

Abstract
Cancer stem cells (CSCs) have been reported to be associated with the recurrence and drug resistance of liver cancer. In the present study, stem cell-like HepG2 cell spheres were enriched using stem cell conditioned culture medium. As expected, stem-like HepG2 cell spheres exhibited increased resistance to sorafenib. Metformin, a common drug used to treat type 2 diabetes mellitus, reduced the diameters and numbers of stem-like HepG2 spheres, and increased their sensitivity to sorafenib. Western blotting confirmed that low doses of metformin reversed the epithelial-mesenchymal transformation (EMT) process of HepG2 spheres. These results suggested that metformin enhanced sensitivity to sorafenib, which was probably through reversal of the EMT process of sphere-forming cells and by reducing the formation of CSCs.
Keywords: liver cancer, cancer stem cell, metformin, sorafenib, epithelial-mesenchymal transformation
Introduction
Liver cancer is one of the most lethal cancer types worldwide and its incidence is steadily rising (1). The overall 5-year survival rate for liver cancer is 17% (2) and drops to only 3% in patients with advanced malignancy. At diagnosis, <20% of patients with hepatic carcinoma have early-stage tumors that are potentially curable with surgery. For the majority of patients with non-resectable tumors, the treatments are largely palliative. Sorafenib is a multi-targeted tyrosine kinase inhibitor, and the only first-line drug for the clinical management of primary liver cancer (3). However, concerns have been raised about sorafenib therapy, including acquired drug resistance (3). Therefore, it is of great significance to elucidate the underlying mechanism and effective approaches to enhance sensitivity to sorafenib in patients with liver cancer.
Cancer stem cells (CSCs) are stem cell-like cells that are self-renewing and differentiating in tumors. The existence of tumor stem cells has been identified in liver cancer and various solid tumors (4). It was recently hypothesized that CSCs were correlated with occurrence, metastasis and drug resistance in many cancer types (5). The existence of liver CSCs is directly associated with resistance to cisplatin and 5-fluorouracil through regulation of RAC-α serine/threonine-protein kinase (Akt), transforming growth factor (TGF)-β and other signaling pathways (6). Liver CSCs may develop resistance to sorafenib through diverse mechanisms (7). These previous studies indicated that if sorafenib-resistant CSCs are eliminated, the incidence of drug resistance may be reduced.
Biguanide drugs are commonly used in the treatment of type 2 diabetes mellitus (T2DM). A recent study reported that metformin may reduce the incidence and mortality rates of multiple tumors in patients with T2DM (8). Metformin also suppressed CSC formation in breast cancer (9), glioblastoma (10), colon cancer (11), pancreatic cancer (12), prostate cancer (13) and osteosarcoma (14). Metformin was reported to improve overall survival by reducing the proportion of CSCs in oral squamous cell carcinoma (15). Metformin may improve CSC sensitivity to chemotherapeutic drugs, reduce the proportion of CSCs in liver cancer and increase liver cancer cell sensitivity to sorafenib (16). According to these results, it was hypothesized that metformin may be a useful therapeutic agent in enhancing sensitivity to sorafenib in T2DM, in addition to in patients without T2DM, by targeting CSCs.
Sphere-forming cell subpopulations isolated from human hepatoma cell lines possess properties that define CSCs (17). In the present study, cancer stem-like HepG2 spheres were generated using stem cell conditioned culture medium, and decreased sensitivity to sorafenib was observed in these stem-like spheres. Low doses of metformin reduced the formation by reversing the epithelial-mesenchymal transformation (EMT) process of HepG2 stem-like spheres.
Materials and methods
Cell culture
The HepG2 cell line was purchased from the Cell Resource Center of the Shanghai Institutes for Biological Science, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) in a humidified atmosphere at 37°C with 5% CO2.
Enrichment of stem cell-like HepG2 spheres
HepG2 cells were digested with Accutase cell detachment solution (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and washed with PBS (Gibco; Thermo Fisher Scientific, Inc.) twice. Subsequently, cells were seeded at a density of 1×104 cells/100 mm Petri dish, and cultured in stem cell conditioned culture medium [RPMI 1640 medium containing 2% B27 supplement (Gibco; Thermo Fisher Scientific, Inc.), 20 ng/ml recombinant human epidermal growth factor (PeproTech, Inc., Rocky Hill, NJ, USA) and 20 ng/ml recombinant human insulin-like growth factor (PeproTech, Inc.)]. Cells were maintained in a humidified incubator at 37°C with 5% CO2. Culture medium was half-replaced every other day. The diameters of the spheres reached 90–100 µm after 8 days. Following culturing for 8 days, the spheres were collected, dissociated into a single cell suspension and resuspended in fresh medium for serial subcultivation every 6 days. The morphology of the spheres was recorded with an inverted microscope (×200 magnification; Olympus IX73; Olympus Corporation, Tokyo, Japan) every 3 days. The total number and diameter of spheres >50 µm in three typical fields in each well or dish were counted, and the mean values were calculated accordingly. For cell passages, single cell suspensions derived from the original spheroids were obtained using Accutase cell detachment solution and placed into a new culture dish or plate, and the spheres grew to 100 µm in diameter within 9 days.
Flow cytometry
In order to detect the expression of the stem cell surface markers cluster of differentiation (CD) 133 and CD90, spheres at day 7 were digested into single cells with Accutase cell detachment solution. A total of ~1.0×106 cells were washed with cold PBS twice and resuspended in 400 µl PBS. A volume of 10 µl rabbit anti-human CD133/1 (AC133)-phycoerythrin (PE; cat. no. 130098826; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) or 2 µl rabbit anti-human CD90 (REA897)-fluorescein isothiocyanate (FITC; cat. no. 130114901; Miltenyi Biotec GmbH) was added and incubated in the dark for 30 min at 4°C. Following two washes with PBS, the markers were determined using a FACSCalibur system running BD CellQuest™ Pro software version 3.3 (BD Biosciences, San Jose, CA, USA) and the data were analyzed using FlowJo version 7.6 (FlowJo LLC, Ashland, OR, USA). Isotype controls of PE and FITC were used to eliminate false positive expression.
Cell viability assay
HepG2 spheres and parental HepG2 cells were seeded into a 96-well plate at a density of 600 cells/well for 3 days. Cells were treated with sorafenib at concentrations of 5, 10, 15, 20 or 25 µM, alone or combined with 1, 3 or 5 mM metformin and incubated at 37°C for 48 h. Cell proliferation analysis was conducted using a Cell Counting Kit-8 (CCK8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) assay, according to the manufacturer's protocol. In brief, CCK8 solution was added into the culture and incubated for 2 h. The absorbance at 450 nm [optical density (OD) value] was measured using an auto-microplate reader (BERTHOLD TECHNOLOGIES GmbH & Co. KG, Bad Wildbad, Germany). The inhibition percentage was calculated using the following equation: Inhibition percentage=(1-ODexperiment/ODcontrol) ×100. The IC50 value was determined by plotting the inhibition percentage values. The resistance index (RI) was calculated using the following equation: RI=half-maximal inhibitory concentration (IC50) spheres/IC50HepG2.
Western blotting
HepG2 cells and spheres were treated with sorafenib, metformin or a combination of sorafenib and metformin for 48 h. Cells were collected and lysed in Cell Lysis Buffer (cat. no. P0013) containing 1 mM phenylmethylsulfonyl fluoride (cat. no. ST506-2; both Beyotime Institute of Biotechnology, Haimen, China) for 30 min on ice. The lysate was centrifuged at 12,000 × g for 15 min at 4°C, and the supernatant was collected. The protein concentration was determined using a Bicinchoninic Acid Protein Assay kit (cat. no. P0012; Beyotime Institute of Biotechnology). A total of 30 µg of protein lysate was boiled in SDS-PAGE Sample Loading Buffer (cat. no. P0015; Beyotime Institute of Biotechnology), resolved by electrophoresis on 10% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes (cat. no. FFP33; Beyotime Institute of Biotechnology). The membranes were blocked with 5% skimmed milk in TBS/Triton-X-100 at room temperature for 90 min. The membranes were probed overnight at 4°C with primary antibodies against human zinc finger protein SNAI1 (Snail; rabbit monoclonal antibody; cat. no. 3879T; Cell Signaling Technology, Inc., Danvers, MA, USA), E-cadherin (mouse monoclonal antibody; cat. no. AF0138; Beyotime Institute of Biotechnology), vimentin (mouse monoclonal antibody; cat. no. AF0318; Beyotime Institute of Biotechnology) and twist-related protein 1 (Twist1; rabbit polyclonal antibody; cat. no. 21642; Signalway Antibody LLC, College Park, MD, USA), with GAPDH (rabbit monoclonal antibody; cat. no. 5174; Cell Signaling Technology, Inc.) as the control. All primary antibodies were diluted 1:1,000 with Antibody Dilution Buffer (cat. no. P0023A; Beyotime Institute of Biotechnology). Horseradish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin (Ig)G (H+L; cat. no. A0216; Beyotime Institute of Biotechnology) and HRP-labeled goat anti-rabbit IgG (H+L; cat. no. A0208; Beyotime Institute of Biotechnology) diluted 1:5,000 with Antibody Dilution Buffer, were used to detect specific proteins. Finally, the bands were detected using Enhanced Chemiluminescent Substrates (http://www.bio-kits.cn; Biokits Technologies Inc., Beijing, China).
Statistics
Data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). Experiments were performed at least in triplicate, and data are presented as the mean ± standard deviation. Data were analyzed by Bonferroni analysis (least significant difference-t test). α risk was set at 0.05 and β risk was set at 0.95. P<0.05 was considered to indicate a statistically significant difference.
Results
Stem-like HepG2 spheres are enriched by stem-cell enrichment medium
The human liver cancer cell line HepG2 was cultured with stem cell enrichment medium as described above. The cells detached and formed small spheres from the 3rd day (Fig. 1A), with a diameter of ~34.32±1.07 µm (Fig. 1B). There were ~100±21.2 small spheres in three fields on day 3. The diameters of the spheres reached ~81.97±1.87 µm on day 9. The numbers of spheroids first increased to 145±19.1 on day 6, although they decreased to 91±5.1 on day 9 as the small spheres joined to become a large sphere (Fig. 1B and C).
Figure 1.
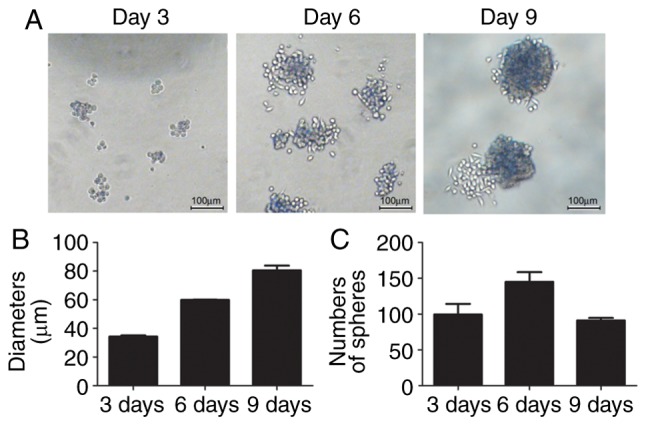
Suspension sphere formation of HepG2 cells in stem cell conditioned medium. (A) Typical bright-field image of HepG2 cells cultured in stem cell conditioned medium on days 3, 6 and 9. Scale bar, 100 µm. (B) Average diameters of suspended spheres on days 3, 6 and 9. (C) Numbers of spheres on days 3, 6 and 9. Data are expressed as the mean ± standard deviation.
CD133 and CD90 are the most recognized liver CSC markers. To examine the stemness of the enriched spheres, the proportion of CD133- or CD90-positive cells in the spheres was detected with flow cytometry. As expected, few cells expressed CD133 or CD90 in parental HepG2 cells. In spheres passaged three times (P3), ~4.43±2.48 and 10.33±1.23% cells expressed CD133 and CD90, respectively (Fig. 2A and B). The differences were statistically significant (P<0.05).
Figure 2.

Stem cell markers expressed in HepG2 spheres. CD133 and CD90 expression in parental HepG2 cells and spheres on day 12 was examined by flow cytometry. Spheroid cells incubated with isotype antibodies were used as a negative control. (A) Representative flow cytometry dot plots of CD133 (upper panel) and CD90 (lower panel) staining. Percentage of (B) CD133- and (C) CD90-positive cells in parental HepG2 cells and spheres. All experiments were repeated at least three times and the data are presented as the mean ± standard deviation. *P<0.05, ***P<0.001. CD, cluster of differentiation; PE, phycoerythrin; FITC, fluorescein isothiocyanate.
Stem cell-like HepG2 spheres are less susceptible to sorafenib
Single cell suspensions of HepG2 sphere cells and HepG2 cells were seeded in 96-well plates at 600 cells/well. After 3 days, cells were treated with sorafenib at 5, 10, 20 and 25 µM for 24 and 48 h, respectively. The CCK8 cell proliferation activity assay demonstrated that the inhibition rate of sorafenib on HepG2 spheres was significantly lower compared with that on parental HepG2 cells at 48 h post-treatment (Fig. 3). The IC50 value of HepG2 spheres (14.84 µM) was significantly higher than that of parental HepG2 cells (9.4 µM) (P<0.05; data not shown).
Figure 3.
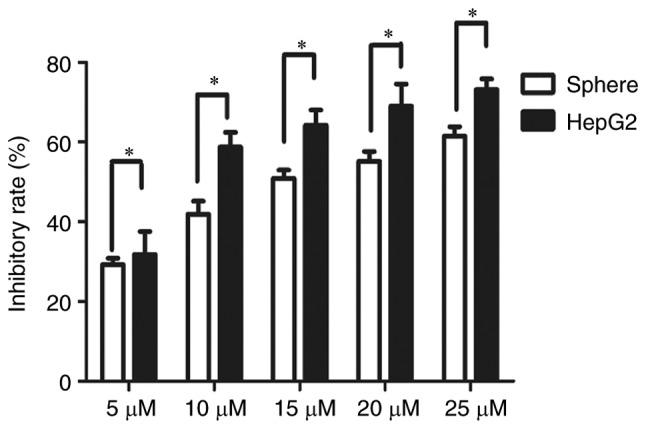
Increased resistance to sorafenib in HepG2 spheres. Spheres were treated with 5, 10, 15, 20 or 25 µM sorafenib for 48 h and the inhibition rate was calculated according to cell viability, as assessed by Cell Counting Kit-8 assay. Each experiment was repeated in triplicate and the data are expressed as the mean ± standard deviation. *P<0.05.
Metformin increases the sensitivity to sorafenib of HepG2 spheres
As there was no suggestion for the dose of metformin for non-T2DM patients, the present study sought to analyze whether lower doses of metformin have effects on enhancing drug sensitivity. Thus, HepG2 sphere cells were treated with 0, 1, 3 and 5 mM metformin combined with 5 µM sorafenib. As expected, 1 mM metformin and 5 µM sorafenib significantly inhibited the cell viability of HepG2 spheres (Fig. 4A and B). As the concentration of metformin increased, the diameter of the suspension spheres was significantly reduced (Fig. 4A and C). The number of spheres increased when 1 mM metformin was added, although it decreased when the concentration of metformin increased (Fig. 4D).
Figure 4.

Metformin enhances the sensitivity of HepG2 spheres to sorafenib. (A) Cells were treated with 5 µM sorafenib alone or combined with 1, 3 or 5 µM metformin. Non-treated cells were used as a control. The typical bright-field images from three independent experiments are presented. Scale bar, 200 µm. (B) The inhibition rate was calculated according to the cell viability, as analyzed by Cell Counting Kit-8 assay. (C) The average diameter and (D) numbers of spheres were recorded. All experiments were repeated at least three times independently and the data are expressed as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001. CON, control; SORA, sorafenib; MET, metformin.
Metformin inhibits EMT and reverses the formation of HepG2 spheres
EMT is associated with the formation of CSCs. From previous results, it was identified that alterations in the diameters and numbers of spheres following treatment with metformin indicated that metformin may reverse the formation of stem-like spheres. As demonstrated in Fig. 5A, the diameters and numbers of spheres were markedly reduced by 1 and 5 mM metformin. As expected, expression of the epithelial marker E-cadherin was significantly reduced in stem-like spheres compared with parental HepG2 cells, while the expression of the mesenchymal markers vimentin, Snail and Twist1 were significantly elevated in spheres. Metformin reversed the expression of these EMT markers in a dose-dependent manner (Fig. 5B-F).
Figure 5.

Metformin reverses HepG2 sphere formation by reducing epithelial-mesenchymal transformation. (A) Typical bright-field images of spheroids treated with 0 (CON), 1 or 5 mM metformin for 24 h. Scale bar, 200 µm. (B) Expression of E-cadherin, vimentin, Snail and Twist1 in parental HepG2 cells and spheroids treated with metformin was detected by western blotting. GAPDH was used as a loading control. Semi-quantification of protein expression was performed for (C) E-cadherin, (D) vimentin, (E) Snail and (F) Twist1. All experiments were repeated at least three times independently and the data are expressed as the mean ± standard deviation. *P<0.05, **P<0.01, ***P<0.001. CON, control; sph, spheroid; met, metformin; Snail, zinc finger protein SNAI1; Twist1, twist-related protein 1.
Discussion
In this study, stem cell-like HepG2 spheres were enriched using stem cell enrichment medium. It was demonstrated that those spheres expressed increased levels of the stem cell markers CD133 and CD90 compared with parental HepG2 cells, and were more resistant to sorafenib. Metformin reduced the diameters and numbers of spheres, and reversed their resistance to sorafenib. In addition, it was observed that these effects were associated with the metformin-induced reduction in EMT and stemness.
Researchers have identified that the mechanism of tumor resistance to sorafenib is multifaceted. The anti-tumor activity of sorafenib was largely attributed to the inhibition of growth factor signaling pathways, including vascular endothelial growth factor receptor and platelet-derived growth factor receptor, and the downstream RAF proto-oncogene serine/threonine-protein kinase (RAF)/dual specificity mitogen-activated protein kinase kinase 1 (MEK)/extracellular signal-regulated kinase (ERK) pathway. The activation of an escape pathway from RAF/MEK/ERK may result in chemoresistance (7). Certain studies have demonstrated that upregulation of hypoxia inducible factor-2α induced by sorafenib contributes to the resistance of hypoxic liver cancer cells by activating the TGF-α/epidermal growth factor receptor pathway (18). Emerging evidence has indicated that the activation of the EMT process, the emergence of CSCs and the activation of compensatory pathways leading to sorafenib have been implicated (18–20). In response to treatment with sorafenib, MHCC97H cells develop a mesenchymal phenotype and resistance is conferred (19). The formation of CSCs is also thought to serve an important role in chemotherapy resistance to multiple drugs, including sorafenib (7). A recent study reported that CSCs were enriched in tumor tissues following treatment with sorafenib (21). An increased proportion of CD133+ CSCs may cause resistance to sorafenib. In the present study, stem-cell like HepG2 spheres were enriched using stem cell conditioned enrichment medium. Compared with parental HepG2 cells, there were significantly more CD133+ cells in enriched spheres, and the sensitivity of spheres to sorafenib was significantly decreased. These results supported the hypothesis that stem cells are partially responsible for sorafenib resistance.
A recent study suggested that EMT may be a predictive marker for the development of resistance to sorafenib (21). Following long-term exposure to sorafenib, liver cancer cells exhibit EMT and resistance to sorafenib (22). EMT-associated serum response factor was reported to induce HLE cells to acquire a mesenchymal phenotype, which leads to resistance against a sorafenib-mediated apoptotic effect (20). Multi-drug resistance (MDR) is a downstream molecular event of EMT and is also responsible for sorafenib resistance. Silencing Snail with small interfering RNA inhibits EMT and partially reverses MDR, thereby markedly decreasing invasion and metastasis in sorafenib-resistant liver cancer cells (22). In the present study, it was observed that in the stem-cell like HepG2 spheres, expression of the markers of EMT indicated that they acquired the mesenchymal phenotype during enrichment, which may explain the increased resistance to sorafenib compared with parental HepG2 cells.
Metformin is the most popular drug used to treat T2DM, and functions through inhibition of the mitochondrial electron transport chain complex I and activation of 5′-adenosine monophosphate-activated protein kinase (AMPK)-serine/threonine-protein kinase mTOR (mTOR) signaling. The anti-tumor effect of metformin has received extensive attention (8). Metformin also exhibits great potential in the prevention and treatment of liver cancer, as the incidence of liver cancer reduces with increasing doses of metformin (23). Metformin may reduce the incidence of liver cancer in patients with T2DM (16). Metformin promotes apoptosis in a variety of malignancies (24), and increases the sensitivity of sorafenib to MHCC97H cells (24). The present study demonstrated that the combination of metformin and sorafenib at low concentrations significantly inhibited the cell viability of stem-like spheres.
The anti-tumor mechanism of metformin remains unclear. Previous studies suggested that metabolic reprograming was important in maintaining the stemness of CSCs; metformin may interfere with oxidative phosphorylation by inhibiting the mitochondrial electron transport chain complex, and has been reported to delay tumor progression by reducing the formation of CSCs (7,9,10,12,13,15,16). Certain studies have reported that drug resistance is associated with EMT in CSCs. Metformin inhibits tumor stem cell EMT to enhance sensitivity to chemotherapeutic drugs (11–14,25). Metformin at a low concentration may inhibit EMT in ovarian CSCs (26). It has been suggested that metformin may enhance the sensitivity of hepatoma cells to sorafenib by inhibiting the production of CSCs, and inhibiting EMT to reduce the formation of drug-resistant stem cells (16,19). It was identified that metformin reduced the diameters and numbers of HepG2 spheres. Metformin reversed the alterations in E-cadherin and vimentin expression in a suspension of HepG2 sphere-forming cells, and reversed the expression of the stem cell-associated transcription factors Twist1 and Snail in a dose-dependent manner. The above results suggested that metformin may reverse resistance to sorafenib by attenuating EMT and stem cell enrichment.
Metformin may enhance the sensitivity of cells to sorafenib though pathways which also regulate EMT (27), including TGF-β signaling, mothers against decapentaplegic homolog (SMAD)-dependent signaling, the ERK-Jun pathway and the Akt-mTOR pathway. For example, the combination of sorafenib and metformin enhances the inhibitory effect of metformin on the mTOR complex 1 and ERK signaling pathways (28). Metformin activates the AMPK pathway, which suppresses EMT by modulating Akt-E3 ubiquitin-protein ligase Mdm2-forkhead box protein O3 signaling axis (29) and the TGF-β-SMAD2/3 pathway (30). Thus, it was hypothesized that metformin may affect a number of crosslinked pathways, and further study was required clarify this.
However, not all cancer cells respond well to metformin or other biguanide drugs. Hepatoma cells that have a higher mitochondrial respiration rate are more sensitive to these treatments; whereas tumor cells that exhibit increased glycolysis are more resistant. Altered energy metabolism in hepatoma cells, from glycolysis to mitochondrial respiration, improves cellular sensitivity to biguanides (31). Since metformin is a mitochondrial complex enzyme inhibitor, this may explain why it only affects tumor cells with enhanced oxidative phosphorylation.
In conclusion, in the present study, human liver cancer cell line HepG2 stem cell-like cells were enriched with tumor stem cell conditioned medium suspension culture, and it was verified that the sensitivity of stem cell-like spheres to sorafenib was significantly reduced. Metformin enhanced sensitivity to sorafenib, possibly by inhibiting EMT in the suspension spheres and reducing the formation of stem cell-like cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science Foundation of China (grant nos. 8156049 and 81360347) and the Guangxi Nanning Qingxiu District Science and Technology Development Project (grant no. 2015S14).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
YF, XG and XLL conceived and designed the study. YF, XG, XPH, MYW, XL and SSW acquired, analyzed or interpreted the data. YF and XG drafted the manuscript. XLL revised and approved the final version of the manuscript to be published.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Gauthier A, Ho M. Role of sorafenib in the treatment of advanced hepatocellular carcinoma: An update. Hepatol Res. 2013;43:147–154. doi: 10.1111/j.1872-034X.2012.01113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clevers H. The cancer stem cell: Premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 5.Oikawa T. Cancer stem cells and their cellular origins in primary liver and biliary tract cancers. Hepatology. 2016;64:645–651. doi: 10.1002/hep.28485. [DOI] [PubMed] [Google Scholar]
- 6.Ji J, Wang XW. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. Semin Oncol. 2012;39:461–472. doi: 10.1053/j.seminoncol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nishida N, Kitano M, Sakurai T, Kudo M. Molecular mechanism and prediction of sorafenib chemoresistance in human hepatocellular carcinoma. Dig Dis. 2015;33:771–779. doi: 10.1159/000439102. [DOI] [PubMed] [Google Scholar]
- 8.Safe S, Nair V, Karki K. Metformin-induced anticancer activities: Recent insights. Biol Chem. 2018;399:321–335. doi: 10.1515/hsz-2017-0271. [DOI] [PubMed] [Google Scholar]
- 9.Shi P, Liu W, Tala, Wang H, Li F, Zhang H, Wu Y, Kong Y, Zhou Z, Wang C, et al. Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell Discov. 2017;3:17010. doi: 10.1038/celldisc.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gritti M, Würth R, Angelini M, Barbieri F, Peretti M, Pizzi E, Pattarozzi A, Carra E, Sirito R, Daga A, et al. Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget. 2014;5:11252–11268. doi: 10.18632/oncotarget.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E, Majumdar AP. Metformin: A potential therapeutic agent for recurrent colon cancer. PLoS One. 2014;9:e84369. doi: 10.1371/journal.pone.0084369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chai X, Chu H, Yang X, Meng Y, Shi P, Gou S. Metformin increases sensitivity of pancreatic cancer cells to gemcitabine by reducing CD133+ cell populations and suppressing ERK/P70S6K signaling. Sci Rep. 2015;5:14404. doi: 10.1038/srep14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayer MJ, Klotz LH, Venkateswaran V. Metformin and prostate cancer stem cells: A novel therapeutic target. Prostate Cancer Prostatic Dis. 2015;18:303–309. doi: 10.1038/pcan.2015.35. [DOI] [PubMed] [Google Scholar]
- 14.Shang D, Wu J, Guo L, Xu Y, Liu L, Lu J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int J Oncol. 2017;50:1848–1856. doi: 10.3892/ijo.2017.3950. [DOI] [PubMed] [Google Scholar]
- 15.Siddappa G, Kulsum S, Ravindra DR, Kumar VV, Raju N, Raghavan N, Sudheendra HV, Sharma A, Sunny SP, Jacob T, et al. Curcumin and metformin-mediated chemoprevention of oral cancer is associated with inhibition of cancer stem cells. Mol Carcinog. 2017;56:2446–2460. doi: 10.1002/mc.22692. [DOI] [PubMed] [Google Scholar]
- 16.Saito T, Chiba T, Yuki K, Zen Y, Oshima M, Koide S, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, et al. Metformin, a diabetes drug, eliminates tumor-initiating hepatocellular carcinoma cells. PLoS One. 2013;8:e70010. doi: 10.1371/journal.pone.0070010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu F, Cao X, Liu Z, Guo H, Ren K, Quan M, Zhou Y, Xiang H, Cao J. Casticin suppresses self-renewal and invasion of lung cancer stem-like cells from A549 cells through down-regulation of pAkt. Acta Biochim Biophys Sin (Shanghai) 2014;46:15–21. doi: 10.1093/abbs/gmt123. [DOI] [PubMed] [Google Scholar]
- 18.Zhao D, Zhai B, He C, Tan G, Jiang X, Pan S, Dong X, Wei Z, Ma L, Qiao H, et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014;26:1030–1039. doi: 10.1016/j.cellsig.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 19.You A, Cao M, Guo Z, Zuo B, Gao J, Zhou H, Li H, Cui Y, Fang F, Zhang W, et al. Metformin sensitizes sorafenib to inhibit postoperative recurrence and metastasis of hepatocellular carcinoma in orthotopic mouse models. J Hematol Oncol. 2016;9:20. doi: 10.1186/s13045-016-0253-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae JS, Noh SJ, Kim KM, Jang KY, Chung MJ, Kim DG, Moon WS. Serum response factor induces epithelial to mesenchymal transition with resistance to sorafenib in hepatocellular carcinoma. Int J Oncol. 2014;44:129–136. doi: 10.3892/ijo.2013.2154. [DOI] [PubMed] [Google Scholar]
- 21.Mir N, Jayachandran A, Dhungel B, Shrestha R, Steel JC. Epithelial-to-mesenchymal transition: A mediator of sorafenib resistance in advanced hepatocellular carcinoma. Curr Cancer Drug Targets. 2017;17:698–706. doi: 10.2174/1568009617666170427104356. [DOI] [PubMed] [Google Scholar]
- 22.Dong J, Zhai B, Sun W, Hu F, Cheng H, Xu J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS One. 2017;12:e0185088. doi: 10.1371/journal.pone.0185088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen HP, Shieh JJ, Chang CC, Chen TT, Lin JT, Wu MS, Lin JH, Wu CY. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: Population-based and in vitro studies. Gut. 2013;62:606–615. doi: 10.1136/gutjnl-2011-301708. [DOI] [PubMed] [Google Scholar]
- 24.Guo Z, Cao M, You A, Gao J, Zhou H, Li H, Cui Y, Fang F, Zhang W, Song T, et al. Metformin inhibits the prometastatic effect of sorafenib in hepatocellular carcinoma by upregulating the expression of TIP30. Cancer Sci. 2016;107:507–513. doi: 10.1111/cas.12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang R, Zhang P, Wang H, Hou D, Li W, Xiao G, Li C. Inhibitory effects of metformin at low concentration on epithelial-mesenchymal transition of CD44(+)CD117(+) ovarian cancer stem cells. Stem Cell Res Ther. 2015;6:262. doi: 10.1186/s13287-015-0249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(re8) doi: 10.1126/scisignal.2005189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling S, Song L, Fan N, Feng T, Liu L, Yang X, Wang M, Li Y, Tian Y, Zhao F, et al. Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int J Oncol. 2017;50:297–309. doi: 10.3892/ijo.2016.3799. [DOI] [PubMed] [Google Scholar]
- 29.Chou CC, Lee KH, Lai IL, Wang D, Mo X, Kulp SK, Shapiro CL, Chen CS. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014;74:4783–4795. doi: 10.1158/0008-5472.CAN-14-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin H, Li N, He H, Ying Y, Sunkara S, Luo L, Lv N, Huang D, Luo Z. AMPK inhibits the stimulatory effects of TGF-β on Smad2/3 activity, cell migration, and epithelial-to-mesenchymal transition. Mol Pharmacol. 2015;88:1062–1071. doi: 10.1124/mol.115.099549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu CC, Wu LC, Hsia CY, Yin PH, Chi CW, Yeh TS, Lee HC. Energy metabolism determines the sensitivity of human hepatocellular carcinoma cells to mitochondrial inhibitors and biguanide drugs. Oncol Rep. 2015;34:1620–1628. doi: 10.3892/or.2015.4092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.