

Abstract
Background
An important issue in microarray data is to select, from thousands of genes, a small number of informative differentially expressed (DE) genes which may be key elements for a disease. If each gene is analyzed individually, there is a big number of hypotheses to test and a multiple comparison correction method must be used. Consequently, the resulting cut-off value may be too small. Moreover, an important issue is the selection’s replicability of the DE genes. We present a new method, called ORdensity, to obtain a reproducible selection of DE genes. It takes into account the relation between all genes and it is not a gene-by-gene approach, unlike the usually applied techniques to DE gene selection.
Results
The proposed method returns three measures, related to the concepts of outlier and density of false positives in a neighbourhood, which allow us to identify the DE genes with high classification accuracy. To assess the performance of ORdensity, we used simulated microarray data and four real microarray cancer data sets. The results indicated that the method correctly detects the DE genes; it is competitive with other well accepted methods; the list of DE genes that it obtains is useful for the correct classification or diagnosis of new future samples and, in general, it is more stable than other procedures.
Conclusions
ORdensity is a new method for identifying DE genes that avoids some of the shortcomings of the individual gene identification and it is stable when the original sample is changed by subsamples.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2318-8) contains supplementary material, which is available to authorized users.
Keywords: Differentially expressed gene, Multivariate statistics, Outlier, Quantile
Background
Analysis of gene expression data arising from microarray or RNA-Seq technologies is a very important task and a major advance in biomedical research. In this kind of experiments, the main goal is to identify a small number of informative genes whose patterns of expression differ according to the experimental conditions. These genes, selected from thousands, are differentially expressed, between two possible conditions, as control and treatment groups or between two groups of patients. This gene discovery is challenging, because there is a large number of genes, a relatively small number of samples and it is important to identify which genes, independently of the sample studied of the same disease, are selected as differentially expressed genes. The selection of relevant genes to differentiate these two conditions has two main objectives for researchers. On the one hand, to select a small number of genes so that the information given by these genes is not redundant, and the classification or diagnostic of new samples which lead to lower prediction error. On the other hand, a large number of selected genes are related to others that have the same function and that are highly correlated. It is clear that, in order to obtain a list of genes that allows a good diagnosis for future samples, a combination of these two objectives is desirable. It is also necessary to know the relation and function of the selected genes.
As, in general, the expression levels of genes are dependent on each other because genes are involved in complex regulatory pathways and networks [1], it seems convenient to consider the joint distribution of genes. However, methods to identify DE (differentially expressed) genes that are based on a gene-by-gene approach, ignoring the dependences between genes, are widely used. Maybe, the most popular is the t-test, but it has some restrictions [2, 3]. To solve the problem of unstable variances, the Significance Analysis of Microarrays (SAM) [2] was introduced. It works with a modified t-test introducing a factor to minimize the effect that small per-gene variances could make genes, with small differences between the expression conditions, statistically significant. An integrated solution for analyzing data from gene expression experiments is provided by limma [4, 5] an R package for Bioconductor [6]. limma fits a linear model for each gene and uses an empirical Bayes (eBayes) method for assessing differential expression. The empirical Bayes method (eBayes) [7] also uses moderated t-statistics, where instead of the global or single gene estimated variances, a weighted average of the global and single-gene variances is used. In the gene-by-gene approaches, as a large number of hypothesis tests are carried out, multiple testing procedures must be applied to assess the overall significance, controlling the family-wise error rate (FWER) or the false positive rate (FDR). The FWER is a very stringent criterion which measures the probability of at least one false positive in the set of significant genes, and most investigators accept a FWER of 5% [8]. A more liberal criterion is the FDR [9], which is the expected proportion of false positives among the significant genes. However, all the p-value adjustment methods lose sensitivity as they have a reduced chance of detecting true positives.
As different statistical selection methods may capture different statistical aspects of expression changes, they may give different lists of selected genes. Nevertheless, these inconsistent gene lists could be rather functionally consistent [10–12]. However, it is clear that one desirable property for a proper statistical method is that it detects true differentially expressed genes and has the ability to maintain a consistent list of differentially expressed genes within a single data set, that is, with samples based on subsets of the same data. In this direction, in [3] an empirical evaluation of consistency and accuracy for different methodologies was presented, concluding that for smaller sample sizes, moderated versions of the t-test can generally be recommended, while for large data sets, the method may involve a compromise between consistency and power.
A different approach was introduced in [13, 14]. In these works, the authors presented a statistic, called OR, which is useful to identify extreme observations or outliers in high-dimensional data sets. They presented the possibility to use the OR statistic as a tool to detect differentially expressed genes. The idea is that in expression studies, there are a large number of genes, and very few are expected to be important for the disease development. Thus, the important genes (which are DE) should show a different behaviour to those that are non-important. For this reason, the important genes could be considered as outliers in a background population of non-important genes.
One more fruitful endeavour than searching for lists of differentially expressed genes might be to search for the best way in which the two groups under study can be distinguished. Suppose that a new sample is considered, and we wish to decide which of the two groups it belongs to. The best set of differentially expressed genes will be the one that leads to the smallest probability of misclassifying this new sample, that is, the set of differentially expressed genes that leads to the smallest error rate of all future allocations of new samples. Thus, it is very interesting to analyze if a procedure obtains lists of differentially expressed genes useful for classification of future samples. It is therefore important to recognize the two objectives: to maximize separation between the groups of available samples, and to minimize the misclassification rate over all possible future allocations.
In this article, we present a new method which uses the OR statistics and two new measures which, together, are useful to obtain consistent lists of true differentially expressed genes, and which allows the correct classification of future samples. This novel approach, called ORdensity, takes into account the relation between all genes and it is not a gene-by-gene approach.
In the “Methods” section, we detail the basic ideas, the new concepts, the description of the new method, a small example to aid the comprehension of the new methodology, as well as the simulation studies and gene expression data from four public cancer studies, that were used to evaluate the behaviour of the procedure. In the “Results” and “Discussion” sections we show the usefulness of our approach. We close this paper with some conclusions.
Methods
The new ORdensity procedure has two main steps: finding potential differentially expressed genes and identifying differentially expressed genes. Next, we detail these two steps of the method and Fig. 1 resumes the general outline of the approach. In order to better understand the procedure development, a small artificial example is also included.
Fig. 1.

General outline of the proposed ORdensity approach. In green the first step of the method and in red the second step of the method
Let E={e1,…,eG} be a set of expression level values for G genes such that each eg is a vector eg=(egX,egY)′ giving the expression of the g-gene in two conditions X and Y (e.g., treatment/control or two patient groups). Then, and , nX+nY=n, are vectors of values giving the expression of the g-gene in the j-sample under condition X and Y, respectively. Each eg can then be considered a point in a continuous n-dimensional gene expression space S⊂Rn. Let Xg and Yg be the random variables representing the expression level of gene g in conditions X and Y, respectively (g=1,…,G).
The proposed approach focuses on the differences of quantiles between samples: , p∈Cp where Cp is a set of probabilities. For instance, Cp={0.25,0.5,0.75} is an adequate set for small sample sizes. A gene, g, whose expressions in conditions X and Y are considered not differentially expressed (see Fig. 2 left) would verify that , where F is the cumulative distribution function and p∈[0,1]. Otherwise, gene g is differentially expressed (DE) or it is important (see Fig. 2 right). Broadly speaking, matrix V=(vgp) with vgp=, for g=1,…,G and p∈Cp must contain small values corresponding to the major number of no DE genes. However, the most differentially expressed genes should show a different behaviour, and for this reason they can be considered as outliers in V. Thus, our approach attempts, in two main steps, to find outliers in V which can be identified as differentially expressed genes.
Fig. 2.

Visualization of differences for p∈Cp={0.25,0.5,0.75} for two genes. In the left side, a gene whose expressions in conditions X and Y are not differentially expressed (No DE gene); in the right side, a gene that is differentially expressed in conditions X and Y (DE gene)
First step: finding potential differentially expressed genes
Let V=(vgp) the G×P matrix with , p∈Cp where Cp is a set of probabilities (P=#Cp). As DE genes are expected to be outliers based on the values, the procedure computes the robust index OR of outlyingness [13, 14] as follows. Define dgh the Euclidean distance between vg and vh. For a fixed gene g, the OR statistic is given by:
In this ratio, the numerator gives the median value of all (squared) distances of the gene of interest with respect to all the other genes; the denominator gives (half) the median of all (squared) distances among all genes. As a consequence, given a set of G genes, OR gives a ranking of genes, so that genes with a large value of OR will be genes that are further away from the set of all genes and therefore they are possible outliers (i.e., important genes). In this way, the original G×n data matrix is firstly transformed in a G×P matrix with the vgp differences between Cp-quantiles and, secondly, in a G dimensional vector with the OR values OR=(OR(g1),…,OR(gG))′. As the distribution of OR is unknown, the procedure considers permuted samples in order to generate values associated with genes which are not differentially expressed. Thus, the expression values of each gene are permuted B times, that is, each sample has its corresponding label and actually the permutation is carried out on the labels. Once the labels of the samples are reassigned by permutation (B times), the expressions of the genes are classified according to those two classes. Then, the procedure computes matrix and vector ORb=(ORb(g1),…,ORb(gG))′ for each permutation b, (b=1,…,B). Given a fixed α∈(0,1), it calculates the percentile (1−α) 100% of all the elements of the matrix with the permuted samples ORb. Let us denote this value by cα. In this way, the method excludes those genes, g, with OR(g)≤cα. Then, DE genes must be in the subset of genes Sα={g | OR(g)>cα}. We call them potential genes.
Second step: identifying differentially expressed genes
By construction, there are α×G×B cases where ORb(h)>cα among the permuted samples and we call Rα={h | ORb(h)>cα, b=1,…B} the set of those cases. That is, in Rα there are all permuted cases h with ORb(h)>cα for some b. These cases are related to genes that are not DE but have large OR values. The corresponding values of , reflect the behaviour of these genes in relation to the differences between quantiles. Hence, we have, on the one hand, differences for potential differentially expressed genes and on the other hand, we know that the differences represent the behaviour of cases that are false positives with a misleadingly large value of OR. Therefore, the analysis of the differences and similarities between vg and will provide a way to discriminate the truly DE genes among the set Sα of potential DE genes.
To this aim, consider matrix randomly divided into k-folds. Then, consider the union of set Sα with the ith fold Rα,i={h∈Rα| ith fold}, that is, Ui=Sα∪Rα,i. In order to understand what happens in Ui, consider the following small artificial example.
Small artificial example: We simulated [15] 1000 genes under two conditions X and Y with 30 samples for each condition and 60 of these genes were generated as differentially expressed. We considered Cp={0.25,0.5,0.75} to build the (1000×3)-matrix V of differences between the quartiles and these differences were weighted with {0.25,0.5,0.25}, respectively. The procedure obtained the matrix V of differences between the weighted quartiles and it calculates the OR statistic for the 1000 original genes, OR=(OR(g1),…,OR(g1000))′. Next, for B=100 permuted samples of X and Y, we computed matrix of differences between the weighted quartiles and we computed their OR values ORb=(ORb(g1),…,ORb(g1000)), b=1,…,B. For a fixed α=0.05, the percentile (1−α) 100% of ORb on the B=100 permuted samples was c0.05=6.27 and the set S0.05={g | OR(g)>c0.05}={g | OR(g)>6.27} contained 100 genes, which were potential DE genes. Furthermore, between the 1000×100 permuted observations, 5000 of them had an OR value above the threshold c0.05=6.27. Next, we considered a 10-fold partition. For fold 1, the Fig. 3a shows the results of the two first principal components analysis on the matrix , with 99.3% of explained variability. As can be observed, a potential gene g that is not really differentially expressed was closely surrounded by cases from Rα,i, that is, by false positive permuted cases. On the contrary, a gene g that is really differentially expressed was not surrounded by cases from Rα,i. That holds for every fold and clearly shows the intuitive idea behind the proposed methodology.
Fig. 3.
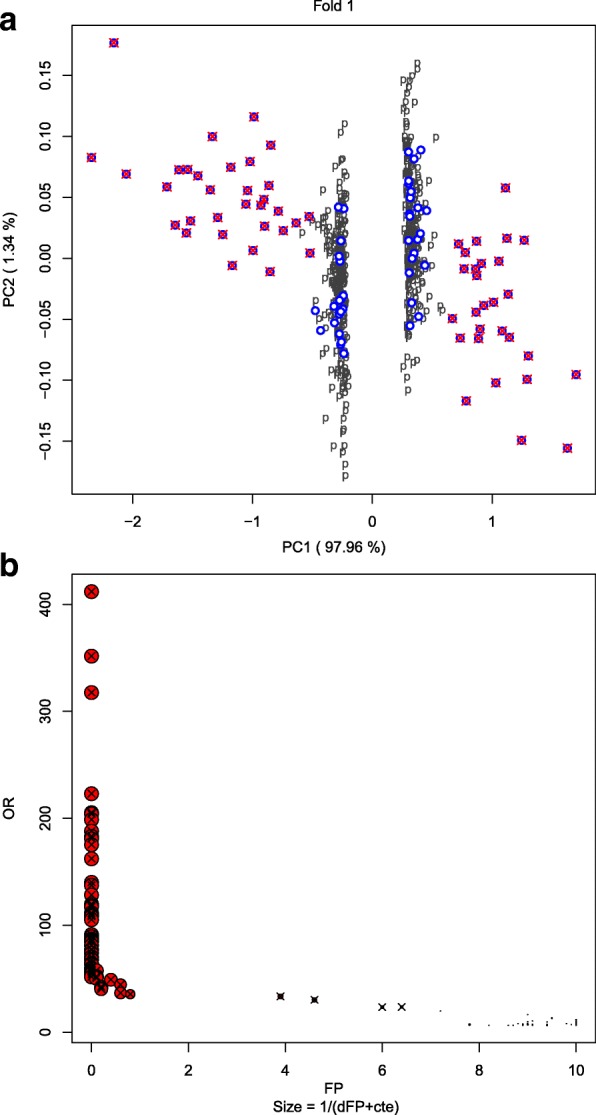
Illustrative example. a First two principal components of data corresponding to fold 1 (99.3% of explained variability), and there are represented: the potential genes (genes in S0.05) by circles; the false positives genes (genes in Rα,i) by “p"s, and the differentially expressed genes (genes generated as truly DE genes) by crosses. b Representation of the potential genes based on OR (vertical axis), FP (horizontal axis) and dFP (size of the circle is inversely proportional to its value). Truly DE genes are marked with a cross; in red and blue, genes belonging to cluster 1 and cluster 2, respectively
Following with the method, let us call (fi,fi0) the proportion of potential genes and permuted cases in set Ui (i=1,…,k). As we have observed in the small artificial example, if a potential gene is genuinely DE, its behavior should be different from those cases in Rα,i and the other way round; if a potential gene presents a similar behavior to those cases in Rα,i, then it should be considered as not truly DE. So, for each gene g in Sα, its 10-Nearest-Neighbourhood (NNi(g)) in Ui is considered. We calculate two indicators in this neighbourhood: the number of cases from Rα,i, FPi(g)=#{j∈NNi(g)|j∈Rα,i} (number of false positive permuted cases) and the density . The denominator will rarely be 0 since it would involve 10 tied nearest distances for g in NNi(g), but if it is equal to 0 then dFPi(g)=0 if FPi(g)=0 and dFPi(g)=NaN if FPi(g)>0.
Gathering the k folds, we obtain for each g∈Sα the average number of false positive permuted cases (FP) and the average density (dFP) of FP in the neighbourhood: and .
Thus, to discriminate DE genes among those in set Sα, we have to look for those g with low value of FP(g) and dFP(g) along with high values of OR(g).
Under this criterion, two types of differentially expressed gene selection can be made:
ORdensity strong selection: take as differentially expressed genes those with a large OR value and with FP and dFP equal to 0.
ORdensity relaxed selection: take as differentially expressed genes those with a large OR value and with small FP and dFP values. The average proportion of potential genes and the permuted cases among the k folds ( and respectively) are a good reference to look for small values of FP.
Furthermore, beyond the inspection of individual genes, we tackled the selection of DE genes by clustering the genes in Sα based on the input variables OR, FP and dFP. This can be useful as it offers different patterns of genes based on their importance.
Small artificial example (cont.): Using the Partition Around Medoids (PAM) clustering method [16] on variables OR,FP and dFP, and selecting two clusters as indicated in the silhouette analysis [17], the variables presented the following basic characteristics (Table 1) and the two clusters are represented in Fig. 3b. It is worth mentioning that the average distribution of potential genes and permuted cases in sets Ui was . It means that if the distribution were random, an average of 8.3 permuted cases would be expected in the 10-NN. Clearly, in cluster 1 the FP values are below this value. Finally, we checked the distribution of real DE genes across the two clusters, and the 60 genes in Cluster 1 are exactly the 60 real DE genes.
Table 1.
Illustrative example
| Cluster 1 (n1=60) | Cluster 2 (n2=40) | |||||
|---|---|---|---|---|---|---|
| OR | FP | dFP | OR | FP | dFP | |
| Min | 23.5 | 0.0 | 0.00 | 6.3 | 7.1 | 8.13 |
| Q 1 | 53.8 | 0.0 | 0.00 | 6.7 | 9.0 | 24.78 |
| Q 2 | 79.3 | 0.0 | 0.00 | 7.3 | 9.1 | 29.13 |
| Mean | 104.5 | 0.4 | 0.25 | 8.5 | 9.2 | 28.51 |
| Q 3 | 122.1 | 0.0 | 0.00 | 9.6 | 9.7 | 32.78 |
| Max | 412.0 | 6.7 | 5.22 | 19.8 | 10.0 | 43.21 |
Basic description for the OR,FP and dFP values in the two clusters obtained using PAM and silhouette procedures
Note 1: When the variability of genes among different types of samples is different, it is advisable to scale the differences between quantiles, for instance, , with RI(Xg) and RI(Yg) the interquartile ranges in samples X and Y, respectively.
Note 2: As the method considers the differences between quantiles, it may be interesting to give greater importance to some of them, such as the median. Simultaneously, robustness can be obtained avoiding the possible fluctuation of the estimations of the percentiles. Therefore, different weights to the quantiles can be introduced in the procedure.
Note 3: We have considered for each gene g in Sα its 10-Nearest-Neighbourhood. This is a parameter of the method that could be set in different ways, and it is considered to obtain better estimations of the proportion of potential genes and permuted cases in set Ui. In this sense, a small number of neighbors as 5 does not seem adequate since the percentage of false positive permuted cases that could be detected would be very unstable. On the other hand, note that as the method takes into account the mean value of the false positive density, the possible effect of the number of chosen neighbors is minimized.
Experimental setup
To evaluate the usefulness of the ORdensity procedure, we simulated multiple gene expression data sets using different parameter settings. Furthermore, we applied the procedure to four real cancer data sets. All computations were performed using the R language and Environment for Statistical Computing (R) 3.3.1 [18, 19] in combination with Bioconductor 3.3 [6]. As the proposed method may depend on the quantiles estimation of each gene expression for different conditions, different sample size situations including the case of small samples were considered in the simulated studies. Moreover, for both simulated and actual data, the method was compared with other well-recognized methods in order to assess whether it could compete with them.
Simulation study 1
We assumed a total of 1000 genes, among which 50, 100 or 200 were differentially expressed (DE) genes. On the one hand, the expression levels of all no DE genes were generated by and distributions in conditions X and Y, respectively. On the other hand, the DE genes were generated using the and distributions for conditions X and Y, respectively, with |μg|=Δg max{αg,βg}. Parameter Δg sets the importance of gene g, being gene g more important as long as Δg is bigger. Within this general setting, three different scenarios were considered:
Scenario 1: All genes had equal variability (σg = γg = 1,αg = βg=1), and all DE genes are equally important under this scenario, i.e., Δg=Δ, with Δ in {1.5,2,3}.
Scenario 2: All genes had not necessarily equal variability (σg≠γg,αg≠βg). These variabilities were randomly selected among {1,1.2,1.5,2}. All DE genes are equally important, i.e., Δg=Δ for all g, with Δ in {1.5,2,3}.
Scenario 3: All genes had not necessarily equal variability (σg≠γg,αg≠βg) neither the importance of DE genes is the same (Δg≠Δh, g≠h). Variability parameters were randomly selected in {1,1.2,1.5,2} as in the previous scenario and Δg values were randomly selected among {1.5,2,3}.
To better understand the performance of our approach, we simulated the data for the three scenarios assuming equal sample sizes for X and Y (nX=nY=30) and different sample sizes (nX=30, nY=10). For the most general situation, scenario 3, we also considered the case nX=nY=10 in order to evaluate the procedure for small sample sizes.
Using the area under the ROC curve, for the three scenairos and for nX=nY=30, nX=30, nY=10 and nX=nY=10, the ORdensity results were compared with those obtained using other well-known methods in this field, such as Significant Analysis of Microarrays [2] and Linear models with Empirical Bayes statistic (limma [4, 5]). Furthermore, for each situation 100 replicates were performed.
The Significant Analysis of Microarrays (SAM) method is a modification of the t-statistic and it was introduced to avoid the effect of the small per-gene variances that can make small fold-changes statistically significant. This modification adds a value which is calculated from the distribution of gene-specific standard errors. SAM was applied using the package samr for Bioconductor [6] in R language and Environment for Statistical Computing [18, 19].
The Empirical Bayes statistic (eBayes) is equivalent to shrinking the estimated sample variances towards a pooled estimate, resulting in far more stable inference when the number of arrays is small. The linear model with empirical Bayes statistic (called limma in the following) was applied using the limma package for Bioconductor [6] in R language and Environment for Statistical Computing [18, 19].
Simulation study 2
The simulation was set with blocks of DE genes [20] which are correlated within each block. The data was simulated from a multivariate normal distribution, with all genes having variance 1 and correlation 0.9 between genes within each block. It means that the variance-covariance matrix was a block-diagonal matrix such as:
where σblock=(σgh)gh with σgg=1 and σgh=0.9g≠h.
We considered 1, 2 or 3 blocks, and each block with 5, 20 or 100 DE genes. Following [20], the difference between mean values of conditions X and Y was set depending on the number of blocks:
One block: μA=−1.65, μB=1.65
Two blocks: μA=(−1.18,−1.18)′, μB=(1.18,1.18)′
Three blocks: μA=(−0.98,−0.98,−0.98)′, μB=(0.98,0.98,0.98)′
Once the DE genes were generated, 4000 variables representing no DE genes were added: 2000 following a N(0,1) distribution and 2000 following a distribution.
For the different number of blocks, we considered equal sample sizes for X and Y (nX=nY=30) and different sample sizes (nX=30, nY=10). For each situation 100 replicates were done and using the area under the ROC curve, the OR results were compared with those obtained by limma and SAM.
In all the above simulations, in order to obtain comparable results throughout the 100 runs, we always considered 3 clusters determined by PAM [16] clustering procedure. Obviously, to be absolutely accurate it would have been necessary to determine the number of clusters in each dataset.
Actual data sets: lymphoma, Golub, colon and prostate cancer
We considered four publicly available cancer data sets: a well-known lymphoma data set which is in the R package spls, and post-processed Golub, colon and prostate data sets that were downloaded from [21]. With these data sets, we compared the ORdensity results with SAM and limma.
The standard rule used for selecting a gene as DE with limma was to present an adjusted p-value smaller than 0.05. To compute the adjusted p-values for gene ranking we used a very stringent method (Bonferroni), and a more liberal procedure (BH, [9]). For SAM, the considered rule was to present a q-value [22] equal to 0.
For the ORdensity procedure, the Partition Around Medoids (PAM) [16] clustering and the silhouette analysis [17] were performed in order to establish the number of clusters and both the strong and relaxed selection were considered.
We evaluated the obtained results considering three different perspectives: the agreement between the three methods of the selected gene lists; the ability to maintain consistent lists with samples based on subsets of 80% of the original data selected at random, and the leave-one-out cross-validation correct classification rate for future classifications obtained when a Weighted Distance Based Discriminant analysis (WDB-discriminant) using Euclidean distance was performed [23]. Weighted Distance Based Discriminant (WDB-discriminant) is an improvement of the Distance Base rule [24, 25] which takes into account the statistical depth of the units. The WDB-discriminant was applied using the WeDiBaDis package available at https://github.com/ItziarI/WeDiBaDis.
Lymphoma cancer data set:
This data set [26] contains the gene expression of 1095 genes measured on 42 adults with large B-cell lymphoma (DLBCL), which can present two different molecular forms denoted by DLBCL1 and DLBCL2, respectively. Half of the samples presented the form DLBCL1 and the other half the form DLBCL2.
Golub data set:
The data set [27] contains the gene expression of 7129 genes. There are 72 samples with two different types of leukaemia, 47 acute lymphoblastic leukaemia (ALL) and 25 with acute myeloblastic leukaemia (AML).
Colon cancer data set:
This colon cancer data study [28] consists of 6000 genes measured on 62 patients, 40 of them diagnosed with colon cancer and 22 of them are healthy.
Prostate cancer data set:
This study [29] considered 12,626 genes and 102 samples, 50 of which were non-tumour prostate samples and 52 of which were prostate tumours.
In all the experiments, simulated or actual data sets, we considered the differences between the three quartiles, that is, Cp={0.25,0.5,0.75}. Moreover, we scaled these differences and weighted them by {1/4,1/2,1/4} respectively, making the most important difference the one between the medians (See Note 1 and 2, in the “Methods” section). In the case of the Golub data set, these differences were not scaled because for some of the genes the interquartile range was null.
Results
Next, we present the results obtained with the simulated data, as well as the actual cancer data sets.
With the simulated microarray data, we evaluated the behavior of the method in relation to the correct selection of DE genes since, in this case, we know which genes are actually differentially expressed. We evaluated the general behavior of the procedure in relation with the proportion of False Positive among the selected genes as DE and using the area under the ROC curve value, we compared the ORdensity results with those obtained using the alternative approaches, SAM and limma. Furthermore, we present a detail evaluation of both, the first and the second step of the method.
With the actual microarray data, we evaluated and compared our procedure with the alternative approaches SAM and limma, measuring the ability of the method in preserving predictive accuracy classifying the samples, and the stability of the lists of selected genes when a fraction of the samples (20%) was eliminated at random.
Simulation study 1
General behavior
As a general summary, the results indicated that the three variables that the method builds, OR, FP and dFP, are good discriminative variables, separating correctly the DE genes from the not DE genes. On the one hand, the number of DE genes not detected by the method was very small and always related with the less DE genes (see “Detail evaluation of step 1” subsection). On the other hand, with the strong selection no False Positives were detected. With the more relaxed selection and considering the partition in three clusters for all the runs, the results indicated that, in all situations, the method only considers as DE genes those from clusters 1 and 2. The False Positives were mostly in cluster 2 and the worst results were obtained with 50 simulated genes and for small sample sizes (equal to 10). However, for the majority of the situations the average number of False Positives was 0 in cluster 1 and very small in cluster 2. It is worth to mention that partition in 3 clusters is not necessarily the best partition that could be obtained in each of the 100 runs. As a consequence, the variability of the percentage of False Positives in cluster 2 was very high for some cases. That is, for some of the runs, cluster 2 was formed by DE genes but not for other runs, for which a partition with different number of clusters would have probably been more appropriate (see Detail evaluation of step 2 subsection).
Detail evaluation of step 1
Regarding this step, it is necessary to evaluate the number of simulated DE genes that the method did not consider as potential genes and therefore were missed. The results indicated that as the number of simulated DE genes increases, it is easy to not include at least one DE gene in the set of potential DE genes. Moreover, when the sample size for one or the two conditions was small (equal to 10), the sensibility to include among the potential all the simulated DE genes decreases. Nevertheless, the proportion of DE genes considered as potential DE genes was always very large, and DE genes not considered as potential genes were very few and always associated with Δ=1.5.
With more detail, on the simulated data sets with equal sample sizes for the two conditions (nX=nY=30), and in scenarios 1 and 2, when all the simulated DE genes were related to the same value of the parameter Δg (Δg=Δ for all DE genes), we can observe that as the number of simulated DE genes increases, it is easy to not include at least one DE gene in the set of potential DE genes (Table 2, Fig. 4 and Additional file 1: Table S13, Figure S8). However, it is clear that, in any case, the proportion of DE genes considered as potential DE genes is very large, being always higher than 95% and only in two cases presents a lower value (78.80% and 85.24%, respectively). Moreover, these worst results were associated with the lowest value of Δ, specifically Δ=1.5. In scenario 3, where the importance of DE genes is different and set by their values Δg selected at random in {1.5,2,3}, it is interesting to analyze what is the importance of the genes that are not considered as potential in set Sα. For instance, in the case of 50 DE genes, on average 99.32% of them are included in S0.01. Moreover, the DE genes not detected as potential had Δg=1.5, the lowest value. Thus, all the DE genes not considered as potential in S0.01 are between the less DE. Similar results are observed in the rest of the Table (Additional file 1: Table S14, and Figure S9).
Table 2.
Simulation study 1, scenario 1 with nX=nY=30 and 100 runs
| Δ=1.5 | ||||
| Nb. of | α | |||
| DE genes | 0.1 | 0.05 | 0.01 | |
| 50 | 0.08 | 0.20 | 0.80 | |
| % | 99.84 (0.54) | 99.56 (0.92) | 97.20 (2.2) | |
| 100 | 0.13 | 0.45 | 0.99 | |
| % | 99.87 (0.34) | 99.34 (0.66) | 95.20 (2.3) | |
| 200 | 0.80 | 0.99 | 1.00 | |
| % | 99.27 (0.56) | 96.70 (1.5) | 78.80 (4.0) | |
| Δ=2 | ||||
| Nb. of | α | |||
| DE genes | 0.1 | 0.05 | 0.01 | |
| 50 | 0.00 | 0.00 | 0.00 | |
| % | 100 | 100 | 100 | |
| 100 | 0.00 | 0.00 | 0.05 | |
| % | 100 | 100 | 99.95 (0.22) | |
| 200 | 0.01 | 0.03 | 0.95 | |
| % | 100 | 99.98 (0.09) | 98.50 (0.83) | |
| Δ=3 | ||||
| Nb. of | α | |||
| DE genes | 0.1 | 0.05 | 0.01 | |
| 50 | 0.00 | 0.00 | 0.00 | |
| % | 100 | 100 | 100 | |
| 100 | 0.00 | 0.00 | 0.00 | |
| % | 100 | 100 | 100 | |
| 200 | 0.00 | 0.00 | 0.00 | |
| % | 100 | 100 | 100 |
Evaluation of the first step of the ORdensity method using different values of α. The Table shows the estimated probability, , of no considering as potential DE gene at least one gene that it really is, and the mean proportion of DE genes (row named “%”) that the procedure considered as potential DE genes. Corresponding standard deviations are in brackets
Fig. 4.
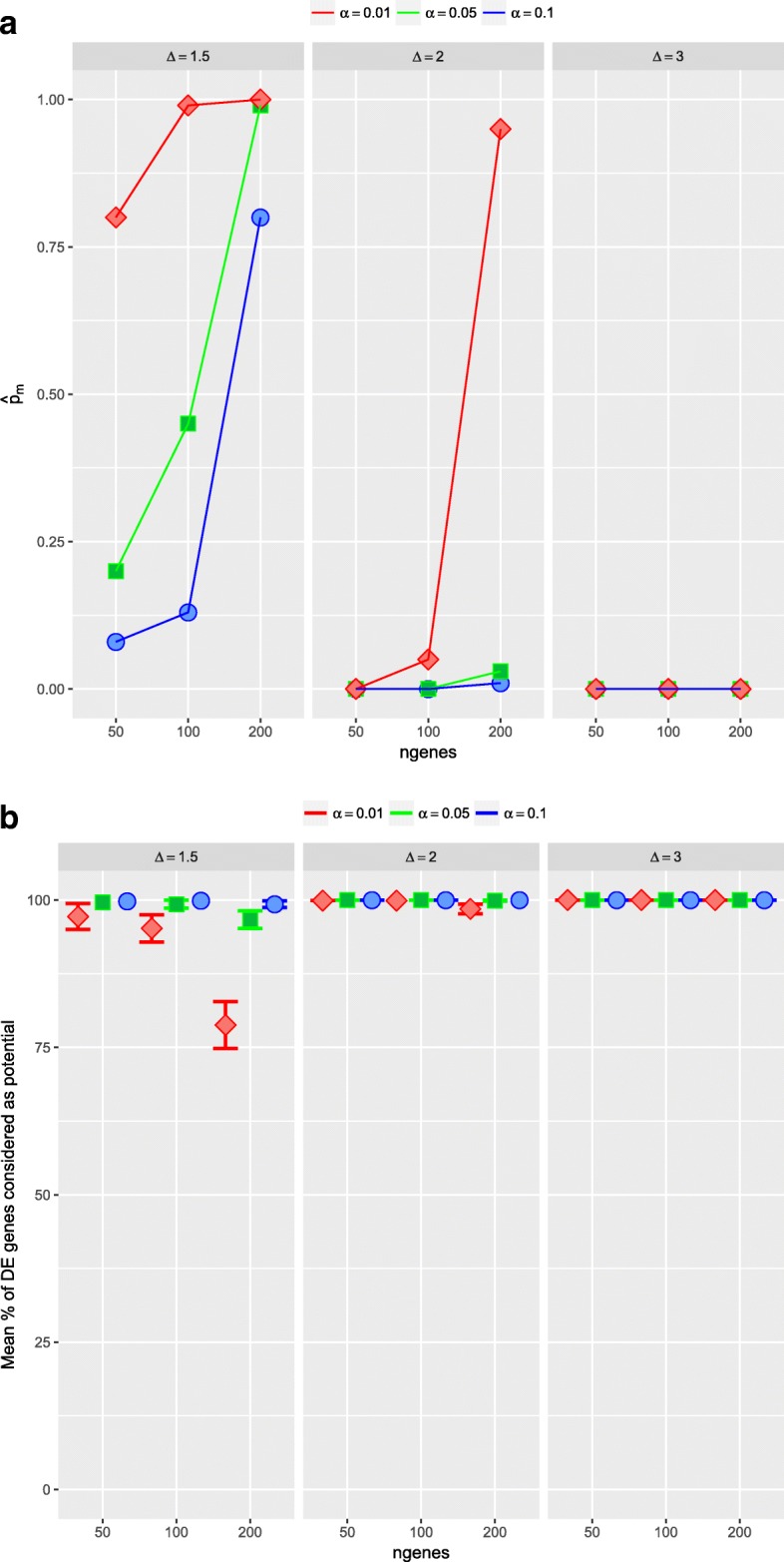
Simulation study 1, scenario 1 with nX=nY=30 and 100 runs. Evaluation of the first step of the ORdensity method using different values of α. Top: in x axis number of DE genes; in y axis estimated probability, , of no considering as potential DE gene at least one gene that it really is. Bottom: in x axis number of DE genes; in y axis mean proportion of DE genes that the procedure considered as potential DE genes
In the case where the sample sizes for the two conditions were different (nX=30,nY=10) and for the three scenarios, as we can observe, the results were very similar. As the sample size in one condition is smaller, the sensibility to include among the potential all the DE genes decreases, that is, the probability of not considering as potential DE gene at least one DE gene really increases (Table 3, Fig. 5 and Additional file 1: Tables S15 and S16, Figures S10 and S11).
Table 3.
Simulation study 1, scenario 1 with nX=30, nY=10 and 100 runs
| Δ=1.5 | ||||
| Nb. of | α | |||
| DE genes | 0.1 | 0.05 | 0.01 | |
| 50 | 0.85 | 1 | 1 | |
| % | 97.2 (2.8) | 91.1 (2.1) | 69.6 (6.7) | |
| 100 | 1 | 1 | 1 | |
| % | 93.7 (2.5) | 86.0 (3.5) | 60.1 (5.0) | |
| 200 | 1 | 1 | 1 | |
| % | 84.9 (2.6) | 70.5 (3.5) | 34.9 (3.4) | |
| Δ=2 | ||||
| Nb. of | α | |||
| DE genes | 0.1 | 0.05 | 0.01 | |
| 50 | 0.13 | 0.33 | 0.98 | |
| % | 99.7 (0.7) | 99.2 (1.2) | 93.2 (3.6) | |
| 100 | 0.38 | 0.88 | 1 | |
| % | 99.6 (0.6) | 98.4 (1.0) | 88.4 (3.2) | |
| 200 | 0.99 | 1 | 1 | |
| % | 97.8 (1.1) | 92.1 (1.9) | 65.3 (4.0) | |
| Δ=3 | ||||
| Nb. of | α | |||
| DE genes | 0.1 | 0.05 | 0.01 | |
| 50 | 0.00 | 0.00 | 0.02 | |
| % | 100 | 100 | 96.0 (0.3) | |
| 100 | 0.00 | 0.00 | 0.14 | |
| % | 100 | 100 | 99.8 (0.5) | |
| 200 | 0.01 | 0.11 | 0.97 | |
| % | 100.0 (0.1) | 99.9 (0.2) | 98.1 (1.1) |
Evaluation of the first step of the ORdensity method using different values of α. The Table shows the estimated probability, , of no considering as a potential DE gene at least one gene that it really is, and the mean proportion of DE genes (row named “%”) that the procedure considered as potential DE genes. Corresponding standard deviations are in brackets
Fig. 5.
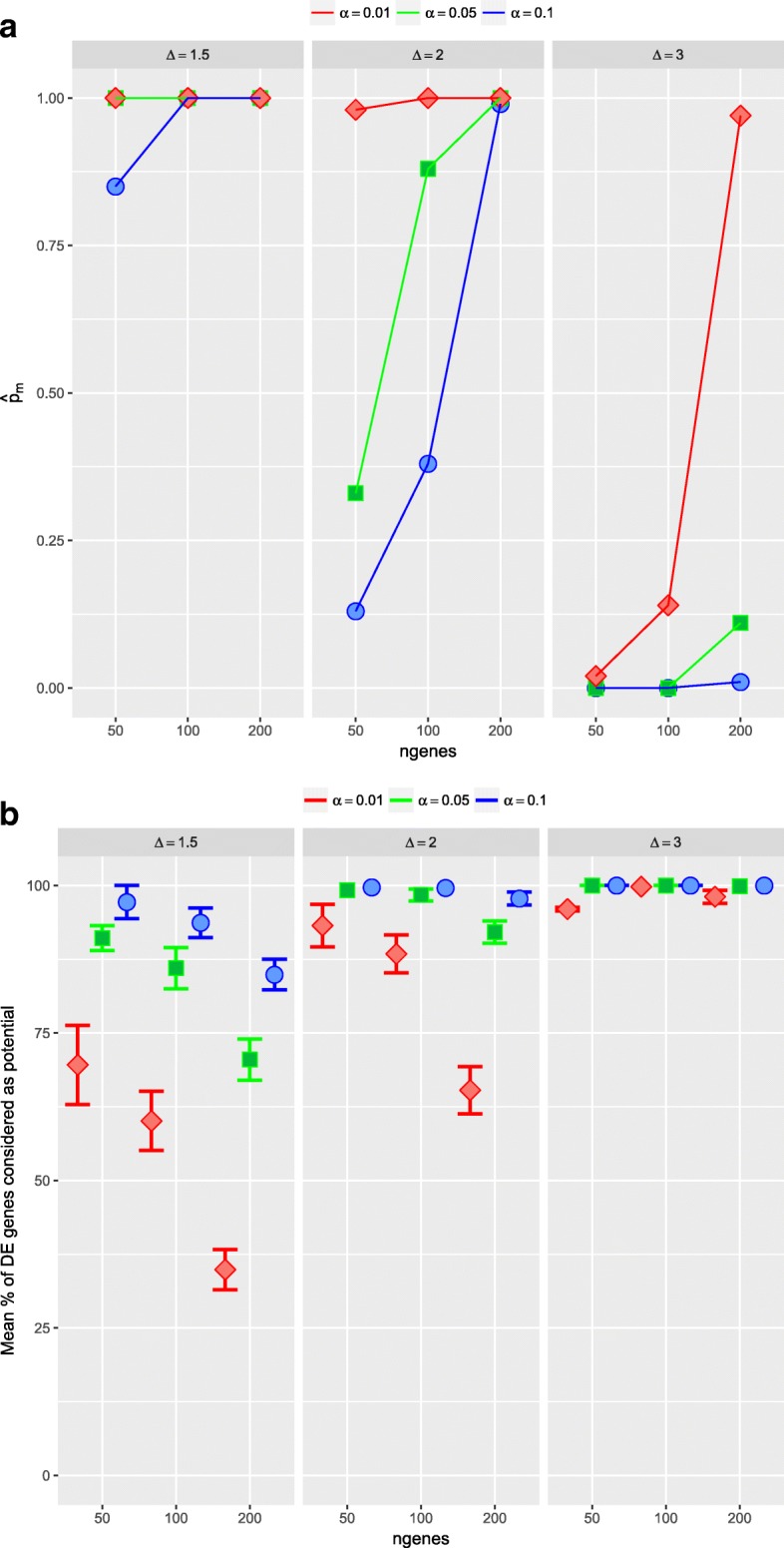
Simulation study 1, scenario 1 with nX=30, nY=10 and 100 runs. Evaluation of the first step of the ORdensity method using different values of α. Top: in x axis number of DE genes; in y axis estimated probability, , of no considering as potential DE gene at least one gene that it really is. Bottom: in x axis number of DE genes; in y axis mean proportion of DE genes that the procedure considered as potential DE genes
For scenario 3, when both sample sizes are small (nX=nY=10) similar results were obtained. Nevertheless, the mean proportion of DE genes that were included in the potential was notably lower. Again, the missed DE genes were mostly related to Δg = 1.5 (Additional file 1: Table S17 and Figure S12).
Detail evaluation of step 2
In the second step of the procedure, the interest lies in evaluating the number of False Positive genes selected by the method. As a general summary, with the strong selection no False Positive were selected. When the method used the more relaxed selection it retained as DE, in all cases, genes in clusters 1 and 2. The False Positive genes were mostly in cluster 2 and the worst results were obtained with 50 simulated genes and for small sample sizes (equal to 10). As mentioned before, partition in 3 clusters, throughout all runs, may lead to a high variability in cluster 2.
Focusing on the details, it can be seen that in scenario 1, nX=nY=30, Δ=1.5, and for any number of simulated DE genes, by chance an average of 8.5 False Positive permuted cases would be expected in the 10-NN. For 50 simulated DE genes, clearly, in clusters 1 and 2 the mean FP value is below 8.3. Then, the method considered genes in clusters 1 and 2 as DE, with on average less than one False Positive gene in cluster 1 (0.2% in cluster 1) and an average of 4.5 False Positive genes in cluster 2 (20.7% in cluster 2). When the number of simulated DE genes increases, a small number of False Positive genes were obtained. For 100 simulated DE genes, again the method considered those in clusters 1 and 2 as DE genes, with 0 False Positive in cluster 1 and on average less than one False Positive gene in cluster 2 (1.6%). Similar results were obtained for 200 simulated DE genes. For greater values of Δ, and for any number of simulated DE genes, the average number of False Positives per cluster is 0 or very small, being in the worst case equal to 1.6 (Table 4, Fig. 6). With equal sample sizes for the two conditions (nX=nY=30), in scenarios 2 and 3, again, small values of the average number of False Positives per cluster were obtained, being 0 or less than one in the majority of the situations. In the worst situation related to 50 simulated DE genes, the average number of False Positives per cluster was 1.47.
Table 4.
Simulation study 1, scenario 1 with nX=nY=30 and 100 runs
| Δ | DE | 10f0 | C i | |||||
|---|---|---|---|---|---|---|---|---|
| (sd) | (sd) | (sd) | (sd) | (sd) | ||||
| 30.7 | 45.7 | 0.5 | 0.4 | 0.2 | ||||
| (7.9) | (6.7) | (0.3) | (0.3) | (0.9) | ||||
| 50 | 8.5 | 21.8 | 19.4 | 5.4 | 7.5 | 20.7 | ||
| (5.9) | (5.0) | (2.1) | (4.4) | (20.9) | ||||
| C 3 | 32.9 | 8.4 | 9.2 | 29.0 | 93.7 | |||
| (6.3) | (0.6) | (0.2) | (3.0) | (5.8) | ||||
| 34.1 | 46.5 | 0.0 | 0.0 | 0.0 | ||||
| (11.3) | (6.3) | (0.1) | (0.1) | (0.2) | ||||
| 1.5 | 100 | 8.5 | 56.2 | 22.5 | 1.5 | 1.8 | 1.6 | |
| (10.3) | (2.7) | (0.67) | (9.0) | (2.0) | ||||
| C 3 | 32.1 | 9.1 | 8.7 | 21.5 | 68.9 | |||
| (5.6) | (0.5) | (0.35) | (2.0) | (8.9) | ||||
| 59.3 | 29.5 | 0.0 | 0.0 | 0.0 | ||||
| (12.5) | (2.8) | (0.0) | (0.0) | (0) | ||||
| 200 | 8.5 | 113.0 | 14.8 | 0.5 | 0.8 | 0.4 | ||
| (13.0) | (1.0) | (0.2) | (0.2) | (0.6) | ||||
| C 3 | 25.3 | 8.0 | 6.0 | 9.9 | 14.9 | |||
| (6.5) | (0.5) | (0.9) | (1.8) | (8.2) | ||||
| 22.3 | 86.3 | 0.0 | 0.0 | 0.0 | ||||
| (9.2) | (17.1) | (0.1) | (0.0) | (0) | ||||
| 50 | 8.0 | 27.8 | 42.0 | 1.4 | 1.5 | 5.8 | ||
| (6.9) | (9.2) | (2.0) | (3.3) | (18.0) | ||||
| C 3 | 36.0 | 9.1 | 9.2 | 27.8 | 97.0 | |||
| (5.3) | (0.61) | (0.19) | (2.0) | (2.7) | ||||
| 35.5 | 74.7 | 0.0 | 0.0 | 0.0 | ||||
| (7.9) | (8.1) | (0.0) | (0.0) | (0) | ||||
| 2 | 100 | 8.0 | 63.1 | 37.3 | 0.4 | 0.4 | 0.3 | |
| (7.9) | (3.1) | (0.2) | (0.2) | (0.7) | ||||
| C 3 | 26.0 | 8.8 | 9.3 | 24.9 | 93.7 | |||
| (4.7) | (0.6) | (0.3) | (1.9) | (4.5) | ||||
| 69.2 | 46.6 | 0.0 | 0.0 | 0.0 | ||||
| (14.1) | (3.9) | (0.0) | (0.0) | (0) | ||||
| 200 | 8.0 | 122.9 | 23.8 | 0.2 | 0.2 | 0.0 | ||
| (17.0) | (2.4) | (0.1) | (0.1) | (0.3) | ||||
| C 3 | 13.1 | 8.9 | 8.1 | 13.0 | 58.0 | |||
| (17.3) | (2.4) | (2.0) | (4.1) | (23.1) | ||||
| 18.9 | 191.2 | 0.0 | 0.0 | 0.0 | ||||
| (6.6) | (22.7) | (0.0) | (0.0) | (0) | ||||
| 50 | 7.2 | 31.5 | 99.9 | 0.29 | 0.4 | 2.1 | ||
| (4.9) | (16.1) | (1.2) | (2.6) | (14.1) | ||||
| C 3 | 37.0 | 9.0 | 9.2 | 27.7 | 99.8 | |||
| (5.1) | (0.6) | (0.19) | (1.5) | (0.8) | ||||
| 38.6 | 155.7 | 0.0 | 0.0 | 0.0 | ||||
| (10.0) | (15.1) | (0.0) | (0.0) | (0) | ||||
| 3 | 100 | 7.1 | 61.3 | 83.4 | 0.0 | 0.0 | 0.0 | |
| (10.0) | (6.6) | (0.1) | (0.0) | (0) | ||||
| C 3 | 25.1 | 8.7 | 9.3 | 25.8 | 99.6 | |||
| (4.5) | (0.49) | (0.26) | (1.7) | (1.1) | ||||
| 74.1 | 95.8 | 0 | 0 | 0.0 | ||||
| (16.4) | (8.2) | (0) | (0) | (0) | ||||
| 200 | 7.1 | 115.5 | 53.1 | 0.0 | 0.0 | 0.0 | ||
| (21.3) | (6.6) | (0.0) | (0.0) | (0) | ||||
| C 3 | 16.1 | 12.5 | 8.3 | 16.4 | 86.0 | |||
| (24.8) | (11.9) | (3.2) | (6.8) | (33.6) |
Evaluation of the second step of the ORdensity method with α=0.05. In the first two columns, delta (Δ) values and number of total simulated DE genes. In column 3, the 10×f0 values where f0 is the average proportion of permuted cases in sets Ui. In column 4, the “*” indicates the clusters considered by the procedure. Columns 5–8 contain for each cluster: the mean number of genes (), the mean of OR values (), the mean of FP values (), the mean of dFP values (). In the last column the mean of False Positives genes per cluster in % (). Corresponding standard deviations are in brackets
Fig. 6.
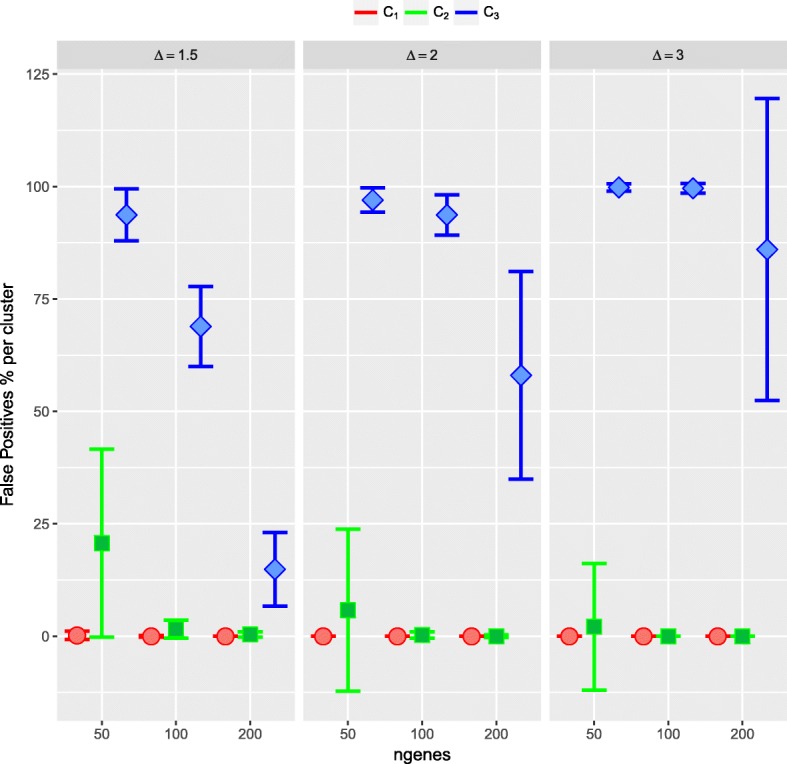
Simulation study 1, scenario 1 with nX=nY=30 and 100 runs. Evaluation of the second step of the ORdensity method with α=0.05. In x axis number of DE genes; in y axis the mean of False Positives genes per cluster in % (). In red cluster 1 (C1), in green cluster 2 (C2) and in blue cluster 3 (C3)
With different samples sizes (nX=30,nY=10) and even small sample sizes (nX=nY=10), the average number of False Positive genes was again very small. The worst cases were obtained when only 50 simulated DE genes and Δ=1.5 were considered. In the majority of the other situations, the average number of False Positive genes was 0 or less than one (Table 4, Fig. 6 and in Additional file 1: Tables S18–S23, Figures S13–S18).
Moreover, for scenario 3, where DE genes were associated with different values of Δg, observing the distribution of DE genes within each cluster in relation with their corresponding Δg values, we obtained that the most important DE genes, which are related to Δg=3, are mainly in cluster 1, and that the DE genes included in cluster 3 are the less important since they are related most principally to Δg=1.5 and never with Δg=3. It is worth noting that even for a small number of samples (nX=nY=10) similar results were obtained (Additional file 1: Tables S19, S22 and S23 last column and Figures S14, S17 and S18).
Simulation study 2
General behavior
As in the above simulation study, the method identified correctly the DE genes. With the strong selection, again, no False Positive genes were obtained. With the more relaxed selection, the worst results were obtained with only 5 simulated DE genes. In the majority of situations the method only considers as DE genes those in cluster 1, with 0 or a very small number of False Positive genes.
Evaluation of the area under the ROC curve
The mean AUC values were very large and similar to those obtained by SAM and limma even when the sample sizes are small (Table 5, and Additional file 1: Tables S24 and S25).
Table 5.
AUC mean values for Simulation study 1, scenario 1 and 100 runs. In first column: n indicates the number of DE gens; Δ the Δ values; A the ORdensity method; B the limma method and C the SAM method
| nX=nY=30 | ||||||
| n | 50 | 100 | ||||
| Δ | 1.5 | 2 | 3 | 1.5 | 2 | 3 |
| A | 0.998 | 0.998 | 0.997 | 0.998 | 0.997 | 0.995 |
| (0.002) | (0.001) | (0.002) | (0.001) | (0.002) | (0.003) | |
| B | 0.995 | 0.993 | 0.993 | 0.997 | 0.993 | 0.993 |
| (0.002) | (0.000) | (0.000) | (0.003) | (0.000) | (0.000) | |
| C | 0.994 | 0.993 | 0.993 | 0.996 | 0.993 | 0.993 |
| (0.003) | (0.000) | (0.000) | (0.002) | (0.000) | (0.000) | |
| nX=nY=30 | nX=30, nY=10 | |||||
| n | 200 | 50 | ||||
| Δ | 1.5 | 2 | 3 | 1.5 | 2 | 3 |
| A | 0.996 | 0.997 | 0.993 | 0.974 | 0.996 | 0.998 |
| (0.000) | (0.000) | (0.000) | (0.001) | (0.001) | (0.001) | |
| B | 0.997 | 0.992 | 0.992 | 0.967 | 0.996 | 0.993 |
| (0.003) | (0.001) | (0.000) | (0.011) | (0.003) | (0.001) | |
| C | 0.998 | 0.992 | 0.992 | 0.940 | 0.994 | 0.993 |
| (0.002) | (0.001) | (0.000) | (0.016) | (0.003) | (0.001) | |
| nX=30, nY=10 | ||||||
| n | 100 | 200 | ||||
| Δ | 1.5 | 2 | 3 | 1.5 | 2 | 3 |
| A | 0.973 | 0.994 | 0.998 | 0.959 | 0.992 | 0.997 |
| (0.001) | (0.002) | (0.003) | (0.000) | (0.000) | (0.000) | |
| B | 0.981 | 0.997 | 0.993 | 0.993 | 0.998 | 0.992 |
| (0.006) | (0.003) | (0.000) | (0.002) | (0.002) | (0.000) | |
| C | 0.946 | 0.995 | 0.993 | 0.960 | 0.996 | 0.992 |
| (0.010) | (0.003) | (0.000) | (0.007) | (0.002) | (0.000) | |
| nX=nY=10 | ||||||
| n | 50 | 100 | ||||
| Δ | 1.5 | 2 | 3 | 1.5 | 2 | 3 |
| A | 0.925 | 0.974 | 0.998 | 0.921 | 0.971 | 0.997 |
| (0.014) | (0.011) | (0.011) | (0.010) | (0.001) | (0.008) | |
| B | 0.894 | 0.980 | 0.994 | 0.931 | 0.989 | 0.994 |
| (0.023) | (0.009) | (0.001) | (0.013) | (0.004) | (0.002) | |
| C | 0.859 | 0.959 | 0.994 | 0.865 | 0.960 | 0.994 |
| (0.023) | (0.015) | (0.001) | (0.016) | (0.009) | (0.002) | |
| nX=nY=10 | ||||||
| n | 200 | |||||
| Δ | 1.5 | 2 | 3 | |||
| A | 0.900 | 0.956 | 0.996 | |||
| (0.002) | (0.001) | (0.001) | ||||
| B | 0.968 | 0.996 | 0.993 | |||
| (0.007) | (0.001) | (0.003) | ||||
| C | 0.876 | 0.974 | 0.995 | |||
| (0.017) | (0.007) | (0.003) | ||||
Detail evaluation of step 1
Concerning the results of the first step, we can observe that as a general behaviour, the method conrectly included the DE genes in the set of potential genes. With an equal number of samples (nX=nY=30), the method included all the DE genes in the set of potential genes, being 99.8% the worst result in the case of 3 blocks (Table 6). When the number of samples in one condition is small (nX=30, nY=10) and there is only one block of correlated genes, the same behaviour can be observed. With 2 or 3 blocks of correlated genes some of the DE can be missed, however, the proportion of DE genes that are actually in Sα is very large, being 87.5% in the worst case (Additional file 1: Table 26, Figure 19).
Table 6.
Simulation study 2 with nX=nY=30 and 100 runs
| Number of | 1 block | 2 blocks | |||||
| DE genes | α | α | |||||
| per block | 0.1 | 0.05 | 0.01 | 0.1 | 0.05 | 0.01 | |
| 5 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) | |
| 20 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) | |
| 100 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) | |
| Number of | 3 blocks | ||||||
| DE genes | α | ||||||
| per block | 0.1 | 0.05 | 0.01 | ||||
| 5 | 0 | 0 | 0 | ||||
| % | 100 (0) | 100 (0) | 100 (0) | ||||
| 20 | 0 | 0 | 0.01 | ||||
| % | 100 | 100 | 99.9 (0.7) | ||||
| 100 | 0 | 0.01 | 0.04 | ||||
| % | 100 (0) | 100.0 (0.0) | 99.8 (1.9) | ||||
Evaluation of the first step of the ORdensity method using different values of α. The Table shows the estimated probability, , of no considering as a potential DE gene at least one gene that it really is, and the mean proportion of DE genes (row named “%”) that the procedure considered as potential DE genes. Corresponding standard deviations are in brackets
Detail evaluation of step 2
As a general comment, one can see a strong correlation/correspondence between being a cluster with high OR and low false positiveness in the neighbourhood (FP, dFP), along with a small number of False Positive genes in the cluster. This holds for a different number of blocks and a different number of genes in the blocks. With nX=nY=30 and one block (Table 7, Fig. 7), the worst results were obtained for 5 simulated DE genes. In this case, the method only considered those in cluster 1 as DE genes, but the size of cluster 1 varied between 5 and 58 genes. This high variability in the number of genes in cluster 1 is probably due to always considering 3 clusters. For 20 or 100 simulated DE genes, again the procedure only considers genes in cluster 1 and not one False Positive gene was detected. For two blocks, with 5 and 20, again only cluster 1 was considered and no False Positives were obtained. With 100 simulated DE genes in each block, clusters 1 and 2 were considered by the method with 0 and 15.9 False Positives genes, respectively. With three blocks, independently of the number of simulated DE genes, no False Positives were found. Similar results were obtained for nX=30,nY=10 (Additional file 1: Table 27, Figure 20).
Table 7.
Simulation study 2, with nX=nY=30 and 100 runs
| B | DE | 10f0 | C i | |||||
|---|---|---|---|---|---|---|---|---|
| (sd) | (sd) | (sd) | (sd) | (sd) | ||||
| 27.9 | 160.4 | 6.6 | 8.1 | 32.0 | ||||
| (31.2) | (102.2) | (1.1) | (10.8) | (43.9) | ||||
| 5 | 9.1 | C 2 | 92.1 | 10.1 | 9.0 | 24.1 | 100 | |
| (25.5) | (0.8) | (0.4) | (4.0) | (0) | ||||
| C 3 | 82.5 | 7.8 | 9.4 | 41.6 | 100 | |||
| (17.9) | (0.3) | (0.3) | (2.2) | (0) | ||||
| 20.0 | 233.0 | 0.0 | 0.0 | 0.0 | ||||
| (0.0) | (35.1) | (0.0) | (0.0) | (0) | ||||
| 1 | 20 | 9.1 | C 2 | 96.5 | 10.4 | 9.0 | 22.0 | 100 |
| (12.7) | (0.5) | (0.1) | (1.5) | (0) | ||||
| C 3 | 90.1 | 7.7 | 9.2 | 41.1 | 100 | |||
| (14.2) | (0.2) | (0.1) | (1.4) | (0) | ||||
| 100.0 | 209.5 | 0.0 | 0.0 | 0.0 | ||||
| (0.0) | (32.9) | (0.0) | (0.0) | (0) | ||||
| 100 | 8.9 | C 2 | 73.7 | 10.8 | 9.1 | 20.7 | 100 | |
| (10.9) | (0.59) | (0.2) | (1.5) | (0) | ||||
| C 3 | 69.1 | 8.2 | 9.2 | 37.5 | 100 | |||
| (11.2) | (0.25) | (0.2) | (1.6) | (0) | ||||
| 9.9 | 119.7 | 1.0 | 0.2 | 0.0 | ||||
| (0.6) | (16.9) | (0.2) | (0.0) | (0.0) | ||||
| 10 | 9.1 | C 2 | 102.2 | 10.5 | 9.0 | 22.1 | 99.9 | |
| (14.2) | (0.7) | (0.1) | (1.5) | (0.7) | ||||
| C 3 | 94.2 | 7.7 | 9.2 | 41.7 | 100 | |||
| (13.2) | (0.2) | (0.1) | (1.4) | (0.0) | ||||
| 40.0 | 118.6 | 0.0 | 0.0 | 0.0 | ||||
| (0.1) | (20.0) | (0.0) | (0.0) | (0.0) | ||||
| 2 | 40 | 9.1 | C 2 | 97.0 | 10.3 | 9.1 | 22.2 | 100 |
| (12.0) | (0.5) | (0.1) | (0.4) | (0.0) | ||||
| C 3 | 87.4 | 7.7 | 9.2 | 41.2 | 100 | |||
| (12.9) | (0.2) | (0.1) | (1.2) | (0.0) | ||||
| 115.9 | 120.1 | 0.0 | 0.0 | 0.0 | ||||
| (38.9) | (18.9) | (0.0) | (0.0) | (0.0) | ||||
| 200 | 8.6 | 95.3 | 72.7 | 1.6 | 3.7 | 16.7 | ||
| (15.5) | (31.3) | (3.5) | (8.2) | (37.8) | ||||
| C 3 | 118.5 | 8.9 | 9.3 | 31.2 | 100 | |||
| (28.3) | (0.5) | (0.1) | (3.2) | (0.0) | ||||
| 14.6 | 86.2 | 0.26 | 0.09 | 0.0 | ||||
| (1.14) | (14.1) | (0.31) | (0.11) | (0) | ||||
| 15 | 9.0 | C 2 | 103.9 | 9.0 | 8.4 | 22.1 | 99.6 | |
| (11.8) | (0.13) | (0.48) | (1.5) | (1.1) | ||||
| C 3 | 92.8 | 7.2 | 9.2 | 41.6 | 100 | |||
| (10.7) | (0.77) | (0.13) | (1.4) | (0.0) | ||||
| 59.9 | 81.2 | 0.03 | 0.03 | 0.0 | ||||
| (0.29) | (12.1) | (0.07) | (0.06) | (0) | ||||
| 3 | 60 | 8.9 | C 2 | 93.8 | 10.3 | 9.1 | 22.3 | 99.9 |
| (12.3) | (0.43) | (0.13) | (1.4) | (0.3) | ||||
| C 3 | 87.2 | 7.7 | 9.2 | 41.3 | 100 | |||
| (11.4) | (0.16) | (0.13) | (1.3) | (0.0) | ||||
| 142.4 | 81.0 | 0.00 | 0.00 | 0.0 | ||||
| (31.0) | (13.7) | (0.00) | (0.00) | (0) | ||||
| 300 | 8.4 | 157.5 | 54.6 | 0.03 | 0.04 | 0.0 | ||
| (31.0) | (11.1) | (0.06) | (0.09) | (0) | ||||
| C 3 | 119.2 | 8.9 | 9.4 | 30.3 | 99.9 | |||
| (11.4) | (0.28) | (0.10) | (0.96) | (0.02) |
Evaluation of the second step of the ORdensity method with α=0.05. In the first column number of blocks. In column 2, the number of total simulated DE genes. In column 3, the 10×f0 values where f0 is the average proportion of permuted cases in sets Ui. In column 4, the “*” indicates the clusters considered by the procedure. Columns 5–8 contain for each cluster: the mean number of genes (), the mean of OR values (), the mean of FP values (), the mean of dFP values (). In the last column the mean of False Positives genes per cluster in % (). Corresponding standard deviations are in brackets
Fig. 7.
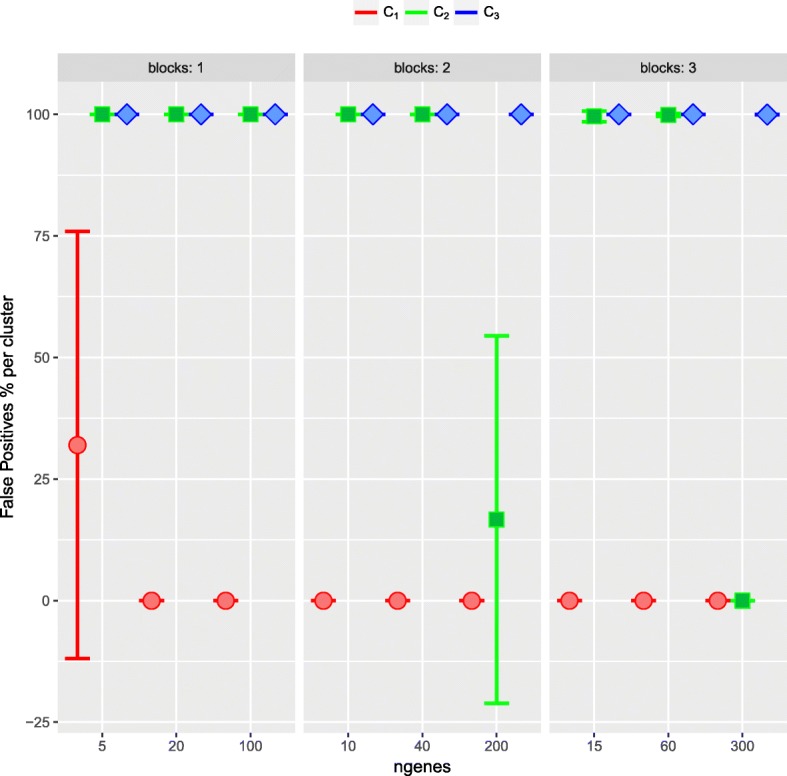
Simulation study 2 with nX=nY=30 and 100 runs. Evaluation of the second step of the ORdensity method with α=0.05. In x axis number of DE genes; in y axis the mean of False Positives genes per cluster in % (). In red cluster 1 (C1), in green cluster 2 (C2) and in blue cluster 3 (C3)
Evaluation of the area under the ROC curve
For nX=nY=30, the mean AUC values were very large and somewhat better than those obtained with limma and SAM, especially for one and two blocks (Table 8). Similar results were found for nX=30, nY=10.
Table 8.
AUC mean values for Simulation study 2, with nX=nY=30 and 100 runs
| nX=nY=30 | ||||||
| 1 block | 2 blocks | |||||
| Nb. DE genes | 5 | 20 | 100 | 5 | 20 | 50 |
| ORdensity | 0.995 | 0.995 | 0.980 | 0.993 | 0.998 | 0.985 |
| (0.000) | (0.000) | (0.000) | (0.000) | (0.000) | (0.000) | |
| limma | 0.701 | 0.854 | 0.903 | 0.796 | 0.789 | 0.917 |
| (0.0004) | (0.001) | (0.001) | (0.000) | (0.001) | (0.045) | |
| SAM | 0.701 | 0.854 | 0.902 | 0.796 | 0.789 | 0.918 |
| (0.001) | (0.001) | (0.00002) | (0.000) | (0.0003) | (0.045) | |
| 3 block | ||||||
| Nb. DE genes | 5 | 20 | 50 | |||
| ORdensity | 0.983 | 0.997 | 0.990 | |||
| (0.002) | (0.000) | (0.000) | ||||
| limma | 0.982 | 0.997 | 0.990 | |||
| (0.001) | (0.003) | (0.069) | ||||
| SAM | 0.825 | 0.994 | 0.876 | |||
| (0.000) | (0.003) | (0.070) | ||||
In summary, the results with simulated data showed that ORdensity correctly detects the DE genes and is competitive with other well-known methods.
Actual data: lymphoma, Golub, colon and prostate cancer data sets
For the three procedures, the number of genes selected under the standard criterion varies, being much larger for SAM and limma.
For the lymphoma data set (1095 genes), the ORdensity procedure selected 96 potential DE genes (#S0.05=96), distributed in two clusters with 19 and 77 genes, respectively. However, the strong selection could not be used since no gene had FP and dFP values equal to 0. The limma method produced a list with 24 and 88 genes using Bonferroni and BH procedures, respectively. The SAM procedure produced a list containing 64 genes. When the Golub data set (with 7129 genes) was considered, the procedure selected 556 potential DE genes, distributed in two groups with 291 and 265 genes, respectively. Among the genes in cluster 1, only 4 had FP and dFP values equal to zero. Thus, with the ORdensity, the most restricted criterion gave a list with only 4 genes and the relaxed criterion gave a list with 291 genes. The limma method produced a list with 193 and 938 genes using, respectively, Bonferroni and BH methods, and the SAM procedure produced a list with 403 genes.
For the colon data set (6000 genes), the ORdensity procedure selected 186 genes as DE potential genes (#S0.05=186), distributed in three clusters with 59, 88 and 39 genes, respectively. Among the 59 genes of cluster 1, twelve of them had no false positive permuted cases in their neighbourhood, with FP and dFP values equal to zero. Thus, with the ORdensity, the most restricted criterion gave a list with only 12 genes and the relaxed criterion gave a list with 59 genes. The limma method produced a list with 49 and 366 genes using Bonferroni and BH, respectively, and the SAM procedure produced a list containing 166 genes.
In the case of the prostate data set (12626 genes), 1531 potential DE genes (#S0.05=1531) were detected belonging to two clusters with 990 and 541 genes, respectively. Out of the 990 genes in cluster 1, 131 had no false positive permuted cases in their neighbourhood, with FP and dFP values equal to zero. Different number of selected genes were considered: the 131 produced by the more restricted ORdensity; the 990 for the relaxed ORdensity; the 1531 total candidate genes; the 263 and 2684 selected by limma rule under Bonferroni and BH, respectively, and the 3322 genes selected under SAM procedure.
The results obtained with these actual data sets are shown in Tables 9, 10, 11 and 12, respectively. The leave-one-out cross-validation correct classification rate (rows I in Tables 9, 10, 11 and 12) indicates that ORdensity does not lead to overfitting, and can achieve the objectives of reducing the set of selected genes and reaching high leave-one-out cross-validation correct classification accuracy rates. With the lymphoma data set, the ORdensity, with the 19 genes selected, reached 100% of leave-one-out cross-validation correct classification rate and this result was matched by SAM and limma using 19 or 24 genes, respectively. With the Golub data set and using only 4 genes, a correct classification rate of 90.28% was reached and with the 291 genes selected by the relaxed selection the maximum value of 97.22% was obtained. With limma or SAM, using the first 4 selected genes, the classification rate was improved (97.22 versus 90.28), but using limma or SAM there was not any objective criterion to select 4 genes. A similar situation happened with the colon and prostate data sets.
Table 9.
Lymphoma cancer data set
| Ns | ||||||
|---|---|---|---|---|---|---|
| 19 | 24 | 64 | 88 | 96 | ||
| I | ORdensity | 100 | 100 | 100 | 100 | 100 |
| limma | 95.24 | 100 | 100 | 100 | - | |
| SAM | 100 | 100 | 100 | - | - | |
| II | A vs B | 13 | 17 | 14 | 17 | - |
| A vs C | 14 | 16 | 42 | 63 | - | |
| B vs C | 17 | 23 | 58 | - | - | |
| III | ORdensity | 10.2 | 13.2 | 40.4 | 61.4 | 65.3 |
| (1.48) | (2.84) | (2.01) | (2.27) | (3.13) | ||
| limma | 7.9 | 9.7 | 27.4 | 36.7 | - | |
| (0.87) | (1.57) | (2.17) | (1.34) | - | ||
| SAM | 14.2 | 17.3 | 51.4 | - | - | |
| (2.82) | (3.97) | (8.85) | - | - | ||
Results for different number (Ns) of selected genes: 19 with ORdensity relaxed selection; 24 with limma and Bonferroni; 64 with SAM; 88 with limma and BH, and 96 total potential DE genes. Rows I present the leave-one-out cross-validation correct classification rate. In bold, the results for the genes selected under standard criteria for ORdensity, limma and SAM procedures; in rows II, number of common selected genes between the ORdensity, limma and SAM approaches; in rows III, mean and standard deviation (in brackets) of the number of genes that for 10 subsamples were always kept selected
Table 10.
Golub cancer data set
| Ns | ||||
|---|---|---|---|---|
| 4 | 193 | 291 | ||
| I | ORdensity (A) | 90.28 | 97.22 | 97.22 |
| limma (B) | 97.22 | 97.22 | 97.22 | |
| SAM (C) | 97.22 | 97.22 | 97.22 | |
| II | A vs B | 1 | 127 | 206 |
| A vs C | 1 | 130 | 213 | |
| B vs C | 4 | 175 | 265 | |
| III | ORdensity | 0.5 (0.71) | 150.4 (3.89) | 241.8 (6.94) |
| limma | 0 | 12.3 (2.26) | 4.3 (1.83) | |
| SAM | 3.3 (0.48) | 151.8 (24.18) | 220.7 (35.88) | |
| Ns | ||||
| 403 | 556 | 938 | ||
| I | ORdensity (A) | 97.22 | 97.22 | - |
| limma (B) | 97.22 | 97.22 | 97.22 | |
| SAM (C) | 97.22 | - | - | |
| II | A vs B | 274 | 358 | - |
| A vs C | 280 | - | - | |
| B vs C | 368 | - | - | |
| III | ORdensity | 334.1 (6.03) | 473.3 (9.56) | - |
| limma | 029.2 (2.44) | 55.5 (3.10) | 149.3 (6.36) | |
| SAM | 316.3 (50.06) | - | - | |
Results for different number (Ns) of selected genes: 4 with ORdensity strong selection; 193 with limma and Bonferroni; 291 with ORdensity relaxed selection; 403 with SAM; 556 total potential DE genes and 938 with limma and BH. Rows I present the leave-one-out cross-validation correct classification rate. In bold, the results for the genes selected under the standard criteria for ORdensity, limma and SAM procedures; in rows II, number of common selected genes between the ORdensity, limma and SAM approaches; in rows III, mean and standard deviation (in brackets) of the number of genes that for 10 subsamples were always kept selected
Table 11.
Colon cancer data set
| Ns | ||||
|---|---|---|---|---|
| 12 | 49 | 59 | ||
| I | ORdensity (A) | 88.71 | 90.32 | 90.32 |
| limma (B) | 91.94 | 88.71 | 88.71 | |
| SAM (C) | 85.48 | 88.71 | 88.71 | |
| II | A vs B | 7 | 32 | 38 |
| A vs C | 0 | 14 | 22 | |
| B vs C | 2 | 26 | 29 | |
| III | ORdensity | 7.5 (1.27) | 35.4 (4.09) | 43.0 (3.56) |
| limma | 0.1 (0.32) | 3.5 (0.85) | 4.1 (0.57) | |
| SAM | 1.8 (1.14) | 23.7 (2.98) | 29.4 (3.57) | |
| Ns | ||||
| 166 | 186 | 366 | ||
| I | ORdensity (A) | 87.10 | 87.10 | - |
| limma (B) | 87.10 | 87.10 | 87.10 | |
| SAM (C) | 87.10 | - | - | |
| II | A vs B | 119 | 134 | - |
| A vs C | 118 | - | - | |
| B vs C | 155 | - | - | |
| III | ORdensity | 127.2 (4.49) | 142.5 (4.84) | - |
| limma | 13.1 (1.37) | 16.7 (2.21) | 72.1 (4.72) | |
| SAM | 136.7 (5.23) | - | - | |
Results for different number (Ns) of selected genes: 12 with ORdensity strong selection; 49 with limma and Bonferroni; 59 with ORdensity relaxed selection; 166 with SAM; 186 total potential DE genes and 366 with limma and BH. Rows I present the leave-one-out cross-validation correct classification rate. In bold, the results for the genes selected under standard criteria for ORdensity, limma and SAM procedures; in rows II, number of common selected genes between the ORdensity, limma and SAM approaches. In rows III, mean and standard deviation (in brackets) of the number of genes that for 10 subsamples were always kept selected
Table 12.
Prostate cancer data set
| Ns | ||||
|---|---|---|---|---|
| 131 | 263 | 990 | ||
| I | ORdensity (A) | 75.49 | 74.51 | 70.59 |
| limma (B) | 86.27 | 82.35 | 71.57 | |
| SAM (C) | 82.35 | 76.47 | 70.59 | |
| II | A vs B | 64 | 170 | 691 |
| A vs C | 64 | 159 | 746 | |
| B vs C | 54 | 211 | 791 | |
| III | ORdensity | 72.4 (7.12) | 184.1 (23.56) | 787.6 (29.40) |
| limma | 108.6 (4.99) | 220.2 (10.07) | 827.2 (39.17) | |
| SAM | 0.7 (0.67) | 4.4 (0.84) | 66.1 (4.38) | |
| Ns | ||||
| 1531 | 2684 | 3322 | ||
| I | ORdensity (A) | 66.67 | - | - |
| limma (B) | 70.59 | 68.63 | - | |
| SAM (C) | 70.59 | 66.67 | 65.69 | |
| II | A vs B | 961 | - | - |
| A vs C | 1035 | - | - | |
| B vs C | 1321 | 2422 | - | |
| III | ORdensity | 1215.4 (99.66) | - | - |
| limma | 1297.9 (50.64) | 2299.5 (86.19) | - | |
| SAM | 165.1 (6.15) | 563 (19.87) | 862.2 (19.41) | |
Results for different number (Ns) of selected genes: 131 with ORdensity strong selection; 263 with limma and Bonferroni; 990 with ORdensity relaxed selection; 1531 total potential DE genes; 2684 with limma and BH, and 3322 with SAM. Rows I present the leave-one-out cross-validation correct classification rate. In bold, the results for the genes selected under standard criteria for ORdensity, limma and SAM procedures; in rows II, number of common selected genes between the ORdensity, limma and SAM approaches; in rows III, mean and standard deviation (in brackets) of the number of genes that for 10 subsamples were always kept selected
As can be observed (rows II in Tables 9, 10, 11 and 12), the three methods only share some of the genes in their respective lists, as is usual in these type of procedures. Furthermore, the method with which our analysis shares more genes varies according to each data set.
These results indicated that ORdensity returns a small number of crucial genes, that are strongly related to the disease, since the values of leave-one-out cross-validation correct classification rates are large. As a general trend, SAM and limma consider a larger number of DE genes, but nevertheless it does not guarantee to obtain better leave-one-out cross-validation correct classification rates.
It is important to note that ORdensity identifies several genes, not detected by the other methods, that are biologically relevant. For instance, consider the strong selection with the leukemia data set. Interestingly, genes selected only by our method could be fundamental to explain some traits of the leukemia. For example, our method recognizes genes that codify for cyclin D2 (protein involved in cell cycle), neprilysin (an enzyme common in acute lymphoblastic leukemia), a protein-tyrosine phosphatase of T-cells and a protein similar to phorbolin-1 (that can be expressed in leukocytes).
Finally, we evaluated the stability of the procedure in order to assess how often a gene, selected as DE with the original sample, was selected again when a fraction of the 20% of the original data was eliminated at random. In order to mitigate any effect of selection bias, the process was repeated 10 times. Note that (rows III in Tables 9, 10, 11 and 12) ORdensity procedure was, in general, the method that best kept a coherent list with DE genes.
From a biomedical point of view, all the above results indicated that our method is very valuable to detect a small group of genes with a large effect in a particular disease. Therefore, this information can be used to develop new in vivo and in vitro studies.
Discussion
In this paper, a novel procedure, ORdensity, is proposed for the detection of differentially expressed genes. The proposed method is not a gene-by-gene procedure, and it takes into account the relationships between genes. The procedure obtains a ranking of importance of genes based in three measures, which reflect if the differences between quantiles for a gene under the two considered experimental conditions are important enough in order to consider the gene as a DE gene and if, in its neighbourhood, there are permuted false positives or not. We have presented an exhaustive evaluation of the performance of ORdensity using simulated microarray data, and results indicated that the new method correctly detects the DE genes. Furthermore, the simulation study showed that the procedure is useful even with small samples and it is competitive with other well established procedures, such as SAM and limma. The results, obtained with actual cancer microarray data sets, showed that ORdensity is very useful to obtain a small number of DE genes with high correct classification rate by the leave-one-out cross-validation approach and it is, in general, more stable than other well accepted procedures when the original sample is substituted with a subsample.
The main advantages of the proposed method are, therefore, that it returns very small sets of genes that retain a high predictive accuracy; the selected gene list is stable; it avoids the classical multiple comparison restrictions; as it takes into consideration the tails of distributions, it can detect outlying genes that only exhibit differential qualities in the tails, and, moreover, as we can cluster the genes using the three discriminative values OR, FP and dFP, different patterns of genes can be obtained. The results generated by our new procedure could be of extreme interest to biomedical research, because they can focus on a short, but crucial number of genes. Thus, these small numbers of genes could act as the corner stones to understand the origin and development of several serious diseases. The idea that lies beneath the proposed methodology seems applicable to RNA-Sequencing data. However, although it is usual to model RNA-Seq data as both negative binomial distribution and as normal distribution by ln-transforming normalized count data, because the properties of RNA-Seq data have not yet been fully established, additional research is needed.
Conclusions
Here we present a new method to identify differentially expressed genes that avoids losing sensitivity due to correction by multiple comparisons. This method is able to identify the nucleus of the genes that are candidates to explain a particular disease or pathology. With this information, further biomedical studies can be developed, focusing the attention in these candidate genes.
Additional file
This file contains Tables 13-27 and Figures 8-20 cited in the text. (PDF 533 kb)
Acknowledgements
We express our sincere thanks to the participants of the Data Science, Statistics and Visualisation (DSSV 2017) a Satellite Conference of the 61th World Statistics Congress (Lisboa, 2017) whose comments and suggestions led to substantial improvements in the content and organization of the paper. We also thank to the geneticist Prof. F. Mestres (University of Barcelona, Spain) his help in the interpretation of the biological function of the genes.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article. This study was partially supported: II by the Spanish Ministerio de Economia y Competitividad (TIN2015-64395-R) and by the Basque Government Research Team Grant (IT313-10) SAIOTEK ProjectSA-2013/00397 and by the University of the Basque Country UPV/EHU (Grant UFI11/45 (BAILab). CA by the Spanish Ministerio de Economia y Competitividad (SAF2015-68341-R), by the Spanish Ministerio de Economia y Competitividad (TIN2015-64395-R) and by Grant 2014 SGR 464 (GRBIO) from the Departament d’Economia i Coneixement de la Generalitat de Catalunya. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Availability of data and materials
The authors declare that all data has been previously published and the corresponding articles has been cited in the text.
Abbreviations
- ALL
Acute lymphoblastic leukemia
- AML
Acute myelooblastic leukemia
- AUC
Area under the ROC curve
- BH
Benjamini and Hochberg adjustment method
- DE
Differentially expressed
- DLBCL
Large B cell lymphoma
- dFP
Average density of false positive permuted cases in 10-NN
- FDR
False discovery Rate
- FP
Average number of false positive permuted cases in 10-NN
- FPC
Percentage of false positive genes per cluster
- FWER
Family wise error rate
- limma
Linear models with empirical Bayes statistic
- OR
Outlier robust index
- PAM
Partition around Medoids clustering method
- ROC
Receiver operating characteristic
- SAM
The significant analysis of microarrays
- WDB-discriminant
weighted distance based discriminant
Authors’ contributions
II and CA conceived the procedure, conducted and analyzed the results, wrote and reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Authors’ information
Concepcion Arenas received a PhD degree in mathematics, spezializing in statistics, from the University of Barcelona (Spain), where she is now a research professor in the Department of Genetics, Microbiology and Statistics, Statistics section. Her research interests include multivariate analysis as applied to bioinformatics, specifically DNA sequence analysis and microarray interpretation. She also works in biomedical statistics. Itziar Irigoien received a PhD degree in informatics from the University of the Basque Country, (UPV/EHU), Donostia, Spain, where she is now a research professor in the Department of Computation Science and Artificial Intelligence. Her research interests include the development of new statistical methods and software to solve bioinformatics and biomedical questions.
Contributor Information
Itziar Irigoien, Email: itziar.irigoien@ehu.eus.
Concepción Arenas, Email: carenas@ub.edu.
References
- 1.Quackenbush J. Microarray analysis and tumor classification. N Engl J Med. 2006;354(23):2463–72. doi: 10.1056/NEJMra042342. [DOI] [PubMed] [Google Scholar]
- 2.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;90(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang D, Parrish RS, Brock GN. Empirical evaluation of consistency and accuracy of methods to detect differentially expressed genes based on microarray. Comput Biol Med. 2014;46:1–10. doi: 10.1016/j.compbiomed.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3(1):1–25. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 5.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Efron B, Tibshirani R. Empirical bayes methods and false discovery rates for microarrays. Genet Epidemiol. 2002;23(1):70–86. doi: 10.1002/gepi.1124. [DOI] [PubMed] [Google Scholar]
- 8.Allison DB, Cui X, Page GP, Sabripour M. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7(1):55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 9.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 10.Guo L, Lobenhofer EK, Wang C, Shippy R, Harris SC, Zhang L, Mei N, Chen T, Herman D, Goodsaid FM, et al. Rat toxicogenomic study reveals analytical consistency across microarray platforms. Nat Biotechnol. 2006;24(9):1162–9. doi: 10.1038/nbt1238. [DOI] [PubMed] [Google Scholar]
- 11.Zhu J, Wang J, Guo Z, Zhang M, Yang D, Li Y, Wang D, Xiao G. GO-2D: identifying 2-dimensional cellular-localized functional modules in gene ontology. BMC Genomics. 2007;8(1):30. doi: 10.1186/1471-2164-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang M, Zhang L, Zou J, Yao C, Xiao H, Liu Q, Wang J, Wang D, Wang C, Guo Z. Evaluating reproducibility of differential expression discoveries in microarray studies by considering correlated molecular changes. Bioinformatics. 2009;25(13):1662–8. doi: 10.1093/bioinformatics/btp295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arenas C, Toma C, Cormand B, Irigoien I. Identifying extreme observations, outliers and noise in clinical and genetic data. Curr Bioinform. 2017;12(2):101–17. doi: 10.2174/1574893611666160606161031. [DOI] [Google Scholar]
- 14.Arenas C, Irigoien I, Mestres F, Toma C, Cormand B. Extreme observations in biomedical data. In: Ainsbury EA, Calle ML, Cardis E, editors. Extended Abstracts Fall 2015. Trends in Mathematics vol 7. Birkhäuser, Cham: Springer; 2017. [Google Scholar]
- 15.Dembélé D. A flexible microarray data simulation model. Microarrays. 2013;2(2):115–30. doi: 10.3390/microarrays2020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaufman L, Rousseeuw P. Clustering by Means of Medoids. Amsterdam: North-Holland; 1987. [Google Scholar]
- 17.Rousseeuw PJ. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J Comp Appl Stat. 1987;20(1):53–65. doi: 10.1016/0377-0427(87)90125-7. [DOI] [Google Scholar]
- 18.R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 19.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comp Graph Stat. 1996;5(3):299–314. [Google Scholar]
- 20.Díaz-Uriarte R, Alvarez De Andres S. Gene selection and classification of microarray data using random forest. BMC Bioinformatics. 2006;7(1):3. doi: 10.1186/1471-2105-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeffery IB, Higgins DG, Culhane AC. Comparison and Evaluation of Microarray Feature Selection Methods. http://www.bioinf.ucd.ie/people/ian/. Accessed 27 Aug 2018.
- 22.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irigoien I, Mestres F, Arenas C. Weighted distance based discriminant analysis: The R package WeDiBaDis. R J. 2016;8(2):434–50. [Google Scholar]
- 24.Anderson MJ, Robinson J. Generalized discriminant analysis based on distances. Aust NZ J Stat. 2003;45(3):301–18. doi: 10.1111/1467-842X.00285. [DOI] [Google Scholar]
- 25.Cuadras CM, Fortiana J, Oliva F. The proximity of an individual to a population with applications in discriminant analysis. J Classif. 1997;14(1):117–36. doi: 10.1007/s003579900006. [DOI] [Google Scholar]
- 26.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 27.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286(5439):531–7. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 28.Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci U S A. 1999;96(12):6745–50. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, Tamayo P, Renshaw AA, D’Amico AV, Richie JP, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1(2):203–9. doi: 10.1016/S1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file contains Tables 13-27 and Figures 8-20 cited in the text. (PDF 533 kb)
Data Availability Statement
The authors declare that all data has been previously published and the corresponding articles has been cited in the text.