the CD3ζ CAR T cells without co-stimulation, which is likely due to the absence of autocrine IL-2 stimulation.
To prove whether IL-2 is involved in mediating resistance of CD28-ζ CAR T cells toward TGF-β-mediated suppression, T cells with the CD28ΔLCK-ζ CAR were stimulated by cognate antigen with or without added IL-2 in the presence of TGF-β. The suppressed amplification of CD28ΔLCK-ζ CAR T cells was reverted by adding IL-2 (Figure 3A); CAR T cells amplified as efficiently as in the absence of TGF-β.
Figure 3.
CD28-Induced IL-2 Is Involved in the Resistance of CD28-ζ CAR T Cells toward TGF-β-Mediated Suppression
(A) T cells were engineered with the CD28ΔLCK-ζ CAR, labeled with CFSE, and incubated (2.5 × 104 T cells per well) with added TGF-β (10 ng/mL) or TGF-β plus IL-2 (500 U/mL) for 4 days on plates coated with the mAb BW2064/36 (1.5 μg/mL) as CAR antigen. Incubation without cytokines (w/o) served as control. T cell amplification was monitored by CSFE dilution and recorded by flow cytometry; CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. (B) T cells with CAR and with or without engineered constitutive IL-2 release were incubated for 2 days (2.5 × 104 CAR T cells per well) on BW2064/36 mAb (1.5 μg/mL)-coated plates. Plates coated with an isotype matched mouse IgG1 and T cells without CAR (w/o) served as controls. IFN-γ and IL-2 in the culture supernatants were recorded by ELISA. (C) T cells engineered with the CD28ΔLCK-ζ CAR without or with transgenic IL-2 release were labeled with CFSE and incubated (2.5 × 104 T cells per well) with or without TGF-β (10 ng/mL) for 4 days on plates coated with the anti-idiotypic mAb BW2064/36 (1.5 μg/mL). T cell amplification indicated by CSFE dilution was recorded by flow cytometry; CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. Data represent the mean of triplicates ± SD. The assays were repeated three times; a representative assay is shown. Statistical analyses were performed using a two-tailed Student’s t test (**p < 0.01; n.s., not significant).
To confirm that T cell-released IL-2 promotes T cell amplification in the presence of TGF-β, we engineered CD28ΔLCK-ζ CAR T cells to constitutively release IL-2; the level of released IFN-γ after CAR-mediated stimulation did not change (Figure 3B). However, the IL-2 released by CD28ΔLCK-ζ CAR T cells compensated for the TGF-β-mediated repression of T cell amplification (Figure 3C). Data indicated that IL-2 induced by CD28 signaling-mediated resistance of CD28-ζ CAR T cells to TGF-β repression.
IL-7 Can Replace IL-2 in Mediating TGF-β Resistance of CAR T Cells
We asked whether other γ-cytokines than IL-2 can also counteract TGF-β repression. The question is of particular relevance since we previously showed that CAR T cell-released IL-2 negatively impacts the anti-tumor activity through sustaining survival and function of Treg cells.11 Therefore, we asked whether other γ-cytokines like IL-7 or IL-15 are capable of replacing IL-2 in counteracting TGF-β-mediated suppression. IL-7 is of particular interest in this context, since tumor-infiltrating Treg cells do not express the IL-7 receptor12 and are therefore not stimulated by IL-7 released by CAR T cells. CD28ΔLCK-ζ CAR T cells were stimulated by cognate antigen in the presence of TGF-β with or without added IL-7 or IL-15. While CAR T cell amplification was suppressed by TGF-β, added IL-7 and IL-15, respectively, made the cells resistant to repression, allowing CAR T cell amplification at the same magnitude as without TGF-β (Figure 4A).
Figure 4.

IL-7 and IL-15 Overcome TGF-β-Mediated Suppression in T Cell Amplification
(A) CD28ΔLCK-ζ CAR T cells were labeled with CFSE and incubated (2.5 × 104 T cells per well) with TGF-β or TGF-β plus IL-7 or IL-15 (each 10 ng/mL) for 4 days on BW2064/36 mAb (1.5 μg/mL)-coated plates for CAR stimulation. Incubation without cytokines (w/o) served as control. T cell amplification was monitored by CSFE dilution and recorded by flow cytometry; CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. (B) CAR T cells with or without constitutive release of transgenic IL-7 or IL-15 (2.5 × 104 CAR T cells per well) were stimulated through the CAR by incubating for 2 days on 96-well plates coated with the mAb BW2064/36 (1.5 μg/mL). Plates coated with an isotype-matched IgG1 antibody as well as T cells without CAR (w/o) served as controls. IFN-γ, IL-2, IL-7, and IL-15 were recorded in the culture supernatants by ELISA. (C) Amplification of CD28ΔLCK-ζ CAR T cells with or without constitutive release of transgenic IL-7 or IL-15 (2.5 × 104 T cells per well) was recorded by flow cytometric recording of CSFE dilution. Cells were incubated with or without TGF-β (10 ng/mL) on 96-well plates coated with the mAb BW2064/36 (1.5 μg/mL). CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. (D) CAR T cells (0.125 − 4 × 104 CAR T cells per well) were incubated with CEA+ LS174T cells (2 × 104 cells per well) in the presence or absence of TGF-β (10 ng/mL) for 2 days, and specific cytotoxicity was determined by the XTT-based viability assay. Data represent the mean of triplicates ± SD. The assays were repeated three times; a representative assay is shown. Statistical analyses were performed using a two-tailed Student’s t test (*p < 0.05; **p < 0.01; n.s., not significant).
To accumulate IL-7 or IL-15 at the tumor site upon CAR stimulation, CD28ΔLCK-ζ CAR T cells were additionally engineered to express IL-7 and IL-15, respectively. Modified CAR T cells released IL-7 and IL-15 into the supernatants upon CAR engagement of cognate antigen; incubation with irrelevant antigen did not induce cytokine release (Figure 4B).
CD28ΔLCK-ζ CAR T cells secreting IL-7 amplified upon engagement of cognate antigen in the presence of TGF-β (Figure 4C). CAR T cells with transgenic IL-15 showed TGF-β resistance to the same extent. TGF-β did not impact the cytolytic activity of those CAR T cells toward CEA+ LS174T cells (Figure 4D), while the cytolytic activity of CD28ΔLCK-ζ CAR T cells was reduced in the presence of TGF-β (Figure 2E). Taken together, CD28ΔLCK-ζ CAR T cells engineered for the γ-cytokines IL-7 or IL-15 release resisted TGF-β suppression.
A Hybrid IL-7Rα/IL-2Rβ Receptor Mediates TGF-β Resistance Independently of IL-2
Although both IL-7 and IL-15 counteracted TGF-β suppression, IL-15 stimulates Treg cells, as does IL-2. Therefore, we further explored IL-7 to improve CAR T cell activity. However, the IL-7 receptor is rapidly downregulated by effector and central memory T cells upon ligand binding,13, 14 which would desensitize CD28ΔLCK-ζ CAR T cells to transgenic IL-7 in the long-term and would lose TGF-β resistance. To prolong the protective effect of IL-7, we engineered a hybrid receptor molecule composed of the physiologic IL-7 receptor α chain in the extracellular moiety and the IL-2 receptor β chain in the intracellular moiety (Figure 5A). The hybrid IL-7Rα/IL-2Rβ receptor was designed to convert IL-7 receptor binding by IL-7 into an IL-2 receptor signal in order to overcome TGF-β repression.
Figure 5.

Signaling through the IL-7Rα-IL-2Rβ Receptor Conveys Resistance to TGF-β Repression
(A) Schematic representation of the expression cassette encoding the anti-CEA CAR and the IL-7Rα-IL-2Rβ hybrid receptor, which is composed of the extracellular domain of the IL-7 receptor α chain and of the intracellular IL-2 receptor β chain. (B and C) T cells were engineered with the CD28ΔLCK-ζ CAR with the hybrid IL-7Rα-IL-2Rβ receptor, stimulated through the CAR by immobilized BW2064/36 mAb (1.5 μg/mL) and incubated (2.5 × 104 CAR T cells per well) in the presence of TGF-β with or without added IL-7 (10 ng/mL each). Incubating cells on plates coated with IgG (1.5 μg/mL) as irrelevant antigen as well as incubation without TGF-β or IL-7 served as controls. IFN-γ (B) and IL-2 (C) secreted into the culture supernatants were recorded by ELISA. (D) T cells were engineered with a CD28ΔLCK-ζ CAR and the IL-7Rα-IL-2β receptor. Cells were labeled with CFSE, stimulated through the CAR by incubation on immobilized BW2064/36 mAb (1.5 μg/mL) (2.5 × 104 CAR T cells per well) in the presence of TGF-β or TGF-β plus IL-7 (10 ng/mL each) for 4 days. Incubation without cytokines (w/o) served as controls. Amplification of CAR T cells was recorded by flow cytometric recording of CFSE dilution. CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. (E) Engineered CAR T cells (0.125 − 4 × 104 CAR T cells per well) were incubated with CEA+ LS174T cells (2 × 104 cells per well) in the presence of TGF-β, IL-7, or both (10 ng/mL each) for 2 days, and specific cytotoxicity was determined by the XTT-based viability assay. Incubation without cytokines (w/o) served as control. Data represent the mean of triplicates ± SD. The assays were repeated three times; a representative assay is shown. Statistical analyses were performed using a two-tailed Student’s t test (**p < 0.01; n.s., not significant).
The IL-7Rα/IL-2Rβ receptor was co-expressed with the CAR in T cells and was activated upon IL-7 binding as indicated by increased IFN-γ release (Figure 5B). IL-2 was not produced due to the lack of LCK activation through the CD28ΔLCK-ζ CAR (Figure 5C). Consequently, the T cell proliferation was repressed in the absence of IL-7. In contrast, addition of IL-7 counteracted the TGF-β suppression of proliferation (Figure 5D) and further enhanced the cytolytic capacity of CD28ΔLCK-ζ CAR T cells (Figure 5E), indicating a robust resistance of the CAR T cells to TGF-β repression.
Finally, we combined the transgenic IL-7 release with the co-expressed hybrid IL-7Rα/IL-2Rβ receptor in CD28ΔLCK CAR T cells (Figure 6A). IFN-γ and IL-7 were released by those cells upon CAR engagement of antigen (Figure 6B). In the presence of TGF-β, such CAR T cells amplified and executed specific cytolysis as efficiently as in the absence of TGF-β, which is in contrast to CAR T cells without the synthetic autocrine loop (Figures 6C and 6D). Data demonstrate that CAR T cells can be made resistant to TGF-β repression by signaling through an engineered IL-2 receptor β chain, which can be triggered by transgenic IL-7 in an autocrine fashion.
Figure 6.

IL-7Rα-IL-2Rβ Receptor Can Be Triggered by Transgenic IL-7 to Improve the Efficiency in Autocrine Fashion
(A) Schematic representation of the expression cassette encoding the CD28ΔLCK-ζ CAR, the IL-7Rα-IL-2Rβ hybrid receptor, and transgenic IL-7. (B) T cells were engineered with the expression vector shown in (A), and 2.5 × 104 CAR T cells per well were stimulated through the CAR by the immobilized BW2064/36 mAb (1.5 μg/mL). Incubation on plates coated with IgG (1.5 μg/mL) as irrelevant antigen as well as T cells with CD28-ζ CAR or without CAR (w/o) served as controls. IFN-γ and IL-7 secreted into the culture supernatants were recorded by ELISA. (C) CAR T cells from (A) were labeled with CFSE and stimulated through the CAR by incubation on immobilized BW2064/36 mAb (1.5 μg/mL) (2.5 × 104 T cells per well) in the presence or absence of TGF-β (10 ng/mL) for 2 days. Amplification of CAR T cells was recorded by flow cytometric recording of CSFE dilution. CAR T cells were identified by staining with the PE-conjugated anti-IgG antibody. Comparison to CAR without hybrid receptor and without IL-7 shows suppression by TGF-β in a comparative setting. (D) CAR T cells (0.125 − 4 × 104 cells per well) were incubated with CEA+ LS174T cells (2 × 104 cells per well) in the presence or absence of TGF-β (10 ng/mL) for 2 days. Specific cytotoxicity was determined by the XTT-based viability assay. Incubation without cytokines (w/o) served as control. Data represent the mean of triplicates ± SD. The assays were repeated three times; a representative assay is shown. Statistical analyses were performed using a two-tailed Student’s t test (**p < 0.01; ***p < 0.001; n.s., not significant).
CD28ΔLCK-ζ CAR T Cells with Hybrid IL-7Rα/IL-2Rβ Receptor and Transgenic IL-7 Show Superior Activity in the Presence of TGF-β in the Long-Term
We asked whether the engineered CAR T cells with the IL-7 autocrine loop resist TGF-β under conditions of repetitive antigen stimulation. CD28ΔLCK-ζ CAR T cells with the hybrid IL-7Rα/IL-2Rβ receptor and transgenic IL-7 was repetitively stimulated by adding cancer cells every 2 days (Figure 7A). As summarized in Figure 7B, CAR T cells exhibited superior cytolytic activities compared with the unmodified CD28-ζ CAR T cells. Production of granzyme B in the presence of TGF-β was found the same in T cells engineered with the wild-type and modified CARs, respectively (Figure 7C). We think the improved elimination of cancer cells in the long-term due to improved survival of the modified CAR T cells in the presence of TGF-β since the number of apoptotic CD28ΔLCK-ζ CAR T cells with the hybrid IL-7Rα/IL-2Rβ receptor and transgenic IL-7 was substantially lower than of T cells with the non-modified CAR (Figure 7D). In addition, early antigen-mediated T cell activation was indicated by STAT5 phosphorylation superior; after 30 min of cognate antigen engagement, the CD28ΔLCK-ζ CAR T cells with the hybrid receptor and IL-7 showed an increase in pSTAT5, which was not the case for CAR T cells without the IL-7 autocrine loop (Figure 7E).
Figure 7.

Comparative Analysis of CAR T Cells with the CD28ΔLCK-ζ CAR and Hybrid IL-7Rα-IL-2Rβ Receptor and Transgenic IL-7 versus CD28ΔLCK-ζ CAR T Cells
(A) Schematic timeline of the serial-killing assay demonstrating the addition of target cells and the cytotoxicity and cytokine readings. (B) Specific cytotoxicity of CAR-modified T cells after serial killing of effector cells. CAR T cells (2 × 104 CAR+ T cells per well) were incubated with CEA+ LS174T cells (2 × 104 cells per well) for 2 days, and specific cytotoxicity was determined by the XTT-based viability assay. After 2 days, the cells from a parallel assay were harvested and incubated again with fresh CEA+ LS174T cells (2 × 104 cells per well) for 2 days; the same procedure was repeated every 2 days until day 6. ***p < 0.001 CAR T cells with IL-7 loop compared with CD28-ζ CAR T cells. IFN-γ and IL-2 were measured in the culture supernatants by ELISA. (C) Intracellular staining of granzyme B. T cells with CAR were incubated on plates (2.5 × 104 CAR+ T cells per well) coated with the anti-idiotypic mAb BW2064/36 or mouse IgG1 (1.5 μg/mL each) as an isotype control in the presence or absence of TGF-β (10 ng/mL) for 2 days. CAR+ T cells were identified by staining with a PE-conjugated anti-human IgG antibody; granzyme B was stained by a FITC-conjugated anti-granzyme B antibody. (D) Apoptotic and living cells were identified by staining with APC-conjugated AnnexinV and 7-AAD. Cells were gated on CAR+ cells; the percentages represent the cell numbers in each quadrant. (E) Intracellular staining of pSTAT5. T cells with or without (w/o) CAR were incubated on plates (3.5 × 105 CAR T cells per well) coated with the anti-idiotypic mAb BW2064/36 (1.5 μg/mL each) for 30 min or 16 hr. For pSTAT5 detection, a PE-conjugated anti-pSTAT5 antibody was used. T cells without CAR (w/o) or with the CD28-ζ CAR served as controls. The histograms show the pSTAT5-PE staining of the respective cells. Data represent the mean of triplicates ± SD. Statistical analyses were performed using a two-tailed Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001).
Improved Anti-tumor Activity of CD28ΔLCK-ζ CAR T Cells with Autocrine IL-7/IL-2Rβ Loop
To assay the anti-tumor activity of such modified CAR T cells, TGF-β+ CEA+ tumors were induced by subcutaneously inoculating C15A3 cells; tumors were grown for 16 days to the size of about 200 mm3. Treatment of mice by one injection of CD28ΔLCK-ζ CAR T cells with IL-7Rα/IL-2Rβ receptor and IL-7 release more substantially reduced tumor progression in comparison to treatment with CD28ΔLCK-ζ CAR T cells alone (Figures 8A and 8B); CAR T cells without the IL-7 loop had no major effect on advanced TGF-β+ tumors. The observation is consistent with the improved persistence of CAR T cells in the tumor tissue compared with CD28ΔLCK-ζ CAR T cells (Figures 8C and 8D). For control, there are no differences in the number of TGF-β+ and CEA+ cancer cells in the tumor tissue (Figures 8D and 8E). We concluded that CAR T cells with an engineered autocrine IL-7 triggered IL-2Rβ signaling loop are highly efficacious against advanced TGF-β+ tumors.
Figure 8.

Progression of TGF-β+ Tumors Is Slowed Down by CD28ΔLCK-ζ CAR T Cells with Hybrid IL-7Rα-IL-2Rβ Receptor and Transgenic IL-7
(A) TGF-β+ CEA+ C15A3 tumor cells (106 cells per mouse) were subcutaneously inoculated into Rag2−/−γc−/− mice (four mice per group). T cells were engineered with the CD28ΔLCK-ζ CAR or additionally with the hybrid IL-7Rα/IL-2Rβ receptor and transgenic IL-7. Engineered T cells were applied by intravenous injection at day 16 when the tumor size reached about 200 mm3 (1.5 × 106 CAR T cells per mouse). Mice without adoptively transferred T cells (w/o T cells) or with T cells without CAR (w/o CAR) served as controls. Tumor growth after T cell injection was weekly monitored. (B) Relative tumor growth of mice at day 14 after T cell injection. Tumor growth at day 14 was referred to the tumor size at the day of treatment. Statistical analyses were performed using the two-tailed Student’s t test (*p < 0.05). (C) Tumor tissue slides were recorded for CAR+ T cells. (D) CAR+ T cells in tumor tissue were detected by staining with Alexa Fluor 555-conjugated anti-human IgG antibody (dilution 1:250), tumor cells by staining with Alexa Fluor 488-conjugated anti-CEA (αCD66a/c/e) antibody (dilution 1:50); cell nuclei were stained with “Reddot2” (dilution 1:200). Scale bar represents 100 μm. (E) TGF-β in tumor tissue was detected using the anti-TGF-β antibody (dilution 1:50) and Alexa Fluor 555-labeled anti-mouse IgG (H+L) antibody (dilution 1:200); tumor cells were identified with the Alexa Fluor 488-conjugated anti-CEA (αCD66a/c/e) antibody (dilution 1:50); cell nuclei were stained with DAPI. Scale bar represents 100 μm.
Discussion
The success of adoptive cell therapy for the treatment of solid tumors is currently limited due to immune suppression mediated by TGF-β.15, 16, 17, 18 Our previous work demonstrated that CD28 co-stimulation, but not 4-1BB co-stimulation, makes CAR T cells resistant to TGF-β-mediated repression; CD28-ζ CAR T cells act superior in eliminating TGF-β+ tumors.8 We revealed here that CD28 activation of LCK and autocrine signaling through released IL-2 are essential to induce the effect. Our conclusion is based on the observation that abolishing IL-2 secretion by abrogating LCK activation through mutation of the LCK binding site in the CD28 endodomain makes CD28ΔLCK-ζ CAR T cells sensitive to TGF-β suppression; adding back IL-2 reconstitutes TGF-β resistance. Other γ-cytokines like IL-7 and IL-15 can replace IL-2 in order to overcome TGF-β repression.
While the CD28-induced IL-2 autocrine loop is effective and can be used for CD28-ζ CAR T cell therapy, the strategy has substantial limitations in the application for targeting solid tumor lesions, since accumulating IL-2 in the tumor tissue will sustain tumor-infiltrating Treg cells, which are a major source of TGF-β. In line with the strategy, we replaced IL-2 with IL-7 and engineered a hybrid receptor that allows binding of IL-7 to the extracellular IL-7 receptor α chain (CD127) and provides IL-2 receptor β chain signaling to T cells. In this scenario, the T cell becomes activated upon binding to antigen by the CAR, amplifies, executes cytolysis, and releases cytokines like IFN-γ, however, is deficient in releasing IL-2; co-released transgenic IL-7 binds to the hybrid receptor, which provides the IL-2 receptor signal to the CAR T cell required to resist TGF-β repression. Treg cells lack the IL-7Rα chain and thereby do not benefit from transgenic IL-7 release.12 Although IL-15 can also replace IL-2 in this context, we think IL-15 less suitable, since IL-15 stimulates Treg cells19 in contrast to IL-7. In addition, locally accumulating IL-7 has the advantage to promote resident tumor-infiltrating T cells (TILs) in sustaining their anti-tumor response.20
We think co-release of IL-7 by CAR T cells is likely safe in the clinical situation, since systemic administration of IL-7 to cancer patients in the treatment of lymphopenia did not produce severe side effects but provided the benefit to increase CD4+ and CD8+ cell numbers and to decrease Treg cells.21 CAR T cells with inducible IL-7 release, nick-named IL-7 TRUCKs (T cells redirected for unrestricted cytokine mediated killing), deposit IL-7 upon CAR activation preferentially in the targeted tumor tissue; off-tumor IL-7 release and systemic IL-7 effects are not expected.22 Although under control of a constitutive promoter in the transducing expression vector, the transgenic IL-7 is only released upon CAR activation, which we think is due to the fact that IL-7 is released from intracellular stores in an activation-dependent fashion.
IL-7 not only counteracts TGF-β repression, it also augments amplification of activated T cells as long as the IL-7 receptor is expressed. However, transcription of the IL-7 receptor is downregulated in effector T cells in response to IL-7 signaling,13, 14 making the cells less sensitive to IL-7 stimulation. The hybrid IL-7Rα/IL-2Rβ receptor circumvents the situation by constitutively expressing the receptor and translating extracellular IL-7 binding into intracellular IL-2 signaling. Accordingly, CAR T cells with IL-7 release and hybrid IL-7Rα/IL-2Rβ receptor showed improved survival over a prolonged period.
Our observation that IL-2 and other γ-cytokines can overcome the suppressive effect of TGF-β points to an integrating crossover of the TGF-β- and γ-cytokine-signaling pathways; such crossover may represent a valuable target for specifically shaping the T cell activity. TGF-β represses the response of activated T cells through inhibiting c-myc transcription,23 finally resulting in the suppression of T cell amplification and pro-inflammatory cytokine release.24 On the other hand, IL-2 signaling is transmitted through the STAT5 pathway to accumulate MYC protein25 and to activate BCL-2 and BCL-x to prevent apoptosis,26 implying c-myc as a central regulator. Signaling through IL-7 feeds into the same final pathway,27, 28 which makes IL-7 a good candidate to replace IL-2 in this context.
Other strategies to overcome the suppressive effect of TGF-β were reported; for instance, the expression of a dominant-negative TGF-β receptor,3, 4, 29, 30 which acts as a decoy receptor to reduce TGF-β-mediated downstream signaling, an improved modification of the dnTGF-β receptor, was recently reported.31 The strategy may theoretically be limited by high TGF-β concentrations, which will also bind to the physiological TGF-β receptor, providing some repressive signals even in the presence of the dnTGF-β receptor. Other alternatives are small-molecule inhibitors of the TGF-β signaling pathway, repression of TGF-β expression by antisense oligo-nucleotides, or neutralization by blocking monoclonal antibodies.32, 33, 34 In these cases, the risk of systemic side effects is high, as observed in an uncontrolled and lethal immune response in a mouse model.35
In contrast, our concept is based on overcoming TGF-β signaling in a CAR T cell-intrinsic fashion. This is realized by an inducible and locally provided activating signal that is leaving the systemic immune regulation untouched. By revealing γ-cytokines like IL-2 or IL-7 as mediators to convey resistance to TGF-β and by applying synthetic biology, we here present a strategy that has the potential to make cell therapy of solid tumor lesions with expectedly high TGF-β levels and infiltrating Treg cells feasible. Key features are CAR T cells, which are deficient in IL-2 release but release transgenic IL-7 and co-express the IL-7Rα/IL-2β hybrid receptor to provide cell-intrinsic IL-2 signaling to persist through IL-7 release as long as the CAR engages cognate antigen. Due to the dependency on CAR signaling, off-target auto-stimulatory activation is not expected, making the strategy suitable for systemic clinical application.
Materials and Methods
Blood Samples, Cell Lines, and Reagents
All studies involving human blood cells were approved by the Uniklinik Köln Institutional Review Board (reference no. 01-090). Human T cells were isolated from the peripheral blood of healthy donors by density gradient centrifugation and stimulated by the agonistic anti-CD3 antibody OKT3 (50 ng/mL) and IL-2 (500 U/mL) for 48 hr. HEK293T cells (ATCC CRL-11268) are human embryonic kidney cells that express the SV40 large T antigen. LS174T (ATCC CCL 188) is a CEA-expressing human colon carcinoma cell line. C15A3 cells (kindly provided by Dr M. Neumaier, Universität Heidelberg-Mannheim) were derived from mouse MC38 fibrosarcoma cells by transfection with a CEA-encoding plasmid. OKT3 (ATCC CRL 8001) is a hybridoma cell line producing the agonistic anti-CD3 monoclonal antibody (mAb) OKT3. BW2064/36 is an internal image anti-idiotypic antibody directed against the anti-CEA single-chain fragment of variable region (scFv) antibody BW431/26.36 T cells and hybridoma cell lines were cultured in RPMI 1640 medium (Invitrogen Life Technologies, Karlsruhe, Germany) and 10 mM HEPES; adherent cells were cultured in DMEM (Invitrogen Life Technologies), both media supplemented with 10% (v/v) fetal calf serum (FCS) (PAN-Biotech, Aidenbach, Germany) and 100 IU/mL penicillin, streptomycin (PAN-Biotech). OKT3 and BW2064/36 mAbs were affinity purified from hybridoma supernatants using goat anti-mouse immunoglobulin G1 (IgG1) antibody (SouthernBiotech, Birmingham, AL, USA) immobilized on N-hydroxysuccinimide ester-activated Sepharose (Amersham Biosciences, Freiburg, Germany). The following antibodies against human proteins were used: fluoresceine isothiocyanate (FITC)-conjugated anti-CD3 mAb (Miltenyi Biotec, Bergisch Gladbach, Germany), phycoerythrin (PE)-conjugated goat anti-IgG1 antibody F(ab’)2 (SouthernBiotech), anti-IFN-γ antibody NIB42, biotinylated anti-IFN-γ antibody 4S.B3, anti-IL-2 antibody 5344-111, biotinylated anti-IL-2 antibody B33-2 (all from BD Bioscience, San Jose, CA, USA). The anti-mouse IL-7 antibody and the biotinylated anti-mouse IL-7 antibody (mouse IL-7 DuoSet ELISA, DY407), the anti-human IL-15 antibody, and the biotinylated anti-human IL-15 antibody (human IL-15 DuoSet ELISA, DY247) were purchased from R&D Systems (Minneapolis, MN, USA) for ELISA use. Recombinant human IL-2 was purchased from Novartis (Basel, Switzerland), recombinant IL-7, IL-15, and TGF-β from Miltenyi Biotec.
CARs and T Cell Modification
The CEA-specific CARs BW431/26scFv-Fc-ζ, BW431/26scFv-Fc-CD28-ζ,37 BW431/26scFv-Fc-4-1BB-ζ,38 and BW431/26scFv-Fc-CD28ΔLCK-ζ11 were described previously. BW431/26scFv-Fc-CD28ΔPI3K-ζ and BW431/26scFv-Fc-CD28ΔLCKΔPI3K-ζ were engineered by site-directed mutagenesis.9 Co-expression of IL-2, IL-7, and IL-15, respectively, was achieved by linking the respective cytokine encoding sequence to the CAR expression cassette by P2A. The DNA encoding the extracellular part of the IL-7 receptor α chain and the transmembrane and intracellular part of the IL-2 receptor β chain were produced as “gBlocks” by Intergrated DNA Technologies (IDT, Coralville, IO, USA) and assembled in an expression vector by routine techniques. CAR encoding γ-retroviral vectors were produced by 293T cells after “PEIpro” (Polyplus-transfection, Illkirch, France)-mediated transfection, and T cells were transduced by spinfection as described previously.39 CAR expression was recorded by flow cytometry using the PE-conjugated F(ab’)2 anti-IgG1 antibody (SouthernBiotech), which detects the common extracellular spacer; T cells were identified by the FITC-conjugated anti-CD3 mAb (Miltenyi Biotec). Data were recorded using a fluorescence-activated cell sorting (FACS) Canto II cytofluorometer equipped with the FACS-Diva software (Becton Dickinson, Mountain View, CA, USA).
CSFE Labeling
T cells with or without a CAR were washed two times with PBS and stained with 0.5 μM 5-carboxylfluorescein diacetate succinimidyl ester (CFSE) (Invitrogen Life Technologies) for 10 min at room temperature; staining was stopped by adding 4–5 volumes of cold culture medium and incubated on ice for 5 min. The cells were washed twice before subjecting to the assay. CFSE staining was recorded by flow cytometry.
Intracellular Staining
Detection of pLCK
CAR T cells were stimulated through the CAR by incubating with the immobilized BW2064/36 mAb for 5 min. The cells were harvested, fixed, and permeabilized using the “Intrasure Kit” (BD Bioscience) and stained by the PE-conjugated anti-phosphoLCK antibody (BD Bioscience).
Detection of Granzyme B
CAR T cells were stimulated through the CAR by incubating with the immobilized BW2064/36 mAb for 48 hr with or without TGF-β (10 ng/mL). The cells were harvested, fixed, and permeabilized using the “Cytofix/Cytoperm” Kit (BD Bioscience) and stained by the FITC-conjugated granzyme B antibody (BD Bioscience).
Detection of pSTAT5
CAR T cells were stimulated through the CAR by incubating with the immobilized BW2064/36 mAb for 30 min or 16 hr. The cells were harvested, fixed using IC Fixation Buffer (eBioscience/Thermo Fisher Scientific, Waltham, MA USA), permeabilized using ice-cold methanol (99.9%), and stained by the PE-conjugated anti-phosphoSTAT5 antibody (eBioscience). Cells were recorded by flow cytometry.
CAR-Redirected T Cell Activation
Anti-CEA CAR-engineered T cells were stimulated through their CAR by incubating on 96-well round-bottom plates coated with the BW2064/36 mAb, which is a surrogate antigen for the anti-CEA CAR. Culture supernatants were analyzed for IFN-γ, IL-2, IL-7, and IL-15 by ELISA using the matched-pairs capture and biotinylated detection antibodies. The reaction products were visualized by peroxidase-streptavidin (1:10,000) and ABTS (Roche Diagnostics GmbH, Mannheim, Germany). Proliferation assay was performed by incubating CFSE-labeled CAR T cells on 96-well round-bottom plates coated with the BW2064/36 mAb for 4 days in the presence or absence of TGF-β (10 ng/mL), IL-2 (500 U/mL), IL-7, or IL-15 (10 ng/mL). Apoptotic and dead cells in the CAR+ T cell population were identified by staining with APC-conjugated AnnexinV and 7-aminoactinomycin-D (7-AAD); 7-AAD was added 10 min before flow cytometry. To monitor the cytolytic activity, CAR T cells were co-incubated with tumor cells for 48 hr in 96-well round-bottom plates, and the viability was monitored by the 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide salt (XTT)-based colorimetric assay (Cell Proliferation Kit II, Roche Diagnostics). In serial-killing assay, fresh tumor cells were added to the CAR T cells in every 2 days until day 6. Maximal reduction of XTT to formazan was determined as the mean of 12 wells containing target cells only and the background as the mean of 12 wells containing culture medium. T cell-mediated formation of formazan was determined by recording triplicate wells containing T cells in same numbers as in the corresponding experimental wells. CAR T cell-mediated specific cytotoxicity was calculated as follows: cytotoxicity (%) = (1 − OD [experimental wells − corresponding number of T cells]/OD [tumor cells without T cells − medium]) × 100, where OD is optical density.
Assay for Tumor Growth
Rag2−/−γc−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All animal experiments were performed according to the Animal Experiments Committee regulations and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz, Recklinghausen, Germany (K17/35-05). TGF-β secreting CEA+ C15A3 tumor cells were subcutaneously injected into Rag2−/−γc−/− mice (106 cells per mouse; four mice per group). Mice received CAR-modified human T cells or non-modified T cells (1.5 × 106 CAR T cells per mouse) at day 16 when the tumor size has reached about 200 mm3. Tumor growth was weekly monitored; statistical analyses were performed using the Student’s t test.
Immuno-histological Analysis
Tumor tissues were stained with the Alexa Fluor 488-conjugated anti-human CD66/a/c/e (anti-CEA clone, ASL-32) antibody (BioLegend, San Diego, CA USA) (dilution 1:50) to detect the tumor cells, with the Alexa Fluor 555-conjugated goat F(ab’)2 anti-human IgG antibody (SouthernBiotech) (dilution 1:250) to detect the CAR T cells and with “Reddot2 nuclear dye” (dilution 1:200) (Biotium, Fremont, CA, USA) or with DAPI (IS Mounting Medium DAPI, dianova, Hamburg, Germany) to detect the nuclei. TGF-β staining was performed with the anti-TGF-β antibody (mouse monoclonal IgG1, 3C11) (dilution 1:50) (Santa Cruz Biotechnology, Dallas, TX, USA) and Alexa Fluor 555-conjugated goat anti-mouse IgG (heavy and light [H+L]) antibody (dilution 1:200) (Invitrogen). Imaging was performed by using the Olympus IX81 microscope and the Olympus Fluoview (FV1000) software (Olympus, Center Valley, PA, USA).
Statistics
Statistical analyses were performed using a two-tailed Student’s t test. Data are presented as mean ± SD.
Author Contributions
V.G.-N. conceived and performed the experiments, interpreted the data, and wrote the paper. J.K. designed the cloning strategies. A.A.H. and H.A. designed experiments. H.A. interpreted the data and wrote the paper.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Dr. Gunter Rappl for his technical advice and Petra Hofmann and Nicole Riet for their technical assistances. The work was supported by the Wilhelm Sander-Stiftung, Munich, Else Kröner-Fresenius Stiftung, Bad Homburg v.d.H., Deutsche Krebshilfe, Bonn, the German Israeli Foundation (GIF), Deutsche Forschungsgemeinschaft (DFG) and the Medical Faculty of the University of Cologne through the Fortune Program.
Footnotes
Supplemental Information includes one figure and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.07.005.
Supplemental Information
References
- 1.Abken H. Driving CARs on the Highway to Solid Cancer: Some Considerations on the Adoptive Therapy with CAR T Cells. Hum. Gene Ther. 2017;28:1047–1060. doi: 10.1089/hum.2017.115. [DOI] [PubMed] [Google Scholar]
- 2.Pickup M., Novitskiy S., Moses H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L., Yu Z., Muranski P., Palmer D.C., Restifo N.P., Rosenberg S.A., Morgan R.A. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013;20:575–580. doi: 10.1038/gt.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foster A.E., Dotti G., Lu A., Khalil M., Brenner M.K., Heslop H.E., Rooney C.M., Bollard C.M. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J. Immunother. 2008;31:500–505. doi: 10.1097/CJI.0b013e318177092b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bollard C.M., Tripic T., Cruz C.R., Dotti G., Gottschalk S., Torrano V., Dakhova O., Carrum G., Ramos C.A., Liu H. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J. Clin. Oncol. 2018;36:1128–1139. doi: 10.1200/JCO.2017.74.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mohammed S., Sukumaran S., Bajgain P., Watanabe N., Heslop H.E., Rooney C.M., Brenner M.K., Fisher W.E., Leen A.M., Vera J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017;25:249–258. doi: 10.1016/j.ymthe.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkie S., Burbridge S.E., Chiapero-Stanke L., Pereira A.C.P., Cleary S., van der Stegen S.J.C., Spicer J.F., Davies D.M., Maher J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010;285:25538–25544. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koehler H., Kofler D., Hombach A., Abken H. CD28 costimulation overcomes transforming growth factor-beta-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 2007;67:2265–2273. doi: 10.1158/0008-5472.CAN-06-2098. [DOI] [PubMed] [Google Scholar]
- 9.Tai X., Cowan M., Feigenbaum L., Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 10.Lovatt M., Filby A., Parravicini V., Werlen G., Palmer E., Zamoyska R. Lck regulates the threshold of activation in primary T cells, while both Lck and Fyn contribute to the magnitude of the extracellular signal-related kinase response. Mol. Cell. Biol. 2006;26:8655–8665. doi: 10.1128/MCB.00168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kofler D.M., Chmielewski M., Rappl G., Hombach A., Riet T., Schmidt A., Hombach A.A., Wendtner C.M., Abken H. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory t cells which can be overcome by preventing Lck activation. Mol. Ther. 2011;19:760–767. doi: 10.1038/mt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perna S.K., Pagliara D., Mahendravada A., Liu H., Brenner M.K., Savoldo B., Dotti G. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin. Cancer Res. 2014;20:131–139. doi: 10.1158/1078-0432.CCR-13-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghazawi F.M., Faller E.M., Sugden S.M., Kakal J.A., MacPherson P.A. IL-7 downregulates IL-7Rα expression in human CD8 T cells by two independent mechanisms. Immunol. Cell Biol. 2013;91:149–158. doi: 10.1038/icb.2012.69. [DOI] [PubMed] [Google Scholar]
- 14.Henriques C.M., Rino J., Nibbs R.J., Graham G.J., Barata J.T. IL-7 induces rapid clathrin-mediated internalization and JAK3-dependent degradation of IL-7Ralpha in T cells. Blood. 2010;115:3269–3277. doi: 10.1182/blood-2009-10-246876. [DOI] [PubMed] [Google Scholar]
- 15.Beavis P.A., Slaney C.Y., Kershaw M.H., Gyorki D., Neeson P.J., Darcy P.K. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Semin. Immunol. 2016;28:64–72. doi: 10.1016/j.smim.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Ganesh K., Massagué J. TGF-β Inhibition and Immunotherapy: Checkmate. Immunity. 2018;48:626–628. doi: 10.1016/j.immuni.2018.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mariathasan S., Turley S.J., Nickles D., Castiglioni A., Yuen K., Wang Y., Kadel E.E., III, Koeppen H., Astarita J.L., Cubas R. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tauriello D.V.F., Palomo-Ponce S., Stork D., Berenguer-Llergo A., Badia-Ramentol J., Iglesias M., Sevillano M., Ibiza S., Cañellas A., Hernando-Momblona X. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 19.Xu S., Sun Z., Sun Y., Zhu J., Li X., Zhang X., Shan G., Wang Z., Liu H., Wu X. IL-15 and dendritic cells induce proliferation of CD4+CD25+ regulatory T cells from peripheral blood. Immunol. Lett. 2011;140:59–67. doi: 10.1016/j.imlet.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Ditonno P., Tso C.L., Sakata T., deKernion J.B., Belldegrun A. Regulatory effects of interleukin-7 on renal tumor infiltrating lymphocytes. Urol. Res. 1992;20:205–210. doi: 10.1007/BF00299718. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg S.A., Sportès C., Ahmadzadeh M., Fry T.J., Ngo L.T., Schwarz S.L., Stetler-Stevenson M., Morton K.E., Mavroukakis S.A., Morre M. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J. Immunother. 2006;29:313–319. doi: 10.1097/01.cji.0000210386.55951.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chmielewski M., Kopecky C., Hombach A.A., Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 23.Ruegemer J.J., Ho S.N., Augustine J.A., Schlager J.W., Bell M.P., McKean D.J., Abraham R.T. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J. Immunol. 1990;144:1767–1776. [PubMed] [Google Scholar]
- 24.Yoshimura A., Wakabayashi Y., Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-β. J. Biochem. 2010;147:781–792. doi: 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou C., Egawa T. Myc or no Myc, that is the question. EMBO J. 2015;34:1990–1991. doi: 10.15252/embj.201592267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lord J.D., McIntosh B.C., Greenberg P.D., Nelson B.H. The IL-2 receptor promotes lymphocyte proliferation and induction of the c-myc, bcl-2, and bcl-x genes through the trans-activation domain of Stat5. J. Immunol. 2000;164:2533–2541. doi: 10.4049/jimmunol.164.5.2533. [DOI] [PubMed] [Google Scholar]
- 27.Rani A., Murphy J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2016;36:226–237. doi: 10.1089/jir.2015.0054. [DOI] [PubMed] [Google Scholar]
- 28.Fry T.J., Mackall C.L. Interleukin-7: from bench to clinic. Blood. 2002;99:3892–3904. doi: 10.1182/blood.v99.11.3892. [DOI] [PubMed] [Google Scholar]
- 29.Bollard C.M., Rössig C., Calonge M.J., Huls M.H., Wagner H.-J., Massague J., Brenner M.K., Heslop H.E., Rooney C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 30.Lacuesta K., Buza E., Hauser H., Granville L., Pule M., Corboy G., Finegold M., Weiss H., Chen S.Y., Brenner M.K. Assessing the safety of cytotoxic T lymphocytes transduced with a dominant negative transforming growth factor-beta receptor. J. Immunother. 2006;29:250–260. doi: 10.1097/01.cji.0000192104.24583.ca. [DOI] [PubMed] [Google Scholar]
- 31.Kim S.K., Barron L., Hinck C.S., Petrunak E.M., Cano K.E., Thangirala A., Iskra B., Brothers M., Vonberg M., Leal B. An engineered transforming growth factor β (TGF-β) monomer that functions as a dominant negative to block TGF-β signaling. J. Biol. Chem. 2017;292:7173–7188. doi: 10.1074/jbc.M116.768754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagaraj N.S., Datta P.K. Targeting the transforming growth factor-β signaling pathway in human cancer. Expert Opin. Investig. Drugs. 2010;19:77–91. doi: 10.1517/13543780903382609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kemaladewi D.U., Pasteuning S., van der Meulen J.W., van Heiningen S.H., van Ommen G.-J., Ten Dijke P., Aartsma-Rus A., ’t Hoen P.A., Hoogaars W.M. Targeting TGF-β Signaling by Antisense Oligonucleotide-mediated Knockdown of TGF-β Type I Receptor. Mol. Ther. Nucleic Acids. 2014;3:e156. doi: 10.1038/mtna.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hau P., Jachimczak P., Schlingensiepen R., Schulmeyer F., Jauch T., Steinbrecher A., Brawanski A., Proescholdt M., Schlaier J., Buchroithner J. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201–212. doi: 10.1089/oli.2006.0053. [DOI] [PubMed] [Google Scholar]
- 35.Yingling J.M., Blanchard K.L., Sawyer J.S. Development of TGF-β signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004;3:1011–1022. doi: 10.1038/nrd1580. [DOI] [PubMed] [Google Scholar]
- 36.Hombach A., Schneider C., Sent D., Koch D., Willemsen R.A., Diehl V., Kruis W., Bolhuis R.L., Pohl C., Abken H. An entirely humanized CD3 zeta chain signaling receptor that directs peripheral blood t cells to specific lysis of carcinoembryonic antigen-positive tumor cells. Int. J. Cancer. 2000;88:115–120. [PubMed] [Google Scholar]
- 37.Hombach A., Wieczarkowiecz A., Marquardt T., Heuser C., Usai L., Pohl C., Seliger B., Abken H. Tumor-specific T cell activation by recombinant immunoreceptors: CD3 zeta signaling and CD28 costimulation are simultaneously required for efficient IL-2 secretion and can be integrated into one combined CD28/CD3 zeta signaling receptor molecule. J. Immunol. 2001;167:6123–6131. doi: 10.4049/jimmunol.167.11.6123. [DOI] [PubMed] [Google Scholar]
- 38.Hombach A.A., Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer. 2011;129:2935–2944. doi: 10.1002/ijc.25960. [DOI] [PubMed] [Google Scholar]
- 39.Golumba-Nagy V., Kuehle J., Abken H. Genetic Modification of T Cells with Chimeric Antigen Receptors: A Laboratory Manual. Hum. Gene Ther. Methods. 2017;28:302–309. doi: 10.1089/hgtb.2017.083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.