

Abstract
Antipsychotic drugs, including both typical such as haloperidol and atypical such as clozapine, remain the current standard for schizophrenia treatment. These agents are relatively effective in treating hallucinations and delusions. However, cognitive deficits are at present essentially either persistent or exacerbated following chronic antipsychotic drug exposure. This underlines the need of new therapeutic approaches to improve cognition in treated schizophrenia patients. Our previous findings suggested that upregulation of histone deacetylase 2 (HDAC2) expression upon chronic antipsychotic treatment may lead to negative effects on cognition and cortical synaptic structure. Here we tested different phenotypes of psychosis, synaptic plasticity, cognition and antipsychotic drug action in HDAC2 conditional knockout (HDAC2-cKO) mice and controls. Conditional depletion of HDAC2 function in glutamatergic pyramidal neurons led to a protective phenotype against behavior models induced by psychedelic and dissociative drugs, such as DOI and MK801, respectively. Immunoreactivity toward synaptophysin, which labels presynaptic terminals of functional synapses, was decreased in the frontal cortex of control mice chronically treated with clozapine - an opposite effect occurred in HDAC2-cKO mice. Chronic treatment with the class I and class II HDAC inhibitor SAHA prevented via HDAC2 the disruptive effects of MK801 on recognition memory. Additionally, chronic SAHA treatment affected transcription of numerous plasticity-related genes in the frontal cortex of control mice, an effect that was not observed in HDAC2-cKO animals. Together, these findings suggest that HDAC2 may represent a novel target to improve synaptic plasticity and cognition in treated schizophrenia patients.
Keywords: Histone deacetylase (HDAC), HDAC2, vorinostat (SAHA), antipsychotics, clozapine, schizophrenia
INTRODUCTION
Schizophrenia is a severe brain disorder that usually produces a lifetime of disability (Sawa and Snyder 2002; van Os and Kapur 2009). The symptoms of schizophrenia can be divided into three broad categories: psychotic or positive symptoms (e.g., hallucinations and delusions), negative symptoms (e.g., loss or decrease in the ability to initiate plans, express emotion, or find pleasure) and cognitive symptoms (e.g., confused and disordered speech, poor executive functioning, trouble with logical thinking, and difficulties to pay attention). First generation or typical (e.g., chlorpromazine and haloperidol) and second generation or atypical (e.g., clozapine and risperidone) antipsychotics remain the current standard for psychotic disorders including schizophrenia (Lieberman et al., 2008; Miyamoto et al., 2012; Meltzer 2013). In some patients with schizophrenia, typical and atypical antipsychotic drugs produce complete remission of psychotic symptoms. However, antipsychotic drugs are currently ineffective against cognitive deficits (Ibrahim and Tamminga 2012; Millan et al., 2012), and hence treated schizophrenia patients present either small improvement or even deterioration in several cognitive domains, including working memory and executive function (Goldberg et al., 1993; Fervaha et al., 2015; Nielsen et al., 2015; Husa et al., 2016).
These limitations of antipsychotic treatment are further highlighted by recent clinical trials in which it was found that three-fourths of schizophrenia patients stop using antipsychotic medication within 18 months of starting therapy. The reasons given for discontinuing prescribed drugs included reduced efficacy, poor tolerability, and severe side effects (Lieberman et al., 2005; Liu-Seifert et al., 2005; Snyder and Murphy 2008). Thus, adherence to an antipsychotic drug regime is currently a significant issue in the clinical management of this psychiatric condition. Furthermore, although recent genome-wide association studies (GWAS) show convincing evidence that alterations caused by interactions of several genes involved in signaling, synaptic plasticity and neurodevelopmental processes contribute substantially to the disorder (Consortium. 2014; Fromer et al., 2014; Purcell et al., 2014; Sekar et al., 2016), the primary target of currently used antipsychotic medications is restricted to monoaminergic neurotransmitter systems. The current antipsychotic arsenal in the clinic can be reduced to two drug-classes based on their pharmacological profile: while both typical and atypical antipsychotics target the dopaminergic system, the differentiating feature is a more prominent antagonism/inverse agonism at the serotonin 5-HT2A receptor in the mechanism of action of the latter (Lieberman et al., 2008; Miyamoto et al., 2012; Meltzer 2013). The need for novel and more efficient therapeutic targets to address the complexity of schizophrenia and its treatment is demonstrated by the poor mechanistic repertoire of current antipsychotics and their limited clinical performance.
Histone deacetylases (HDACs) are critical elements in the modulation of chromatin structure (Abel and Zukin 2008; Graff and Tsai 2013; Ibi and Gonzalez-Maeso 2015). They remove acetyl groups from histone tails, an epigenetic modification that correlates with transcriptional repression. Preclinical assays in rodents suggest that both peripheral and local administration of HDAC inhibitors in brain regions, such as frontal cortex and nucleus accumbens, affect behavioral responses in paradigms of memory function, depression, and sensory gating (Covington et al., 2009; Guan et al., 2009; Graff et al., 2012; Kurita et al., 2012; Morris et al., 2013; Graff et al., 2014). Clinical studies also demonstrate that valproate, a drug that inhibits HDACs among many other actions (Nalivaeva et al., 2009), improves the clinical efficacy of antipsychotic drugs, particularly in aspects related to sensorimotor and cognitive deficits (Casey et al., 2003; Citrome et al., 2004; Kelly et al., 2006; Suzuki et al., 2009). However, this clinical improvement was not observed in certain clinical trials (Casey et al., 2009; Meltzer et al., 2011).
Our previous work showed that chronic treatment with atypical antipsychotic drugs, such as clozapine and risperidone, but not with the typical antipsychotic haloperidol, targets a non-canonical pathway that leads to up-regulation of HDAC2 both in mice and in postmortem human frontal cortex tissue samples (Kurita et al., 2012; Kurita et al., 2013b; Ibi et al., 2017). Since it has been shown before that HDAC2 negatively regulates transcription of genes involved in synaptic plasticity and memory (Guan et al., 2009; Kurita et al., 2012; Ibi et al., 2017), we previously proposed that the above-mentioned up-regulation of frontal cortex HDAC2 might be responsible for at least part of the negative effects of chronic atypical antipsychotic treatment on synaptic plasticity and cognitive processes. We also suggested that inhibition of this pathway may represent a new approach to improve schizophrenia treatment (Kurita et al., 2013a; Ibi et al., 2017).
In order to further test the role of HDAC2 in phenotypes related to psychosis and antipsychotic drug action, the current study focused on three main goals. The first goal was to further validate the functional role of HDAC2 in the negative effects of chronic clozapine treatment on synaptic plasticity. To do so, we tested synaptophysin as a presynaptic marker of active synapses (Calhoun et al., 1996; Fischer et al., 2007) in mice with genetic deletion of HDAC2 (HDAC2 conditional knockout, cKO, mice). The second goal was to test behavior models of psychosis and memory in HDAC2-cKO mice and controls. Specifically, we aimed to investigate paradigms that included head-twitch behavior, which is induced by hallucinogenic 5-HT2A receptor agonists such as lysergic acid diethylamide (LSD) and DOI (Hanks and Gonzalez-Maeso 2013), and hyperlocomotor activity induced by the dissociative drug MK801 (a non-competitive NMDA receptor antagonist) (Moreno and Gonzalez-Maeso 2013); along with a behavior model of recognition memory. The third goal aimed to examine whether HDAC2 is necessary for the effects chronic treatment with the HDAC inhibitor SAHA, a selective inhibitor of class I and class II HDACs (Ibi and Gonzalez-Maeso 2015), on antipsychotic-related behaviors. We also employed microarray analysis protocols to compare the effect of chronic SAHA treatment on gene expression profiles in the frontal cortex of HDAC2-cKO and control mice.
EXPERIMENTAL PROCEDURES
Materials and Drug Administration
1-(2,5-Dimethoxy-4-iodophenyl)-2-aminopropane (DOI) and (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate (dizocilpine, (+)-MK801) were purchased from Sigma-Aldrich. Clozapine and haloperidol were obtained from Tocris Cookson Inc. Suberoylanilide hydroxamic acid (SAHA; vorinostat) was purchased from Cayman Chemical. The injected doses (i.p.) were DOI, 1.0 mg/kg; clozapine, 10 mg/kg; haloperidol, 1 mg/kg; and SAHA, 20 mg/kg; unless otherwise indicated. DOI was dissolved in saline. MK801, SAHA and haloperidol were injected after suspension in minimal amount of DMSO, and made up to volume with saline. Clozapine was dissolved in DMSO supplemented with a minimal amount of acetic acid and suspended in saline. For chronic treatment with clozapine or haloperidol, or vehicle, mice were injected (once daily for 21 days), as assays were carried out one day after the last injection. To test the effect of chronic SAHA treatment, or vehicle, on frontal cortex gene expression, mice were injected (once daily for 21 days), and frontal cortex tissue samples were collected one day after the last injection. To test the effect of chronic SAHA treatment, or vehicle, on behavior, mice were injected (once daily for 10 days), and novel object recognition was assayed one day after the last injection. Doses and route of administration were selected based on previous findings (Kurita et al., 2012; Ibi et al., 2017).
Animals
Experiments were performed on adult (10–20 weeks old) male C57BL/6 mice. Animals were housed at 12 h light/dark cycle at 23°C with food and water ad libitum, except during behavioral testing. Experiments were conducted in accord with NIH guidelines, and were approved by the Virginia Commonwealth University Animal Care and Use Committee. All efforts were made to minimize animal suffering and the number of animals used. Behavioral testing took place between 9:00 a.m. and 6:00 p.m. (i.e., during the light phase of the light/dark cycle).
Our previous findings show that homozygosity of the Hdac2-null allele (The Jackson Laboratory stock number: 022625) results in either embryonic lethality of partial lethality during the first few days postnatal as a result of proliferation defects and impaired development (Ibi et al., 2017). These findings are consistent with some (Montgomery et al., 2007; Morris et al., 2013), but not all (Guan et al., 2009), of the prior descriptions of global Hdac2 gene deletion. We therefore deleted HDAC2 function specifically in forebrain glutamatergic pyramidal neurons. This particular brain network and neural population was selected because our previous findings demonstrate that cortical pyramidal HDAC2 plays a fundamental role in synaptic structure and synaptic plasticity, as well as in certain behavioral paradigms that model perception and cognition (Kurita et al., 2012; Kurita et al., 2013b; Ibi et al., 2017). To delete HDAC2 function in this particular population of neurons, we bred homozygous HDAC2loxP/loxP mice (C57BL/6 background) containing exons 2 through 4 of the Hdac2 gene flanked by loxP sites (Montgomery et al., 2007) to the CaMKUα-Cre transgenic line (C57BL/6 background) in which the CaMKIlα promoter precedes Cre recombinase (Cabungcal et al., 2013). The genotype denoted as HDAC2 conditional knock-out (HDAC2-cKO) corresponds to HDAC2loxP/loxP:CaMKIIα-Cre+/− born at near expected Mendelian ratios from the outlined bred. The selective deletion of Hdac2 in CaMKIΙα-expressing neurons was validated elsewhere (Ibi et al., 2017).
Control group denotes HDAC2loxP/loxP .CaMKUα-Cre−/− genotype for experiments involving behavioral testing, and wild-type genotype for immunofluorescence and mRNAexpression assays. Pilot assayed demonstrated that HDAC2loxp/loxp:CaMK7/a-Cre−/− mice show normal laminar distribution in the cerebral cortex measured by Nissl staining, as compared to wild-type mice (data not shown). Similarly, pilot assays demonstrated that HDAC2loxP/loxP:CaMKIIa-Cre−/− mice do not show alterations in paradigms such as exploratory behavior and novel object recognition test, as compared to wild-type mice (data not shown).
For immunoblot, immunohistochemistry, microarray and quantitative real-time PCR assays, the day of the experiment mice were sacrificed by cervical dislocation, and bilateral frontal cortex (bregma 1.90 to 1.40 mm) was dissected and frozen at either −80°C or immediately processed. These assays were conducted on behaviorally naïve mice.
Immunoblotting
Western blot experiments were performed as previously reported with minor modifications (Ibi et al., 2017). Briefly, samples were loaded onto polyacrylamide gel (10–12%) and submitted to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. After transfer to nitrocellulose membranes, blocking with 5% nonfat dry milk and 0.5% BSA in TBST buffer (Tris-buffered saline and 0.05 or 1 % Tween 20) was followed by overnight incubation in primary antibody at 4°C or one hour at room temperature. HDAC2 (mouse brain: Abeam ab32117, 1:1000), α-tubulin (mouse brain: Abeam ab7291, 1:3000) Incubation with the secondary antibody (1:5000–20000) coupled to peroxidase (Amersham Biosciences) was performed at room temperature for 90 min, followed by repeated washing with TBST. Immunoreactive proteins were visualized with enhanced chemiluminiscence (Thermo Scientific) according to the manufacturer’s instructions. In each case, the blots were stripped and re-probed for a control protein to control loading amounts.
Quantitative immunofluorescence
Quantitative immunofluorescence assays were performed as previously reported with minor changes (Ibi et al., 2017). Briefly, the animals were deeply anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (12 mg/kg) administered intraperitoneally. Transcardiac perfusion was performed with 10 ml PBS, followed by 30 ml of freshly prepared 4% paraformaldehyde (PFA) in PBS at room temperature. Brains were removed and immersion-fixed in 4% PFA in PBS at 4°C (overnight), and stored at 30% sucrose in PBS at 4°C for at least 48 h. Brains were cut to 20 pm-thick coronal sections on a vibratome (Leica VT1000S). Free-floating sections were transferred to 24-well dishes containing PBS. Coronal brain sections were washed with PBS and incubated in 5% bovine serum albumin with 0.1% Triton X-100 in PBS for 60 min at room temperature. The sections were then incubated overnight in the same solution containing synaptophysin antibody (Sigma-Aldrich S5768, 1:1000). The sections were rinsed 5 times in PBS for 10 min and incubated for 1 h with 568 dye-conjugated goat anti-mouse antibody (Invitrogen, A11004, 1:2000). Following incubation, the sections were washed three times with PBS, after which immunostained sections were examined by epifluorescence microscopy (Carl Zeiss Axiolmager A1). Counterstaining with DAPI allowed the determination of cortical areas and laminar borders.
For the quantification of synaptophysin immunoreactivity, the mean signal intensity of synaptophysin immunoreactivity in the different layers of the somatosensory cortex (bregma 0.70 to −2.10 mm) and regions of the hippocampus (DAPI) was measured in both hemispheres of treated mice and controls.
Head-twitch behavior
Head twitch behavioral response was performed as previously reported (Gonzalez-Maeso et al., 2007). Briefly, animals were injected (i.p.) with DOI or vehicle and, 15 min later, they were placed into the center of a Plexiglas cage (28 × 18 × 15 cm) for 30 min, during which they were videotaped at close range by a video camcorder positioned directly above the cage. Videotapes were scored for head-twitches by an experienced observer blind to genotype and treatment. Testing cages were thoroughly cleaned after each animal was tested to eliminate odor cues.
Locomotor activity
Locomotor activity was assessed using a computerized three-dimensional activity monitoring system (Omnitech) as previously reported (Ibi et al., 2017). Briefly, the system determines ambulatory activity based on frequency of interruptions to infrared beams traversing the x, y and z planes. For center time experiments, the proportion of time spent in the center of the arena was taken as a measure of anxiety (dimensions of the arena: 41 × 41 × 30 cm). For experiments limited to study locomotor activity, mice were monitored for 90 min (dimensions of the arena: 27 × 27 × 21 cm). For modulation of locomotor activity upon drug administration, mice were left to habituate in the locomotor box for 90 min before injection of MK801, or vehicle, to exclude novelty of the environment as a confounding factor, and monitored for 2 h after injection. Locomotor activity was automatically determined from the interruptions of beams in the horizontal and vertical planes. Experiments were conducted in dim light.
Novel object recognition (NOR) test
Novel object recognition test was assessed as previously reported (Ibi et al., 2017). Briefly, mice were habituated for 10 min to the NOR arena for three consecutive days before the first NOR test. On the days of testing, mice were given a 10-min acquisition trial and a 5-min recognition trial, separated by a 24 h inter-trial return to their home cage. During the acquisition trial, the animals were allowed to explore two different objects (A and B). During the recognition trial, the animals explored a familiar object (A) from the acquisition trial and a novel object (C). Behavior was recorded on video for blind scoring of object exploration. Object exploration is defined as an animal licking, sniffing, or touching the object with the forepaws while sniffing. The exploration time (s) of each object was recorded manually by the use of two stopwatches. The exploratory preference [100 × (time spent exploring the novel object / total exploration time)] was then calculated for retention trials. If the exploration time in the acquisition or retention trials to either objects was < 5 s, the data were excluded from analysis. This rarely occurred and did not affect the ability to complete the analysis using the data from the remaining animals of that group.
Spontaneous alternation behavior in a Y-maze test
Short-term spatial recognition memory was tested using a Y-maze, as we have previously reported (Ibi et al., 2017). Briefly, each mouse was placed individually at the center of the apparatus and allowed to move freely through the maze during an 8-min session. The number of arm entries was recorded visually. Alternation was defined as successive entries into the three arms on overlapping triplet sets. The alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus 2) multiplied by 100. Spontaneous alternation (%), defined as successive entries into the three arms on overlapping triplet sets, is associated with spatial short-term memory (Hughes 2004).
Microarray study
Two groups of mice with three independent biological replicates per group were used for the microarray study, totaling 6 microarrays. Mice were chronically (21 days) injected with SAHA (20 mg/kg) or vehicle, and sacrificed one day after the last injection. The day of the experiment, mice were sacrificed for analysis by cervical dislocation, and bilateral frontal cortex was dissected (see above) and frozen at −80°C until RNA extraction. All animals were handled, treated, and sacrificed at the same time, under the same conditions. As well, all RNA and array processing was performed at the same time. RNA was extracted from mouse frontal cortex using the RNeasy lipid tissue mini kit (Qiagen). Labelling and hybridization of the samples to Mouse Gene 1.0 ST expression chip (Affymetrix) were performed as previously described with minor modifications (Gonzalez-Maeso et al., 2003). Data quality control was performed using the Affymetrix Expression Console software. Areas under the Receiver Operating Characteristic curve (ROC) discriminate between positive control probesets and negative control probesets (pos.vs.neg.auc) (Howard et al., 2009). All samples were found to be of very high quality, with pos.vs.neg.auc metric > 0.89 (0.5 being no better than chance and 1.0 being perfect distinction), and no outlier was detected. Probe set summarization, background correction and normalization were then carried out in the same software using default settings. Normalized data were analyzed in MultiExperiment Viewer (Saeed et al., 2003) software and differential gene list was generated using the Significance Analysis of Microarrays (Tusher et al., 2001) algorithm; Cutoff was chosen so that the false discovery rate does not exceed 12%. The microarray data discussed in this paper have been deposited in NCBI’s Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo — accession number GSE29419). The data analysis criteria used for our study are recommended by the MicroArray Quality Control project, and these criteria have been validated to provide a high degree of intersite reproducibility and inter- and intra-platform reproducibility (for methods regarding Microarray data analysis, see Shi et al., 2006; Shi et al., 2010).
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) assays were carried out in quadruplicate as previously described (Ibi et al., 2017) using a QuantStudio 6 Flex Real-Time PCR System (ThermoFisher Scientific). Mouse qRT-PCR primer pairs (genBank accesion number): Adam 8 (NM_007403), fwd GCAGGACCATTGCCTCTACC, rev TGGACCCAACT CGGAAAAAGC and fwd CACCACTCCCAGTTCCTGTT, rev AAGGTTGGCTTGACCTGCT; Ankrd9 (NM_175207.4), fwd CCTGGCAGTGCGAGATCAG, rev GCTTCGCTGGCACGTATGT; Cxcr3 (NM_009910), fwd GGTTAGTGAACGTCAAGTGCT, rev CCCCATAATCGTAGGGAGAGGT; Cdkn2c (NM_007671), fwd GGGGCATCGGAACCATAAGG, rev CCTCCATCAGGCTAATGACCT; Tbkbpl (NM_198100.2), fwd AGTCAATGTGTGTGCCGTCTTC, rev CTTGGTCGACCTCCAGGAAA; Zdhhc12 (NM_025428), fwd CTGTGTGGGTGAACGCAAC, rev CACTAACGCGAAGAAGGAGAG; rpS3 (NM_012052), fwd AGGTTGTGGTGTCTGGGAAG, rev GAGGCTTCTTGGGACCAATC.
Statistical analysis
Statistical analyses were performed with GraphPad Prism software version 6. For all mRNA data, fold changes relative to controls were determined using the corrected Ct method (Gonzalez-Maeso et al., 2003). Immunohistochemical images were acquired using epifluorescence microscope (see above) at identical settings for each of the conditions. Immunoblot and immunohistochemical images were quantified using NIH image 1.63 software by an experimenter blind to treatment and genotype groups. In immunoblot assays in mouse frontal cortex, the theoretical amount of protein in each sample was obtained following standard protocols, as previously reported (Kurita et al., 2012; Ibi et al., 2017). For the quantification of synaptophysin immunoreactivity after chronic clozapine treatment, the mean signal intensity of synaptophysin immunoreactivity in the different layers of the frontal cortex and regions of the hippocampus (DAPI) was measured in both hemispheres of treated mice and controls. Animals were randomly allocated into the different experimental groups. Statistical significance of experiments involving three or more groups and two or more treatments was assessed by two-way ANOVA followed by Tukey’s or Fisher’s uncorrected least square difference (LSD) post hoc test, and three-way ANOVA followed by Tukey’s post hoc test. Statistical significance of experiments involving three or more groups was assessed by one-way ANOVA followed by Tukey’s post hoc test. Statistical significance of experiments involving two groups was assessed by Student’s t-test. The level of significance was chosen at p = 0.05.
RESULTS
Chronic clozapine treatment up-regulates HDAC2 in mouse frontal cortex
We previously showed that up-regulation of HDAC2 (mRNA and immunoreactivity) occurs upon chronic treatment with atypical antipsychotics, but not with the typical ones. This was observed via immunohistochemical assays in frontal cortex tissue sections and qRT-PCR assays (Kurita et al., 2012; Ibi et al., 2017). We observed here similar findings using immunoblot assays. Thus, consistent with our previous studies, western blot assays showed up-regulation (F2,15 = 9.203, p < 0.01) of HDAC2 protein in the frontal cortex of mice chronically treated with clozapine (p < 0.01), but not with haloperidol (p > 0.05) (Fig. 1A and Fig. 1B).
Fig 1.
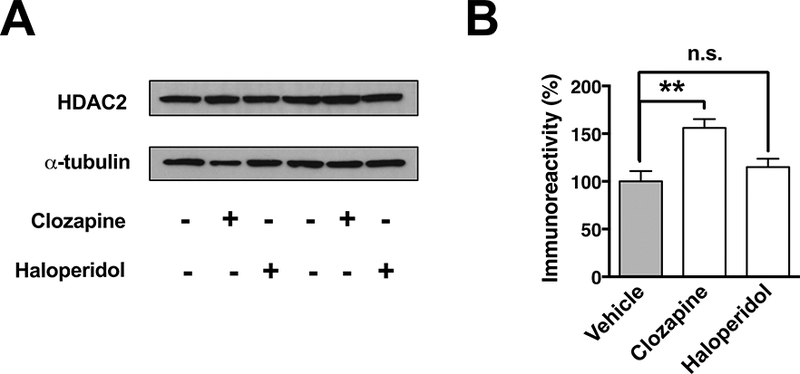
Chronic treatment with the atypical antipsychotic clozapine, but not the typical antipsychotic haloperidol, induces up-regulation of HDAC2 immunoreactivity in the mouse frontal cortex. Immunoblotting showing the effect of chronic antipsychotic treatment on expression of HDAC2 (A) and quantification of immunoreactivity (B). Values plotted are mean ± S.E.M. (n = 6 per group). One-way ANOVA with Tukey’s post hoc test (**p < 0.01; n.s. not significant).
Genetic deletion of HDAC2 protects again pro-psychotic insults and improves short-term recognition memory
In order to study the effects of chronic atypical antipsychotic treatment on HDAC2 transcription in cortical pyramidal neurons, we recently used a genetic strategy to selectively suppress HDAC2 expression in CaMKIIα-positive glutamatergic neurons (Ibi et al., 2017). Other than this selective suppression of HDAC2, our previous data showed that these HDAC2-cKO mice showed undistinguishable level of expression of other Hdac tested compared to control littermates (Ibi et al., 2017).
The exposure of mice to a novel environment triggers an exploratory behavior generally reflected as increased locomotor activity. Interestingly, HDAC2-cKO mice showed an undistinguishable horizontal and vertical locomotor activity compared to control littermates (Fig. 2A and Fig. 2B) (F1, 15= 0.0840, p > 0.05; F1,15 = 0.0426, p > 0.05; respectively). Center-avoidance in an illuminated open field is correlated with anxiety-like behaviors (Weisstaub et al., 2006). On a separate experiment (see experimental details in the methods section), HDAC2-cKO animals did not show any differences in the time spent in the center of the arena compared to control mice (Fig. 2C) (F1,81 = 0.0889, p > 0.05). Taken together, our data indicate that deletion of HDAC2 in glutamatergic pyramidal neurons does not affect spontaneous exploratory locomotion activity or center-avoidance.
Fig 2.
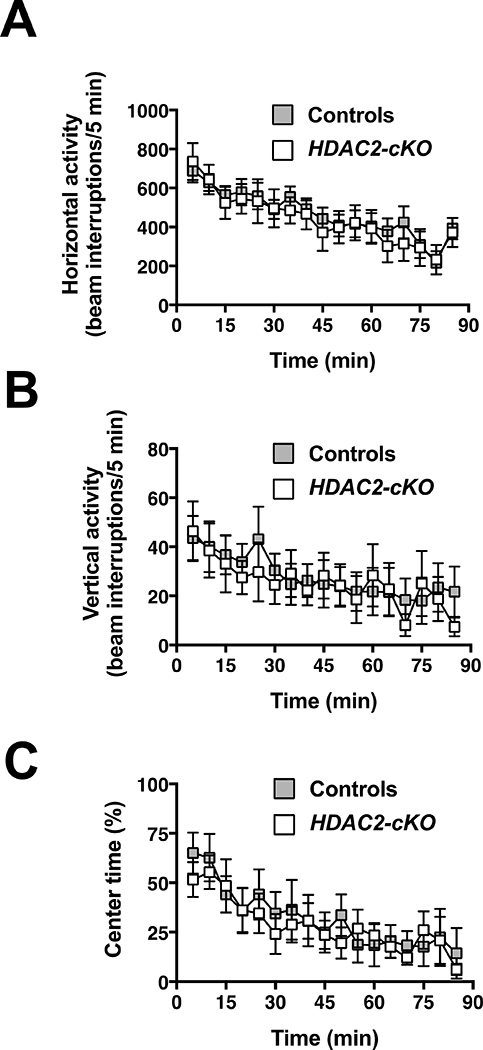
Horizontal activity (A) and vertical activity (B) are unaffected in HDAC2-cKO mice. Time spent in the center of the open field relative to total ambulatory time is comparable on both genotypes (C). Values plotted are mean ± S.E.M shown in 5-min blocks for HDAC2-cKO and control mice. (A and B, controls n = 9, HDAC2-cKO n = 8; C, controls n = 5, HDAC2-cKO n = 5). Two-way ANOVA (p > 0.05).
We next interrogated the effect of conditional FIDAC2 deletion on drug-based models of psychosis. Compared to control littermates, the head-twitch response elicited by the hallucinogen 5-ΗΤ2Α receptor agonist DOI was diminished in HDAC2-cKO (Fig. 3A) (t6=2.974, p < 0.05). Additionally, the HDAC2-cKO genotype was phenotypically resistant to the hyperlocomotive effects of the dissociative drug MK801, as shown in the time course of horizontal activity during 5-min fractions (Fig. 3B) (F1,7 = 5.927, p < 0.05) and total horizontal (Fig. 3C) (t7 = 2.435, p < 0.05) and vertical (Fig. 3D) (t8 = 2.817, p < 0.05) locomotor activity as summation of events during from t = 5 to t = 60 min. Together, these findings suggest that deletion of FIDAC2 in glutamatergic pyramidal neurons results in a protective phenotype against pro-psychotic insults.
Fig 3.
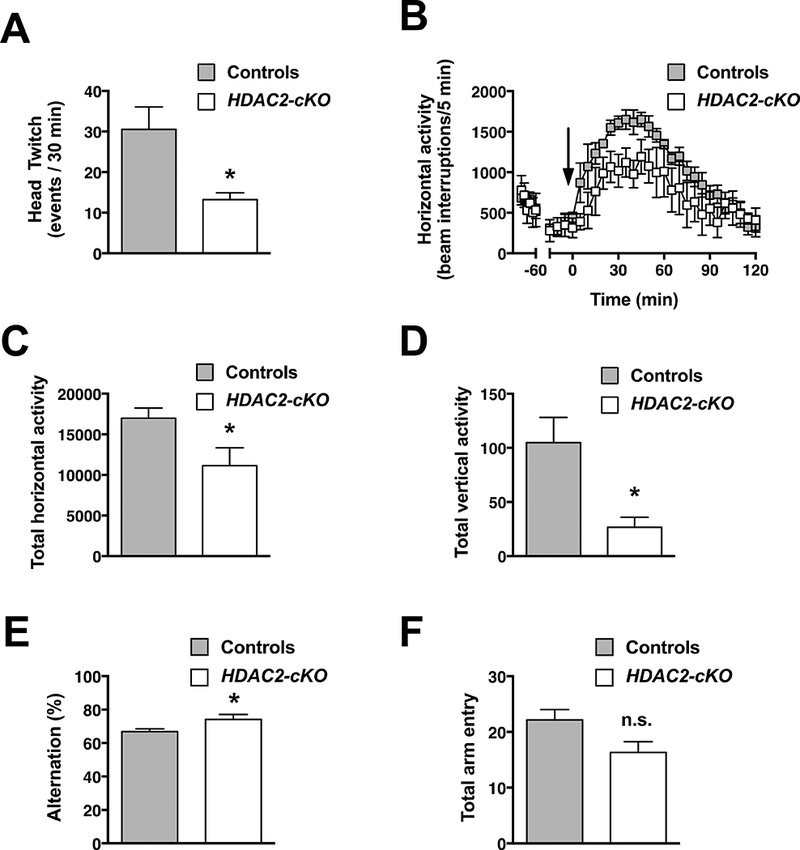
HDAC2-cKO mice are less responsive to DOI-induced stereotyped behavior. Quantification of head-twitch response induced by the psychedelic drug DOI (1 mg/kg) in HDAC2-cKO and control mice (A). HDAC2-cKO show decreased sensitivity to MK801-induced hyperlocomotion. HDAC2-cKO and control mice were allowed to freely-explore the test chamber for 90 min before the administration of MK801 (0.3 mg/kg) at t = 0. Horizontal activity measured as beam breaks shown in 5-min blocks over the 120-min test period (B) and quantified from t = 5 to t = 60 min (C). Vertical activity measured as beams broken upon rearing quantified over the same period of time (D). HDAC2-cKO showed a greater degree of alternation compared to controls in the Y-maze paradigm (E). Total number of arm entries (F). Data shown as mean ± S.E.M. (A - D, controls n = 5, HDAC2-cKO n = 4; E and F, controls n = 18, HDAC2-cKO n = 9). Time of injection is indicated by arrow (B). Two-tailed f-test (A,C,D); two-way ANOVA (B); (*p < 0.05; n.s., not significant).
Spontaneous alternation in the Y-maze reflects spatial memory retention. This test has been widely used as surrogate of cognitive performance sensitive to both pharmacological and genetic interventions (Flughes 2004). Compared to control mice, HDAC2-cKO showed a significant increase in arm alternation (Fig. 3E) (t25 = 2.324, p < 0.05), along with a decrease trend in the amount of total arm entries (Fig. 3F) (t25 =1.948,p=0.06
Chronic clozapine treatment negatively affects cortical active synapses via HDAC2
We previously reported that chronic treatment with clozapine induces an HDAC2-dependent decrease in the density of mature spines in frontal cortex pyramidal neurons (Ibi et al., 2017). Synaptophysin is a synaptic vesicle glycoprotein specifically localized in presynaptic terminals - its immunoreactivity is employed as a surrogate measure of active synapses (Calhoun et al., 1996; Fischer et al., 2007). We found here that immunoreactivity against synaptophysin was significantly reduced in somatosensory cortex layers ll/lll, IV and V (Fig. 4A and Fig. 4B) (Layer I, t80 = 1.87, p > 0.05; Layer II/III, t80 = 2.086, p < 0.05; Layer IV, t80 = 2.286, p < 0.05; Layer V, t80 = 2.224, p < 0.05; Layer VI, t80 = 1.522, p > 0.05). In opposition to the effect of chronic clozapine on synaptophysin immunoreactivity in control mice, we also found that HDAC2-cKO mice treated with chronic clozapine showed a generalized increased synaptophysin immunoreactivity through different somatosensory cortical layers as compared to HDAC2-cKO mice chronically treated with vehicle (Fig. 4C and Fig 4D) (Layer I, t98 = 8.788, p < 0.001; Layer II/III, t98 = 6.701, p < 0.001; Layer IV, t98 = 5.850, p < 0.001; Layer V, t98 = 6.685, p < 0.001; Layer VI, t98 = 3.578, p < 0.001 ). Three-way ANOVA analysis of immunoreactivity per cortical layer revealed significant individual effects for genotype (Controls vs. HDAC2-cKO, F1,182 = 132.89, p < 0.001), chronic treatment (Vehicle vs. Clozapine, F1,182 = 17.50, p < 0.001), and cortical layer (I through VI, F4,182 = 14.41, p < 0.001), as well as a two-way interaction Genotype: Treatment (F1,182 = 7.158, p < 0.001). Three-way interaction was absent. Immunoreactivity against synaptophysin was not significantly affected in the dentate gyrus of the hippocampus of control animals treated chronically with clozapine (Fig. 4E and Fig 4F) (Hilus, t23 = 0.827, p > 0.05; Granular, t23 = 0.4021, p > 0.05; Molecular, t23 = 0.6653, p > 0.05).
Fig 4.
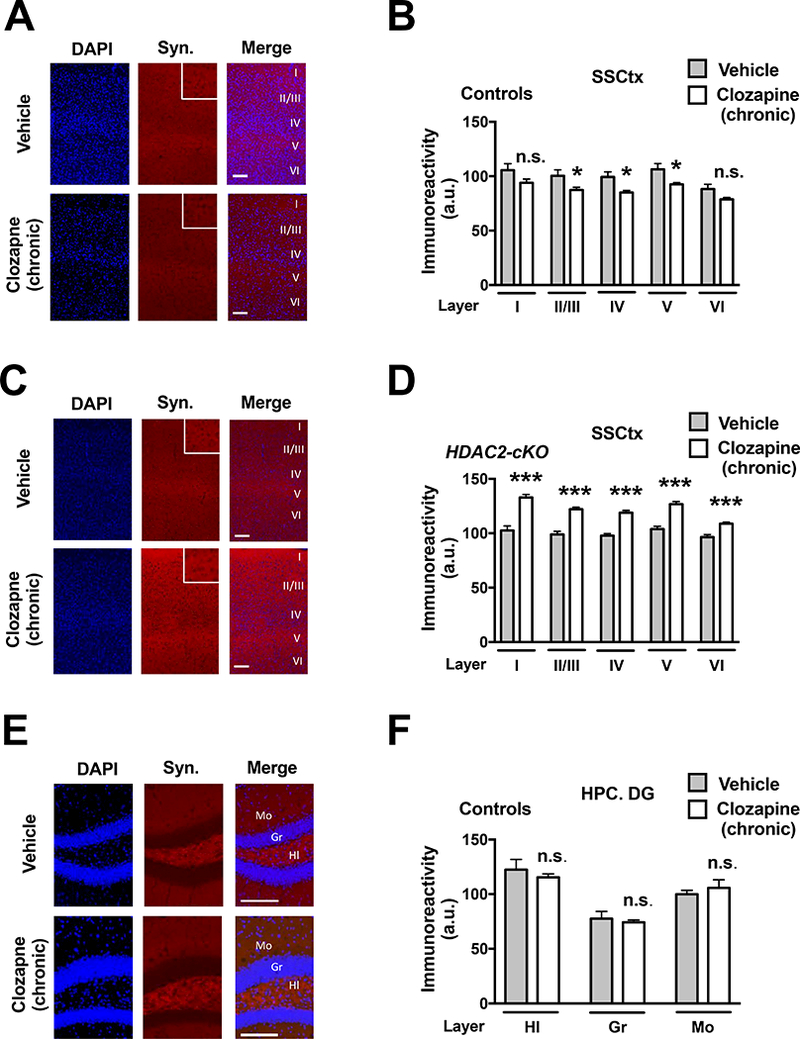
Effect of chronic clozapine treatment on synaptophysin immunoreactivity in HDAC2-cKO mice and controls. Representative immunohistochemical images of somatosensory cortex (A) and hippocampus (E) from control mice chronically treated with clozapine or vehicle. Quantification of the signal in the layers I through VI of the cortex (B) and dentate gyrus of the hippocampus (F). Representative immunohistochemical images of somatosensory cortex from HDAC2-cKO mice chronically treated with clozapine or vehicle (C). Quantification of the signal in the different layers of the cortex (D). SSCtx: somatosensory cortex, HPC: hippocampus, DG: dentate gyrus, Mo: molecular layer, Gr: granular layer, Hl: hilus. Scale bars 100 μm. Higher magnification insets (2x) of cortical layer IV (A and C). Values plotted are mean ± S.E.M. Two-tailed f-test (*p < 0.05; ***p < 0.001; n.s., not significant). Data shown as mean ± S.E.M. (B, vehicle n = 10, clozapine n = 8; D, vehicle 10, clozapine 12; F, vehicle n = 5, clozapine n = 5).
Chronic clozapine treatment reduces drug-induced psychosis-like behavior
Our current (see Fig. 4, above) and previous (Ibi et al., 2017) findings suggest that chronic clozapine treatment induces negative effects on synaptic plasticity and cognition. Based on this, we next aimed to interrogate the effect of chronic clozapine treatment on drug-induced models of psychosis. In agreement with our previous findings (Moreno et al., 2013), we found that chronic clozapine treatment was able to significantly reduce the count of head-twitch responses elicited by DOI (Fig. 5A) (t10 = 5.924, p < 0.001 ). While psychedelic 5-HT2A receptor agonists, such as DOI, remain the classic paradigm of pharmacologically-induced psychotic-like effects (Hanks and Gonzalez-Maeso 2013), non-competitive NMDA receptor antagonist, such as PCP and MK801, appear to recapitulate a wider array of core positive symptoms and cognitive deficits of schizophrenia (Moreno and Gonzalez-Maeso 2013). Mice chronically treated with chronic clozapine, or vehicle, were tested for the hyper-locomotive effects induced by MK801 (0.5 mg/kg) one day after the last dose of the antipsychotic. The marked increase in locomotor activity observed in mice previously treated chronically with vehicle was significantly reduced in mice previously treated chronically with clozapine, as shown both in horizontal activity during 5-min fractions (Fig. 5B) (F1,14 = 4.654, p < 0.05) and summation of locomotor activity events between t = 5 to t = 60 min (Fig. 5C)(t14 = 2.157, p < 0.05) after administration of MK801.
Fig 5.
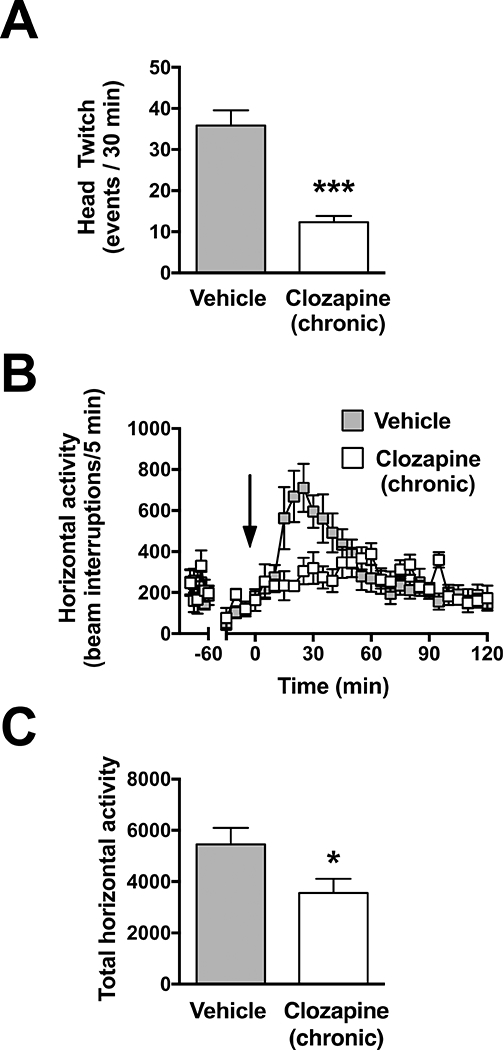
Chronic clozapine treatment reduces the action of the psychedelic DOI on stereotyped behavior. Quantification of head-twitch response to DOI (1 mg/kg) administered 24h after the last administration of clozapine or vehicle (A). Twenty-four hours after the last injection of clozapine or vehicle, mice were allowed to freely-explore the test chamber for 90 min administration of MK801 (0.5 mg/kg) at t = 0. Horizontal activity measured as beam breaks shown in 5-min blocks over the 120 min test period and quantified from t = 5 to t = 60 min (C). Time of injection is indicated by arrow. Values plotted are mean ± S.E.M. (A, n = 6 per group; B and C, vehicle n = 9, clozapine (chronic) n = 7). Two-tailed t-test (A,C); two-way ANOVA (B), (*p < 0.05; ***p < 0.001).
Additionally, we quantified the basal activity prior to administration of MK801 (Fig 5B (t = − 90 min to t = − 30 min) mean beam breaks ± S.E.M. from; Controls: 2011+280, Clozapine (chronic): 2038 ± 362). No differences were observed in the treated group relative to vehicle (t8 = 0.298, p > 0.05) thus suggesting that the repressive effect of chronic clozapine treatment on MK801-induced hyperlocomotor activity does not occur as a consequence of chronic clozapine treatment on basal exploratory behavior.
Chronic SAHA treatment prevents cognitive deficits induced by MK801
Previous findings have shown that chronic treatment with SAHA prevents the disruptive effects elicited by MK801 on the T-maze test as a mouse model of working memory (Kurita et al., 2012). Here we tested in HDAC2-cKO mice and control littermates the effect of chronic SAHA treatment on prevention of MK801 (0.1 mg/kg)-induced deficits in the novel object recognition test as an additional model of cognitive performance. (Fig. 6A and Fig. 6B).
Fig 6.
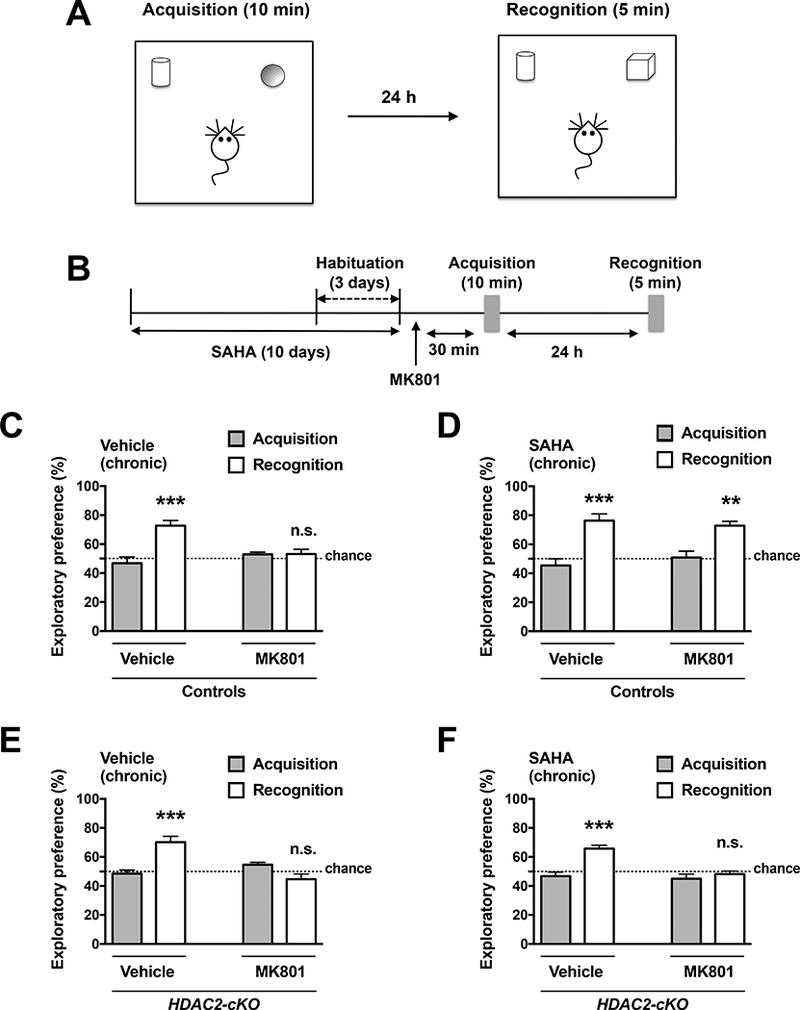
Chronic pharmacological inhibition of HDAC2 prevents the amnesic effects of MK801 in the novel object recognition test. Experimental setup of the novel object recognition test (A) and timeline of the experiment (B). The animals were injected with MK801 (0.1 mg/kg) one day after the last SAHA (20 mg/kg) injection. Novel object recognition test was conducted 30 min after MK801 injection. In the acquisition phase (10 min), mice were exposed to two different objects, and after a 24h interval, they were allowed to explore a duplicate of the familiar object and a novel object. Effect of MK801 on novel object exploratory preference in control mice chronically treated with vehicle or SAHA (D). Effect of MK801 on novel object exploratory preference in HDAC2-cKO mice chronically treated with vehicle (E) or SAHA (F). Values plotted are mean ± S.E.M. (n = 5 – 8 per group). Two-way ANOVA with Bonferroni post hoc test (**p < 0.01; ***p < 0.001; n.s., not significant). Dashed line indicates chance performance.
Control mice showed preference exploring the novel object, an effect that was prevented by previous administration of MK801 (Fig. 6C) (Treatment effect: F1,22 = 3.958, p > 0.05; post hoc test, Vehicle (acquisition vs. recognition) p < 0.001; MK801 (acquisition vs. recognition) p > 0.05). We also found that chronic SAHA treatment prevented the effects of MK801-induced deficits in novel object recognition (Fig. 6D) (Treatment effect: F1,18 = 0.0483, p > 0.05; post hoc test, Vehicle (acquisition vs. recognition) p < 0.01; MK801 (acquisition vs. recognition) p < 0.01). Importantly, this therapeutic-related effect of chronic SAHA treatment was absent in HDAC2-cKO mice (Fig. 6E and Fig. 6F). (Fig 6E; Treatment effect: F1,18 = 9.494, p < 0.01; post hoc test, Vehicle (acquisition vs. recognition) p < 0.001; MK801 (acquisition vs. recognition) p > 0.05) (Fig 6F. Treatment effect: F1,18 = 13.89, p < 0.01; post hoc test, Vehicle (acquisition vs. recognition) p < 0.001; MK801 (acquisition vs. recognition) p > 0.05). Additionally, HDAC2-cKO mice chronically treated with vehicle did not show alterations in the novel object recognition test as compared to control littermates chronically treated with vehicle (Fig. 6C and Fig. 6E). No significant differences were found in the total exploratory time (Figs. 7A-D). (Fig 7A. chronic treatment effect: F1,24 = 4.031, p < 0.05; post hoc test, chronic vehicle (vehicle vs. MK801) p > 0.05; chronic SAHA (vehicle vs. MK801) p > 0.05); (Fig. 7B, chronic treatment: F1,18 = 1.324, p > 0.05; post hoc test, chronic vehicle (vehicle vs. MK801 p > 0.05, chronic SAHA (vehicle vs. MK801) p > 0.05); (Fig. 7C, chronic treatment: F1,24 = 0.1426, p > 0.05; post hoc test, chronic vehicle (vehicle vs. MK801) p > 0.05; chronic SAHA (vehicle vs. MK801) p > 0.05); (Fig. 7D, chronic treatment: F1,18 = 1.273, p > 0.05; post hoc test, chronic vehicle (vehicle vs. MK801) p > 0.05; chronic SAHA (vehicle vs. MK801) p > 0.05).
Fig 7.
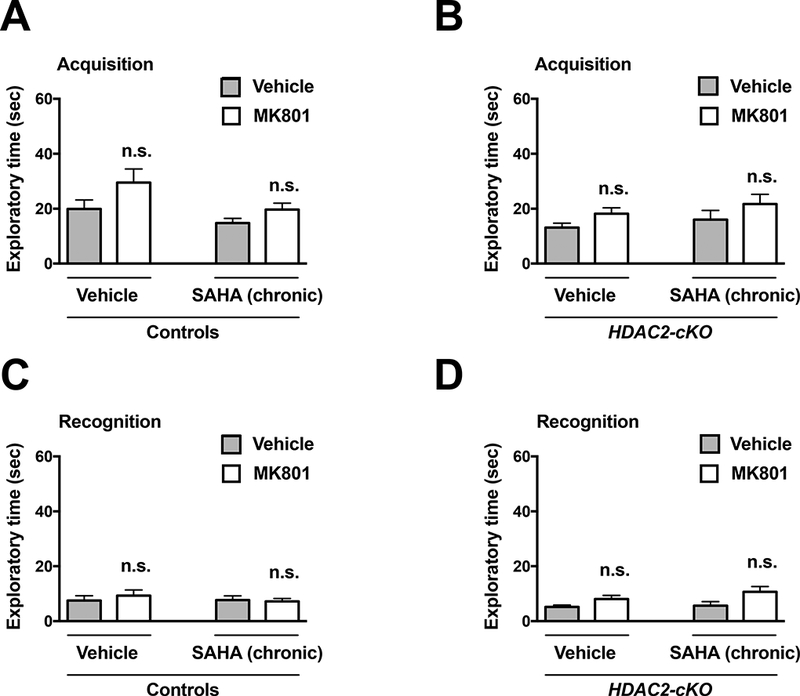
Exploration times are unaffected by MK801 (0.1 mg/kg). Total exploration times in the acquisition (A) and recognition (C) sessions of control animals chronically treated with vehicle or SAHA (20 mg/kg). Total exploration times in the acquisition (B) and recognition (D) sessions of HDAC2-cKO animals chronically treated with vehicle or SAHA (20 mg/kg). Values plotted are mean ± S.E.M. (n = 11 – 15 per group). Two-way ANOVA with Bonferroni post hoc test (n.s., not significant).
Three-way ANOVA analysis of exploratory preference in the recognition phase also revealed significant individual effects for genotype (control vs. HDAC2-cKO, F1,43 = 15.917, p < 0.001 ), chronic treatment (vehicle vs. SAHA, F1,43 = 7.145, p < 0.05), and acute treatment (saline vs. MK801, F 1,43 = 44.761, p < 0.001 ), as well as a two-way interaction Genotype: Chronic (F1,43 = 7.158, p < 0.05) and Chronic: Acute (F1,43 = 6.574, p < 0.05). Tukey’s post-hoc analysis of Genotype: Chronic interactions showed significant differences between vehicle vs. SAHA subgroups within the control group (Control (Vehicle vs. SAHA), p < 0.001), but not in HDAC2-cKO mice (HDAC2-cKO (Vehicle vs. SAHA), p > 0.05).
Transcriptome regulation by chronic SAHA treatment via HDAC2
We next employed microarray analysis to examine gene expression profiles induced upon chronic SAHA treatment in the frontal cortex of HDAC2-cKO and control mice. Control animals that received chronic SAHA treatment displayed a dramatically increased gene expression in comparison to vehicle-treated mice. As a result, we obtained a final list of 284 probe sets, corresponding to 124 RefSeq genes that are associated with chronic SAHA treatment (Fig. 8A and Table 1). We confirmed the significant regulation of representative genes that were selected from our microarray analysis based on their involvement in synaptic plasticity and connectivity: Adam8, Ankrd9, Cxcr3, Cdkn2c, Tbkbp1 and Zdhhc12 (Li et al., 2007; Bajova et al., 2008; Liebau et al., 2009; Bartsch et al.,; Fukata and Fukata). Control mice chronically treated with SAHA showed frontal cortex upregulation of these genes in control mice chronically treated with SAHA, however, these changes were not observed in SAHA-treated HDAC2-cKO littermates (Fig. 8B) (Two-way ANOVA Fisher’s LSD post hoc test; Adam8, Control (Saline vs. SAHA) p < 0.01, HDAC2-cKO (Saline vs. SAHA) p > 0.05; Ankrd9, Control (Saline vs. SAHA) p < 0.001, HDAC2-cKO (Saline vs. SAHA) p > 0.05; Cdkn2c, Control (Saline vs. SAHA) p<0.01, HDAC2-cKO (Saline vs. SAHA) p>0.05; Cxcr3, Control (Saline vs. SAHA) p < 0.05, HDAC2-cKO (Saline vs. SAHA) p> 0.05; Tbkbp1, Control (Saline vs. SAHA) p < 0.05, HDAC2-cKO (Saline vs. SAHA) p> 0.05; Zdhhc12, Control (Saline vs. SAHA) p < 0.01, HDAC2-cKO (Saline vs. SAHA) p>0.05). Housekeeping gene Rps3 was not affected by either treatment or genotype (not shown) (Two-way ANOVA Fisher’s LSD post hoc test; Rps3, Control (Saline vs. SAHA) p >0.05, HDAC2-cKO (Saline vs. SAHA) p > 0.05).
Fig 8.
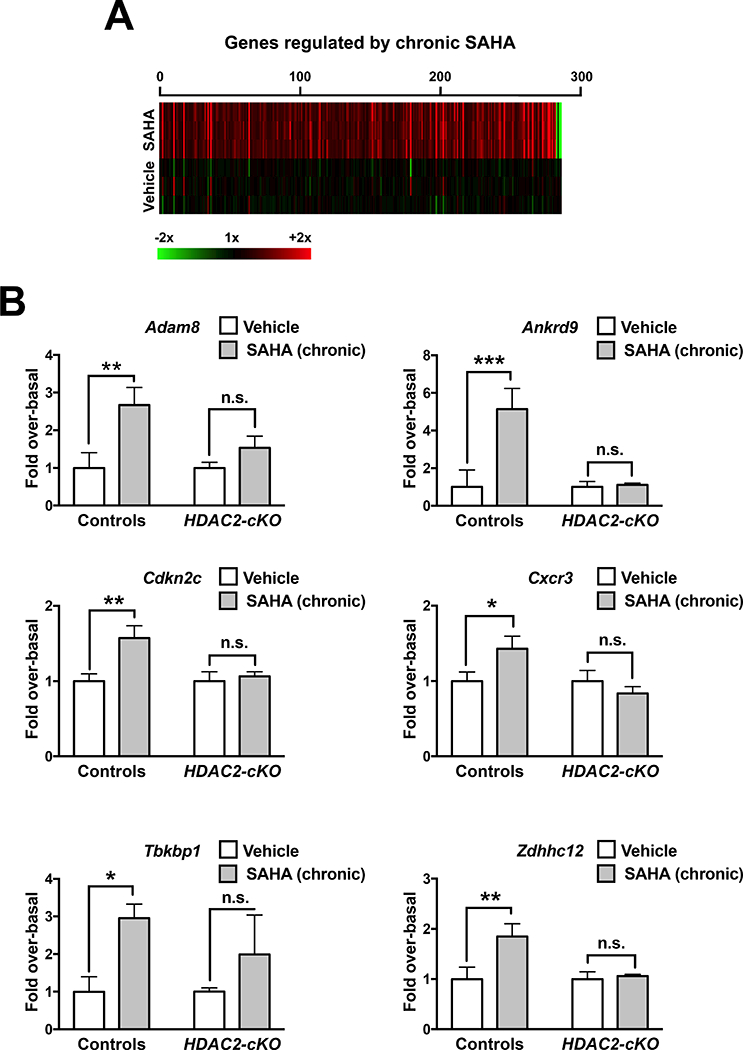
Comparative heat map displaying genes differentially expressed in the frontal cortex of mice chronically treated with SAHA (20 mg/kg) versus vehicle (n = 3 mice per library) (A). See also Table 1 for a complete list of top 100 genes up-regulated by chronic SAHA treatment. Validation of mRNA expression of gene transcripts by qRT-PCR in the frontal cortex of HDAC2-cKO mice and controls (B) (n = 5 – 6 per group) chronically treated with SAHA or vehicle. Values plotted are mean ± S.E.M. Two-way ANOVA with Fisher’s LSD post hoc test (*p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant).
Table 1.
Top 100 genes up-regulated by chronic SAHA treatment in the frontal cortex of control mice
| Gene Name | GenBank | Fold change (Unlogged) | q-value (%) |
|---|---|---|---|
| Vmn2r9 | NM_001104621 | 1.75 | 10.91 |
| Ripk4 | NM_023663 | 1.52 | 10.75 |
| Akap3 | NM_009650 | 1.50 | 10.91 |
| Zdhhc12 | NM_025428 | 1.48 | 11.59 |
| Csf2rb2 | NM_007781 | 1.48 | 10.75 |
| Hbq1 | NM_175000 | 1.48 | 10.53 |
| Rhoh | NM_001081105 | 1.48 | 10.72 |
| Olfr745 | NM_146299 | 1.48 | 11.72 |
| Sftpd | NM_009160 | 1.48 | 10.16 |
| Gm11564 | NM_001100614 | 1.47 | 11.59 |
| Cyp2d40 | NM_023623 | 1.46 | 10.91 |
| Prp2 | NM_031499 | 1.44 | 10.13 |
| Olfr1463 | NM_001011840 | 1.43 | 10.62 |
| Atp6v1g3 | NM_177397 | 1.43 | 10.62 |
| Lcn8 | NM_033145 | 1.42 | 10.91 |
| Il9 | NM_008373 | 1.42 | 10.16 |
| Tsnaxip1 | NM_024445 | 1.42 | 10.48 |
| S100z | NM_001081159 | 1.40 | 10.75 |
| BC089491 | NM_175033 | 1.40 | 10.91 |
| Olfr234 | NM_001001807 | 1.39 | 11.59 |
| Ceacam14 | NM_025957 | 1.38 | 10.91 |
| 2210415F13Rik | NM_001083884 | 1.37 | 10.91 |
| V1rc5 | NM_053235 | 1.37 | 11.59 |
| Ankrd9 | NM_175207 | 1.36 | 10.91 |
| Olfr830 | NM_146566 | 1.36 | 11.59 |
| Krtap5–1 | NM_015808 | 1.36 | 10.91 |
| Khdc1a | NM_183322 | 1.36 | 11.59 |
| Pbp2 | NM_029595 | 1.36 | 11.59 |
| Olfr1010 | NM_207149 | 1.36 | 10.91 |
| Zfp513 | NM_175311 | 1.35 | 10.91 |
| Qrfp | NM_183424 | 1.35 | 10.43 |
| Efnb1 | NM_010110 | 1.35 | 10.62 |
| Tnfsf11 | NM_011613 | 1.34 | 10.91 |
| Olfr1219 | NM_146899 | 1.34 | 10.75 |
| Oxt | NM_011025 | 1.34 | 11.59 |
| Tulp1 | NM_021478 | 1.34 | 10.91 |
| Bhlhb8 | NM_010800 | 1.34 | 10.91 |
| Olfr12 | NM_206896 | 1.34 | 10.91 |
| Pgc | NM_025973 | 1.33 | 11.85 |
| Lpar2 | NM_020028 | 1.33 | 11.59 |
| Aqp1 | NM_007472 | 1.33 | 10.91 |
| Zdhhc11 | NM_027704 | 1.33 | 11.59 |
| Pnliprp2 | NM_011128 | 1.33 | 10.91 |
| Olfr1105 | NM_001011825 | 1.32 | 10.75 |
| Gpr31c | NM_001013832 | 1.32 | 10.91 |
| Sctr | NM_001012322 | 1.32 | 11.72 |
| Plb1 | NM_001081407 | 1.32 | 10.75 |
| Apoc2 | NM_009695 | 1.32 | 11.59 |
| D430042O09Rik | NM_001081022 | 1.32 | 11.59 |
| Mesp2 | NM_008589 | 1.32 | 10.91 |
| Krt2 | NM_010668 | 1.31 | 10.62 |
| EG574081 | NM_001025351 | 1.31 | 10.62 |
| Col22a1 | XM_907370 | 1.31 | 10.91 |
| Mrgprg | NM_203492 | 1.31 | 10.13 |
| Cpz | NM_153107 | 1.31 | 10.91 |
| Olfr1036 | NM_207142 | 1.31 | 10.91 |
| Tas2r113 | NM_207018 | 1.30 | 11.59 |
| Gpr31c | NM_001013832 | 1.30 | 10.73 |
| Gm428 | NM_001081644 | 1.30 | 10.91 |
| Tbkbp1 | NM_198100 | 1.30 | 10.16 |
| Uts2r | NM_145440 | 1.30 | 10.16 |
| BC023744 | NM_001033311 | 1.30 | 10.62 |
| Scarf2 | NM_153790 | 1.30 | 10.91 |
| Tcl1b4 | NM_013774 | 1.30 | 10.62 |
| Nkx2–5 | NM_008700 | 1.30 | 10.91 |
| BC055004 | NM_001013773 | 1.30 | 11.59 |
| Hemgn | NM_053149 | 1.29 | 10.16 |
| Cxcr3 | NM_009910 | 1.29 | 11.85 |
| Olfr1451 | NM_146705 | 1.29 | 10.72 |
| Cypt3 | NM_173367 | 1.28 | 11.59 |
| AI747448 | NM_001033199 | 1.28 | 10.16 |
| Shank2 | XR_034431 | 1.28 | 10.91 |
| Mustn1 | NM_181390 | 1.28 | 10.13 |
| Pnma5 | NM_001100461 | 1.28 | 10.16 |
| Defb25 | NM_001039122 | 1.28 | 10.91 |
| Sall4 | NM_201395 | 1.28 | 10.16 |
| Atp4b | NM_009724 | 1.27 | 11.46 |
| Tbc1d10c | NM_178650 | 1.27 | 10.73 |
| Muc4 | NM_080457 | 1.27 | 10.78 |
| Cd70 | NM_011617 | 1.26 | 10.16 |
| Oxct2b///Oxct2a | NM_181859 | 1.26 | 11.59 |
| Gm815 | NM_001033407 | 1.26 | 10.78 |
| Olfr319 | NM_146500 | 1.26 | 10.75 |
| Olfr769 | NM_146267 | 1.25 | 10.91 |
| Lgals7 | NM_008496 | 1.25 | 10.91 |
| Ros1 | NM_011282 | 1.25 | 10.13 |
| Cd7 | NM_009854 | 1.25 | 10.16 |
| Chit1 | NM_027979 | 1.24 | 10.91 |
| Tchhl1 | NM_027762 | 1.24 | 10.16 |
| Fcgbp | NM_001122603 | 1.24 | 10.91 |
| 1110028A07Rik | NM_026808 | 1.23 | 10.62 |
| Rhbdl2 | NM_183163 | 1.23 | 10.13 |
| Col4a1 | NM_009931 | 1.23 | 11.59 |
| Col9a2 | NM_007741 | 1.22 | 10.62 |
| Adam8 | NM_007403 | 1.22 | 10.16 |
| Slc25a43 | NM_001085497 | 1.22 | 10.16 |
| BC048671 | NM_177738 | 1.22 | 10.13 |
| Cdkn2c | NM_007671 | 1.20 | 10.16 |
| Nlrp10 | NM_175532 | 1.20 | 10.62 |
DISCUSSION
Results from the present study suggest that either pharmacological or genetic inhibition of HDAC2 leads to behavioral phenotypes that model therapeutic-related action on both psychosis-like states and cognition. Consistent with previous reports of upregulation of HDAC2 expression in the frontal cortex of mice chronically treated with atypical antipsychotics, but not with haloperidol (Kurita et al., 2011; Ibi et al., 2017), we also validate this particular effect of chronic clozapine treatment on frontal cortex HDAC2 immunoreactivity with the use of independent experimental approaches. Additionally, we show that chronic administration of clozapine reduces the number of active synapses in the frontal cortex, as defined by immunostaining against synaptophysin. However, an opposite effect was observed in the frontal cortex of HDAC2-cKO mice treated chronically with clozapine, suggesting that augmentation of HDAC2 expression, which would consequently boost HDAC2-dependent epigenetic function, may repress some of the therapeutic effects of chronic clozapine treatment. This is further supported by results showing that chronic treatment with the class I and class II HDAC inhibitor SAHA leads to HDAC2-dependent changes in expression of genes associated to signaling pathways involved in synaptic plasticity and synaptic connectivity.
Positive psychotic symptoms in schizophrenia patients, including hallucinations and delusions, represent the most devastating consequence of this psychiatric condition (Sawa and Snyder 2002; van Os and Kapur 2009). Pharmacological blockade of the 5-HT2A receptor has been proposed as one of the potential mechanism underlying the therapeutic properties of antipsychotic medications on psychosis (Lieberman et al., 2008; Miyamoto et al., 2012; Meltzer 2013). This is further supported by the relatively recent clinical use of the highly selective 5-HT2A receptor antagonist/inverse agonist pimavanserin for the treatment of Parkinson’s disease psychosis (Sahli and Tarazi 2017). Many articles describe improvement of psychotic symptoms hours/days immediately after antipsychotic drug administration (Wright et al., 2001; Agid et al., 2003). However, psychiatric disorders in general, and schizophrenia in particular, are often characterized by continuous drug administration (weeks, months or even years of sustained drug treatment). This does not question the validity of antipsychotic-like behavior models of acute antipsychotic drug administration, but rather validate the importance of understanding the consequences of long-term antipsychotic treatment.
Accordingly, our current data showed that chronic clozapine treatment prevents psychosis-like behavioral events induced by pharmacological tools such as the psychedelic drug DOI and the dissociative drug MK801. This validates the therapeutic-related activity of chronic clozapine treatment using psychosis-related behaviors. Notably, we also demonstrated that DOI-induced head-twitch behavior and MK801-induced hyperlocomotor activity are significantly decreased in HDAC2-cKO mice as compared to controls. Despite the inherent limitations of rodent models of psychosis have limitations (Fernando and Robbins 2011; Forrest et al., 2014), our findings establish a parallelism between the phenotypes emerging upon blockade of HDAC2 and previous preclinical models of classic antipsychotic drug action. Prospectively, these findings suggest that pharmacological inhibition of HDAC2 may serve as a new approach to improve the clinical efficacy of currently available antipsychotics on hallucinations and delusions. Nevertheless, further work will be necessary to establish the precise molecular link between genetic deletion of forebrain HDAC2 and decreased sensitivity to hallucinogens and dissociative drugs.
Our results show that chronic administration of the HDAC inhibitor SAHA modulates expression of genes in mouse frontal cortex involved in signaling networks that regulate neuronal morphogenesis and synaptic plasticity, including Tbkbp1, Adam8, Cxcr3, Cdkn2c, and Zdhhc12 (Li et al., 2007; Bajova et al., 2008; Liebau et al., 2009; Bartsch et al.,; Fukata and Fukata). We also demonstrate that HDAC2 is involved in modulating transcription of these genes. HDAC inhibitors including trichostatin A (TSA), sodium butyrate, and SAHA improve learning consolidation and enhance synaptic plasticity (Lattal et al., 2007; Bredy and Barad 2008; Fontan-Lozano et al., 2008; Kilgore et al., 2010; Kurita et al., 2012). It has also been shown that HSV mediated over-expression of HDAC2 in the frontal cortex results in behaviors that are associated with negative regulation of sensorimotor gating of the startle reflex and working memory impairments (Kurita et al. 2012). Our previous data also suggested that chronic treatment with clozapine induces 5-HT2A receptor-dependent augmentation of HDAC2 expression (Kurita et al., 2012; Ibi et al., 2017). HDAC2 has been shown to negatively affect transcription of genes involved in synaptic structure, synaptic plasticity and memory (Guan et al., 2009). Considering that we also reported that chronic clozapine treatment induced HDAC2-dependent repression of genes involved in cell morphogenesis, neuron projection and synapse structure (Ibi et al., 2017), together these findings suggest that either absence of effect or negative outcome of chronic antipsychotic administration on cognitive function may represent a consequence of compensatory pathways induced after long-lasting and repeated pharmacological blockade of 5-HT2A receptor-dependent function. This hypothesis is further supported by our current findings showing that chronic clozapine treatment also led to a decrease in cortical synaptophysin immunoreactivity. Notably, and opposite to what occurred in control mice treated chronically with clozapine, synaptophysin immunoreactivity was increased in the somatosensory cortex of HDAC2-cKO mice treated chronically with clozapine. Together, these results suggest that inhibition of HDAC2 may represent a new approach to improve cognitive capabilities in medicated schizophrenia patients.
Using a mouse model of maternal stress during pregnancy, previous findings convincingly demonstrate that prenatal environmental insults induce adult offspring changes in frontal cortex DNA methylation at the promoter region of genes such as Gad1, Reln, and Bdnf, and that these epigenetic changes were reversed by a short treatment (5 days) with clozapine, but not with haloperidol (Dong et al., 2016). Our current findings suggest that up-regulation of frontal cortex HDAC2 immunoreactivity occurs in mice chronically (21 days) treated with clozapine, but not with haloperidol. Importantly, we previously showed that this effect is not observed after sub-chronic (2 days) treatment with clozapine (Ibi et al., 2017). Together, these findings suggest that the HDAC2-dependent negative effects of chronic clozapine treatment on synaptic plasticity and behavior require a long-term regimen of medication.
An important finding was the specificity of the effect of chronic treatment with clozapine and other atypical antipsychotics, but not with haloperidol, on HDAC2 expression. These data, however, do not exclude the existence of alternative signaling pathways affected by haloperidol and other typical antipsychotics that might be responsible for their negative effects in terms of serious movement disorders as well as cognitive capabilities.
It is well recognized that gender differences have an impact on mental health and, in particular, on the course of schizophrenia (Crawford and DeLisi 2016). The current analysis focus on male mice, and most of the subjects included in our previous studies in postmortem human brain samples were male (Kurita et al., 2012; Ibi et al., 2017). Future work should extend these findings to females to determine the generalizability and specificity of epigenetic mechanisms of antipsychotic drug action.
One of the limitations of the Cre-lox system in rodent models is that related to unexpected recombination events resulting from transient ectopic expression of Cre driver genes during early development in cell lines different from the targeted population (Song and Palmiter 2018). Using the Cre-lox system approach, we and others have previously validated the selective deletion of HDAC2 expression in CaMKIlα-positive glutamatergic cortical and hippocampal pyramidal neurons of HDAC2loxP/loxP:CaMKIIα-Cre+/− (HDAC2-cKO) mice. This was achieved using either HDAC2loxP/loxP:CaMKIIα:Cre−/− (Ibi et al., 2017) or wild-type (Morris et al., 2013) littermates as the control group. Although we cannot completely eliminate the possibility of unexpected expression of Cre, which would create nonspecific results, based on our previous data characterizing conditional deletion of HDAC2 in glutamatergic pyramidal neurons (Ibi et al., 2017), we are confident of appropriately restricted expression of the Cre recombinase. Another concerns related to the use of Cre-lox technology is the potential toxicity related to non-specific recombinase activity in the host genomic DNA (Schmidt-Supprian and Rajewsky 2007), further experiments will address the potentially deleterious effect of Cre recombinase on gene expression and synaptic plasticity in HDAC2-cKO and control mice.
CONCLUSIONS
Here we show that selective deletion of HDAC2 function in forebrain pyramidal glutamatergic neurons operates as a protective factor against pro-psychotic insults elicited by psychedelic and dissociative drugs as well as against the deleterious effect of chronic clozapine treatment on active synapses. Additionally, our results based on microarray analysis and behavioral models of recognition memory support the notion that HDAC2 inhibitors may serve as a new pharmacological tool to ameliorate deficits related to perception, cognition and memory function in patients with neuropsychiatric disorders such as schizophrenia.
Highlights.
Inhibition or genetic deletion of HDAC2 results in antipsychotic-like phenotypes
HDAC2 underlies the negative effects of chronic clozapine on cortical synapses
Chronic HDAC inhibition augments expression of plasticity-related genes
HDAC2 may represent a new epigenetic target for the treatment of schizophrenia
ACKNOWLEDGMENTS
The authors would like to thank Hirofumi Morishita for the donation of CaMKIIα-Cre mice; and Eric Olson, Rhonda Bassel-Duby and Eric Nestler for their gift of floxed HDAC2 mice. NIH R01 MH084894 (J.G.M.), MH111940 (J.G.M.), and the Japan Society for the Promotion of Science (JSPS) 15H06719 and 16K19786 (D.l.) participated in the funding of this study. R.T. was recipient of undergraduate fellowship from Pierre et Marie Curie University. D.l. was recipient of postdoctoral fellowships from JSPS (Young Scientists JSPS 23–3454) and the Uehara Memorial Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
M.d.l.F.R., D.I. and J.G.M. designed experiments and analyzed data. M.d.l.F.R. and D.I. performed experiments. J.G.M. supervised the research. M.d.l.F.R. and J.G.M. wrote the manuscript. J.M.S., T.C., M.K.I., R.T., M.K. and T.H. assisted with experiments. mice. L.S., J.S. and M.G.D. performed biostatistical analysis. All authors contributed to the revision of the manuscript.
CONFLICT OF INTERESTS
The authors declare no conflict of interests
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abel T, Zukin RS (2008) Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol 8:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agid O, Kapur S, Arenovich T, Zipursky RB (2003) Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry 60:1228–1235. [DOI] [PubMed] [Google Scholar]
- Bajova H, Nelson TE, Gruol DL (2008) Chronic CXCL10 alters the level of activated ERK1/2 and transcriptional factors CREB and NF-kappaB in hippocampal neuronal cell culture. J Neuroimmunol 195:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch JW, Wildeboer D, Koller G, Naus S, Rittger A, Moss ML, Minai Y, Jockusch H (2010) Tumor necrosis factor-alpha (TNF-alpha) regulates shedding of TNF-alpha receptor 1 by the metalloprotease-disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J Neurosci 30:12210–12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Barad M (2008) The histone deacetylase inhibitor valproic acid enhances acquisition, extinction, and reconsolidation of conditioned fear. Learn Mem 15:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, Do KQ (2013) Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci U S A 110:9130–9135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Jucker M, Martin LJ, Thinakaran G, Price DL, Mouton PR (1996) Comparative evaluation of synaptophysin-based methods for quantification of synapses. J Neurocytol 25:821–828. [DOI] [PubMed] [Google Scholar]
- Casey DE, Daniel DG, Tamminga C, Kane JM, Tran-Johnson T, Wozniak P, Abi-Saab W, Baker J, Redden L, Greco N, Saltarelli M (2009) Divalproex ER combined with olanzapine or risperidone for treatment of acute exacerbations of schizophrenia. Neuropsychopharmacology 34:1330–1338. [DOI] [PubMed] [Google Scholar]
- Casey DE, Daniel DG, Wassef AA, Tracy KA, Wozniak P, Sommerville KW (2003) Effect of divalproex combined with olanzapine or risperidone in patients with an acute exacerbation of schizophrenia. Neuropsychopharmacology 28:182–192. [DOI] [PubMed] [Google Scholar]
- Citrome L, Casey DE, Daniel DG, Wozniak P, Kochan LD, Tracy KA (2004) Adjunctive divalproex and hostility among patients with schizophrenia receiving olanzapine or risperidone. Psychiatr Serv 55:290–294. [DOI] [PubMed] [Google Scholar]
- Consortium. SWGotPG (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ (2009) Antidepressant actions of histone deacetylase inhibitors. J Neurosci 29:11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford MB, DeLisi LE (2016) Issues related to sex differences in antipsychotic treatment. Curr Opin Psychiatry 29:211–217. [DOI] [PubMed] [Google Scholar]
- Dong E, Tueting P, Matrisciano F, Grayson DR, Guidotti A (2016) Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl Psychiatry 6:e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando AB, Robbins TW (2011) Animal Models of Neuropsychiatric Disorders. Annu Rev Clin Psychol DOI 10.1146/annurev-clinpsy-032210-104454 [DOI] [PubMed] [Google Scholar]
- Fervaha G, Takeuchi H, Lee J, Foussias G, Fletcher PJ, Agid O, Remington G (2015) Antipsychotics and amotivation. Neuropsychopharmacology 40:1539–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH (2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447:178–182. [DOI] [PubMed] [Google Scholar]
- Fontan-Lozano A, Romero-Granados R, Troncoso J, Munera A, Delgado-Garcia JM, Carrion AM (2008) Histone deacetylase inhibitors improve learning consolidation in young and in KA-induced-neurodegeneration and SAMP-8-mutant mice. Mol Cell Neurosci 39:193–201. [DOI] [PubMed] [Google Scholar]
- Forrest AD, Coto CA, Siegel SJ (2014) Animal Models of Psychosis: Current State and Future Directions. Curr Behav Neurosci Rep 1:100–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L,
- Rees E, Palta P, Ruderfer DM, Carrera N, Humphreys I, Johnson JS, Roussos P, Barker DD, Banks E, Milanova V, Grant SG, Hannon E, Rose SA, Chambert K, Mahajan M,
- Scolnick EM, Moran JL, Kirov G, Palotie A, McCarroll SA, Holmans P, Sklar P, Owen MJ, Purcell SM, O’Donovan MC (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature 506:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Fukata M (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci 11:161–175. [DOI] [PubMed] [Google Scholar]
- Goldberg TE, Greenberg RD, Griffin SJ, Gold JM, Kleinman JE, Pickar D, Schulz SC, Weinberger DR (1993) The effect of clozapine on cognition and psychiatric symptoms in patients with schizophrenia. Br J Psychiatry 162:43–48. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Weisstaub NV, Zhou M, Chan P, Ivic L, Ang R, Lira A, Bradley-Moore M, Ge Y, Zhou Q, Sealfon SC, Gingrich JA (2007) Hallucinogens Recruit Specific Cortical 5-HT(2A) Receptor-Mediated Signaling Pathways to Affect Behavior. Neuron 53:439–452. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Maeso J, Yuen T, Ebersole BJ, Wurmbach E, Lira A, Zhou M, Weisstaub N, Hen R, Gingrich JA, Sealfon SC (2003) Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J Neurosci 23:8836–8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Joseph NF, Horn ME, Samiei A, Meng J, Seo J, Rei D, Bero AW, Phan TX, Wagner F, Holson E, Xu J, Sun J, Neve RL, Mach RH, Haggarty SJ, Tsai LH (2014) Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell 156:261–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Tsai LH (2013) The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol 53:311–330. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459:55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks JB, Gonzalez-Maeso J (2013) Animal models of serotonergic psychedelics. ACS Chem Neurosci 4:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard BE, Sick B, Heber S (2009) Unsupervised assessment of microarray data quality using a Gaussian mixture model. BMC Bioinformatics 10:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes RN (2004) The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev 28:497–505. [DOI] [PubMed] [Google Scholar]
- Husa AP, Moilanen J, Murray GK, Marttila R, Haapea M, Rannikko I, Barnett JH, Jones PB, Isohanni M, Remes AM, Koponen H, Miettunen J, Jaaskelainen E (2016) Lifetime antipsychotic medication and cognitive performance in schizophrenia at age 43 years in a general population birth cohort. Psychiatry Res 247:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, de la Fuente Revenga M, Kezunovic N, Muguruza C, Saunders JM, Gaitonde SA, Moreno JL, Ijaz MK, Santosh V, Kozlenkov A, Holloway T, Seto J, Garcia-Bea A, Kurita M, Mosley GE, Jiang Y, Christoffel DJ, Callado LF, Russo SJ, Dracheva S, Lopez-Gimenez JF, Ge Y, Escalante CR, Meana JJ, Akbarian S, Huntley GW, Gonzalez-Maeso J (2017) Antipsychotic-induced Hdac2 transcription via NF-kappaB leads to synaptic and cognitive side effects. Nat Neurosci 20:1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, Gonzalez-Maeso J (2015) Epigenetic signaling in schizophrenia. Cell Signal DOI 10.1016/j.cellsig.2015.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim HM, Tamminga CA (2012) Treating impaired cognition in schizophrenia. Curr Pharm Biotechnol 13:1587–1594. [DOI] [PubMed] [Google Scholar]
- Kelly DL, Conley RR, Feldman S, Yu Y, McMahon RP, Richardson CM (2006) Adjunct divalproex or lithium to clozapine in treatment-resistant schizophrenia. Psychiatr Q 77:81–95. [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G (2010) Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35:870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, Heshmati M, Golden SA, Kennedy PJ, Takahashi N, Dietz DM, Mocci G, Gabilondo AM, Hanks J, Umali A, Callado LF, Gallitano AL, Neve RL, Shen L, Buxbaum JD, Han MH, Nestler EJ, Meana JJ, Russo SJ, Gonzalez-Maeso J (2012) HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci 15:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Garcia-Bea A, Moreno JL, Kozlenkov A, Heshmati M, Golden S, Dietz DM, Kennedy PJ, Umali A, Callado LF, Neve RL, Nestler EJ, Meana JJ, Russo SJ, Gonzalez-Maeso J (2011) HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. The New York Academy of Sciences 24: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Gonzalez-Maeso J (2013a) HDAC2 as a new target to improve schizophrenia treatment. Expert Rev Neurother 13:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Moreno JL, Holloway T, Kozlenkov A, Mocci G, Garcia-Bea A, Hanks JB, Neve R, Nestler EJ, Russo SJ, Gonzalez-Maeso J (2013b) Repressive Epigenetic Changes at the mGlu2 Promoter in Frontal Cortex of 5-HT2A Knockout Mice. Mol Pharmacol 83:1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Barrett RM, Wood MA (2007) Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci 121:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Li X, Chen W, Yu S, Chen J, Wang H, Ruan D (2007) The different roles of cyclinD1-CDK4 in STP and mGluR-LTD during the postnatal development in mice hippocampus area CA1. BMC Dev Biol 7:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau S, Proepper C, Schmidt T, Schoen M, Bockmann J, Boeckers TM (2009) ProSAPiP2, a novel postsynaptic density protein that interacts with ProSAP2/Shank3. Biochem Biophys Res Commun 385:460–465. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Bymaster FP, Meltzer HY, Deutch AY, Duncan GE, Marx CE, Aprille JR, Dwyer DS, Li XM, Mahadik SP, Duman RS, Porter JH, Modica-Napolitano JS, Newton SS, Csernansky JG (2008) Antipsychotic drugs: comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol Rev 60:358–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK (2005) Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353:1209–1223. [DOI] [PubMed] [Google Scholar]
- Liu-Seifert H, Adams DH, Kinon BJ (2005) Discontinuation of treatment of schizophrenic patients is driven by poor symptom response: a pooled post-hoc analysis of four atypical antipsychotic drugs. BMC Med 3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer HY (2013) Update on typical and atypical antipsychotic drugs. Annu Rev Med 64:393–406. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Bonaccorso S, Bobo WV, Chen Y, Jayathilake K (2011) A 12-month randomized, open-label study of the metabolic effects of olanzapine and risperidone in psychotic patients: influence of valproic acid augmentation. J Clin Psychiatry 72:1602–1610. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Agid Y, Brune M, Bullmore ET, Carter CS, Clayton NS, Connor R, Davis S, Deakin B, DeRubeis RJ, Dubois B, Geyer MA, Goodwin GM, Gorwood P, Jay TM, Joels M, Mansuy IM, Meyer-Lindenberg A, Murphy D, Rolls E, Saletu B, Spedding M, Sweeney J, Whittington M, Young LJ (2012) Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov 11:141–168. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA (2012) Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry 17:1206–1227. [DOI] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21:1790–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JL, Gonzalez-Maeso J (2013) Preclinical models of antipsychotic drug action. Int J Neuropsychopharmacol 16:2131–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JL, Holloway T, Umali A, Rayannavar V, Sealfon SC, Gonzalez-Maeso J (2013) Persistent effects of chronic clozapine on the cellular and behavioral responses to LSD in mice. Psychopharmacology (Berl) 225:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MJ, Mahgoub M, Na ES, Pranav H, Monteggia LM (2013) Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J Neurosci 33:6401–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalivaeva NN, Belyaev ND, Turner AJ (2009) Sodium valproate: an old drug with new roles. Trends Pharmacol Sci 30:509–514. [DOI] [PubMed] [Google Scholar]
- Nielsen RE, Levander S, Kjaersdam Telleus G, Jensen SO, Ostergaard Christensen T, Leucht S (2015) Second-generation antipsychotic effect on cognition in patients with schizophrenia-a meta-analysis of randomized clinical trials. Acta Psychiatr Scand DOI 10.1111/acps.12374 [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O’Dushlaine C, Chambert K, Bergen SE, Kahler A, Duncan L, Stahl E, Genovese G, Fernandez E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PK, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SG, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll SA, Sklar P (2014) A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovi ch E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378. [DOI] [PubMed] [Google Scholar]
- Sahli ZT, Tarazi FI (2017) Pimavanserin: novel pharmacotherapy for Parkinson’s disease psychosis. Expert Opin Drug Discov DOI 10.1080/17460441.2018.13948381-8. [DOI] [PubMed] [Google Scholar]
- Sawa A, Snyder SH (2002) Schizophrenia: diverse approaches to a complex disease. Science 296:692–695. [DOI] [PubMed] [Google Scholar]
- Schmidt-Supprian M, Rajewsky K (2007) Vagaries of conditional gene targeting. Nat Immunol 8:665–668. [DOI] [PubMed] [Google Scholar]
- Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, Tooley K, Presumey J, Baum M, Van Doren V, Genovese G, Rose SA, Handsaker RE, Schizophrenia Working Group of the Psychiatric Genomics C, Daly MJ, Carroll MC, Stevens B, McCarroll SA (2016) Schizophrenia risk from complex variation of complement component 4. Nature 530:177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Campbell G, Jones WD, Campagne F, Wen Z, Walker SJ, Su Z, Chu TM, Goodsaid FM, Pusztai L, Shaughnessy JD Jr, Oberthuer A, Thomas RS, Paules RS, Fielden M, Barlogie B, Chen W, Du P, Fischer M, Furlanello C, Gallas BD, Ge X, Megherbi DB, Symmans WF, Wang MD, Zhang J, Bitter H, Brors B, Bushel PR, Bylesjo M, Chen M, Cheng J, Chou J, Davison TS, Delorenzi M, Deng Y, Devanarayan V, Dix DJ, Dopazo J, Dorff KC, Elloumi F, Fan J, Fan S, Fan X, Fang H, Gonzaludo N, Hess KR, Hong H, Huan J, Irizarry RA, Judson R, Juraeva D, Lababidi S, Lambert CG, Li L, Li Y, Li Z, Lin SM, Liu G, Lobenhofer EK, Luo J, Luo W, McCall MN, Nikolsky Y, Pennello GA, Perkins RG, Philip R, Popovici V, Price ND, Qian F, Scherer A, Shi T, Shi W, Sung J, Thierry-Mieg D, Thierry-Mieg J, Thodima V, Trygg J, Vishnuvajjala L, Wang SJ, Wu J, Wu Y, Xie Q, Yousef WA, Zhang L, Zhang X, Zhong S, Zhou Y, Zhu S, Arasappan D, Bao W, Lucas AB, Berthold F, Brennan RJ, Buness A, Catalano JG, Chang C, Chen R, Cheng Y, Cui J, Czika W, Demichelis F, Deng X, Dosymbekov D, Eils R, Feng Y, Fostel J, Fulmer-Smentek S, Fuscoe JC, Gatto L, Ge W, Goldstein DR, Guo L, Halbert DN, Han J, Harris SC, Hatzis C, Herman D, Huang J, Jensen RV, Jiang R, Johnson CD, Jurman G, Kahlert Y, Khuder SA, Kohl M, Li J, Li M, Li Qz , Li S, Liu J, Liu Y, Liu Z, Meng L, Madera M, Martinez-Murillo F, Medina I, Meehan J, Miclaus K, Moffitt RA, Montaner D, Mukherjee P, Mulligan GJ, Neville P, Nikolskaya T, Ning B, Page GP, Parker J, Parry RM, Peng X, Peterson RL, Phan JH, Quanz B, Ren Y, Riccadonna S, Roter AH, Samuelson FW, Schumacher MM, Shambaugh JD, Shi Q, Shippy R, Si S, Smalter A, Sotiriou C, Soukup M, Staedtler F, Steiner G, Stokes TH, Sun Q, Tan PY, Tang R, Tezak Z, Thorn B, Tsyganova M, Turpaz Y, Vega SC, Visintainer R, von Frese J, Wang C, Wang E, Wang J, Wang W, Westermann F, Willey JC, Woods M, Wu S, Xiao N, Xu J, Xu L, Yang L, Zeng X, Zhang M, Zhao C, Puri RK, Scherf U, Tong W, Wolfinger RD (2010) The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat Biotechnol 28:827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, de Longueville F, Kawasaki ES, Lee KY, Luo Y, Sun YA, Willey JC, Setterquist RA, Fischer GM, Tong W, Dragan YP, Dix DJ, Frueh FW, Goodsaid FM, Herman D, Jensen RV, Johnson CD, Lobenhofer EK, Puri RK, Schrf U, Thierry-Mieg J, Wang C, Wilson M, Wolber PK, Zhang L, Amur S, Bao W, Barbacioru CC, Lucas AB, Bertholet V, Boysen C, Bromley B, Brown D, Brunner A, Canales R, Cao XM, Cebula TA, Chen JJ, Cheng J, Chu TM, Chudin E, Corson J, Corton JC, Croner LJ, Davies C, Davison TS, Delenstarr G, Deng X, Dorris D, Eklund AC, Fan XH, Fang H, Fulmer-Smentek S, Fuscoe JC, Gallagher K, Ge W, Guo L, Guo X, Hager J, Haje PK, Han J, Han T, Harbottle HC, Harris SC, Hatchwell E, Hauser CA, Hester S, Hong H, Hurban P, Jackson SA, Ji H, Knight CR, Kuo WP, LeClerc JE, Levy S, Li QZ, Liu C, Liu Y, Lombardi MJ, Ma Y, Magnuson SR, Maqsodi B, McDaniel T, Mei N, Myklebost O, Ning B, Novoradovskaya N, Orr MS, Osborn TW, Papallo A, Patterson TA, Perkins RG, Peters EH, Peterson R, Philips KL, Pine PS, Pusztai L, Qian F, Ren H, Rosen M, Rosenzweig BA, Samaha RR, Schena M, Schroth GP, Shchegrova S, Smith DD, Staedtler Su Z, Sun H, Szallasi Z, Tezak Z, Thierry-Mieg D, Thompson KL, Tikhonova I, Turpaz Y, Vallanat B, Van C, Walker SJ, Wang SJ, Wang Y, Wolfinger R, Wong A, Wu J, Xiao C, Xie Q, Xu J, Yang W, Zhong S, Zong Y, Slikker W Jr. (2006) The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Murphy MR (2008) Schizophrenia therapy: beyond atypical antipsychotics. Nat Rev Drug Discov 7:471–472. [DOI] [PubMed] [Google Scholar]
- Song AJ, Palmiter RD (2018) Detecting and Avoiding Problems When Using the Cre-lox System. Trends Genet 34:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Uchida H, Takeuchi H, Nakajima S, Nomura K, Tanabe A, Yagi G, Watanabe K, Kashima H (2009) Augmentation of atypical antipsychotics with valproic acid. An open-label study for most difficult patients with schizophrenia. Hum Psychopharmacol 24:628–638. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Os J, Kapur S (2009) Schizophrenia. Lancet 374:635–645. [DOI] [PubMed] [Google Scholar]
- Weisstaub NV, Zhou M, Lira A, Lambe E, Gonzalez-Maeso J, Hornung JP, Sibille E, Underwood M, Itohara S, Dauer WT, Ansorge MS, Morelli E, Mann JJ, Toth M, Aghajanian Sealfon SC, Hen R, Gingrich JA (2006) Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science 313:536–540. [DOI] [PubMed] [Google Scholar]
- Wright P, Birkett M, David SR, Meehan K, Ferchland I, Alaka KJ, Saunders JC, Krueger J, Bradley P, San L, Bernardo M, Reinstein M, Breier A (2001) Double-blind, placebo-controlled comparison of intramuscular olanzapine and intramuscular haloperidol in the treatment of acute agitation in schizophrenia. Am J Psychiatry 158:1149–1151. [DOI] [PubMed] [Google Scholar]