

Dopamine-melanin, polydopamine molecular assembly by cation-π bonds.
Abstract
Biological functions depend on biomolecular assembly processes. Assemblies of lipid bilayers, actins, microtubules, or chromosomes are indispensable for cellular functions. These hierarchical assembly processes are reasonably predictable by understanding chemical structures of the defined building blocks and their interactions. However, biopigment assembly is rather fuzzy and unpredictable because a series of covalently coupled intermediates from catecholamine oxidation pathways progressively form a higher-level hierarchy. This study reports a different yet unexplored type of assembly process named “cation-π progressive assembly.” We demonstrated for the first time that the cation-π is the primary mechanism for intermolecular assembly in dopamine-melanin biopigment. We also found that the self-assembled products physically grow and chemically gain new functions “progressively” over time in which cation-π plays important roles. The progressive assembly explains how biological systems produce wide spectra of pigment colors and broad wavelength absorption through energy-efficient processes. Furthermore, we also demonstrate surface-independent wettability control using cation-π progressive assembly.
INTRODUCTION
Molecular self-assembly is the process of building hierarchical supramolecular structures through multiple levels of noncovalent interactions such as hydrogen bonding, hydrophobic interactions, van der Waals forces, π-π stacking, and electrostatic bonds. An essential feature of self-assembly is that all participating building blocks are structurally defined small molecules or macromolecules. Lipid bilayers (1) and various vesicles, including liposomes (2) and exosomes (3), are assembled from phospholipids, cholesterols, and membrane-bound proteins. Chromosomes are constructed mostly from DNA and histone self-assembly (4). Actin and microtubules undergo linear and directional assembly from their corresponding monomeric proteins (5), and protein quaternary structures self-assemble from monomeric proteins (6, 7). In biological self-assembly, the structurally defined building blocks noncovalently associate to perform defined functions. Another research area, artificial self-assembly, such as in the case of alkanethiol self-assembled monolayers (8), polymeric layer-by-layer deposition (9, 10), DNA origami (11, 12), peptides (13, 14), and peptide amphiphiles (15), uses defined small molecules and macromolecules.
In contrast, there is also the uncontrolled assembly of heterogeneous derivatives sequentially generated through reaction/conversion pathways. This is common in melanin biochemistry (16, 17) and dopamine-melanin surface chemistry (18), and the understanding of these catecholamine compounds remains poor. Dopamine-melanin, also known as polydopamine, follows a progressive assembly mechanism, a concept explained in this study. Dopamine (C8H11O2N) is continuously oxidized to dopamine-quinone→dopamine-chrome→5,6-dihydroxyindole (DHI)→5,6-dihydroxyindole-quionone (DHIq)→dimer of DHI→diverse oligomerizations (19, 20). At any time and stage, the molecular intermediates and oligomer species in the oxidation pathway can participate in assembly processes, forming nanoscale thin films on solid substrates (18, 21, 22) and spherical melanin micro-/nanoparticles in the absence of substrates (23). This fuzzy assembly displays a progressive nature in that the assembly simultaneously occurs when a molecule undergoes a series of intra- and intermolecular reactions. Thus, progressive assembly is a fundamentally different concept from programmed self-assemblies, which are mediated by noncovalent interactions between “programmed” defined molecular building blocks (9–15). These phenomena are not artificially created by scientists but rather occur naturally in the skin, as with eumelanin, and in the brain, as with neuromelanin (24–27).
This study reports that cation-π interactions are the main intermolecular chemistry for the progressive assembly of dopamine-melanin/polydopamine formation. In 1990, Dougherty and Stauffer (28) suggested that the binding mechanism of acetylcholine (ACh) to its receptor was based on a new type of noncovalent binding called a “cation-π interaction” between hydrophobic aromatic rings and cations instead of electrostatic attractions between anions and cations (28). The first crystal structure of the ACh receptor protein in 1991 confirmed the existence of 14 aromatic residues in the binding site, strongly supporting the presence of cation-π interactions (29). Similar to ACh, dopamine binding to its receptor was confirmed by the proximity of aromatic residues and protonated dopamine (30). A cation-π interaction is a quadruple-monopole noncovalent binding interaction between an electron-rich π system and alkali metals (K+ and Na+) or nitrogenous cations (–NH3+ and –NMe3+) that is now widely accepted as an important binding interaction that regulates molecular recognition and signaling in nature (31). Extensive theoretical calculations and various experimental studies have suggested the characteristic binding energy of cation-π interactions, which are largely distinct from classical noncovalent interactions in similar atomic proximity ranges. The binding energy of a cation with an aromatic compound was higher than that with an aliphatic compound in a gas phase; −ΔH° for Na+/benzene = 28 kcal/mol, whereas −ΔH° for Na+/ethylene = 12 kcal/mol (32). Moreover, the distance dependency has proven to be different from classical ion-quadruple interactions: 1/r3, where r is the distance between the ion and the quadruple. In contrast, the distance relationship in cation-π interactions more likely follows the Columbic interaction: 1/r (33). There are large differences between cation-π interactions and classical salt bridges particularly in an aqueous medium. In the case of methylammonium/acetate salt bridge, desolvation and neutralization penalty dramatically decrease the binding energy between cations and anions from a gas phase (−125.5 kcal/mol) to a water phase (−2.2 kcal/mol). However, in the case of methylammonium/benzene cation-π interaction in water, its binding energy in aqueous media is in fact higher than that in a salt bridge (−5.5 kcal/mol) because the aromatic group is poorly solvated by water, which has a large benefit in desolvation energy (34).
Our study also demonstrates that the biological roles of cation-π interactions are not limited to ligand-receptor or ion-ion channel binding but further expand to biopigment assembly, polydopamine surface chemistry (18, 21, 22, 35, 36), and polydopamine nanoparticle technology (23, 37, 38). The data shown herein demonstrate that cation-π interactions ubiquitously exist among protonated amines of uncyclized dopamine/dopamine-quinone and the π system indole groups of DHI and that their subsequent oligomers are the unexplored driving force for the progressive assembly process. Understanding cation-π interactions during progressive assembly opens a new surface wettability control method for material surfaces to exhibit superhydrophilicity through the sandwich intercalation of quaternary ammonium. Furthermore, understanding cation-π interactions will allow us to determine how biology achieves broad wavelength absorption (instead of placing a number of chromophore arrays in the skin) and the detoxification of a broad spectrum of toxic molecules through small microscale melanin particles.
RESULTS AND DISCUSSION
We would like to practically define two terms. First, dopamine-melanin is a collective term describing assembled structures naturally occurring in vivo in the absence of material surfaces. Second, polydopamine is a nearly exchangeable term with dopamine-melanin, but it forms as a nanoscale coating layer in the presence of substrates. Here, polydopamine will be used unless otherwise mentioned because nearly all experiments were performed on substrates. The current polydopamine chemical model is clearly described down to the formation of DHI (Fig. 1, left large background arrow). However, the downward pathways in DHI oxidation are largely speculative. Several models have been proposed. The earlier models commonly assumed that polydopamine was a long-chain polymer of covalently cross-linked DHIs (18). However, later studies have consistently reported a limited degree of DHI oligomerization. Okuda et al. (39) established an equation based on the ratio of two DHI-melanin degradation products, pyrrole-2,3,5-tricarboxylic acid and pyrrole-2,3-dicarboxylic acid, demonstrating that the degree of oligomerization is limited up to eight units of DHI oligomers coexisting as tetramers and pentamers (39). A recent study by Chen et al. (40) investigated nearly 3000 possible DHI oligomers through a set of computational methods including brute-force algorithmic generation of isomers, molecular dynamics simulations, and density functional theory calculations (40). They also consistently suggested that oligomers with high molecular weights are less likely to form and that a group of tetramers are notably stable, which can be the majority of polydopamine. In addition to the DHI basis, other types of oligomers have also been suggested. Liebscher et al. (41) suggested a complexed form of linear-shaped oligomers consisting of DHI, indoledione, and unoxidized open-chain dopamines formed by covalent couplings via benzene rings (41). A considerable amount of uncyclized catecholamine units and pyrrolecarboxylic acid moieties arising from the partial degradation of DHIs have also been proposed by d’Ischia et al. (42–44). Along with covalent coupling oxidation, studies suggest that oligomers and monomers (for example, dopamine and DHI) are assembled via physical interactions to form a polydopamine layer. We previously published that there is a sufficient amount of uncyclized dopamine monomer physically entrapped in a (dopamine)2/DHI physical trimer form (45). Computational studies showed that hydrogen bonds and π-π stacking are driving forces for assembly (Fig. 1, right lower box) (39, 44, 46). Very recently, cation-π interactions have been suggested to be an unexplored molecular interaction that can clarify the synergistic relationship between aromatic and cationic amino acids for the reinforcement of underwater adhesion (47). The presence of cation-π interactions was confirmed by measurement of the surface force apparatus and by solid-state nuclear magnetic resonance spectroscopy, in which there exists a considerable amount of uncyclized, protonated amine groups under mild basic (Md-Bc) pH conditions. Thus, we hypothesized that deletion of the protonated cationic species by increasing the pH might result in dissociation of the entire polydopamine layer if the cation-π bonds are the dominant force for the progressive assembly.
Fig. 1. A newly proposed progressive assembly for polydopamine coating.
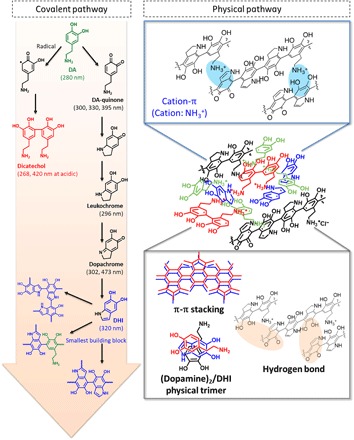
Dopamine is sequentially oxidized to various heterogeneous derivatives (dopamine-quinone, leukochrome, dopachrome, DHI, and dicatechol) via covalent bonds (pink arrow). The covalently bonded oligomers are physically assembled via hydrogen bonds, π-π stacking, van der Waals interactions (gray box), and cation-π interactions (blue box), forming the polydopamine coating.
To show the effect of neutralizing the dopamine cationic amine group by increasing the pH, we prepared a variety of polydopamine-coated substrates [titanium oxide (TiO2), silicon oxide (SiO2), gold (Au), stainless steel 304 (SS304), glass, poly(dimethyl siloxane), poly(ethylene terephthalate) (PET), thermoplastic polyurethane (TPU), poly(tetrafluoroethylene) (PTFE), cellulose nitrate (CN), polyester (PE), and nylon] and then applied either a strong basic (St-Bc; pH 9.8) or Md-Bc (pH 8.8) solution to the substrates (Fig. 2A). As shown in the representative photos in Fig. 2A, we coated all the tested substrates with polydopamine in the Md-Bc solution (first column), whereas a polydopamine layer was not formed on all the tested substrates in a St-Bc solution (pH 9.8); this was evident by the original color or transparency of the substrates [St-Bc (second column) versus bare substrate (third column)]. Ellipsometry experiments quantitatively showed the thickness of the polydopamine coated on the TiO2 substrates with a range from 20 to 100 nm under Md-Bc conditions between pH 7.4 and 8.8. The triggering of the polydopamine coating under Md-Bc pH (<8.8) can be explained by the pKa1 (= ~8.7) of the catechol moiety 3′-hydroxyl group (Fig. 2B). In contrast, the substrate treated with St-Bc solution (pH > 10) exhibited nearly no polydopamine residues on the surfaces (Fig. 2B). The thickness of the polydopamine coating was unexpectedly decreased in the reaction conditions above pH 10, although DHI- and dopamine-based oligomers still formed under the St-Bc conditions; these oligomers were identified by high-performance liquid chromatography–mass spectrometry (HPLC-MS) equipped with a C18 column (fig. S1). The peak at approximately 5.5 to 6 min of elution time was assigned to a mixture of both the dopamine monomer and dopamine-dopamine dimer. The hydrophilicities of both species are similar, resulting in similar retention times, and their mass spectra are identical ([dopamine monomer + H]+ = [dopamine dimer + 2H]2+/2 = 154 m/z). However, the two species display distinct light absorption characteristics because the π-conjugation system in the dimer is absorbed at a 420-nm wavelength (fig. S1A, red line). We assigned another peak at approximately 6.5 to 7 min as a DHI-dopamine dimer, which was detected by its ultraviolet (UV) absorbance at both 280 nm (catechol) and 320 nm (DHI) with a mass spectrum of [M + H]+ = 301 m/z (fig. S1A, blue line). The DHI-dopamine dimer is a major intermediate formed during conventional polydopamine coating (Md-Bc, pH 8.5). In addition, the previously reported characteristic physical trimer (45), (dopamine)2/DHI, was also formed under St-Bc pH conditions, which was confirmed by the peak appearance at approximately 14 min of elution time with UV absorbance at both 280 nm (catechol) and 320 nm (DHI) (fig. S1B, blue box). We also detected other trimers, ranging from 420 to 444 m/z (fig. S1B, red box). We assumed that these are covalent trimers because they exhibited absorption at 420 nm due to π-conjugation system extension through covalent coupling. Plausible structures for these trimers may include partially opened/degraded DHI units; these were newly suggested on the basis of their mass spectra, hydrophilicity (interpreted from their elution time), and UV absorbance characteristics (fig. S1C, red box). Even after polydopamine coating formation, further addition of sodium hydroxide triggered immediate destruction (that is, disassembly) of the polydopamine coating layer as shown in Fig. 2C. Figure 2D shows that a transparent TPU film was coated with polydopamine in a Md-Bc buffer (pH 8.8) for 24 hours. Subsequently, the polydopamine-coated TPU film was treated with 0.1 N NaOH (pH 12), which triggered polydopamine amine group deprotonation. The outmost polydopamine layer was rapidly delaminated within 5 s by NaOH treatment, and the entire polydopamine coating layer was disassembled and peeled off from the substrate within 150 s. Subsequently, the disassembled polydopamines were dispersed into the solution by shaking, showing that the polydopamine-coated TPU film recovered its transparency (Fig. 2D). The data shown in Fig. 2 collectively indicate that a strong base triggers dopamine amine group deprotonation, which affects the unique properties of the material-independent surface coatings. The pKa value of the dopamine amine groups is approximately 10, which eliminates cationic species in the assembled polydopamine. Removal of the cation might cause the observed instant dissociation of polydopamine fragments, potentially indicating the importance of cation-π bonds. In principle, it has been shown that the cation-π binding force becomes large for K+-π compared with Na+-π (48, 49). Thus, recovery of cation-π interactions by adding potassium ions can be reasonably hypothesized because the presence of the potassium ion can compensate for the loss of amine cationic species in polydopamine caused by St-Bc conditions (Fig. 3A). This condition, K+ dissolved in pH 9.8 solution, was denoted by St-Bc/K+. Similarly, the Md-Bc condition at pH 8.8 was named as Md-Bc. As shown in Fig. 3B, the progressive thickness evolution properties of the polydopamine layer were regained by adding potassium ions, with a thickness of 34.0 nm for St-Bc/K+ with 20 mM KCl, 63.2 nm for St-Bc/K+ with 50 mM KCl, and 97.3 nm for St-Bc/K+ with 100 mM KCl (green bars), even after polydopamine layer delamination by St-Bc treatment. In other words, the potassium ion returned the polydopamine coating thickness to a value similar to that shown under the Md-Bc conditions (94.4 nm, blue bar). Not only did the addition of potassium ions contribute to polydopamine reassembly on the surfaces via cation-π bonds, but the ionic intervention between polydopamine molecules also resulted in hydrophilic surfaces. The water contact angle of polydopamine-coated TiO2 under Md-Bc conditions was 32.8°, while the angle under St-Bc conditions was 43.9° (Fig. 2B, photos). However, the polydopamine contact angles deposited under St-Bc/K+ (20 mM) were 32.0° and 21.5° for 50 mM and below 5° for 100 mM KCl (Fig. 3C). On the basis of the morphology of St-Bc/K+ analyzed by SEM (Fig. 3C), we observed that the roughness was increased as a function of KCl concentration (20 to 100 mM) in the coating solution. The roughness changes might attribute to the superhydrophilicity. X-ray photoelectron spectroscopy (XPS) further confirmed the intercalation of potassium ions under St-Bc/K+ as a surrogate for protonated amines (–NH3+) under Md-Bc (Fig. 3D). The polydopamine coating obtained under Md-Bc exhibited a high amount of protonated amine species (401.6 eV) from dopamine or DHI (blue line, left). In contrast, the protonated amine peak decreased for the polydopamine layers prepared under St-Bc and St-Bc/K+; instead, deprotonated amines were detected (red lines shown for St-Bc and St-Bc/K+). The polydopamine prepared under St-Bc/K+ showed a clear and strong potassium peak (K2p, 292.9 eV), which replaced the sodium peak detected in the St-Bc sample. As we used NaOH (not KOH) for the pH titration, Na+ is ubiquitously present in both solutions, suggesting that the XPS peak change from Na1s (St-Bc) to K2p (St-Bc/K+) demonstrated the importance of K+-π interactions for progressive assembly and that the interaction force is larger than that for Na+-π. To further demonstrate the strong K+-π intercalations, we immersed polydopamine-coated TiO2 surfaces either in NaOH (0.1 N, pH 11) or in KOH (0.1 N, pH 11) solution for 1 hour. No polydopamine layer delamination was expected in the KOH solution. XPS analysis showed that the polydopamine layer on TiO2 was destroyed in the NaOH solution, showing the appearance of the underlying substrate Ti2p (458.2 eV) (fig. S2, top left). In contrast, the K2p peak (292.9 eV) appeared to be simply added to a typical polydopamine XPS pattern in the survey spectrum (fig. S2, top right). In addition to the TiO2 surface, we also used TPU substrates onto which polydopamine was coated in a conventional pH 8.8 solution, as the TPUs were suitable for movie demonstration because of their optical transparency. We immersed the coated TPU substrates in NaOH (pH 12), KOH (pH 12), and double-distilled water (DDW). The polydopamine assembly on the TPU surface was stably maintained in the St-Bc KOH solution (fig. S3 and movie S1). The TPU film maintained its dark brown surface color that originated from the polydopamine layer. From the series of experiments in Fig. 3, cation-π bonds play an essential role in polydopamine progressive assembly, as schematically shown in Fig. 3E. This result is in contrast with the conventional belief, wherein π-π interactions and hydrogen bonds have been experimentally (41, 45, 46) and theoretically (50, 51) suggested to be the responsible mechanisms in polydopamine assembly. To the best of our knowledge, cation-π bonds have not been reported as a driving force in molecular assembly of catecholamines for surface chemistry and synthetic pigment generation.
Fig. 2. Delamination of polydopamine (dopamine-melanin) coating under St-Bc solutions.
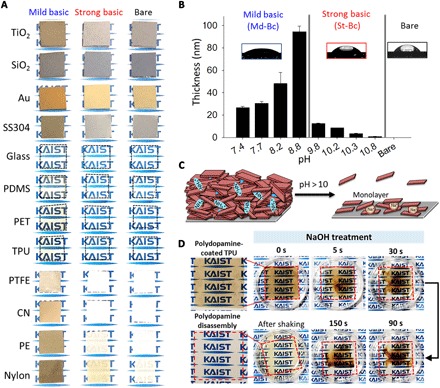
(A) Photos show the color changes before and after polydopamine coating at different pH values [Md-Bc, pH 8.8 (left); St-Bc, pH 9.8 (middle); and unmodified (right)]. PDMS, poly(dimethyl siloxane). (B) The thickness of the polydopamine coating on TiO2 substrates at different pH values and the corresponding contact angle images of the polydopamine-coated substrate immersed in Md-Bc solutions (7.4 < pH < 8.8; left) or St-Bc solutions (9.8 < pH < 10.8; middle) and the bare TiO2 substrate (right). (C) Delamination scheme for the polydopamine coating by potential deprotonation in strong alkaline solutions. (D) The polydopamine coating on TPU film was peeled off in 0.1 N NaOH (pH 12) solution.
Fig. 3. Polydopamine coating through K+ assistance in delaminating conditions consisting of strong alkaline pH.
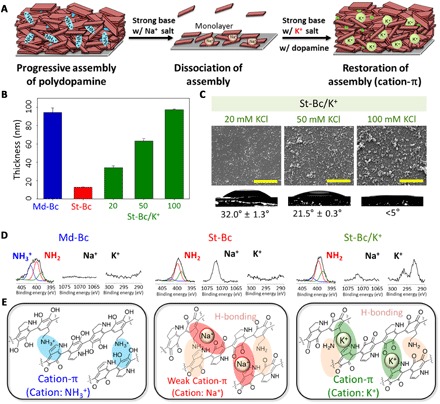
(A) A schematic description for resuming polydopamine coating in the presence of potassium ions after delamination in a strong alkaline condition. (B) The polydopamine coating thickness at pH 8.8 [a method described in (19)] (Md-Bc, blue bar), pH 9.8 (St-Bc, red bar), and pH 9.8 with 20, 50, or 100 mM KCl (St-Bc/K+, green bars). (C) Surface morphology by scanning electron microscopy (SEM) and the static water K+/St-Bc contact angles in increasing KCl concentration [20 mM (left), 50 mM (middle), and 100 mM (right)]. Scale bars, 2 μm. (D) XPS analysis of nitrogen (N1s, –NH3+ in blue, –NH2 in red, and –NH– in green), sodium (Na1s), and potassium (K2p) elements showing cation-to-neutral (–NH3+→–NH2) charge conversion from Md-Bc to St-Bc transfer (that is, elimination of cation-π). The surface-bound Na+ and K+ ions were also detected. (E) Assembly force transitions from strong cation-π interactions between the protonated amines and π system (left, Md-Bc conditions) to weak cation-π interactions between Na+ and the π system due to pH-induced deprotonation (middle, St-Bc conditions). Finally, cation-π interactions are resumed by the addition of K+ under the deprotonation conditions (right, St-Bc/K+ conditions).
Among the variety of cation candidates (for example, Li+, Na+, K+, R-NH3+, and NR4+), the quaternary ammonium ion (that is, R-NMe3+) has an innately high affinity for various hydrophobic aromatic rings, resulting in nearly the strongest cation-π interactions (49). In nature, quaternary ammoniums [that is, tetraethylammonium (TEA; (CH3CH2)4N+) and ACh ((CH3)3(CH3COOCH2CH2)N+)] are active in several biological functions, such as the potassium channel blocker TEA or the neurotransmitter ACh. In Fig. 3, we observed that surface wettability was determined to be superhydrophilic due to the K+-π intercalation in the polydopamine layer. Thus, the quaternary ammonium-π intercalation can also be expected in polydopamine formation, which might also result in polydopamine-mediated superhydrophilic surfaces. Incorporation of quaternary ammonium enables to achieve superhydrophilic surfaces by all organic molecule–based coatings instead of K+ ionic species (Fig. 4A). Superhydrophilic surfaces have been applied in a variety of fields, such as oil/water separations (52), anti-fogging (53), and cell/protein adhesion (54). For this reason, material-independent superhydrophilic surface engineering mediated by polydopamine can be a useful toolkit. The various surfaces were simply functionalized with dopamine and tetramethylammonium chloride salt (NMe4+Cl−) under St-Bc conditions (pH 9.8). We denoted this surface coating condition as St-Bc/NMe4+. After 24 hours of coating, we observed material-independent superhydrophilic wettability conversion. The initial different surface contact angles, including those for TiO2 (62.9°), SiO2 (56.2°), Au (56.7°), acrylate (82.4°), polyether ether ketone (92.2°), PET (72.2°), and polystyrene (77.4°), were changed to be very low static contact angles below 5° (Fig. 4B). Tian and Jiang (55) reported the relationship between wettability and roughness in which a flat hydrophilic surface becomes a superhydrophilic one with increased roughness. Similarly, hydrophobicity is also enhanced by generating surface roughness (55). Ponzio et al. (56) demonstrated that polydopamine coating by various concentrations of NaIO4 showed different surface morphology and achieved superhydrophilic surfaces by increasing roughness. We confirmed that the surface roughness prepared by St-Bc/NMe4+ conditions was also enhanced as the concentration of NMe4+ became increased (fig. S6), similar to the case of St-Bc/K+ in Fig. 3C. XPS analysis was performed as a function of NMe4+ concentration (0.1, 0.5, and 1.0 M). The quaternary ammonium N1s peak appeared at 402.4 eV, and its intensity became higher with increasing NMe4+ concentration (Fig. 4C, pink line). The N1s peak detected in 1.0 M NMe4+ was slightly higher than that shown in 0.5 M NMe4+. However, we observed a notable difference in the surface hydrophilic wettability. For the St-Bc/NMe4+ (1.0 M) polydopamine coating, the static contact angle value became zero in the 5-hour sample (TiO2). In contrast, for the St-Bc/NMe4+ (0.5 M) coating, the angle was 25.4° ± 0.5° (Fig. 4D). For 12 hours after coating, the contact angle for the sample prepared under the St-Bc/NMe4+ (0.5 M) conditions did not exhibit superhydrophilic wettability (13.2° ± 0.6°).
Fig. 4. An application of cation-π–mediated progressive assembly on material-independent superhydrophilic coatings.
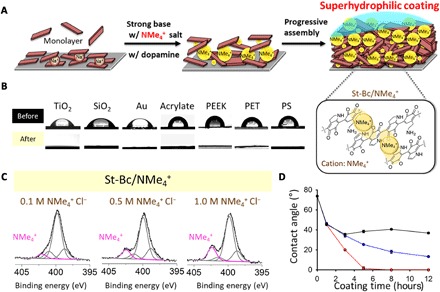
(A) Plausible substructures of polydopamine with ammonium cations (that is, tetramethylammonium, NMe4+) via cation-π interactions between the polydopamine progressive building block indole ring and NMe4+ under St-Bc conditions, resulting in a superhydrophilic coating. (B) The static water contact angle for a variety of substrates without any treatment (top row) and one with a polydopamine/NMe4+ coating on the surface as a superhydrophilic modifier (bottom row). PEEK, polyether ether ketone; PS, polystyrene. (C) The chemical status of nitrogen elements in the polydopamine coating at St-Bc pH in the presence of NMe4+ (concentrations of 0.1, 0.5, or 1.0 M). The amount of the intercalated ammonium (indicated as a signature peak at 402.3 eV) increased with the presence of coatings with higher concentrations of NMe4+. (D) Changes in the water contact angle according to the NMe4+ Cl− concentrations and coating times (red line for 1 M NMe4+, blue line for 0.5 M NMe4+, and black line for 0.1 M NMe4+). A water contact angle below 5° is defined as superhydrophilic.
We further investigated the contribution of NMe4+ in Md-Bc in addition to the St-Bc condition. Related to this, our new hypothesis is that there are two cationic species competitively participating in cation-π interactions in Md-Bc: R-NH3+ in dopamine and NMe4+ can competitively bind to the indole ring. The initial growth of polydopamine on a transparent glass substrate was monitored by the UV absorption at 400 nm as the surface color became dark brown progressively. NMe4+ showed minimal influence in the coating rate in Md-Bc as shown by the blue lines in fig. S4A, which is different from the case for St-Bc, where the initial growth of the coating dramatically increased in the presence of NMe4+ (red lines in fig. S4A). Chemical composition analysis of the nitrogen species by XPS indicated that there was minimal intercalation of NMe4+ in Md-Bc, and most of cationic amines on the coated surface were R-NH3+ (area ratio, 0.21; fig. S4B) rather than NR4+ (area ratio, 0.1). From this investigation, in Md-Bc, we suggest that the main driving force in polydopamine coating is achieved by the contribution of the protonated R-NH3+ in dopamine even in the presence of excess of quaternary ammonium. As there was no significant difference in the chemical composition of the coating in the absence and presence of NMe4+, the surface wettability also was not altered by adding NMe4+ in Md-Bc (fig. S4C). Similar to NMe4+, we tested another cationic small molecule with a quaternary ammonium named choline as a surrogate for NMe4+ in polydopamine coating. Intercalation of choline (a precursor to neurotransmitter ACh) also resulted in superhydrophilic surfaces in the St-Bc pH condition as shown in fig. S5.
We demonstrate in Figs. 3 and 4 that cation-π interactions are the primary intermolecular binding forces in polydopamine coating. Many studies use polydopamine as a synthetic mimetic material for dopamine-melanin (23, 42). At this point, the following question may arise. Why does nature use unusual cation-π intermolecular interactions in the progressive assembly of catecholamine? The first reason is the enhanced water solubility in aromatic π configurations. The representative function of melanin is strong light absorption due to its various aromatic ring structures. However, molecules with rich aromatic rings are essentially water-insoluble. Most bioactive polyphenol compounds, such as catechin, tannin, and quercetin, are also water-insoluble (57). Therefore, nature chose to add a cationic group to the aromatic system to enhance the water solubility and not use pure aromatic systems. The second reason is to achieve multifunctionality via the progressive intermolecular assembly of various oxidized catecholamine products. As aforementioned, most polyphenols are water-insoluble, but nature wants to gain multifunctionality by assembling various phenolic compounds. For this purpose, the cation serves as a permanent bridge for the further assembly of catecholamine species that are newly generated in the catecholamine oxidation pathway. In addition, eumelanin in skin and eyes can effectively gain light absorption for almost the entire UV to near-infrared (IR) light spectrum because of the cation-π progressive assembly of heterogeneous chromophores rather than a simple unassembled array of a number of chromophores (Fig. 5A) (58). Figure 5B explains the light absorption spectra broadening during polydopamine progressive assembly similar to melanogenesis in nature. Up to 12 hours, the absorption of the wide range of light from UV to near-IR was continuously increased: (i) dopamine only absorbed UV light at 280 nm in the initial stage prior to the oxidative reaction initiation (T = 0 hours), (ii) the absorption of the wide range of wavelengths from 300 to 700 nm appeared (T = 1 hour), and (iii) the absorption function from 700 to 900 nm was newly gained by the further progressive assembly in time, covering the entire range of wavelengths from 300 to 900 nm (T = 10 hours). Although it is difficult to define individual chromophores, we understand that different types of monomeric species, such as dopamine-quinone, DHI, indolequinone, and quinone-imine, can be covalently combined into a variety of oligomers with different oxidative states and progressively assembled simultaneously, contributing to the broadening of light absorption over a wide range of wavelengths (Fig. 5C) (59).
Fig. 5. “Function gaining” by progressive cation-π assembly.
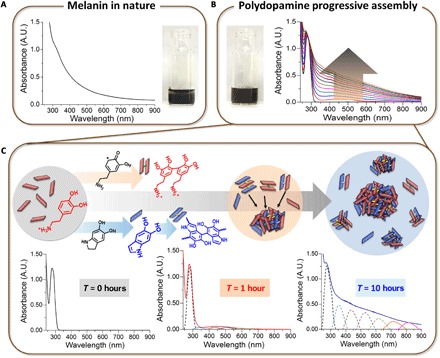
(A) UV-visible (vis) absorbance of eumelanin from Sepia officinalis dissolved in tris buffer (pH 8.5). A.U., arbitrary units. (B) Continuous monitoring of polydopamine formation in tris buffer (pH 8.5) up to 12 hours via the UV-vis absorption spectra. The reaction solution became dark brown to black after 12 hours (the arrow). (C) Covalent and noncovalent progressive assemblies of eumelanin-mimetic polydopamine to gain light protection functions over time.
CONCLUSIONS
Finally, the multifunctionality gained progressively over time cannot be achieved for other scenarios of chemical structures. For “phenol systems only” without cationic amines, such as catechol, gallol, and others, oxidation-induced phenol-to-phenol coupling results in increases in molecular weight, which easily triggers precipitation. For “phenol-anion systems,” such as l-DOPA, caffeic acid, and others, there might be two competitive bindings. One is the electrostatic binding between the carboxylic anion and incoming cation salt (K+, etc.). The other is cation-π interactions. Thus, the well-known fuzzy and broad spectral absorption functions shown by melanin can only be achieved from a “phenol-cation system” through cation-π–mediated progressive assembly. In summary, the results shown in our study on cation-π–mediated progressive assembly provide new insights into biological molecular assembly regarding how molecules gradually obtain their functions over time.
MATERIALS AND METHODS
Materials
Dopamine hydrochloride (DA; H8502), potassium chloride (P9333), tetramethylammonium chloride (T19526), potassium hydroxide (P1767), melanin from S. officinalis (M2659), and Sylgard 184 were purchased from Sigma-Aldrich Chemical Co. Sodium hydroxide (39155-0350) was purchased from Junsei Chemical Co. Silicon wafers were obtained from TASCO Inc. Titanium wafers were prepared by thermal evaporation of titanium (200 nm) onto Si wafers, and Au wafers were prepared by thermal evaporation of Au (200 nm) onto Ti-deposited (20 nm) Si wafers. SS304 was purchased from Shinsan ENG Co. Ltd. TPU was obtained from SK F&C Co. Ltd. Microscope slides from Marienfeld Superior were used as glass substrates. CN membranes with a porosity of 0.45 μm (Whatman Inc.) were used. PET was purchased from Kumjeong Inc. PE and nylon fabrics were obtained from Kolon Industries Inc. PTFE membranes with a pore size of 0.2 μm were purchased from Scilab Korea Co. Ltd.
Methods
Preparation of polydopamine and polydopamine/cation coatings
DA (2 mg/ml) was dissolved in 4 ml of DDW and mixed with different amounts of 0.2 N NaOH solution (20, 40, 60, 120, 240, 300, 360, or 420 μl). Each DA/NaOH solution was measured to determine its pH value, with the values being as follows: pH 7.4 (20 μl of NaOH), pH 7.7 (40 μl of NaOH), pH 8.2 (60 μl of NaOH), pH 8.8 (120 μl of NaOH), pH 9.8 (240 μl of NaOH), pH 10.2 (300 μl of NaOH), pH 10.3 (360 μl of NaOH), and pH 10.8 (420 μl of NaOH). Various substrates with a 1 × 1–cm2 size were placed in six-well plates with 3 ml of DA/NaOH solution. After 24 hours of shaking at 70 rpm, the polydopamine-coated surfaces were washed with DDW and then dried by blowing nitrogen gas. Polydopamine/cation coating was conducted under St-Bc conditions (pH 9.8). Potassium chloride (KCl) and tetramethylammonium chloride (NMe4+Cl−) were dissolved in DDW. DA (2 mg/ml) was dissolved in 4 ml of different concentrations of KCl solution (20, 50, or 100 mM) or NMe4+Cl− solution (0.1, 0.5, or 1 M) and then added to 240 μl of 0.2 N NaOH (pH 9.8). Various substrates with a 1 × 1–cm2 size were placed in six-well plates with 3 ml of DA/cation solution and then shaken at 70 rpm for 1, 3, 5, 8, 12, or 24 hours. After coating, the polydopamine-coated substrates were washed with DDW and dried by blowing nitrogen gas.
Characterization of polydopamine-coated surfaces
The thickness of the polydopamine coatings was measured by a spectroscopic ellipsometer (M2000D, JA Woollam Co. Inc.) with a He-Ne laser (632.8 nm) at a 70° incidence angle. The atomic composition of the surfaces was analyzed using XPS (K-Alpha, Thermo VG Scientific). The surface morphologies were observed with a scanning electron microscope (Inspect F50, FEI). The static water contact angles of the surfaces were compared before and after coating with a Phoenix 300 goniometer (Surface Electro Optics Co. Ltd.) by dropping 5 μl of DDW on the surfaces.
Delamination of polydopamine coating via base treatment
TPU films and TiO2 wafers were coated with polydopamine under the Md-Bc condition (pH 8.8) for 24 hours. The polydopamine-coated TPU films with a 3 × 3–cm2 size were dipped into 5 ml of 0.1 N NaOH (pH 12), DDW, or 0.1 N KOH (pH 12) solution for 1 hour, which was recorded as a video. The polydopamine coating in NaOH was delaminated from the TPU film, whereas those in KOH and DDW remained. Each TiO2 wafer with a 1 × 1–cm2 size was treated with 3 ml of either 0.1 N NaOH or 0.1 N KOH (pH 11) for 1 hour. After base treatment, TiO2 wafers were washed with DDW and dried by blowing N2 gas. The changes in the atomic compositions of the polydopamine-coated TiO2 surfaces were analyzed by XPS after base treatment.
UV-vis absorption analyses for DA and natural melanin polymerization
In total, DA (0.2 mg/ml) was dissolved in 10 mM tris buffer (pH 8.5). The absorbance of both DA solutions was monitored for 12 hours using a UV-vis spectrophotometer (HP8453, Hewlett Packard). Next, melanin (2 mg/ml) from S. officinalis was dissolved in 10 mM tris buffer and centrifuged. The UV-vis absorbance of the supernatant solution was measured.
HPLC-MS analyses with the polydopamine solution
DA (2 mg/ml) was oxidized for 24 hours under Md-Bc (pH 8.8) and St-Bc (pH 9.8) conditions by adjusting the pH values with 0.2 N NaOH. Both polydopamine solutions were filtered (0.45-μm pore size; Minisart, Sartorious Stedim Biotech), and then, 100 μl of each solution was injected into the HPLC equipment (Agilent 1260 Series). The solution passed through a C18 column (3-μm particle size, Supelco Analytical) at a flow rate of 0.5 ml/min, and the eluents were detected using UV-vis spectroscopy (280, 320, and 420 nm). The gradient conditions were as follows: 5 to 90% acetonitrile in 0.1% trifluoroacetic acid over periods ranging from 5 to 35 min, 95% acetonitrile at 40 min, and 5% acetonitrile at 43 min.
Supplementary Material
Acknowledgments
Funding: This research was supported by the National Research Foundation of Republic of Korea (NRF) Grant funded by the Ministry of Science, ICT and Future Planning for convergent research: Development program for convergence R&D over traditional culture and current technology (NRF-2016M3C1B5906485; to H.L.), Mid-career Researcher Program (NRF-2017R1A2A1A05001047; to H.L.), and Center for multiscale chiral structures (2018R1A5A1025208; to H.L.). This research was also supported by Basic Science Research Program through the NRF funded by the Ministry of Education (NRF-2018R1D1A1B07045249; to S.H.). S.H. thanks the Faculty Start-Up Fund (2018010108) of Daegu Gyeongbuk Institute of Science and Technology for support. Author contributions: S.H., Y.W., and H.L. conceived and designed the experiments. S.H. and Y.W. performed the experiments, analyzed the data, and prepared the figures. S.H., Y.W., and H.L. wrote the paper. S.Y.P. provided critical comments and necessary experimental results during the revision process. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaat7457/DC1
Fig. S1. Chemical structure of oligomers formed during polydopamine coating under St-Bc pH conditions analyzed by HPLC-MS with a C18 column.
Fig. S2. XPS analyses of polydopamine-coated TiO2 surfaces after 1 hour of strong base treatment.
Fig. S3. Maintenance of polydopamine coating on a TPU film in 0.1 N KOH (pH 12) solution for 1 hour.
Fig. S4. Intercalation of NMe4+ on polydopamine coating with pH variations.
Fig. S5. Wettability of polydopamine-coated substrates in Md-Bc and St-Bc conditions in the presence and absence of 1 M choline, a precursor to neurotransmitter ACh.
Fig. S6. Surface morphologies of the NMe4+-intercalated polydopamine functionalized surfaces at St-Bc with 0.1 M NMe4+ (top) and 1 M NMe4+ (bottom) by SEM and atomic force microscopy analyses.
Movie S1. Stability of polydopamine coating in NaOH, DDW, and KOH solution.
REFERENCES AND NOTES
- 1.Israelachvili J. N., Mitchell D. J., Ninham B. W., Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 2 72, 1525–1568 (1976). [Google Scholar]
- 2.Allen T. M., Cullis P. R., Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Théry C., Zitvogel L., Amigorena S., Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2, 569–579 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Kornberg R. D., Lorch Y., Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98, 285–294 (1999). [DOI] [PubMed] [Google Scholar]
- 5.Conde C., Cáceres A., Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 10, 319–332 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Janin J., Bahadur R. P., Chakrabarti P., Protein-protein interaction and quaternary structure. Q. Rev. Biophys. 41, 133–180 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Levy E. D., Erba E. B., Robinson C. V., Teichmann S. A., Assembly reflects evolution of protein complexes. Nature 453, 1262–1265 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Love J. C., Estroff L. A., Kriebel J. K., Nuzzo R. G., Whitesides G. M., Self-assembled monolayers of thiolates on metals as a form of nanotechnology. Chem. Rev. 105, 1103–1170 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Li Y., Wang X., Sun J., Layer-by-layer assembly for rapid fabrication of thick polymeric films. Chem. Soc. Rev. 41, 5998–6009 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Kharlampieva E., Kozlovskaya V., Sukhishvili S. A., Layer-by-layer hydrogen-bonded polymer films: From fundamentals to applications. Adv. Mater. 21, 3053–3065 (2009). [Google Scholar]
- 11.He Y., Ye T., Su M., Zhang C., Ribbe A. E., Jiang W., Mao C., Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nature 452, 198–201 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Castro C. E., Kilchherr F., Kim D.-N., Shiao E. L., Wauer T., Wortmann P., Bathe M., Dietz H., A primer to scaffolded DNA origami. Nat. Methods 8, 221–229 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Reches M., Gazit E., Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 300, 625–627 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Kwon S., Jeon A., Yoo S. H., Chung I. S., Lee H.-S., Unprecedented molecular architectures by the controlled self-assembly of a β-peptide foldamer. Angew. Chem. Int. Ed. 122, 8408–8412 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Hartgerink J. D., Beniash E., Stupp S. I., Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 294, 1684–1688 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Lerner A. B., Fitzpatrick T. B., Biochemistry of melanin formation. Physiol. Rev. 30, 91–126 (1950). [DOI] [PubMed] [Google Scholar]
- 17.Simon J. D., Peles D., Wakamatsu K., Ito S., Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 22, 563–579 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Lee H., Dellatore S. M., Miller W. M., Messersmith P. B., Mussel-inspired surface chemistry for multifunctional coatings. Science 318, 426–430 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pezzella A., Panzella L., Natangelo A., Arzillo M., Napolitano A., d’Ischia M., 5,6-Dhydroxyindole tetramers with “anomalous” interunit bonding patterns by oxidative coupling of 5,5’,6,6’-tetrahydroxy-2,7’-biindolyl: Emerging complexities on the way toward an improved model of eumelanin buildup. J. Org. Chem. 72, 9225–9230 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Bisaglia M., Mammi S., Bubacco L., Kinetic and structural analysis of the early oxidation products of dopamine: Analysis of the interactions with α-synuclein. J. Biol. Chem. 282, 15597–15605 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Kang S. M., Hwang N. S., Yeom J., Park S. Y., Messersmith P. B., Choi I. S., Langer R., Anderson D. G., Lee H., One-step multipurpose surface functionalization by adhesive catecholamine. Adv. Funct. Mater. 22, 2949–2955 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei Q., Zhang F., Li J., Li B., Zhao C., Oxidant-induced dopamine polymerization for multifunctional coatings. Polym. Chem. 1, 1430–1433 (2010). [Google Scholar]
- 23.Ju K.-Y., Lee Y., Lee S., Park S. B., Lee J.-K., Bioinspired polymerization of dopamine to generate melanin-like nanoparticles having an excellent free-radical-scavenging property. Biomacromolecules 12, 625–632 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Clancy C. M. R., Nofsinger J. B., Hanks R. K., Simon J. D., A hierarchical self-assembly of eumelanin. J. Phys. Chem. B 104, 7871–7873 (2000). [Google Scholar]
- 25.d’Ischia M., Napolitano A., Pezzella A., Meredith P., Sarna T., Chemical and structural diversity in eumelanins: Unexplored bio-optoelectronic materials. Angew. Chem. Int. Ed. 48, 3914–3921 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito S., Encapsulation of a reactive core in neuromelanin. Proc. Natl. Acad. Sci. U.S.A. 103, 14647–14648 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sulzer D., Zecca L., Intraneuronal dopamine-quinone synthesis: A review. Neurotox. Res. 1, 181–195 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Dougherty D. A., Stauffer D. A., Acetylcholine binding by a synthetic receptor: Implications for biological recognition. Science 250, 1558–1560 (1990). [DOI] [PubMed] [Google Scholar]
- 29.Sussman J. L., Harel M., Frolow F., Oefner C., Goldman A., Toker L., Silman I., Atomic structure of acetylcholinesterase from Torpedo californica: A prototypic acetylcholine-binding protein. Science 253, 872–879 (1991). [DOI] [PubMed] [Google Scholar]
- 30.Torrice M. M., Bower K. S., Lester H. A., Dougherty D. A., Probing the role of the cation-π interaction in the binding sites of GPCRs using unnatural amino acids. Proc. Natl. Acad. Sci. U.S.A. 106, 11919–11924 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dougherty D. A., The cation−π interaction. Acc. Chem. Res. 46, 885–893 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D., Hu S., Tarakeshwar P., Kim K. S., Cation-π interactions: A theoretical investigation of the interaction of metallic and organic cations with alkenes, arenes, and heteroarenes. J. Phys. Chem. A 107, 1228–1238 (2003). [Google Scholar]
- 33.Dougherty D. A., Cation-π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 271, 163–168 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Gallivan J. P., Dougherty D. A., A computational study of cation−π interactions vs salt bridges in aqueous media: Implications for protein engineering. J. Am. Chem. Soc. 122, 870–874 (2000). [Google Scholar]
- 35.Hong S., Kim K. Y., Wook H. J., Park S. Y., Lee K. D., Lee D. Y., Lee H., Attenuation of the in vivo toxicity of biomaterials by polydopamine surface modification. Nanomedicine 6, 793–801 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Ye Q., Zhou F., Liu W., Bioinspired catecholic chemistry for surface modification. Chem. Soc. Rev. 40, 4244–4258 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Ho C.-C., Ding S.-J., The pH-controlled nanoparticles size of polydopamine for anti-cancer drug delivery. J. Mater. Sci. Mater. Med. 24, 2381–2390 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Farnad N., Farhadi K., Voelcker N. H., Polydopamine nanoparticles as a new and highly selective biosorbent for the removal of copper (II) ions from aqueous solutions. Water Air Soil Pollut. 223, 3535–3544 (2012). [Google Scholar]
- 39.Okuda H., Yoshino K., Wakamatsu K., Ito S., Sota T., Degree of polymerization of 5,6-dihydroxyindole-derived eumelanin from chemical degradation study. Pigment Cell Melanoma Res. 27, 664–667 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Chen C.-T., Martin-Martinez F. J., Jung G. S., Buehler M. J., Polydopamine and eumelanin molecular structures investigated with ab initio calculations. Chem. Sci. 8, 1631–1641 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liebscher J., Mrówczyński R., Scheidt H. A., Filip C., Hădade N. D., Turcu R., Bende A., Beck S., Structure of polydopamine: A never-ending story? Langmuir 29, 10539–10548 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Della Vecchia N. F., Avolio R., Alfè M., Errico M. E., Napolitano A., d’Ischia M., Building-block diversity in polydopamine underpins a multifunctional eumelanin-type platform tunable through a quinone control point. Adv. Funct. Mater. 23, 1331–1340 (2013). [Google Scholar]
- 43.Alfieri M. L., Micillo R., Panzella L., Crescenzi O., Oscurato S. L., Maddalena P., Napolitano A., Ball V., d’Ischia M., Structural basis of polydopamine film formation: Probing 5,6-dihydroxyindole-based eumelanin type units and the porphyrin issue. ACS Appl. Mater. Interfaces 10, 7670–7680 (2018). [DOI] [PubMed] [Google Scholar]
- 44.d’Ischia M., Napolitano A., Ball V., Chen C.-T., Buehler M. J., Polydopamine and eumelanin: From structure–property relationships to a unified tailoring strategy. Acc. Chem. Res. 47, 3541–3550 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Hong S., Na Y. S., Choi S., Song I. T., Kim W. Y., Lee H., Non-covalent self-assembly and covalent polymerization co-contribute to polydopamine formation. Adv. Funct. Mater. 22, 4711–4717 (2012). [Google Scholar]
- 46.Dreyer D. R., Miller D. J., Freeman B. D., Paul D. R., Bielawski C. W., Elucidating the structure of poly(dopamine). Langmuir 28, 6428–6435 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Gebbie M. A., Wei W., Schrader A. M., Cristiani T. R., Dobbs H. A., Idso M., Chmelka B. F., Waite J. H., Israelachvili J. N., Tuning underwater adhesion with cation-π interactions. Nat. Chem. 9, 473–479 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Davis M. R., Dougherty D. A., Cation-π interactions: Computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation. Phys. Chem. Chem. Phys. 17, 29262–29270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu Q., Oh D. X., Lee Y., Jho Y., Hwang D. S., Zeng H., Nanomechanics of cation-π interactions in aqueous solution. Angew. Chem. Int. Ed. 52, 3944–3948 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Prampolini G., Cacelli I., Ferretti A., Intermolecular interactions in eumelanins: A computational bottom-up approach. I. Small building blocks. RSC Adv. 5, 38513–38526 (2015). [Google Scholar]
- 51.Watt A. A. R., Bothma J. P., Meredith P., The supramolecular structure of melanin. Soft Matter 5, 3754–3760 (2009). [Google Scholar]
- 52.Xue Z., Wang S., Lin L., Chen L., Liu M., Feng L., Jiang L., A novel superhydrophilic and underwater superoleophobic hydrogel-coated mesh for oil/water separation. Adv. Mater. 23, 4270–4273 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Thompson C. S., Fleming R. A., Zou M., Transparent self-cleaning and antifogging silica nanoparticle films. Sol. Energy Mater. Sol. Cells 115, 108–113 (2013). [Google Scholar]
- 54.Ishizaki T., Saito N., Takai O., Correlation of cell adhesive behaviors on superhydrophobic, superhydrophilic, and micropatterned superhydrophobic/superhydrophilic surfaces to their surface chemistry. Langmuir 26, 8147–8154 (2010). [DOI] [PubMed] [Google Scholar]
- 55.Tian Y., Jiang L., Wetting: Intrinsically robust hydrophobicity. Nat. Mater. 12, 291–292 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Ponzio F., Barthes J., Bour J., Michel M., Bertani P., Hemmerlé J., d’Ischia M., Ball V., Oxidant control of polydopamine surface chemistry in acids: A mechanism-based entry to superhydrophilic-superhydophobic coatings. Chem. Mater. 28, 4697–4705 (2016). [Google Scholar]
- 57.Haslam E., Natural polyphenols (vegetable tannins) as drugs: Possible modes of action. J. Nat. Prod. 59, 205–215 (1996). [DOI] [PubMed] [Google Scholar]
- 58.Tran M. L., Powell B. J., Meredith P., Chemical and structural disorder in eumelanins: A possible explanation for broadband absorbance. Biophys. J. 90, 743–752 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaxiras E., Tsolakidis A., Zonios G., Meng S., Structural model of eumelanin. Phys. Rev. Lett. 97, 218102 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/9/eaat7457/DC1
Fig. S1. Chemical structure of oligomers formed during polydopamine coating under St-Bc pH conditions analyzed by HPLC-MS with a C18 column.
Fig. S2. XPS analyses of polydopamine-coated TiO2 surfaces after 1 hour of strong base treatment.
Fig. S3. Maintenance of polydopamine coating on a TPU film in 0.1 N KOH (pH 12) solution for 1 hour.
Fig. S4. Intercalation of NMe4+ on polydopamine coating with pH variations.
Fig. S5. Wettability of polydopamine-coated substrates in Md-Bc and St-Bc conditions in the presence and absence of 1 M choline, a precursor to neurotransmitter ACh.
Fig. S6. Surface morphologies of the NMe4+-intercalated polydopamine functionalized surfaces at St-Bc with 0.1 M NMe4+ (top) and 1 M NMe4+ (bottom) by SEM and atomic force microscopy analyses.
Movie S1. Stability of polydopamine coating in NaOH, DDW, and KOH solution.