

Abstract
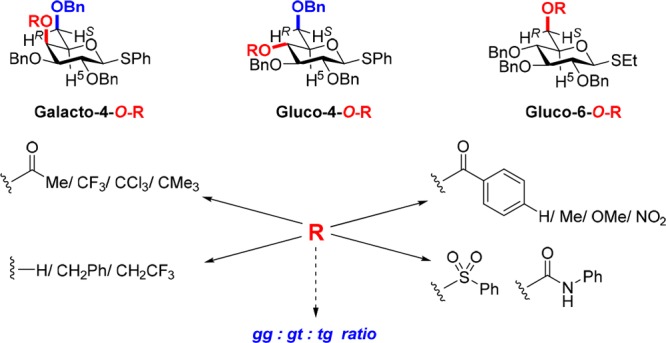
The synthesis and conformational analysis of a series of phenyl 2,3,6-tri-O-benzyl-β-d-thio galacto- and glucopyranosides and their 6S-deuterio isotopomers, with systematic variation of the protecting group at the 4-position, are described. For the galactopyranosides, replacement of a 4-O-benzyl ether by a 4-O-alkanoyl or aroyl ester results in a small but measurable shift in side chain population away from the trans,gauche conformation and in favor of the gauche,trans conformer. In the glucopyranoside series on the other hand, replacement of a 4-O-benzyl ether by a 4-O-alkanoyl or aroyl ester results in a small but measurable increase in the population of the trans,gauche conformer at the expense of the gauche,gauche conformer. The possible modulating effect of these conformational changes on the well-known changes in the anomeric reactivity of glycosyl donors as a function of protecting group is discussed, raising the possibility that larger changes may be observed at the transition state for glycosylation. A comparable study with a series of ethyl 2,3,4-tri-O-benzyl-β-d-thioglucopyranosides reveals that no significant influence in side chain population is observed on changing the O6 protecting group.
Introduction
In carbohydrate chemistry, it is widely understood that anomeric reactivity is strongly influenced by the relative configuration of the complete set of stereogenic centers in the backbone.1 Thus, for example, galactopyranosides undergo both acid-catalyzed and spontaneous hydrolysis more rapidly than their gluco isomers (Figure 1);2−5 the same pattern of reactivity is found in glycosylation reactions with a series of comparably protected thioglycosides.6 The influence of protecting groups on the anomeric reactivity of glycosyl donors also is broadly appreciated, with the more electron-withdrawing (or disarming) esters retarding reaction rates compared to the less electron-withdrawing (or arming) ethers.6−11
Figure 1.

Relative rates of spontaneous hydrolysis of galacto- and glucopyranosides in water at 37 °C.5
Ring conformation is another important factor in anomeric reactivity. Thus, for any given configuration, ring conformations that maximize the number of axial (or pseudoaxial) C–O bonds generally exhibit the greatest anomeric reactivity (Figure 2).12−15 The influence of configuration and ring conformation on anomeric reactivity is best explained by the ability of axial C–O bonds to stabilize nascent positive charge at the anomeric center as compared to their equatorial counterparts.16−19
Figure 2.

Influence of ring conformation on the hydrolysis of axial methyl glucosides in 2 M HCl at 60 °C.4,15
The conformation of the side chain, defined as gauche,gauche (gg), gauche,trans (gt), or trans,gauche (tg) where the first and second terms refer to the position of O6 relative to O5 and C4, respectively,20−22 is also increasingly recognized as influencing anomeric reactivity. Thus, the trans,gauche conformation exerts maximum retardation due to the strongly electron-withdrawing antiperiplanar relationship of the C6–O6 and the C5–O5 bonds (Figure 3).23−25 The interplay between the conformation of the side chain and the glycosidic bond is further apparent from the work of Vázquez and co-workers in which it is demonstrated by CD and NMR methods that both the anomeric configuration and the nature of the aglycone influence the population of the different side chain conformers.26−30
Figure 3.

Influence of the gauche,gauche (gg), gauche,trans (gt), and trans,gauche (tg) side chain conformations on the relative rates of spontaneous hydrolysis of 2,4-dinitrophenyl glycosides in water at 37 °C.23,24
In this Article, we begin to explore the possibility that protecting groups, in addition to their well-known influence on glycoside reactivity as a function of their electron-withdrawing ability,1,7−11 also exert an indirect influence on anomeric reactivity by modulating the conformation of the side chain. To this end, we describe the preparation of a series of galacto and gluco thiopyranosides and, to facilitate spectral assignment, their 6S-deuterio isotopomers and study the conformation of the side chain as a function of protecting group at either the 4- or the 6-position. We show that the side chain population in a series of phenyl 2,3,6-tri-O-benzyl-β-d-thiogalacto- and glycopyranosides does indeed vary in a systematic manner on changing the functionality at the 4-position from a hydroxyl group to an ether and to an ester, albeit in a different manner in the two configurations. While these protecting-group-induced changes in conformation are small, they open the possibility that larger changes might arise at the transition state for glycosylation and thus open alternative avenues for the explanation of remote protecting group effects.
Results and Discussion
Experimental Design and Synthesis
In this investigation,
we focus on the interplay between the protecting groups at O4 and
O6 in the gluco- and galactopyranosides as the strongest candidates
for observation of any changes in side chain conformation due to these
interactions. Judging that the interaction in question could be probed
through the variation of the O4 protecting group in the presence of
a fixed O6 protecting group, or the inverse, we prepared phenyl 2,3,6-tri-O-benzyl-β-d-thiogalactopyranoside (1),31 phenyl 2,3,6-tri-O-benzyl-β-d-thioglucopyranoside (2),32 and ethyl 2,3,4-tri-O-benzyl-β-d-thioglucopyranoside (3)33 by standard means. The use of the ethyl thioglycoside
in 3 as opposed to the phenyl thioglycosides 1 and 2 was a matter of experimental convenience and
was not expected to affect the analysis of side chain conformations,
as is borne out by the subsequent results.
As the rigorous assignment of the diastereotopic pro-R and pro-S hydrogens at the 6-position of 1–3 and their derivatives is critical to the correct conformational analysis of their side chains,20 we also prepared 6S-deuterio 1–3 as outlined in Schemes 1 and 2. Thus, preferring a longer but unambiguous route based on exoselective quenching of radicals at the 6-position of 1,6-anhydropyranoses34−38 over shorter routes involving asymmetric reduction of 6-aldehydo-sugars,39−41 2,3,4-tri-O-acetyl-1,6-anhydro-d-galactose (4) was subjected to white-light-mediated bromination with N-bromosuccinimide to give the exo-bromide 5 in good yield. This transformation follows the literature description36 with the exception that the original solvent, tetrachloromethane, was replaced by the more environmentally friendly α,α,α-trifluoromethylbenzene42 as described previously for the gluco series.43 Reductive debromination with tributyltin deuteride, prepared according to Neumann,44 then gave the 6S-deuterio anhydrogalactose (6) in 77% yield. A series of standard transformations were then applied to convert 6 via intermediates 7–10 to the desired 6S-d-1 uneventfully (Scheme 1). The 6S-deuterio analogue of 2 was prepared by inversion of 6S-d-1 by triflate formation, displacement with sodium benzoate, and saponification as reported in detail in the Experimental Section.
Scheme 1. Synthesis of Phenyl 2,3,6-Tri-O-benzyl-6S-deuterio-β-d-thiogalactopyranoside (6S-d-1).

Scheme 2. Synthesis of Ethyl 2,3,4-Tri-O-benzyl-6S-deuterio-β-d-thioglucopyranoside (6S-d-3).

The 6S-deuterio-1,6-anhydroglucose derivative (11)34,35,43 was the starting material for the preparation of 6S-d-3. Thus, 11 was converted to the thioglycoside 12 by cleavage of the 1,6-anhydro bridge with trimethylsilyl ethanethiol in the presence of zinc iodide45 followed by acetylation (Scheme 2). A series of standard reactions were then employed to convert 12 to the tribenzyl ether 6S-d-3 as described previously for the nondeuterated isotopomer33 (Scheme 2).
The thiogalactoside 1 and its 6S-monodeuterio isotopomer were converted to a series of esters 13–20 at the 4-position as well as to the benzyl ether 21 by standard methods as described in the Experimental Section. The 4-O-(2,2,2-trifluoroethyl) ether (22) and its 6S-deuterio analogue (6S-d-22) were obtained from 2 and 6S-deuterio 2 by triflation followed by displacement with sodium trifluoroethoxide in DMF (Scheme 3). The thioglucoside 2 and its 6S-monodeuterio isomer were also converted to the derivatives 23–27 by standard means, as described in the Experimental Section. Similarly, the thioglucoside 3 and its 6S-monodeuterio isotopomer were converted to the 6-O-esters 28–34, the 6-O-carbamate 35, and the 6-O-benzyl ether 36 by standard means, as described in the Experimental Section.
Scheme 3. Synthesis of Phenyl 2,3,6-Tri-O-benzyl-4-O-(2,2,2-trifluoroethyl)-β-d-thiogalactopyranoside (22) and Its 6S-Deuterio Analogue (6S-d-22).

Measurement of NMR Spectra, Influence of Solvent, and Estimation of Errors in Coupling Constants
The 1H NMR spectra
of 1–3 and 13–36 were recorded in CDCl3 and deuteriobenzene and
fully assigned by the usual array of 1D- and 2D-NMR methods, with
the distinction between the 6-pro-R and pro-S resonances, herein after H6R and H6S, made on the basis of comparison
with the selectively 6S-deuterated analogues. All
first order couplings were analyzed directly. For second order spectra,
including those complicated by the presence of virtual coupling, the
spin simulation tool in the MestReNova 9.0 suite of programs was used
to extract first order coupling constants. The chemical shifts and
so-obtained 3J coupling constants in the
H5, H6R, and H6S spin systems are presented in Table 1 for the 4-O-substituted
galacto series of compounds, in Table 2 for the corresponding 4-O-substituted
gluco compounds, and in Table 3 for the 6-O-substituted gluco series of
compounds. For ease of comparison, in each of Tables 1–3, the alcohols
are listed first, followed by the ethers, and then the esters grouped
according to patterns in the side chain conformations.
Table 1. Pertinent 1H Chemical Shifts, 3J Coupling Constants, and Side Chain Populations for 1 and 13–22 in CDCl3 and C6D6.
| chem
shifta (ppm) |
3J5,6a (Hz) |
populationa (%) |
||||||
|---|---|---|---|---|---|---|---|---|
| 4-O-Subs | H6R | H6S | J5,6R | J5,6S | gg | gt | tg | |
| 1 | H | 3.78 (3.72) | 3.81 (3.75) | 5.8 (6.1) | 5.7 (5.8) | 25.6 (21.9) | 31.8 (34.4) | 42.6 (43.7) |
| 21 | PhCH2 | 3.65 (3.57) | 3.67 (3.66) | nd (5.7) | nd (7.4) | nd (13.2) | nd (22.6) | nd (64.2) |
| 22 | CF3CH2 | 3.69 (3.55) | 3.74 (3.68) | 5.5 (5.7) | 7.7 (7.6) | 12.8 (11.7) | 19.1 (21.7) | 68.0 (66.7) |
| 13 | Ac | 3.64 (3.47) | 3.55 (3.42) | 6.0 (6.1) | 6.7 (6.5) | 15.8 (16.4) | 29.0 (31.0) | 55.2 (52.6) |
| 14 | Me3CCO | 3.62 (3.49) | 3.49 (3.42) | 6.1 (6.0) | 6.7 (6.8) | 14.8 (15.0) | 30.1 (28.6) | 55.1 (56.4) |
| 17 | PhCO | 3.69 (3.53) | 3.59 (3.49) | 5.8 (6.1) | 6.8 (6.6) | 17.0 (15.6) | 26.5 (30.5) | 56.5 (53.9) |
| 18 | p-MeC6H4CO | 3.68 (3.56) | 3.57 (3.52) | 5.9 (6.2) | 6.8 (6.5) | 16.0 (15.4) | 27.5 (32.0) | 56.5 (52.5) |
| 19 | p-MeOC6H4CO | 3.69 (3.57) | 3.59 (3.54) | 6.1 (6.2) | 6.5 (6.4) | 16.4 (16.2) | 31.0 (32.5) | 52.6 (51.3) |
| 20 | p-O2NC6H4CO | 3.69 (3.47) | 3.56 (3.42) | 5.7 (5.6) | 7.2 (7.0) | 14.8 (17.3) | 23.6 (23.5) | 61.6 (59.1) |
| 15 | CF3CO | 3.67 (3.39) | 3.49 (3.36) | 5.6 (5.7) | 8.3 (8.1) | 7.1 (7.7) | 17.3 (19.2) | 75.6 (73.0) |
| 16 | Cl3CCO | 3.71 (3.43) | 3.59 (3.46) | 5.7 (nd) | 8.2 (nd) | 6.9 (nd) | 18.8 (nd) | 74.3 (nd) |
Chemical shift, coupling constants, and populations in CDCl3 (chemical shift, coupling constants, and populations in C6D6).
Table 2. Pertinent 1H Chemical Shifts, 3J Coupling Constants, and Side Chain Populations for 2 and 23–27 in CDCl3 and C6D6.
| chem
shifta (ppm) |
3J5,6a (Hz) |
populationa (%) |
||||||
|---|---|---|---|---|---|---|---|---|
| 4-O-Subs | H6R | H6S | J5,6R | J5,6S | gg | gt | tg | |
| 2 | H | 3.76 (3.60) | 3.80 (3.64) | 5.3 (5.3) | 4.1 (3.7) | 43.1 (46.2) | 34.5 (36.4) | 22.4 (17.4) |
| 27 | PhCH2 | 3.73 (3.63) | 3.80 (3.63) | 4.8 (nd) | 1.9 (nd) | 65.2 (nd) | 40.0 (nd) | –5.3 (nd) |
| 23 | Ac | 3.58 (3.54) | 3.58 (3.58) | nd (5.8) | nd (3.2) | nd (45.3) | nd (43.9) | nd (10.8) |
| 25 | PhCO | 3.65 (3.58) | 3.65 (3.62) | nd (6.0) | nd (2.9) | nd (45.7) | nd (47.3) | nd (7.0) |
| 24 | CF3CO | 3.58 (3.30) | 3.61 (3.37) | 4.7 (4.3) | 3.6 (3.5) | 52.9 (57.6) | 30.8 (27.2) | 16.3 (15.2) |
| 26 | PhSO2 | 3.51 (3.69) | 3.71 (3.81) | 5.6 (5.3) | 2.1 (2.0) | 55.9 (59.6) | 47.1 (44.6) | –3.0 (−4.2) |
Chemical shift, coupling constants, and populations in CDCl3 (chemical shift, coupling constants, and populations in C6D6).
Table 3. Pertinent 1H Chemical Shifts and 3J Coupling Constants for 3 and 28–36 in CDCl3 and C6D6.
| chem
shifta (ppm) |
3J5,6a (Hz) |
populationa (%) |
||||||
|---|---|---|---|---|---|---|---|---|
| 6-O-Subs | H6R | H6S | J5,6R | J5,6S | gg | gt | tg | |
| 3 | H | 3.71 (3.58) | 3.87 (3.69) | 4.8 (nd) | 2.7 (nd) | 59.0 (nd) | 36.1 (nd) | 4.9 (nd) |
| 36 | PhCH2 | 3.69 (3.61) | 3.76 (3.61) | 5.0 (nd) | 1.9 (nd) | 63.3 (nd) | 42.0 (nd) | –5.3 (nd) |
| 28 | Ac | 4.20 (4.26) | 4.33 (4.43) | 5.4 (5.4) | 2.4 (2.2) | 55.5 (57.0) | 43.7 (44.6) | 0.9 (−1.7) |
| 29 | Me3CCO | 4.12 (4.23) | 4.44 (4.55) | 5.6 (5.7) | 1.8 (2.1) | 58.2 (54.9) | 48.6 (48.2) | –6.8 (−3.0) |
| 31 | PhCO | 4.46 (4.51) | 4.65 (4.65) | nd (5.6) | nd (2.3) | nd (54.3) | nd (46.2) | nd (−0.5) |
| 32 | p-MeC6H4CO | 4.40 (4.68) | 4.59 (4.54) | 5.5 (5.7) | 2.2 (2.2) | 56.1 (54.1) | 45.6 (47.7) | –1.7 (−1.8) |
| 33 | p-MeOC6H4CO | 4.58 (4.56) | 4.41 (4.69) | 5.5 (5.6) | 2.3 (2.2) | 55.3 (55.1) | 45.2 (46.7) | –0.4 (−1.7) |
| 34 | p-O2NC6H4CO | 4.44 (4.40) | 4.62 (4.56) | 5.4 (5.6) | 2.2 (2.3) | 57.0 (54.3) | 44.6 (46.2) | –1.7 (−0.5) |
| 30 | CF3CO | 4.32 (4.05) | 4.54 (4.24) | 6.3 (6.3) | 2.1 (2.2) | 49.0 (48.2) | 54.2 (53.8) | –3.3 (−2.0) |
| 35 | PhNHCO | 4.37 (4.46) | 4.40 (4.43) | nd (4.6) | nd (2.5) | nd (62.5) | nd (35.1) | nd (2.4) |
Chemical shift, coupling constants, and populations in CDCl3 (chemical shift, coupling constants, and populations in C6D6).
Inspection of the coupling constant data in Tables 1–3 reveals no systematic difference in coupling constants for any given system on changing from CDCl3 to C6D6 consistent with an earlier study with rigid bicyclic models of galacto- and glucopyranosides,46 from which we conclude that the side chain conformation is unaffected by the nature of the solvent (CDCl3 or C6D6). It follows that data acquired in one of the two solvents can be extrapolated to the other solvent in cases for which resolution was insufficient to enable the determination of coupling constants in both solvents. Small nonsystematic differences between solvents are found and permit the estimation of errors in the 3J5,6R and 3J5,6S coupling constants. For the 18 compounds (and hence 36 sets of coupling constants) in which both values could be measured in both solvents, differences in a given coupling constant are 0.4 Hz or less consistent with the digital resolution of 0.38 Hz, leading us to adopt 0.4 Hz as the error limit in these measurements. The differences in the 3J5,6R and 3J5,6S coupling constants of the phenyl and ethyl glycosides of 2,3,4,6-tetra-O-benzyl-β-d-thioglucopyranoside (27 and 36) (Tables 2 and 3, respectively) are less than the error limit and so substantiate the use of the different aglycones in the two series.
Calculation of Side Chain Populations and Estimation of Errors
With the aid of limiting 3JR,gg–3JS,tg coupling constants for the pure gg, gt, and tg conformers (Table 4), determined using a series of rigid bicyclic models,46 the side chain populations (fgg–ftg) of all compounds were determined with the aid of eqs 1–3 in the usual manner and reported in Tables 1–3.47−49 Further inspection of Tables 1–3 reveals that an error of 0.4 Hz in one of the coupling constants results in a maximal change of 5% in the population of any given conformer. Therefore, in the discussion that follows, we adopt 5% as the error limit for any given conformer. In two series of compounds (Tables 2 and 3), small negative populations of the tg conformer are computed for some derivatives, which have no physical significance. In view of the 5% error, these negative populations are either indistinguishable from zero or so close to it as not to warrant further discussion.
| 1 |
| 2 |
| 3 |
Table 4. Limiting Coupling Constants for the Pure gg, gt, and tg Staggered Conformers.
|
3JH5,H6R |
3JH5,H6S |
||||
|---|---|---|---|---|---|
| JR,gg | JR,gt | JR,tg | JS,gg | JS,gt | JS,tg |
| 1.0 | 11.0 | 4.8 | 2.2 | 2.5 | 10.2 |
Corrections for the Influence of Electronegative Groups on the Magnitude of Coupling Constants
The derivation of side chain populations from experimental NMR vicinal coupling constants requires that the magnitude of the coupling constants not be affected by differences in electronegativity of the substituents across the series. It is well-known that vicinal coupling constants are reduced by the presence of electronegative substituents in the coupled system,50−53 but more subtle differences on replacement of ether groups by esters are less appreciated.51,54,55 Consideration of the 3J3,4 coupling constants in the series of gluco- and galactopyranoside derivatives in Figure 4 indicates that replacement of a single ether group in a vicinal diether by an ester results in an increase of approximately 0.5 Hz in the coupling constant, whether the coupled spins have a fixed trans or gauche relationship irrespective of the solvent, CDCl3 or C6D6. However, it is known that in freely rotating systems that more closely approximate the C5–C6 bond in the 6-O-substituted series 3 and 28–36 the difference in coupling constants on replacing an ether by an ester substituent is only 0.1 Hz.54 As this is within the error limit, no correction for the change in substituents is required for the coupling constants in Table 3.
Figure 4.

Differing influences of ether and ester substituents on vicinal coupling constants.
The influence of replacing an ether substituent by an ester in the β-position to the coupled spins is expected to be smaller, as is borne out by the constant magnitude of the 3J2,3 coupling constants in the gluco- and galactopyranosides of Figure 4 regardless of the substituent at the 4-position. The 3J5,6 coupling constants in the galactopyranosides 1 and 13–22 (Table 1) and the glucopyranosides 2 and 23–27 (Table 2) therefore do not require correction for the nature of the substituent at the 4-position.
Comparison of the 3J2,3 coupling constants in the galactopyranosides reveals them to be ∼0.4 Hz larger than the corresponding coupling constants in the glucopyranosides (Figure 4), as we have noted previously in a series of rigid bicyclic models.46 This is a manifestation of the Altona and Haasnoot β-effect wherein the vicinal coupling constant between a pair of coupled antiperiplanar spins is increased by approximately 0.5 Hz when one of the coupled spins is also antiperiplanar to an electron-withdrawing substituent at the β-position.56 A comparable relationship exists between H6R, H5, and O4 in the gt conformer and between H6S, H5, and O4 in the tg conformer of the galactopyranosides (Figure 5) but not in the glucopyranosides, albeit in a series of rigid bicyclic models, no significant difference was found in the H5–H6 coupling constants between the galacto and gluco configurations,46 suggesting that the effect does not extend to this spin system. Moreover, as the magnitude of the β-effect is not influenced by the switch from an ether to an ester (Figure 4), any residual influence will be of a systematic nature and affect all derivatives to a similar extent. The result of any small systematic β-effect simply will be to overestimate the population of the gt and tg conformers in the galactopyranoside series (Table 1) and underestimate that of the gg conformer correspondingly, with respect to the glucopyranosides (Table 2).
Figure 5.

Vicinal coupling constants subject to the Altona–Haasnoot β-effect.
In the final analysis, no corrections to the diagnostic coupling constants used for conformational analysis of the side chain arising from changes in electronegativity of the substituents or the Altona and Haasnoot β-effect were deemed necessary.
Influence of Substituents at the Galactopyranose 4-Position
Comparison of alcohol 1 with ethers 21 and 22 in Table 1 reveals that, while the two ethers have the same population distribution for the side chain given the 5% error, converting the 4-hydroxy group to a benzyl or trifluoroethyl ether has a significant influence on the side chain conformation. Thus, the side chain population of the two ethers of 21 and 22 consists of ∼13% of the gg conformer, ∼21% of the gt conformer, and ∼66% of the tg conformer, whereas the alcohol 1 has a much greater population of the gg conformer (∼24%) and a greater population of the gt conformer (∼33%), which are balanced by a significantly lower occupancy of the tg conformer (∼43%). These differences reflect either the destabilizing influence of the steric bulk at the 4-position on the gg conformer or the stabilization of the gg conformer in the alcohol 1 by a favorable intramolecular hydrogen bond to O6. Because of the differences in conformation between the alcohol 1 and the ethers 21 and 22 and because glycosyl donors typically do not have unprotected hydroxy groups, we retain the benzyl ether 21 as the standard for further comparisons.
Esterification of 1 gives a series of 4-O-esters, alkanoyl 13 and 14, or aroyl 17–20, that all adopt the same side chain population, but one that differs significantly from that of the benzyl ether 21. With similar proportions of the gg conformer in the ether 21 and the esters 13, 14, and 17–20, the change in the overall side chain population can be described as one of an approximately 10% decrease in the population of the tg conformer in favor of the gt conformer on going from the ethers to the esters. Installation of the more electron-withdrawing trifluoroacetyl and trichloroacetyl groups affords a separate set of two esters 15 and 16, respectively, whose side chain populations exhibit a pronounced shift away from the gg conformer toward the tg conformer. This change in population is also accompanied by a reduction in the population of the gt conformer with respect to the more standard alkanoyl and aroyl esters such that the tg conformer dominates the equilibrium and accounts for ∼74% of the population. Although the effect is smaller, the p-nitrobenzoate 20 exhibits a shift in side chain population away from that of the more electron-rich benzoates 17–19 in the same direction as that observed with the trifluoro- and trichloroacetates 15 and 16, suggesting that this change is a function of the electron-withdrawing nature of these esters.
Influence of Substituents at the Glucopyranose 4-Position
In the glucopyranose series, there is also a significant change in the side chain conformation when the alcohol 2 is converted to the benzyl ether 27. Specifically, benzylation results in a drop in the population of the tg conformer that is compensated by an increase in the population of the gg conformer and a smaller one in that of the gt conformer (Table 2). This effect parallels that seen in the galactopyranose series, in that it is the conformer in which O4 and O6 have a syn-pentane-type relationship (gg in galactose and tg in glucose) whose population is reduced on benzylation (Figure 6), suggesting that this change in conformation is caused by the loss of a favorable hydrogen bond or increased steric interactions in both cases.
Figure 6.
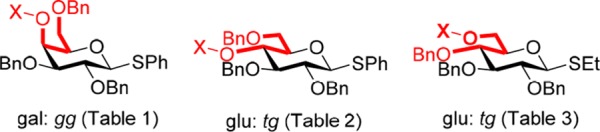
syn-Pentane conformations of the galacto- and glucopyranoses destabilized on replacement of a hydroxy group (X = H) by an ether (X = R).
As in the galactopyranose series, we adopt the benzyl ether as the standard for the subsequent comparisons with the influence of alternative protecting groups at the 4-position. The 4-O-acetate 23 and the benzoate 25 adopt very similar conformations in which the gg conformer is populated to a noticeably lower extent than in the benzyl ether 27, while the population of the tg conformer increases. With the more electron-withdrawing trifluoroacetate 24, the population of the tg conformer increases further, but it is balanced by a reduction in the population of both the gg and gt conformers when compared to the benzyl ether 27. The population of the side chain conformation of the benzenesulfonate 26 is anomalous insofar as, unlike the other esters, the tg conformation is not occupied, presumably for steric reasons arising from an increased syn-pentane interaction. This minimal population of the tg conformation in 26 results in a higher population of the gg conformer than in the acetal and benzoate esters 23 and 25.
Influence of Substituents at the Glucopyranose 6-Position
In contrast to the differences in side chain population brought about by changing protecting groups at the 4-position in the galacto- and glucopyranose series, changes in the protecting group at the 6-position of the 2,3,4-tri-O-benzyl glucopyranosides have a minimal influence on the side chain conformation (Table 3). It is noteworthy, however, that two derivatives, the 6-alcohol 3 and the 6-carbamate 35, have a small population of the tg conformation, with the syn-pentane conformation (Figure 6), suggesting that this conformer is stabilized by hydrogen bonding. The only other noteworthy feature from this series of compounds is the increased population of the gt conformer at the expense of the gg conformer on the installation of the strongly electron-withdrawing trifluoroacetyl group. In view of the relatively small changes in side chain conformation observed in the glucopyranose series with variation in the O6 protecting group, we did not undertake a parallel study in the galactopyranose series.
Discussion
The observed changes in side chain conformation with protecting groups at the 4-position for both the galacto- and glucopyranose systems are summarized in Figure 7. These changes are small, worth <1 kcal·mol–1, difficult to compute accurately with electronic structure calculations,57−60 and insufficient to account alone for the changes in anomeric reactivity and selectivity seen with such comparable changes in protecting group.6,61−66 As the effects are small, we make no attempt here to rationalize them in terms of stereoelectronic or other phenomena, other than to note that they are certainly related among other things to the distinct conformational preferences of esters and ethers.67−69 Nevertheless, such changes can be considered to modulate the larger effects on anomeric reactivity arising from replacing an arming with a disarming protecting group.
Figure 7.

Summary of changes in side chain conformation with protecting groups at the 4-position of (a) galactopyranosides and (b) glucopyranosides.
In both the galacto- and glucopyranosyl systems, the replacement of an arming8 ether protecting group at the 4-position by an ester group results in diminished anomeric reactivity, whether under standard conditions for the activation of thioglycosides6 or in SN2-displacements of anomeric bromides by chloride.9 This change in reactivity is usually understood in terms of the increased electron-withdrawing ability of the ester destabilizing nascent positive or partial positive charge at the reaction center (Scheme 4).1,5,6,8,17,70 The results presented in Table 1 and summarized in Figure 7a indicate that this effect will be moderated by the change in side chain conformation in the galactopyranoside series. Thus, the increase in the gt conformation with its intermediate reactivity at the expense of the less reactive tg conformation on replacement of a benzyl ether by an alkanoyl or aroyl ester will partially offset the added electron-withdrawing effect of the ester group. In the glucopyranosyl series on the other hand (Table 2 and Figure 7b), the main effect of the replacement of a benzyl ether by an alkanoyl or aroyl ester is to reduce the population of the most reactive gg conformer in favor of population of the least reactive tg conformer, thereby complementing the increased electron-withdrawing effect of the ester.
Scheme 4. Abbreviated General Glycosylation Mechanism and Influence of Protecting Groups and Side Chain Conformation.

In both the galacto- and glucopyranosyl series (Tables 1 and 2, Figure 7), on replacement of a benzyl ether at the 4-position by the strongly electron-withdrawing trifluoroacetyl group the trichloroacetyl group investigated in the galactose series, Table 1, the population of the most strongly electron-withdrawing tg conformer is increased. Thus, in both the galacto- and glucopyranosides, the change in side chain conformation on installation of a trifluoroacetyl group will reinforce the diminution of anomeric reactivity occasioned by the increased electron-withdrawing effect.
The sulfonyl protecting group, initially explored as a strongly electron-withdrawing group at the 2-position capable of stabilizing manno- and rhamnopyranosyl triflates and other sulfonates,71−73 and subsequently employed with varying degrees of success at the 4-position of 2,6-dideoxyglycopyranosyl donors,74 and at the 3-, 4-, and 6-positions of other pyranosyl donors,75,76 does not change the population of the side chain when replacing a benzyl ether at the 4-position of a glucosyl donor (Table 2, Figure 7b). The influence of the 4-O-sulfonyl group on glucopyranosylation can therefore be interpreted solely in terms of the change in electron-withdrawing ability.
The small changes in side chain conformation in a series of 4-O-alkanoyl and aroyl esters in both the galacto- and glucopyranosyl series do not provide strong support for a protecting-group-induced change in side chain population as the basis for the changes in anomeric selectivity and previously seen in the series of 17–2077 and related systems,78 previously explained by the controversial61−66 concept of stereodirecting participation by remote groups. Similarly, the consistent side chain conformation observed with numerous protecting groups at the 6-position in the 2,3,4-tri-O-benzyl glucopyranosides (Table 3) does not support a role of the side chain conformation in the changes in anomeric reactivity and glycosylation selectivity reported in such series of compounds.79−81
Conclusion
Replacement of a benzyl ether at the 4-position of phenyl 2,3,6-tri-O-benzyl-β-d-thio-galactopyranosides by either an alkanoyl or aroyl ester results in a small but consistent change in the population of the three staggered conformers of the side chain in which the proportion of the less reactive tg conformer is reduced in favor of the gt conformer. This suggests that the reduction in anomeric reactivity occasioned by the benzyl ether–ester switch seen with glycosyl donors, and attributed to the increased electron-withdrawing ability of the ester, is moderated by a change in side chain conformation in the galactose series. In the corresponding glucopyranosyl series, on the other hand, the same ether–ester change results in an increased population of the less reactive tg conformer, indicating that the change in conformation reinforces the effect of the increased electron-withdrawing ability of the ester group. In both the galacto- and glucopyranosyl series, the installation of a trifluoroacetyl group at the 4-position results in an enhanced population of the less reactive tg conformer. While the variations in side chain conformation with changes in protecting group recorded here are small, it must be understood that they are for the unactivated glycosyl donor. In view of the partial positive charge on the ring oxygen during glycosylation, it is likely that such changes are accentuated at the transition state for glycosylation reactions.. This possibility is under active investigation in our laboratory.
Experimental Section
General Experimental Section
Commercial reagents were used without further purification unless otherwise stated. NMR spectra were recorded in CDCl3 solution unless otherwise stated at 400, 500, or 600 MHz. 13C NMR spectra were recorded in CDCl3 solution unless otherwise stated at 100, 125, or 150 MHz. Mass spectra were recorded in the +ve ion mode using electrospray ionization (ESI-TOF). ESI-HRMS were recorded with a Waters LCT Premier Xe time-of-flight mass spectrometer. Specific rotations were recorded in dichloromethane solution at room temperature.
Phenyl 2,3,6-Tri-O-benzyl-1-thio-β-d-galactopyranoside (1)
Compound 1 was synthesized as described in the literature.311H NMR (600 MHz, CDCl3) δ 7.59–7.57 (m, 2H), 7.42–7.41 (m, 2H), 7.36–7.21 (m, 13H), 7.25–7.23 (m, 3H), 4.83 (d, J = 10.3 Hz, 1H), 4.75 (d, J = 10.3 Hz, 1H), 4.73 (d, J = 11.6 Hz, 1H), 4.69 (d, J = 11.6 Hz, 1H), 4.64 (d, J = 9.9 Hz, 1H), 4.57 (br s, 2H), 4.11 (d, J = 3.2 Hz, 1H), 3.81 (dd, J = 5.7, 10.0 Hz, 1H), 3.78 (dd, J = 5.8, 10.0 Hz, 1H), 3.76 (t, J = 9.5 Hz, 1H), 3.61–3.60 (m, 1H), 3.58 (dd, J = 3.3, 9.5 Hz, 1H), 2.55 (s, 1H). 1H NMR (600 MHz, C6D6) δ 7.68 (dd, J = 8.2, 1.0 Hz, 2H), 7.40 (d, J = 7.6 Hz, 2H), 7.23 (d, J = 7.6 Hz, 2H), 7.17–6.96 (m, 13H), 6.91 (t, J = 7.4 Hz, 1H), 4.85 (d, J = 10.8 Hz, 1H), 4.67 (d, J = 10.8 Hz, 1H), 4.57 (d, J = 9.8 Hz, 1H), 4.34–4.26 (m, 4H), 3.83 (t, J = 9.3 Hz, 1H), 3.79 (d, J = 2.7 Hz, 1H), 3.75 (dd, J = 9.8, 5.8 Hz, 1H), 3.72 (dd, J = 9.8, 6.1 Hz, 1H), 3.25 (t, J = 5.9 Hz, 1H), 3.20 (dd, J = 8.9, 3.2 Hz, 1H), 2.39 (s, 1H). IR (neat) ν 3480 cm–1 (O–H).
Phenyl (6S)-[6-2H1]-2,3,6-Tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-1)
Compound 6S-d-1 (0.40 g, 72%) was synthesized from compound 10 analogously to 1. 1H NMR (600 MHz, CDCl3) δ 7.59–7.57 (m, 2H), 7.42–7.40 (m, 2H), 7.36–7.21 (m, 13H), 7.25–7.23 (m, 3H), 4.83 (d, J = 10.3 Hz, 1H), 4.75 (d, J = 10.3 Hz, 1H), 4.73 (d, J = 11.6 Hz, 1H), 4.69 (d, J = 11.6 Hz, 1H), 4.64 (d, J = 9.9 Hz, 1H), 4.56 (br s, 2H), 4.10 (d, J = 3.1 Hz, 1H), 3.76 (d, J = 2.9 Hz, 1H), 3.75 (t, J = 9.5 Hz, 1H), 3.60–3.59 (m, 1H), 3.58 (dd, J = 3.2, 9.3 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.69–7.68 (m, 2H), 7.41–7.40 (m, 2H), 7.23–6.89 (m, 16H), 4.84 (d, J = 10.8 Hz, 1H), 4.66 (d, J = 10.8 Hz, 1H), 4.57 (d, J = 9.8 Hz, 1H), 4.31 (d, J = 11.9 Hz, 1H), 4.29–4.27 (m, 3H), 3.82 (t, J = 9.2 Hz, 1H), 3.78 (d, J = 3.1 Hz, 1H), 3.70 (d, J = 6.2 Hz, 1H), 3.23–3.22 (m, 1H), 3.19 (dd, J = 8.9, 3.2 Hz, 1H). HRMS (ESI) m/z calcd for C33H33DO5SNa [M + Na]+, 566.2087; found, 566.2079.
Phenyl 2,3,6-Tri-O-benzyl-1-thio-β-d-glucopyranoside (2)
Compound 25 (0.01 g, 0.015 mmol) was dissolved in anhydrous methanol (0.5 mL) and cooled to 0 °C before it was treated with Na (catalytic amount). The reaction mixture was stirred under an argon atmosphere at room temperature overnight, quenched with Amberlyst-15 (pH ∼ 4), filtered, and concentrated to dryness. The crude residue was purified by silica gel column chromatography, eluting with hexane/ethyl acetate (19:1 to 4:1) to afford compound 2 (0.005 g, 62%) with spectral data consistent with the literature.821H NMR (600 MHz, CDCl3) δ 7.58–7.55 (m, 2H), 7.43–7.24 (m, 18H), 4.92 (d, J = 11.4 Hz, 1H), 4.92 (d, J = 10.2 Hz, 1H), 4.79 (d, J = 11.4 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H,), 4.70 (d, J = 9.6 Hz, 1H), 4.60 (d, J = 11.9 Hz, 1H), 4.56 (d, J = 11.9 Hz, 1H), 3.80 (dd, J = 10.4, 4.1 Hz, 1H), 3.76 (dd, J = 10.4, 5.3 Hz, 1H), 3.66 (dd, J = 9.6, 8.9 Hz, 1H), 3.54 (dd, J = 8.9, 8.8 Hz, 1H), 3.50 (ddd, J = 9.6, 5.3, 4.1 Hz, 1H), 3.49 (dd, J = 9.6, 8.8 Hz, 1H), 2.54 (br s, 1H). 1H NMR (600 MHz, C6D6) δ 7.69–7.65 (m, 2H), 7.45–7.42 (m, 2H), 7.31–6.94 (m, 16H), 4.94 (d, J = 10.6 Hz, 1H), 4.87 (d, J = 11.7 Hz, 1H), 4.76 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 10.6 Hz, 1H), 4.64 (d, J = 9.7 Hz, 1H), 4.33 (d, J = 11.9 Hz, 1H), 4.30 (d, J = 11.9 Hz, 1H), 3.66 (dd, J = 9.8, 8.9 Hz, 1H), 3.64 (dd, J = 10.4, 3.7 Hz, 1H), 3.60 (dd, J = 10.4, 5.3 Hz, 1H), 3.51 (dd, J = 9.7, 8.7 Hz, 1H), 3.43 (dd, J = 8.9, 8.7 Hz, 1H), 3.22 (ddd, J = 9.8, 5.3, 3.7 Hz, 1H), 2.23 (s, 1H). IR (neat) ν 3441 cm–1 (O–H).
Phenyl (6S)-[6-2H1]-2,3,6-Tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-2)
Compound 6S-d-2 (0.011 g, 60%) was synthesized from 6S-d-25 (21.7 mg, 0.034 mmol) analogously to 2. 1H NMR (600 MHz, CDCl3) δ 7.58–7.53 (m, 1H), 7.44–7.22 (m, 18H), 4.91 (d, J = 11.4 Hz, 1H), 4.91 (d, J = 10.4 Hz, 1H), 4.78 (d, J = 11.4 Hz, 1H), 4.74 (d, J = 10.4 Hz, 1H), 4.69 (d, J = 9.6 Hz, 1H), 4.59 (d, J = 12.0 Hz, 1H), 4.56 (d, J = 12.0 Hz, 1H), 3.74 (d, J = 5.4 Hz, 1H), 3.66 (dd, J = 9.6, 8.8 Hz, 1H), 3.54 (t, J = 8.8 Hz, 1H), 3.48 (dd, J = 9.6, 8.8 Hz, 1H), 3.48 (dd, J = 9.6, 5.4 Hz, 1H), 2.53 (br s, 1H). 1H NMR (600 MHz, C6D6) δ 7.71–7.64 (m, 2H), 7.46–7.41 (m, 2H), 7.30–6.93 (m, 16H), 4.94 (d, J = 10.6 Hz, 1H), 4.87 (d, J = 11.7 Hz, 1H), 4.75 (d, J = 11.7 Hz, 1H), 4.69 (d, J = 10.6 Hz, 1H), 4.64 (d, J = 9.7 Hz, 1H), 4.32 (d, J = 12.2 Hz, 1H), 4.30 (d, J = 12.2 Hz, 1H), 3.65 (ddd, J = 9.7, 8.8, 2.9 Hz, 1H), 3.57 (d, J = 5.3 Hz, 1H), 3.51 (dd, J = 9.7, 8.7 Hz, 1H), 3.42 (t, J = 8.8 Hz, 1H), 3.22 (dd, J = 9.7, 5.3 Hz, 1H), 2.19 (d, J = 2.9 Hz, 1H). HRMS (ESI) m/z calcd for C33H33DO5SNa [M + Na]+, 566.2087; found, 566.2083.
Ethyl 2,3,4-Tri-O-benzyl-1-thio-β-d-glucopyranoside (3)
Compound 3 was synthesized as described in the literature.331H NMR (600 MHz, CDCl3) δ 7.40–7.26 (m, 15H), 4.93 (d, J = 11.0 Hz, 1H), 4.92 (d, J = 10.2 Hz, 1H), 4.87 (d, J = 11.0 Hz, 1H), 4.86 (d, J = 10.9 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.66 (d, J = 10.9 Hz, 1H), 4.51 (d, J = 9.9 Hz, 1H), 3.87 (dd, J = 12.2, 2.7 Hz, 1H), 3.71 (dd, J = 9.1, 8.8 Hz, 1H), 3.70 (dd, J = 12.2, 4.8 Hz, 1H), 3.58 (dd, J = 9.6, 9.1 Hz, 1H), 3.41 (dd, J = 9.9, 8.8 Hz, 1H), 3.38 (ddd, J = 9.6, 4.8, 2.7 Hz, 1H), 2.81–2.70 (m, 2H), 1.82 (br s, 1H), 1.33 (t, J = 7.4 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 7.45–7.42 (m, 2H), 7.35–7.31 (m, 2H), 7.26–7.23 (m, 2H), 7.19–7.05 (m, 9H), 4.99 (d, J = 10.6 Hz, 1H), 4.93 (d, J = 11.2 Hz, 1H), 4.85 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.2 Hz, 1H), 4.74 (d, J = 10.6 Hz, 1H), 4.61 (d, J = 11.2 Hz, 1H), 4.36 (d, J = 9.7 Hz, 1H), 3.77–3.71 (m, 1H), 3.66–3.56 (m, 3H), 3.43 (dd, J = 9.7, 8.4 Hz, 1H), 3.13–3.09 (m, 1H), 1.60 (br s, 1H) 2.56–2.42 (m, 2H), 1.11 (t, J = 7.4 Hz, 3H). IR (neat) ν 3484 cm–1 (O–H).
Ethyl (6S)-[6-2H1]-2,3,4-Tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-3)
A solution of compound 12 (0.105 g, 0.27 mmol) in anhydrous methanol (1.0 mL) was cooled to 0 °C, treated with Na (cat), and stirred under an argon atmosphere at room temperature for 1.5 h. Then, the reaction was quenched with Amberlyst-15 resin (pH ∼ 7) and filtered through cotton before it was concentrated to dryness. The crude residue was dissolved in anhydrous pyridine (0.8 mL) and treated with trityl chloride (84 μL, 0.32 mmol) at room temperature. The reaction mixture was stirred in the dark at room temperature for 3 days. The reaction was then quenched with methanol (∼70 μL), stirred for 1 h, and concentrated to dryness. The crude residue was dissolved in chloroform and washed with cold aqueous saturated NaHCO3 and cold brine. The organic layer was dried over anhydrous Na2SO4 and concentrated to dryness. The crude residue was dissolved in anhydrous DMF (0.9 mL) and cooled to 0 °C before it was treated with 60% NaH in mineral oil (48 mg, 1.2 mmol) followed by benzyl bromide (141 μL, 1.2 mmol). The reaction mixture was stirred under an argon atmosphere at room temperature for 6 h. The reaction mixture was cooled to 0 °C before it was quenched with water. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous Na2SO4, and concentrated to dryness. The crude residue was passed through a short pad of silica, eluting with hexane/ethyl acetate (7:3), and concentrated. The residue was dissolved in a mixture of glacial acetic acid and water (4:1, 1.3 mL) and heated at 80 °C for 2.5 h. The reaction mixture was concentrated to dryness and purified using silica gel column chromatography, eluting with hexane/ethyl acetate (4:1 to 7:2), obtaining compound 6S-d-3 (0.045 g, 35%). 1H NMR (400 MHz, CDCl3) δ 7.40–7.25 (m, 15H), 4.94 (d, J = 10.9 Hz, 1H), 4.92 (d, J = 10.2 Hz, 1H), 4.87 (d, J = 10.9 Hz, 1H), 4.87 (d, J = 10.9 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.66 (d, J = 10.9 Hz, 1H), 4.51 (d, J = 9.8 Hz, 1H), 3.72 (t, J = 9.0 Hz, 1H), 3.68 (d, J = 4.9 Hz, 1H), 3.58 (dd, J = 9.7, 9.0 Hz, 1H), 3.41 (dd, J = 9.8, 8.8 Hz, 1H), 3.38 (dd, J = 9.7, 4.9 Hz, 1H), 2.84–2.69 (m, 2H), 1.33 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z calcd for C29H33DO5SNa [M + Na]+, 518.2087; found, 518.2066.
(6S)-1,6-Anhydro-2,3,4-tri-O-acetyl-6-bromo-β-d-galactopyranose (5)
A mixture of l,6-anhydro-2,3,4-tri-O-acetyl-β-d-galactopyranose 4(83) (0.77 g, 2.67 mmol) and N-bromosuccinimide (1.9 g, 10.6 mmol)) in trifluorotoluene (40 mL) was refluxed over a 300 W heat lamp for 8 h. After 8 h, the solvent was evaporated under reduced pressure and the crude product dissolved in EtOAc (50 mL). The solution was successively washed with aqueous saturated Na2S2O3, aqueous saturated NaHCO3, and dried over Na2SO4. Evaporation of the solvent under reduced pressure gave a crude product which was purified through silica gel column chromatography (eluent: 20% EtOAc in hexane) to give 5 (0.73g, 75%) as a yellowish oil. [α]D22 = −63.6 (c 1.50, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 6.60 (s, 1H), 5.81 (s, 1H), 5.29–5.20 (m, 2H), 4.74 (dd, J = 4.0, 1.3 Hz, 1H), 4.71 (br s, 1H), 2.14 (s, 3H), 2.13 (s, 3H), 2.05 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.2, 169.1, 168.9, 101.7, 82.6, 79.6, 69.3, 66.9, 64.4, 20.7 (2), 20.5. HRMS (ESI) m/z calcd for C12H1579BrO8Na [M + Na]+, 388.9848; found, 388.9849; calcd for C12H1581BrO8Na [M + Na]+, 390.9828; found, 390.9828.
(6S)-[6-2H1]-1,6-Anhydro-2,3,4-tri-O-acetyl-β-d-galactopyranose (6)
To a solution of 5 (2.3 g, 6.26 mmol) and azobis(isobutyronitrile) (0.1 g, 0.63 mmol) in toluene (170 mL) was added freshly prepared tri-n-butyltin deuteride44 (5.5 g, 18.79 mmol), and the reaction mixture was refluxed for 0.5 h. The cooled reaction mixture was evaporated in vacuo to give a crude product which was purified by silica gel column chromatography (eluent: 40% EtOAc in hexane) to give 6 (1.4 g, 77%) as a colorless oil. [α]D22 = −10.0 (c 1.15, CH2Cl2). 1H NMR (400 MHz, CDCl3) δ 5.42 (s, 1H), 5.29–5.20 (m, 2H), 4.75 (s, 1H), 4.45 (d, J = 2.7 Hz, 1H), 4.31 (s, 1H), 2.12 (s, 6H), 2.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 169.5, 169.3, 169.2, 98.9, 72.0, 71.0, 67.4, 64.2 (t, J = 23.5 Hz), 20.8, 20.7, 20.5. HRMS (ESI) m/z calcd for C12H15DO8Na [M + Na]+, 312.0806; found, 312.0818.
(6S)-[6-2H1]-1,2,3,4,6-Penta-O-acetyl-α-d-galactopyranose (7)
To a solution of compound 6 (1.4 g, 4.84 mmol) in MeOH (20 mL) was added Na (catalytic amount) slowly and the reaction mixture was stirred for 7 h, then neutralized with Amberlite IR120, filtered, and concentrated under reduced pressure to give (6S)-[6-2H1]-1,6-anhydrogalactose. A solution of (6S)-[6-2H1]-1,6-anhydrogalactose (0.73 g, 4.47 mmol) in Ac2O (20 mL) was treated with conc. H2SO4 (0.35 mL) at 0 °C and stirred for 3 h. The reaction mixture was poured into a saturated aqueous NaOAc solution (50 mL) and extracted with CHCl3 (3 × 20 mL), washed with brine (10 mL), dried over Na2SO4, and concentrated under a high vacuum to give the crude product. Column chromatography (eluent: 30% EtOAc in hexane) on silica gel yielded 7 (1.5 g, 81% over two steps) as a colorless syrup with spectral data consistent with the literature.361H NMR (400 MHz, CDCl3) δ 6.29 (d, J = 2.2 Hz, 1H), 5.42 (s, 1H), 5.29–5.19 (m, 2H), 4.27 (d, J = 6.4 Hz, 1H), 3.99 (d, J = 6.5 Hz, 1H), 2.09 (s, 3H), 2.08 (s, 3H), 1.96 (d, J = 2.5 Hz, 3H), 1.95 (d, J = 3.9 Hz, 3H), 1.93 (s, 3H).
Phenyl (6S)-[6-2H1]-2,3,4,6-Tetra-O-acetyl-1-thio-β-d-galactopyranoside (8)
To a stirred solution of (6S)-[6-2H1]-1,2,3,4,6-penta-O-acetyl-α-d-galactopyranose 7 (1.5 g, 3.83 mmol) and thiophenol (047 mL, 4.59 mmol) in CH2Cl2 was added BF3·Et2O (1.9 mL, 7.66 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 22 h, then quenched with aqueous saturated NaHCO3 solution (50 mL) and extracted with CH2Cl2 (3 × 25 mL), dried over Na2SO4, and concentrated under a high vacuum. The crude product was purified by silica gel column (eluent: 20% EtOAc in hexane) chromatography to give 8(84) (900 mg, 56%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.51–7.48 (m, 2H), 7.30–7.28 (m, 3H), 5.40 (d, J = 2.8 Hz, 1H), 5.22 (t, J = 10.0 Hz, 1H), 5.03 (dd, J = 9.9, 3.3 Hz, 1H), 4.70 (d, J = 10.0 Hz, 1H), 4.17 (d, J = 6.5 Hz, 1H), 3.92 (d, J = 6.8 Hz, 1H), 2.09 (s, 3H), 2.08 (s, 3H), 2.02 (s, 3H), 1.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 170.3, 170.2, 170.0, 169.4, 132.5, 132.4, 128.9, 128.1, 86.6, 74.3, 72.0, 67.2, 61.3 (t, J = 22.2 Hz), 20.8, 20.6, 20.6, 20.6. HRMS (ESI) m/z calcd for C20H23DO9SNa [M + Na]+, 464.1101; found, 464.1114.
Phenyl 2,3-Di-O-benzyl-4,6-O-benzylidene-(6S)-[6-2H1]-1-thio-β-d-galactopyranoside (10)
To a stirred solution of 8 (0.9 g, 2.04 mmol) in MeOH (10.0 mL) was added Na metal (catalytic amount) slowly. The reaction mixture was stirred for 2 h, then neutralized with Amberlite IR120, filtered, and concentrated under reduced pressure to give a crude thiogalactoside. The crude residue was dissolved in dry CH3CN (30 mL) and treated with benzaldehyde dimethylacetal (0.41 mL, 2.75 mmol) followed by camphorsulfonic acid (4.2 mg, 0.18 mmol) at room temperature. The reaction mixture was stirred for 2.5 h, then neutralized with triethylamine (0.5 mL), and concentrated under reduced pressure to give a crude compound 9. After filtration through a short silica gel column, compound 9 (0.52 g) was dissolved in dry DMF (10 mL) and then treated with NaH (0.17 g, 4.15 mmol) and BnBr (2.27 g, 1.6 mL, 13.3 mmol) at 0 °C. The resulting solution was warmed to room temperature and stirred for 0.5 h. Upon completion of the reaction (TLC), the excess NaH was quenched using sat. NH4Cl solution. The product was extracted with EtOAc (3 × 25 mL), and the combined organic layer was washed with brine (10 mL), dried over Na2SO4, and concentrated under a high vacuum. The crude product was purified via silica gel column chromatography (30% EtOAc in hexane) to give 10 (0.63 g, 81%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.72–7.71 (m, 2H), 7.55–7.53 (m, 2H), 7.43–7.18 (m, 16H), 5.50 (s, 1H), 4.74–4.68 (m, 4H), 4.62 (d, J = 9.5 Hz, 1H), 4.16 (d, J = 3.2 Hz, 1H), 3.98 (br s, 1H), 3.91 (t, J = 9.4 Hz, 1H), 3.63 (dd, J = 9.2, 3.3 Hz, 1H), 3.42 (br s, 1H). 13C NMR (150 MHz, CDCl3) δ 138.6, 138.2, 138.0, 132.9, 132.8, 129.2, 129.0, 128.6, 128.5, 128.4, 128.3, 128.0, 127.9, 127.6, 126.8, 101.5, 86.7, 81.5, 75.6, 75.5, 73.8, 72.0, 69.9, 69.3 (t, J = 21.7 Hz). HRMS (ESI) m/z calcd for C33H31DO5SNa [M + Na]+, 564.1931; found, 564.1927.
Ethyl (6S)-[6-2H1]-2,3,4,6-Tetra-O-acetyl-1-thio-β-d-glucopyranoside (12)
Compound 11(43) (0.1 g, 0.346 mmol) was dissolved in anhydrous dichloroethane (10.5 mL) and treated with (ethylthio)trimethylsilane (167 μL, 1.03 mmol) followed by ZnI2 (332 mg, 1.03 mmol). The reaction mixture was stirred under an argon atmosphere at room temperature for 3 h before it was diluted with ethyl acetate and filtered through Celite. The organic layer was washed with aqueous saturated NaHCO3 and brine, dried over anhydrous Na2SO4, and concentrated to dryness. The crude residue was dissolved in a mixture of THF:H2O (1:1, 20 mL), treated with K2CO3 (200 mg, 1.44 mmol), and stirred for 15 min. The reaction mixture was then diluted with ethyl acetate, washed with water and brine, and treated with anhydrous Na2SO4 before it was concentrated to dryness. Then, the crude residue was passed through a short pad of silica gel, eluting with hexane/ethyl acetate (1:1), and the eluent was concentrated to dryness. The residue (60 mg) was dissolved in pyridine (1.4 mL), treated with acetic anhydride (0.7 mL), and stirred overnight under an argon atmosphere at room temperature. The reaction mixture was then concentrated to dryness, and the crude reaction mixture was purified by silica gel column chromatography, eluting with hexane/ethyl acetate (7:3), to obtain 12 (0.051 g, 40%). 1H NMR (400 MHz, CDCl3) δ 5.22 (t, J = 9.4 Hz, 1H), 5.08 (dd, J = 10.0, 9.4 Hz, 1H), 5.03 (dd, J = 10.0, 9.4 Hz, 1H), 4.49 (d, J = 10.0 Hz, 1H), 4.22 (d, J = 5.0 Hz, 1H), 3.70 (dd, J = 10.0, 5.0 Hz, 1H), 2.78–2.63 (m, 2H), 2.07 (s, 3H), 2.06 (s, 3H), 2.02 (s, 3H), 2.00 (s, 3H), 1.27 (t, J = 7.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.6, 170.2, 169.4, 83.5, 75.9, 73.9, 69.8, 68.3, 61.9 (t, J = 22.8 Hz), 24.2, 20.7, 20.6, 20.6, 14.8. HRMS (ESI) m/z calcd for C16H23DO9SNa [M + Na]+, 416.1102; found, 416.1098.
Phenyl 4-O-Acetyl-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (13).77
Compound 13 was synthesized as described in the literature.76 NMR (600 MHz, CDCl3) δ 7.60–7.58 (m, 2H), 7.42–7.40 (m, 2H), 7.37–7.25 (m, 16H), 5.65 (br s, 1H), 4.78 (d, J = 11.3 Hz, 1H), 4.77 (d, J = 10.3 Hz, 1H), 4.75 (d, J = 10.0 Hz, 1H), 4.71–4.67 (m, 1H), 4.57 (d, J = 11.7 Hz, 1H), 4.50 (d, J = 11.0 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 3.77–3.76 (m, 1H), 3.67–3.66 (m, 2H), 3.64 (dd, J = 9.5, 6.0 Hz, 1H), 3.55 (dd, J = 9.5, 6.7 Hz, 1H), 2.09 (s, 3H). 1H NMR (600 MHz, C6D6) δ 7.65–7.61 (m, 2H), 7.39 (t, J = 7.6 Hz, 2H), 7.31 (d, J = 7.3 Hz, 2H), 7.25 (d, J = 7.4 Hz, 2H), 7.17–7.01 (m, 10H), 6.99–6.91 (m, 2H), 5.58 (dd, J = 3.3, 1.1 Hz, 1H), 4.80 (d, J = 10.7 Hz, 1H), 4.75 (d, J = 10.7 Hz, 1H), 4.66 (d, J = 11.2 Hz, 1H), 4.56 (d, J = 9.7 Hz, 1H), 4.28 (d, J = 11.6 Hz, 2H), 4.17 (d, J = 11.9 Hz, 1H), 3.80 (t, J = 9.5 Hz, 1H), 3.47 (dd, J = 9.4, 6.1 Hz, 1H), 3.42 (dd, J = 9.4, 6.5 Hz, 1H), 3.33 (dd, J = 9.2, 3.3 Hz, 1H), 3.32 (ddd, J = 6.5, 6.1, 1.1 Hz, 1H), 1.65 (s, 3H). IR (neat) ν 1742 cm–1 (C=O).
Phenyl 4-O-Acetyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-13)
Compound 6S-d-13 (0.021 g, 81%) was synthesized from 6S-d-1 analogously to 13. 1H NMR (600 MHz, CDCl3) δ 7.60–7.57 (m, 2H), 7.41–7.23 (m, 18H), 5.63 (d, J = 1.6 Hz, 1H), 4.79 (d, J = 11.1 Hz, 1H), 4.78 (d, J = 10.2 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.72–4.68 (m, 1H), 4.55 (d, J = 11.7 Hz, 1H), 4.49 (d, J = 11.0 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 3.76 (d, J = 6.1 Hz, 1H), 3.68–3.66 (m, 2H), 3.61 (d, J = 6.0 Hz, 1H), 2.10 (s, 3H). 1H NMR (600 MHz, C6D6) δ 7.65–7.62 (m, 2H), 7.40 (d, J = 7.6 Hz, 2H), 7.31 (d, J = 7.5 Hz, 2H), 7.24 (t, J = 10.8 Hz, 2H), 7.18–6.91 (m, 12H), 5.58 (d, J = 3.2 Hz, 1H), 4.80 (d, J = 10.7 Hz, 1H), 4.75 (d, J = 10.7 Hz, 1H), 4.66 (d, J = 11.2 Hz, 1H), 4.56 (d, J = 9.7 Hz, 1H), 4.28 (d, J = 11.4 Hz, 2H), 4.17 (d, J = 11.9 Hz, 1H), 3.79 (t, J = 9.4 Hz, 1H), 3.47–3.45 (m, 1H), 3.35–3.26 (m, 1H), 1.65 (s, 3H). HRMS (ESI) m/z calcd for C35H35DO6SNa [M + Na]+, 608.2193; found, 608.2191.
Phenyl 2,3,6-Tri-O-benzyl-4-O-pivolyl-1-thio-β-d-galactopyranoside (14).77
A solution of 1 (0.050 g, 2.77 mmol) in dry pyridine (0.5 mL) was treated with pivaloyl chloride (0.25 mL) at room temperature and stirred at 80 °C for 1 h. The solvents were evaporated under a high vacuum to give a crude residue. The crude product was purified by silica gel column chromatography (eluent: 20% ethyl acetate in hexane) to afford 14 (0.045 g, 78%) as a colorless oil. [α]D22 = +6.6 (c 0.50, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.59–7.57 (m, 2H), 7.37–7.24 (m, 18H), 5.63 (d, J = 2.9 Hz, 1H), 4.73 (d, J = 10.6 Hz, 1H), 4.71 (d, J = 10.6 Hz, 1H), 4.67 (d, J = 10.3 Hz, 1H), 4.63 (d, J = 9.5 Hz, 1H), 4.54 (d, J = 11.7 Hz, 1H), 4.47 (d, J = 11.8 Hz, 1H), 4.45 (d, J = 11.0 Hz, 1H), 3.81 (t, J = 6.3 Hz, 1H), 3.65 (dd, J = 9.2, 3.3 Hz, 1H), 3.62 (dd, J = 9.5, 6.1 Hz, 1H), 3.57 (t, J = 9.2 Hz, 1H), 3.49 (dd, J = 9.5, 6.7 Hz, 1H), 1.14 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 177.4, 138.1, 137.8, 137.6, 133.2, 132.3, 128.8, 128.4, 128.3, 128.2, 128.2, 128.1, 127.9, 127.8, 127.7, 127.6, 127.4, 87.0, 81.4, 76.3, 76.1, 75.6, 73.7, 71.6, 68.2, 66.3, 39.0, 27.1. 1H NMR (600 MHz, C6D6) δ 7.66–7.63 (m, 2H), 7.40 (d, J = 8.0 Hz, 2H), 7.30–7.24 (m, 4H), 7.18–6.91 (m, 12H), 5.56 (d, J = 3.2 Hz, 1H), 4.78 (d, J = 10.8 Hz, 1H), 4.76 (d, J = 10.9 Hz, 1H), 4.58 (d, J = 11.0 Hz, 1H), 4.49 (d, J = 9.9 Hz, 1H), 4.30 (d, J = 11.9 Hz, 1H), 4.22 (d, J = 11.9 Hz, 1H), 4.20 (d, J = 11.8 Hz, 1H), 3.69 (t, J = 9.4 Hz, 1H), 3.49 (dd, J = 9.2, 6.0 Hz, 1H), 3.42 (dd, J = 9.2, 6.8 Hz, 1H), 3.37–3.35 (m, 1H), 3.27 (dd, J = 9.0, 3.2 Hz, 1H), 1.08 (s, 9H). IR (neat) ν 1732 cm–1 (C=O). HRMS (ESI) m/z calcd for C38H42O6SNa [M + Na]+, 649.2600; found, 649.2584.
Phenyl 4-O-Pivaloyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-14)
Compound 6S-d-14 (0.011 g, 90%) was synthesized from 6S-d-1 analogously to 14. 1H NMR (600 MHz, CDCl3) δ 7.59–7.57 (m, 2H), 7.37–7.23 (m, 18H), 5.63 (d, J = 3.1 Hz, 1H), 4.73 (d, J = 10.4 Hz, 1H), 4.70 (d, J = 10.4 Hz, 1H), 4.66 (d, J = 10.3 Hz, 1H), 4.64 (d, J = 9.7 Hz, 1H), 4.54 (d, J = 11.7 Hz, 1H), 4.48 (d, J = 11.8, 1H), 4.44 (d, J = 11.0 Hz, 1H), 3.80 (d, J = 6.1 Hz, 1H), 3.64 (dd, J = 9.1, 3.2 Hz, 1H), 3.60 (d, J = 6.1 Hz, 1H), 3.56 (t, J = 9.3 Hz, 1H), 1.14 (s, 9H). 1H NMR (600 MHz, C6D6) δ 7.66–7.6 (m, 2H), 7.40 (d, J = 7.1 Hz, 2H), 7.30–7.25 (m, 4H), 7.17–6.91 (m, 12H), 5.56 (d, J = 3.2 Hz, 1H), 4.79 (d, J = 10.9 Hz, 1H), 4.76 (d, J = 10.9 Hz, 1H), 4.58 (d, J = 11.0 Hz, 1H), 4.49 (d, J = 9.7 Hz, 1H), 4.30 (d, J = 11.9 Hz, 1H), 4.21 (d, J = 11.5 Hz, 1H), 4.20 (d, J = 11.5 Hz, 1H), 3.70 (t, J = 9.4 Hz, 1H), 3.47 (d, J = 6.0 Hz, 1H), 3.36 (d, J = 5.9 Hz, 1H), 3.27 (dd, J = 9.0, 3.3 Hz, 1H), 1.08 (s, 9H). HRMS (ESI) m/z calcd for C38H41DO6SNa [M + Na]+, 650.2663; found, 650.2662.
Phenyl 2,3,6-Tri-O-benzyl-4-O-trifluoroacetyl-1-thio-β-d-galactopyranoside (15).77
To a stirred solution of 1 (0.05 g, 0.092 mmol) in dry CH2Cl2 (1.0 mL) were added pyridine (0.030 mL, 0.18 mmol), trifluoroacetic anhydride (0.026 mL, 0.18 mmol), and DMAP (1.0 mg, 0.009 mmol) at 0 °C. The reaction mixture was stirred for 0.5 h before it was quenched with water (5 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined extracts were washed with 1 N HCl (5.0 mL) solution, dried over Na2SO4, and concentrated under a high vacuum. Silica gel column chromatography (eluent: 10% ethyl acetate in hexane) afforded 15 (0.038 g, 65%) as a colorless oil. [α]D22 = −6.9 (c 0.80, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.56–7.52 (m, 2H), 7.37–7.23 (m, 18H), 5.74 (dd, J = 3.2, 0.9 Hz, 1H), 4.75 (d, J = 11.2 Hz, 1H), 4.68 (d, J = 10.2 Hz, 1H), 4.65 (d, J = 9.5 Hz, 1H), 4.64 (d, J = 9.9 Hz, 1H), 4.52 (d, J = 11.3 Hz, 1H), 4.50 (d, J = 11.7 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 3.82 (ddd, J = 8.2, 5.6, 1.0 Hz, 1H), 3.70 (dd, J = 9.1, 3.2 Hz, 1H), 3.67 (dd, J = 9.3, 5.6 Hz, 1H), 3.61 (t, J = 9.5 Hz, 1H), 3.49 (dd, J = 9.3, 8.3 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 156.8 (q, J = 42.7 Hz, COCF3), 137.9, 137.1, 137.1, 132.8, 132.1, 128.9, 128.5, 128.4, 128.3, 128.2, 128.0, 128.0, 127.9, 127.8, 127.7, 114.5 (q, J = 285.8 Hz, CF3CO), 87.4, 80.5, 76.0, 75.7, 74.7, 73.8, 72.3, 71.3, 67.0. 19F NMR (376 MHz, CDCl3) δ −74.7 (COCF3). 1H NMR (600 MHz, C6D6) δ 7.59 (d, J = 7.6 Hz, 2H), 7.32 (d, J = 7.4 Hz, 2H), 7.19–6.91 (m, 16H), 5.57 (d, J = 3.1 Hz, 1H), 4.63 (d, J = 10.7 Hz, 1H), 4.53 (d, J = 10.8 Hz, 1H), 4.45 (d, J = 11.4 Hz, 1H), 4.37 (d, J = 9.5 Hz, 1H), 4.19 (d, J = 11.4 Hz, 1H), 4.15 (d, J = 11.7 Hz, 1H), 4.06 (d, J = 11.7 Hz, 1H), 3.71 (t, J = 9.4 Hz, 1H), 3.34 (dd, J = 9.1, 5.7 Hz, 1H), 3.31 (dd, J = 9.1, 8.1 Hz, 1H), 3.24 (dd, J = 9.1, 3.2 Hz, 1H), 3.19–3.15 (m, 1H). IR (neat) ν 1790 cm–1 (C=O). HRMS (ESI) m/z calcd for C35H33O6F3SNa [M + Na]+, 661.1848; found, 661.1844.
Phenyl 4-O-Trifluoroacetyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-15)
Compound 6S-d-15 (0.021 g, 87%) was synthesized from 6S-d-1 analogously to 15. 1H NMR (600 MHz, CDCl3) δ 7.56–7. 52 (m, 2H), 7.37–7.24 (m, 18H), 5.74 (d, J = 3.0 Hz, 1H), 4.75 (d, J = 11.2 Hz, 1H), 4.68 (d, J = 10.2 Hz, 1H), 4.65 (d, J = 9.5 Hz, 1H), 4.64 (d, J = 9.9 Hz, 1H), 4.52 (d, J = 11.6 Hz, 1H), 4.50 (d, J = 11.6 Hz, 1H), 4.45 (d, J = 11.6 Hz, 1H), 3.81 (d, J = 5.6 Hz, 1H), 3.70 (dd, J = 9.1, 3.2 Hz, 1H), 3.65 (d, J = 5.6 Hz, 1H), 3.61 (t, J = 9.4 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.61–7.58 (m, 2H), 7.33 (d, J = 7.2 Hz, 2H), 7.21–6.92 (m, 16H), 5.58 (d, J = 3.0 Hz, 1H), 4.63 (d, J = 10.8 Hz, 1H), 4.53 (d, J = 10.8 Hz, 1H), 4.45 (d, J = 11.4 Hz, 1H), 4.38 (d, J = 9.7 Hz, 1H), 4.19 (d, J = 11.4 Hz, 1H), 4.16 (d, J = 11.8 Hz, 1H), 4.06 (d, J = 11.8 Hz, 1H), 3.71 (t, J = 9.4 Hz, 1H), 3.32 (d, J = 5.6 Hz, 1H), 3.25 (dd, J = 9.1, 3.2 Hz, 1H), 3.17 (d, J = 5.7 Hz, 1H). HRMS (ESI) m/z calcd for C35H32F3DO6SNa [M + Na]+, 662.1910; found, 662.1905.
Phenyl 2,3,6-Tri-O-benzyl-4-O-trichloroacetyl-1-thio-β-d-galactopyranoside (16).77
Compound 16 (0.058 g, 92%) was synthesized from 1 analogously to 15, as a colorless oil. [α]D22 = −8.0 (c 1.80, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.59–7.52 (m, 2H), 7.39–7.21 (m, 18H), 5.71 (d, J = 2.9 Hz, 1H), 4.80 (d, J = 11.4 Hz, 1H), 4.65 (d, J = 10.6 Hz, 1H), 4.64 (d, J = 9.9 Hz, 1H), 4.62 (d, J = 10.3 Hz, 1H), 4.54 (d, J = 11.6 Hz, 1H), 4.51 (d, J = 11.4 Hz, 1H), 4.49 (d, J = 11.4 Hz, 1H), 3.87 (dd, J = 7.9, 6.0 Hz, 1H), 3.74–3.72 (m, 1H), 3.71 (dd, J = 9.2, 5.7 Hz, 1H), 3.68 (t, J = 9.3 Hz, 1H), 3.59 (dd, J = 9.2, 8.2 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 161.3, 137.9, 137.3, 137.3, 132.6, 132.3, 129.0, 128.6, 128.3, 128.3, 128.3, 128.1, 128.0, 127.9, 127.8, 127.6, 89.9, 87.1, 80.9, 75.6, 75.6, 75.2, 73.9, 72.2, 72.1, 67.3. 1H NMR (600 MHz, C6D6) δ 7.62–7.60 (m, 2H), 7.33 (d, J = 7.4 Hz, 2H), 7.24–7.19 (m, 4H), 7.16–7.07 (m, 11H), 6.95–6.91 (m, 1H), 5.53 (d, J = 3.0 Hz, 1H), 4.69 (d, J = 10.8 Hz, 1H), 4.66 (d, J = 10.8 Hz, 1H), 4.52 (d, J = 11.4 Hz, 1H), 4.38 (d, J = 9.9 Hz, 1H), 4.23 (d, J = 11.7 Hz, 1H), 4.19 (d, J = 11.4 Hz, 1H), 4.18 (d, J = 11.7 Hz, 1H), 3.79 (t, J = 9.4 Hz, 1H), 3.46 (m, 1H), 3.43 (m, 1H), 3.29–3.28 (m, 1H), 3.28–3.25 (m, 1H). IR (neat) ν 1769 cm–1 (C=O). HRMS (ESI) m/z calcd for C35H33O6Cl3SNa [M + Na]+, 709.0961; found, 709.0955.
Phenyl 4-O-Trichloroacetyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-16)
Compound 6S-d-16 (0.029 g, 90%) was synthesized from 6S-d-1 analogously to 1. 1H NMR (600 MHz, CDCl3) δ 7.59–7.52 (m, 2H), 7.39–7.21 (m, 18H), 5.70 (d, J = 2.9 Hz, 1H), 4.79 (d, J = 11.2 Hz, 1H), 4.65 (d, J = 10.9 Hz, 1H), 4.63 (d, J = 9.9 Hz, 1H), 4.62 (d, J = 10.6 Hz, 1H), 4.53 (t, J = 11.6 Hz, 1H), 4.52 (d, J = 11.6 Hz, 1H), 4.49 (d, J = 11.4 Hz, 1H), 3.86 (d, J = 5.7 Hz, 1H), 3.72 (dd, J = 9.1, 3.0 Hz, 1H), 3.69 (d, J = 5.7, 1H), 3.68 (t, J = 9.3 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.63–7.59 (m, 2H), 7.33 (d, J = 7.4 Hz, 2H), 7.23–7.20 (m, 4H), 7.17–6.90 (m, 12H), 5.53 (d, J = 3.0 Hz, 1H), 4.69 (d, J = 10.8 Hz, 1H), 4.66 (d, J = 10.8 Hz, 1H), 4.52 (d, J = 11.3 Hz, 1H), 4.39 (d, J = 9.7 Hz, 1H), 4.22 (d, J = 11.7 Hz, 1H), 4.19 (d, J = 11.4 Hz, 1H), 4.17 (d, J = 11.8 Hz, 1H), 3.79 (t, J = 9.4 Hz, 1H), 3.43 (d, J = 5.7 Hz, 1H), 3.29–3.26 (m, 2H). HRMS (ESI) m/z calcd for C35H32Cl3DO6SNa [M + Na]+, 710.1024; found, 710.1016.
Phenyl 4-O-Benzoyl-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (17).77
Compound 17 was synthesized as described in the literature.761H NMR (600 MHz, CDCl3) δ 8.01–7.93 (m, 2H), 7.66–7.65 (m, 2H), 7.61–7.59 (m, 1H), 7.47–7.45 (m, 2H), 7.40–7.39 (m, 2H), 7.35–7.22 (m, 16H), 5.91 (d, J = 3.0 Hz 1H), 4.87 (d, J = 11.4 Hz, 1H), 4.74 (br s, 2H), 4.70 (d, J = 9.5 Hz, 1H), 4.54 (d, J = 11.0 Hz, 1H), 4.52 (dd, J = 10.6 Hz, 1H), 4.46 (d, J = 10.6 Hz, 1H), 3.92–3.89 (m, 1H), 3.77 (d, J = 9.2, 3.3 Hz, 1H), 3.72 (t, J = 9.4 Hz, 1H), 3.69 (dd, J = 9.6, 5.8 Hz, 1H), 3.59 (dd, J = 9.6, 6.8 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 8.11 (d, J = 8.1 Hz, 2H), 7.70 (dd, J = 12.2, 5.2 Hz, 2H), 7.39 (d, J = 7.9 Hz, 2H), 7.26 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 7.9 Hz, 2H), 7.17–6.95 (m, 15H), 5.87 (d, J = 3.1 Hz, 1H), 4.75 (d, J = 11.4 Hz, 1H), 4.72 (d, J = 10.9 Hz, 1H), 4.67 (d, J = 10.9 Hz, 1H), 4.54 (d, J = 9.7 Hz, 1H), 4.31 (d, J = 11.4 Hz, 1H), 4.19 (d, J = 11.8 Hz, 1H), 4.12 (d, J = 11.9 Hz, 1H), 3.83 (t, J = 9.4 Hz, 1H), 3.53 (dd, J = 9.3, 6.1 Hz, 1H), 3.49 (dd, J = 9.3, 6.6 Hz, 1H), 3.42 (t, J = 4.8 Hz, 1H), 3.42–3.40 (m, 1H). IR (neat) ν 1722 cm–1 (C=O).
Phenyl 4-O-Benzoyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-17)
Compound 6S-d-17 (0.021 g, 84%) was synthesized analogously as 17 from 6S-d-1. 1H NMR (600 MHz, CDCl3) δ 8.02–7.99 (m, 2H), 7.67–7.64 (m, 2H), 7.61–7.59 (m, 1H), 7.47–7.45 (m, 2H), 7.40–7.39 (m, 2H), 7.37–7.21 (m, 16H), 5.91 (d, J = 3.1 Hz, 1H), 4.87 (d, J = 11.2 Hz, 1H), 4.74 (br s, 2H), 4.71 (d, J = 9.5 Hz, 1H), 4.54 (d, J = 11.2 Hz, 1H), 4.52 (d, J = 11.8 Hz, 1H), 4.46 (d, J = 11.8 Hz, 1H), 3.90 (d, J = 6.0 Hz, 1H), 3.77 (dd, J = 9.1, 3.2 Hz, 1H), 3.72 (t, J = 9.3 Hz, 1H), 3.69 (d, J = 6.0 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 8.11 (d, J = 7.7 Hz, 2H), 7.70 (d, J = 7.3 Hz, 2H), 7.39 (d, J = 7.5 Hz, 2H), 7.26 (d, J = 7.4 Hz, 2H), 7.20 (d, J = 7.5 Hz, 2H), 7.14 (t, J = 7.6 Hz, 2H), 7.09–6.94 (m, 13H), 5.87 (d, J = 3.1 Hz, 1H), 4.75 (d, J = 11.4 Hz, 1H), 4.72 (d, J = 10.9 Hz, 1H), 4.67 (d, J = 10.9 Hz, 1H), 4.54 (d, J = 9.7 Hz, 1H), 4.31 (d, J = 11.4 Hz, 1H), 4.19 (d, J = 11.9 Hz, 1H), 4.12 (d, J = 11.9 Hz, 1H), 3.83 (t, J = 9.4 Hz, 1H), 3.51 (d, J = 6.0 Hz, 1H), 3.42–3.40 (m, 2H). HRMS (ESI) m/z calcd for C40H37DO6SNa [M + Na]+, 670.2350; found, 670.2344.
Phenyl 4-O-(p-Methylbenzoyl)-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (18).77
Compound 18 (0.057 g, 95%) was synthesized from 1 analogously to 19, as a colorless oil. [α]D22 = +20.9 (c 0.55, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.89–7.88 (m, 2H), 7.64–7.63 (m, 2H), 7.38–7.21 (m, 20H), 5.87 (d, J = 3.0 Hz, 1H), 4.84 (d, J = 11.2 Hz, 1H), 4.71 (br s, 2H), 4.69 (d, J = 9.5 Hz, 1H), 4.53 (d, J = 11.0 Hz, 1H), 4.51 (d, J = 11.7 Hz, 1H), 4.45 (d, J = 11.7 Hz, 1H), 3.89–3.87 (m, 1H), 3.74 (dd, J = 9.1, 3.2 Hz, 1H), 3.69 (t, J = 9.2 Hz, 1H), 3.68 (dd, J = 9.5, 5.9 Hz, 1H), 3.57 (dd, J = 9.5, 6.8 Hz, 1H), 2.44 (s, 3H). 13C NMR (150 MHz, CDCl3) δ 165.7, 143.9, 138.3, 137.6, 132.9, 132.8, 130.0, 129.1, 128.8, 128.4, 128.3, 128.3, 128.2, 128.2, 127.9, 127.7, 127.7, 127.6, 127.0, 87.1, 81.4, 76.5, 76.4, 75.7, 73.7, 71.7, 68.5, 67.1, 21.7. 1H NMR (600 MHz, C6D6) δ 8.09 (d, J = 8.0 Hz, 2H), 7.74–7.70 (m, 2H), 7.39 (d, J = 7.6 Hz, 2H), 7.28 (d, J = 7.6 Hz, 2H), 7.21 (t, J = 8.6 Hz, 2H), 7.15 (dd, J = 15.7, 8.0 Hz, 2H), 7.12–6.95 (m, 10H), 6.84 (d, J = 8.0 Hz, 2H), 5.88 (d, J = 3.2 Hz, 1H), 4.77 (d, J = 11.4 Hz, 1H), 4.73 (d, J = 10.9 Hz, 1H), 4.67 (d, J = 10.9 Hz, 1H), 4.55 (d, J = 9.7 Hz, 1H), 4.32 (d, J = 11.8 Hz, 1H), 4.20 (d, J = 11.8 Hz, 1H), 4.14 (d, J = 10.8 Hz, 1H), 3.86 (t, J = 9.4 Hz, 1H), 3.56 (dd, J = 9.4, 6.2, Hz, 1H), 3.52 (dd, J = 9.4, 6.5 Hz, 1H), 3.45–3.43 (m, 1H), 3.42–3.41 (m, 1H) 1.90 (s, 3H). IR (neat) ν 1720 cm–1 (C=O). HRMS (ESI) m/z calcd for C41H40O6SNa [M + Na]+, 683.2443; found, 683.2446.
Phenyl 4-O-(p-Methylbenzoyl)-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-18)
Compound 6S-d-18 (0.016 g, 65%) was synthesized from 6S-d-1 analogously to 18. 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 8.1 Hz, 2H), 7.64–7.63 (m, 2H), 7.39–7.19 (m, 20H), 5.86 (d, J = 3.0 Hz, 1H), 4.83 (d, J = 11.1 Hz, 1H), 4.70 (br s, 2H), 4.68 (d, J = 9.5 Hz, 1H), 4.50 (d, J = 11.1 Hz, 1H), 4.49 (d, J = 11.8 Hz, 1H), 4.43 (d, J = 11.8 Hz, 1H), 3.87 (d, J = 5.9 Hz, 1H), 3.74 (dd, J = 9.1, 3.2 Hz, 1H), 3.68 (t, J = 9.3 Hz, 1H), 3.66 (d, J = 6.0 Hz, 1H), 2.44 (s, 3H). 1H NMR (600 MHz, C6D6) δ 8.09 (d, J = 8.2 Hz, 2H), 7.75–7.67 (m, 2H), 7.39 (d, J = 7.2 Hz, 2H), 7.28 (d, J = 7.2 Hz, 2H), 7.22 (d, J = 7.2 Hz, 1H), 7.14 (t, J = 7.6 Hz, 2H), 7.11–6.95 (m, 10H), 6.84 (d, J = 8.1 Hz, 2H), 5.88 (d, J = 3.1 Hz, 1H), 4.77 (d, J = 11.4 Hz, 1H), 4.73 (d, J = 10.9 Hz, 1H), 4.67 (d, J = 10.8 Hz, 1H), 4.55 (d, J = 9.7 Hz, 1H), 4.32 (d, J = 11.4 Hz, 1H), 4.19 (d, J = 11.9 Hz, 1H), 4.14 (d, J = 11.9 Hz, 1H), 3.87 (t, J = 9.4 Hz, 1H), 3.54 (d, J = 6.2 Hz, 1H), 3.44–3.41 (m, 2H), 1.90 (s, 3H). HRMS (ESI) m/z calcd for C41H39DO6SNa [M + Na]+, 684.2506; found, 684.2499.
Phenyl 4-O-(p-Methoxybenzoyl)-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (19).77
To a stirred solution of 1 (0.050 g, 0.092 mmol) in dry pyridine (1.0 mL) was added 4-methoxy benzoyl chloride (0.025 mL, 0.184 mmol) and DMAP (1.0 mg, 0.009 mmol) at room temperature. The reaction mixture was stirred for 8 h before the solvents were evaporated under a high vacuum to give a crude product, which was dissolved in CH2Cl2 (10 mL) and washed with 1 N HCl (5 mL) solution, followed by sat. NaHCO3 solution, dried over Na2SO4, and concentrated under a high vacuum. Silica gel column chromatography (eluent: 20% ethyl acetate in hexane) afforded 19(61) (0.050 g, 80%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.97–7.95 (m, 2H), 7.66–7.62 (m, 2H), 7.40–7.39 (m, 2H), 7.36–7.20 (m, 16H), 6.94–6.92 (m, 2H), 5.87 (d, J = 3.0 Hz, 1H), 4.86 (d, J = 11.4 Hz, 1H), 4.74 (br s, 2H), 4.71 (d, J = 9.2 Hz, 1H), 4.53 (d, J = 11.0, 1H), 4.50 (d, J = 11.7 Hz, 1H), 4.46 (d, J = 11.7 Hz, 1H), 3.89 (s, 3H), 3.89–3.87 (m, 1H), 3.75 (dd, J = 9.2, 2.9 Hz, 1H), 3.71 (t, J = 9.2 Hz, 1H), 3.69 (dd, J = 9.6, 6.1 Hz, 1H), 3.59 (dd, J = 9.6, 6.5 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 8.11 (d, J = 8.7 Hz, 2H), 7.71 (d, J = 7.4 Hz, 2H), 7.40 (d, J = 7.4 Hz, 2H), 7.29 (d, J = 7.4 Hz, 2H), 7.23 (d, J = 7.3 Hz, 2H), 7.17–6.93 (m, 12H), 6.56 (d, J = 8.8 Hz, 2H), 5.88 (d, J = 3.0 Hz, 1H), 4.79 (d, J = 11.4 Hz, 1H), 4.76 (d, J = 10.8 Hz, 1H), 4.71 (d, J = 10.8 Hz, 1H), 4.57 (d, J = 9.7 Hz, 1H), 4.33 (d, J = 11.4 Hz, 1H), 4.21 (d, J = 11.9 Hz, 1H), 4.15 (d, J = 11.8 Hz, 1H), 3.87 (t, J = 9.4 Hz, 1H), 3.57 (dd, J = 9.4, 6.2 Hz, 1H), 3.54 (dd, J = 9.4, 6.4 Hz, 1H), 3.47–3.41 (m, 2H), 3.08 (s, 3H). IR (neat) ν 1728 cm–1 (C=O).
Phenyl 4-O-(p-Methoxybenzoyl)-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-19)
Compound 6S-d-19 (0.009 g, 71%) was synthesized from 6S-d-1 analogously to 19. 1H NMR (600 MHz, CDCl3) δ 7.98–7.91 (m, 2H), 7.69–7.58 (m, 2H), 7.39–7.18 (m, 18H), 6.91 (d, J = 8.9 Hz, 2H), 5.85 (d, J = 3.1 Hz, 1H), 4.83 (d, J = 11.2 Hz, 1H), 4.72 (br s, 2H), 4.68 (d, J = 9.5 Hz, 1H), 4.51 (d, J = 11.6 Hz), 4.49 (d, J = 11.6 Hz, 1H), 4.44 (d, J = 11.7 Hz, 1H), 3.89 (s, 3H), 3.87–3.85 (m, 1H), 3.73 (dd, J = 9.2, 3.2 Hz, 1H), 3.69 (t, J = 9.3 Hz, 1H), 3.66 (d, J = 6.2 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 8.11 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 7.3 Hz, 2H), 7.40 (d, J = 7.5 Hz, 2H), 7.29 (d, J = 7.4 Hz, 2H), 7.23 (d, J = 7.3 Hz, 2H), 7.17–6.93 (m, 12H), 6.55 (d, J = 8.8 Hz, 2H), 5.88 (d, J = 3.1 Hz, 1H), 4.79 (d, J = 11.4 Hz, 1H), 4.76 (d, J = 10.8 Hz, 1H), 4.71 (d, J = 10.8 Hz, 1H), 4.57 (d, J = 9.7 Hz, 1H), 4.33 (d, J = 11.4 Hz, 1H), 4.21 (d, J = 11.9 Hz, 1H), 4.15 (d, J = 11.9 Hz, 1H), 3.87 (t, J = 9.4 Hz, 1H), 3.55 (d, J = 6.1 Hz, 1H), 3.45–3.41 (m, 2H), 3.08 (s, 3H). HRMS (ESI) m/z calcd for C41H39DO7SNa [M + Na]+, 700.2455; found, 700.2449.
Phenyl 4-O-(p-Nitrobenzoyl)-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (20).77
Compound 20 (0.053 g, 84%) was synthesized from 1 analogously to 19, as a colorless oil. [α]D22 = +19.5 (c 1.95, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 8.26–8.24 (m, 2H), 8.07–8.06 (m, 2H), 7.65–7.63 (m, 2H), 7.39–7.19 (m, 18H), 5.90 (d, J = 2.3 Hz, 1H), 4.83 (d, J = 10.6 Hz, 1H), 4.78 (d, J = 10.3 Hz, 1H), 4.73 (d, J = 10.3 Hz, 1H), 4.70 (d, J = 9.5 Hz, 1H), 4.54 (d, J = 11.0 Hz, 1H), 4.52 (d, J = 11.7 Hz, 1H), 4.43 (d, J = 11.7 Hz, 1H), 3.91 (t, J = 6.1 Hz, 1H), 3.78 (dd, J = 9.2, 3.3 Hz, 1H), 3.69 (dd, J = 9.5, 5.7 Hz, 1H), 3.64 (t, J = 9.4 Hz, 1H), 3.56 (dd, J = 9.5, 7.2 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 163.8, 150.6, 138.1, 137.3, 137.3, 135.1, 133.3, 132.5, 130.9, 128.8, 128.4, 128.2, 128.1, 127.9, 127.9, 127.9, 127.8, 123.5, 87.1, 81.3, 76.5, 75.8, 75.7, 73.7, 72.1, 68.5, 67.9. 1H NMR (600 MHz, C6D6) δ 7.70–7.65 (m, 4H), 7.59–7.55 (m, 2H), 7.41 (d, J = 7.3 Hz, 2H), 7.25 (d, J = 7.2 Hz, 2H), 7.15 (dd, J = 16.1, 7.8 Hz, 4H), 7.08–6.91 (m, 10H), 5.79 (d, J = 3.1 Hz, 1H), 4.85 (d, J = 10.7 Hz, 1H), 4.77 (d, J = 10.8 Hz, 1H), 4.67 (d, J = 11.1 Hz, 1H), 4.52 (d, J = 9.7 Hz, 1H), 4.28 (d, J = 11.0 Hz, 1H), 4.24 (d, J = 11.0 Hz, 1H), 4.13 (d, J = 11.9 Hz, 1H), 3.73 (t, J = 9.3 Hz, 1H), 3.47 (dd, J = 8.9, 5.6 Hz, 1H), 3.42 (dd, J = 8.9, 7.0 Hz, 1H), 3.41–3.39 (m, 1H), 3.38–3.37 (m, 1H). IR (neat) ν 1729 cm–1 (C=O). HRMS (ESI) m/z calcd for C40H37NO8SNa [M + Na]+, 714.2138; found, 714.2120.
Phenyl 4-O-(p-Nitrobenzoyl)-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-20)
Compound 6S-d-20 (0.010 g, 83%) was synthesized from 6S-d-1 analogously to 20. 1H NMR (600 MHz, CDCl3) δ 8.26–8.24 (m, 2H), 8.07–8.06 (m, 2H), 7.65–7.64 (m, 2H), 7.44–7.18 (m, 18H), 5.90 (d, J = 3.1 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 4.78 (d, J = 10.8 Hz, 1H), 4.73 (d, J = 10.3 Hz, 1H), 4.70 (d, J = 9.7 Hz, 1H), 4.54 (d, J = 11.0 Hz, 1H), 4.52 (d, J = 11.7 Hz, 1H), 4.43 (d, J = 11.8 Hz, 1H), 3.91 (d, J = 5.7 Hz, 1H), 3.78 (dd, J = 9.1, 3.1 Hz, 1H), 3.67 (d, J = 5.7 Hz, 1H), 3.64 (t, J = 9.4 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.69–7.65 (m, 4H), 7.59–7.55 (m, 2H), 7.41 (d, J = 7.3 Hz, 2H), 7.25 (d, J = 7.3 Hz, 2H), 7.18–6.90 (m, 14H), 5.78 (d, J = 3.1 Hz, 1H), 4.85 (d, J = 10.7 Hz, 1H), 4.77 (d, J = 10.8 Hz, 1H), 4.67 (d, J = 11.1 Hz, 1H), 4.51 (d, J = 9.7 Hz, 1H), 4.28 (d, J = 11.1 Hz, 1H), 4.24 (d, J = 12.0 Hz, 1H), 4.12 (d, J = 11.9 Hz, 1H), 3.72 (t, J = 9.4 Hz, 1H), 3.45 (d, J = 5.7 Hz, 1H, H), 3.40–3.35 (m, 2H). HRMS (ESI) m/z calcd for C40H36NDO8SNa [M + Na]+, 715.2200; found, 715.2206.
Phenyl 2,3,4,6-Tetra-O-benzyl-1-thio-β-d-galactopyranoside (21)
Compound 21 was synthesized as described in the literature.851H NMR (600 MHz, CDCl3) δ 7.58–7.56 (m, 2H), 7.39–7.27 (m, 21H), 7.20–7.18 (m, 2H), 4.98 (d, J = 11.7 Hz, 1H), 4.79 (d, J = 10.3 Hz, 1H), 4.75 (d, J = 11.7 Hz, 1H), 4.74–4.73 (m, 1H), 4.73 (d, J = 11.7 Hz, 1H), 4.65 (d, J = 9.9 Hz, 1H), 4.61 (d, J = 11.7 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 4.43 (d, J = 11.7 Hz, 1H), 3.99 (dd, J = 2.8, 1.0 Hz, 1H), 3.95 (t, J = 9.5 Hz, 1H), 3.67 (m, 1H), 3.65 (m, 1H), 3.63–3.62 (m, 1H), 3.61 (dd, J = 2.8, 9.2 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.70–7.67 (m, 2H), 7.37 (d, J = 7.2 Hz, 2H), 7.30 (d, J = 7.2 Hz, 2H), 7.21 (d, J = 7.2 Hz, 2H), 7.18 (d, J = 7.1 Hz, 2H), 7.14–7.09 (m, 8H), 7.05 (dt, J = 14.7, 7.4 Hz, 4H), 6.99–6.89 (m, 3H), 4.95 (d, J = 11.4 Hz, 1H), 4.79 (d, J = 10.7 Hz, 1H), 4.64 (d, J = 10.8 Hz, 1H), 4.59 (d, J = 9.6 Hz, 1H), 4.52 (d, J = 11.4 Hz, 1H), 4.42 (d, J = 11.9 Hz, 1H), 4.37 (d, J = 11.9 Hz, 1H), 4.22 (d, J = 11.8 Hz, 1H), 4.16 (d, J = 11.8 Hz, 1H), 4.11 (t, J = 9.4 Hz, 1H), 3.78 (dd, J = 2.9, 1.1 Hz, 1H), 3.66 (dd, J = 9.1, 7.4 Hz, 1H), 3.57 (dd, J = 9.1, 5.7 Hz, 1H), 3.32 (ddd, J = 7.3, 5.7, 1.0 Hz, 1H), 3.29 (dd, J = 9.2, 2.8 Hz, 1H).
Phenyl (6S)-[6-2H1]-2,3,4,6-Tetra-O-benzyl-1-thio-β-d-galactopyranoside (6S-d-21)
Compound 6S-d-21 (0.012 g, 70%) was synthesized from 6S-d-1 analogously to 21. 1H NMR (600 MHz, CDCl3) δ 7.58–7.56 (m, 2H), 7.40–7.26 (m, 21H), 7.21–7.16 (m, 2H), 4.97 (d, J = 11.5 Hz, 1H), 4.79 (d, J = 10.2 Hz, 1H), 4.75 (d, J = 11.7 Hz, 1H), 4.74–4.73 (m, 1H), 4.72 (d, J = 11.7 Hz, 1H), 4.65 (d, J = 9.7 Hz, 1H), 4.61 (d, J = 11.5 Hz, 1H), 4.48 (d, J = 11.7 Hz, 1H), 4.42 (d, J = 11.7 Hz, 1H), 3.99 (d, J = 2.6 Hz, 1H), 3.94 (t, J = 9.5 Hz, 1H), 3.65 (d, J = 5.8 Hz, 1H, H), 3.62–3.61 (m, 2H). 1H NMR (600 MHz, C6D6) δ 7.70–7.67 (m, 2H), 7.37 (d, J = 7.6 Hz, 2H), 7.30 (d, J = 7.5 Hz, 2H), 7.21–7.16 (m, 4H), 7.15–7.02 (m, 12H), 6.99–6.89 (m, 3H), 4.95 (d, J = 11.4 Hz, 1H), 4.79 (d, J = 10.7 Hz, 1H), 4.64 (d, J = 10.8 Hz, 1H), 4.59 (d, J = 9.6 Hz, 1H), 4.52 (d, J = 11.4 Hz, 1H), 4.42 (d, J = 11.9 Hz, 1H), 4.37 (d, J = 11.9 Hz, 1H), 4.22 (d, J = 11.9 Hz, 1H), 4.16 (d, J = 11.8 Hz, 1H), 4.11 (t, J = 9.4 Hz, 1H), 3.78 (d, J = 2.6 Hz, 1H), 3.55 (d, J = 5.6 Hz, 1H), 3.33–3.26 (m, 2H). HRMS (ESI) m/z calcd for C40H39DO5SNa [M + Na]+, 656.2557; found, 656.2548.
Phenyl 2,3,6-Tri-O-benzyl-4-O-(1′,1′,1′-trifluoroethyl)-1-thio-β-d-galactopyranoside (22).77
To a solution of phenyl 2,3,6-tri-O-benzyl-1-thio-d-glucopyranoside (2) (0.05 g, 0.092 mmol) in dry CH2Cl2 was added pyridine (0.03 mL, 0.37 mmol) and trifluoromethanesulfonic anhydride (0.02 mL, 0.12 mmol) at 0 °C. The reaction mixture was stirred for 1 h before it was quenched with water. The reaction mixture was diluted with CH2Cl2 (10 mL), washed with brine (5.0 mL), dried over Na2SO4, and concentrated under a high vacuum. Without further purification, the triflate was dissolved in dry DMF (1.0 mL) and freshly prepared sodium trifluoroethoxide (0.03 g, 0.27 mmol) was added at room temperature. The reaction mixture was stirred for 4 h at room temperature before TLC (10% ethyl acetate in hexane) showed completion. The reaction mixture was quenched with saturated aqueous NH4Cl solution, extracted with ethyl acetate (2 × 10 mL), dried over Na2SO4, and concentrated. Silica gel column chromatography (eluent: 20% EtOAc in hexane) afforded 22 (0.035 g, 61% over two steps) as a semisolid. [α]D22 = −11.0 (c 0.55, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.56–7.55 (m, 2H), 7.37–7.21 (m, 18H), 4.79 (d, J = 10.2 Hz, 1H), 4.75 (d, J = 11.7 Hz, 1H), 4.68–4.67 (m, 1H), 4.66 (d, J = 10.3 Hz, 1H), 4.61 (d, J = 9.5 Hz, 1H), 4.52 (d, J = 11.7 Hz, 1H), 4.49 (d, J = 11.7 Hz, 1H), 4.28 (dq, J = 12.1, 8.9 Hz, 1H), 3.93 (d, J = 2.2 Hz, 1H), 3.92–3.88 (m, 1H), 3.85 (t, J = 9.5 Hz, 1H), 3.74 (d, J = 9.2, 7.7 Hz, 1H), 3.69 (dd, J = 9.2, 5.5 Hz, 1H), 3.61–3.59 (m, 1H), 3.57 (dd, J = 9.2, 2.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 138.1, 137.8, 137.7, 133.8, 131.5, 129.7, 128.9, 128.8, 128.5, 128.4, 128.4, 128.3, 128.0, 127.9, 127.9, 127.8, 127.7, 127.5, 127.2, 124.7, 122.8, 87.7, 83.7, 77.1, 76.6, 76.0, 75.6, 73.6, 73.4, 69.4 (q, J = 34.1 Hz, CF3CH2), 68.1. 19F NMR (376 MHz, CDCl3) δ −74.7 (t, J = 8.7 Hz, CF3CH2O). 1H NMR (600 MHz, C6D6) δ 7.66 (d, J = 8.2 Hz, 2H), 7.35 (d, J = 7.6 Hz, 2H), 7.19 (d, J = 7.6 Hz, 2H), 7.16–7.00 (m, 13H), 6.93 (dd, J = 10.6, 4.2 Hz, 1H), 4.80 (d, J = 10.7 Hz, 1H), 4.54 (d, J = 10.7 Hz, 1H), 4.48 (d, J = 9.9 Hz, 1H), 4.38 (d, J = 11.1 Hz, 1H), 4.23–4.19 (m, 3H), 4.14 (dt, J = 17.8, 8.9 Hz, 1H), 3.95 (t, J = 9.4 Hz, 1H), 3.68 (dd, J = 9.1, 7.6 Hz, 1H), 3.66–3.60 (m, 1H), 3.57 (d, J = 1.7 Hz, 1H), 3.55 (dd, J = 9.1, 5.7 Hz, 1H), 3.22–3.18 (m, 1H), 3.15 (dd, J = 9.2, 2.6 Hz, 1H). HRMS (ESI) m/z calcd for C35H35O5F3SNa [M + Na]+, 647.2055; found, 647.2067.
Phenyl (6S)-[6-2H1]-2,3,6-Tri-O-benzyl-4-O-(1′,1′,1′-trifluoroethyl)-1-thio-β-d-galactopyranoside (6S-d-22)
Compound 6S-d-22 (0.005 g, 30% over two steps) was synthesized from 6S-d-1 analogously to 22. 1H NMR (600 MHz, CDCl3) δ 7.56–7.55 (m, 2H), 7.37–7.21 (m, 18H), 4.78 (d, J = 10.1 Hz, 1H), 4.75 (d, J = 11.1 Hz, 1H), 4.67 (d, J = 11.8 Hz, 1H), 4.65 (d, J = 10.2 Hz, 1H), 4.60 (d, J = 9.7 Hz, 1H), 4.51 (d, J = 11.7 Hz, 1H), 4.49 (d, J = 11.6 Hz, 1H), 4.27 (dq, J = 12.0, 9.0 Hz, 1H), 3.93 (d, J = 2.3 Hz, 1H), 3.92–3.87 (m, 1H), 3.84 (t, J = 9.4 Hz, 1H), 3.66 (d, J = 5.5 Hz, 1H), 3.60–3.59 (m, 1H), 3.57 (dd, J = 9.5, 2.6 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.67 (d, J = 7.4 Hz, 2H), 7.35 (d, J = 7.4 Hz, 2H), 7.21–7.19 (m, 2H), 7.16–6.90 (m, 14H), 4.80 (d, J = 10.7 Hz, 1H), 4.54 (d, J = 10.6 Hz, 1H), 4.48 (d, J = 9.6 Hz, 1H), 4.37 (d, J = 11.8 Hz, 1H), 4.22–4.19 (m, 3H), 4.18–4.10 (m, 1H), 3.95 (t, J = 9.4 Hz, 1H), 3.66–3.60 (m, 1H), 3.57 (d, J = 2.3 Hz, 1H), 3.53 (d, J = 5.6 Hz, 1H, H), 3.19 (d, J = 6.0 Hz, 1H), 3.15 (dd, J = 9.2, 2.7 Hz, 1H). HRMS (ESI) m/z calcd for C35H34DF3O5SNa [M + Na]+, 648.2118; found, 648.2102.
Phenyl 4-O-Acetyl-2,3,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (23)
Compound 23 was synthesized as described in the literature.861H NMR (600 MHz, CDCl3) δ 7.59–7.55 (m, 2H), 7.41–7.21 (m, 18H), 5.01 (dd, J = 9.4, 8.7 Hz, 1H), 4.88 (d, J = 10.3 Hz, 1H), 4.81 (d, J = 11.4 Hz, 1H), 4.70 (d, J = 10.3 Hz, 1H), 4.69 (d, J = 9.8 Hz, 1H), 4.64 (d, J = 11.4 Hz, 1H), 4.51 (s, 2H), 3.67 (dd, J = 9.4, 8.8 Hz, 1H), 3.61–3.57 (m, 3H), 3.55 (dd, J = 9.8, 8.8 Hz, 1H), 1.85 (s, 3H). 1H NMR (600 MHz, C6D6) δ 7.69–7.64 (m, 2H), 7.43–7.39 (m, 2H), 7.33–7.29 (m, 2H), 7.28–7.24 (m, 2H), 7.21–6.94 (m, 12H), 5.33 (dd, J = 10.6, 9.2 Hz, 1H), 4.86 (d, J = 10.9 Hz, 1H), 4.76 (d, J = 11.8 Hz, 1H), 4.64 (d, J = 9.6 Hz, 1H), 4.59–4.57 (m, 2H), 4.31 (s, 2H), 3.58 (dd, J = 10.6, 3.2 Hz, 1H), 3.54 (dd, J = 10.6, 5.8 Hz, 1H), 3.52 (dd, J = 9.2, 8.7 Hz, 1H), 3.48 (dd, J = 9.6, 8.7 Hz, 1H), 3.36 (ddd, J = 10.1, 5.8, 3.2 Hz, 1H), 1.54 (s, 3H). IR (neat) ν 1745 cm–1 (C=O).
Phenyl 2,3,6-Tri-O-benzyl-4-O-trifluoroacetyl-1-thio-β-d-glucopyranoside (24)
Compound 2 (0.009 g, 0.016 mmol) was dissolved in anhydrous pyridine (0.05 mL) and cooled to 0 °C before the addition of trifluoroacetic anhydride (4.5 μL, 0.032 mmol). The reaction mixture was then stirred for 4 h under an argon atmosphere and concentrated to dryness. The crude residue was purified by silica gel column chromatography, eluting with hexane/ethyl acetate (7:3), to obtain 24 (0.008 g, 74%) as a colorless oil. [α]D22 = +37.0 (c 0.33, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.57–7.54 (m, 2H), 7.41–7.17 (m, 18H), 5.24 (dd, J = 9.8, 9.6 Hz, 1H), 4.91 (d, J = 10.2 Hz, 1H), 4.81 (d, J = 10.8 Hz, 1H), 4.70 (d, J = 10.2 Hz, 1H), 4.69 (d, J = 9.8 Hz, 1H), 4.62 (d, J = 10.8 Hz, 1H), 4.54 (d, J = 11.6 Hz, 1H), 4.47 (d, J = 11.6 Hz, 1H), 3.77 (dd, J = 9.6, 8.8 Hz, 1H), 3.69 (ddd, J = 9.8, 4.7, 3.6 Hz, 1H), 3.62 (dd, J = 10.7, 3.6 Hz, 1H), 3.58 (dd, J = 10.7, 4.7 Hz, 1H), 3.58 (dd, J = 9.8, 8.8 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 156.5 (d, J = 42.5 Hz), 137.8, 137.6, 137.5, 133.2, 132.4, 129.2, 128.6, 128.5, 128.3, 128.2, 128.1, 128.01, 127.95, 127.9, 114.6 (d, J = 285.8 Hz), 87.9, 83.2, 80.8, 76.5, 75.9, 75.7, 74.7, 73.9, 69.1. 19F NMR (376 MHz, CDCl3) δ −75.0 (OCOCF3). 1H NMR (600 MHz, C6D6) δ 7.61–7.55 (m, 2H), 7.40–7.36 (m, 2H), 7.27–6.94 (m, 16H), 5.40 (dd, J = 10.0, 9.5 Hz, 1H), 4.83 (d, J = 10.6 Hz, 1H), 4.71 (d, J = 11.3 Hz, 1H), 4.55 (d, J = 11.3 Hz, 1H), 4.50 (d, J = 10.6 Hz, 1H), 4.46 (d, J = 9.7 Hz, 1H), 4.26 (d, J = 12.0 Hz, 1H), 4.18 (d, J = 12.0 Hz, 1H), 3.43 (dd, J = 9.5, 8.7 Hz, 1H), 3.37 (dd, J = 10.6, 3.5 Hz, 1H), 3.34 (dd, J = 9.7, 8.7 Hz, 1H), 3.30 (dd, J = 10.6, 4.3 Hz, 1H), 3.00 (ddd, J = 10.0, 4.3, 3.5 Hz, 1H). IR (neat) ν 1793 cm–1 (C=O). HRMS (ESI) m/z calcd for C35H33O6SF3Na [M + Na]+, 661.1848; found, 661.1843.
Phenyl 4-O-Benzoyl-2,3,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (25)
Compound 1 (0.010 g, 0.018 mmol) was dissolved in anhydrous dichloromethane (0.5 mL) and cooled to 0 °C. Anhydrous pyridine (15 μL, 0.18 mmol) was added followed by Tf2O (10 μL, 0.060 mmol) and the reaction mixture stirred for 1.5 h before it was quenched with water at 0 °C. The reaction mixture was diluted with water and extracted with dichloromethane. The combined organic layers were dried over anhydrous Na2SO4 and concentrated to dryness. The crude residue was then dissolved in anhydrous DMF (0.15 mL), and sodium benzoate (7.9 mg, 0.055 mmol) was added. The reaction mixture was stirred under an argon atmosphere at room temperature for 3 h and then diluted with water and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous Na2SO4, and concentrated to dryness. The crude residue was purified by column chromatography over silica gel, eluting with hexane/ethyl acetate (8:2) to obtain 25 (0.008 g, 71%) with spectral data consistent with the literature.761H NMR (600 MHz, CDCl3) δ 7.98–7.93 (m, 2H), 7.64–7.53 (m, 3H), 7.44–7.04 (m, 20H), 5.29 (dd, J = 10.1, 9.4 Hz, 1H), 4.90 (d, J = 10.2 Hz, 1H), 4.77 (d, J = 9.8 Hz, 1H), 4.75 (d, J = 11.0 Hz, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.62 (d, J = 11.0 Hz, 1H), 4.49 (d, J = 11.6 Hz, 1H), 4.46 (d, J = 11.6 Hz, 1H), 3.83 (dd, J = 9.4, 8.7 Hz, 1H), 3.79–3.75 (m, 1H), 3.66–3.64 (m, 2H), 3.62 (dd, J = 9.8, 8.7 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 8.07–8.04 (m, 2H), 7.73–7.68 (m, 2H), 7.45–7.42 (m, 2H), 7.27–6.94 (m, 19H), 5.65 (dd, J = 10.0, 9.4 Hz, 1H), 4.89 (d, J = 10.8 Hz, 1H), 4.72 (d, J = 9.8 Hz, 1H), 4.72 (d, J = 11.4 Hz, 1H), 4.62 (d, J = 11.4 Hz, 1H), 4.61 (d, J = 10.8 Hz, 1H), 4.26–4.20 (m, 2H), 3.68 (dd, J = 9.4, 8.6 Hz, 1H), 3.62 (dd, J = 10.7, 2.9 Hz, 1H), 3.58 (dd, J = 10.7, 6.0 Hz, 1H), 3.54 (dd, J = 9.8, 8.6 Hz, 1H), 3.48 (ddd, J = 10.0, 6.0, 2.9 Hz, 1H). IR (neat) ν 1726 cm–1 (C=O).
Phenyl 4-O-Benzoyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-25)
Compound 6S-d-25 (0.030 g, 74%) was synthesized from 6S-d-2 analogously to 25. 1H NMR (600 MHz, CDCl3) δ 7.97–7.93 (m, 2H), 7.62–7.54 (m, 3H), 7.45–7.04 (m, 20H), 5.29 (dd, J = 10.0, 9.4 Hz, 1H), 4.90 (d, J = 10.2 Hz, 1H), 4.77 (d, J = 9.8 Hz, 1H), 4.77 (d, J = 10.9 Hz, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.62 (d, J = 10.9 Hz, 1H), 4.49 (d, J = 11.7 Hz, 1H), 4.46 (d, J = 11.7 Hz, 1H), 3.83 (dd, J = 9.4, 8.8 Hz, 1H), 3.76 (dd, J = 10.0, 6.4 Hz, 1H), 3.65–3.60 (m, 2H). 1H NMR (600 MHz, C6D6) δ 8.07–8.03 (m, 2H), 7.73–7.68 (m, 2H), 7.46–7.41 (m, 2H), 7.27–6.92 (m, 19H), 5.64 (dd, J = 10.0, 9.4 Hz, 1H), 4.89 (d, J = 10.7 Hz, 1H), 4.72 (d, J = 9.8 Hz, 1H), 4.72 (d, J = 11.4 Hz, 1H), 4.62 (d, J = 11.4 Hz, 1H), 4.61 (d, J = 10.7 Hz, 1H), 4.25 (d, J = 11.8 Hz, 1H), 4.22 (d, J = 11.8 Hz, 1H), 3.68 (dd, J = 9.4, 8.7 Hz, 1H), 3.56 (d, J = 6.1 Hz, 1H), 3.54 (dd, J = 9.8, 8.7 Hz, 1H), 3.47 (dd, J = 10.0, 6.1 Hz, 1H). HRMS (ESI) m/z calcd for C40H37DO6SNa [M + Na]+, 670.2350; found, 670.2354.
Phenyl 4-O-Benzenesulfonyl-2,3,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (26)
Compound 2 (0.007 g, 0.014 mmol) and 4-dimethylaminopyridine (catalytic amount) were dissolved in anhydrous pyridine (0.1 mL) and cooled to 0 °C. Benzenesulfonyl chloride (5 μL, 0.039 mmol) was added at 0 °C, and the reaction mixture was stirred overnight under an argon atmosphere at room temperature. The reaction mixture was concentrated to dryness, and the crude residue was dissolved in ethyl acetate and filtered through a cotton plug before the organic layer was concentrated to dryness. The crude residue was then purified by silica gel column chromatography, eluting with hexane/ethyl acetate (9:1 to 4:1) to obtain 26 (0.005 g, 57%) as a colorless oil. [α]D22 = +13.3 (c 0.15, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.88–7.81 (m, 2H), 7.59–7.16 (m, 23H), 4.82 (dd, J = 9.9, 9.2 Hz, 1H), 4.81 (d, J = 10.2 Hz, 1H), 4.70 (d, J = 11.2 Hz, 1H), 4.63 (d, J = 9.8 Hz, 1H), 4.60 (d, J = 10.2 Hz, 1H), 4.55 (d, J = 11.2 Hz, 1H), 4.49 (d, J = 11.6 Hz, 1H), 4.46 (d, J = 11.6 Hz, 1H), 3.71 (dd, J = 10.8, 2.1 Hz, 1H), 3.66 (dd, J = 9.2, 8.7 Hz, 1H), 3.62 (ddd, J = 9.9, 5.6, 2.1 Hz, 1H), 3.54 (dd, J = 9.8, 8.7 Hz, 1H), 3.51 (dd, J = 10.8, 5.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ 133.7, 132.2, 129.1, 129.1, 128.5, 128.5, 128.3, 128.1, 127.8, 127.7, 127.6, 127.5, 87.5, 83.7, 81.0, 78.0, 77.8, 75.6, 75.4, 73.5, 68.9. 1H NMR (600 MHz, C6D6) δ 7.83–7.77 (m, 2H), 7.67–7.64 (m, 2H), 7.43–6.60 (m, 23H), 5.08 (dd, J = 9.9, 9.1 Hz, 1H), 4.80 (d, J = 10.4 Hz, 1H), 4.64 (d, J = 11.7 Hz, 1H), 4.49 (d, J = 9.4 Hz, 1H), 4.47 (d, J = 11.7 Hz, 1H), 4.44 (d, J = 10.4 Hz, 1H), 4.38 (d, J = 11.4 Hz, 1H), 4.36 (d, J = 11.4 Hz, 1H), 3.81 (dd, J = 10.8, 2.0 Hz, 1H), 3.69 (dd, J = 10.8, 5.3 Hz, 1H), 3.45–3.35 (m, 2H), 3.24 (ddd, J = 9.9, 5.3, 2.0 Hz, 1H). IR (neat) ν 1360 cm–1 (S=O), 1188 cm–1 (S=O). HRMS (ESI) m/z calcd for C39H38O7S2Na [M + Na]+, 705.1957; found, 705.1954.
Phenyl 4-O-Benzenesulfonyl-(6S)-[6-2H1]-2,3,6-tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-26)
Compound 6S-d-26 (0.005 mg, 62%) was synthesized from 6S-d-2 analogously to 26. 1H NMR (600 MHz, CDCl3) δ 7.87–7.81 (m, 2H), 7.60–7.17 (m, 23H), 4.81 (dd, J = 9.9, 9.2 Hz, 1H), 4.81 (d, J = 10.1 Hz, 1H), 4.70 (d, J = 10.8 Hz, 1H), 4.64 (d, J = 9.7 Hz, 1H), 4.60 (d, J = 10.1 Hz, 1H), 4.55 (d, J = 10.8 Hz, 1H), 4.49 (d, J = 11.8 Hz, 1H), 4.45 (d, J = 11.8 Hz, 1H), 3.66 (dd, J = 9.2, 8.7 Hz, 1H), 3.61 (dd, J = 9.9, 5.7 Hz, 1H), 3.54 (dd, J = 9.7, 8.7 Hz, 1H), 3.50 (d, J = 5.7 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.82–7.79 (m, 2H), 7.67–7.64 (m, 2H), 7.43–6.62 (m, 21H), 5.07 (dd, J = 9.9, 9.0 Hz, 1H), 4.80 (d, J = 10.6 Hz, 1H), 4.64 (d, J = 11.6 Hz, 1H), 4.49 (d, J = 9.5 Hz, 1H), 4.47 (d, J = 11.6 Hz, 1H), 4.45 (d, J = 10.6 Hz, 1H), 4.39 (d, J = 11.5 Hz, 1H), 4.36 (d, J = 11.5 Hz, 1H), 3.67 (d, J = 5.3 Hz, 1H), 3.44–3.36 (m, 2H), 3.24 (dd, J = 9.9, 5.3 Hz, 1H). HRMS (ESI) m/z calcd for C39H37DO7S2Na [M + Na]+, 706.2019; found, 706.2014.
Phenyl 2,3,4,6-Tetra-O-benzyl-1-thio-β-d-glucopyranoside (27)
Compound 27 was synthesized as described in the literature.871H NMR (600 MHz, CDCl3) δ 7.61–7.58 (m, 2H), 7.41–7.18 (m, 23H), 4.90 (d, J = 10.9 Hz, 1H), 4.89 (d, J = 10.1 Hz, 1H), 4.85 (d, J = 10.9 Hz, 1H), 4.83 (d, J = 10.9 Hz, 1H), 4.73 (d, J = 10.1 Hz, 1H), 4.67 (d, J = 9.8 Hz, 1H), 4.61 (d, J = 12.0 Hz, 1H), 4.60 (d, J = 10.9 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 3.80 (dd, J = 10.8, 1.9 Hz, 1H), 3.73 (dd, J = 10.8, 4.8 Hz, 1H), 3.71 (dd, J = 8.9, 8.8 Hz, 1H), 3.66 (dd, J = 9.6, 8.9 Hz, 1H), 3.52 (dd, J = 9.8, 8.8 Hz, 1H), 3.51 (ddd, J = 9.6, 4.8, 1.9 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.73–7.70 (m, 2H), 7.46–7.42 (m, 2H), 7.35–7.27 (m, 4H), 7.23–6.95 (m, 17H), 4.95 (d, J = 10.7 Hz, 1H), 4.89 (d, J = 11.3 Hz, 1H), 4.83–4.79 (m, 2H), 4.72 (d, J = 10.7 Hz, 1H), 4.65 (d, J = 9.6 Hz, 1H), 4.56 (d, J = 11.3 Hz, 1H), 4.44 (d, J = 12.0 Hz, 1H), 4.35 (d, J = 12.0 Hz, 1H), 3.70 (dd, J = 9.8, 9.0 Hz, 1H), 3.64–3.62 (m, 2H), 3.61 (dd, J = 9.0, 8.7 Hz, 1H), 3.56 (dd, J = 9.6, 8.7 Hz, 1H), 3.27–3.23 (m, 1H).
Phenyl (6S)-[6-2H1]-2,3,4,6-Tetra-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-27)
A solution of compound 6S-d-2 (0005 g, 0.008 mmol) in anhydrous DMF (0.2 mL) was cooled to 0 °C and treated with NaH (60%, 1 mg, 0.025 mmol) and benzyl bromide (5.1 μL, 0.025 mmol), respectively. The reaction mixture was then stirred for 3.5 h under an argon atmosphere at room temperature and quenched with water at 0 °C. The aqueous layer was extracted with ethyl acetate, and the organic layer was washed with brine, treated with anhydrous Na2SO4, and concentrated to dryness. The crude residue was purified by silica gel column chromatography, eluting with hexane/ethyl acetate (19:1 to 4:1) to obtain 6S-d-27 (0.004 g, 80%). 1H NMR (600 MHz, CDCl3) δ 7.60–7.56 (m, 2H), 7.41–7.17 (m, 23H), 4.90 (d, J = 10.9 Hz, 1H), 4.89 (d, J = 10.3 Hz, 1H), 4.85 (d, J = 11.0 Hz, 1H), 4.83 (d, J = 10.8 Hz, 1H), 4.73 (d, J = 10.3 Hz, 1H), 4.67 (d, J = 9.8 Hz, 1H), 4.61 (d, J = 12.0 Hz, 1H), 4.59 (d, J = 10.8 Hz, 1H), 4.55 (d, J = 12.0 Hz, 1H), 3.73–3.69 (m, 2H), 3.67–3.61 (m, 1H), 3.51 (dd, J = 9.8, 8.7 Hz, 1H), 3.50 (dd, J = 9.7, 4.9 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.75–7.67 (m, 2H), 7.46–7.41 (m, 2H), 7.35–6.93 (m, 21H), 4.96 (d, J = 10.5 Hz, 1H), 4.89 (d, J = 11.4 Hz, 1H), 4.83–4.78 (m, 2H), 4.72 (d, J = 10.7 Hz, 1H), 4.65 (d, J = 9.6 Hz, 1H), 4.56 (d, J = 11.4 Hz, 1H), 4.43 (d, J = 12.0 Hz, 1H), 4.36 (d, J = 12.0 Hz, 1H), 3.70 (dd, J = 9.7, 9.2 Hz, 1H), 3.63–3.59 (m, 2H), 3.56 (dd, J = 9.6, 8.8 Hz, 1H), 3.25 (dd, J = 9.7, 4.5 Hz, 1H). HRMS (ESI) m/z calcd for C40H39DO5SNa [M + Na]+, 656.2557; found, 656.2559.
Ethyl 6-O-Acetyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (28)
Compound 28 was synthesized as described in the literature.331H NMR (600 MHz, CDCl3) δ 7.39–7.24 (m, 15H), 4.95 (d, J = 10.9 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 4.86 (d, J = 10.8 Hz, 1H), 4.85 (d, J = 10.9 Hz, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.57 (d, J = 10.8 Hz, 1H), 4.47 (d, J = 9.8 Hz, 1H), 4.33 (dd, J = 11.9, 1.9 Hz, 1H), 4.20 (dd, J = 11.9, 5.1 Hz, 1H), 3.71 (dd, J = 8.8, 8.7 Hz, 1H), 3.54 (dd, J = 9.8, 8.7 Hz, 1H), 3.51 (ddd, J = 9.8, 5.1, 1.9 Hz, 1H), 3.44 (dd, J = 9.8, 8.8 Hz, 1H), 2.81–2.70 (m, 2H), 2.04 (s, 3H), 1.33 (t, J = 7.4 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 7.43–7.40 (m, 2H), 7.34–7.32 (m, 2H), 7.27–7.25 (m, 2H), 7.20–7.05 (m, 9H), 4.99 (d, J = 10.6 Hz, 1H), 4.95 (d, J = 11.2 Hz, 1H), 4.81 (d, J = 11.2 Hz, 1H), 4.81 (d, J = 11.2 Hz, 1H), 4.70 (d, J = 10.6 Hz, 1H), 4.49 (d, J = 11.2 Hz, 1H), 4.43 (dd, J = 11.9, 2.2 Hz, 1H), 4.35 (d, J = 9.7 Hz, 1H), 4.26 (dd, J = 11.9, 5.4 Hz, 1H), 3.61 (dd, J = 8.9, 8.8 Hz, 1H), 3.48 (dd, J = 9.8, 8.8 Hz, 1H), 3.47 (dd, J = 9.7, 8.9 Hz, 1H), 3.26 (ddd, J = 9.8, 5.4, 2.2 Hz, 1H), 2.59 (dq, J = 12.6, 7.4 Hz, 1H), 2.49 (dq, J = 12.6, 7.4 Hz, 1H), 1.64 (s, 3H), 1.13 (t, J = 7.4 Hz, 3H). IR (neat) ν 1742 cm–1 (C=O).
Ethyl 2,3,4-Tri-O-benzyl-6-O-pivolyl-1-thio-β-d-glucopyranoside (29)
Compound 3(33) (0.008 g, 0.016 mmol) was dissolved in anhydrous pyridine (0.05 mL) and treated with pivolyl chloride (2.5 μL, 0.020 mmol). The reaction mixture was then stirred under an argon atmosphere at room temperature overnight and concentrated to dryness. The crude residue was purified by silica gel column chromatography, eluting with hexane/ethyl acetate (4:1 to 7:3) to obtain 29 (0.007 g, 92%) as a colorless oil. [α]D22 = +14.0 (c 0.57, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.39–7.25 (m, 15H), 4.94 (d, J = 10.8 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 4.88 (d, J = 10.7 Hz, 1H), 4.85 (d, J = 10.8 Hz, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.58 (d, J = 10.7 Hz, 1H), 4.48 (d, J = 9.8 Hz, 1H), 4.44 (dd, J = 11.9, 1.8 Hz, 1H), 4.12 (dd, J = 11.9, 5.6 Hz, 1H), 3.71 (dd, J = 8.8, 8.7 Hz, 1H), 3.53 (ddd, J = 9.9, 5.6, 1.8 Hz, 1H), 3.50 (dd, J = 9.9, 8.7 Hz, 1H), 3.44 (dd, J = 9.8, 8.8 Hz, 1H), 2.78 (dq, J = 12.7, 7.4 Hz, 1H), 2.70 (dq, J = 12.7, 7.4 Hz, 1H), 1.32 (t, J = 7.4 Hz, 3H), 1.21 (s, 9H). 13C NMR (150 MHz, CDCl3) δ 178.2, 138.4, 138.0, 137.8, 128.7, 128.6, 128.6, 128.4, 128.2, 128.0, 128.0, 127.9, 86.7, 84.8, 81.9, 78.2, 76.0, 75.7, 75.4, 63.5, 27.4, 24.9, 15.4. 1H NMR (600 MHz, C6D6) δ 7.45–7.41 (m, 2H), 7.35–7.31 (m, 2H), 7.27 (dd, J = 8.5, 1.7 Hz, 2H), 7.19–7.05 (m, 9H), 4.99 (d, J = 10.6 Hz, 1H), 4.95 (d, J = 11.2 Hz, 1H), 4.85 (d, J = 11.1 Hz, 1H), 4.82 (d, J = 11.2 Hz, 1H), 4.71 (d, J = 10.6 Hz, 1H), 4.55 (dd, J = 11.9, 2.1 Hz, 1H), 4.50 (d, J = 11.1 Hz, 1H), 4.34 (d, J = 9.7 Hz, 1H), 4.23 (dd, J = 11.9, 5.7 Hz, 1H), 3.60 (dd, J = 9.0, 8.8 Hz, 1H), 3.48 (dd, J = 9.7, 8.8 Hz, 1H), 3.46 (dd, J = 9.8, 9.0 Hz, 1H), 3.26 (ddd, J = 9.8, 5.7, 2.1 Hz, 1H), 2.63 (dq, J = 12.7, 7.4 Hz, 1H), 2.49 (dq, J = 12.7, 7.4 Hz, 1H), 1.18 (s, 9H), 1.16 (t, J = 7.4 Hz, 3H). IR (neat) ν 1731 cm–1 (C=O). HRMS (ESI) m/z calcd for C34H42O6SNa [M + Na]+, 601.2600; found, 601.2602.
Ethyl 2,3,4-Tri-O-benzyl-6-O-trifluoroacetyl-1-thio-β-d-glucopyranoside (30)
Compound 30 was synthesized using the same procedure as described for the synthesis of compound 24 from compound 3 (0.007 g, 0.014 mmol) and trifluoroacetic anhydride (3.8 μL, 0.028 mmol) in anhydrous pyridine (0.05 mL). After purification by silica gel column chromatography, eluting with hexane/ethyl acetate (7:3), 30 (6.0 mg, 72%) was obtained as a colorless oil. [α]D22 = +16.6 (c 0.31, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.39–7.23 (m, 15H), 4.96 (d, J = 10.9 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 4.90 (d, J = 11.1 Hz, 1H), 4.85 (d, J = 10.9 Hz, 1H), 4.74 (d, J = 10.2 Hz, 1H), 4.56 (d, J = 11.1 Hz, 1H), 4.54 (dd, J = 11.6, 2.1 Hz, 1H), 4.48 (d, J = 9.8 Hz, 1H), 4.32 (dd, J = 11.6, 6.3 Hz, 1H), 3.72 (dd, J = 8.9, 8.8 Hz, 1H), 3.58 (ddd, J = 9.9, 6.3, 2.1 Hz, 1H), 3.49 (dd, J = 9.9, 8.9 Hz, 1H), 3.45 (dd, J = 9.8, 8.8 Hz, 1H), 2.75 (dq, J = 12.8, 7.4 Hz, 1H), 2.68 (dq, J = 12.8, 7.4 Hz, 1H), 1.30 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 157.27 (q, J = 42.5 Hz), 138.3, 137.9, 137.5, 128.8, 128.7, 128.6, 128.4, 128.3, 128.2, 128.1, 127.98, 127.95, 114.6 (q, J = 285.4 Hz), 86.7, 85.2, 81.8, 76.3, 76.0, 75.7, 75.3, 66.7, 25.1, 15.2. 19F NMR (376 MHz, CDCl3) δ −74.9(COCF3). 1H NMR (600 MHz, C6D6) δ 7.42–7.39 (m, 2H), 7.32–7.28 (m, 2H), 7.20–7.06 (m, 11H), 4.95 (d, J = 10.6 Hz, 1H), 4.92 (d, J = 11.2 Hz, 1H), 4.75 (d, J = 11.4 Hz, 1H), 4.75 (d, J = 11.2 Hz, 1H), 4.65 (d, J = 10.6 Hz, 1H), 4.34 (d, J = 11.4 Hz, 1H), 4.24 (dd, J = 11.3, 2.4 Hz, 1H), 4.23 (d, J = 9.7 Hz, 1H), 4.05 (dd, J = 11.5, 6.3 Hz, 1H), 3.51 (dd, J = 8.9, 8.8 Hz, 1H), 3.37 (dd, J = 9.7, 8.8 Hz, 1H), 3.26 (dd, J = 9.9, 8.9 Hz, 1H), 3.07 (ddd, J = 9.9, 6.3, 2.2 Hz, 1H), 2.55 (dq, J = 12.8, 7.4 Hz, 1H), 2.43 (dq, J = 12.8, 7.4 Hz, 1H), 1.12 (t, J = 7.4 Hz, 3H). IR (neat) ν 1790 cm–1 (C=O). HRMS (ESI) m/z calcd for C31H33O6SF3Na [M + Na]+, 613.1848; found, 613.1850.
Ethyl 6-O-Benzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (31)
Compound 31 was synthesized as described in the literature.881H NMR (600 MHz, CDCl3) δ 8.07–8.04 (m, 2H), 7.60–7.56 (m, 1H), 7.49–7.40 (m, 4H), 7.39–7.24 (m, 13H), 4.99 (d, J = 10.8 Hz, 1H), 4.97 (d, J = 10.2 Hz, 1H), 4.91 (d, J = 10.8 Hz, 1H), 4.90 (d, J = 10.8 Hz, 1H), 4.79 (d, J = 10.2 Hz, 1H), 4.66–4.63 (m, 1H), 4.64 (d, J = 10.8 Hz, 1H), 4.55 (d, J = 9.8 Hz, 1H), 4.48–4.44 (m, 1H), 3.78 (t, J = 8.8 Hz, 1H), 3.69–3.67 (m, 2H), 3.52 (dd, J = 9.8, 8.8 Hz, 1H), 2.79 (dq, J = 12.7, 7.5 Hz, 1H), 2.73 (dq, J = 12.7, 7.5 Hz, 1H), 1.32 (d, J = 7.5 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 8.18–8.16 (m, 2H), 7.43–7.40 (m, 2H), 7.34–7.31 (m, 2H), 7.28–7.25 (m, 2H), 7.20–7.01 (m, 12H), 4.98 (d, J = 10.6 Hz, 1H), 4.96 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.2 Hz, 1H), 4.81 (d, J = 11.1 Hz, 1H), 4.71 (d, J = 10.6 Hz, 1H), 4.65 (dd, J = 11.8, 2.3 Hz, 1H), 4.52 (d, J = 11.1 Hz, 1H), 4.51 (dd, J = 11.8, 5.6 Hz, 1H), 4.38 (d, J = 9.7 Hz, 1H), 3.64 (dd, J = 9.0, 8.8 Hz, 1H), 3.56 (dd, J = 9.8, 9.0 Hz, 1H), 3.51 (dd, J = 9.6, 8.8 Hz, 1H), 3.36 (ddd, J = 9.8, 5.6, 2.3 Hz, 1H), 2.59 (dq, J = 12.7, 7.4 Hz, 1H), 2.46 (dq, J = 12.7, 7.5 Hz, 1H), 1.11 (d, J = 7.5 Hz, 3H). IR (neat) ν 1721 cm–1 (C=O).
Ethyl (6S)-[6-2H1]-6-O-Benzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-31)
Compound 6S-d-31 (0.007 g, 60%) was synthesized from 6S-d-3 analogously to 31. 1H NMR (600 MHz, CDCl3) δ 8.04–8.01 (m, 2H), 7.58–7.54 (m, 1H), 7.46–7.24 (m, 17H), 4.97 (d, J = 10.8 Hz, 1H), 4.94 (d, J = 10.2 Hz, 1H), 4.89 (d, J = 10.7 Hz, 1H), 4.87 (d, J = 10.8 Hz, 2H), 4.76 (d, J = 10.2 Hz, 1H), 4.62 (d, J = 10.7 Hz, 1H), 4.53 (d, J = 9.8 Hz, 1H), 4.42–4.40 (m, 1H), 3.76 (dd, J = 8.8, 8.7 Hz, 1H), 3.66–3.64 (m, 2H), 3.49 (dd, J = 9.7, 8.8 Hz, 1H), 2.77 (dq, J = 12.7, 7.5 Hz, 1H), 2.70 (dq, J = 12.7, 7.5 Hz, 1H), 1.29 (t, J = 7.5 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 8.19–8.16 (m, 2H), 7.44–7.41 (m, 2H), 7.35–7.32 (m, 2H), 7.28–7.25 (m, 2H), 7.20–7.01 (m, 12H), 4.99 (d, J = 10.6 Hz, 1H), 4.96 (d, J = 11.3 Hz, 1H), 4.83 (d, J = 11.3 Hz, 1H), 4.81 (d, J = 11.1 Hz, 1H), 4.71 (d, J = 10.6 Hz, 1H), 4.52 (d, J = 11.1 Hz, 1H), 4.49 (d, J = 5.6 Hz, 1H), 4.37 (d, J = 9.7 Hz, 1H), 3.64 (dd, J = 8.9, 8.8 Hz, 1H), 3.55 (dd, J = 9.8, 8.9 Hz, 1H), 3.51 (dd, J = 9.6, 8.8 Hz, 1H), 3.35 (dd, J = 9.8, 5.6 Hz, 1H), 2.59 (dq, J = 12.8, 7.5 Hz, 1H), 2.46 (dq, J = 12.8, 7.5 Hz, 1H), 1.11 (t, J = 7.5 Hz, 3H). HRMS (ESI) m/z calcd for C36H37DO6SNa [M + Na]+, 622.2350; found, 622.2358.
Ethyl 2,3,4-Tri-O-benzyl-6-O-p-methylbenzoyl-1-thio-β-d-glucopyranoside (32).
Compound 3 (0.015 g, 0.030 mmol) was dissolved in anhydrous pyridine (0.1 mL) and cooled to 0 °C before the addition of p-methylbenzoyl chloride (5.6 μL, 0.061 mmol). The reaction mixture was then warmed up to room temperature and stirred overnight under an argon atmosphere. The reaction mixture was then diluted with ethyl acetate and washed with aqueous hydrochloric acid (1 M), saturated aqueous NaHCO3, and brine. The organic layer was separated and dried over anhydrous Na2SO4 and concentrated to dryness. The resultant crude residue was purified using silica gel column chromatography, eluting with hexane/ethyl acetate (6:1 to 4:1), to afford compound 32 (0.018 g, 95%) as a white semisolid. [α]D22 = +20.5 (c 0.6, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.92–7.90 (m, 2H), 7.39–7.37 (m, 2H), 7.35–7.21 (m, 17H), 4.95 (d, J = 10.8 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 4.87 (d, J = 10.7 Hz, 1H), 4.86 (d, J = 10.8 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.60 (d, J = 10.7 Hz, 1H), 4.59 (dd, J = 12.0, 1.6 Hz, 1H), 4.51 (d, J = 9.8 Hz, 1H), 4.40 (dd, J = 12.0, 5.2 Hz, 1H), 3.74 (t, J = 8.8 Hz, 1H), 3.67–3.63 (m, 2H), 3.48 (dd, J = 9.8, 8.8 Hz, 1H), 2.76 (dq, J = 12.8, 7.4 Hz, 1H), 2.69 (dq, J = 12.8, 7.4 Hz, 1H), 2.40 (s, 3H), 1.28 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 166.4, 143.9, 138.4, 138.0, 137.7, 129.8, 129.2, 128.7, 128.6, 128.4, 128.2, 128.2, 128.1, 128.0, 128.0, 127.4, 86.8, 85.2, 82.0, 78.1, 77.3, 76.1, 75.7, 75.4, 63.8, 25.2, 21.8, 15.3. 1H NMR (600 MHz, C6D6) δ 8.17–8.14 (m, 2H), 7.44–7.41 (m, 2H), 7.35–7.32 (m, 2H), 7.29–7.27 (m, 2H), 7.19–7.03 (m, 9H), 6.89–6.86 (m, 2H), 4.99 (d, J = 10.6 Hz, 1H), 4.96 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.2 Hz, 1H), 4.82 (d, J = 11.0 Hz, 1H), 4.71 (d, J = 10.6 Hz, 1H), 4.68 (dd, J = 11.8, 2.2 Hz, 1H), 4.54 (dd, J = 11.8, 5.7 Hz, 1H), 4.53 (d, J = 11.0 Hz, 1H), 4.38 (d, J = 9.7 Hz, 1H), 3.64 (dd, J = 8.9, 8.8 Hz, 1H), 3.56 (dd, J = 9.8, 8.9 Hz, 1H), 3.51 (dd, J = 9.7, 8.8 Hz, 1H), 3.37 (ddd, J = 9.8, 5.7, 2.2 Hz, 1H), 2.61 (dq, J = 12.7, 7.4 Hz, 1H), 2.48 (dq, J = 12.7, 7.4 Hz, 1H), 1.95 (s, 3H), 1.13 (t, J = 7.4 Hz, 1H). IR (neat) ν 1718 cm–1 (C=O). HRMS (ESI) m/z calcd for C37H40O6SNa [M + Na]+, 635.2443; found, 635.2474.
Ethyl (6S)-[6-2H1]-6-O-p-Methylbenzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-32)
Compound 6S-d-32 (0.003 g, 76%) was synthesized from 6S-d-3 analogously to 32. 1H NMR (600 MHz, CDCl3) δ 8.00–7.85 (m, 2H), 7.42–7.20 (m, 19H), 4.99–4.92 (m, 2H), 4.90–4.84 (m, 2H), 4.76 (d, J = 10.1 Hz, 1H), 4.61 (d, J = 10.7 Hz, 1H), 4.52 (d, J = 9.7 Hz, 1H), 4.39 (d, J = 5.0 Hz, 1H), 3.75 (t, J = 8.8 Hz, 1H), 3.68–3.61 (m, 2H), 3.49 (dd, J = 9.7, 8.8 Hz, 1H), 2.82–2.65 (m, 2H), 2.41 (s, 3H), 1.29 (t, J = 7.4 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 8.18–8.14 (m, 2H), 7.44–7.41 (m, 2H), 7.35–7.26 (m, 4H), 7.20–7.02 (m, 9H), 6.89–6.86 (m, 2H), 4.99 (d, J = 10.5 Hz, 1H), 4.96 (d, J = 11.0 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 4.82 (d, J = 11.1 Hz, 1H), 4.71 (d, J = 10.5 Hz, 1H), 4.53 (d, J = 11.1 Hz, 1H), 4.51 (d, J = 5.8 Hz, 1H, H), 4.38 (d, J = 9.7 Hz, 1H), 3.64 (t, J = 8.8 Hz, 1H), 3.56 (dd, J = 9.8, 8.8 Hz, 1H), 3.51 (dd, J = 9.7, 8.8 Hz, 1H), 3.37 (dd, J = 9.8, 5.8 Hz, 1H), 2.65–2.57 (m, 1H), 2.51–2.45 (m, 1H), 1.95 (s, 3H), 1.13 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z calcd for C37H39DO6SNa [M + Na]+, 636.2506; found, 636.2508.
Ethyl 2,3,4-Tri-O-benzyl-6-O-p-methoxybenzoyl-1-thio-β-d-glucopyranoside (33)
Compound 33 was synthesized by the same procedure as that described for compound 32 from compound 3 (0.015 g, 0.030 mmol) and p-methoxybenzoyl chloride (0.010 g, 0.061 mmol) in anhydrous pyridine (0.1 mL). After purification by silica gel column chromatography, eluting with hexane/ethyl acetate (6:1 to 4:1), compound 33 (0.015 g, 77%) was obtained as a white semisolid. [α]D22 = +23.4 (c 0.7, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 8.00–7.97 (m, 2H), 7.41–7.37 (m, 2H), 7.36–7.23 (m, 13H), 6.93–6.90 (m, 2H), 4.96 (d, J = 10.8 Hz, 1H), 4.94 (d, J = 10.2 Hz, 1H), 4.88 (d, J = 10.8 Hz, 1H), 4.87 (d, J = 10.8 Hz, 1H), 4.76 (d, J = 10.2 Hz, 1H), 4.61 (d, J = 10.8 Hz, 1H), 4.58 (dd, J = 11.8, 1.7 Hz, 1H), 4.52 (d, J = 9.8 Hz, 1H), 4.41 (dd, J = 11.8, 5.2 Hz, 1H), 3.87 (s, 3H), 3.75 (dd, J = 8.9, 8.8 Hz, 1H), 3.67–3.63 (m, 2H), 3.49 (dd, J = 9.8, 8.8 Hz, 1H), 2.80–2.67 (m, 2H), 1.30 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 166.1, 163.6, 138.4, 138.0, 137.7, 131.7, 128.6, 128.6, 128.4, 128.3, 128.1, 128.1, 128.0, 128.0, 122.5, 113.7, 86.8, 85.2, 82.0, 78.1, 77.3, 76.1, 75.7, 75.4, 63.7, 55.6, 25.2, 15.3. 1H NMR (600 MHz, C6D6) δ 8.21–8.18 (m, 2H), 7.44–7.41 (m, 2H), 7.35–7.32 (m, 2H), 7.31–7.28 (m, 2H), 7.20–7.02 (m, 9H), 6.65–6.60 (m, 2H), 5.00 (d, J = 10.6 Hz, 1H), 4.96 (d, J = 11.3 Hz, 1H), 4.84 (d, J = 11.3 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 4.73 (d, J = 10.6 Hz, 1H), 4.69 (dd, J = 11.8, 2.2 Hz, 1H), 4.56 (dd, J = 11.8, 5.6 Hz, 1H), 4.55 (d, J = 11.0 Hz, 1H), 4.39 (d, J = 9.7 Hz, 1H), 3.65 (dd, J = 8.9, 8.7 Hz, 1H), 3.59 (dd, J = 9.8, 8.9 Hz, 1H), 3.54 (dd, J = 9.7, 8.7 Hz, 1H), 3.38 (ddd, J = 9.8, 5.6, 2.2 Hz, 1H), 3.13 (s, 3H), 2.62 (dq, J = 12.7, 7.4 Hz, 1H), 2.49 (dq, J = 12.7, 7.4 Hz, 1H), 1.13 (t, J = 7.4 Hz, 3H). IR (neat) ν 1714 cm–1 (C=O). HRMS (ESI) m/z calcd for C37H40O7SNa [M + Na]+, 651.2392; found, 651.2399.
Ethyl (6S)-[6-2H1]-6-O-p-Methoxybenzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-33)
Compound 6S-d-33 (0.004 g, 60%) was synthesized from 6S-d-3 analogously to 33. 1H NMR (600 MHz, CDCl3) δ 8.01–7.96 (m, 2H), 7.44–7.22 (m, 15H), 6.94–6.88 (m, 2H), 4.96 (d, J = 10.8 Hz, 1H), 4.94 (d, J = 10.2 Hz, 1H), 4.88 (d, J = 10.8 Hz, 1H), 4.87 (d, J = 10.8 Hz, 1H), 4.76 (d, J = 10.2 Hz, 1H), 4.61 (d, J = 10.8 Hz, 1H), 4.52 (d, J = 9.8 Hz, 1H), 4.39 (d, J = 5.0 Hz, 1H), 3.87 (s, 3H), 3.75 (t, J = 8.8 Hz, 1H), 3.68–3.61 (m, 2H), 3.49 (dd, J = 9.7, 8.8 Hz, 1H), 2.77 (dq, J = 12.7, 7.4 Hz, 1H), 2.70 (dq, J = 12.7, 7.4 Hz, 1H), 1.30 (t, J = 7.4 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 8.22–8.17 (m, 2H), 7.45–7.40 (m, 2H), 7.35–7.32 (m, 2H), 7.30–7.28 (m, 2H), 7.20–7.02 (m, 9H), 6.64–6.60 (m, 2H), 5.00 (d, J = 10.8 Hz, 1H), 4.96 (d, J = 11.2 Hz, 1H), 4.84 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 4.72 (d, J = 11.0 Hz, 1H), 4.55 (d, J = 10.8 Hz, 1H), 4.53 (d, J = 5.6 Hz, 1H), 4.39 (d, J = 9.7 Hz, 1H), 3.65 (dd, J = 8.9, 8.7 Hz, 1H), 3.58 (dd, J = 9.7, 8.9 Hz, 1H), 3.53 (dd, J = 9.7, 8.7 Hz, 1H), 3.38 (dd, J = 9.7, 5.7 Hz, 1H), 3.13 (s, 3H,), 2.62 (dq, J = 12.7, 7.4 Hz, 1H), 2.49 (dq, J = 12.7, 7.4 Hz, 1H), 1.13 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z calcd for C37H39DO7SNa [M + Na]+, 652.2455; found, 652.2468.
Ethyl 6-O-p-Nitrobenzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (34)
Compound 34 was synthesized using the same procedure as that described for the synthesis of compound 32 from compound 3 (0.015 g, 0.030 mmol) and p-nitrobenzoyl chloride (0.011 g, 0.061 mmol) in anhydrous pyridine (0.1 mL). After chromatographic purification using silica gel (hexane/ethyl acetate 6:1 to 4:1), compound 34 (0.019 g, 96%) was obtained as a white semisolid. [α]D22 = +29.7 (c 1.2, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 8.29–8.26 (m, 2H), 8.17–8.14 (m, 2H), 7.41–7.20 (m, 15H), 4.98 (d, J = 10.8 Hz, 1H), 4.95 (d, J = 10.2 Hz, 1H), 4.90 (d, J = 11.0 Hz, 1H), 4.88 (d, J = 10.8 Hz, 1H), 4.77 (d, J = 10.2 Hz, 1H), 4.62 (dd, J = 11.8, 2.2 Hz, 1H), 4.62 (d, J = 11.0 Hz, 1H), 4.53 (d, J = 9.8 Hz, 1H), 4.44 (dd, J = 11.8, 5.4 Hz, 1H), 3.77 (t, J = 8.8 Hz, 1H), 3.66 (ddd, J = 9.8, 5.4, 2.2 Hz, 1H), 3.61 (dd, J = 9.8, 8.8 Hz, 1H), 3.49 (dd, J = 9.8, 8.8 Hz, 1H), 2.76 (dq, J = 12.7, 7.4 Hz, 1H), 2.70 (dq, J = 12.7, 7.4 Hz, 1H, 1H), 1.29 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 164.4, 150.7, 138.3, 137.9, 137.6, 135.4, 130.9,128.7, 128.6, 128.4, 128.2, 128.2, 128.1, 128.0, 123.6, 86.8, 85.4, 81.9, 77.6, 76.9, 76.1, 75.7, 75.2, 64.8, 25.3, 15.3. 1H NMR (600 MHz, C6D6) δ 7.76–7.73 (m, 2H), 7.66–7.63 (m, 2H), 7.45–7.41 (m, 2H), 7.36–7.33 (m, 2H), 7.26–7.23 (m, 2H), 7.20–6.97 (m, 9H), 5.02 (d, J = 10.6 Hz, 1H), 5.00 (d, J = 11.1 Hz, 1H), 4.85 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.1 Hz, 1H), 4.74 (d, J = 10.6 Hz, 1H), 4.56 (dd, J = 11.8, 2.3 Hz, 1H), 4.51 (d, J = 11.2 Hz, 1H), 4.40 (dd, J = 11.8, 5.6 Hz, 1H), 4.37 (d, J = 9.7 Hz, 1H), 3.66 (t, J = 8.8 Hz, 1H), 3.53 (dd, J = 9.7, 8.8 Hz, 1H), 3.50 (dd, J = 9.8, 8.8 Hz, 1H), 3.31 (ddd, J = 9.8, 5.6, 2.3 Hz, 1H), 2.57 (dq, J = 12.7, 7.4 Hz, 1H), 2.46 (dq, J = 12.7, 7.4 Hz, 1H), 1.09 (t, J = 7.4 Hz, 3H). IR (neat) ν 1726 cm–1 (C=O). HRMS (ESI) m/z calcd for C36H37NO8SNa [M + Na]+, 666.2138; found, 666.2148.
Ethyl (6S)-[6-2H1]-6-O-p-Nitrobenzoyl-2,3,4-tri-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-34)
Compound 6S-d-34 (0.004 g, 100%) was synthesized from 6S-d-3 analogously to 34. 1H NMR (600 MHz, CDCl3) δ 8.31–8.25 (m, 2H), 8.18–8.12 (m, 2H), 7.44–7.19 (m, 15H), 4.98 (d, J = 10.8 Hz, 1H), 4.95 (d, J = 10.2 Hz, 1H), 4.90 (d, J = 11.0 Hz, 1H), 4.87 (d, J = 10.8 Hz, 1H), 4.76 (d, J = 10.2 Hz, 1H), 4.61 (d, J = 11.0 Hz, 1H), 4.52 (d, J = 9.8 Hz, 1H), 4.42 (d, J = 5.4 Hz, 1H), 3.76 (t, J = 8.8 Hz, 1H), 3.65 (dd, J = 9.8, 5.4 Hz, 1H), 3.60 (dd, J = 9.8, 8.8 Hz, 1H), 3.48 (dd, J = 9.7, 8.8 Hz, 1H), 2.76 (dq, J = 12.7, 7.4 Hz, 1H), 2.70 (dq, J = 12.7, 7.4 Hz, 1H), 1.29 (t, J = 7.4 Hz, 1H). 1H NMR (600 MHz, C6D6) δ 7.76–7.72 (m, 2H), 7.66–7.63 (m, 2H), 7.45–7.41 (m, 2H), 7.36–7.33 (m, 2H), 7.26–7.22 (m, 2H), 7.20–6.97 (m, 9H), 5.02 (d, J = 10.7 Hz, 1H), 5.00 (d, J = 11.2 Hz, 1H), 4.85 (d, J = 11.3 Hz, 1H), 4.83 (d, J = 11.2 Hz, 1H), 4.74 (d, J = 10.7 Hz, 1H), 4.51 (d, J = 11.3 Hz, 1H), 4.38 (d, J = 5.5 Hz, 1H), 4.37 (d, J = 9.6 Hz, 1H), 3.66 (t, J = 8.8 Hz, 1H), 3.53 (dd, J = 9.6, 8.8 Hz, 1H), 3.50 (dd, J = 9.8, 8.8 Hz, 1H), 3.30 (dd, J = 9.8, 5.5 Hz, 1H), 2.57 (dq, J = 12.8, 7.4 Hz, 1H), 2.46 (dq, J = 12.8, 7.4 Hz, 1H), 1.09 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z calcd for C36H36DNO8SNa [M + Na]+, 667.2200; found, 667.2196.
Ethyl 2,3,4-Tri-O-benzyl-6-O-(N-phenylcarbamoyl)-1-thio-β-d-glucopyranoside (35)
Compound 3 (0.010 g, 0.020 mmol) was dissolved in anhydrous pyridine (0.4 mL) and cooled to 0 °C before the addition of phenyl isothiocyanate (13.3 μL, 0.120 mmol). The reaction mixture was then stirred for 48 h under an argon atmosphere and concentrated to dryness. The crude reaction mixture was dissolved in ethyl acetate and filtered through cotton before it was concentrated to dryness. The crude residue was purified over silica gel column chromatography, eluting with hexane/ethyl acetate (9:1 to 4:1), to afford the compound 35 (0.011 g, 87%) as a white solid with m.p. 119–121 0 °C. [α]D22 = +3.5 (c 0.2, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.39–7.25 (m, 19H), 7.09–7.06 (m, 1H), 6.57 (br s, 1H), 4.96 (d, J = 10.9 Hz, 1H), 4.93 (d, J = 10.2 Hz, 1H), 4.87 (d, J = 10.9 Hz, 1H), 4.86 (d, J = 10.7 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.63 (d, J = 10.7 Hz, 1H), 4.49 (d, J = 9.8 Hz, 1H), 4.41–4.35 (m, 2H), 3.73 (t, J = 8.8 Hz, 1H), 3.58–3.52 (m, 2H), 3.45 (dd, J = 9.8, 8.8 Hz, 1H), 2.81–2.70 (m, 2H), 1.31 (t, J = 7.4 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ 153.1, 138.5, 138.0, 137.8, 137.7, 129.2, 128.7, 128.63, 128.6, 128.5, 128.2, 128.1, 127.9, 123.7, 118.7, 86.8, 85.4, 81.9, 77.2, 75.9, 75.7, 75.2, 63.9, 29.9, 25.3, 15.2. 1H NMR (600 MHz, C6D6) δ 7.47–7.40 (m, 2H), 7.39–7.30 (m, 4H), 7.29–7.01 (m, 13H), 6.86–6.79 (m, 1H), 6.06 (br s, 1H), 5.01 (d, J = 10.6 Hz, 1H), 4.98 (d, J = 11.4 Hz, 1H), 4.86 (d, J = 11.4 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 4.73 (d, J = 10.6 Hz, 1H), 4.58 (d, J = 11.0 Hz, 1H), 4.46 (dd, J = 11.7, 4.6 Hz, 1H), 4.43 (dd, J = 11.8, 2.5 Hz, 1H), 4.39 (d, J = 9.7 Hz, 1H), 3.66 (dd, J = 9.0, 8.8 Hz, 1H), 3.57–3.52 (m, 2H), 3.29 (ddd, J = 9.8, 4.6, 2.5 Hz, 1H), 2.62 (dq, J = 12.6, 7.4 Hz, 1H), 2.52 (dq, J = 12.6, 7.4 Hz, 1H), 1.13 (t, J = 7.4 Hz, 3H). IR (neat) ν 3329 cm–1 (N—H), 1716 cm–1 (C=O). HRMS (ESI) m/z calcd for C36H39NO6SNa [M + Na]+, 636.2396; found, 636.2374.
Ethyl 2,3,4-Tri-O-benzyl-(6S)-[6-2H1]-6-O-(N-phenylcarbamoyl)-1-thio-β-d-glucopyranoside (6S-d-35)
Compound 6S-d-35 (0.004 g, 100%) was synthesized from 6S-d-3 analogously to 35. 1H NMR (600 MHz, CDCl3) δ 7.31 (m, 19H), 7.10–7.05 (m, 1H), 6.57 (br s, 1H), 4.96 (d, J = 10.8 Hz, 1H), 4.93 (d, J = 10.0 Hz, 1H), 4.87 (d, J = 10.8 Hz, 1H), 4.86 (d, J = 10.6 Hz, 1H), 4.75 (d, J = 10.0 Hz, 1H), 4.62 (d, J = 10.6 Hz, 1H), 4.49 (d, J = 9.8 Hz, 1H), 4.35 (d, J = 4.5 Hz, 1H), 3.72 (t, J = 8.8 Hz, 1H), 3.59–3.50 (m, 2H), 3.44 (dd, J = 9.8, 9.0 Hz, 1H), 2.87–2.66 (m, 2H), 1.31 (t, J = 7.4 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 7.45–7.41 (m, 2H), 7.37–7.32 (m, 4H), 7.30–7.02 (m, 13H), 6.84–6.79 (m, 1H), 6.04 (br s, 1H), 5.01 (d, J = 10.5 Hz, 1H), 4.98 (d, J = 11.2 Hz, 1H), 4.86 (d, J = 11.2 Hz, 1H), 4.83 (d, J = 11.0 Hz, 1H), 4.73 (d, J = 10.5 Hz, 1H), 4.58 (d, J = 11.0 Hz, 1H), 4.44 (d, J = 4.9 Hz, 1H), 4.38 (d, J = 9.6 Hz, 1H), 3.66 (t, J = 8.7 Hz, 1H), 3.56–3.52 (m, 2H), 3.29 (dd, J = 9.8, 4.9 Hz, 1H), 2.62 (dq, J = 12.6, 7.4 Hz, 1H), 2.52 (dd, J = 12.6, 7.4 Hz, 1H), 1.13 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z calcd for C36H38DNO6SNa [M + Na]+, 637.2459; found, 637.2466.
Ethyl 2,3,4,6-Tetra-O-benzyl-1-thio-β-d-glucopyranoside (36)
Compound 36 was synthesized as described in the literature.871H NMR (600 MHz, CDCl3) δ 7.40–7.26 (m, 18H), 7.19–7.16 (m, 2H), 4.93 (d, J = 10.9 Hz, 1H), 4.92 (d, J = 10.1 Hz, 1H), 4.86 (d, J = 10.9 Hz, 1H), 4.82 (d, J = 10.7 Hz, 1H), 4.75 (d, J = 10.1 Hz, 1H), 4.61 (d, J = 12.1 Hz, 1H), 4.57 (d, J = 10.7 Hz, 1H), 4.56 (d, J = 12.1 Hz, 1H), 4.47 (d, J = 9.8 Hz, 1H), 3.76 (dd, J = 10.8, 1.9 Hz, 1H), 3.69 (dd, J = 9.0, 8.9 Hz, 1H), 3.69 (dd, J = 10.8, 5.0 Hz, 1H), 3.62 (dd, J = 9.8, 9.0 Hz, 1H), 3.48 (ddd, J = 9.8, 5.0, 1.9 Hz, 1H), 3.45 (dd, J = 9.8, 8.9 Hz, 1H), 2.81 (dd, J = 12.7, 7.5 Hz, 1H), 2.75 (dd, J = 12.7, 7.5 Hz, 1H), 1.34 (d, J = 7.5 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 7.40–7.00 (m, 20H), 4.94 (d, J = 10.7 Hz, 1H), 4.89 (d, J = 11.3 Hz, 1H), 4.81 (d, J = 11.2 Hz, 1H), 4.79 (d, J = 11.3 Hz, 1H), 4.67 (d, J = 10.7 Hz, 1H), 4.55 (d, J = 11.2 Hz, 1H), 4.41 (d, J = 12.2 Hz, 1H), 4.36 (d, J = 9.7 Hz, 1H), 4.34 (d, J = 12.2 Hz, 1H), 3.67 (dd, J = 9.7, 9.1 Hz, 1H), 3.61–3.60 (m, 2H), 3.60 (dd, J = 9.1, 8.7 Hz, 1H), 3.46 (dd, J = 9.7, 8.7 Hz, 1H), 3.28–3.24 (m, 1H), 2.59 (dq, J = 12.6, 7.4 Hz, 1H), 2.48 (dq, J = 12.6, 7.4 Hz, 1H), 1.11 (t, J = 7.4 Hz, 3H).
Ethyl (6S)-[6-2H1]-2,3,4,6-Tetra-O-benzyl-1-thio-β-d-glucopyranoside (6S-d-36)
Compound 6S-d-36 was synthesized, by the same procedure as that described for 6S-d-27 from 6S-d-3 (0.010 g, 0.020 mmol), NaH (1.0 mg, 0.025 mmol), and benzyl bromide (2.8 μL, 0.024 mmol) in anhydrous DMF (0.1 mL). After purification by silica gel column chromatography, eluting with hexane/ethyl acetate (7:1 to 7:3) gave the title compound (0.011 g, 94%). 1H NMR (600 MHz, CDCl3) δ 7.40–7.26 (m, 18H), 7.19–7.15 (m, 2H), 4.93 (d, J = 11.0 Hz, 1H), 4.92 (d, J = 10.2 Hz, 1H), 4.85 (d, J = 10.9 Hz, 1H), 4.82 (d, J = 10.8 Hz, 1H), 4.75 (d, J = 10.2 Hz, 1H), 4.60 (d, J = 12.2 Hz, 1H), 4.57 (d, J = 10.8 Hz, 1H), 4.56 (d, J = 12.2 Hz, 1H), 4.47 (d, J = 9.8 Hz, 1H), 3.69 (dd, J = 9.0, 8.9 Hz, 1H), 3.66 (d, J = 5.0 Hz, 1H), 3.61 (dd, J = 9.8, 9.0 Hz, 1H), 3.47 (dd, J = 9.8, 5.0 Hz, 1H), 3.45 (dd, J = 9.8, 8.9 Hz, 1H), 2.80 (dd, J = 12.7, 7.5 Hz, 1H), 2.75 (dd, J = 12.7, 7.5 Hz, 1H), 1.34 (d, J = 7.5 Hz, 3H). 1H NMR (600 MHz, C6D6) δ 7.39–7.00 (m, 20H), 4.94 (d, J = 10.7 Hz, 1H), 4.89 (d, J = 11.3 Hz, 1H), 4.81 (d, J = 11.3 Hz, 1H), 4.79 (d, J = 11.3 Hz, 1H), 4.67 (d, J = 10.7 Hz, 1H), 4.55 (d, J = 11.3 Hz, 1H), 4.41 (d, J = 12.2 Hz, 1H), 4.36 (d, J = 9.7 Hz, 1H), 4.35 (d, J = 12.2 Hz, 1H), 3.67 (dd, J = 9.7, 9.1 Hz, 1H), 3.60 (dd, J = 9.1, 8.7 Hz, 1H), 3.59 (d, J = 4.6 Hz, 1H), 3.46 (dd, J = 9.7, 8.7 Hz, 1H), 3.26 (dd, J = 9.7, 4.6 Hz, 1H), 2.59 (dq, J = 12.6, 7.4 Hz, 1H), 2.48 (dq, J = 12.6, 7.4 Hz, 1H), 1.11 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z calcd for C36H39DO5SNa [M + Na]+, 608.2557; found, 608.2551.
Acknowledgments
We thank the NIH (GM62160) for support of this work, and we acknowledge support from the NSF (MRI-084043) for the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b01459.
Copies of the 1H and 13C NMR spectra of all new compounds (PDF)
Author Contributions
† S.D., H.A.: These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Adero P. O.; Amarasekara H.; Wen P.; Bohé L.; Crich D. The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev. 2018, 10.1021/acs.chemrev.8b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edward J. T. Stability of Glycosides toward Acid Hydrolysis: A Conformational Analysis. Chem. Ind. 1955, 1102–1104. [Google Scholar]
- Foster A. B.; Overend W. G. The Acidic Hydrolysis of O-Glycosides. Chem. Ind. 1955, 566–567. [Google Scholar]
- Overend W. G.; Rees C. W.; Sequeira J. S. Reactions at Position 1 of Carbohydrates. The Acid-catalyzed Hydrolysis of Glycosides. J. Chem. Soc. 1962, 3429–3440. 10.1039/jr9620003429. [DOI] [Google Scholar]
- Namchuk M. N.; McCarter J. D.; Becalski A.; Andrews T.; Withers S. G. The Role of Sugar Substituents in Glycoside Hydrolysis. J. Am. Chem. Soc. 2000, 122, 1270–1277. 10.1021/ja992044h. [DOI] [Google Scholar]
- Zhang Z.; Ollmann I. R.; Ye X.-S.; Wischnat R.; Baasov T.; Wong C.-H. Programmable One-Pot Oligosaccharide Synthesis. J. Am. Chem. Soc. 1999, 121, 734–753. 10.1021/ja982232s. [DOI] [Google Scholar]
- Glaudemans C. P. J.; Fletcher H. G. Long Range Effects of Acyl Groups in the Solvolysis of Glycofuranosyl Halides. The Synthesis of 2,3-Di-O-benzyl-5-O-p-nitrobenzoyl-α-D-arabinofuranosyl Chloride and of 2-O-Benzyl-3,5-di-O-p-nitrobenzoyl-α-D-arabinofuranosyl Chloride. J. Am. Chem. Soc. 1965, 87, 4636–4641. 10.1021/ja00948a041. [DOI] [Google Scholar]
- Mootoo D. R.; Konradsson P.; Udodong U.; Fraser-Reid B. ″Armed″ and ″Disarmed″ n-Pentenyl Glycosides in Saccharide Couplings Leading to Oligosaccharides. J. Am. Chem. Soc. 1988, 110, 5583–5584. 10.1021/ja00224a060. [DOI] [Google Scholar]
- Paulsen H.; Richter A.; Sinnwell V.; Stenzel W. Darstellung Selektiv Blockierter 2-Azido-2-desoxy-D-gluco und Galactopyranosylhalogenide: Reaktivitat und 13C-NMR-Spektren. Carbohydr. Res. 1978, 64, 339–362. 10.1016/S0008-6215(00)83722-2. [DOI] [Google Scholar]
- Douglas N. L.; Ley S. V.; Lucking U.; Warriner S. L. Tuning Glycoside Reactivity: New Tool for Efficient Oligosaccharide Synthesis. J. Chem. Soc., Perkin Trans. 1 1998, 51–65. 10.1039/a705275h. [DOI] [Google Scholar]
- Fraser-Reid B.; López J. C. Armed-Disarmed Effects in Carbohydrate Chemistry: History, Synthetic and Mechanistic Studies. Top. Curr. Chem. 2010, 301, 1–29. 10.1007/128_2010_105. [DOI] [PubMed] [Google Scholar]
- Pedersen C. M.; Nordstrom L. U.; Bols M. ″Super Armed″ Glycosyl Donors: Conformational Arming of Thioglycosides by Silylation. J. Am. Chem. Soc. 2007, 129, 9222–9235. 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]
- Pedersen C. M.; Bols M. On the Nature of the Electronic Effect of Multiple Hydroxyl Groups in the 6-Membered Ring – The Effects Are Additive But Steric Hindrance Plays A Role Too. Org. Biomol. Chem. 2017, 15, 1164–1173. 10.1039/C6OB02427K. [DOI] [PubMed] [Google Scholar]
- Bols M.; Pedersen C. M. Silyl-Protective Groups Influencing the Reactivity and Selectivity in Glycosylations. Beilstein J. Org. Chem. 2017, 13, 93–105. 10.3762/bjoc.13.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell C.; López O.; Murphy P. V.; Fernández Bolaños J. G.; Hazell R. G.; Bols M. Conformational Effects on Glycoside Reactivity: Study of the High Reactive Conformer of Glucose. J. Am. Chem. Soc. 2004, 126, 12374–12385. 10.1021/ja047476t. [DOI] [PubMed] [Google Scholar]
- Miljkovic M.; Yeagley D.; Deslongchamps P.; Dory Y. L. Experimental and Theoretical Evidence of Through-Space Electrostatic Stabilization of the Incipient Oxocarbenium Ion by an Axially Oriented Electronegative Substituent During Glycopyranoside Acetolysis. J. Org. Chem. 1997, 62, 7597–7604. 10.1021/jo970677d. [DOI] [Google Scholar]
- Jensen H. H.; Bols M. Stereoelectronic Substituent Effects. Acc. Chem. Res. 2006, 39, 259–265. 10.1021/ar050189p. [DOI] [PubMed] [Google Scholar]
- Walvoort M. T. C.; Dinkelar J.; van den Bos L. J.; Lodder G.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. The Impact of Oxacarbenium Ion Conformers on the Stereochemical Outcome of Glycosylations. Carbohydr. Res. 2010, 345, 1252–1263. 10.1016/j.carres.2010.02.027. [DOI] [PubMed] [Google Scholar]
- Heuckendorff M.; Pedersen C. M.; Bols M. Quantifying Electronic Effects of Common Carbohydrate Protecting Groups in a Piperidine Model System. Chem. - Eur. J. 2010, 16, 13982–13994. 10.1002/chem.201002313. [DOI] [PubMed] [Google Scholar]
- Bock K.; Duus J. O. A Conformational Study of Hydroxymethyl Groups in Carbohydrates Investigated by 1H NMR Spectroscopy. J. Carbohydr. Chem. 1994, 13, 513–543. 10.1080/07328309408011662. [DOI] [Google Scholar]
- Grindley T. B. In Glycoscience: Chemistry and Chemical Biology; Fraser-Reid B., Tatsuta K., Thiem J., Eds.; Springer: Berlin, 2001; Vol. 1, pp 3–51. [Google Scholar]
- Rao V. S. R.; Qasba P. K.; Balaji P. V.; Chandrasekaran R.. Conformation of Carbohydrates; Harwood Academic Publishers: Amsterdam, The Netherlands, 1998. [Google Scholar]
- Jensen H. H.; Nordstrøm L. U.; Bols M. The Disarming Effect of the 4,6-Acetal Group on Glycoside Reactivity: Torsional or Electronic. J. Am. Chem. Soc. 2004, 126, 9205–9213. 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- Moumé-Pymbock M.; Furukawa T.; Mondal S.; Crich D. Probing the Influence of a 4,6-O-Acetal on the Reactivity of Galactopyranosyl Donors: Verification of the Disarming Influence of the trans-gauche Conformation of C5-C6 Bonds. J. Am. Chem. Soc. 2013, 135, 14249–14255. 10.1021/ja405588x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kancharla P. K.; Crich D. Influence of Side Chain Conformation and Configuration on Glycosyl Donor Reactivity and Selectivity as Illustrated by Sialic Acid Donors Epimeric at the 7-Position. J. Am. Chem. Soc. 2013, 135, 18999–19007. 10.1021/ja410683y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales E. Q.; Padron J. I.; Trujillo M.; Vázquez J. T. CD and 1H NMR Study of the Rotational Population Dependence of the Hydroxymethyl Group in β-Glucopyranosides on the Aglycon and Its Absolute Configuration. J. Org. Chem. 1995, 60, 2537–2548. 10.1021/jo00113a038. [DOI] [Google Scholar]
- Padrón J. I.; Morales E. Q.; Vázquez J. T. Alkyl Galactopyranosides: Rotational Population Dependence of the Hydroxymethyl Group on the Aglycon and Its Absolute Configuration and on the Anomeric Configuration. J. Org. Chem. 1998, 63, 8247–8258. 10.1021/jo981002t. [DOI] [Google Scholar]
- Nobrega C.; Vázquez J. T. Conformational Study of the Hydroxymethyl Group in D-Mannose Derivatives. Tetrahedron: Asymmetry 2003, 14, 2793–2801. 10.1016/S0957-4166(03)00623-2. [DOI] [Google Scholar]
- Roën A.; Padron J. I.; Vázquez J. T. Hydroxymethyl Rotamer Populations in Disaccharides. J. Org. Chem. 2003, 68, 4615–4630. 10.1021/jo026913o. [DOI] [PubMed] [Google Scholar]
- Dey S.; Jayaraman N. Glycosidic Bond Hydrolysis in Septanosides: A Comparison of Mono-, Di-, and 2-Chloro-2-deoxy-septanosides. Carbohydr. Res. 2014, 399, 49–56. 10.1016/j.carres.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Faltin F.; Fehring V.; Miethchen R. Chiral Crown Ethers Based on Galactopyranosides. Synthesis 2002, 2002, 1851–1856. 10.1055/s-2002-33916. [DOI] [Google Scholar]
- Motawia M. S.; Olsen C. E.; Enevoldsen K.; Marcussen J.; Moeller B. L. Chemical Synthesis of 6′-α-Maltosyl-maltotriose, A Branched Oligosaccharide Representing the Branch Point of Starch. Carbohydr. Res. 1995, 277, 109–123. 10.1016/0008-6215(95)00203-6. [DOI] [PubMed] [Google Scholar]
- Ray A. K.; Roy N. Synthesis of the Tetrasaccharide Repeating Unit of the Polysaccharide from Klebsiella Type 23. Carbohydr. Res. 1990, 196, 95–100. 10.1016/0008-6215(90)84108-7. [DOI] [PubMed] [Google Scholar]
- Ohrui H.; Horiki H.; Kishi H.; Meguro H. The Synthesis of D-(6R)- and (6S)-D-Glucose-6-2H through Stereospecific Photobromination of 1,6-Anhydro-β-D-glucopyranose Derivative. Agric. Biol. Chem. 1983, 47, 1101–1106. [Google Scholar]
- Ichikawa Y.; Kuzuhara H. Synthesis of 1,6-Anhydro-2,3-di-O-benzoyl-4-O-(Methyl 2,3,4-tri-O-benzoyl-α-L-iodpyranosyluronate)-β-D-glucopyranose from Cellobiose. Carbohydr. Res. 1983, 115, 117–129. 10.1016/0008-6215(83)88140-3. [DOI] [Google Scholar]
- Ohrui H.; Nishida Y.; Meguro H. The Synthesis of D-(6R)- and (6S)-(6-2H1)-Galactose. Agric. Biol. Chem. 1984, 48, 1049–1053. 10.1080/00021369.1984.10866239. [DOI] [Google Scholar]
- Nishida Y.; Ohrui H.; Meguro H. 1H-NMR Studies of 6R- and 6S-Deuterated D-Hexoses: Assignment of the Preferred Rotamers about C5-C6 Bond of D-Glucose and D-Galactose Derivatives in Solutions. Tetrahedron Lett. 1984, 25, 1575–1578. 10.1016/S0040-4039(01)90014-0. [DOI] [Google Scholar]
- Ohrui H.; Nishida Y.; Watanabe M.; Hori H.; Meguro H. 1H-NMR Studies of 6R- and 6S-Deuterated 1,6-Linked Disaccharides: Assignment of the Preferred Rotamers about C5-C6 Bond of 1,6-Disaccharides in Solutions. Tetrahedron Lett. 1985, 26, 3251–3254. 10.1016/S0040-4039(00)98164-4. [DOI] [Google Scholar]
- Midland M. M.; Asirwatham G.; Cheng J. C.; Miller J. A.; Morel L. A. Synthesis and Conformational Analysis of (6R)-[6-2H1]-1,2:3,4-Di-O-isopropylidene-α-D-galactopyranose. NMR and Molecular Modeling Studies. J. Org. Chem. 1994, 59, 4438–4442. 10.1021/jo00095a019. [DOI] [Google Scholar]
- Falcone-Hindley M. L.; Davis J. T. Stereoselective Preparation of Deuterium-Labeled Sugars: (6R)-(6-2H1)-N-Acetylglucosamine Derivatives. J. Org. Chem. 1998, 63, 5555–5561. 10.1021/jo980781a. [DOI] [Google Scholar]
- Xu L.; Price N. P. J. Stereoselective Synthesis of Chirally Deuterated (S)-D-(6-2H1)-Glucose. Carbohydr. Res. 2004, 339, 1173–1178. 10.1016/j.carres.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Ogawa A.; Curran D. P. Benzotrifluoride: A Useful Alternative Solvent for Organic Reactions Currently Conducted in Dichloromethane and Related Solvents. J. Org. Chem. 1997, 62, 450–451. 10.1021/jo9620324. [DOI] [PubMed] [Google Scholar]
- Kato T.; Vasella A.; Crich D. Stereospecific Synthesis of Methyl 2-Amino-2-deoxy-(6S)-deuterio-α,β-D-glucopyranoside and Methyl 2,6-Diamino-2,6-dideoxy-(6R)-deuterio-α,β-D-glucopyranoside: Side Chain Conformations of the 2-Amino-2-deoxy and 2,6-Diamino-2,6-Dideoxyglucopyranosides. Carbohydr. Res. 2017, 448, 10–17. 10.1016/j.carres.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert H. J.; Neumann W. P. A Convenient Preparation of Tributyltin Deuteride and Some Other Triorganotin Deuterides. Synthesis 1980, 1980, 942–943. 10.1055/s-1980-29283. [DOI] [Google Scholar]
- Wang L.-X.; Sakairi N.; Kuzuhara H. 1,6-Anhydro-β-D-glucopyranose Derivatives as Glycosyl Donors for Thioglycosidation Reactions. J. Chem. Soc., Perkin Trans. 1 1990, 1677–1682. 10.1039/P19900001677. [DOI] [Google Scholar]
- Amarasekara H.; Dharuman S.; Kato T.; Crich D. Synthesis of Conformationally-Locked cis- and trans-Bicyclo[4.4.0] Mono-, Di- and Trioxadecane Modifications of Galacto- and Glucopyranose. Experimental Limiting 3JH,H Coupling Constants for the Estimation of Carbohydrate Side Chain Populations and Beyond. J. Org. Chem. 2018, 83, 881–897. 10.1021/acs.joc.7b02891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudemer A. In Stereochemistry: Fundamentals and Methods; Kagan H. B., Ed.; Georg Thieme: Stuttgart, Germany, 1977; Vol. 1, pp 44–136. [Google Scholar]
- Eliel E. L.; Wilen S. H.. Stereochemistry of Organic Compounds; Wiley: New York, 1994. [Google Scholar]
- Tvaroška I.; Gajdoš J. Angular Dependence of Vicinal Carbon-Proton Coupling Constants for Conformational Studies of the Hydroxymethyl Group in Carbohydrates. Carbohydr. Res. 1995, 271, 151–162. 10.1016/0008-6215(95)00046-V. [DOI] [Google Scholar]
- Haasnoot C. A. G.; De Leeuw F. A. A. M.; Altona C. The Relationship Between Proton-Proton NMR Coupling Constants and Substituent Electronegativities-I. An empirical Generalization of the Karplus Equation. Tetrahedron 1980, 36, 2783–2792. 10.1016/0040-4020(80)80155-4. [DOI] [Google Scholar]
- Altona C. In Encyclopedia of NMR; Harris R. K., Wasylishen R. E., Eds.; Wiley: Chichester, U.K., 2012; Vol. 9, pp 5364–5378. [Google Scholar]
- Coxon B. Developments in the Karplus Equation as They Relate to the NMR Coupling Constants of Carbohydrates. Adv. Carbohydr. Chem. Biochem. 2009, 62, 17–82. 10.1016/S0065-2318(09)00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duus J. O.; Gotfredsen C. H.; Bock K. Carbohydrate Structural Determination by NMR Spectroscopy: Modern Methods and Limitations. Chem. Rev. 2000, 100, 4589–4614. 10.1021/cr990302n. [DOI] [PubMed] [Google Scholar]
- Altona C.; Ippel J. H.; Westra H.; Aldert J. A.; Erkelens C.; Groesbeek M.; Donders L. A. Relationship Between Proton-Proton NMR Coupling Constants and Substituent Electronegativities. V. Empirical Substituent Constants Deduced from Ethanes and Propanes. Magn. Reson. Chem. 1989, 27, 564–576. 10.1002/mrc.1260270609. [DOI] [Google Scholar]
- Altona C.; Francke R.; de Haan R.; Ippel J. H.; Daalmans G. J.; Hoekzema A. J. A. W.; van Wijk J. Empirical Group Electronegativities for Vicinal NMR Proton-Proton Couplings Along a C-C bond: Solvent Effects and Reparameterization of the Haasnoot Equation. Magn. Reson. Chem. 1994, 32, 670–678. 10.1002/mrc.1260321107. [DOI] [Google Scholar]
- Altona C.; Haasnoot C. A. G. Prediction of anti and gauche Vicinal Proton-Proton Coupling Constants in Carbohydrates: A Simple Additivity Rule for Pyranose Rings. Org. Magn. Reson. 1980, 13, 417–429. 10.1002/mrc.1270130606. [DOI] [Google Scholar]
- Steinmetz M.; Hansen A.; Ehrlich S.; Risthaus T.; Grimme S. Accurate Thermochemistry for Large Molecules with Modern Density Functionals. Top. Curr. Chem. 2014, 365, 1–23. 10.1007/128_2014_543. [DOI] [Google Scholar]
- Schwabe T.; Grimme S. Theoretical Thermodynamics for Large Molecules: Walking the Thin Line between Accuracy and Computational Cost. Acc. Chem. Res. 2008, 41, 569–579. 10.1021/ar700208h. [DOI] [PubMed] [Google Scholar]
- Řezáč J.; Hobza P. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. Chem. Rev. 2016, 116, 5038–5071. 10.1021/acs.chemrev.5b00526. [DOI] [PubMed] [Google Scholar]
- Brauer B.; Kesharwani M. K.; Kozuch S.; Martin J. M. L. The S66 × 8 Benchmark for Noncovalent Interactions Revisited: Explicitly Correlated ab initio Methods and Density Functional Theory. Phys. Chem. Chem. Phys. 2016, 18, 20905–20925. 10.1039/C6CP00688D. [DOI] [PubMed] [Google Scholar]
- Crich D.; Hu T.; Cai F. Does Neighboring Group Participation by Non-Vicinal Esters Play a Role in Glycosylation Reactions? Effective Probes for the Detection of Bridging Intermediates. J. Org. Chem. 2008, 73, 8942–8953. 10.1021/jo801630m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y.; Lian G.; Li Y.; Yu B. Identification of 3,6-Di-O-acetyl-1,2,4-O-orthoacetyl-α-D-glucopyranose as a Direct Evidence for the 4-O-Acyl Group Participation in Glycosylation. Chem. Commun. 2011, 47, 7515–7517. 10.1039/c1cc11680k. [DOI] [PubMed] [Google Scholar]
- Komarova B. S.; Ustyuzhanina N. E.; Tsvetkov Y. E.; Nifantiev N. E. In Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates; Werz D. B., Vidal S., Eds.; Wiley: Weinheim, Germany, 2014; pp 125–160. [Google Scholar]
- Wen P.; Crich D. Absence of Stereodirecting Participation by 2-O-Alkoxycarbonylmethyl Ethers in 4,6-O-Benzylidene-Directed Mannosylation. J. Org. Chem. 2015, 80, 12300–12310. 10.1021/acs.joc.5b02203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova B. S.; Tsvetkov Y. E.; Nifantiev N. E. Design of α-Selective Glycopyranosyl Donors Relying on Remote Anchimeric Assistance. Chem. Rec. 2016, 16, 488–506. 10.1002/tcr.201500245. [DOI] [PubMed] [Google Scholar]
- Yao D.; Liu Y.; Yan S.; Li Y.; Hu C.; Ding N. Evidence of Robust Participation by an Equatorial 4-O Group in Glycosylation on a 2-Azido-2-Deoxyglucopyranosyl Donor. Chem. Commun. 2017, 53, 2986–2989. 10.1039/C7CC00274B. [DOI] [PubMed] [Google Scholar]
- Schweizer W. B.; Dunitz J. D. Structural Characteristics of the Carboxylic Ester Group. Helv. Chim. Acta 1982, 65, 1547–1554. 10.1002/hlca.19820650528. [DOI] [Google Scholar]
- González-Outeiriño J.; Nasser R.; Anderson J. E. Conformation of Acetate Derivatives of Sugars and Other Cyclic Alcohols. Crystal Structures, NMR Studies, and Molecular Mechanics Calculations of Acetates. When is the Exocyclic C-O Bond Eclipsed?. J. Org. Chem. 2005, 70, 2486–2493. 10.1021/jo048295c. [DOI] [PubMed] [Google Scholar]
- Dale J. The Conformational Consequences of Replacing Methylene Groups by Ether Oxygen. Tetrahedron 1974, 30, 1683–1694. 10.1016/S0040-4020(01)90690-8. [DOI] [Google Scholar]
- van Boeckel C. A. A.; Beetz T.; van Aelst S. F. Substituent Effects on Carbohydrate Coupling Reactions Promoted by Insoluble Silver Salts. Tetrahedron 1984, 40, 4097–4107. 10.1016/0040-4020(84)85091-7. [DOI] [Google Scholar]
- Srivastava V. K.; Schuerch C. Synthesis of β-D-Mannopyranosides and β-L-Rhamnopyranosides by Glycosidation at C-1. J. Org. Chem. 1981, 46, 1121–1126. 10.1021/jo00319a016. [DOI] [Google Scholar]
- Crich D.; Picione J. Direct Synthesis of the ß-L-Rhamnopyranosides. Org. Lett. 2003, 5, 781–784. 10.1021/ol0340890. [DOI] [PubMed] [Google Scholar]
- Crich D.; Hutton T. K.; Banerjee A.; Jayalath P.; Picione J. Disarming, Non-participating 2-O-Protecting Groups in Manno- and Rhamnopyranosylation: Scope and Limitations of Sulfonates, Vinylogous Esters, Phosphates, Cyanates, and Nitrates. Tetrahedron: Asymmetry 2005, 16, 105–119. 10.1016/j.tetasy.2004.11.032. [DOI] [Google Scholar]
- Tanaka H.; Yoshizawa A.; Takahashi T. Direct and Stereoselective Synthesis of β-Linked 2,6-Deoxyoligosaccharides. Angew. Chem., Int. Ed. 2007, 46, 2505–2507. 10.1002/anie.200604031. [DOI] [PubMed] [Google Scholar]
- Baek J. Y.; Lee B.-Y.; Jo M. G.; Kim K. S. β-Directing Effect of Electron-Withdrawing Groups at O-3, O-4, and O-6 Positions and α-Directing Effect by Remote Participation of 3-O-Acyl and 6-O-Acetyl Groups of Donors in Mannopyranosylations. J. Am. Chem. Soc. 2009, 131, 17705–17713. 10.1021/ja907252u. [DOI] [PubMed] [Google Scholar]
- Baek J. Y.; Kwon H.-W.; Myung S. J.; Park J. J.; Kim M. Y.; Rathwell D. C. K.; Jeon H. B.; Seeberger P. H.; Kim K. S. Directing Effect by Remote Electron-Withdrawing Protecting Groups at O-3 or O-4 Position of Donors in Glucosylations and Galactosylations. Tetrahedron 2015, 71, 5315–5320. 10.1016/j.tet.2015.06.014. [DOI] [Google Scholar]
- Demchenko A. V.; Rousson E.; Boons G.-J. Stereoselective 1,2-cis-Galactosylation Assisted by Remote Neighboring Group Participation and Solvent Effects. Tetrahedron Lett. 1999, 40, 6523–6536. 10.1016/S0040-4039(99)01203-4. [DOI] [Google Scholar]
- Huang W.; Zhou Y.-Y.; Pan X.-L.; Zhou X.-Y.; Lei J.-C.; Liu D.-M.; Chu Y.; Yang J. S. Stereodirecting Effect of C5-Carboxylate Substituents on the Glycosylation Stereochemistry of 3-Deoxy-D-manno-oct-2-ulosonic Acid (Kdo) Thioglycoside Donors: Stereoselective Synthesis of α- and β-Kdo Glycosides. J. Am. Chem. Soc. 2018, 140, 3574–3582. 10.1021/jacs.7b09461. [DOI] [PubMed] [Google Scholar]
- Fréchet J. M.; Schuerch C. Solid-Phase Synthesis of Oligosaccharides. 11. Steric Control by C-6 Substituents in Glucoside Syntheses. J. Am. Chem. Soc. 1972, 94, 604–609. 10.1021/ja00757a047. [DOI] [Google Scholar]
- Kronzer F. J.; Schuerch C. The Methanolysis of Some Derivatives of 2,3,4-Tri-O-Benzyl-α-D-Glucopyranosyl Bromide in the Presence and Absence of Silver Salts. Carbohydr. Res. 1973, 27, 379–390. 10.1016/S0008-6215(00)81320-8. [DOI] [Google Scholar]
- Lourenço E. C.; Ventura M. R. Improvement of the Stereoselectivity of the Glycosylation Reaction with 2-Azido-2-deoxy-1-thioglucoside Donors. Carbohydr. Res. 2016, 426, 33–39. 10.1016/j.carres.2016.03.021. [DOI] [PubMed] [Google Scholar]
- Fujita S.; Oka N.; Matsumura F.; Wada T. Synthesis of Oligo(α-D-glycosyl phosphate) Derivatives by a Phosphoramidite Method via Boranophosphate Intermediates. J. Org. Chem. 2011, 76, 2648–2659. 10.1021/jo102584g. [DOI] [PubMed] [Google Scholar]
- Goodwin J. C. Amine-Catalyzed Transformation of Enolic Nonenzymatic Browning Products, Isomaltol Glycopyranosides into 1,6-Anhydro-β-D-hexopyranoses. Carbohydr. Res. 1985, 143, 61–68. 10.1016/S0008-6215(00)90695-5. [DOI] [Google Scholar]
- Xu C.; Liu H.; Li X. Thioglycosylation of 1,2-cis-Glycosyl Acetates: A Long Standing Overlooked Issue in Preparative Carbohydrate Chemistry. Carbohydr. Res. 2011, 346, 1149–1153. 10.1016/j.carres.2011.03.033. [DOI] [PubMed] [Google Scholar]
- Ohlsson J.; Magnusson G. Galabiosyl Donors: Efficient Synthesis from 1,2,3,4,6-Penta-O-acetyl-β-D-galactopyranose. Carbohydr. Res. 2000, 329, 49–55. 10.1016/S0008-6215(00)00154-3. [DOI] [PubMed] [Google Scholar]
- Damager I.; Olsen C. E.; Møller B. L.; Motawia M. S. Chemical Synthesis of 6‴-α-Maltotriosyl-maltohexaose as Substrate for Enzymes in Starch Biosynthesis and Degradation. Carbohydr. Res. 1999, 320, 19–30. 10.1016/S0008-6215(99)00131-7. [DOI] [PubMed] [Google Scholar]
- Kitowski A.; Jiménez-Moreno E.; Salvadó M.; Mestre J.; Castillón S.; Jiménez-Osés G.; Boutureira O.; Bernardes G. J. L. Oxidative Activation of C–S Bonds with an Electropositive Nitrogen Promoter Enables Orthogonal Glycosylation of Alkyl over Phenyl Thioglycosides. Org. Lett. 2017, 19, 5490–5493. 10.1021/acs.orglett.7b02886. [DOI] [PubMed] [Google Scholar]
- van Straten N. C. R.; Kriek N. M. A. J.; Timmers C. M.; Wigchert S. C. M.; van der Marel G. A.; van Boom J. H. Synthesis of a Trisaccharide Fragment Corresponding to the Lipopolysaccharide Region of Vibrio parahaemolyticus. J. Carbohydr. Chem. 1997, 16, 947–966. 10.1080/07328309708006550. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.