

Abstract
Lipoprotein lipase (LPL) catalyzes the breakdown of circulating triglycerides in muscle and fat. LPL is inhibited by several proteins, including angiopoietin-like 4 (ANGPTL4), and may be cleaved by members of the proprotein convertase subtilisin/kexin (PCSK) family. Here, we aimed to investigate the cleavage of LPL in adipocytes by PCSKs and study the potential involvement of ANGPTL4. A substantial portion of LPL in mouse and human adipose tissue was cleaved into N- and C-terminal fragments. Treatment of different adipocytes with the PCSK inhibitor decanoyl-RVKR-chloromethyl ketone markedly decreased LPL cleavage, indicating that LPL is cleaved by PCSKs. Silencing of Pcsk3/furin significantly decreased LPL cleavage in cell culture medium and lysates of 3T3-L1 adipocytes. Remarkably, PCSK-mediated cleavage of LPL in adipocytes was diminished by Angptl4 silencing and was decreased in adipocytes and adipose tissue of Angptl4−/− mice. Differences in LPL cleavage between Angptl4−/− and WT mice were abrogated by treatment with decanoyl-RVKR-chloromethyl ketone. Induction of ANGPTL4 in adipose tissue during fasting enhanced PCSK-mediated LPL cleavage, concurrent with decreased LPL activity, in WT but not Angptl4−/− mice. In adipocytes, after removal of cell surface LPL by heparin, levels of N-terminal LPL were still markedly higher in WT compared with Angptl4−/− adipocytes, suggesting that stimulation of PCSK-mediated LPL cleavage by ANGPTL4 occurs intracellularly. Finally, treating adipocytes with insulin increased full-length LPL and decreased N-terminal LPL in an ANGPTL4-dependent manner. In conclusion, ANGPTL4 promotes PCSK-mediated intracellular cleavage of LPL in adipocytes, likely contributing to regulation of LPL in adipose tissue. Our data provide further support for an intracellular action of ANGPTL4 in adipocytes.
Keywords: lipoprotein metabolism, adipocyte, furin, lipase, human, ANGPTL4, lipoprotein lipase, angiopoietin-like 4, PCSK, triglycerides
Introduction
Elevated levels of plasma triglycerides are increasingly recognized as an important causal risk factor for coronary artery disease (1). Plasma triglyceride levels are determined by the balance between the secretion of triglycerides by the liver and small intestine and the clearance of triglycerides in muscle and fat tissue. The rate of triglyceride clearance is determined by the enzyme lipoprotein lipase (LPL),4 which catalyzes the hydrolysis of triglycerides at the capillary endothelium (2). LPL is produced by myocytes and adipocytes and is transported toward the endothelial cell surface by the protein GPIHBP1 (glycosylphosphatidylinositol-anchored high-density lipoprotein–binding protein 1) (3). To match the tissue uptake of fatty acids in accordance with the local needs, the activity of LPL is tightly regulated, primarily at the post-translational level (4).
Angiopoietin-like 3 (ANGPTL3), angiopoietin-like 4 (ANGPTL4), and angiopoietin-like 8 (ANGPTL8) are three members of the angiopoietin-like protein family that are involved in the post-translational regulation of LPL (5). ANGPTL3 and ANGPTL8 are secreted by the liver as hepatokines and cooperate to inhibit the activity of LPL in oxidative tissues, such as heart and brown fat, with a primary action in the fed state (6–9). By contrast, ANGPTL4 is produced by several tissues and appears to mainly function locally in a tissue-specific manner (5). Studies have shown that ANGPTL4 plays a major role in the regulation of LPL activity during various physiological conditions, such as exercise, fasting, and cold exposure (10–12). ANGPTL4 inactivates LPL by promoting the unfolding of the protein, leading to the conversion of the catalytically active LPL dimer into inactive monomers (13, 14). Although ANGPTL4 is able to inhibit LPL activity in the subendothelial spaces and on the endothelial surface (15, 16), recently, we provided evidence that ANGPTL4 and LPL also interact intracellularly, causing the degradation of LPL (17). However, the specific steps involved in the intracellular degradation of LPL by ANGPTL4 remain unclear.
Members of the proprotein convertase subtilisin/kexin (PCSK) protein family (PCSK1–7, SKI-1/S1P, and PCSK9) are calcium-dependent serine endopeptidases that convert proproteins into their active forms by cleavage (18, 19). PCSK1–7 recognize and cleave substrates at specific lysine- and/or arginine-containing basic amino acid sequences (18). Despite overlap in substrate recognition, their functions are tissue-specific and dependent on their cellular localization. For example, PCSK3 (furin) is primarily found in the trans-Golgi network, in the endosomes, and on the cell surface, whereas PCSK5 (PC5/6) and PCSK6 (PACE4) are primarily present on the cell surface (19). PCSKs have been repeatedly implicated in the regulation of lipoprotein metabolism. The best-known example is PCSK9, which raises plasma low-density lipoprotein (LDL) cholesterol levels. Specifically, PCSK9 promotes the degradation of the LDL receptor in endosomes/lysosomes, resulting in a reduced clearance of LDL cholesterol (20). Other examples include the cleavage of ANGPTL3 and ANGPTL4 by PCSKs in vitro and in vivo (21–24). Interestingly, it has been demonstrated that LPL is also subject to cleavage by PCSKs, at least in vitro (25, 26). Specifically, a cleavage site was identified at residues 321–324, which is in the middle of a stretch of 60 amino acids that is 100% conserved between mouse and human LPL (26), yielding an N- and C-terminal LPL cleavage fragment. However, to what extent LPL is cleaved in vivo in adipocytes has remained unclear. In addition, the potential role of ANGPTL4 has not been investigated. Accordingly, the aim of the present study was to investigate the mechanism underlying the cleavage of LPL in adipocytes and to explore the potential role of ANGPTL4.
Results
LPL is cleaved in human and mouse adipose tissue
To examine whether LPL is cleaved in vivo in white adipose tissue, we performed Western blotting for LPL in human adipose tissue using antibodies directed against the N- or C-terminal portion of human LPL (27). Both antibodies gave rise to two bands, corresponding to full-length LPL (slightly above 50 kDa), and the N-terminal or C-terminal LPL cleavage fragment at around 30 kDa or 20–25 kDa, respectively (Fig. 1A). These data indicate that LPL is cleaved in human adipose tissue. Using two different antibodies directed against the C-terminal part of human LPL, it was observed that treatment with endoglycosidase H (EndoH), an enzyme that cleaves asparagine-linked mannose-rich oligosaccharides, did not alter the migration of full-length and C-terminal LPL (Fig. 1B). By contrast, treatment with PNGase, which removes most N-linked oligosaccharides, altered the migration of full-length LPL but not C-terminal LPL (Fig. 1B). Adipose tissue of different human subjects always showed a substantial proportion of LPL in the truncated form (Fig. 1C). Similarly, using an antibody directed against the N-terminal portion of mouse LPL, immunoblotting of mouse adipose tissue yielded two bands, corresponding to full-length LPL and the N-terminal LPL cleavage fragment) (Fig. 1D) (28). Whereas part of mouse full-length LPL changed migration after treatment with EndoH, the N-terminal LPL cleavage product did not change migration. Treatment with PNGase altered the migration of both full-length and N-terminal LPL (Fig. 1D). LPL was also found to be cleaved in mouse heart but not in mouse bone marrow–derived macrophages (Fig. 1E). Overall, these data indicate that 1) LPL is cleaved into N- and C-terminal cleavage fragments in human and mouse adipose tissue and 2) human and mouse LPL is glycosylated in the N-terminal region (26, 29, 30, 31).
Figure 1.

Cleavage of LPL in human and mouse adipose tissue. A, Western blotting of lysates of human adipose tissue using antibodies against the N-terminal (Y-20) and C-terminal (88b8) portion of hLPL. B, Western blotting of lysates of human adipose tissue treated with EndoH or PNGase, using two different antibodies against hLPL, 88b8, and 5D2 (27). C, Western blotting of lysates of human adipose tissue of different subjects enrolled in the MONDIAL study, using an antibody against hLPL (88b8). D, Western blotting of lysates of mouse adipose tissue treated with EndoH or PNGase, using an antibody against mLPL (28). E, Western blotting of lysates of mouse heart and bone marrow–derived macrophages. Molecular weight markers are indicated. l.e., long exposure; s.e., short exposure.
LPL is cleaved in adipose tissue by PCSKs
Previously, members of the PCSK family were suggested to cleave LPL at residues 321–324 to yield nearly complete N-terminal and C-terminal domains (26). To examine whether the observed N-terminal LPL was the result of PCSK-mediated cleavage, mature 3T3-L1 and 3T3-F442a adipocytes as well as primary adipocytes and white adipose tissue explants from mice were treated with the inhibitor decanoyl-RVKR-chloromethyl ketone (dec-CMK) to block PCSK activity (32). In all adipocyte models, incubation with dec-CMK resulted in the almost complete disappearance of N-terminal LPL in cell culture medium and cell lysates, indicating that LPL is cleaved by PCSKs in adipocytes (Fig. 2).
Figure 2.

LPL is cleaved by PCSKs in adipocytes. Shown are Western blots of cell culture medium (top panels) and cell lysates (bottom panels) of mature 3T3-L1 adipocytes, mature 3T3-F442a adipocytes, primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from mice, and adipose tissue explants from mice that were treated with 50 μm dec-CMK for 9 h. Western blots were probed with antibodies against mLPL and HSP90 (as loading control). Coomassie Blue staining was performed as loading control for cell culture medium, primary adipocytes, and white adipose tissue (WAT) explants. FL, full-length; N-ter, N-terminal.
PCSK3 is expressed in adipose tissue and cleaves adipocyte LPL
To assess whether adipocyte LPL is cleaved by PCSKs intracellularly and/or upon secretion, we treated 3T3-L1 adipocytes with heparin to release LPL bound to heparin sulfate proteoglycans (HSPGs) from the cell surface. As expected, heparin treatment resulted in a pronounced increase of LPL in the cell culture medium, along with a reduction of LPL in the cell lysates (Fig. 3). In line with the notion that binding of LPL to HSPG is mainly mediated by C-terminal and not N-terminal LPL (29, 33), heparin did not influence the amount of N-terminal LPL in the cell culture medium and cell lysates (Fig. 3). These data indicate that little or no N-terminal LPL is bound to HSPGs on the cell surface and that N-terminal LPL in cell lysates must originate from intracellular LPL cleavage, from where it is secreted.
Figure 3.
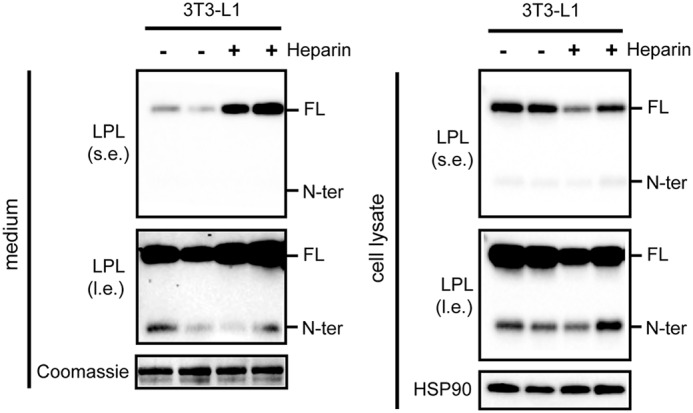
N-terminal LPL cannot be released from the cell surface by heparin. Shown are Western blots of cell culture medium (left) or cell lysates (right) of mature 3T3-L1 adipocytes that were treated with 10 IU/ml heparin for 20 min. Western blots were probed with antibodies against mLPL and HSP90 (as loading control). Coomassie Blue staining was performed as loading control for cell culture medium. l.e., long exposure; s.e., short exposure. FL, full-length; N-ter, N-terminal.
Given that LPL is cleaved intracellularly by PCSKs, a promising candidate that may catalyze LPL cleavage in adipocytes is PCSK3, as PCSK3 is activated and active intracellularly in the trans-Golgi (19). Expression profiling showed that PCSK3 is most highly expressed in liver and kidney, with comparatively low but clearly detectable PCSK3 expression in adipose tissue (Fig. 4A). To assess whether PCSK3 might be involved in LPL cleavage in adipocytes, we silenced Pcsk3 in mature 3T3-L1 adipocytes by means of siRNA. siRNA-mediated silencing resulted in a 90% reduction in Pcsk3 expression levels (Fig. 4B). In line with a role for PCSK3 in LPL cleavage, silencing of Pcsk3 significantly reduced the amount of LPL cleavage in cell culture medium and cell lysates (Fig. 4C). These data are in line with a previous finding that PCSK3 is able to cleave LPL in a stable HEK293 cell line expressing LPLmyc (26).
Figure 4.
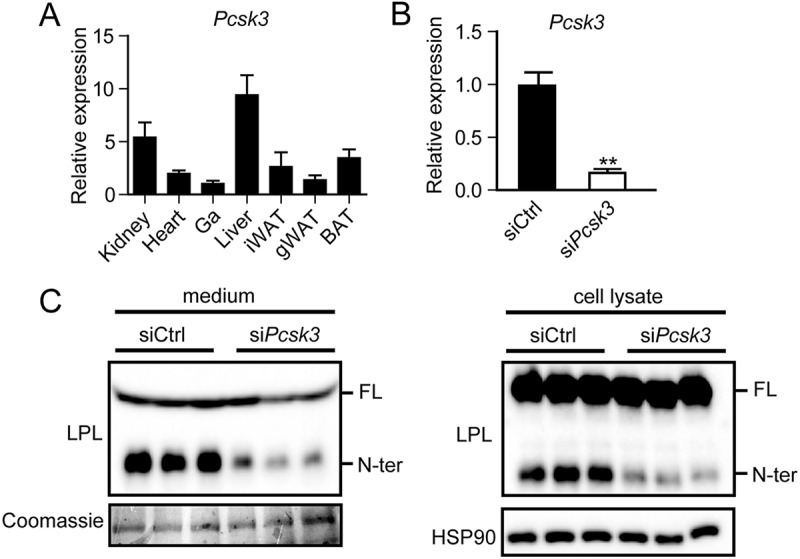
PCSK3 is well-expressed in adipose tissue and cleaves adipocyte LPL. A, Pcsk3 mRNA levels in different tissues from C57BL/6 mice (n = 4). B, Pcsk3 mRNA levels in fully differentiated 3T3-L1 adipocytes that were trypsinized, replated at 70% confluence, and treated with siPcsk3 or siCtrl for 48 h. **, significantly different from siCtrl according to Student's t test; p < 0.01. C, Western blots of cell culture medium (left) and cell lysates (right) of fully differentiated 3T3-L1 adipocytes that were trypsinized, replated at 70% confluence, and treated with siPcsk3 or siCtrl for 48 h. Western blots were probed with antibodies against mLPL and HSP90 (as loading control). Coomassie Blue staining was performed as loading control for cell culture medium. FL, full-length; N-ter, N-terminal. Error bars, S.E.
N-terminal LPL is cleared by the lysosomes
Previously, PCSK-mediated cleavage was shown to promote the inactivation of endothelial lipase, a family member of LPL (26). Similarly, cleavage of LPL by PCSKs might also serve to inactivate LPL (26). To assess the fate of cleaved LPL and to study whether it may be further subject to lysosomal degradation in adipocytes, we incubated mature 3T3-L1 adipocytes and primary adipocytes with bafilomycin A1 (BafA1) to inhibit lysosomal degradation. In agreement with previous studies, treatment of adipocytes with BafA1 significantly increased protein levels of full-length LPL in cell lysates and to a lesser extent in the cell culture medium (Fig. 5, A and B) (34). Interestingly, in both 3T3-L1 and primary adipocytes, BafA1 also increased the amount of N-terminal LPL in cell lysates and medium, suggesting that at least part of the N-terminal LPL is cleared by lysosomes (Fig. 5, A and B). The increase in LPL protein abundance in primary and 3T3-L1 adipocytes upon BafA1 treatment cannot be attributed to an increase in Lpl mRNA (Fig. 5, C and D).
Figure 5.

N-terminal LPL is cleared by the lysosomes. A, Western blots of cell culture medium (left) and cell lysates (right) of mature 3T3-L1 adipocytes that were treated with 100 nm bafilomycin A1 (BafA1) for 10 h. B, Western blots of cell culture medium (left) and cell lysates (right) of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue that were treated with 100 nm bafilomycin A1 for 10 h. Western blots were probed with antibodies against LPL and HSP90. Coomassie Blue staining was performed as loading control for cell culture medium. C, Lpl mRNA in mature 3T3-L1 adipocytes treated with 100 nm bafilomycin A1 for 10 h. D, Lpl mRNA in primary mouse adipocytes treated with 100 nm bafilomycin A1 for 10 h. FL, full-length; N-ter, N-terminal. Error bars, S.D.
ANGPTL4 promotes PCSK-mediated cleavage of adipocyte LPL
An important regulator of LPL in adipose tissue is ANGPTL4. Indeed, ANGPTL4 is highly abundant in mouse adipose tissues, where it exists as a glycosylated protein (Fig. 6A). Treatment with PNGase, which removes most N-linked glycans, and a protein deglycosylation mix, which removes N-linked glycans as well as simple and some complex O-linked glycans, altered the mobility of ANGPTL4, indicating removal of carbohydrate side chains (Fig. 6A). However, EndoH, which removes N-linked mannose-rich glycans, had no effect, suggesting that most of the carbohydrate side chains on ANGPTL4 contain complex sugars (22). Recently, we found that ANGPTL4 promotes the intracellular degradation of LPL in adipocytes (17). To explore the connection between PCSK-mediated cleavage of LPL and ANGPTL4-induced LPL degradation, we investigated whether ANGPTL4 is able to enhance PCSK-mediated LPL cleavage. To that end, we assessed the effect of siRNA-mediated Angptl4 silencing on LPL cleavage in 3T3-L1 adipocytes. The ∼80% reduction in Angptl4 mRNA (Fig. 6B) was accompanied by a marked decrease in N-terminal LPL in 3T3-L1 cell lysate and a modest increase in full-length LPL in cell lysates and medium (Fig. 6C). These results suggest that ANGPTL4 may promote LPL cleavage in adipocytes. To further investigate this notion, we determined the accumulation of N-terminal LPL in adipose tissue of Angptl4−/− and WT mice. Corroborating our previous findings, levels of full-length LPL were notably higher in adipose tissue of Angptl4−/− mice compared with WT mice (Fig. 7A) (17). Consistent with a stimulatory effect of ANGPTL4 on cleavage of LPL, levels of N-terminal LPL were significantly lower in adipose tissue of Angptl4−/− mice (Fig. 7A). Of note, LPL activity was significantly higher in adipose tissue of Angptl4−/− mice compared with WT mice, confirming the increase in full-length LPL protein (Fig. 7B).
Figure 6.
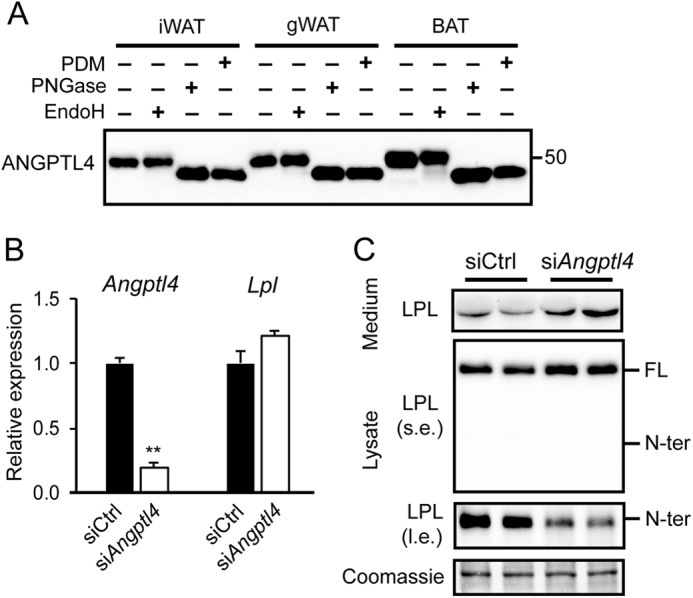
Angptl4 silencing reduces LPL cleavage. A, Western blotting of lysate of inguinal white (iWAT), gonadal white (gWAT), and interscapular brown (BAT) adipose tissue treated with protein deglycosylation mix (PDM), EndoH, or PNGase, using an antibody against mANGPTL4. B, Lpl and Angptl4 mRNA levels in fully differentiated 3T3-L1 adipocytes that were treated with siAngptl4 or siCtrl for 48 h. **, significantly different from siCtrl according to Student's t test; p < 0.01. C, Western blots of cell culture medium and cell lysates of mature 3T3-L1 adipocytes that were treated with siAngptl4 or siCtrl for 48 h, using an antibody against mLPL. Coomassie Blue staining served as loading control. l.e., long exposure; s.e., short exposure. FL, full-length; N-ter, N-terminal. Error bars, S.D.
Figure 7.

ANGPTL4 regulates LPL cleavage in adipose tissue during fasting. A, Western blotting of cell lysates of gonadal adipose tissue (gWAT) from Angptl4−/− and WT mice, using an antibody against mLPL. Coomassie Blue staining was performed as loading control. B, LPL activity in gonadal adipose tissue of Angptl4−/− mice (n = 7) and WT mice (n = 14). ***, significantly different from Angptl4−/− mice according to Student's t test (p < 0.001). Samples were from ad libitum fed mice. C, Western blotting of cell lysates of gonadal adipose tissue of fasted and refed WT mice treated with protein deglycosylation mix (PDM), EndoH, or PNGase, using an antibody against mANGPTL4. D, Western blotting of cell lysates of gonadal adipose tissue of fasted and refed Angptl4−/− and WT mice treated with EndoH, using an antibody against mLPL. Separate boxes for Angptl4−/− and WT mice indicates that all samples were run on the same gel with the same exposure time but that intermediate lanes were removed. Part of this Western blotting at different exposure has been published previously (17). R, EndoH-resistant LPL (complex oligosaccharides: Golgi and cell surface LPL). S, EndoH-sensitive LPL (high-mannose oligosaccharides, ER LPL). l.e., long exposure; s.e., short exposure. FL, full-length; N-ter, N-terminal. Error bars, S.E.
To examine whether levels of N-terminal LPL respond to changes in endogenous ANGPTL4 production, we measured ANGPTL4 and LPL under conditions of fasting/refeeding. Samples were treated with EndoH to distinguish between EndoH-resistant LPL (LPL in the Golgi and the cell surface) and EndoH-sensitive LPL (LPL in the ER). As previously observed (17, 35), ANGPTL4 levels in white adipose tissue were elevated in the fasted state compared with the refed state (Fig. 7C), which was accompanied by a decrease in full-length (EndoH-resistant) LPL and an increase in N-terminal LPL (Fig. 7D). Strikingly, levels of N-terminal LPL were lower in Angptl4−/− mice. Moreover, the difference in levels of N-terminal LPL between the fasted and refed state was abolished in Angptl4−/− mice (Fig. 7D).
To further understand the mechanism underlying the differential abundance of N-terminal LPL in adipose tissue of WT and Angptl4−/− mice, we shifted our experiments to primary adipocytes. Similar to whole adipose tissue, primary adipocytes of Angptl4−/− mice also showed higher levels of full-length LPL and lower levels of N-terminal LPL as compared with WT adipocytes (Fig. 8A). Treatment with the PCSK inhibitor dec-CMK abrogated these differences, indicating that PCSK-mediated LPL cleavage mediates the differences in N-terminal LPL between Angptl4−/− and WT mice (Fig. 8B). To investigate whether stimulation of PCSK-mediated LPL cleavage by ANGPTL4 may initiate the complete degradation of LPL, we treated primary adipocytes with cycloheximide to arrest protein synthesis and measured the decline in full-length and N-terminal LPL by Western blotting. The rate of decline of N-terminal LPL and to a lesser extent full-length LPL was notably lower in Angptl4−/− adipocytes than in WT adipocytes, indicating that ANGPTL4 accelerates the degradation of LPL (Fig. 8C). These data suggest that induction of PCSK-mediated LPL cleavage by ANGPTL4 is part of the degradation pathway of LPL.
Figure 8.

ANGPTL4 promotes PCSK-mediated cleavage of adipocyte LPL. A, Western blotting of cell lysates of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from Angptl4−/− and WT mice, using an antibody against mLPL. Coomassie Blue staining was performed as loading control. B, Western blots of cell culture medium (top panels) and cell lysates (bottom panels) of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from Angptl4−/− and WT mice that were treated with 50 μm dec-CMK for 9 h. Western blots were probed with antibodies against mLPL and HSP90. Coomassie Blue staining was performed as loading control for cell culture medium. C, Western blotting of cell lysates of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from Angptl4−/− and WT mice. Adipocytes were treated with cycloheximide (Cyclo) for 0, 30, or 60 min. Adipocyte lysates were treated with EndoH. Western blots were probed with antibodies against mLPL and HSP90. R, EndoH-resistant LPL (complex oligosaccharides: Golgi and cell surface LPL). S, EndoH-sensitive LPL (high-mannose oligosaccharides, ER LPL). l.e., long exposure; s.e., short exposure. FL, full-length; N-ter, N-terminal.
Induction of PCSK-mediated LPL cleavage by ANGPTL4 occurs intracellularly
Finally, to unequivocally establish that stimulation of PCSK-mediated LPL cleavage by ANGPTL4 occurs intracellularly, we determined the levels of full-length and N-terminal LPL in medium and lysate of WT and Angptl4−/− adipocytes treated with heparin for 20 min. As shown previously (17), the heparin-induced release of full-length LPL into the medium was higher in Angptl4−/− than in WT adipocytes (Fig. 9A). In concordance with these data, the reduction in cellular LPL content by heparin was more pronounced in Angptl4−/− than in WT adipocytes. Consistent with the inability of N-terminal LPL to bind to HSPG, heparin treatment did not cause any release of N-terminal LPL into the medium. Importantly, even after removal of the heparin-releasable pool of LPL, the levels of N-terminal LPL were still markedly higher in WT than in Angptl4−/− adipocytes (Fig. 9B). These data indicate that the stimulation of PCSK-mediated LPL cleavage by ANGPTL4 happens inside the cell and not on the cell surface.
Figure 9.

ANGPTL4 promotes LPL cleavage inside adipocytes. Shown are Western blots of cell culture medium (A) and cell lysates (B) of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from Angptl4−/− and WT mice treated with 10 IU/ml heparin for 20 min. Western blots were probed with antibodies against mLPL and HSP90. Coomassie Blue staining was performed as loading control for cell culture medium. l.e., long exposure; s.e., short exposure. FL, full-length; N-ter, N-terminal.
As a final experiment, we studied the effect of insulin. Injection of insulin was previously shown to decrease Angptl4 mRNA and increase LPL activity in adipose tissue of rats (12). To study whether down-regulation of Angptl4 by insulin leads to corresponding changes in the different LPL forms, we treated primary adipocytes with insulin and determined the levels of full-length and N-terminal LPL in the lysates. Insulin reduced Angptl4 mRNA in primary adipocytes by about 50% and did not have a noticeable effect on Lpl and Pcsk3 mRNA (Fig. 10A). Interestingly, whereas insulin treatment increased levels of full-length LPL, it modestly decreased levels of N-terminal LPL (Fig. 10B). These effects were observed specifically in WT adipocytes, but not Angptl4−/− adipocytes. The increase in full-length LPL in response to insulin and in Angptl4−/− adipocytes was accounted for by an increase in EndoH-resistant LPL (Fig. 10C). These data indicate that the down-regulation of Angptl4 by insulin in adipocytes leads to reduced LPL cleavage.
Figure 10.

Down-regulation of Angptl4 by insulin reduces LPL cleavage. A, expression of Angptl4 and Lpl in primary mouse adipocytes treated with insulin (500 nm) for 12 h. **, significantly different from control according to Student's t test; p < 0.01. B, Western blotting of cell lysates of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from WT and Angptl4−/− mice treated with 500 nm insulin for 12 h. C, Western blotting of cell lysates of primary adipocytes differentiated from the stromal vascular fraction of adipose tissue from WT and Angptl4−/− mice treated with 500 nm insulin for 12 h. Adipocyte lysates were treated with EndoH. Western blots were probed with antibodies against mLPL. Coomassie Blue staining was performed as loading control. R, EndoH-resistant LPL (complex oligosaccharides: Golgi and cell surface LPL). S, EndoH-sensitive LPL (high-mannose oligosaccharides, ER LPL). FL, full-length; N-ter, N-terminal. Error bars, S.D.
Discussion
Little is known about the cleavage of LPL in adipocytes. Here, we provide in vitro and in vivo evidence that LPL undergoes substantial cleavage in mouse and human adipocytes to yield N- and C-terminal fragments. The cleavage of LPL in adipocytes is at least partly mediated by PCSK3 (furin) and likely represents an initial step in the intracellular degradation of LPL. Importantly, we find that ANGPTL4 stimulates the intracellular cleavage of LPL by PCSKs. Induction of ANGPTL4 levels in adipose tissue during fasting enhanced PCSK-mediated LPL cleavage, concurrent with decreased LPL levels and activity, suggesting that stimulation of LPL cleavage by ANGPTL4 contributes to suppression of LPL levels in adipocytes during fasting. Conversely, suppression of ANGPTL4 by insulin in adipocytes reduced PCSK-mediated LPL cleavage, concurrent with increased LPL levels. Induction of PCSK-mediated LPL cleavage by ANGPTL4 occurs inside the cell, thereby providing further support for an intracellular mode of action of ANGPTL4 in adipocytes.
ANGPTL4 is a well-established inhibitor of LPL that mediates the reduction in LPL activity in adipose tissue during fasting (12, 36). Through this action, ANGPTL4 reduces uptake of plasma triglyceride–derived fatty acids into adipose tissue during fasting, thereby raising plasma triglyceride levels (12, 36). Genetic studies strongly support a role of ANGPTL4 in regulating LPL activity in humans (37, 38). Specifically, carriers of an inactivating variant of the ANGPTL4 gene have lower plasma triglyceride levels than noncarriers and a lower risk of coronary artery disease (37, 38). At the biochemical level, ANGPTL4 promotes the unfolding of LPL (14), leading to the dissociation of the catalytically active, dimeric form of LPL to catalytically inactive monomers (13). Previously, we showed that ANGPTL4 and LPL interact already before being secreted by the cell, at least in adipocytes, causing the intracellular degradation of LPL (17). Specifically, we observed that Angptl4−/− adipocytes secrete markedly more LPL compared with WT adipocytes. Our studies also indicated that the increase in ANGPTL4 protein during fasting diverts LPL away from secretion toward degradation, strongly suggesting that ANGPTL4 is the long sought after factor responsible for the redistribution of LPL mass in adipocytes during fasting (39). The data in the present paper suggest that induction of LPL cleavage in adipocytes by ANGPTL4 represents an intermediate stage in the degradation pathway of LPL triggered by ANGPTL4 and activated during fasting (Fig. 11). As a consequence, less LPL is available for transport to the endothelium to carry out the hydrolysis of circulating triglycerides. At the molecular level, it can be hypothesized that the ANGPTL4-induced unfolding of LPL and dissociation of LPL dimers unmasks the PCSK cleavage site at residues 321–324, thereby rendering LPL more susceptible to cleavage and subsequent degradation (13, 14, 26).
Figure 11.

Schematic model of the role of ANGPTL4 in intracellular LPL processing. ANGPTL4 causes the intracellular unfolding of LPL and dissociation of LPL dimers into monomers, rendering LPL more prone to cleavage by PCSK3. Cleaved LPL is further subject to degradation in lysosomes. This action of ANGPTL4 leads to reduced secretion of LPL and reduced delivery of LPL to the endothelial surface by GPIHBP1. Besides being subject to further intracellular degradation, the N- and C-terminal LPL cleavage fragments can also be secreted. Cleavage of LPL may also occur extracellularly (25, 26). PCSK3, proprotein convertase subtilisin/kexin type 3; GPIHBP1, glycosylphosphatidylinositol-anchored high-density lipoprotein–binding protein 1.
Besides being subject to further intracellular degradation, the N- and C-terminal LPL cleavage fragments can also be secreted (17). It is possible that C-terminal LPL is retained on the cell surface via HSPG. This is not the case for N-terminal LPL, which lacks the ability to bind HSPG. Cleavage of LPL may also occur extracellularly (25, 26) (Fig. 11).
As indicated above, the induction of PCSK-mediated LPL cleavage by ANGPTL4 occurs intracellularly, strengthening the notion that ANGPTL4 is able to act inside the cell. Preliminary evidence from our group indicates that ANGPTL4 may also influence the secretion of other proteins. There is evidence that the intracellular mode of action of ANGPTL4 extends to ANGPTL3 as well (40) and as such may provide a new mechanistic framework underlying certain functional properties of this class of proteins.
Interestingly, a previous report indicated that liver-derived ANGPTL3 also enhances the cleavage of LPL, possibly by serving as a cofactor for PCSKs (25). This action was suggested to lead to inactivation of LPL and the release of LPL from the endothelial cell surface. Because ANGPTL3 and LPL are not produced in the same cell, the effect of ANGPTL3 on PCSK-mediated LPL cleavage is likely extracellular. A previous study in the Huh7 human liver cell line hinted at a similar stimulatory effect on LPL cleavage by ANGPTL4 (21). Together, these data suggest that induction of PCSK-mediated LPL cleavage may be a common property of ANGPTL3 and ANGPTL4. An interesting twist to the story is that PCSKs not only cleave LPL but also ANGPTL4 and ANGPTL3, releasing their N-terminal domain (21, 22, 24, 41). Because the N-terminal domains of ANGPTL4 and ANGPTL3 mediate the inhibition of LPL, the cleavage of ANGPTL4 and ANGPTL3 may potentiate LPL inhibition. Altogether, these data raise the possibility that PCSKs might reduce LPL activity by two complementary cleavage mechanisms (21, 24).
We found that PCSK3 is for a large part responsible for the intracellular cleavage of LPL in mouse adipocytes. Previously, PCSK3 was already found to be the primary convertase responsible for cleavage of endothelial lipase in hepatocytes (24). In contrast to PCSK3, which localizes to the trans-Golgi and the endosomes, other PCSKs, such as PCSK5 (PC5/6) and PCSK6 (PACE4), are mainly activated and present on the cell surface, anchored to HSPGs (19). Similar to PCSK3, PCSK5 and PCSK6 are widely expressed, recognize similar target sequences, and share multiple target proteins (19). Hence, it is possible that the observed extracellular cleavage of LPL, leading to the release of cleavage products in the cell culture medium, partly reflects the action of PCSK5 and PCSK6. Indeed, in vitro transfection studies have shown that LPL can also be cleaved by PCSK5 and PCSK6 (26, 25). Interestingly, in our experiments, PCSK inhibition did not lead to an increase in full-length LPL. The reason remains unclear.
An interesting question concerns the rationale behind PCSK3-mediated cleavage of LPL. PCSK3 catalyzes the maturation of a variety of proproteins, including growth factors and pathogen-recognizing proteins (42), and is essential for proper embryonal development (43). Whereas the action of PCSK3 toward certain proproteins results in their activation, the PCSK-mediated cleavage of endothelial lipase results in the inactivation of the protein (24, 26). Similarly, it is likely that the PCSK3-mediated cleavage of LPL serves to inactivate LPL rather than to activate LPL. Consistent with this notion, cleaved LPL products that were found in human pre-heparin plasma had little LPL activity (44). Furthermore, mutations G409R and E410V in the human LPL gene that are characterized by enhanced cleavage of LPL in vitro cause a severe chylomicronemia (45). Our data support the idea that PCSK-mediated cleavage of LPL serves to inactivate LPL and indicate that N-terminal LPL is efficiently cleared by the lysosomes.
Besides being part of the degradation of LPL, it can be hypothesized that PCSK-mediated cleavage of LPL may serve to separate the N- and C-terminal fragments of LPL, which each might have specific physiological functions that complement or extend beyond LPL's capacity to hydrolyze triglycerides. For example, the C-terminal domain of LPL was previously found to mediate the margination of triglyceride-rich lipoproteins to the endothelium (46). However, it is questionable whether the C-terminal domain alone is capable of translocating to the endothelium via GPIHBP1 (47). Likewise, the separate fragments are unlikely to bind to HSPGs on the cell surface, as both monomeric LPL and C-terminal LPL have a much lower affinity for HSPGs compared with full-length dimeric LPL (48, 49). Overall, there is insufficient evidence that the N- and C-terminal fragments of LPL carry specific biological functions and thus that the cleavage of LPL serves any purpose other than LPL degradation.
In conclusion, we demonstrate that LPL is cleaved in adipocytes by PCSK3 and that this cleavage is stimulated by the LPL inhibitor ANGPTL4. We propose that the stimulation of LPL cleavage by ANGPTL4 is part of the degradation pathway of LPL in adipocytes during fasting.
Experimental procedures
All animal experiments were performed in accordance with Directive 2010/63/EU from the European Union. All animal studies were reviewed and approved by the Animal Ethics Committee of Wageningen University.
Chemicals
5-Aminoimidazole-4-carboxamide ribonucleotide, dexamethasone, isobutylmethylxanthine, insulin, rosiglitazone, and intralipid were from Sigma. dec-RVKR-CMK was from Cayman Chemicals (via Sanbio, Uden, The Netherlands).
Human adipose tissue
Human adipose tissue was obtained from the cross-sectional MONDIAL cohort (50). Briefly, the MONDIAL (markers of organ health in nondiabetic and diabetics; intestine, adipose tissue, and liver) study was a cross-sectional study in patients undergoing bariatric surgery at Rijnstate Hospital/Vitalys clinics in Arnhem, The Netherlands. In this study, tissue samples were obtained from residual biological material from patients undergoing either a primary laparoscopic Roux-en-Y gastric bypass or a primary laparoscopic gastric sleeve procedure. The study was approved by the local ethics committee of Rijnstate hospital.
Mouse studies
WT and Angptl4−/− mice on a C57Bl/6 background were used that were bred and maintained in the same facility for more than 20 generations (51). Angptl4−/− mice were generated via homologous recombination of embryonic stem cells and lack part of the Angptl4 gene, resulting in a nonfunctional ANGPTL4 protein. The Angptl4−/− mice were imported to our animal facility in 2006 as strain B6.129P2-Lp139tm1 N10 from Taconic (Germantown, NY) (kind gift of Dr. Anja Köster, Lilly). Mice were individually housed in temperature- and humidity-controlled specific pathogen–free conditions. Mice had ad libitum access to food and water. For the fasting experiment, male WT and Angptl4−/− mice between the ages of 4 and 6 months were used. Fasted mice were fasted from 15:00 h and sacrificed the next day between 9:00 and 10:00 h. Refed mice were fasted from 15:00 h, refed with chow the next day at 6:00 h, and sacrificed between 9:00 and 10:00 h. Mice were anesthetized with a mixture of isoflurane (1.5%), nitrous oxide (70%), and oxygen (30%). Blood was collected by orbital puncture into EDTA tubes. Mice were euthanized by cervical dislocation, after which tissues were excised and directly frozen in liquid nitrogen. For the tissue panel, WT mice (n = 4) were euthanized, and multiple tissues were harvested, snap-frozen in liquid nitrogen, and stored at −80 °C until further analyses.
Cell culture
3T3-L1 fibroblasts (P7–P16) were maintained in DMEM (Lonza, Verviers, Belgium) supplemented with 10% newborn calf serum (Lonza) and 1% penicillin/streptomycin (P/S) (Lonza). Two days post-confluence, cells were switched to DMEM, supplemented with 10% fetal bovine serum (FBS), 1% P/S, 0.5 mm isobutylmethylxanthine, 10 μm dexamethasone, 5 μg/ml insulin for 2 days. Subsequently, cells were maintained in DMEM supplemented with 10% FBS, 1% P/S, and 5 μg/ml insulin for 6 days and switched to DMEM with 10% FBS and 1% P/S for 3 days, after which experiments were performed as indicated in the figure legends (52). To examine the accumulation of N-terminal LPL on the cell surface, mature 3T3-L1 adipocytes were incubated with 10 IU/ml heparin for 20 min.
3T3-F442a cells (P8–P14; Sigma) were maintained in DMEM (Lonza), supplemented with 10% newborn calf serum and 1% P/S. At confluence, cells were switched to DMEM (Lonza) supplemented with 10% FBS, 1% P/S, and 5 μg/ml insulin (Sigma) to stimulate differentiation. During differentiation, medium was changed every 2–3 days. After 10 days, cells were switched back to regular medium and used for experiments 2–3 days later.
For isolation and differentiation of primary adipocytes, inguinal white adipose tissue was removed from Angptl4−/− and WT mice and placed in DMEM supplemented with 1% P/S and 1% BSA (Sigma-Aldrich) (17). Material from 2–3 mice was pooled, minced with scissors, and digested in collagenase-containing medium (DMEM with 3.2 mm CaCl2, 1.5 mg/ml collagenase type II (C6885, Sigma-Aldrich), 10% FBS, 0.5% BSA, and 15 mm HEPES) for 1 h at 37 °C. Following digestion, the cells were filtered through a 100-μm cell strainer (Falcon) to remove remaining cell clumps. The cell suspension was centrifuged at 1600 rpm for 10 min, after which the supernatant was removed, and the pellet was resuspended in erythrocyte lysis buffer (155 mm NH4Cl, 12 mm NaHCO3, 0.1 mm EDTA). Upon incubation for 2–3 min at room temperature, cells were centrifuged at 1200 rpm for 5 min, and the pelleted cells were resuspended in DMEM + 10% FCS + 1% P/S and plated. Upon confluence, the cells were differentiated according to the protocol as described above for 3T3-L1 cells, with the addition of 1 μm rosiglitazone during the initial differentiation step.
For adipose tissue explants, gonadal white adipose tissue was taken from WT and Angptl4−/− mice and placed in DMEM supplemented with 1% P/S and 1% BSA. Fat pads were minced into small pieces and divided into small mounds of adipose tissue (∼50–100 mg of tissue). Adipose explants were placed into wells containing medium (DMEM with 1% P/S and 10% FCS) and incubated as indicated in the figure legends. Next, the medium was harvested, and explant weights were determined. Explants were immediately lysed to prepare protein extracts.
siRNA-mediated knockdown
Dharmacon ON-TARGETplus SMARTpool siRNAs against mouse Pcsk3 and Angptl4 were purchased from Thermo Fisher Scientific (Landsmeer, The Netherlands). siRNAs were diluted in Dharmacon 1× siRNA buffer (final concentration 20 mm KCl, 6 mm HEPES, pH 7.5, 0.2 mm MgCl2). Transfections for Pcsk3 were performed with Lipofectamine RNAiMAX transfection reagent (Life Technologies, Bleiswijk, The Netherlands) at a concentration of 10 nm siRNA and 1.5 μl of Lipofectamine for a 24-well plate. Transfections for Angptl4 were performed at a concentration of 25 nm siRNA and 2 μl of Lipofectamine in a 24-well plate. To examine the impact of either knockdown on LPL cleavage, mature 3T3-L1 adipocytes were washed with PBS, trypsinized, and collected in DMEM. Following centrifugation at 1250 rpm for 5 min, pelleted cells were resuspended and filtered through a 70-μm cell strainer. Adipocytes were plated at 70% confluence, and 2 h later, siRNAs complexed to Lipofectamine were added. Cleavage of LPL was examined after 48 h of incubation.
Western blots
Mouse or human fat pads, adipose tissue explants, differentiated primary adipocytes, and differentiated 3T3-L1 and 3T3-F442a adipocytes were lysed in RIPA buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, and 0.1% SDS; Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors (Roche Diagnostics, Almere, The Netherlands). Following homogenization, lysates were placed on ice for 30 min and centrifuged 2–3 times at 13,000 rpm for 10 min at 4 °C to remove fat and cell debris. Concentration of protein lysates was determined by using a bicinchoninic acid assay (Thermo Fisher Scientific). Protein lysates (10–30 μg of protein/lane) or medium aliquots (10–15 μl) were loaded onto 8–16% or 10% Criterion gels (Bio-Rad, Veenendaal, The Netherlands). Next, proteins were transferred onto a polyvinylidene difluoride membrane using the Transblot Turbo System (Bio-Rad). Membranes were probed with a goat anti-mouse LPL antibody (28), mouse anti-human LPL antibodies 88b8 and 5D2 (27), a goat anti-human LPL antibody (Y-20, Santa Cruz Biotechnology), a rabbit anti-mouse HSP90 antibody (Cell Signaling Technology, catalog no. 4874), a rat anti-mouse ANGPTL4 antibody (Kairos 142-2, Adipogen), a mouse anti-mouse β-TUBULIN (sc-5274, Santa Cruz Biotechnology), or a rabbit anti-mouse PCSK3 (Abcam, ab183495) at 1:5000 (mLPL), 1:2000 (HSP90), or 1:1000 (PCSK3, ANGPTL4, hLPL, and β-TUBULIN) dilutions. Blocking and the incubation of primary and secondary antibodies were all done in TBS, pH 7.5, 0.1% Tween 20 (TBS-T), and 5% (w/v) skimmed milk. In between, membranes were washed in TBS-T. Quantification was performed with the ChemiDoc MP system (Bio-Rad) and Clarity ECL substrate (Bio-Rad). Equal loading of medium samples was verified by Coomassie Blue staining.
Glycosylation of ANGPTL4 and LPL
Glycosylation of ANGPTL4 and LPL was analyzed by Western blotting, as described above, after digestion of 10–20 μg with EndoH, PNGase, or Protein Deglycosylation Mix (New England Biolabs), according to the manufacturer's instructions.
RNA isolation and quantitative PCR
To isolate RNA, tissues or cells were homogenized using TRIzol (Thermo Fisher Scientific) in a Qiagen Tissue Lyser II (Qiagen, Venlo, The Netherlands) or by pipetting up and down. RNA was reverse-transcribed using the First Strand cDNA synthesis kit (Thermo Fisher Scientific). Quantitative PCR analyses were done on a CFX384 real-time PCR platform (Bio-Rad) with the SensiMix PCR mix from Bioline (GC Biotech, Alphen aan de Rijn, The Netherlands).
Author contributions
W. D., P. M. R., and S. K. conceptualization; W. D., P. M. R., and L. J. O. formal analysis; W. D. and S. K. supervision; W. D., P. M. R., and L. J. O. validation; W. D., P. M. R., L. J. O., and S. K. investigation; W. D., P. M. R., L. J. O., and S. K. visualization; W. D., P. M. R., and L. J. O. methodology; W. D. writing-original draft; P. M. R., L. J. O., and S. K. writing-review and editing; S. K. funding acquisition; S. K. project administration.
Acknowledgments
We kindly thank Anne Beigneux and Katsuyuki Nakajima for providing the antibodies 88b8 and 5D2 against human LPL, André Bensadoun for providing the antibody against mouse LPL, and Merel Defour for cDNA of the mouse tissue panel. We thank Sophie Schutte and Lydia Afman for the permission to use human adipose tissue material from the MONDIAL cohort.
This study was supported by Fondation Leducq Grant 12CVD04. The authors declare that they have no conflicts of interest with the contents of this article.
- LPL
- lipoprotein lipase
- mLPL
- mouse LPL
- hLPL
- human LPL
- PCSK
- proprotein convertase subtilisin/kexin
- LDL
- low-density lipoprotein
- EndoH
- endoglycosidase H
- PNGase
- peptide:N-glycosidase
- dec-CMK
- decanoyl-RVKR-chloromethyl ketone
- HSPG
- heparin sulfate proteoglycan
- ER
- endoplasmic reticulum
- DMEM
- Dulbecco's modified Eagle's medium
- P/S
- penicillin/streptomycin
- FBS
- fetal bovine serum.
References
- 1. Musunuru K., and Kathiresan S. (2016) Surprises from genetic analyses of lipid risk factors for atherosclerosis. Circ. Res. 118, 579–585 10.1161/CIRCRESAHA.115.306398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bensadoun A. (1991) Lipoprotein lipase. Annu. Rev. Nutr. 11, 217–237 10.1146/annurev.nu.11.070191.001245 [DOI] [PubMed] [Google Scholar]
- 3. Davies B. S. J., Beigneux A. P., Barnes R. H. 2nd, Tu Y., Gin P., Weinstein M. M., Nobumori C., Nyrén R., Goldberg I., Olivecrona G., Bensadoun A., Young S. G., and Fong L. G. (2010) GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 12, 42–52 10.1016/j.cmet.2010.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kersten S. (2014) Physiological regulation of lipoprotein lipase. Biochim. Biophys. Acta 1841, 919–933 10.1016/j.bbalip.2014.03.013 [DOI] [PubMed] [Google Scholar]
- 5. Dijk W., and Kersten S. (2016) Regulation of lipid metabolism by angiopoietin-like proteins. Curr. Opin. Lipidol. 27, 249–256 10.1097/MOL.0000000000000290 [DOI] [PubMed] [Google Scholar]
- 6. Wang Y., Quagliarini F., Gusarova V., Gromada J., Valenzuela D. M., Cohen J. C., and Hobbs H. H. (2013) Mice lacking ANGPTL8 (betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 110, 16109–16114 10.1073/pnas.1315292110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y., McNutt M. C., Banfi S., Levin M. G., Holland W. L., Gusarova V., Gromada J., Cohen J. C., and Hobbs H. H. (2015) Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 112, 11630–11635 10.1073/pnas.1515374112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haller J. F., Mintah I. J., Shihanian L. M., Stevis P., Buckler D., Alexa-Braun C. A., Kleiner S., Banfi S., Cohen J. C., Hobbs H. H., Yancopoulos G. D., Murphy A. J., Gusarova V., and Gromada J. (2017) ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J. Lipid Res. 58, 1166–1173 10.1194/jlr.M075689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chi X., Britt E. C., Shows H. W., Hjelmaas A. J., Shetty S. K., Cushing E. M., Li W., Dou A., Zhang R., and Davies B. S. J. (2017) ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Mol. Metab. 6, 1137–1149 10.1016/j.molmet.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Catoire M., Alex S., Paraskevopulos N., Mattijssen F., Evers-van Gogh I., Schaart G., Jeppesen J., Kneppers A., Mensink M., Voshol P. J., Olivecrona G., Tan N. S., Hesselink M. K. C., Berbée J. F., Rensen P. C. N., et al. (2014) Fatty acid-inducible ANGPTL4 governs lipid metabolic response to exercise. Proc. Natl. Acad. Sci. U.S.A. 111, E1043–E1052 10.1073/pnas.1400889111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dijk W., Heine M., Vergnes L., Boon M. R., Schaart G., Hesselink M. K. C., Reue K., van Marken Lichtenbelt W. D., Olivecrona G., Rensen P. C. N., Heeren J., and Kersten S. (2015) ANGPTL4 mediates shuttling of lipid fuel to brown adipose tissue during sustained cold exposure. Elife 4, e08428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kroupa O., Vorrsjö E., Stienstra R., Mattijssen F., Nilsson S. K., Sukonina V., Kersten S., Olivecrona G., and Olivecrona T. (2012) Linking nutritional regulation of Angptl4, Gpihbp1, and Lmf1 to lipoprotein lipase activity in rodent adipose tissue. BMC Physiol. 12, 13 10.1186/1472-6793-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sukonina V., Lookene A., Olivecrona T., and Olivecrona G. (2006) Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. U.S.A. 103, 17450–17455 10.1073/pnas.0604026103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mysling S., Kristensen K. K., Larsson M., Kovrov O., Bensadouen A., Jørgensen T. J., Olivecrona G., Young S. G., and Ploug M. (2016) The angiopoietin-like protein ANGPTL4 catalyzes unfolding of the hydrolase domain in lipoprotein lipase and the endothelial membrane proteinGPIHBP1 counteracts this unfolding. Elife 5, e20958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Makoveichuk E., Vorrsjö E., Olivecrona T., and Olivecrona G. (2013) Inactivation of lipoprotein lipase in 3T3-L1 adipocytes by angiopoietin-like protein 4 requires that both proteins have reached the cell surface. Biochem. Biophys. Res. Commun. 441, 941–946 10.1016/j.bbrc.2013.11.013 [DOI] [PubMed] [Google Scholar]
- 16. Desai U., Lee E.-C., Chung K., Gao C., Gay J., Key B., Hansen G., Machajewski D., Platt K. A., Sands A. T., Schneider M., Van Sligtenhorst I., Suwanichkul A., Vogel P., Wilganowski N., Wingert J., et al. (2007) Lipid-lowering effects of anti-angiopoietin-like 4 antibody recapitulate the lipid phenotype found in angiopoietin-like 4 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11766–11771 10.1073/pnas.0705041104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dijk W., Beigneux A. P., Larsson M., Bensadoun A., Young S. G., and Kersten S. (2016) Angiopoietin-like 4 (ANGPTL4) promotes intracellular degradation of lipoprotein lipase in adipocytes. J. Lipid Res. 57, 1670–1683 10.1194/jlr.M067363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Turpeinen H., Ortutay Z., and Pesu M. (2013) Genetics of the first seven proprotein convertase enzymes in health and disease. Curr. Genomics 14, 453–467 10.2174/1389202911314050010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seidah N. G., Sadr M. S., Chrétien M., and Mbikay M. (2013) The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 288, 21473–21481 10.1074/jbc.R113.481549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lagace T. A. (2014) PCSK9 and LDLR degradation. Curr. Opin. Lipidol. 25, 387–93 10.1097/MOL.0000000000000114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lei X., Shi F., Basu D., Huq A., Routhier S., Day R., and Jin W. (2011) Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. J. Biol. Chem. 286, 15747–15756 10.1074/jbc.M110.217638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mandard S., Zandbergen F., Tan N. S., Escher P., Patsouris D., Koenig W., Kleemann R., Bakker A., Veenman F., Wahli W., Müller M., and Kersten S. (2004) The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 279, 34411–34420 10.1074/jbc.M403058200 [DOI] [PubMed] [Google Scholar]
- 23. Chomel C., Cazes A., Faye C., Bignon M., Gomez E., Ardidie-Robouant C., Barret A., Ricard-Blum S., Muller L., Germain S., and Monnot C. (2009) Interaction of the coiled-coil domain with glycosaminoglycans protects angiopoietin-like 4 from proteolysis and regulates its antiangiogenic activity. FASEB J. 23, 940–949 10.1096/fj.08-115170 [DOI] [PubMed] [Google Scholar]
- 24. Essalmani R., Susan-Resiga D., Chamberland A., Asselin M.-C., Canuel M., Constam D., Creemers J. W., Day R., Gauthier D., Prat A., and Seidah N. G. (2013) Furin is the primary in vivo convertase of angiopoietin-like 3 and endothelial lipase in hepatocytes. J. Biol. Chem. 288, 26410–26418 10.1074/jbc.M113.501304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu J., Afroza H., Rader D. J., and Jin W. (2010) Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J. Biol. Chem. 285, 27561–27570 10.1074/jbc.M110.144279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin W., Fuki I. V., Seidah N. G., Benjannet S., Glick J. M., and Rader D. J. (2005) Proprotein convertases [corrected] are responsible for proteolysis and inactivation of endothelial lipase. J. Biol. Chem. 280, 36551–36559 10.1074/jbc.M502264200 [DOI] [PubMed] [Google Scholar]
- 27. Allan C. M., Larsson M., Hu X., He C., Jung R. S., Mapar A., Voss C., Miyashita K., Machida T., Murakami M., Nakajima K., Bensadoun A., Ploug M., Fong L. G., Young S. G., and Beigneux A. P. (2016) An LPL-specific monoclonal antibody, 88B8, that abolishes the binding of LPL to GPIHBP1. J. Lipid Res. 57, 1889–1898 10.1194/jlr.M070813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weinstein M. M., Yin L., Beigneux A. P., Davies B. S. J., Gin P., Estrada K., Melford K., Bishop J. R., Esko J. D., Dallinga-Thie G. M., Fong L. G., Bensadoun A., and Young S. G. (2008) Abnormal patterns of lipoprotein lipase release into the plasma in GPIHBP1-deficient mice. J. Biol. Chem. 283, 34511–34518 10.1074/jbc.M806067200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Davis R. C., Wong H., Nikazy J., Wang K., Han Q., and Schotz M. C. (1992) Chimeras of hepatic lipase and lipoprotein lipase: domain localization of enzyme-specific properties. J. Biol. Chem. 267, 21499–21504 [PubMed] [Google Scholar]
- 30. Ben-Zeev O., Stahnke G., Liu G., Davis R. C., and Doolittle M. H. (1994) Lipoprotein lipase and hepatic lipase: the role of asparagine-linked glycosylation in the expression of a functional enzyme. J. Lipid Res. 35, 1511–1523 [PubMed] [Google Scholar]
- 31. Chajek-Shaul T., Friedman G., Knobler H., Stein O., Etienne J., and Stein Y. (1985) Importance of the different steps of glycosylation for the activity and secretion of lipoprotein lipase in rat preadipocytes studied with monensin and tunicamycin. Biochim. Biophys. Acta 837, 123–134 10.1016/0005-2760(85)90235-8 [DOI] [PubMed] [Google Scholar]
- 32. Garten W., Hallenberger S., Ortmann D., Schäfer W., Vey M., Angliker H., Shaw E., and Klenk H. D. (1994) Processing of viral glycoproteins by the subtilisin-like endoprotease furin and its inhibition by specific peptidylchloroalkylketones. Biochimie 76, 217–225 10.1016/0300-9084(94)90149-X [DOI] [PubMed] [Google Scholar]
- 33. Lutz E. P., Merkel M., Kako Y., Melford K., Radner H., Breslow J. L., Bensadoun A., and Goldberg I. J. (2001) Heparin-binding defective lipoprotein lipase is unstable and causes abnormalities in lipid delivery to tissues. J. Clin. Invest. 107, 1183–1192 10.1172/JCI11774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cisar L. A., Hoogewerf A. J., Cupp M., Rapport C. A., and Bensadoun A. (1989) Secretion and degradation of lipoprotein lipase in cultured adipocytes: binding of lipoprotein lipase to membrane heparan sulfate proteoglycans is necessary for degradation. J. Biol. Chem. 264, 1767–1774 [PubMed] [Google Scholar]
- 35. Kersten S., Mandard S., Tan N. S., Escher P., Metzger D., Chambon P., Gonzalez F. J., Desvergne B., and Wahli W. (2000) Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 275, 28488–28493 10.1074/jbc.M004029200 [DOI] [PubMed] [Google Scholar]
- 36. Cushing E. M., Chi X., Sylvers K. L., Shetty S. K., Potthoff M. J., and Davies B. S. J. (2017) Angiopoietin-like 4 directs uptake of dietary fat away from adipose during fasting. Mol. Metab. 6, 809–818 10.1016/j.molmet.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dewey F. E., Gusarova V., O'Dushlaine C., Gottesman O., Trejos J., Hunt C., Van Hout C. V., Habegger L., Buckler D., Lai K.-M. V., Leader J. B., Murray M. F., Ritchie M. D., Kirchner H. L., Ledbetter D. H., et al. (2016) Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med. 374, 1123–1133 10.1056/NEJMoa1510926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators, Stitziel N. O., Stirrups K. E., Masca N. G. D., Erdmann J., Ferrario P. G., König I. R., Weeke P. E., Webb T. R., Auer P. L., Schick U. M., Lu Y., Zhang H., Dube M.-P., Goel A., et al. (2016) Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N. Engl. J. Med. 374, 1134–1144 10.1056/NEJMoa1507652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doolittle M. H., Ben-Zeev O., Elovson J., Martin D., and Kirchgessner T. G. (1990) The response of lipoprotein lipase to feeding and fasting: evidence for posttranslational regulation. J. Biol. Chem. 265, 4570–7 [PubMed] [Google Scholar]
- 40. Xu Y.-X., Redon V., Yu H., Querbes W., Pirruccello J., Liebow A., Deik A., Trindade K., Wang X., Musunuru K., Clish C. B., Cowan C., Fizgerald K., Rader D., and Kathiresan S. (2018) Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis 268, 196–206 10.1016/j.atherosclerosis.2017.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ono M., Shimizugawa T., Shimamura M., Yoshida K., Noji-Sakikawa C., Ando Y., Koishi R., and Furukawa H. (2003) Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. J. Biol. Chem. 278, 41804–41809 10.1074/jbc.M302861200 [DOI] [PubMed] [Google Scholar]
- 42. Molloy S. S., Anderson E. D., Jean F., and Thomas G. (1999) Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 9, 28–35 10.1016/S0962-8924(98)01382-8 [DOI] [PubMed] [Google Scholar]
- 43. Roebroek A. J., Umans L., Pauli I. G., Robertson E. J., van Leuven F., Van de Ven W. J., and Constam D. B. (1998) Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase Furin. Development 125, 4863–4876 [DOI] [PubMed] [Google Scholar]
- 44. Kern P. A., Martin R. A., Carty J., Goldberg I. J., and Ong J. M. (1990) Identification of lipoprotein lipase immunoreactive protein in pre- and postheparin plasma from normal subjects and patients with type I hyperlipoproteinemia. J. Lipid Res. 31, 17–26 [PubMed] [Google Scholar]
- 45. Gin P., Goulbourne C. N., Adeyo O., Beigneux A. P., Davies B. S. J., Tat S., Voss C. V., Bensadoun A., Fong L. G., and Young S. G. (2012) Chylomicronemia mutations yield new insights into interactions between lipoprotein lipase and GPIHBP1. Hum. Mol. Genet. 21, 2961–2972 10.1093/hmg/dds127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goulbourne C. N., Gin P., Tatar A., Nobumori C., Hoenger A., Jiang H., Grovenor C. R. M., Adeyo O., Esko J. D., Goldberg I. J., Reue K., Tontonoz P., Bensadoun A., Beigneux A. P., Young S. G., and Fong L. G. (2014) The GPIHBP1-LPL complex is responsible for the margination of triglyceride-rich lipoproteins in capillaries. Cell Metab. 19, 849–860 10.1016/j.cmet.2014.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mysling S., Kristensen K. K., Larsson M., Beigneux A. P., Gårdsvoll H., Fong L. G., Bensadouen A., Jørgensen T. J., Young S. G., and Ploug M. (2016) The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. Elife 5, e12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lookene A., Nielsen M. S., Gliemann J., and Olivecrona G. (2000) Contribution of the carboxy-terminal domain of lipoprotein lipase to interaction with heparin and lipoproteins. Biochem. Biophys. Res. Commun. 271, 15–21 10.1006/bbrc.2000.2530 [DOI] [PubMed] [Google Scholar]
- 49. Lookene A., Chevreuil O., Ostergaard P., and Olivecrona G. (1996) Interaction of lipoprotein lipase with heparin fragments and with heparan sulfate: stoichiometry, stabilization, and kinetics. Biochemistry 35, 12155–12163 10.1021/bi960008e [DOI] [PubMed] [Google Scholar]
- 50. Dijk W., Schutte S., Aarts E. O., Janssen I. M. C., Afman L., and Kersten S. (2018) Regulation of angiopoietin-like 4 and lipoprotein lipase in human adipose tissue. J. Clin. Lipidol. 12, 773–783 10.1016/j.jacl.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 51. Köster A., Chao Y. B., Mosior M., Ford A., Gonzalez-DeWhitt P. A., Hale J. E., Li D., Qiu Y., Fraser C. C., Yang D. D., Heuer J. G., Jaskunas S. R., and Eacho P. (2005) Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology 146, 4943–4950 10.1210/en.2005-0476 [DOI] [PubMed] [Google Scholar]
- 52. Alex S., Lange K., Amolo T., Grinstead J. S., Haakonsson A. K., Szalowska E., Koppen A., Mudde K., Haenen D., Al-Lahham S., Roelofsen H., Houtman R., van der Burg B., Mandrup S., Bonvin A. M. J. J., et al. (2013) Short-chain fatty acids stimulate angiopoietin-like 4 synthesis in human colon adenocarcinoma cells by activating peroxisome proliferator-activated receptor γ. Mol. Cell. Biol. 33, 1303–1316 10.1128/MCB.00858-12 [DOI] [PMC free article] [PubMed] [Google Scholar]