

Abstract
SYNGAP1 loss-of-function variants are causally associated with intellectual disability, severe epilepsy, autism spectrum disorder and schizophrenia. While there are hundreds of genetic risk factors for neurodevelopmental disorders (NDDs), this gene is somewhat unique because of the frequency and penetrance of loss-of-function variants found in patients combined with the range of brain disorders associated with SYNGAP1 pathogenicity. These clinical findings indicate that SYNGAP1 regulates fundamental neurodevelopmental processes that are necessary for brain development. Here, we describe four phenotypic domains that are controlled by Syngap1 expression across vertebrate species. Two domains, the maturation of cognitive functions and maintenance of excitatory-inhibitory balance, are defined exclusively through a review of the current literature. Two additional domains are defined by integrating the current literature with new data indicating that SYNGAP1/Syngap1 regulates innate survival behaviors and brain structure. These four phenotypic domains are commonly disrupted in NDDs, suggesting that a deeper understanding of developmental Syngap1 functions will be generalizable to other NDDs of known or unknown etiology. Therefore, we discuss the known molecular and cellular functions of Syngap1 and consider how these functions may contribute to the emergence of disease-relevant phenotypes. Finally, we identify major unexplored areas of Syngap1 neurobiology and discuss how a deeper understanding of this gene may uncover general principles of NDD pathobiology.
Keywords: Syngap1, SynGAP, Intellectual Disability, Autism Spectrum Disorder, Neurodevelopment, Circuits, Synapse, Microcephaly, Cognitive impairment, Epilepsy
SYNGAP1 gene function is important in health and disease
The advent of genomic sequencing in previously undefined patients with NDDs has demonstrated that there is a subset of autosomal genes that confer near 100% risk for developing ID, ASD and/or epilepsy (Deciphering Developmental Disorders, 2015, 2017; Epi et al., 2013). There is considerable interest in understanding the molecular and cellular mechanisms regulated by this subset of high-impact genetic risk factors (Hoischen et al., 2014; Zhu et al., 2014). In-depth study of animal models harboring pathogenic variants common to patient populations is a powerful approach for determining linkages between molecular and cellular functions of distinct disease risk factors, and perhaps more importantly, how possible convergent molecular mechanisms contribute to disease-relevant phenotypes. Through in-depth biological investigations of highly-penetrant risk genes in model systems, it may be possible to identify common molecular mechanisms that converge to influence disease-relevant phenotypes. With such knowledge, therapeutic approaches developed through modeling of one gene may be successfully applied to other NDDs of known or unknown etiology.
The NDD risk-factor, SYNGAP1, is a major cause of genetically-defined childhood brain disorders and an attractive candidate for in-depth investigations that span multiple model systems. The SYNGAP1 gene has emerged as a high-risk locus for neuropsychiatric disorders that cross diagnostic barriers (Hoischen et al., 2014; Zhu et al., 2014). Indeed, causal rare variants are found in enriched populations with ID (Deciphering Developmental Disorders, 2015, 2017; Hamdan et al., 2009; Rauch et al., 2012), ASD (Hamdan et al., 2011; O'Roak et al., 2014), severe epilepsy (Carvill et al., 2013; von Stulpnagel et al., 2015) and schizophrenia (Purcell et al., 2014). Severe de novo variants in SYNGAP1 resulting in haploinsufficiency lead to a defined phenotype characterized by ID with epilepsy [termed Mental Retardation- Type 5(MRD5); OMIM#603384] that may explain up to 1% of ID cases (Berryer et al., 2013; Deciphering Developmental Disorders, 2015, 2017). A recent study has found that there are no severe or obvious pathogenic SYNGAP1 variants in more than 60,000 subjects lacking any known neuropsychiatric conditions, solidifying the notion that pathogenic SYNGAP1 loss-of-function variants are both highly penetrant and sufficient to cause NDDs (Kosmicki et al., 2017).
SynGAP proteins, the products encoded by the SYNGAP1/Syngap1 gene (Chen et al., 1998; Kim et al., 1998), lie at a critical intersection of protein signaling networks strongly linked to a spectrum of NDDs. Mechanisms that drive brain dysfunction associated with neuropsychiatric disorders intersect at excitatory synapse regulation in glutamatergic neurons. For example, the NMDA receptor signaling complex within dendritic spine synapses is enriched with proteins encoded by a high-proportion of genes with pathogenic variants linked to a range of neuropsychiatric disorders marked by cognitive impairment (Bayes et al., 2014; Volk et al., 2015). SynGAP is both a core postsynaptic density (PSD) protein and a major constituent of the NMDA receptor signaling complex (Bayes et al., 2012; Chen et al., 1998; Kim et al., 1998). SynGAP protein-protein interactions are believed to promote organization of macromolecular complexes within dendritic spines (Walkup et al., 2016; Zeng et al., 2017). Moreover, SynGAP also regulates mRNA translation machinery (Barnes et al., 2015; Wang et al., 2013) that regulates excitatory synapse plasticity, which is a cellular process believed to contribute to ASD pathogenesis (Huber et al., 2015; Richter et al., 2015).
Species-Aligned Phenotypic Domains Regulated by Syngap1 Function
A relatively homogenous human phenotype emerges in patients harboring pathogenic variants causing SYNGAP1 haploinsufficiency (Berryer et al., 2013; Mignot et al., 2016; Parker et al., 2015). Indeed, ∼85% of known patients with pathogenic SYNGAP1 variants have rare, loss-of-function variants predicted to cause reduced protein expression or function (Mignot et al., 2016). The high proportion of loss-of-function variants, combined with a relatively homogenous core phenotype in humans, indicates that SYNGAP1 has essential natural functions during brain development.
Here, we discus known human phenotypes (Berryer et al., 2013; Mignot et al., 2016; Parker et al., 2015) in the context of conservation across several vertebrate species to highlight the fundamental importance of SYNGAP1/Syngap1 in sculpting brain function. Studies performed across species demonstrate that this gene has retained functions throughout vertebrate evolution to promote cognitive functions, excitatory-inhibitory (E-I) balance, brain structure, and innate behavioral adaptations (see following sub-sections). The alignment of SYNGAP1/Syngap1 phenotypes across vertebrate species suggests that it controls fundamental cellular processes that promote the assembly and function of neural circuits that underlie behavior and cognition. Thus, in-depth study of Syngap1 phenotypes in animal models may provide molecular insight into the shared pathobiology of NDDs.
Phenotype 1: Cognitive function
Reduced cognitive function is a common feature of many NDDs, including intellectual disability, ASD, epilepsy and schizophrenia. In humans, proper SYNGAP1 expression is essential for the development of cognitive abilities. SYNGAP1 haploinsufficiency leads to an intellectual disability disorder characterized by severe cognitive impairment (Berryer et al., 2013; Mignot et al., 2016; Parker et al., 2015). Most patients have reduced capacity for language and are non-verbal. IQ is usually <50 and patients have impaired executive functions. Animal models with disrupted Syngap1 expression also display behaviors and neurophysiological abnormalities consistent with cognitive impairment (Guo et al., 2009; Komiyama et al., 2002; Muhia et al., 2010; Ozkan et al., 2014). For instance, Syngap1 heterozygous KO mice (Hets) express disruptions in various forms of learning and memory, including impaired spatial learning, altered spatial working memory, weakened social memory, and deficits in remote contextual memory consolidation. These learning and memory phenotypes are highly reproducible in mice with Syngap1 haploinsufficiency, having been observed across several laboratories using independently generated Syngap1 knockout lines (Guo et al., 2009; Komiyama et al., 2002; Muhia et al., 2010; Ozkan et al., 2014).
Syngap1 also regulates synaptic plasticity in the same brain regions that support memory and cognition. Disruptions to this cellular process may contribute to the cognitive impairments displayed by Syngap1 haploinsufficient animals. Indeed, reduced germline Syngap1 expression causes deficits in various forms of hippocampal synaptic plasticity. Long-term potentiation (LTP) is a cellular correlate of learning and memory and the synaptic strengthening that accompanies LTP is believed to be a mechanism for storing newly acquired information within neural circuits (Lynch et al., 2007; Nicoll, 2017). Alterations in this cellular process is, therefore, an attractive substrate for cognitive impairments commonly observed in NDDs. Consistent with this, many animal models of NDD risk gene pathogenicity display impaired synaptic plasticity (Araujo et al., 2017; Lauterborn et al., 2007; Lee et al., 2014; Li et al., 2016), including LTP impairments in Syngap1 Het mice (Clement et al., 2013; Kim et al., 2003; Komiyama et al., 2002; Ozkan et al., 2014). Similar to learning and memory impairments, several laboratories using independently generated Syngap1 Het mouse lines and distinct induction protocols have routinely observed severely impaired LTP.
LTP deficits in Syngap1 mice are associated with alterations in NMDAR-activated Ras signaling dynamics in dendritic spines (Ozkan et al., 2014). SynGAP is a GTPase activating protein (GAP) (Chen et al., 1998; Kim et al., 1998), which inactivates several small GTPases from the Ras superfamily, including Ras, Rap1/2 and Rab5 (Krapivinsky et al., 2004; Pena et al., 2008; Tomoda et al., 2004). SynGAP content within spines is dynamically reduced in response to neuronal activation (Araki et al., 2015). This process promotes a transient elevation in Ras activation, which is known to drive AMPA receptor membrane insertion required for LTP expression (Zhu et al., 2002). Whole brain extracts (Clement et al., 2013; Kim et al., 2003; Komiyama et al., 2002) and even hippocampal dendritic spines (Ozkan et al., 2014) within Syngap1 Het mice display basally elevated Ras signaling, which occludes further Ras activation in response to synaptic stimulation. Restoring normal SynGAP protein levels in adult Syngap1 Het mice rescues both LTP expression and Ras-related signaling impairments (Ozkan et al., 2014). Together, these data support the view that a function of SynGAP protein within dendritic spines is to maintain low basal level Ras-ERK signaling in an unstimulated state, which may be a mechanism to maximize the signal/noise ratio of this pathway upon synaptic stimulation to promote LTP during learning.
Studies in long-term depression (LTD) are consistent with the role of SynGAP to balance Ras-ERK signaling at synapses. LTD is a unique form of synaptic plasticity that weakens neural connections in response to activity and may act to enhance computational flexibility within neural networks (Pinar et al., 2017). In contrast to the clear impact of Syngap1 on LTP, this gene has a more complex role in LTD. For example, LTD in response to a standard input-specific and synaptically-driven induction paradigm in CA1 is normal in Syngap1 Hets (Kim et al., 2003). Additionally, no change was observed in CA1 tissue slices from Syngap1 Hets after LTD induced by bath application of NMDA (Carlisle et al., 2008). In contrast, mGlur5-dependent CA1 LTD was enhanced and resistant to protein synthesis inhibitors (Barnes et al., 2015). This mGlur5-dependent LTD phenotype is similar to what was reported in Fmr1 KO mice (Osterweil et al., 2010), which is an animal model of Fragile X syndrome. In both Syngap1 Het and Fmr1 KO mice, pharmacological targeting of elevated Ras signaling rescued LTD phenotypes (Barnes et al., 2015). This finding demonstrates a form of molecular convergence at the synapse caused by two distinct NDD risk factors and suggests that targeting aberrant Ras signaling may improve behaviors associated with genetically distinct NDDs. Indeed, Ras-targeted therapies improve seizure phenotypes in Fmr1 mice (Osterweil et al., 2013) and memory in models of Neurofibromatosis (Li et al., 2005) and Noonan syndrome (Lee et al., 2014). However, the efficacy of Ras-regulating pharmacological therapies in similar animal-level phenotypes in Syngap1 mice remain unknown. Thus, it remains an open question if elevated Ras-ERK signaling is a causal factor driving impaired cognitive function in animals with Syngap1 heterozygosity.
Phenotype 2: Excitatory balance/Seizure
In humans, SYNGAP1 haploinsufficiency leads to impaired excitability, as nearly all patients exhibit some form of epilepsy (Berryer et al., 2013; Mignot et al., 2016; Parker et al., 2015). Early clinical reports indicated that prevalence in SYNGAP1-related disorders was 70-80%. However, when only MRD5 cases are considered (i.e. cases with clear loss-of-function variants), prevalence is believed to approach 100% (Weldon et al., 2018). Seizure type and age of onset varies among patients. Some cases involve early onset and intractable neonatal seizures (Carvill et al., 2013; Okazaki et al., 2017), including drop attacks and eyelid myoclonus, while other reports involve seizure episodes occurring later in development that are responsive to standard pharmacotherapies (Berryer et al., 2013). EEG abnormalities are common in MRD5 patients. Frequent inter-ictal cortical discharges and high-frequency oscillations are common in these patients (Berryer et al., 2013; Carvill et al., 2013).
In animal models, Syngap1 heterozygosity leads to altered neural circuit excitability, spontaneous seizure, and altered EEG waveforms. Spontaneous seizure-like behaviors are observed in a Zebrafish model of reduced syngap1 expression (Kozol et al., 2015). Similar to what was found in MRD5 patients, generalized, bilateral spike discharges were frequently observed during EEG recordings in Syngap1 Het mice (Ozkan et al., 2014). Consistent with altered neural excitability, the threshold for pharmacological seizure induction was lower in Syngap1 mice (Clement et al., 2012; Guo et al., 2009). Interestingly, restricting Syngap1 haploinsufficiency to cortical glutamatergic neurons and glia was sufficient to lower seizure threshold, while selectively repairing Syngap1 haploinsufficiency within these brain cells protected animals from developing seizure threshold phenotypes (Ozkan et al., 2014). It is important to mention that Syngap1 is also expressed in GABAergic neurons (Moon et al., 2008; Zhang et al., 1999). Restricting Syngap1 haploinsufficiency to GABAergic neurons disrupts oscillatory activity within cortical networks (Berryer et al., 2016), but does not change seizure threshold (Ozkan et al., 2014).
Maintaining a balance between excitation and inhibition (E-I balance) within neural circuits is essential for normal brain function (Deneve and Machens, 2016; Haider et al., 2006). Disrupting this balance is sufficient to impair cognitive functions and behavior (Marlin et al., 2015; Yizhar et al., 2011). As such, impaired E-I balance is a substrate of NDDs (Rubenstein and Merzenich, 2003; Tatti et al., 2017). Syngap1 controls E-I balance in the developing brain. Syngap1 is primarily a negative regulator of excitatory synaptic structure and function in developing neurons, which may explain, in part, how this gene shapes E-I balance in the brain. Downregulation of total SynGAP protein levels within glutamatergic neurons leads to an enhancement of excitatory synaptic function (Kim et al., 2003; Rumbaugh et al., 2006; Vazquez et al., 2004), while overexpression of SynGAP leads to excitatory synapse depression (Rumbaugh et al., 2006), but see (McMahon et al., 2012). Consistent with a predominantly repressive role in excitatory synapses, a larger population of GluR1 positive excitatory synapses are present in hippocampal cultures prepared from Syngap1 knockout animals (Kim et al., 2003; Vazquez et al., 2004). Indeed, Syngap1 represses excitatory synapse function though inhibition of surface AMPAR content and baseline synaptic activity (Araki et al., 2015; Rumbaugh et al., 2006; Vazquez et al., 2004; Zeng et al., 2016). In vivo, the repressive effect of Syngap1 on excitatory synapse function in mice is largely limited to a critical period of postnatal development that spans the first three postnatal weeks (Clement et al., 2012; Clement et al., 2013). Reducing SynGAP protein levels after this critical period has limited effect on excitatory synaptic function in hippocampal neurons. Seizure phenotypes and other behavioral abnormalities are present during these early developmental time periods in Syngap1 Het mice (Clement et al., 2012). Thus, haploinsufficiency of Syngap1 may trigger circuit hyperexcitability, at least in part, through elevated excitatory synapse function, tipping the balance of circuits toward excitation. The intrinsic functions of Syngap1 in interneurons is also consistent with circuit hyperexcitability. However, the role of Syngap1 in these neurons appears to promote the formation of inhibitory synapses onto glutamatergic neurons (Berryer et al., 2016). Thus, loss of SynGAP protein through haploinsufficiency further disrupts E-I balance by reducing a form of synaptic inhibition. It remains unclear how E-I balance and cognitive function are related in Syngap1 mice. There are many possible forms of E-I balance and all of them have yet to be explored experimentally. However, measures of seizure susceptibility can be dissociated from cognitive impairment in mice when Syngap1 haploinsufficiency is induced in adulthood (Ozkan et al., 2014). This indicates at least some cellular processes mediated by SynGAP that suppress seizure are distinct from cellular processes that contribute to cognitive functions.
Syngap1 also represses spine formation and maturation. Precocious dendritic spine maturation from distinct neuronal subtypes in Syngap1 Het mice has been reported by several groups. Dendritic spine formation occurs earlier in cultured neurons from Syngap1 knockout mice (Vazquez et al., 2004). In vivo studies have also shown accelerated spine formation in cortical pyramidal neurons of Syngap1 Het mice and spines that form in cortical neurons are larger than those found in wild type (WT) neurons (Aceti et al., 2015). Syngap1 modulates multiple growth-related pathways regulating protein synthesis, receptor content and cytoskeletal arrangement of dendritic spines, which may underlie alterations in dendritic spine properties in Syngap1 mutants. Reduced SynGAP expression in neurons results in increased growth promoting signaling, such as elevated Ras, mTOR and PAK activity, (Barnes et al., 2015; Carlisle et al., 2008; Komiyama et al., 2002; Rumbaugh et al., 2006; Wang et al., 2013) while decreasing activity of growth limiting pathways such as Rap1/2 and p38 MAPK (Krapivinsky et al., 2004; Rumbaugh et al., 2006).
Phenotype 3: Innate (unlearned) behavior
Recent reports suggest that MRD5 patients express impairments in innate behaviors. Specifically, parents note extreme risk-taking behaviors by their children (Weldon et al., 2018). Children do not typically require prior experience falling from especially high places to avoid such situations, as this is an innate instinct that promotes survival. However, climbing and jumping from high places is reported in MRD5 and is a major challenge for some caregivers. It is unclear what neural domains underlie these abnormal behaviors. ADHD, an impulse control disorder, is a common comorbid diagnosis with SYNGAP1-related disorders (Berryer et al., 2013; Hamdan et al., 2011). ADHD is associated with both impulsivity and risky behaviors (Dekkers et al., 2016), suggesting that behaviors observed in SYNGAP1 patients may be related to impulsivity. However, risk-taking may be due to impairments in their ability to appropriately process visual fields, leading to alterations in the perception of height or other innately dangerous situations.
Syngap1 Het mice also display impaired innate behaviors and increased risk-taking. The elevated plus maze (EPM) can quantify risk assessment in mice, as it challenges animals to balance the drive to explore a novel space with an innate fear of open spaces (Carobrez and Bertoglio, 2005). It is thought that mice generally avoid open arms of the maze because they prefer the safety of vertical walls. Anxiogenic compounds decrease open arm time (OAT) and anxiolytic compounds increase it, indicating that OAT reflects anxiety-like levels elicited from innate fear of open spaces (Walf and Frye, 2007). Syngap1 Het mice have increased OAT in EPM (Berryer et al., 2016; Guo et al., 2009). This phenotype is observed in different labs using distinct Syngap1 lines, indicating that it is highly reproducible. It remains unclear if increased OAT reflects reduced anxiety or increased exploratory drive, or both. Syngap1 mice exhibit increased locomotion in the open field test, but not in a Pavlovian fear conditioning box (Guo et al., 2009). Some groups have found no changes in horizontal locomotion in EPM (Guo et al., 2009), while others have found the opposite (Berryer et al., 2016). Syngap1 mouse activity in the homecage remains an open question.
Changes in OAT in EPM may reflect impaired learning (Bertoglio et al., 2006), suggesting that Syngap1 phenotypes in this test are unrelated to innate fear of open spaces. To confirm that increased OAT occurs through impaired innate processes rather than learning, OAT should be calculated for each one-minute interval of the test (Jurgenson et al., 2010). We reasoned that if deficits in OAT in Syngap1 mice are observed in the first minute, then altered innate processes are likely disrupted. To do this, we performed a re-analysis of EPM data previously published from our lab (Ozkan et al., 2014). In this re-analysis, we calculated open arm entries (OAE) and OAT at each minute of the test in EMX1-Syngap1 Hets, which have heterozygosity of the Syngap1 gene restricted to forebrain excitatory neurons and some glia. We chose this experiment because we observed an overall cumulative increase in OAT and this study had very high statistical power (∼n=30 per genotype). Here, we found that Syngap1 mutant mice had significant overall differences in open arm entries during the duration of the test, but not during the first one-minute bin (Fig. 1A). In contrast, OAT was significantly different over all binned periods (Fig. 1B), including the initial minute of testing, indicating that these Syngap1 mice were innately less fearful of the open arm. This idea was strengthened by observing the aggregate location of animals within the maze during the first minute of the test (Fig. 1C). WT mice entered the open arm, but then quickly moved back to the center. In contrast, Syngap1 Hets were equally likely to enter the open arm, but were more likely to thoroughly explore it before returning to the center. This behavior appeared to be the major factor driving increased OAT between the genotypes in the first minute.
Figure 1. Evidence that Syngap1 heterozygosity disrupts innate fear within the Elevated Plus Maze.
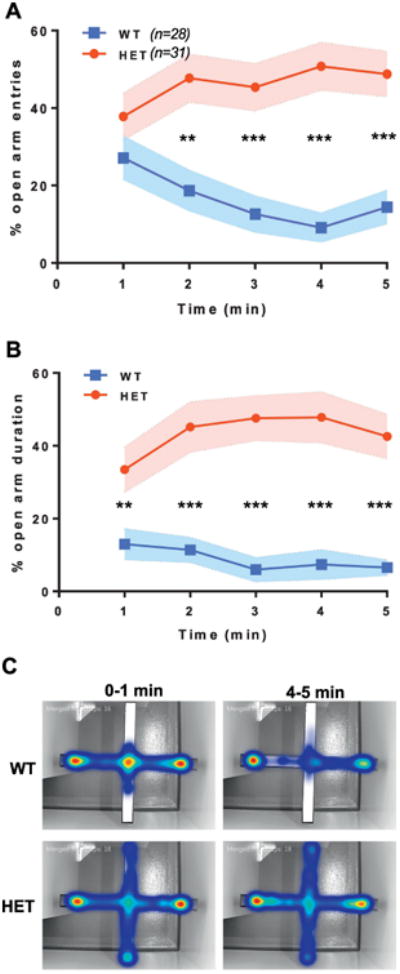
The data shown here reflect a re-analysis of an EPM experiment previously published by our group (Ozkan et al., 2014). Mice were run in a standard 5 min elevated plus maze paradigm. Percent open arm entries were binned for each minute and calculated. A) Open arm entries for EMX1-WT and EMX1-Syngap1 Hets over five one-minute bins. Large overall differences in open arm entries between genotypes (F(1,57)=29.511, p=1.197E-6); RMANOVA. Posthoc comparisons show all bins, except for the first, were significantly different from each other; **p<0.01, ***p<0.001. B) Open arm duration for EMX1-Syngap1 Hets over five one-minute bins. Large overall differences in open arm entries between genotypes (F(1,57)=44.577, p=1.098E-8); RMANOVA. Posthoc comparisons show all bins, including the first, were significantly different from each other; **p<0.01, ***p<0.001. C) Collapsed, integrated heat maps for EMX1-WT and EMX1-Syngap1 Hets showing the cumulative location of animals at two times during the test.
The EPM findings in Syngap1 Het mice suggest that they exhibit a form of reduced innate fear leading to an increase in risk-taking. To further investigate this idea, we utilized the cliff avoidance task (Matsuoka et al., 2005; Yokota et al., 2013) to determine if risky behaviors associated with heights observed in MRD5 patients was evolutionarily conserved in mice with Syngap1 haploinsufficiency. We reasoned that if Syngap1 Het mice were less afraid of heights, then they would be more likely to venture off the elevated platform. We performed two versions of the test. In the first, we reproduced the experimental conditions of Matsuoka and colleagues, and then quantified height-related risk-taking in adult (>PND60) conventional Syngap1 Het mice. We found that Syngap1 Het mice were much more likely to leave the elevated platform (Fig. 2A), consistent with the idea that these mice were less fearful of the apparatus. In the second study, we were interested in assessing innate fear of heights in developing Syngap1 Het mice. Assessing behaviors in young mice (PND21) required modifications of the task (Fig. 2B). This modified apparatus allowed us to measure two distinct behaviors, “edge departures” and “full departures” (Fig. 2C). We found that Syngap1 Het mice ventured over the sides of the platform more often than controls, as measured by increases in edge departures (Fig. 2C-E). Syngap1 Het mice were also more likely to completely leave the platform compared to Syngap1 WT mice (Fig. 2E). It is unlikely that severely impaired vision is related to this Syngap1 Het phenotype. Syngap1 Het mice are able to find the visible platform during Morris Water Maze training (Komiyama et al., 2002) and they have normal object recognition memory (Muhia et al., 2010). Along with increased OAT in EPM, these data support the idea that Syngap1 Het mice are more likely to engage in ethologically-relevant risk-taking behaviors. Prior experience was not necessary to elicit these behaviors in Syngap1 Het mice, indicating they reflect impaired innate, rather than learned, neural functions.
Figure 2. Modeling height-related risk-taking in Syngap1 mice.
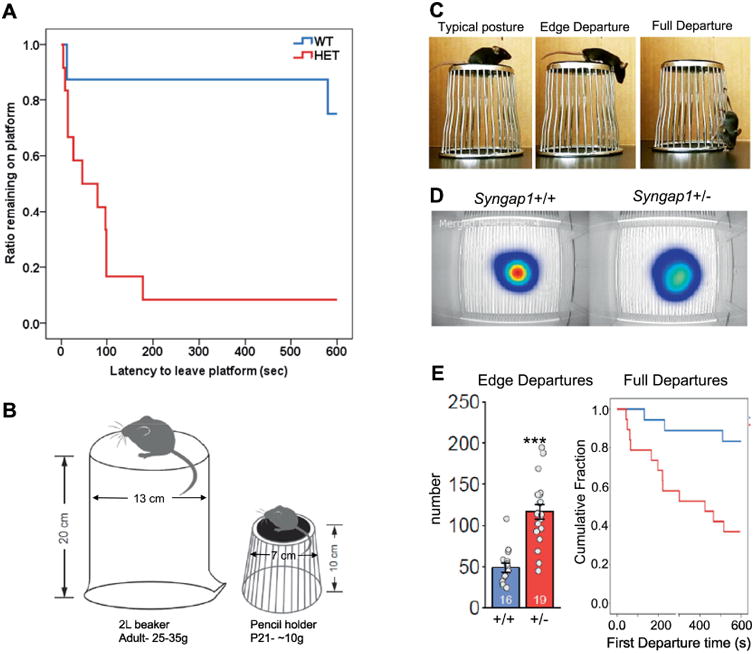
A) Cliff avoidance task in adult conventional Syngap1 animals. We calculated the fraction of animals remaining on the platform over the course of a ten-minute testing period; chi square, X2(1)= 9.061, p<0.003. B) Modifications to cliff avoidance apparatus made for testing young mice. C) Risk-taking platform with quantifiable risk-taking postures in PND21 mice. D) Top-view heat map showing cumulative location of nose around the platform for each genotype. E) Quantification of risk-taking behaviors. left, number of edge departures during the test; t-test, p<0.001. right, cumulative fraction remaining on the apparatus as animals make their first full departure from the platform; chi square, X2(1)= 8.59, p<0.005.
While the data on impaired innate survival behaviors in MRD5 patients remain suggestive (Weldon et al., 2018), the data obtained in animal models support the idea that the Syngap1 gene regulates the function of circuits that control innate survival behaviors. More clinical research in MRD5 is therefore warranted to determine the extent of risk-taking behaviors in these patients. Studies in animal models will be necessary to understand the neural basis of impaired risk-taking. It would be useful if clinical studies assessed depth perception and other forms of visual processing to understand if these behaviors are cognitive or affective in nature. Moreover, studies in Syngap1 animal models will enable in-depth neurobiological studies, such as visualization and possible manipulation of circuits that are activated during execution of behaviors that elicit innate fear of open spaces or height.
Phenotype 4: Gross/Fine brain morphology
Some MRD5 patients have mild microcephaly (Parker et al., 2015; Prchalova et al., 2017), or a reduction in head/brain size. Syngap1 heterozygous mice express a phenotype consistent with this. Small animal MRI measurements revealed a reduction in total absolute brain volume of Het mice compared to WT at PND90 (Fig. 3A-B). The effect on brain size was largely driven by males within this large cohort (Fig. 3C; Supplemental Tables 1-3). We did not measure body weight in these animals. Indeed, it is possible that a change in body weight is a confounding variable in these studies. However, brain size and body weight can vary independently from each other in mice (Ellegood et al., 2013). Moreover, at a regional level, only ∼51% of the 182 anatomically distinct regions were significantly different between male Syngap1 WT and Hets (Supplemental Table 2). This indicates that decreased total volume is not driven by global downscaling of the brain. Thus, if body size is a confound, it is unlikely to be the only factor driving a change in brain size. More studies are needed to determine mechanisms driving the reduced brain size. It is notable that many cortical areas did not reach significance, including the somatosensory cortex (Supplemental Tables 1-2). This result agrees with past literature that reported no changes in somatosensory cortex lamination in developing Syngap1 Hets (Barnett et al., 2006). However, the volume of many cortical areas related to the visual system were changed in Syngap1 Hets. This finding is interesting in the context of risk-taking phenotypes related to innate fear of open spaces or height (Figs. 1-2). It will be important to probe circuit function in visual processing areas of Syngap1 animal models and to understand how these areas interact with circuits that guide impulse control, decision making, and/or emotional valance.
Figure 3. Brain volume measurements in Syngap1 mice.
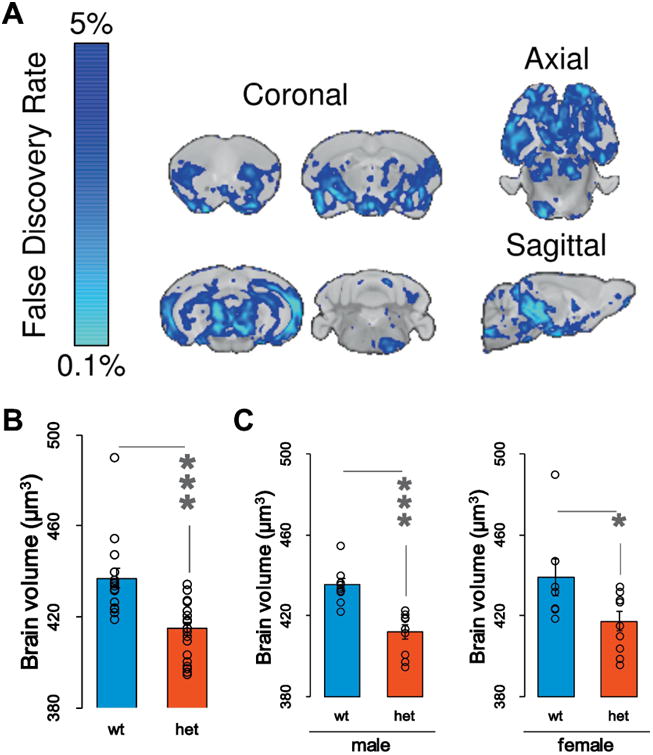
A) Various MRI sections depicting absolute changes in brain volume in Syngap1 Het mice (n=18) relative to Syngap1 WT (n=16) at PND90. Blue color represents significance of reduced volume across brain areas thresholded at 5% FDR. B) Total brain volume in WT and Het mice. Bar graphs and relative data points depicting the absolute volume change in both mutants and wildtypes [t(27.680) = 4.319 p = .0002]; C) Evaluation of total brain volume analyzing male and female separately. Male [t(16) = 4.977, p = .00014]; Female [t(14) = 2.251, p =.041]
In addition to changes in brain volume, prior studies in Syngap1 Het mice demonstrated that there are more fine-grained alterations in brain structures. Syngap1 Het mice demonstrated an impairment in cellular barrel segregation in the somatosensory cortex (Barnett et al., 2006). Syngap1 heterozygosity results in alterations in dendritic morphogenesis. L5 pyramidal neuron dendritic morphogenesis is accelerated during postnatal development in Syngap1 Hets (Aceti et al., 2015). This same dendritic phenotype was not observed in adult animals, indicating that Syngap1 represses the developmental maturation rate of dendritic morphogenesis in this neuronal subtype. A similar phenotype was also observed in deep neurons of the developing prefrontal cortex. Because dendrites are a substrate for synapse formation, altered rates of dendritic arborization are suggestive of impaired assembly of circuits during developmental critical periods. Interestingly, cell type-independent monosynaptic tracing of presynaptic inputs into the prefrontal cortex of Syngap1 Het mice revealed very few changes in anatomical long-range connectivity (Aceti et al., 2015). These findings suggest that altered patterns of circuit assembly in Syngap1 Het mice may be restricted to specific cell types or circuits. More sophisticated anatomical and functional tracing studies are required to determine how changes in dendritic morphogenesis translate into altered circuit assembly. Future studies will also be necessary to understand possible links between reduced brain volume and altered dendritic morphogenesis in Syngap1 Het mice.
Major Unexplored Areas of Syngap1 Neurobiology
A major unexplored area of Syngap1 biology relates to the molecular and/or cellular mechanisms that connect gene function to species-conserved phenotypes. Do diverse phenotypes driven by Syngap1 gene function arise through a single function of SynGAP protein? Alternatively, the Syngap1 gene may produce a range of protein functions, and these distinct functions may contribute to the range of disease-relevant phenotypes. In this latter example, termed the “single-gene multiple-hit” model, Syngap1 controls multiple and distinct molecular and/or cellular processes that converge in either simple or complex ways to produce the wide range of phenotypes observed in animal models. In its most simplistic form, this hypothesis predicts that individual animal-level phenotypes can be assigned to a precise molecular function of SynGAP. A more complex manifestation of this hypothesis is that several distinct molecular functions of SynGAP converge to produce individual phenotypes. These diverse SynGAP functions may interact in a spatial and/or temporal manner to regulate phenotypes. This hypothesis is useful because it provides a framework for drawing links between defined molecular functions driven by this gene (and resultant proteins) to emergent animal-level phenotypes. Research that establishes these links would be expected to further our basic understanding of this critical neurodevelopmental gene. Moreover, identifying these linkages would be therapeutically relevant because many phenotypes are commonly associated with NDDs.
SynGAP is best known as a modulator of glutamatergic synapses through regulation of GTPase signaling associated with the PSD (Araki et al., 2015; Chen et al., 1998; Kim et al., 1998; Ozkan et al., 2014; Rumbaugh et al., 2006; Vazquez et al., 2004). While this function of SynGAP is well accepted, it remains unclear how this, or any, function of SynGAP contributes to species-conserved phenotypes observed in response to Syngap1 loss-of-function. This uncertainty exists because animal models targeting the Syngap1 gene have, thus far, been limited to null mutations. The incorporation of more subtle variants into Syngap1 that disrupt known biological functions of the protein have yet to be carried out. This approach would be useful because it would enable a better understanding of how specific molecular functions of Syngap1 contribute to various species-aligned and disease-relevant phenotypes. The dissociation of key phenotypes from each other in animals expressing more subtle variants would promote a better understanding of how diverse molecular and/or cellular functions of SynGAP proteins contribute to brain function.
Syngap1 gene structure is well understood and the protein has been extensively studied at the biochemical level (Fig. 4A-B). Thus, there are clear strategies to disrupt specific features of the protein. A principle cellular function of SynGAP is regulation of Ras-like GTPases (Chen et al., 1998; Kim et al., 1998). This function arises through activity of the GAP domain, which stimulates the conversion of GTP-to-GDP in small GTPases. GAP function in SynGAP is unique, as it has been shown to also directly regulate Rap1/2 (Krapivinsky et al., 2004) and Rab GTPases (Tomoda et al., 2004). RapGAP activity requires an interaction between the GAP and C2 domains (Pena et al., 2008). The ability to regulate multiple GTPases enables the protein to have a powerful control over different signaling pathways within brain cells (Kennedy et al., 2005). SynGAP is heavily phosphorylated and this process regulates the activity of the GAP domain. CAMKII phosphorylation drives Ras-dominant regulation, while CDK5 phosphorylation biases activity toward inactivation of RAP1 (Walkup et al., 2015). This type of regulation is likely biologically meaningful because Ras and Rap are known to drive opposing cellular process that promote circuit assembly and synaptic plasticity (Zhu et al., 2002). SynGAP also regulates axon growth through regulation of Rab5 (Tomoda et al., 2004). Although it has only been shown in cerebellar granule neurons, SynGAP regulation of Rab5 constitutes a strong candidate for contribution to MRD5 disease mechanisms, due to its influence on early endosome formation. Disruptions in early endosome formation is associated with various brain disorders (Ouyang et al., 2013). Furthermore, this property of SynGAP is unrelated to dendritic spine biology, supporting the “multiple-hit” model of Syngap1 gene function. Animal models with engineered variants that selectively target activity at individual GTPases would help to identify signaling pathways that drive disease-relevant Syngap1 phenotypes.
Figure 4. Syngap1 alternative splicing and resulting isoforms.
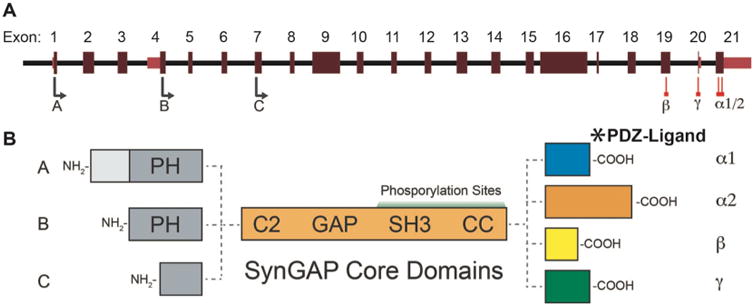
A) Map showing alternative use of exons in N and C-terminal isoforms. N-terminal variants are constituted via use of different start codons in exon 1, 4 or 7. Exon 4 is present only in B-SynGAP. C-terminal isoforms originate from use of different splice acceptors in exon 19 and 21. Exon 20 is included only in γ isoform. B) Schematics of SynGAP isoforms & protein domains. A and B isoforms include full pleckstrin homology (PH) domain. In C-SynGAP, this domain is truncated. Core regions common to all isoforms include C2, GAP, Src Homology 3 (SH3) and coiled-coil (CC) domains. Multiple phosphorylation sites are present downstream of the GAP domain. In the C-terminus, α1 isoforms contain a type-1 PDZ ligand. Structure/function relationships of α2, β, γ isoforms remain largely unknown
It has been suggested that SynGAP performs a structural role in the PSD that is independent of GTPase activity (Dosemeci et al., 2016; Walkup et al., 2016). Certain splice variants of SynGAP contain a C-terminal motif that enables binding to PDZ-containing proteins (Chen et al., 1998; Kim et al., 1998), such as PSD95 and SAP102, which are scaffolding proteins that organize macromolecular structures within glutamatergic synapses that enable plasticity supporting cognitive functions (Feng and Zhang, 2009). Due to its abundance, SynGAP is a dominant binding partner of major PDZ domain-containing proteins in the synapse and may therefore act as a PDZ-blocking molecule within the PSD (Dosemeci et al., 2016; Walkup et al., 2016; Zeng et al., 2016). In the context of this work, this idea is relevant because it implies that there are structural/scaffolding functions of SynGAP protein that are independent from GAP domain activity, which is consistent with our proposed “multiple-hit” hypothesis of Syngap1 gene function. The so-called “slot” hypothesis (Walkup et al., 2016) posits that SynGAP occupies PDZ protein-containing slots in the PSD, and SynGAP occupation of these slots is essential for structural and functional organization of this macromolecular complex. According to this hypothesis, reduced SynGAP within the synapse, which occurs during genetic haploinsufficiency or during LTP-like stimuli (Araki et al., 2015), may enhance synaptic function in a GAP-independent manner. It is argued that when SynGAP is at low abundance in the PSD, more PDZ binding “slots” become available, which become occupied by other proteins with PDZ binding domains. Some of these proteins may stabilize and/or recruit AMPARs at the synapse, leading to an increase in synaptic strength. This hypothesis is supported by data demonstrating that AMPAR stabilizing proteins, such as TARPs, are increased in the PSD of Syngap1 Het mice (Walkup et al., 2016). The general hypothesis that SynGAP acts as a PDZ-blocking molecule is also supported by the recent finding that SynGAP prevents Tau accumulation in the PSD (Bi et al., 2017). This finding broadens the relative importance of the slot hypothesis because SynGAP-mediated prevention of synaptic Tau accumulation is neuroprotective. Multilevel analysis (i.e. biochemical, cell-biological, electrophysiological, and behavioral approaches) in an animal model engineered to have impaired GTPase activity, combined with similar studies in a separate model engineered to selectively disrupt SynGAP-PDZ binding, could in theory test critical aspects of slot hypothesis, while also testing the relative importance of GAP activity on key Syngap1 phenotypes.
Genetic features of Syngap1 also support the “multiple-hit” model. Syngap1 mRNAs are heavily spliced (Fig. 4A-B), leading to at least twelve distinct SynGAP protein isoforms (McMahon et al., 2012). Alternative splicing of the same locus is a common mechanism for generating protein isoforms with completely unique molecular functions (Yang et al., 2016). Thus, the genetic complexity of Syngap1 may yield protein isoforms with distinct functions. C-terminal splicing is of particular importance because it is known to regulate molecular and cellular functions of SynGAP. There are four possible SynGAP C-termini, Alpha1, Alpha2, Beta and Gamma (Li et al., 2001; McMahon et al., 2012), which arise from the alternative splicing of the final three exons of the Syngap1 gene. The Alpha1 spliced event, the only C-terminus studied in any detail, gives rise to a protein with a C-terminal PDZ ligand that regulates binding to PSD proteins (Kim et al., 1998), such as PSD95 and SAP102. This binding is believed to promote PSD organization and may regulate the threshold for activity-dependent AMPA receptor insertion required for LTP (Walkup et al., 2016; Zeng et al., 2017; Zeng et al., 2016).
The most current framework for understanding how Syngap1 functions in the brain is heavily dominated by PDZ-dependent molecular functions of SynGAP proteins (Walkup et al., 2016; Zeng et al., 2017). However, there is evidence that the other C-terminal sequences also regulate SynGAP protein function. For instance, alternative splicing of Exon 21 encodes the Alpha2 C-terminus (Fig. 4A), which reverses the function of SynGAP at glutamatergic synapses. Inclusion of this C-tail in SynGAP promotes synaptic function (McMahon et al., 2012), rather than represses it, as in the case when the Alpha1 C-terminus is present (Rumbaugh et al., 2006). This function of Alpha2 appears to refine SynGAP function at the synapse rather than to drive key synaptic phenotypes. Heterozygosity of Syngap1 leads to enhanced synaptic function in hippocampal neurons on a global scale in vitro (Rumbaugh et al., 2006), as well as during developmental epochs in vivo (Clement et al., 2012; Clement et al., 2013), indicating that non-Alpha2 mechanisms are dominant with respect to synaptic function. However, Alpha2 inclusion within SynGAP molecules may act to balance functions at selected synapses. While this possible role of Alpha2 remains untested, it could arise through synapse-specific regulation of the Alpha1/Alpha2 ratio. Alpha2 transcripts appear to contain unique regulatory elements in the 3′ untranslated region that lead to selective regulation of its expression levels (Yokoi et al., 2017).
The signaling mechanisms that underlie the Alpha isoform-specific differential function of SynGAP at synapses remain unknown. Indeed, it is unclear if the protein sequence of Alpha2 leads to differential signaling within the synapse. This is presumably the case because GTPase function of SynGAP is known to regulate synapse function (Rumbaugh et al., 2006) and Alpha2 leads to a distinct effect on synapse regulation, relative to Alpha1 (McMahon et al., 2012). Moreover, it remains unknown how each of the Alpha isoforms contribute to species-conserved Syngap1 phenotypes. Creating animal models that selectively target Alpha1 versus Alpha2 function or expression at the organismal level, combined with targeted cell biological studies that probe the molecular functions of each isoform, may help to clarify the roles of these two important SynGAP variants as they related to key disease-relevant phenotypes. It would be of interest to understand how loss of PDZ binding of SynGAP, which is encoded through Alpha1 splicing, impacts species-conserved Syngap1 phenotypes. It is assumed that this interaction is crucial for maintaining E/I balance in vivo and for promoting cognitive functions (Walkup et al., 2016; Zeng et al., 2017), though these assumptions have not been tested directly. In addition, it would be of interest to understand how the other major Syngap1 phenotypes, such as brain structure and innate behavioral regulation, are impacted by PDZ-dependent functions of SynGAP.
The clear impact that Alpha1/2 spliced motifs have on SynGAP regulation at the synapse illustrates the importance of splicing on protein function. This would suggest that the other two remaining C-terminal variants may also act to shape SynGAP function. Beta and Gamma isoforms have received the least attention, and there is very little known about how each isoform regulates cellular signaling, neuronal function or relevant animal-level phenotypes. SynGAP Gamma-containing mRNAs, which arise through selective inclusion of Exon 20 (Fig. 4A), can be found in publically available databases containing rodent and human cDNA sequences. However, unlike the other three C-terminal sequences (Kim et al., 1998; McMahon et al., 2012; Moon et al., 2008), there is no specific antibody for these isoforms. Thus, there is essentially nothing known about Gamma isoforms other than that they are likely expressed in mammals. SynGAP-Beta, which arises from alternative splicing of Exon19 (Fig. 4A), has been studied in neurons (Li et al., 2001; Moon et al., 2008). It was found to have a unique subcellular expression pattern in neurons compared to other isoforms, suggesting that it may have unique functions compared to the Alpha isoforms. As with SynGAP-Gamma, there is no information on how Beta isoforms regulate neuronal signaling and synapse function. It will be of considerable interest to determine how their expression contributes to disease-relevant phenotypes. Importantly, these isoforms can be selectively disrupted using standard genetic engineering approaches, which would provide an opportunity to explore potential SynGAP diversity through isoform-specific functions.
Methods
Animals
Syngap1+/- (Syngap1 Het mice) constitutive knockout mice (Kim et al., 2003) were generated and maintained as previously described (Clement et al., 2012). Males and females were used for all studies. Animals were >PND60 for risk-taking studies and >PND90 for MRI studies.
Platform Departures Test of Risk-taking
The risk-taking/impulsivity test in adult Syngap1 mice was carried out using an apparatus and protocol that was similar to published reports (Yokota et al., 2013, Matsuoka et al., 2005). For young (∼PND21) mice, several modifications were made to the published apparatus and protocol. C57BL/6 wild type and mutant mice were weaned the day before the test was conducted. Each mouse, two at a time, was placed on an overturned stainless-steel wire Galaxy pencil/utility cup (7.8cm bottom diameter (“platform”), 10.8cm high, Yang, Silverman, Crawley, 2011) situated on a black granite bench top in a vivarium procedure room within demarcated areas (30×30×27 cm) using box cardboard dividers to separate the two mice from each other and two other sides on the bench with a front open side where a notebook web camera videotaped each 10min session. White duct tape was affixed to the black foam bottom of both cups for easier cleaning between trials and better contrasts with black mice. Each mouse's activities were hand scored thereafter. The number of “edge departures” were the principle measure. This behavior was defined by the mouse forepaws grasping the platform edge or wire mesh that made up the vertical walls exhibiting a forward stance over the edge. The latency to first full departure was also scored. Platform departures occurred when all four paws were the top platform and on the vertical bars. Pilot tests using other cups or beakers of different heights (unpublished observations from our lab) did not alter scoring parameter measures. However, we preferred the wire sided cups because we could measure intended edge departures as well as climbing tendencies not possible with a glass beaker eliminating most unintended loss-of-balance movements from the platform. Experimenters and scorers were blind to group identities with several individual scorers attaining similar parameter measurements.
Small Animal MRI
A multi-channel 7.0 Tesla MRI scanner (Varian Inc., Palo Alto, CA) was used to image the brains within their skulls. Sixteen custom-built solenoid coils were used to image the brains in parallel (Bock et al., 2005).
Anatomical Scan
To detect volumetric changes, we used the following parameters for the MRI scan: T2- weighted, 3-D fast spin-echo sequence, with a cylindrical acquisition of k-space, a TR of 350 ms, and TEs of 12 ms per echo for 6 echoes, field-of-view equaled to 20 × 20 × 25 mm3 and matrix size equaled to 504 × 504 × 630. Our parameters output an image with 0.040 mm isotropic voxels (Spencer Noakes et al., 2017). The total imaging time was 14 hours.
MRI Registration and Analysis
To visualize and compare any changes in the mouse brains the images are linearly (6 followed by 12 parameter) and non-linearly registered together. Registrations were performed with a combination of mni_autoreg tools and ANTS (advanced normalization tools). All scans are then resampled with the appropriate transform and averaged to create a population atlas representing the average anatomy of the study sample. The result of the registration is to have all images deformed into alignment with each other in an unbiased fashion. For the volume measurements, this allows for the analysis of the deformations needed to take each individual mouse's anatomy into this final atlas space, the goal being to model how the deformation fields relate to genotype. The jacobian determinants of the deformation fields are then calculated as measures of volume at each voxel. Significant volume differences can then be calculated by warping a pre-existing classified MRI atlas onto the population atlas, which allows for the volume of 182 different segmented structures encompassing cortical lobes, large white matter structures (i.e. corpus callosum), ventricles, cerebellum, brain stem, and olfactory bulbs (Dorr et al., 2008; Richards et al., 2011; Steadman et al., 2014; Ullmann et al., 2013) to be assessed in all brains. Further, these measurements can be examined on a voxel-wise basis to localize the differences found within regions or across the brain. Multiple comparisons in this study were controlled for using the False Discovery Rate (Genovese et al., 2002).
Supplementary Material
Highlights.
the current understanding of Syngap1 neurobiology is reviewed
four species-conserved Syngap1 phenotypes are described
new findings are presented that strengthen these phenotypes
molecular and cellular functions of Syngap1 are discussed in a phenotypic context
major unexplored areas of Syngap1 neurobiology are discussed
Acknowledgments
The authors would like to thank Dr. Alex Bayes for comments on an earlier version of this manuscript. This work was supported in part by grants (to GR) from the National Institute of Mental Health [MH096847 and MH108408] and the National Institute of Neurological Disorders and Stroke [NS064079]. Murat Kilinc was funded by Autism Speaks Dennis Weatherstone Predoctoral Fellowship [# 10646].
Footnotes
Disclosures: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aceti M, Creson TK, Vaissiere T, Rojas C, Huang WC, Wang YX, Petralia RS, Page DT, Miller CA, Rumbaugh G. Syngap1 Haploinsufficiency Damages a Postnatal Critical period of Pyramidal Cell Structural Maturation Linked to Cortical Circuit Assembly. Biological Psychiatry. 2015 doi: 10.1016/j.biopsych.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL. Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron. 2015;85:173–189. doi: 10.1016/j.neuron.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo DJ, Toriumi K, Escamilla CO, Kulkarni A, Anderson AG, Harper M, Usui N, Ellegood J, Lerch JP, Birnbaum SG, et al. Foxp1 in Forebrain Pyramidal Neurons Controls Gene Expression Required for Spatial Learning and Synaptic Plasticity. J Neurosci. 2017;37:10917–10931. doi: 10.1523/JNEUROSCI.1005-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes SA, Wijetunge LS, Jackson AD, Katsanevaki D, Osterweil EK, Komiyama NH, Grant SG, Bear MF, Nagerl UV, Kind PC, Wyllie DJ. Convergence of Hippocampal Pathophysiology in Syngap+/- and Fmr1-/y Mice. J Neurosci. 2015;35:15073–15081. doi: 10.1523/JNEUROSCI.1087-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett MW, Watson RF, Vitalis T, Porter K, Komiyama NH, Stoney PN, Gillingwater TH, Grant SG, Kind PC. Synaptic Ras GTPase activating protein regulates pattern formation in the trigeminal system of mice. J Neurosci. 2006;26:1355–1365. doi: 10.1523/JNEUROSCI.3164-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes A, Collins MO, Croning MD, van de Lagemaat LN, Choudhary JS, Grant SG. Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS One. 2012;7:e46683. doi: 10.1371/journal.pone.0046683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayes A, Collins MO, Galtrey CM, Simonnet C, Roy M, Croning MD, Gou G, van de Lagemaat LN, Milward D, Whittle IR, et al. Human post-mortem synapse proteome integrity screening for proteomic studies of postsynaptic complexes. Mol Brain. 2014;7:88. doi: 10.1186/s13041-014-0088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryer MH, Chattopadhyaya B, Xing P, Riebe I, Bosoi C, Sanon N, Antoine-Bertrand J, Levesque M, Avoli M, Hamdan FF, et al. Decrease of SYNGAP1 in GABAergic cells impairs inhibitory synapse connectivity, synaptic inhibition and cognitive function. Nat Commun. 2016;7:13340. doi: 10.1038/ncomms13340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryer MH, Hamdan FF, Klitten LL, Moller RS, Carmant L, Schwartzentruber J, Patry L, Dobrzeniecka S, Rochefort D, Neugnot-Cerioli M, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Human mutation. 2013;34:385–394. doi: 10.1002/humu.22248. [DOI] [PubMed] [Google Scholar]
- Bertoglio LJ, Joca SR, Guimaraes FS. Further evidence that anxiety and memory are regionally dissociated within the hippocampus. Behavioural brain research. 2006;175:183–188. doi: 10.1016/j.bbr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Bi M, Gladbach A, van Eersel J, Ittner A, Przybyla M, van Hummel A, Chua SW, van der Hoven J, Lee WS, Muller J, et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun. 2017;8:473. doi: 10.1038/s41467-017-00618-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock NA, Nieman BJ, Bishop JB, Mark Henkelman R. In vivo multiple-mouse MRI at 7 Tesla. Magn Reson Med. 2005;54:1311–1316. doi: 10.1002/mrm.20683. [DOI] [PubMed] [Google Scholar]
- Carlisle HJ, Manzerra P, Marcora E, Kennedy MB. SynGAP regulates steady-state and activity-dependent phosphorylation of cofilin. J Neurosci. 2008;28:13673–13683. doi: 10.1523/JNEUROSCI.4695-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carobrez AP, Bertoglio LJ. Ethological and temporal analyses of anxiety-like behavior: the elevated plus-maze model 20 years on. Neurosci Biobehav Rev. 2005;29:1193–1205. doi: 10.1016/j.neubiorev.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O'Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron. 1998;20:895–904. doi: 10.1016/s0896-6273(00)80471-7. [DOI] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Almonte AG, Miller BH, Wiltgen BJ, Miller CA, et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell. 2012;151:709–723. doi: 10.1016/j.cell.2012.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, Ozkan ED, Aceti M, Miller CA, Rumbaugh G. SYNGAP1 links the maturation rate of excitatory synapses to the duration of critical-period synaptic plasticity. J Neurosci. 2013;33:10447–10452. doi: 10.1523/JNEUROSCI.0765-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders, S. Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders, S. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–438. doi: 10.1038/nature21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers TJ, Popma A, Agelink van Rentergem JA, Bexkens A, Huizenga HM. Risky decision making in Attention-Deficit/Hyperactivity Disorder: A meta-regression analysis. Clin Psychol Rev. 2016;45:1–16. doi: 10.1016/j.cpr.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Deneve S, Machens CK. Efficient codes and balanced networks. Nat Neurosci. 2016;19:375–382. doi: 10.1038/nn.4243. [DOI] [PubMed] [Google Scholar]
- Dorr AE, Lerch JP, Spring S, Kabani N, Henkelman RM. High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. NeuroImage. 2008;42:60–69. doi: 10.1016/j.neuroimage.2008.03.037. [DOI] [PubMed] [Google Scholar]
- Dosemeci A, Weinberg RJ, Reese TS, Tao-Cheng JH. The Postsynaptic Density: There Is More than Meets the Eye. Front Synaptic Neurosci. 2016;8:23. doi: 10.3389/fnsyn.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegood J, Babineau BA, Henkelman RM, Lerch JP, Crawley JN. Neuroanatomical analysis of the BTBR mouse model of autism using magnetic resonance imaging and diffusion tensor imaging. NeuroImage. 2013;70:288–300. doi: 10.1016/j.neuroimage.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KC, Epilepsy Phenome/Genome, P. Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, Zhang M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat Rev Neurosci. 2009;10:87–99. doi: 10.1038/nrn2540. [DOI] [PubMed] [Google Scholar]
- Guo X, Hamilton PJ, Reish NJ, Sweatt JD, Miller CA, Rumbaugh G. Reduced expression of the NMDA receptor-interacting protein SynGAP causes behavioral abnormalities that model symptoms of Schizophrenia. Neuropsychopharmacology. 2009;34:1659–1672. doi: 10.1038/npp.2008.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, Duque A, Hasenstaub AR, McCormick DA. Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. J Neurosci. 2006;26:4535–4545. doi: 10.1523/JNEUROSCI.5297-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Daoud H, Piton A, Gauthier J, Dobrzeniecka S, Krebs MO, Joober R, Lacaille JC, Nadeau A, Milunsky JM, et al. De novo SYNGAP1 mutations in nonsyndromic intellectual disability and autism. Biol Psychiatry. 2011;69:898–901. doi: 10.1016/j.biopsych.2010.11.015. [DOI] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Cote M, Perreau-Linck E, Carmant L, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009;360:599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci. 2014;17:764–772. doi: 10.1038/nn.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Klann E, Costa-Mattioli M, Zukin RS. Dysregulation of Mammalian Target of Rapamycin Signaling in Mouse Models of Autism. J Neurosci. 2015;35:13836–13842. doi: 10.1523/JNEUROSCI.2656-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgenson M, Aonurm-Helm A, Zharkovsky A. Behavioral profile of mice with impaired cognition in the elevated plus-maze due to a deficiency in neural cell adhesion molecule. Pharmacol Biochem Behav. 2010;96:461–468. doi: 10.1016/j.pbb.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Beale HC, Carlisle HJ, Washburn LR. Integration of biochemical signalling in spines. Nat Rev Neurosci. 2005;6:423–434. doi: 10.1038/nrn1685. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee HK, Takamiya K, Huganir RL. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J Neurosci. 2003;23:1119–1124. doi: 10.1523/JNEUROSCI.23-04-01119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, Huganir RL. SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron. 1998;20:683–691. doi: 10.1016/s0896-6273(00)81008-9. [DOI] [PubMed] [Google Scholar]
- Komiyama NH, Watabe AM, Carlisle HJ, Porter K, Charlesworth P, Monti J, Strathdee DJ, O'Carroll CM, Martin SJ, Morris RG, et al. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J Neurosci. 2002;22:9721–9732. doi: 10.1523/JNEUROSCI.22-22-09721.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M, Karczewski KJ, Cutler DJ, Devlin B, Roeder K, et al. Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nat Genet. 2017;49:504–510. doi: 10.1038/ng.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozol RA, Cukier HN, Zou B, Mayo V, De Rubeis S, Cai G, Griswold AJ, Whitehead PL, Haines JL, Gilbert JR, et al. Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum Mol Genet. 2015;24:4006–4023. doi: 10.1093/hmg/ddv138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Medina I, Krapivinsky L, Gapon S, Clapham DE. SynGAP-MUPP1-CaMKII synaptic complexes regulate p38 MAP kinase activity and NMDA receptor-dependent synaptic AMPA receptor potentiation. Neuron. 2004;43:563–574. doi: 10.1016/j.neuron.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J Neurosci. 2007;27:10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Ehninger D, Zhou M, Oh JY, Kang M, Kwak C, Ryu HH, Butz D, Araki T, Cai Y, et al. Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat Neurosci. 2014;17:1736–1743. doi: 10.1038/nn.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15:1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Li W, Okano A, Tian QB, Nakayama K, Furihata T, Nawa H, Suzuki T. Characterization of a novel synGAP isoform, synGAP-beta. J Biol Chem. 2001;276:21417–21424. doi: 10.1074/jbc.M010744200. [DOI] [PubMed] [Google Scholar]
- Li W, Xu X, Pozzo-Miller L. Excitatory synapses are stronger in the hippocampus of Rett syndrome mice due to altered synaptic trafficking of AMPA-type glutamate receptors. Proc Natl Acad Sci U S A. 2016;113:E1575–1584. doi: 10.1073/pnas.1517244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM. LTP consolidation: substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Marlin BJ, Mitre M, D'Amour JA, Chao MV, Froemke RC. Oxytocin enables maternal behaviour by balancing cortical inhibition. Nature. 2015;520:499–504. doi: 10.1038/nature14402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Furuyashiki T, Yamada K, Nagai T, Bito H, Tanaka Y, Kitaoka S, Ushikubi F, Nabeshima T, Narumiya S. Prostaglandin E receptor EP1 controls impulsive behavior under stress. Proc Natl Acad Sci U S A. 2005;102:16066–16071. doi: 10.1073/pnas.0504908102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AC, Barnett MW, O'Leary TS, Stoney PN, Collins MO, Papadia S, Choudhary JS, Komiyama NH, Grant SG, Hardingham GE, et al. SynGAP isoforms exert opposing effects on synaptic strength. Nat Commun. 2012;3:900. doi: 10.1038/ncomms1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot C, von Stulpnagel C, Nava C, Ville D, Sanlaville D, Lesca G, Rastetter A, Gachet B, Marie Y, Korenke GC, et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet. 2016;53:511–522. doi: 10.1136/jmedgenet-2015-103451. [DOI] [PubMed] [Google Scholar]
- Moon IS, Sakagami H, Nakayama J, Suzuki T. Differential distribution of synGAP alpha1 and synGAP beta isoforms in rat neurons. Brain Res. 2008;1241:62–75. doi: 10.1016/j.brainres.2008.09.033. [DOI] [PubMed] [Google Scholar]
- Muhia M, Yee BK, Feldon J, Markopoulos F, Knuesel I. Disruption of hippocampus-regulated behavioural and cognitive processes by heterozygous constitutive deletion of SynGAP. Eur J Neurosci. 2010;31:529–543. doi: 10.1111/j.1460-9568.2010.07079.x. [DOI] [PubMed] [Google Scholar]
- Nicoll RA. A Brief History of Long-Term Potentiation. Neuron. 2017;93:281–290. doi: 10.1016/j.neuron.2016.12.015. [DOI] [PubMed] [Google Scholar]
- O'Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, Vives L, Baker C, Hiatt JB, Nickerson DA, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki T, Saito Y, Hiraiwa R, Saitoh S, Kai M, Adachi K, Nishimura Y, Nanba E, Maegaki Y. Pharmacoresistant epileptic eyelid twitching in a child with a mutation in SYNGAP1. Epileptic Disord. 2017;19:339–344. doi: 10.1684/epd.2017.0922. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77:243–250. doi: 10.1016/j.neuron.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Q, Lizarraga SofiaB, Schmidt M, Yang U, Gong J, Ellisor D, Kauer JulieA, Morrow EricM. Christianson Syndrome Protein NHE6 Modulates TrkB Endosomal Signaling Required for Neuronal Circuit Development. Neuron. 2013;80:97–112. doi: 10.1016/j.neuron.2013.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkan ED, Creson TK, Kramar EA, Rojas C, Seese RR, Babyan AH, Shi Y, Lucero R, Xu X, Noebels JL, et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron. 2014;82:1317–1333. doi: 10.1016/j.neuron.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park SM, Benoit V, et al. De novo, heterozygous, loss-of-function mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am J Med Genet A. 2015;167A:2231–2237. doi: 10.1002/ajmg.a.37189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena V, Hothorn M, Eberth A, Kaschau N, Parret A, Gremer L, Bonneau F, Ahmadian MR, Scheffzek K. The C2 domain of SynGAP is essential for stimulation of the Rap GTPase reaction. EMBO Rep. 2008;9:350–355. doi: 10.1038/embor.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinar C, Fontaine CJ, Trivino-Paredes J, Lottenberg CP, Gil-Mohapel J, Christie BR. Revisiting the flip side: Long-term depression of synaptic efficacy in the hippocampus. Neurosci Biobehav Rev. 2017;80:394–413. doi: 10.1016/j.neubiorev.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Prchalova D, Havlovicova M, Sterbova K, Stranecky V, Hancarova M, Sedlacek Z. Analysis of 31-year-old patient with SYNGAP1 gene defect points to importance of variants in broader splice regions and reveals developmental trajectory of SYNGAP1-associated phenotype: case report. BMC Med Genet. 2017;18:62. doi: 10.1186/s12881-017-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Richards K, Watson C, Buckley RF, Kurniawan ND, Yang Z, Keller MD, Beare R, Bartlett PF, Egan GF, Galloway GJ, et al. Segmentation of the mouse hippocampal formation in magnetic resonance images. NeuroImage. 2011;58:732–740. doi: 10.1016/j.neuroimage.2011.06.025. [DOI] [PubMed] [Google Scholar]
- Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16:595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Adams JP, Kim JH, Huganir RL. SynGAP regulates synaptic strength and mitogen-activated protein kinases in cultured neurons. Proc Natl Acad Sci U S A. 2006;103:4344–4351. doi: 10.1073/pnas.0600084103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer Noakes TL, Henkelman RM, Nieman BJ. Partitioning k-space for cylindrical three-dimensional rapid acquisition with relaxation enhancement imaging in the mouse brain. NMR Biomed. 2017;30 doi: 10.1002/nbm.3802. [DOI] [PubMed] [Google Scholar]
- Steadman PE, Ellegood J, Szulc KU, Turnbull DH, Joyner AL, Henkelman RM, Lerch JP. Genetic effects on cerebellar structure across mouse models of autism using a magnetic resonance imaging atlas. Autism Res. 2014;7:124–137. doi: 10.1002/aur.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatti R, Haley MS, Swanson OK, Tselha T, Maffei A. Neurophysiology and Regulation of the Balance Between Excitation and Inhibition in Neocortical Circuits. Biol Psychiatry. 2017;81:821–831. doi: 10.1016/j.biopsych.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda T, Kim JH, Zhan C, Hatten ME. Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 2004;18:541–558. doi: 10.1101/gad.1151204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann JF, Watson C, Janke AL, Kurniawan ND, Reutens DC. A segmentation protocol and MRI atlas of the C57BL/6J mouse neocortex. NeuroImage. 2013;78:196–203. doi: 10.1016/j.neuroimage.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Vazquez LE, Chen HJ, Sokolova I, Knuesel I, Kennedy MB. SynGAP regulates spine formation. J Neurosci. 2004;24:8862–8872. doi: 10.1523/JNEUROSCI.3213-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk L, Chiu SL, Sharma K, Huganir RL. Glutamate Synapses in Human Cognitive Disorders. Annu Rev Neurosci. 2015 doi: 10.1146/annurev-neuro-071714-033821. [DOI] [PubMed] [Google Scholar]
- von Stulpnagel C, Funke C, Haberl C, Hortnagel K, Jungling J, Weber YG, Staudt M, Kluger G. SYNGAP1 Mutation in Focal and Generalized Epilepsy: A Literature Overview and A Case Report with Special Aspects of the EEG. Neuropediatrics. 2015;46:287–291. doi: 10.1055/s-0035-1554098. [DOI] [PubMed] [Google Scholar]
- Walf AA, Frye CA. The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc. 2007;2:322–328. doi: 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup WG, Mastro TL, Schenker LT, Vielmetter J, Hu R, Iancu A, Reghunathan M, Bannon BD, Kennedy MB. A model for regulation by SynGAP-alpha1 of binding of synaptic proteins to PDZ-domain ‘Slots’ in the postsynaptic density. Elife. 2016;5 doi: 10.7554/eLife.16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup WGt, Washburn L, Sweredoski MJ, Carlisle HJ, Graham RL, Hess S, Kennedy MB. Phosphorylation of synaptic GTPase-activating protein (synGAP) by Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5) alters the ratio of its GAP activity toward Ras and Rap GTPases. J Biol Chem. 2015;290:4908–4927. doi: 10.1074/jbc.M114.614420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Held RG, Hall BJ. SynGAP regulates protein synthesis and homeostatic synaptic plasticity in developing cortical networks. PLoS One. 2013;8:e83941. doi: 10.1371/journal.pone.0083941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldon M, Kilinc M, Lloyd Holder J, Jr, Rumbaugh G. The first international conference on SYNGAP1-related brain disorders: a stakeholder meeting of families, researchers, clinicians, and regulators. J Neurodev Disord. 2018;10:6. doi: 10.1186/s11689-018-9225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, et al. Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell. 2016;164:805–817. doi: 10.1016/j.cell.2016.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi S, Udagawa T, Fujioka Y, Honda D, Okado H, Watanabe H, Katsuno M, Ishigaki S, Sobue G. 3'UTR Length-Dependent Control of SynGAP Isoform alpha2 mRNA by FUS and ELAV-like Proteins Promotes Dendritic Spine Maturation and Cognitive Function. Cell Rep. 2017;20:3071–3084. doi: 10.1016/j.celrep.2017.08.100. [DOI] [PubMed] [Google Scholar]
- Yokota S, Moriya N, Iwata M, Umezawa M, Oshio S, Takeda K. Exposure to diesel exhaust during fetal period affects behavior and neurotransmitters in male offspring mice. J Toxicol Sci. 2013;38:13–23. doi: 10.2131/jts.38.13. [DOI] [PubMed] [Google Scholar]
- Zeng M, Bai G, Zhang M. Anchoring high concentrations of SynGAP at postsynaptic densities via liquid-liquid phase separation. Small GTPases. 2017:1–9. doi: 10.1080/21541248.2017.1320350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, Zhang M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell. 2016;166:1163–1175 e1112. doi: 10.1016/j.cell.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Vazquez L, Apperson M, Kennedy MB. Citron binds to PSD-95 at glutamatergic synapses on inhibitory neurons in the hippocampus. J Neurosci. 1999;19:96–108. doi: 10.1523/JNEUROSCI.19-01-00096.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu X, Need AC, Petrovski S, Goldstein DB. One gene, many neuropsychiatric disorders: lessons from Mendelian diseases. Nat Neurosci. 2014;17:773–781. doi: 10.1038/nn.3713. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.