

Abstract
Arachidonic acid (AA) metabolic network generates a variety of products that mediate or modulate inflammatory reactions. (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate (HOEC), isolated from Incarvillea mairei var. granditlora (Wehrhahn) Grierson, was found as an inhibitor of 5-LOX and 15-LOX in vitro. When evaluated in collagen-induced arthritis (CIA) rats, however, lowdose of HOEC (1 mg/kg) showed better efficacy than that of high dose (10 mg/kg). To study how HOEC interfered the AA metabolic pathway, in this study, we dynamically observed the changes of plasma AA metabolites (LTB4, LTC4, 15-HETE, PGE2, TXB2 and PGD2) in the CIA rats treated with different doses of HOEC by using enzyme-linked immunosorbent assay (ELISA). The results showed that eicosanoids were elevated synchronously at three time points in different treated rats. The incidence of arthritis had a higher correlation with LOX pathway while the COX pathway might be more important in the severity of arthritis. HOEC in all doses could inhibit LOX pathway in the beginning of arthritis while highdose of HOEC could induce the increase of COX metabolites in the later stage of disease. These dynamic changes of eicosanoids, depending on the regulation of metabolic flux, can be interfered by HOEC and thus affect the output of efficacy.
Keywords: Arachidonic acid metabolic network, eicosanoids, quantified dynamic, metabolic flux, rheumatoid arthritis
Introduction
Arachidonic acid (AA) pathway generates bioactive lipid mediators which regulate a diverse range of inflammatory processes [1]. In recent years, researchers turn to target-based drug design as an efficient way to treat inflammation. However, the withdrawal of VIOXX (rofecoxib) has sparked a wave of reexamination of the previous thought. Since the metabolic network of arachidonic acid is a balancing system, over-inhibiting a single metabolic pathway will disturb the balance of the network, which is not advisable. Multi-targeted drug design for anti-inflammation, which is a novel alternative of drug design, making people to explore the quantitative relationship between metabolites of arachidonic acid. Lai et al. proposed the concept of AA metabolic network in 2007. They established a model of arachidonic acid metabolism network of polymorphonuclear leukocyte (PMN), which can be used to predict the effect of multi-target drug [2]. Similar work by Shankar Subramaniam et al. in RAW264.7 and bone marrow derived macrophages (BMDM) provided a model for computer-assisted drug design [3,4]. The further studies of Lai’s lab in 2015 improved the AA metabolic network and provided a new strategy for designing safe and effective anti-inflammatory drugs [5]. However, all the above mentioned works were based on cell-free and cell based systems. The animal based AA metabolism network model is still unestablished due to the complexity of in vivo system.
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by persistent synovial inflammation and cartilage destruction induced articular malformation [6]. A metabolic pathway can produce many lipid mediators (eicosanoids) of inflammation which play important roles in the RA pathogenesis. Lipoxygenase (LOX) pathway and cyclooxygenase (COX) pathway are two major pathways of AA metabolism. Expression levels of both COX-1 and COX-2 levels are markedly increased in RA patients and the COX metabolites increase not only in the inflammatory site but also in the overall system. The levels of PGE2, PGF2α, 6-keto-PGF1α, PGD2 and TXB2 in the synovial fluid of NSAIDs-treated RA patients are lower than those of non-treated [7,8]. The levels of 11-dehydro-TXB2 and 2, 3-dinor-6-keto-PGF1α in RA patients’ urine are higher than that in healthy volunteers [9]. As for LOX pathway, LTB4 is also a key inflammatory factor in RA. Both increase of LTB4 in serum, synovial fluid and synovium of RA patients [10,11] and the enhancement of neutrophil synthesized LTB4 [12] have been reported. However, neither COX nor 5-LOX inhibitor is satisfactory in clinical application. The inhibition of COX induces gastrointestinal adverse reaction and the selective inhibitor of COX-2 causes fatal cardiovascular risk, not to mention that 5-LOX inhibitor and 5-LOX antagonist have little therapeutic effect on RA patients [13,14]. New modes for drug design are urgently needed for RA medications.
Although the pathogenesis of RA is still unclearly demonstrated, it is certain that RA is an autoimmunity disease [15]. Collagen-induced arthritis (CIA) is a common animal model of RA. Collagen II is expressed only in the arthrodial cartilage; thus, heterogeneous collagen II induced rejection reaction will result in damage to joint [16]. Owing to the very similar pathological characteristics to RA including synovitis, pannus, cartilage erosion and bone damage, CIA is generally considered as the most reliable animal model of RA. COX-2 plays a very important role in CIA which is over-expressed in the synovial tissue. The inhibition of COX-2 can reverse pathological changes effectively [17]. In addition, the severity of CIA in COX-2-/- mice is milder than in COX-1-/- and wild type mice, which suggests that COX-2 plays a critical role in the pathogenesis of CIA [18]. mPGES-1, the terminal biosynthetic enzyme of PGE2, is also significantly increased in the arthritis paws [19]. However, PGD2 as another metabolite of COX-2 is probably an anti-inflammatory factor. The injection of exogenous PGD2 can alleviate CIA, PGD2 receptors, i.e. DP1 and DP2, rise and antagonist of DP1 treatment may aggravate the condition [20]. On the other hand, though no good performance of 5-LOX inhibitor is observed in RA, this kind of medicine has some therapeutic effect on CIA [21,22]. 15-HETE is the main metabolite of 15-LOX, however little has been known about its role in RA. 15-HETE is one of the ligands of PPARγ [6], but it can also induce the production of IL-4, IL-1β [23], indicating that 15-HETE may have both pro-inflammatory and anti-inflammatory function.
(+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate (HOEC) is isolated from Incarvillea mairei var. granditlora (Wehrhahn) Grierson. The plants of Incarvillea genus have long been used as folk medicines for the treatment of inflammation-related diseases in China. It has been reported to inhibit 5-LOX (IC50 = 34.6 μM) and 15-LOX (IC50 = 7.8 μM) on the enzyme level and to inhibit 5-LOX in RAW264.7 and human whole blood [24]. In the previous studies, we found that HOEC was effective on CIA rats. However, the efficacy of HOEC in lower dose (1 mg/kg) was better than that in higher dose (10 mg/kg). To explain why higher dose is worse than the lower dose, in this study, we focused on HOEC’s effect on dynamic changes of AA metabolites and how HOEC interfering AA metabolism network, with the aim of finding the cause behind the unusual phenomenon.
Materials and methods
Rats
Male Wistar rats, weighing 130±10 g, were purchased from Slac Laboratory Animal (Shanghai, China). The rats were housed in a temperature (22±1°C) and humidity (55%) - controlled room with a 12-hour light/dark cycle. Water and food were provided ad libitum.
Collagen-induced arthritis
A 1:1 emulsion composed of bovine type II collagen (2 mg/ml, dissolved in 0.05 M acetic acid) (CII, Chondrex, USA) and incomplete Freund’s adjuvant (IFA, Chondrex, USA) was prepared using a high-speed homogenizer (IKA Inc., Germany) on ice. To establish a CIA model, rats were immunized by intradermally injecting the emulsified mixture containing CII (200 mL/rat) at their back base of tails. On day 7, rats received booster injection of 100 mL emulsified CII mixture. In the normal control group, equal volumes of sterilized saline were injected in rats simultaneously when the experimental group was immunized.
Clinical scores were used to evaluate the incidence and severity of arthritis [25]. The scoring criteria, which was based on the swelling of each back paw, was as follows: 0 for no swelling and redness; 1 for swelling of the toe joints; 2 for swelling of the toe joints and footpad; 3 for swelling of the paw below ankle; and 4 for swelling of the entire paw including ankle. The total score of each rat was calculated by adding the scores of two back paws. Scores were recorded every other day and the body weights were recorded every three days both until the end of the experiment.
Treatments of HOEC and drawing blood
Rats (n = 48) were randomly divided into five groups before immunization: a normal control group (n = 8), a CIA model group (n = 10) and three HOEC treated groups (1 mg/kg, n = 10, 3 mg/kg, n = 10, and 10 mg/kg, n = 10). 1 ml of saline or HOEC (1 mg/kg, 3 mg/kg and 10 mg/kg) was intraperitoneally injected into normal control rats, CIA model rats and HOEC treated rats, respectively, once a day from the first day to the end of the experiment.
Blood samples were collected on days 0, 4, 7, 9, 12, 15, 18, 21, 25, 29, 34 (5 control rats and 8 rats of every other groups). The plasma was prepared by 15-min centrifugation at 1000 × g, 4°C and then stored at -80°C for further detection.
On day 36, rats were sacrificed and two back paws of each rat were collected for histological study and immunohistochemistry assay.
Histopathology
After 24 hour’s fixation in 4% paraformaldehyde, the hind paws were decalcified in 10% EDTA for 4 weeks and then embedded in paraffin. Tissue sections (5 μm thick) were stained with haematoxylin-eosin (Beyotime Biotechnology, China) for light microscope (ECLIPSE TS-100, Nikon, Japan) examination.
Ice-cold ethanol was added into 250 μL plasma on a three-times-the-volume basis, and then thoroughly mixed and incubated at 4°C for five minutes. After a 15 min centrifugation (3000 × g, 4°C), the supernatant was transferred into a clean polypropylene tube and the ethanol was evaporated completely under a gentle stream of nitrogen at room temperature. The dry sample was dissolved in 250 μL EIA buffer.
The concentrations of plasmas LTB4, LTC4, 15-HETE, PGE2, TXB2, and PGD2 were assayed using ELISA kit (Cayman Chemical Company, Ann Arbor, MI, USA). Take the LTB4 ELISA Kit for example, the assay is based on the competition between LTB4 and an LTB4-acetyl-cholinesterase (AChE) conjugate (LTB4 Tracer) for a limited amount of LTB4 Antiserum. The concentration of LTB4 tracer is constant and LTB4 in the sample solution competitively binds to the LTB4 Antiserum. Ellman’s Reagent can react with the AChE on the LTB4 Tracer and turn to a yellow color product which can be counted as concentration by reading the absorption intensity of the well. During the experiment, the assay was performed according to instrument instructions. The plate contained two blanks (Blk), two non-specific binding wells (NSB), two maximum binding wells (B0), and an eight points standard curve running in duplicate. As the first step, 100 μL EIA Buffer was added to NSB wells, 50 μL EIA Buffer was added to B0 wells, 50 μL standard 1-8 was added to standard wells, and 50 μL sample per well was added, with each sample having a duplicate well. Then 50 μL LTB4 Tracer was added to each well except the Blk wells and 50 μL LTB4 Antiserum was added to each well except the NSB and the Blk wells. The plate was later incubated overnight at 4°C. Wells were then emptied and rinsed five times with Wash Buffer, and 200 μL Ellmans’s Reagent was added to each well. In the end, the plate was incubated in the dark at 25°C for 90 min and read at a wavelength of 410 nm using a multimode reader (Synergy 2, BioTek, USA). The results of the concentrations were calculated according to the procedure provided by the manufacturer.
Statistical analysis
Data were expressed as mean ± SEM. Statistical significance was analyzed using Student’s t for two-group comparisons, analysis of variance (ANOVA) for multiple group comparisons followed by least significant difference (LSD) post hoc test, and Pearson correlation for correlation between two groups. P < 0.05 was considered significant. Non-parametric test was used when homogeneity of variances had significant difference. Statistical analyses were performed with IBM SPSS Statistics 16.0.
Results
Pharmacodynamics evaluation of HOEC on collagen-induced arthritis
To compare the drug effect of different dosages of HOEC, we set five groups of rats, including normal control, CIA model, and three HOEC doses, that is, 1 mg/kg, 3 mg/kg and 10 mg/kg, respectively. HOEC and equal volumes of solvent of HOEC were administered i.p. to rats in the corresponding group once per day from day 1 to day 36. As shown in Figure 1B, rats in model group were initiated in the early stage of disease (arthritis score of 1.8±1.5 on day 18), and the disease became worse at the maximal arthritis score of 6.6±0.8 by day 26 and then decreased gradually (having a final arthritis score of 5.4±1.0 on day 36). HOEC in different doses could alleviate the clinical symptoms of arthritis. However, the 1 mg/kg and 3 mg/kg HOEC showed better inhibitory effects on arthritis than 10 mg/kg. No obvious difference (maximal arthritis score: 4.7±1.0, 4.5±0.8 and final arthritis score: 3.2±0.9, 3.1±0.6) was observed between 1 mg/kg and 3 mg/kg HOEC treatments. The group of 10 mg/kg had the poorest inhibitory effect compared with other two doses (maximal arthritis score: 5.2±1.0 and final arthritis score: 4.1±0.9).
Figure 1.

Effect of HOEC on collagen induced arthritis in rats. A. Experimental processes of collagen induced arthritis. Rats were primary immunized on day 0 and boost immunized on day 7. HOEC was administered i.p. once per day from day 1 to day 36 and blood samples were collected on days 0, 4, 7, 9, 12, 15, 18, 21, 25, 29, 34. On day 36, rats were sacrificed and their hind paws were cut for histological study and immunohistochemistry assay. B. Arthritis scores of rats in different treatment groups. Rats were divided into five groups: normal control (n = 5), model (n = 5), HOEC 1 mg/kg (n = 8), HOEC 3 mg/kg (n = 6) and HOEC 10 mg/kg (n = 9). Arthritis scores were evaluated every two days. C. Representative image of hind paws from each treatment groups on day 36. Data shown are mean ± SEM. #P < 0.05, ##P < 0.01 for model versus normal control, *P < 0.05 for HOEC 3 mg/kg versus model. (LSD post hoc test).
Histological analysis of different doses of HOEC
On day 36 of the experiment, rats were sacrificed by decapitation. Hind paws from rats of each group with average scores (n = 2) were obtained as representative samples for histological analysis. Typical arthritis features, such as synovial hypeplasia, multiple cartilage destruction and pannus, were observed in model rats (Figure 2B). Meanwhile, rats in HOEC treatment groups showed mild cartilage loss and pannus formation, and rats in HOEC 10 mg/kg group exhibited synovial hyperplasia, which matched the clinical scores (Figure 2C-E).
Figure 2.
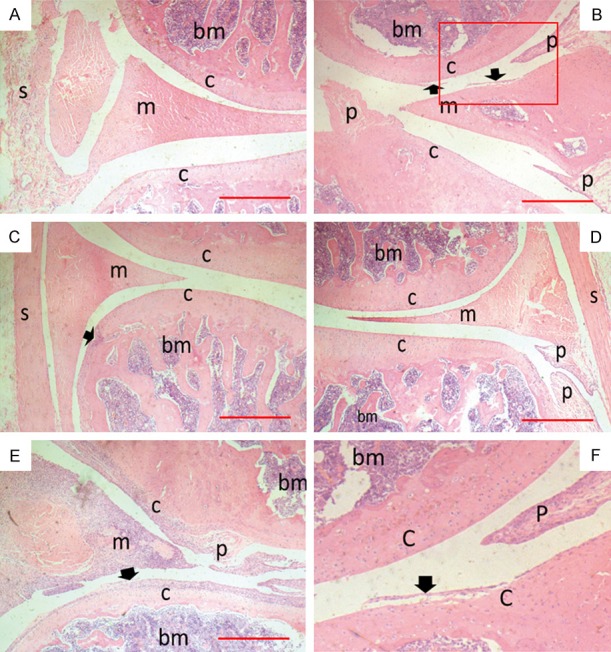
Ankle joint histology from hind paws of CIA rats. Rats were sacrificed on day 36 and two back paws of each rat were collected to be made into tissue paraffin section which were then stained with H&E for microscopic photography. Scale bar = 500 μm. (A-E) Representative tissue sections of control, model, HOEC 1 mg/kg, HOEC 3 mg/kg and HOEC 10 mg/kg treatment groups; (F) Details of red box in (B), where s is synovial; cis cartilage, m is meniscus, bm is bone marrow, p is pannus, and arrow is cartilage destruction. HOEC can reduce damage of joint tissue caused by arthritis, but the effect was weaker in high dose than low and middle doses.
Profile of plasma arachidonic acid metabolites in collagen-induced model rats
To explain the relationship between the distinct effects of HOEC in different doses and the AA metabolism network, we monitored dynamic changes of six major eicosanoids involved in RA in plasma (Figure 3Ea). In symptomatic rats of the model group, all measured metabolites, i.e. LTB4, LTC4 and 15-HETE from LOX pathway and PGE2, TXB2 and PGD2 from COX pathway, increased after the first immunization and reached to the first peak on day 7. The second peak of all measured metabolites appeared on day 15, 8 days after boost immunization. Furthermore, PGE2 and TXB2 had the third peak on day 25. No obvious increases of AA metabolites were observed in rats in the normal control group.
Figure 3.
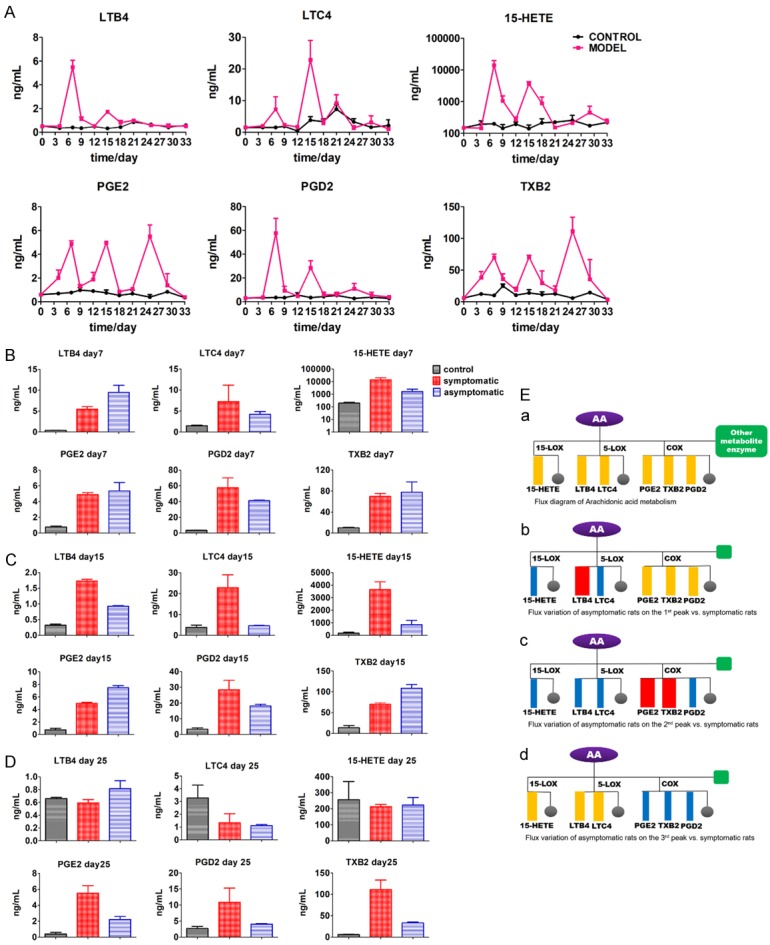
Dynamic changes of arachidonic acid metabolites in normal control and model groups. A. Concentration-time curve of AA metabolites in control and model (symptomatic rats in model group) group rats. Plasmas were collected from rats at different time points (control n = 3, model n = 3) and the samples were assessed by ELISA. B. Comparison of the first peak values of arachidonic acid metabolites between the symptomatic and asymptomatic rats in model group. (control n = 3, symptomatic n = 3, asymptomatic n = 3). C. Comparison of the second peak values of arachidonic acid metabolites between the symptomatic and asymptomatic rats in model group. (control n = 3, symptomatic n = 3, asymptomatic n = 3). D. Comparison of the concentrations of AA metabolites between the symptomatic and asymptomatic rats in model group on day 25. (control n = 3, symptomatic n = 3, asymptomatic n = 3). E. (a) Metabolic pathway flux distribution diagram. Six metabolites we monitored are enumerated. (b) Metabolic flux comparison diagram of symptomatic and asymptomatic rats in model group at the first peak. (c) Metabolic flux comparison diagram of symptomatic and asymptomatic rats in model group at the second peak. (d) Metabolic flux comparison diagram of symptomatic and asymptomatic rats in model group at the third peak. Grey circle represents the other metabolites and green square represents the other metabolic enzymes. Blue rectangle represents the decrease of flux, red rectangle represents the increase of flux, and orange rectangle represents no change on flux, all asymptomatic rats versus symptomatic rats. Data shown are mean ± SEM. #P < 0.05, ##P < 0.01, ###P < 0.001 for model (symptomatic rats) versus control; *P < 0.05, **P < 0.01, ***P < 0.001 for symptomatic rats versus asymptomatic rats. (LSD post hoc test, non-parametric test for TXB2 at the 3rd peak).
Correlation between metabolites’ peaks and the incidence of arthritis
Since some rats in the model group were asymptomatic, we compared the peak values of both symptomatic and asymptomatic rats in the model group, trying to explain the pathological meaning of these peaks. Though the first peaks of all AA metabolites increased markedly on day 7, when compared with asymptomatic rats, the level of COX metabolites, as shown in Figure 3B, had no significant difference in symptomatic rats, suggesting that the rise of PGE2, TXB2 and PGD2 was a general response to stimulation of the first immunization (p value = 0.004, 0.012, LSD post hoc test). However, the level LOX metabolites was different between the symptomatic rats and the asymptomatic rats on day 7. LTB4 had a significant rise in symptomatic rats of the model group compared with the control group (p value = 0.013, LSD post hoc test); moreover, LTB4’s level in asymptomatic rats was higher than that in symptomatic rats (p value = 0.035, LSD post hoc test), and LTC4 and 15-HETE were markedly lower in asymptomatic rats than in symptomatic rats (Figure 3B). The results indicate that the rise of LTB4 on day 7 was not characteristic in symptomatic rats and LTC4’s and 15-HETE’s first peaks might have little association with the incidence of arthritis.
As for the second peak on day 15, the maximal values of LTB4, LTC4 and 15-HETE in symptomatic rats were significantly higher than those in symptomatic rats of the model group (P value = 0, 0.007, 0.005, LSD post hoc test). But the maximal values of PGE2 and TXB2 in symptomatic rats were lower than that in asymptomatic rats (P value = 0, 0.004, LSD post hoc test). Interestingly, the change trend of PGD2 was similar as LTB4, LTC4 and 15-HETE varied (Figure 3C). According to the results, we can speculate that the increase of LOX pathway on day 15 (8 days after the boost immunization) was involved in the incidence of arthritis. As for PGE2 and TXB2 in COX pathway, the rise of metabolites was not distinctive in symptomatic rats; in other words, it suggests that PGE2 and TXB2 had no contribution to the incidence of arthritis. The higher level of PGE2 and TXB2 in asymptomatic rats was probably due to less metabolic flux compared with symptomatic rats of the model group (Figure 3Ec).
On day 25, as shown in Figure 3D, PGE2 and TXB2 in COX pathway emerged to contribute to an obvious third peak (P value = 0.001, LSD post hoc test; P value = 0.009, non-parametric test) and showed a significant difference between symptomatic and asymptomatic rats in the model group as well. Compared with symptomatic rats, the maximal values of PGE2 and TXB2 in asymptomatic rats were much lower (P value = 0.009, LSD post hoc test; P value = 0.05, non-parametric test). PGD2 as another metabolite of COX pathway also had a decline trend in asymptomatic rats compared with symptomatic rats in the model group, indicating that PGE2 and TXB2 were the main inflammatory mediators in later stage of arthritis. However, metabolites’ level of LOX pathway in the model group had no difference compared with the control group, suggesting that the LOX pathway was not active in the later stage of arthritis.
HOEC interfered changes of arachidonic acid metabolic flux in CIA rats
Compared with the model group, the profile of arachidonic acid metabolites in HOEC treatment groups was evidently altered. HOEC markedly decreased the output of products of LOX metabolic pathway, meanwhile the output of products of COX metabolites was significantly increased when different doses of HOEC were used, especially high dose HOEC (10 mg/kg).
To be specific, in LOX pathway, LTB4 was not obviously inhibited by any dose of HOEC. The dose-dependent inhibition was observed at the second peak of LTC4 and 15-HETE. Moreover, the low (1 mg/kg) and high (10 mg/kg) doses of HOEC could decrease the first peak of 15-HETE, and the middle (3 mg/kg) and high doses of HOEC could decrease the second peak of 15-HETE. Interestingly, the middle and high doses of HOEC emerged a third peak on day 25 of 15-HETE while the low dose of HOEC rose a little on day 29 (Figure 4A).
Figure 4.
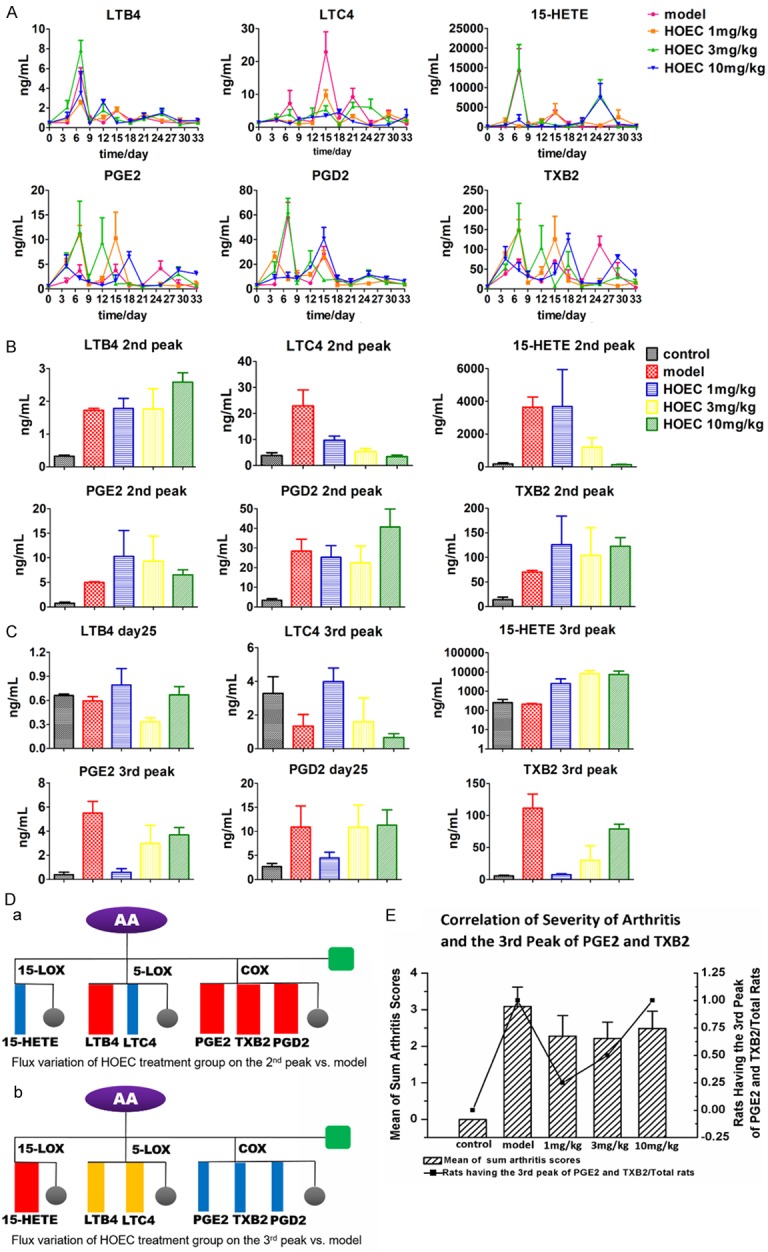
Dynamic changes of arachidonic acid metabolites in model and HOEC treatment group. A. The Concentration-time curve of AA metabolites in model and HOEC treatment group rats (symptomatic). Plasmas were collected from rats at different time points (model n = 3, HOEC 1 mg/kg n = 4, HOEC 3 mg/kg n = 4, HOEC 10 mg/g n = 3) and the samples were assessed by ELISA. B. Comparison of the second peak values of AA metabolites in rats of model group (symptomatic rats) and HOEC treatment group. (model n = 3, HOEC 1 mg/kg n = 4, HOEC 3 mg/kg n = 4, HOEC 10 mg/g n = 3). C. Comparison of the third peak values of AA metabolites in rats of model group (symptomatic rats) and HOEC treatment group. (model n = 3, HOEC 1 mg/kg n = 4, HOEC 3 mg/kg n = 4, HOEC 10 mg/g n = 3). D. (a) Metabolic flux changes diagram of HOEC treatment group at the second peak versus model group. (b) Metabolic flux changes diagram of HOEC treatment group at the third peak versus model group. Grey circle represents the other metabolites and green square represents the other metabolic enzymes. Blue rectangle represents the decrease of flux, red rectangle represents the increase of flux, and orange rectangle represents no change on flux, all HOEC treatment group versus model group. E. Correlation of severity of arthritis and the third peak of PGE2 and TXB2. Data shown are mean ± SEM. #P < 0.05, ##P < 0.01, ###P < 0.001 for model versus control; *P < 0.05, **P < 0.01, ***P < 0.001 for HOEC treatment group versus model. (non-parametric test).
In COX metabolic pathway, the changes of PGE2 and TXB2 were kept in step with each other (Figures 4A, 5). The first and second peaks of PGE2 and TXB2 were enhanced in HOEC treatment groups but the time point on which the peak appeared was delayed or advanced. Compared with the model group and other two doses of HOEC treatment groups, the first peaks of PGE2 and TXB2 shifted forward in 10 mg/kg HOEC treated rats while the second peaks delayed. Most noticeably, the third peaks (on day 25) of PGE2 and TXB2 were completely inhibited by low-dose HOEC while after treated with the middle and high doses of HOEC, the third peaks of PGE2 and TXB2 were delayed even though they were also inhibited. Quantitative results have indicated that the levels of PGE2 and TXB2 were negatively related to the dose of HOEC (Figure 4A, 4C).
Figure 5.

Correlation of eicosanoids in the CIA progress. The scatter diagram of two metabolites at every time point (mean) can reflect the correlation between each other intuitively. LTB4 and 15-HETE, PGE2 and TXB2, LTB4 and PGD2 all have strong correlation with each other. (n = 55).
We found, from the above mentioned results, that the second and third peaks of AA metabolites made more contribution to the arthritis than the first peaks. So we compared the second and the third peaks’ values of every treatment group, trying to find the effect of HOEC in different doses. As a LOX inhibitor, HOEC could dose-dependently inhibit the second peaks of LTC4 (P value = 0.003, 0, 0, non-parametric test) and 15-HETE (P value = 0.05, 0.05, non-parametric test) while LTB4 was little affected, and high dose of HOEC increased the level of LTB4 (P value = 0.05, non-parametric test) (Figure 4B), implying that HOEC dominantly inhibits LTC4 and 15-HETE rather than LTB4 in vivo. Meanwhile, compared with model rats, PGE2 and TXB2 had a rising tendency in HOEC treatment groups, which is similar to the profile of metabolic flux in asymptomatic vs. symptomatic rats of the model group (Figure 4B, 4Da).
As shown in Figure 4C, HOEC could significantly decrease the levels of PGE2 and TXB2 (P value = 0.034, 0.029, non-parametric test), but interestingly with a reversible dose-dependence and the same trend as PGD2 without significance. With respect to LOX pathway, LTB4 and LTC4 in the model and HOEC treatment groups had no much difference with those in the normal control group. However, 15-HETE’s level had an elevation in the HOEC treatment group on day 25 (in both middle and high doses) and day 29 (in low dose), respectively. Though 15-HETE was inhibited at the second peak, the increase of 15-HETE was probably due to the metabolic flux decline in COX pathway (Figure 4Db). The third peaks of PGE2 and TXB2 had a higher correlation with the severity of arthritis because the ratio of rats receiving different treatments and exhibiting the third peaks of PGE2 and TXB2 had a similar trend with the severity of arthritis (Figure 4E).
Correlation of metabolites of arachidonic acid network in CIA
Since the metabolites in AA network are all derived from AA, the quantitative changes of them have some correlation. As shown in Figure 5, in LOX pathway, LTB4 and 15-HETE had a strong correlation (P value = 0, r = 0.828), and in COX pathway, PGE2 and TXB2 had a strong correlation (P value = 0, r = 0.95). Interestingly, PGD2, LTB4, and 15-HETE had a stronger correlation (P value = 0, 0, r = 0.796, 0.779) than PGE2 and TXB2 (P value = 0, 0, r = 0.610, 0.584) while LTC4 had a weak correlation with other eicosanoids we measured.
Discussion
Inflammation is a complex progress where organisms adaptively respond to the trigger of noxious stimuli and conditions. Eicosanoids, as arachidonic acid metabolites, are one kind of lipid mediators of inflammation [26]. Few studies have reported the eicosanoids dynamics during the development of arthritis. In this study, six eicosanoids including LTB4, LTC4, 15-HETE, PGE2, TXB2, and PGD2, which are the major metabolites of arachidonic acid catalyzed by 5-LOX, 15-LOX and COX-2, were measured dynamically during the arthritis process. From the in vivo data, we found that some rats were asymptomatic in different experiments. Thus, we first explored the difference of eicosanoids profile between asymptomatic and symptomatic rats. The peaks of eicosanoids were observed 7 days after the primary immunization (on day 7) and 8 days after the booster immunization (on day 15) (Figure 3A). Since these two peaks emerged subsequently following two immunization processes, we speculate that all these alterations of eicosanoids are related to the stimuli of immunization. In other words, the first peak was induced by the primary collagen injection and the second peaks were induced by the booster. A similar profile of other proinflammatory factors during the collagen-induced arthritis was reported by Griswold et al. They found that serum amyloid P component (SAP), the major acute-phase protein in CIA, reached to the peaks on day 7 and day 35 after the initial collagen injection (7 days after booster immunization) [27]. Nuria Maicas et al. also reported a similar profile of PGD2 which played an anti-inflammatory effect in murine collagen-induced arthritis [28]. In this study, PGD2 increased first and then decreased to the homeostasis both in paw homogenates and serum. Taken together, similar profiles of pro-inflammatory and anti-inflammatory factors might be universal in the pathogenesis of inflammation-related diseases.
Since there was no significant difference in the values of first peak between symptomatic and asymptomatic rats, the first peak seems to be a general reaction to the stimuli of collagen (Figure 3B). However, the profiles of second peaks of eicosanoids were significantly different, in which LTB4, LTC4 and 15-HETE, the metabolites of 5-LOX and 15-LOX pathways, were much higher in symptomatic rats than that in asymptomatic rats, and in COX-2 pathway, PGE2 and TXB2 had an opposite trend compared with metabolites in LOX pathway (Figure 3C, 3Ec) which was probably due to a flux distribution of arachidonic acid metabolism. We speculate that the second peaks of eicosanoids might be more important in the pathogenesis of RA. It also suggests that the rise of LTB4, LTC4 and 15-HETE was closely related to the incidence of arthritis. It was reported that LTB4 receptors, i.e. BLT1 and BLT2, played a critical role in the incidence of mice arthritis model. The loss of BLT1 alone could completely prevent the onset of the collagen induced arthritis [29]. In another study, the BLT2-/- mice showed reduced incidence and severity of K/BxN arthritis [30]. These studies support the pivotal role of 5-LOX-LTB4-BLT1/BLT2 axis in the onset of RA, which coincides with our study.
In the late stage of arthritis (from day 24 to day 33), the levels of LTB4, LTC4 and 15-HETE decreased gradually to the normal. Meanwhile, metabolites of COX pathway got a third peak on day 25 (Figure 3A). Chan & Moore reported that COX-2 mRNA level had three peaks during the process of CIA, which was at the induction, inflammation, and resolution phases of CIA, respectively [31]. This finding is consistent with our data and provides evidence to support three peaks of PGE2, TXB2 and PGD2 in our study. Since TXA2 exerts its effects within a short course of biosynthesis where it is rapidly and non-enzymatically hydrolyzed to form TXB2 which has no bioactivity and usually used to reflect the quantity of TXA2. TXA2 has been reported playing an important role in pathogenesis, in particular in synovial cell proliferation, of RA patients [32]. PGE2 is the most potent pro-inflammatory lipid mediator in COX pathway associated with RA considering that the elevated concentration of PGE2 is observed in the synovial fluid of RA patients [33] and the inflammation pain is mediated by PGE2 [34]. The inhibition of mPGES-1 has been a cure strategy for RA and is even better than the inhibition of COX-2 due to its safety and less side effect [35]. However, recent study has revealed the anti-inflammation of PGE2 [31,36]. In this study, values of the third peak of PGE2 and TXB2 in symptomatic rats were significantly higher than those in asymptomatic rats in the model group, suggesting the pro-inflammatory role of PGE2 and TXB2 at the third peak (Figure 3D).
HOEC is a caffeate that can be easily metabolized into caffeic acid in vivo (Supplementary Materials). Caffeic acid is a commonly used 5-LOX inhibitor for positive control [37-40] and is also used as a high effect inhibitor of 15-LOX [41]. Since the second peak of eicosanoids had a correlation with the arthritis onset, we compared the second peak values of HOEC treatments in 3 different doses with those of the model and normal control groups. At the second peak of metabolites, AA metabolic flux was decreased in LOX pathway (LTC4 and 15-HETE) and increased in COX pathway after HOEC treatment (Figure 4Da). To be specific, HOEC inhibited the generation of LTC4 and 15-HETE dose-dependently but had little inhibition on LTB4. High-dose HOEC even increased LTB4 slightly, which was probably due to the flux compensation of LOX pathway. LTC4, a pro-inflammatory mediator with potent bronchoconstrictor, was previously reported playing a important role in the pathophysiology of asthma but participating less in ongoing RA [42]. As for 15-HETE, there were few studies about 15-HETE in RA and its role in inflammation was not clearly defined. As one of the PPARγ ligands, 15-HETE was reported to have an anti-inflammatory effect [43]. So the over-expression of 15-LOX may have blocked the anti-inflammation effect of 15-HETE, leading to a poor curative effect on arthritis. In addition, 5-LOX and 15-LOX catalyzed the endogenous anti-inflammatory and pro-resolving mediator, lipoxins (LXs) [44], and the over-inhibition of these two enzymes may have induced a contrary anti-inflammatory effect.
As for the effect of HOEC on COX pathway, the influence was significant at the later stage of arthritis. As shown in our study, PGE2 and TXB2 were significantly inhibited by HOEC at the dose of 1 mg/kg and rose with the increase of dosage at the third peak, whose trend was the same as the clinical scores of arthritis (Figure 4E). The reduced AA metabolic flux in COX pathway flowed to the production of 15-HETE to some extent (Figure 4Db), since 15-HETE at the third peak of COX pathway was markedly elevated after HOEC treatment (Figure 4C). AA metabolism network is a system where eicosanoids are metabolized. We venture a guess that when one way is inhibited (shut down), the other (or another) metabolism pathway(s) of arachidonic acid will be turned on. Similar experimental phenomena were reported that in 5LO(-/-) mice’s peritoneal macrophages, PGE2 level was increased by the Ca2+ ionophore-induction compared with the 5LO(+/+) mice [45]. Lai et al. reported that when only the 5-LOX inhibitor was used, the flux of the COX-2 pathway was increased significantly using ordinary differential equations [2]. As for the influence of COX pathway’s change on LOX pathway, closure of COX path by NSAIDs results in opening of arachidonic acid oxidation in 5-LOX pathway and increased CysLTs signaling [46].
In addition, since the alterations of PGE2 and TXB2 kept in step with each other, the correlation was analyzed by SPSS 16.0. There is a strong relativity between PGE2 and TXB2 (Figure 5), suggesting that the generation of PGE2 and TXB2 is synchronous due to the same pathway of COX. As another metabolite of AA in COX pathway, the profile of PGD2, rather than PGE2 and TXB2, was similar to LTB4 and 15-HETE (Figure 5), which was possibly due to the flux compensation of COX pathway.
In conclusion, we report dynamic changes of multiple eicosanoids in the process of CIA in which AA metabolic network in RA animal model and HOEC therapeutics were studied. All eicosanoids increase simultaneously, due to the elevation of released arachidonic acid caused by immune reaction. HOEC can inhibit 5-LOX and 15-LOX in vivo and induce the elevation of COX pathway owing to the shift of flux distribution of arachidonic acid, especially in high dosage. It will be dangerous to inhibit a single target without considering the complete network. This study provides evidence for correctly choosing the dose of anti-inflammatory medicine and helps to propose novel strategies for treatment of infection and inflammation.
Acknowledgements
HL was supported by the National Key Research and Development Program [2016YFA0502304 to H.L.] and National Program for Special Supports of Eminent Professionals and National Program for Support of Top-notch Young Professionals.
Disclosure of conflict of interest
None.
Abbreviations
- AA
arachidonic acid
- CIA
collagen-induced arthritis
- RA
rheumatoid arthritis
- LTB4
leukotriene B4
- LTC4
leukotriene C4
- 15-HETE
15-hydroxyeicosatetraenoic acid
- PGE2
prostaglandin E2
- PGD2
prostaglandin D2
- TXA2
thromboxane A2
- TXB2
thromboxane B2
- LOX
5-lipoxygenase
- COX
cyclooxygenase
- CII
typeII collagen
- EDTA
ethylene diamine tetraacetic acid
- AChE
acetylcholinesterase
Supporting Information
References
- 1.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 2.Yang K, Ma W, Liang H, Ouyang Q, Tang C, Lai L. Dynamic simulations on the arachidonic acid metabolic network. PLoS Comput Biol. 2007;3:e55. doi: 10.1371/journal.pcbi.0030055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gupta S, Maurya MR, Stephens DL, Dennis EA, Subramaniam S. An integrated model of eicosanoid metabolism and signaling based on lipidomics flux analysis. Biophys J. 2009;96:4542–4551. doi: 10.1016/j.bpj.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kihara Y, Gupta S, Maurya Mano R, Armando A, Shah I, Quehenberger O, Glass Christopher K, Dennis Edward A, Subramaniam S. Modeling of eicosanoid fluxes reveals functional coupling between cyclooxygenases and terminal synthases. Biophys J. 2014;106:966–975. doi: 10.1016/j.bpj.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng H, Liu Y, Lai L. Diverse ways of perturbing the human arachidonic acid metabolic network to control inflammation. Acc Chem Res. 2015;48:2242–2250. doi: 10.1021/acs.accounts.5b00226. [DOI] [PubMed] [Google Scholar]
- 6.Korotkova M, Jakobsson PJ. Persisting eicosanoid pathways in rheumatic diseases. Nat Rev Rheumatol. 2014;10:229–241. doi: 10.1038/nrrheum.2014.1. [DOI] [PubMed] [Google Scholar]
- 7.Bombardieri S, Cattani P, Ciabattoni G, Munno O, Pasero G, Patrono C, Pinca E, Pugliese F. The synovial prostaglandin system in chronic inflammatory arthritis: differential effects of steroidal and nonsteroidal anti-inflammatory drugs. Br J Pharmacol. 1981;73:893–901. doi: 10.1111/j.1476-5381.1981.tb08743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egg D. Concentrations of prostaglandins D2, E2, F2 alpha, 6-keto-F1 alpha and thromboxane B2 in synovial fluid from patients with inflammatory joint disorders and osteoarthritis. Z Rheumatol. 1984;43:89–96. [PubMed] [Google Scholar]
- 9.Nakamura H, Hishinuma T, Suzuki N, Chiba S, Tsukamoto H, Takabatake M, Sawai T, Mitomo T, Inoue H, Matsumoto F. Difference in urinary 11-dehydro TXB2 and LTE4 excretion in patients with rheumatoid arthritis. Prostaglandins Leukot Essent Fatty Acids. 2001;65:301–306. doi: 10.1054/plef.2001.0329. [DOI] [PubMed] [Google Scholar]
- 10.Klickstein LB, Shapleigh C, Goetzl EJ. Lipoxygenation of arachidonic acid as a source of polymorphonuclear leukocyte chemotactic factors in synovial fluid and tissue in rheumatoid arthritis and spondyloarthritis. J Clin Invest. 1980;66:1166. doi: 10.1172/JCI109947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davidson E, Rae S, Smith M. Leukotriene B4, a mediator of inflammation present in synovial fluid in rheumatoid arthritis. Ann Rheum Dis. 1983;42:677–679. doi: 10.1136/ard.42.6.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmgreen J, Nielsen O, Ahnfelt-Rønne I. Enhanced capacity for release of leucotriene B4 by neutrophils in rheumatoid arthritis. Ann Rheum Dis. 1987;46:501–505. doi: 10.1136/ard.46.7.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinblatt M, Kremer J, Coblyn J, Helfgott S, Maier A, Petrillo G, Henson B, Rubin P, Sperling R. Zileuton, a 5-lipoxygenase inhibitor in rheumatoid arthritis. J Rheumatol. 1992;19:1537–1541. [PubMed] [Google Scholar]
- 14.Díaz-González F, Alten RH, Bensen WG, Brown JP, Sibley JT, Dougados M, Bombardieri S, Durez P, Ortiz P, de-Miquel G. Clinical trial of a leucotriene B4 receptor antagonist, BIIL 284, in patients with rheumatoid arthritis. Ann Rheum Dis. 2007;66:628–632. doi: 10.1136/ard.2006.062554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chemin K, Klareskog L, Malmström V. Is rheumatoid arthritis an autoimmune disease? Curr Opin Rheumatol. 2016;28:181–188. doi: 10.1097/BOR.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 16.Wooley PH, Chapedelaine JM. Immunogenetics of collagen-induced arthritis. Crit Rev Immunol. 1986;8:1–22. [PubMed] [Google Scholar]
- 17.Gebhard H, Zysk S, Schmitt-Sody M, Jansson V, Messmer K, Veihelmann A. The effects of Celecoxib on inflammation and synovial microcirculation in murine antigen-induced arthritis. Clin Exp Rheumatol. 2005;23:63–70. [PubMed] [Google Scholar]
- 18.Myers LK, Kang AH, Postlethwaite AE, Rosloniec EF, Morham SG, Shlopov BV, Goorha S, Ballou LR. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum. 2000;43:2687–2693. doi: 10.1002/1529-0131(200012)43:12<2687::AID-ANR8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 19.Mancini JA, Blood K, Guay J, Gordon R, Claveau D, Chan CC, Riendeau D. Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J Biol Chem. 2001;276:4469–4475. doi: 10.1074/jbc.M006865200. [DOI] [PubMed] [Google Scholar]
- 20.Maicas N, Ibanez L, Alcaraz MJ, Ubeda A, Ferrandiz ML. Prostaglandin D2 regulates joint inflammation and destruction in murine collagen-induced arthritis. Arthritis Rheum. 2012;64:130–140. doi: 10.1002/art.30656. [DOI] [PubMed] [Google Scholar]
- 21.Nickerson-Nutter CL, Medvedeff ED. The effect of leukotriene synthesis inhibitors in models of acute and chronic inflammation. Arthritis Rheum. 1996;39:515–521. doi: 10.1002/art.1780390320. [DOI] [PubMed] [Google Scholar]
- 22.Griffiths R, Pettipher E, Koch K, Farrell C, Breslow R, Conklyn M, Smith M, Hackman B, Wimberly D, Milici A. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc Natl Acad Sci U S A. 1995;92:517–521. doi: 10.1073/pnas.92.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liagre B, Vergne P, Rigaud M, Beneytout J. Arachidonate 15-lipoxygenase of reticulocyte-type in human rheumatoid arthritis type B synoviocytes and modulation of its activity by proinflammatory cytokines. J Rheumatol. 1999;26:1044–1051. [PubMed] [Google Scholar]
- 24.Li L, Zeng H, Liu F, Zhang J, Yue R, Lu W, Yuan X, Dai W, Yuan H, Sun Q. Target identification and validation of (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate, an anti-inflammatory natural product. European Journal of Inflammation. 2012;10:297–309. [Google Scholar]
- 25.Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nature Protocols. 2007;2:1269–1275. doi: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 26.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 27.Griswold DE, Hillegass LM, Meunier PC, Dimartino MJ, Hanna N. Effect of inhibitors of eicosanoid metabolism in murine collagen-induced arthritis. Arthritis Rheum. 1988;31:1406–1412. doi: 10.1002/art.1780311110. [DOI] [PubMed] [Google Scholar]
- 28.Maicas N, Ibáñez L, Alcaraz MJ, Úbeda A, Ferrándiz ML. Prostaglandin D2 regulates joint inflammation and destruction in murine collagen-induced arthritis. Arthritis Rheum. 2012;64:130–140. doi: 10.1002/art.30656. [DOI] [PubMed] [Google Scholar]
- 29.Shao WH, Del Prete A, Bock CB, Haribabu B. Targeted disruption of leukotriene B4 receptors BLT1 and BLT2: a critical role for BLT1 in collagen-induced arthritis in mice. J Immunol. 2006;176:6254–6261. doi: 10.4049/jimmunol.176.10.6254. [DOI] [PubMed] [Google Scholar]
- 30.Mathis SP, Jala VR, Lee DM, Haribabu B. Nonredundant roles for leukotriene B4 receptors BLT1 and BLT2 in inflammatory arthritis. J Immunol. 2010;185:3049–3056. doi: 10.4049/jimmunol.1001031. [DOI] [PubMed] [Google Scholar]
- 31.Chan MM, Moore AR. Resolution of inflammation in murine autoimmune arthritis is disrupted by Cyclooxygenase-2 inhibition and restored by Prostaglandin E(2)-Mediated Lipoxin A(4) production. J Immunol. 2010;184:6418–6426. doi: 10.4049/jimmunol.0903816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang MJ, Huang Y, Huang RY, Chen XM, Zhou YY, Yu WL, Chu YL, Huang QC. Determination of role of thromboxane A2 in rheumatoid arthritis. DiscovMed. 2015;19:23–32. [PubMed] [Google Scholar]
- 33.Egg D, Günther R, Herold M, Kerschbaumer F. Prostaglandins E2 and F2 alpha concentrations in the synovial fluid in rheumatoid and traumatic knee joint diseases. Z Rheumatol. 1980;39:170–175. [PubMed] [Google Scholar]
- 34.Harvey RJ, Depner UB, Wässle H, Ahmadi S, Heindl C, Reinold H, Smart TG, Harvey K, Schütz B, Abo-Salem OM. GlyR α3: an essential target for spinal PGE2-mediated inflammatory pain sensitization. Science. 2004;304:884–887. doi: 10.1126/science.1094925. [DOI] [PubMed] [Google Scholar]
- 35.Koeberle A, Laufer SA, Werz O. Design and development of microsomal prostaglandin E2 synthase-1 inhibitors: challenges and future directions. J Med Chem. 2016;59:5970–5986. doi: 10.1021/acs.jmedchem.5b01750. [DOI] [PubMed] [Google Scholar]
- 36.Frolov A, Yang L, Dong H, Hammock BD, Crofford LJ. Anti-inflammatory properties of prostaglandin e2: deletion of microsomal prostaglandin e synthase-1 exacerbates non-immune inflammatory arthritis in mice. Prostaglandins Leukot Essent Fatty Acids. 2013;89:351–358. doi: 10.1016/j.plefa.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chowdhury MA, Abdellatif KR, Dong Y, Das D, Suresh MR, Knaus EE. Synthesis of celecoxib analogs that possess a N-hydroxypyrid-2 (1H) one 5-lipoxygenase pharmacophore: Biological evaluation as dual inhibitors of cyclooxygenases and 5-lipoxygenase with anti-inflammatory activity. Bioorg Med Chem Lett. 2008;18:6138–6141. doi: 10.1016/j.bmcl.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Coy ED, Cuca LE, Sefkow M. COX, LOX and platelet aggregation inhibitory properties of Lauraceae neolignans. Bioorg Med Chem Lett. 2009;19:6922–6925. doi: 10.1016/j.bmcl.2009.10.069. [DOI] [PubMed] [Google Scholar]
- 39.Christie MJ, Connor M, Vaughan CW, Ingram SL, Bagley EE. Cellular actions of opioids and other analgesics: implications for synergism in pain relief. Clin Exp Pharmacol Physiol. 2000;27:520–523. doi: 10.1046/j.1440-1681.2000.03291.x. [DOI] [PubMed] [Google Scholar]
- 40.Christie M, Vaughan C, Ingram S. Opioids, NSAIDs and 5-lipoxygenase inhibitors act synergistically in brain via arachidonic acid metabolism. Inflamm Res. 1999;48:1–4. doi: 10.1007/s000110050367. [DOI] [PubMed] [Google Scholar]
- 41.Gleason MM, Rojas CJ, Learn KS, Perrone MH, Bilder GE. Characterization and inhibition of 15-lipoxygenase in human monocytes: comparison with soybean 15-lipoxygenase. Am J Physiol. 1995;268:C1301–C1307. doi: 10.1152/ajpcell.1995.268.5.C1301. [DOI] [PubMed] [Google Scholar]
- 42.Yousefi B, Jadidi-Niaragh F, Azizi G, Hajighasemi F, Mirshafiey A. The role of leukotrienes in immunopathogenesis of rheumatoid arthritis. Mod Rheumatol. 2014;24:225–235. doi: 10.3109/14397595.2013.854056. [DOI] [PubMed] [Google Scholar]
- 43.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 44.Chandrasekharan JA, Sharma-Walia N. Lipoxins: nature’s way to resolve inflammation. J Inflamm Res. 2015;8:181. doi: 10.2147/JIR.S90380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goulet JL, Snouwaert JN, Latour AM, Coffman TM, Koller BH. Altered inflammatory responses in leukotriene-deficient mice. Proc Natl Acad Sci U S A. 1994;91:12852–12856. doi: 10.1073/pnas.91.26.12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15:511. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.