

SUMMARY
Innate lymphoid cells (ILC) are important regulators of early infection at mucosal barriers. ILC are divided into three groups based on expression profiles, and are activated by cytokines and neuropeptides. Yet, it remains unknown if ILC integrate other signals in providing protection. We show that signaling through herpes virus entry mediator (HVEM), a member of the TNF receptor superfamily, in ILC3 is important for host defense against oral infection with the bacterial pathogen Yersinia enterocolitica. HVEM stimulates protective IFNγ secretion from ILC, and mice with HVEM-deficient ILC3 exhibit reduced IFNγ production, higher bacterial burdens and increased mortality. Additionally, IFNγ production is critical as adoptive transfer of wildtype but not IFNγ-deficient ILC3 can restore protection to mice lacking ILC. We identify the TNF superfamily member, LIGHT, as the ligand inducing HVEM signals in ILC. Thus HVEM signaling mediated by LIGHT plays a critical role in regulating ILC3-derived IFNγ production for protection following infection.
Keywords: HVEM, LIGHT, innate lymphoid cells, Yersinia enterocolitica, ileum, IFNγ
Graphical Abstract
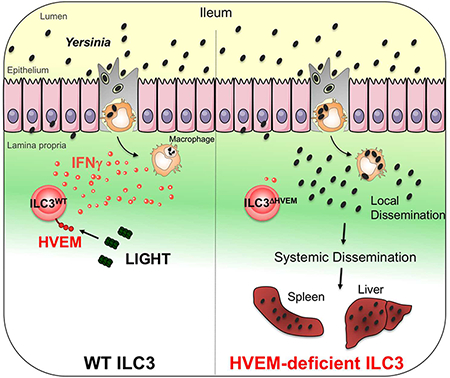
In Brief
Seo et al. find that IFNγ-producing ILC3 in the small intestine are required for host protection against Yersinia enterocolitica infection. HVEM signaling in ILC3, mediated by the ligand LIGHT, is critical for regulating IFNγ production for protection following infection.
INTRODUCTION
HVEM is a member of the tumor necrosis factor receptor superfamily (TNFRSF14). HVEM binds the immunoglobulin (Ig) super family molecules BTLA and CD160 and the TNF superfamily member LIGHT (TNFSF14) (Murphy and Murphy, 2010; Shui and Kronenberg, 2013). LIGHT-HVEM signaling has a number of roles including providing a co-stimulatory signal to T cells (Cohavy et al., 2004; Tamada et al., 2000), and it contributes to the development of dermatitis by keratinocytes (Herro et al., 2018). We have shown that HVEM signaling in epithelial cells contributes to host defense against pathogenic bacteria, but with CD160 involved as the ligand (Shui et al., 2012).
In addition to epithelial cells, HVEM is also expressed by various hematopoietic cells, including B and T lymphocytes, natural killer (NK) cells, dendritic cells (DC) and myeloid cells (Shui and Kronenberg, 2013). Additionally, it has been reported that CD3− ILC express Hvem mRNA (Kim et al., 2006). ILC are heterogeneous lymphoid cells that respond promptly to changes in the microenvironment by secreting effector cytokines and contributing to host defense and tissue homeostasis. ILC are divided into three different groups, defined by patterns of expression of effector cytokines, transcription factors and cell surface markers. Group 1 ILC include NK cells and ILC1 and are analogous to CD4 Th1 cells, while ILC2 and ILC3 share features with Th2 and Th17 cells, respectively (Artis and Spits, 2015). ILC3 can be further divided into subsets distinguished by CCR6 and NKp46 in mice (Klose et al., 2013; Luci et al., 2009; Sanos et al., 2009; Satoh-Takayama et al., 2008). CCR6+ ILC3, which are lymphoid-tissues-inducer cells (LTi) and LTi-like cells, produce IL-22 and IL-17 (Eberl et al., 2004). CCR6− ILC3, including those that do or do not express NKp46, can express T-bet and produce IFNγ in addition to IL-22 (Klose et al., 2013). Although ILC3 are more abundant in the small intestine than in the large intestine (Song et al., 2015), several studies of the functions of ILC3 used infection models, such as Citobacter rodentium (Cella et al., 2009; Satoh-Takayama et al., 2008; Sonnenberg et al., 2011) and Helicobacter hepaticus (Buonocore et al., 2010), which predominantly target the cecum and colon. ILC3 are also important for the response to Salmonella typhimurium (Goto et al., 2014; Klose et al., 2013), and while these bacteria infect M cells in Peyer’s patches, they cause inflammation predominantly in the cecum and colon (Barthel et al., 2003).
To study the function of ILC in the small intestine, we orally infected mice with Yersinia enterocolitica (Y. enterocolitica, YE), a facultative intracellular bacterium that targets the small intestine and causes food borne illness. After oral uptake, YE replicate in the small intestine, invade Peyer’s patches of the distal ileum and disseminate to the spleen and liver (Trulzsch et al., 2007). Frequently the infection is cleared in 1 to 2 weeks (Bottone, 1997; Cover and Aber, 1989), suggesting that innate immune cells could have important roles in protection from YE infection. However, the role of ILC in YE infection remains unknown. Here, we show that ILC populations, in particular CCR6− ILC3, are important for resistance to YE infection. Early production of IFNγ by these cells is, in part, responsible for protection. Furthermore, HVEM signaling by LIGHT is a critical factor that regulates IFNγ production by ILC3 in the small intestine.
RESULTS
ILC are important for defense from YE
To determine if ILC are required for host defense against YE, ILC were depleted from Rag1−/− mice by administering an anti-CD90.2 monoclonal antibody (mAb). The anti-CD90.2 mAb depleted not only ILC but also other cell types, including most NK cells in the small intestinal lamina propria (SI-LP) (Figure S1A). After YE infection, no differences were observed in survival, body weight, bacterial translocation, and splenic yersinosis up to day 7, when comparing Rag1−/− mice, which lack T cells and B cells, to C57BL/6 mice (Figures 1A–1D). These data indicate that innate immune cells, which include ILC, are sufficient to mediate the early defense against YE. Compared to Rag1−/− mice, CD90.2-depleted Rag1−/− mice lacking ILC had a decreased survival rate (Figure 1A), rapid weight loss (Figure 1B), increased bacterial burden in the ileum, spleen and liver (Figure 1C) and large areas of splenic necrosis with intralesional YE colonies (Figure 1D) after oral YE administration. These data demonstrate ILC are important for defense against YE.
Figure 1. ILC are critical for host defense against YE infection.
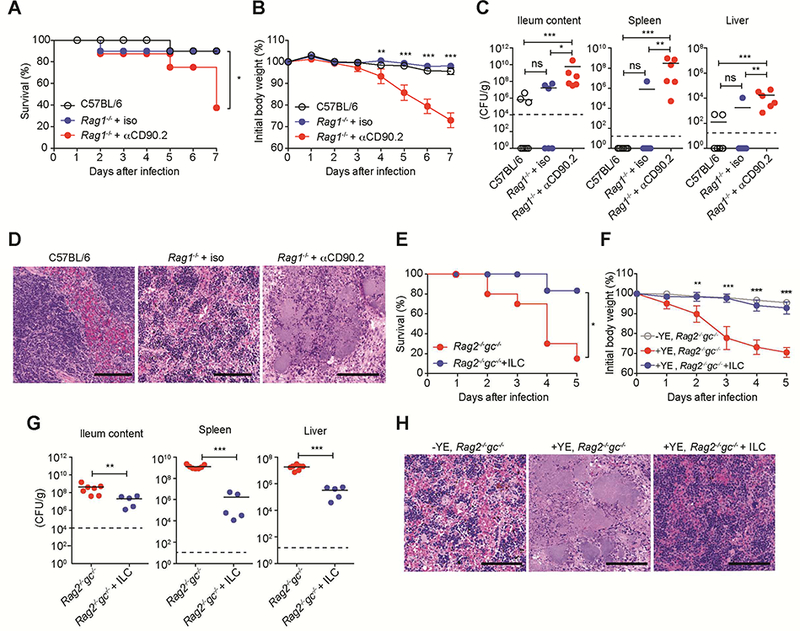
(A-D) Rag1−/− mice were injected i.p. with αCD90.2 mAb or isotype control on days −4, −1, 2, 5. Mice were infected with 1.2 × 108 YE CFU/mouse. (A) Survival curves. (B) Changes in body weight (% of baseline). (C) Bacterial burdens at day 7 p.i. (D) Representative H&E-stained splenic sections from the indicated mice at day 7 p.i. showing pale necrotic areas and bacterial colonies in mice treated with α-CD90.2. Scale bars, 100μm. (E-H) Sorted ILC (CD45+Lin−CD3−CD90.2+NK1.1−; 5 × 105/mouse) from SI-LP of Rag1−/− mice were transferred by retro-orbital injection into Rag2−/−gc−/− recipients at day −1. Control mice were injected with PBS. Mice were infected with 1.4 × 108YE CFU/mouse. (E) Survival curves. (F) Changes in body weight (% of baseline). (G) Bacterial burdens at day 5 p.i. (H) Representative H&E-stained splenic sections from the indicated mice at day 5 p.i. Scale bars, 100μm. Statistical analysis was performed using Log-rank test (A,E), 2 way ANOVA with Bonferroni’s multiple hypothesis correction (B,F), or Mann-Whitney test (C,G). Statistical significance is indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data shown are mean±SEM (B,F). Bars show the mean, symbols represent individual mice. Dotted horizontal line represents the limit of detection (C,G). Data represent pooled results from at least two independent experiments having at least three mice per group in each experiment (n=6–10 mice per group; co-housed). (A-C, E-G). Two independent experiments were carried out yielding similar results (D,H). See also Figure S1.
To confirm if there is a role for ILC during enteric YE infection, Lin−CD3−Thy1.2+NK1.1− ILC, which excludes NK cells and ILC1, were transferred into Rag2−/−gc−/− mice, which lack all ILC subsets. Rag2−/−gc−/− mice infected with YE were rescued from decreased survival, rapid weight loss and increased bacterial translocation by transfer of purified ILC (Figures 1E–1G). Rag2−/−gc−/− mice had multiple, massive necrotic areas in the spleen after oral YE infection, while Rag2−/−gc−/− recipients of ILC exhibited only very small lesions (Figure 1H). We conclude that under these experimental conditions ILC were necessary and sufficient to mediate early host defense against YE.
A role for ILC3 subsets in protection from YE
We identified intestinal ILC subsets via flow cytometry as CD3−, lineage−, CD90+ cells, and based on transcription factor expression (Gronke et al., 2017), we confirmed earlier work revealing that ILC3 were predominant in the small intestine lamina propria (Figures S1B and S1C). In the large intestine lamina propria, ILC1 and ILC2 constituted a larger percentage of the total ILC population (data not shown), and NK cells were infrequent in both locations. To test the role of ILC3 specifically during YE infection, we used Rorc-cre × Ahrfl/fl mice (AR mice), which are selectively deficient for ILC3 (Song et al., 2015). Although AR mice may also have some reduction in the function of Th17 and Th22 cells, we excluded a requirement for T lymphocytes for early host defense for several reasons. First, Rag1−/− mice were protected up to day 7, similar to C57BL/6 mice, emphasizing the early importance of the innate immune response (Figures 1A–1D). Second, as described below, the principal cytokines produced by Th17 and Th22 cells were not essential for survival. We observed that AR mice had marked weight loss and increased mortality following during YE infection (Figures 2A and 2B), implicating a nonredundant function of ILC3 in host defense against YE infection. On the other hand, NKp46-cre × Rorcfl/fl mice (RN mice), which selectively lack NKp46+ILC3 as well as ex-ILC3 (Song et al., 2015), did not have any difference in survival or body weight after YE infection compared to control mice (Figures 2C and 2D), suggesting that the NKp46+ ILC3 subset is not required to mediate early host defense.
Figure 2. An ILC3 subset is essential for protection against enteric bacterial infection.
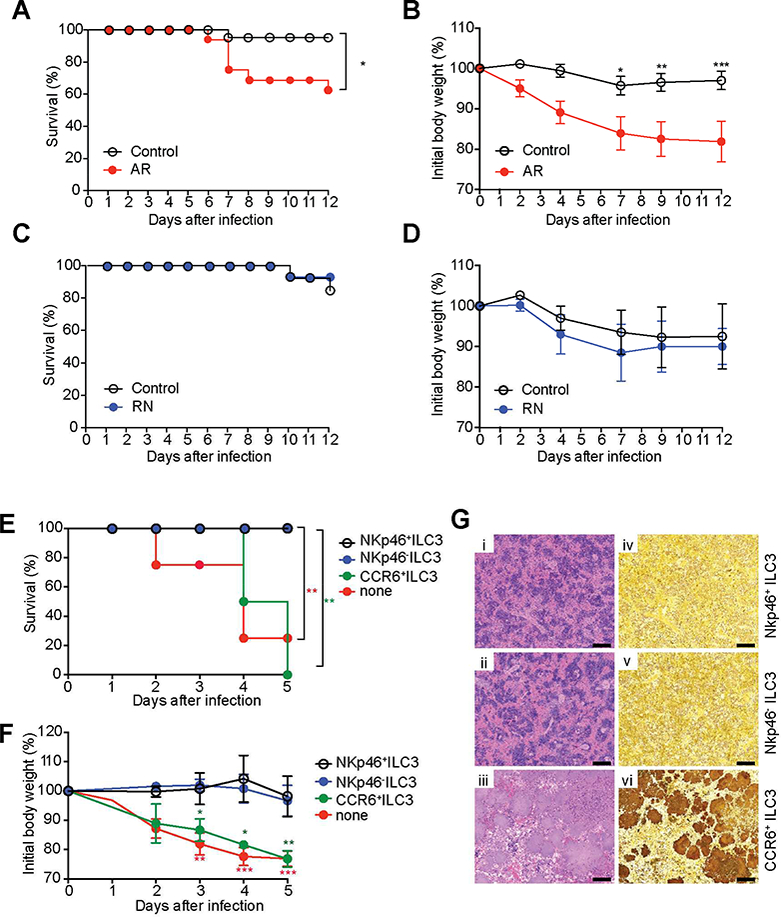
(A-D) Survival curves (n=13–16 per group) (A,C) and changes in body weight (B,D) of AR mice (Rorc-cre × Ahrfl/fl mice, which are selectively deficient for ILC3) and RN mice (NKp46-cre × Rorcfl/fl mice, which selectively lack NKp46+ILC3 as well as ex- ILC3). (E-G) NKp46− ILC3 (Lin−CD3−NK1.1−CD90.2highCD45intCCR6−NKp46−), NKp46+ ILC3 (Lin−CD3−NK1.1−CD90.2highCD45intCCR6−NKp46+), and CCR6+ ILC3 (Lin−CD3− NK1.1 CD90.2highCD45intCCR6+NKp46−) were sorted from SI-LP of Ragr−/− mice. Sorted cells (5 × 105 cells/mouse) were transferred by retro-orbital injection into Rag2−/−gc−/− recipients at day −1, and then mice were infected with 1.2 × 108 YE CFU/mouse (n=6–8 per group; co-housed). (E) Survival curves. (F) Changes in body weight. (G) Representative H&E-stained (i-iii) and Warthin-Starry silver stained (iv-vi) splenic sections, demonstrating large colonies of YE, from the indicated mice at day 5 p.i. Scale bars, 100μm. Two independent experiments were carried out yielding similar results. Statistical analysis was performed using Log-rank test (A,C,E) or 2 way ANOVA with Bonferroni’s multiple hypothesis correction (B,D,E). Statistical significance is indicated by*, p < 0.05; **, p < 0.01; ***, p < 0.001, and statistical significance in E and F is indicated by red stars (NKp46−ILC3 vs none or NKp46+ILC3 vs none) and green stars (NKp46− ILC3 vs CCR6+ILC3 or NKp46− ILC3 vs CCR6+ILC3). Data in B, D and F show mean±SEM. Data represent pooled results from two independent experiments having at least three mice per group in each experiment. See also Figure S2.
To address further which of the ILC3 subsets is essential for protection against YE, CCR6+ ILC3 and CCR6− ILC3, the latter population including NKp46+ ILC3 and NKp46− ILC3, were isolated from the SI of uninfected Rag1−/− mice by cell sorting based on previous criteria (Guo et al., 2016). We selected Lin−CD90highCD45int cells that were mainly RORγt+ILC3 from all subsets, in agreement with a previous report (Guo et al., 2016) (Figure S2A), and then divided these further based on NKp46 and CCR6 expression (Figure S2B). Rag2−/−gc−/− recipients of NKp46−ILC3 infected with YE were rescued from decreased survival, rapid weight loss and large areas of splenic necrosis that contained YE colonies (Figures 2E–2G). The same was true for recipients of equal numbers of NKp46+ ILC3 (Figures 2E–2G), indicating these cells have protective capacity, although they are less numerous and were not required in the presence of other populations (Figures 2C–2D) By contrast, Rag2−/−gc−/− recipients of CCR6+ ILC3 were not protected (Figures 2E–2G).
Intestinal ILC3 secrete protective IFNγ after infection
ILC effector functions are mainly mediated through cytokine secretion, which promotes protection at mucosal barriers. ILC3 have been reported to produce IL-17A, IL-22 and in some instances IFNγ (Klose et al., 2013; Sanos et al., 2009; Satoh-Takayama et al., 2008). To determine which of these cytokines might be important, we examined survival and bacterial translocation in lfng−/−, ll17ra−/−, and ll22−/− mice after oral YE infection. lfng−/− mice showed decreased survival, rapid weight loss, increased bacterial translocation to the spleen and liver and multiple, massive necrotic areas in the spleen after oral YE infection (Figures 3A–3D), in agreement with a previous report (Autenrieth et al., 1996; Echeverry et al., 2010). However, ll17ra−/− mice showed increased bacterial burden in the ileum, but not in the spleen and liver, on day 7 after infection, and the mice did not exhibit decreased survival (Figures S3A and S3B). These results are in contrast to a previous study, in which mice receiving a neutralizing IL-17 antibody showed reduced survival and increased bacterial translocation (DePaolo et al., 2012). ll22−/− mice also exhibited no differences in either survival or bacterial burden (Figures S3C and S3D). Therefore, IFNγ was required for host defense during Y. enterocolitica, whereas, in our colony, IL-17 had a limited role and IL-22 was not essential for survival.
Figure 3. IFNγ provides crucial defense in host against YE infection.
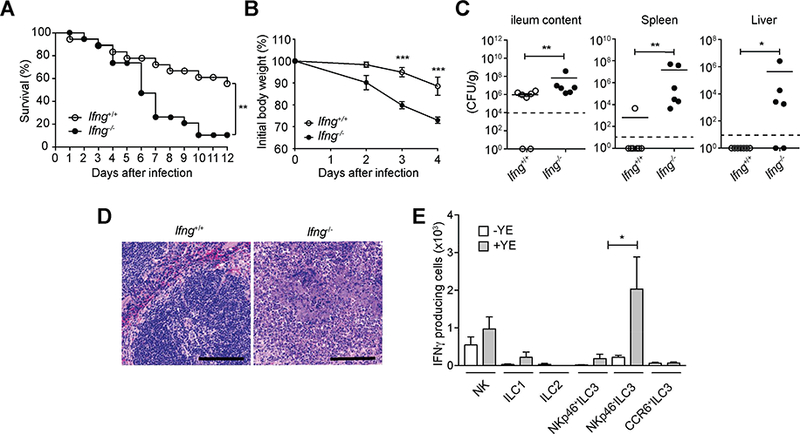
(A-D) lfng−/− mice or control mice were infected orally with 1.5 × 108 YE CFU. (A) Survival curves (n=18–19 per group). (B) Changes in body weight (n=5 per group). (C) Bacterial burdens (n=6–7 per group) at day 7 p.i. Bars show the mean, symbols represent individual mice. Dotted horizontal line represents the limit of detection. (D) Representative H&E-stained splenic sections at day 7 p.i. Scale bars, 100μm. Two independent experiments were carried out yielding similar results. (E) C57BL/6 mice were infected orally with 1.5 × 108 YE CFU (n=5 per group). Absolute numbers of IFNγ-producing cells from ILC subsets from the SI-LP of uninfected (-YE) and Y. enterocolitica infected (+YE) mice were analyzed at day 3 p.i. Cells were stimulated for 4h with PMA and ionomycin (P/I) and brefeldin A (BFA) was added in the last 2h of incubation before analysis by intracellular cytokine staining. Statistical analysis was performed using Log-rank test (A), 2 way ANOVA with Bonferroni’s multiple hypothesis correction (B), or Mann-Whitney test (C,E). Statistical significance is indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data in B and D show mean±SEM. Data represent pooled results from two experiments (A) or representative results of at least two independent experiments with five mice in each experimental group (B-E). See also Figure S3.
We tested IFNγ production by ILC subsets in the SI-LP during oral YE infection to determine how these cells reacted after pathogen exposure. The absolute numbers of ILC subsets in the SI-LP of YE-infected mice were similar to uninfected mice (Figure S3E), indicating that ILC do not expand greatly after YE infection. Consistent with this, only a small percentage of cells in ILC subsets incorporated Ki67 following infection (Figure S3F). Notably, the number of NKp46− ILC3 producing IFNγ in the SI-LP was greater than for other ILC (Figure 3E). There are also some IFNγ-producing NK cells and a smaller number of IFNγ-producing ILC1 and NKp46+ ILC3. By contrast, CCR6+ILC3 did not incorporate Ki67 or produce IFNγ after YE infection (Figures 3E and S3F). These data indicate that CCR6− ILC3, especially NKp46− ILC3, are important for host defense during YE infection.
IFNγ-producing ILC3 provide protection
To confirm that IFNγ production by ILC3 is important for immunity to YE, we performed an adoptive transfer of CCR6− ILC3 from lfng−/− or WT mice into Rag2−/−gc−/− mice. Rag2−/−gc−/− recipients of Ifng−/− ILC3 infected with YE had significantly decreased survival (Figure 4A), rapid weight loss (Figure 4B), and large areas of necrosis and increased bacterial colonies in the spleen and liver (Figure 4C). Therefore, our data indicate that IFNγ-producing ILC3 play an essential role in protective immunity to early YE infection.
Figure 4. IFNγ-producing ILC3 provide protection.
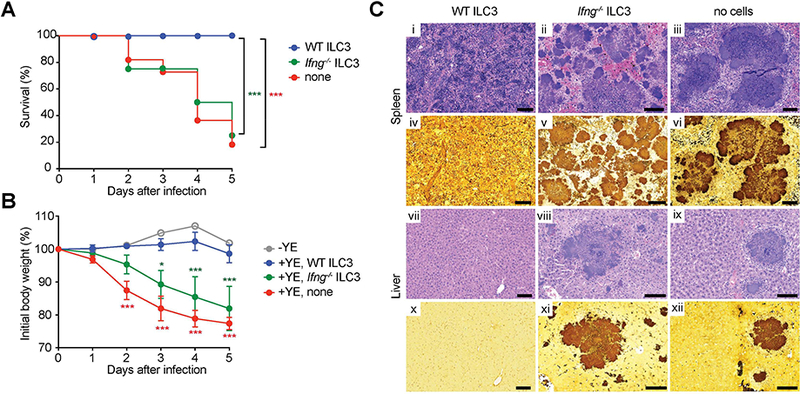
CCR6− ILC3 (Lin−CD3−NK1.1− CD90.2highCD45intCCR6−) were sorted from SI-LP of Rag1−/− Ifng−/− or Rag1−/− mice. Sorted CCR6− ILC3 (~1.5 × 105 cells/mouse) were transferred by retro-orbital injection into Rag2−/−gc−/− recipients at day −1. Groups of mice were infected orally with 1.4 × 108YE CFU (n=5–11 per group; co-housed). (A) Survival curves. (B) Changes in body weight. Data show mean±SEM. (C) Representative H&E stained (i-iii, vii-ix) or Warthin-Starry silver stained (iv-vi, x-xii) tissues from the indicated mice at day 5 p.i. Images of spleen (i-vi) and liver (vii-xii). Scale bars, 100μm. Two independent experiments were carried out yielding similar results. Statistical analysis was performed using Log-rank test (A) or 2 way ANOVA with Bonferroni’s multiple hypothesis correction (B). Statistical significance is indicated by *, p < 0.05; ***, p < 0.001 and statistical significance in A and B is indicated by green stars (WT ILC3 vs Ifng−/−C3) and red stars (WT ILC3 vs none). Data represent pooled results from two independent experiments having at least three mice per group in each experiment.
HVEM expression by RORγt+ ILC3 is important for host defense
Our previous work found a role for HVEM expression in multiple cell types to be important for regulation of the mucosal immune system, including CD8+ memory T cells, epithelial cells, and an unidentified cell type in Rag−/− mice (Shui and Kronenberg, 2013; Steinberg et al., 2013; Steinberg et al., 2008). Therefore, we investigated if HVEM functions in ILC. HVEM was expressed by all small intestine ILC subsets in mice and all ILC in human peripheral blood, and a previous analysis indicated RNA encoding HVEM was present in all ILC subsets from human tonsil (Bjorklund et al., 2016) (Figures S4A–S4C). Mice with a germ line deletion of Hvem had an undiminished absolute number of ILC3 (Figures S4D and S4E), ILC1 or ILC2 (data not shown), in the SI-LP, with similar results in the large intestine (data not shown). In the absence of infection, there was only a very small percentage of IFNγ+ ILC in either the small intestine or large intestine, and Hvem−/− mice were similar to control WT mice (Figure S4F).
To address the role of HVEM expression by ILC3 subsets in the SI-LP during YE infection, we examined the immune response in Rorc-cre mice crossed to Hvemfl/fl mice (Figure S4G), hereafter referred to as HvemᐃRorc. HvemᐃRorc mice exhibited marked loss of HVEM in ILC3 and partially in ILC1, which may reflect the conversion of RORγt- expressing ILC3 to ILC1 (Vonarbourg et al., 2010). There was no deletion in NK cells or ILC2 (Figure S4H). HvemᐃRorc mice also exhibited loss of HVEM expression in T lymphocytes (Figure S4H), because RORγt is expressed in double positive (CD4+CD8+) thymocytes (Eberl and Littman, 2004). To define the role of HVEM expression specifically by ILC, as opposed to T lymphocytes, we also analyzed Cd4-cre × Hvemfl/fl mice (HvemᐃCd4). HvemᐃCd4 mice lacked HVEM expression in all T cells, but not in ILC (Figure S4H).
Hvem−/− mice had reduced survival after oral YE infection (Figure 5A). HvemᐃRorc mice also had reduced survival (Figure 5B), rapid weight loss (Figure 5D), an increased bacterial burden in the ileum, spleen and liver (Figure 5E) and massive necrotic areas containing bacteria in the spleen and liver (Figure 5G) after oral YE infection. HvemᐃCd4 mice had no difference, however, in survival (Figure 5C), weight loss (Figure 5D), bacterial burden (Figure 5F) and histopathology (Figure 5G). Therefore, our data suggest that HVEM expression by ILC3, but not CD4+T cells, is required for protection from early enteric bacterial infection.
Figure 5. HVEM expression by RORγt+ ILC3 is required for host defense.
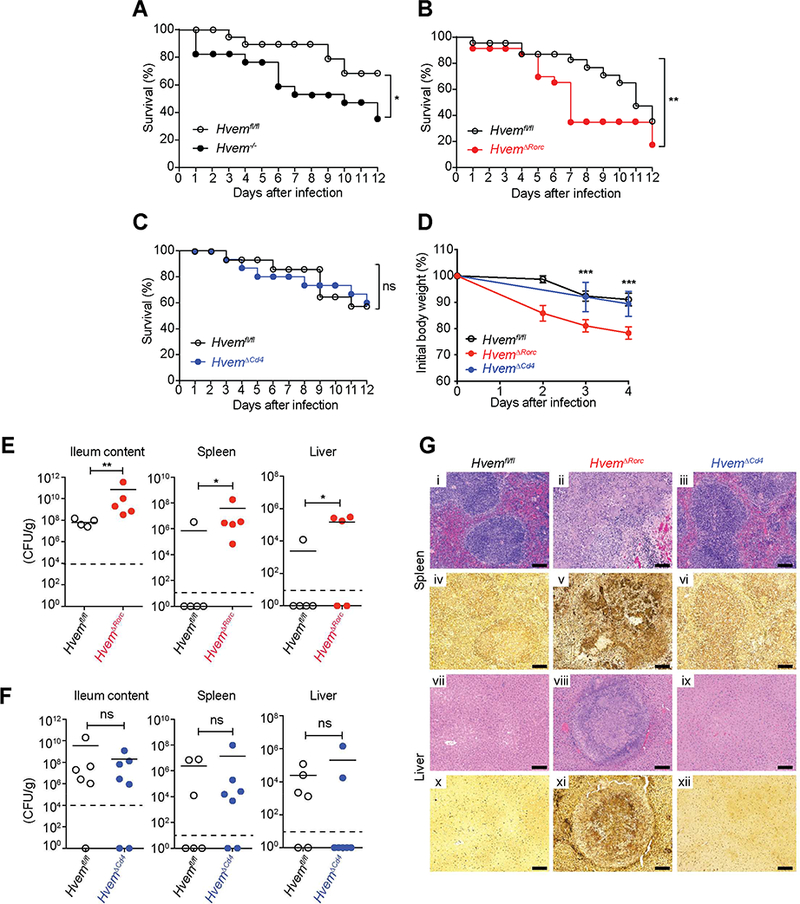
Groups of mice were infected orally with YE. (A-C) Survival curves (A,1×108 YE CFU/mouse; B,1.2×108 YE CFU/mouse; C,1.4×108 YE CFU/mouse; A-C, n=17–23 per group). (D) Changes in body weight (1.1×108 YE CFU/mouse;n=5 per group). Data shown are mean±SEM. (E-F) Bacterial burdens (E, 1.2 × 108 YE CFU/mouse; F, 2 × 108 YE CFU/mouse;n=5 per group) at day 7 p.i. In E and F, bars show the mean, symbols represent individual mice, and dotted horizontal line represents the limit of detection. (G) Representative H&E stained (i-iii,vii-ix) or Warthin-Starry silver stained (iv-vi, x-xii) tissue sections from the indicated mice at day 7 p.i. (1.1 × 108 YE CFU/mouse). Images of spleen (i-vi) and liver (vii-xii). Scale bars, 100μm. Two independent experiments were carried out yielding similar results. Statistical analysis was performed using Log-rank test (A-C), 2 way ANOVA with Bonferroni’s multiple hypothesis correction (D), or Mann- Whitney test (E,F). Statistical significance is indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. Data represent pooled results from at least two experiments (A-C, E-F) or representative results of two independent experiments with at least four mice in each experimental group (D). All mice were co-housed littermates. See also Figure S4.
RORγt+ ILC3 deficient for HVEM are impaired in protective IFNγ production.
To determine if HVEM signaling plays a role in the activation of ILC, we examined IFNγ production by ILC from HvemᐃRorc mice at day 3 and 7 p.i. ILC from HvemᐃRorc mice produced significantly less IFNγ at day 3 (Figures 6A and 6B) and day 7 (Figure S5A) p.i., but cytokine production by CD4+ T cells from HvemᐃRorc mice was not affected at either time (Figures 6C–6D and S5B). Furthermore, ILC and CD4+T cells of HvemᐃCd4 mice did not show any difference in production of IFNγ when compared to Hvemfl/fl mice after infection (Figures S5C and S5D). Therefore, Rorc-mediated deletion of HVEM in ILC in the small intestine diminished their ability to produce protective IFNγ after infection. Furthermore, Rag2−/−gc−/− recipients of Hvem−/− ILC3 infected with YE had significantly decreased survival, massive necrotic areas and increased bacterial colonies in the spleen and liver (Figures 6E and 6F), indicating that HVEM expression by ILC3 plays an essential role in protection during early YE infection.
Figure 6. HVEM deficiency affects IFNγ production by ILC during YE infection.
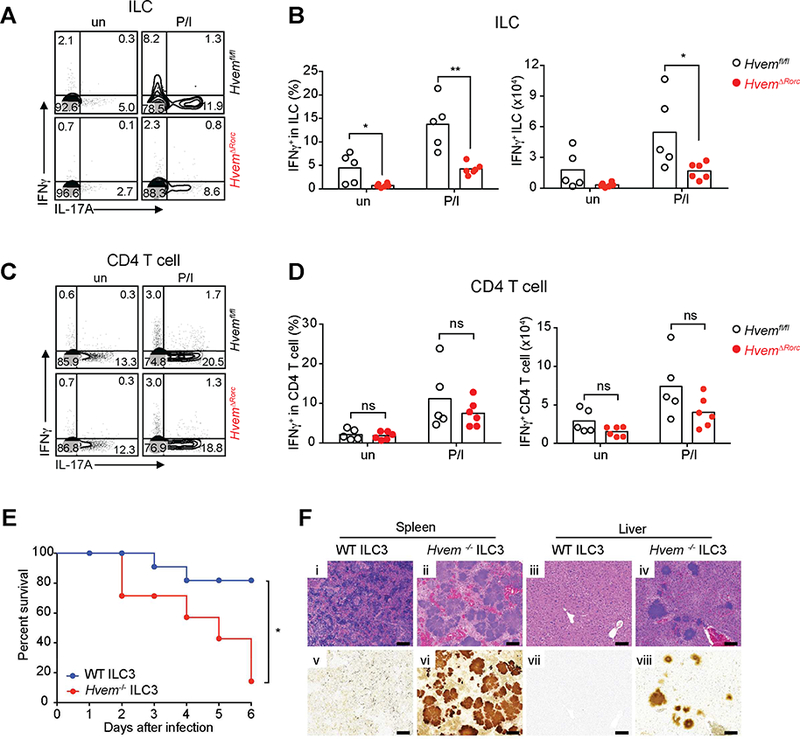
(A,C) Representative plots of IFNγ and IL-17A expression by ILC (CD45+Lin−CD3− CD90.2+)(A) or CD4+ T cells (CD45+Lin−CD3+CD4+)(C). (B,D) Frequencies and absolute numbers of IFNγ expressing ILC (B) and CD4+ T cells (D) from ileal LPL isolated from Hvemfl/fl and HvemᐃRorc mice at day 3 p.i. (2×108 YE CFU/mouse; n=5–6 per group; cohoused littermates). Cells were either unstimulated (un) or stimulated for 4h with P/I and BFA was added in the last 2h of incubation before analysis for intracellular cytokine. (E- F) CCR6− ILC3 (Lin−CD3−NK1.1−CD90.2highCD45intCCR6−) were sorted from SI-LP of Rag1−/− Hvem−/− or Rag1−/− Hvem+/+ mice. Sorted CCR6− ILC3 (~1.5 × 105 cells/mouse) were transferred by retro-orbital injection into Rag2−/−gc−/− recipients at day −1. Groups of mice were infected orally with YE (1.4 × 108 YE CFU/mouse; n=7–11 per group; cohoused). (E) Survival curves. (F) Representative H&E stained (i-iv) or Warthin-Starry silver stained (v-viii) tissues from the indicated mice at day 6 p.i. Images of spleen (i,ii,ν,νi) and liver (iii,iν,νii,viii). Scale bars, 100μm. Two independent experiments were carried out yielding similar results. Statistical analysis was performed using Mann- Whitney test (B,D) or Log-rank test (E). Statistical significance is indicated by*, p < 0.05; **, p < 0.01; ns, not significant. In B and D, bars show the mean and each symbol represents a measurement from a single mouse. Data represent pooled results from two independent experiments having at least three mice per group in each experiment. See also Figure S5.
LIGHT provides the ligand for HVEM in ILC.
To determine if HVEM signals in ILC to induce IFNγ, ILC were stimulated in vitro with the HVEM ligand LIGHT in the presence of IL-7 +/- IL- 23. IFNγ production was increased by soluble LIGHT at both the transcript and protein levels (Figures 7A and 7B). ILC expressed HVEM (Figure S4A), but wild type and Hvem−/− ILC did not express LTβR (Figures S6A and S6B), which also can bind to LIGHT, indicating that LTβR was not involved. BTLA and CD160 did not stimulate IFNγ release by ILC (data not shown), but they bind HVEM as monomers (Cai and Freeman, 2009; Compaan et al., 2005) and therefore this might only reflect their reduced binding strength. Light−/− mice had no difference in the number of ILC3 (Figures S6C and S6D), ILC1 or ILC2 (data not shown) in the SI-LP in the absence of infection.
Figure 7. LIGHT provides the ligand for HVEM in ILC.
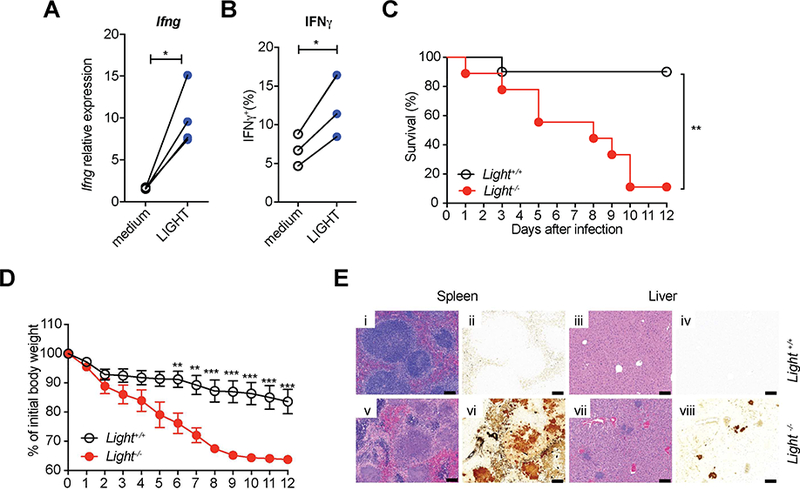
(A) LIGHT induces Ifng expression by ILC. Sorted ILC (CD45+Lin−CD3−CD90.2+) from SI-LP of Rag1−/− mice were cultured with IL-7 and IL-23 in the presence or absence of soluble LIGHT for 18 hours. Ifng transcript was determined by qPCR. (B) IFNγ expression by intracellular cytokine staining. Sorted ILC were cultured with IL-7 in the presence or absence of soluble LIGHT for 18 hours, and then were stimulated for 4h with PI and BFA was added in the last 2h of incubation before analysis. (C-E) Ligh−/− mice and control mice were infected orally with 1.6 × 108 YE CFU (n=9–10 per group; co-housed). (C) Survival curves. (D) Changes in body weight. Data show mean±SEM. (E) Representative H&E stained (i,iii,v,vii) or Warthin-Starry silver stained (ii,iv,vi,viii) tissues from the indicated mice at day 7 p.i. Images of spleen (i,Ν,ν,νi) and liver (iii,iv,vii,viii). Scale bars, 100μm. Two independent experiments were carried out yielding similar results. Statistical analysis was performed using Mann-Whitney test (A,B), Log-rank test (C), or 2 way ANOVA with Bonferroni’s multiple hypothesis correction (D). Statistical significance is indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data show representative results of two independent experiments with at least three mice in each experimental group. See also Figure S6 and S7.
To test if there is an exclusive role for LIGHT engaging HVEM during enteric YE infection, we used two type of mouse colonies. First, wild type and global Light knock out mice from separate breedings were co-housed upon weaning, and, second, Light−/− and Ligh+/- mice littermates were compared. After YE infection, Light−/− mice in both of these experimental setups had decreased survival (Figures 7C and S7A), rapid weight loss (Figures 7D and S7B), and massive necrotic areas and increased bacterial colonies in the spleen and liver (Figures 7E and S7C), similar to HvemᐃRorc mice. Furthermore, combining data from the two experimental setups, RORγt+ ILC3 in the SI-LP from Light−/− mice produced significantly less IFNγ at day 3 p.i., compared to control mice (Figure S7D and S7E). Btla−/− mice and Cd160−/− mice exhibited no differences in survival, body weight, and histopathology of spleen and liver (Figures S7F–S7K). Therefore, our data indicate that LIGHT mediated signaling to HVEM expressed by innate immune cells, especially ILC3 subsets, provides protection from YE.
DISCUSSION
It is established that ILC in the small intestine have a protective role against several mucosal bacterial infections (Buonocore et al., 2010; Cella et al., 2009; Klose et al., 2013). Here, we demonstrated that ILC3 have a crucial function in the innate immune response in protecting the host against YE by secreting IFNγ. Although ILC3-deficient mice (Song et al., 2015) exhibited heightened susceptibility to YE, our analysis demonstrated that the signature ILC3 cytokines, IL-22 and IL-17A, were not important for protection. A possibly similar function of ILC3 that is not dependent on the hallmark cytokines was shown in anti-CD40 induced colitis, which was dependent on ILC and the RORγt/IL-23 axis (Buonocore et al., 2010), but blockade of IL-17A or IL-22 also had no effect (Pearson et al., 2016). Surprisingly, transfer of ILC3 showed that the ability to produce IFNγ was essential. NKp46+ and NKp46− ILC3 previously have been reported to express T-bet and to secrete IFN-γ after infection, with a higher percentage NKp46+ ILC3 capable of secreting this cytokine (Buonocore et al., 2010; Klose et al., 2013). When equal cell numbers were transferred, both NKp46+ and NKp46− ILC3 were protective, while CCR6+ ILC3 were not. However, deletion of NKp46+ ILC3 did not lead to increased susceptibility, likely because the number of IFNγ-secreting NKp46− ILC3 is much greater. ILC1 and NK cells typically secrete IFNγ when activated and released this cytokine after YE infection. Despite the non-redundant role of NKp46− ILC3, there could be a contribution to defense by NK cells, ILC1 and former ILC3. However, our data indicate that ILC3 could not be replaced and provide evidence that IFNγ is an additional cytokine from ILC3 populations that also has an important protective function. IFNγ was previously shown to be a critical cytokine for host defense against YE. However, the effector mechanisms induced by IFNγ for YE clearance are not well defined. It is known that IFNγ activates macrophages that kill extracellular and intracellular pathogens after being engulfed. There might also be an essential role for IFNγ induced chemokines in attracting immune cells.
We followed the infection up to day seven, and therefore it is possible that adaptive immunity plays a larger role in host defense at later times. However, our data demonstrating the importance of the innate immune response are in agreement with the finding that adult CD4 deficient mice were not more susceptible to YE infection (Echeverry et al., 2010), although these mice have CD8+ T cells that also could have contributed to IFNγ secretion and protection.
Because HVEM is expressed by different cell types, and also because it can signal or act as a ligand for signaling receptors BTLA and CD160, it is not surprising that HVEM has multiple functions in mucosal immunity. Regarding bacterial infections, immune defense from C. rodentium infection of mice, a model for enteropathogenic E. coli infection, was dependent on HVEM expression (Shui et al., 2012). Additionally, an effective defense from Clostridium difficile was dependent on the HVEM ligand CD160 (Sadighi Akha et al., 2015). Furthermore, the generation of CD8+ T cell mucosal memory to Listeria monocytogenes was dependent on HVEM (Steinberg et al., 2013). These previous studies prompted us to investigate the role of HVEM in ILC function. We found that Hvem deletion mediated by Rorc-cre led to increased susceptibility to infection and decreased IFNγ secretion by ILC. These results implicated a role for HVEM in either ILC3 or CD4+ T cells, because Rorc-cre also acts in T lymphocytes (Eberl and Littman, 2004). However, mice with a Cd4-cre mediated deletion of Hvem were not more susceptible to YE infection and did not have reduced IFNγ production by ILC after infection, demonstrating a specific role for HVEM in ILC3. ILC are tissue-resident cells that are locally maintained during homeostasis and in some cases expanded during infection. We found that HVEM was not required for ILC differentiation, and during YE infection, ILC subsets did not increase in number in the SI-LP. Instead, the effect of HVEM expression was specifically on cytokine production.
HVEM expression by ILC could have an effect on cytokine production because it can stimulate ILC, or because it serves as a ligand to stimulate an interacting cell type through HVEM ligands that signal, such as BTLA or CD160. These possibilities are not mutually exclusive, but our in vitro data indicated that HVEM could signal in ILC to stimulate cytokine production. The ligand for HVEM stimulating protective ILC cytokine production is LIGHT, a TNF super family member, and not CD160 or BTLA. We did not rule out a role for LTα homotrimers in engaging HVEM, but their affinity for HVEM is weaker than the other ligands (Cai and Freeman, 2009). Furthermore, it is formally possible that LIGHT has additional roles in host protection besides engaging HVEM on ILC3, but the similarity in outcome between global Light knock out mice and the HvemᐃRorc mice suggests this is not the case. It has been shown that LIGHT-HVEM signaling upregulate IFNγ production in CD4 T cells as well as in NK cells (Cohavy et al., 2004; Fan et al., 2006; Tamada et al., 2000). The cellular source of LIGHT is not known, but a previous analysis indicated RNA encoding LIGHT was present in all ILC subsets from the small intestine of mice (Gury-BenAri et al., 2016). Therefore, an autocrine as well as paracrine mechanism of action is possible. Regarding IFNγ, HVEM signals through TRAF proteins to activate nuclear factor-kappaB (NF-κΒ) and AP-1 (Marsters et al., 1997), and it has been shown that transcription binding sites for NF-κΒ and AP-1 are within the Ifng promoter region and increase Ifng transcription (Samten et al., 2008; Sica et al., 1997). Therefore, it is plausible that HVEM signaling mediates increased IFNγ production by ILC through the actions of NF-κΒ and AP-1.
Cytokine secretion by ILC subsets is activated by cytokines from other innate immune cells, such as epithelial or myeloid cells. Recently, there also has been evidence that ILC can be activated by neuropeptides (Cardoso et al., 2017; Ibiza et al., 2016; Klose et al., 2017; Wallrapp et al., 2017). The TNF superfamily of cytokines and receptors play diverse and important roles in regulating both innate and adaptive immune cells, and therefore are good candidates for influencing ILC. It has been shown previously that the DR3 ligand, TNF-like protein 1A (TL1A or TNFSF15), promoted ILC2 cytokine secretion and also the survival and expansion of these cells in allergic responses (Meylan et al., 2014; Yu et al., 2014). Additionally, we demonstrated that human ILC express HVEM and human ILC3 express DR3 and TL1A enhanced their IL-22 production (Ahn et al., 2015). Here, we found that HVEM expression by ILC3 not only regulates cytokine secretion but also has a nonredundant role in host defense. Therefore, an appealing hypothesis is that different members of the TNFR super family will prove to be important regulators of ILC subsets, similar to their role in CD4 and CD8 T cell biology. In doing so, they would provide a third type of signal, in addition to cytokines from innate immune cells and neuropeptides, which ILC must integrate to modulate their responses. If true, this will have implications for the use of agonistic or blocking antibodies to TNFR super family members for augmentation of anti-cancer and other immune responses.
STAR★METHODS
• KEY RESOURCES TABLE
| KEY RESOURCES TABLE | ||
|---|---|---|
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Rat monoclonal anti-mouse GATA-3 (clone TWAJ) | Invitrogen | Cat#46–9966-42; RRID: AB 10804487 |
| Mouse monoclonal anti-mouse NK1.1 (clone PK136) | BioLegend | Cat#108724; RRID: AB 830871 |
| Rat monoclonal anti-mouse NKp46/CD335 (clone 29A1.4) |
BD Bioscience | Cat#561169; RRID: AB 10561840 |
| Rat monoclonal anti-mouse Eomes (clone Dan11 mag) | Thermo Fisher Scientific | Cat#50–4875-82; RRID: AB 2574227 |
| Rat monoclonal anti-mouse CD45 (clone 30.F11) | BD Bioscience | Cat#564225; RRID: AB 2716861 |
| Armenian hamster monoclonal anti-mouse CD3e (clone 145–2C11) | BD Bioscience | Cat#563123; RRID: AB 2687954 |
| Rat monoclonal anti-mouse Thy1.2/CD90.2 (clone 53– 2.1) | BioLegend | Cat#140317; RRID: AB 11203724 |
| Rat monoclonal anti-mouse CD4 (clone RM4–5) | BioLegend | Cat#100559; RRID: AB 2562608 |
| Armenian hamster monoclonal anti-Mouse CD196/CCR6 (clone 29–2L17) |
BioLegend | Cat#129817; RRID: AB 10898320 |
| Rat monoclonal anti-mouse CD19 (clone ID3) | Thermo Fisher Scientific | Cat#25–0193; RRID: AB 657663 |
| Rat monoclonal anti-mouse CD45R / B220 (clone RA3–6B2) | BD Biosciences | Cat#552772; RRID: AB 394458 |
| Armenian hamster monoclonal anti-mouse CD11c (clone N418) | Thermo Fisher Scientific | Cat#25–0114-82; RRID: AB 469590 |
| Rat monoclonal anti-Mouse Ly-6G (Gr-1) (clone RB6– 8C5) | Thermo Fisher Scientific | Cat#25–5931-82; RRID: AB 469663 |
| Rat monoclonal anti-mouse IFNgamma (clone XMG1.2) |
BD Biosciences | Cat#562303; RRID: AB 11153140 |
| Rat monoclonal anti-mouse IL-17A (clone eBio17B7) | Thermo Fisher Scientific | Cat#53–7177-81; RRID: AB 763579 |
| Rat monoclonal anti-mouse RORgt (clone B2D) | Thermo Fisher Scientific | Cat# 12–6981-82; RRID: AB 10807092 |
| Armenian hamster monoclonal anti-mouse HVEM (clone HMHV-1B18) | BioLegend | Cat#136303; RRID: AB 1967105 |
| Mouse monoclonal anti-human CD4 (SK3 (SK-3)) (clone SK3 (SK-3)) |
Thermo Fisher Scientific | Cat#47–0047-42; RRID: AB 10804505 |
| Mouse monoclonal anti-human CD8 (clone RPA-T8) | Thermo Fisher Scientific | Cat#47–0088-42; RRID: AB 1272046 |
| Mouse monoclonal anti-human CD16 (clone eBioCB16 (CB16)) | Thermo Fisher Scientific | Cat#47–0168-42; RRID: AB 11220086 |
| Mouse monoclonal anti-human CD19 (clone HIB19) | Thermo Fisher Scientific | Cat#47–0199-42; RRID: AB 1582230 |
| Mouse monoclonal anti-human CD127 (clone HIL-7R- M21) | BD Bioscience | Cat#560905; RRID: AB 10563899 |
| Mouse monoclonal anti-human CD94 (clone HP-3D9) | BD Bioscience | Trial Ab |
| Mouse monoclonal anti-human CD3 (clone SK7) | BD Bioscience | Cat#563219; RRID: AB 2714001 |
| Mouse monoclonal anti-human CD45 (clone HI30) | BD Bioscience | Cat#562312; RRID: AB 11154590 |
| Mouse monoclonal anti-human CD34 (clone 561) | BioLegend | Cat#343610; RRID: AB 2561358 |
| Mouse monoclonal anti-human HVEM (clone 122) | BioLegend | Cat#318805; RRID: AB 2203704 |
| Mouse IgG 1 kappa isotype control, PE | Thermo Fisher Scientific | Cat#12–4714-81; RRID: AB 470059 |
| Rat monoclonal anti-mouse CD90.2 (clone 30H12) | BioXcell | Cat#BE0066; RRID: AB 1107682 |
| Rat lgG2b isotype control | BioXcell | Cat#BE0090; RRID: AB 1107780 |
| Bacterial and Virus Strains | ||
| Y. enterocolitica strain WA-C (pYV::CM) | Laboratory of Dr. J. Heesemann (Trulzsch et al., 2004) | N/A |
| Biological Samples | ||
| Human PBMC | LJI normal blood donor program | Single donor, male, 44 years old |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse IL-7 | R&D SYSTEMS | Cat# 407-ML |
| Recombinant Mouse IL-23 | BioLegend | Cat# 589002 |
| Recombinant Mouse LIGHT (TNFSF14) | BioLegend | Cat# 557604 |
| LIVE/DEAD™ Fixable Yellow Dead Cell Stain Kit | Thermo Fisher Scientific | Cat# L34959 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: Hvemflox-neo/flox-neo | This paper | N/A |
| Mouse: B6;SJL-Tnfrsf14tm1.1Kro/J (Also known as: Hvemflox) | This paper | RRID: IMSR_JAX:030862 |
| Mouse: Light−/− | This paper | N/A |
| Mouse: Btla−/− | This paper | N/A |
| Mouse: C57BL/6-Cd160tm1Yxf (Also known as: Cd160−/−) | The Jackson Laboratory (Tu et al., 2015) | RRID: IMSR_JAX:028527 |
| Mouse: Ahrtm3.1Bra/J (Also known as: Ahrflor) | The Jackson Laboratory (Walisser et al., 2005) | RRID: IMSR_JAX:006203 |
| Mouse: B6.FVB-Tg(Rorc-cre)1Litt/J (Also known as: Rorc(γt)-cre) | The Jackson Laboratory (Eberl and Littman, 2004) | RRID: IMSR_JAX:022791 |
| Mouse: B6(Cg)-Rorctm3Litt/J (Also Known As: Rorc(γt)f/f) | The Jackson Laboratory (Eberl and Littman, 2004) | RRID: IMSR_JAX:008771 |
| Mouse: NKp46-cre | (Narni-Mancinelli et al., 2011) | N/A |
| Mouse: STOCK Tg(Cd4-cre)1 Cwi/BfluJ (Also Known As: Cd4-cre) | The Jackson Laboratory | RRID: IMSR_JAX:017336 |
| Mouse: B6.129S7-Rag1tm1Mom/J (Also Known As: Rag−/−) | The Jackson Laboratory | RRID: IMSR JAX:002216 |
| Mouse: B10;B6-Rag2tm1Fwa II2rgtm1Wjl (Also Known As: Rag2−/− γc−/−) | Taconic | RRID: IMSR_TAC:4111 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: IMSR JAX:000664 |
| Oligonucleotides | ||
| Primer: lfng(F): AGC TCA TCC GGT GGT CCA C | (Zheng et al., 2008) | |
| Primer: lfng(R): AAA ATT CAA ATA GTG CTG GCA GAA | (Zheng et al., 2008) | |
| Primer: L32(F): GAA ACT GGC GGA AAC CCA | (Mucida et al., 2007) | |
| Primer: L32(R): GGA TCT GGC CCT TGA ACC TT | (Mucida et al., 2007) | |
| Recombinant DNA | ||
| Software and Algorithms | ||
| FlowJo software 10.4.1 | FlowJO, LLC | https://www.flowjo.com/solutions/flowjo |
| Prism softward 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ZEN2.3 (blue edition) | Zeiss | https://www.zeiss.com/microscopy/us/downloads/zen.html#service-packs |
| Other | ||
• Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mitchell Kronenberg (mitch@lji.org).
Experimental Model and Subject Details
Mice
Hvemflox-neo/flox-neo (Hvemfn/fn) mice were generated using C57BL/6NTac embryonic stem (ES) cells by the UC San Diego Health Sciences transgenic mouse core facility. A frt-flanked LacZ reporter cassette was inserted into intron 2 of the HVEM gene and a F3-flanked Neor cassette (for drug selection) was inserted into intron 6 of the Hvem gene by recombineering (Chan et al., 2007). We confirmed that Hvemfn/fn mice (knockout first) were deficient for HVEM expression due to the presence of the Neor cassette and we refer to them as Hvem−/−. Germ line transmitted mice (Hvemfn/fn, knockout first) were crossed with FLPe mice to delete the Neo cassette to generate Hvemfl/fl mice, which have loxP sites flanking exons 3 and 6 of the Tnfrsf14 gene. Hvemfi/fi mice were bred to Cre-expressing mice to generate conditional knockout mice. Light−/− mice were generated using C57BL/6NTac ES cells by the UC San Diego Health Sciences transgenic mouse core facility. We confirmed that Light−/− mice (knockout first) were deficient for Light expression by Southern Blot and long range PCR. Btla−/− generated by crossing B6.C-Tg(CMV-cre)1Cgn/J mice (The Jackson Laboratory, ME), which are useful for deletion of loxp-flanked genes in all tissues, with transgenic Btlafi/fi mice, which have loxp sites flanking exon 4 and 5 of the Btla gene (in preparation). Cd160−/− mice were provided by Dr. Yang-Xin Fu (UT Southwestern, TX) (Tu et al., 2015). AR mice (Rorc-cre × Ahrfi/fi mice) and RN mice (NKp46-cre × Rorcfi/fi mice) have been described previously (Song et al., 2015). Rag2−/−gc−/− mice were purchased from Taconic and bred in-house for all experiments. C57BL/6, Rag1−/−, Rorc-cre and Cd4-cre mice were all purchased from The Jackson Laboratory. All Cre mouse strains were maintained on the C57BL/6 background or were backcrossed to C57BL/6 for at least for 6 generations. Mice of both genders were used at 8–20 weeks of age. Whenever possible, groups of control and gene knockout mice were housed in the same cage to minimize the effect of housing conditions on experimental variation. For tissue or cell analyses, tissues were collected and used for histological analysis and lamina propria cell preparation. Mice were bred and housed under specific pathogen-free conditions in the vivarium of the La Jolla Institute for Allergy & Immunology (LJI) or Washington University, St. Louis. All procedures were approved by the LJI Animal Care and Use Committee or the Washington University Animal Studies Committee.
Bacterial infection
Y. enterocolitica strain WA-C (pYV::CM), which is resistant to chloramphenicol, was obtained from Dr. J. Heesemann (Ludwig Maximilian University of Munich, Germany) (Trulzsch et al., 2004) and used in all infection studies. Bacteria were grown overnight in Luria-Bertani (LB) broth with shaking at 30°C for 1 6h, and the culture was refreshed the next day for 6h. Bacterial density was measured by O.D. 600 and bacterial cultures were diluted with PBS for proper concentration. Individual titers were determined after each experiment by serial dilution. Mice were infected with 1×108 to 2×108 c.f.u. of Y. enterocolitica by oral gavage, and tissues were collected at the indicated time points after infection. For c.f.u. assays, spleen, liver or ileum contents were weighed, homogenized in sterile PBS, serially diluted and plated in chloramphenicol-containing LB agar plates.
Isolation of human PBMCs
Human peripheral blood monocuclear cells (PBMCs) were obtained from donor (male, 44 years old) of LJI normal blood donor program. Blood sample was obtained using standard phlebotomy techniques. Study was approved by La Jolla Institute for Allergy and Immunology Institutional Review Board. PBMCs were isolated from the blood using density-gradient centrifugation (Ficoll).
• Method Details
Depletion of CD90.2+ cells in mice
For depletion of CD90+ cells in Rag1−/− mice, mice were injected with anti-CD90.2 (30H12; BioXCell) (100μg/mouse) or isotype control on days −4, −1, 2, 5.
Adoptive transfer of ILC
Isolated ILC from SI LP from Rag1−/− mice were transferred by retro-orbital injection into Rag2−/−gc−/− recipients at day −1, and then mice were infected with YE. Control mice were injected with PBS. For adoptive transfer of NKp46−ILC3, NKp46+ILC3, and CCR6+ILC3, cells were isolated from the small intestine of uninfected Rag1−/− mice. lfng−/− ILC3 were isolated from the small intestine of uninfected lfng−/−Rag1−/− mice. Hvem−/− ILC3 were isolated from the small intestine of uninfected Hvem−/−Rag1−/− mice.
Preparation of Lamina Propria Lymphocytes
Small intestines were collected from mice. Peyer’s patches were carefully removed, and tissues were cut open longitudinally, briefly washed, and cut into 1.5 cm pieces. The tissue pieces were incubated in 25 ml of HBSS (5% FBS, 25mM HEPES and 1mM DTT) in a shaker at 225 r.p.m., 37°C, for 20 min. After incubation, the cell suspension was filtered through a metal mesh. The tissue debris was saved for Lamina Propria Lymphocytes (LPL) preparation. For LPL preparation, the tissue debris was incubated in 20 ml of HBSS (25mM HEPES and 20mM EDTA) in a shaker at 225 r.p.m., 37°C, 2 times for 15 min to further remove epithelial cells. After that, tissues were placed in 20 ml pre-warmed digestion solution containing 0.5 mg/ml collagenase type VIII (Sigma) and incubated at 37°C for 25 min with rotation. After incubation, digested tissues were filtered through a metal mesh. The flow-through cell suspension was spun down. The cell pellets were then re-suspended in 40% Percoll solution and overlaid above 80% Percoll solution. LPL were collected from the interface, washed once and re-suspended in the complete RPMI-1640 medium. The cells were used immediately for cell counting and staining.
Culture of ILC
For ILC culture, sorted ILC (CD45+Lin−CD3−CD90.2+) (~0.2 × 105/well) from SI-LP of Rag1−/− mice were cultured with IL-7 (100ng/mL) and IL-23 (10ng/mL) in the presence or absence of soluble LIGHT (500ng/mL) for 18 hours. Ifng transcript was determined by qPCR. For intracellular cytokines, sorted ILC (~0.2 × 105/well) were cultured with IL-7 (100ng/mL) in the presence or absence of soluble LIGHT for 18 hours, and then were stimulated for 4h with PI and BFA was added in the last 2h of incubation before analysis for intracellular cytokine.
Analysis of human HVEM expression
For analyzing the protein level of HVEM expression on human ILC, we identified ILC subsets from human PBMC via flow cytometry based on gating strategy from the reference (Simoni et al., 2017). For analyzing the transcript level of Hvem expression in human ILC, single cell RNA-Seq was performed on ILC1, ILC2, ILC3 and NK cells from human tonsils by Bjorkland et al. (Bjorklund et al., 2016). Gene expression was determined in the form of reads per kilobase per million mapped reads (RPKM). Previous reports stated that a minimum RPKM threshold of 10 (or less) adequately eliminates noise associated with sequencing (Glaus et al., 2012; Ramskold et al., 2009).
Flow cytometry
Flow cytometry analysis was performed on an LSRII instrument (BD Biosciences) and data analyzed using FlowJo software (Tree Star).
Histology analysis
Histopathology analysis of spleen and liver samples was performed on zinc formalin (Medical Chemical Corporation)-fixed tissue after hematoxylin and eosin (H&E) stain or Warthin-Starry silver stain.
Real-time PCR analysis
Total RNA was extracted from infected tissues using an RNeasy Kit (Qiagen), according to the manufacturer’s instructions. cDNA synthesis was performed with an iScript cDNA Synthesis Kit (Bio-rad). Quantitative real-time PCR reactions were performed with the SYBR Green I Master Kit and LightCycler 480 system (Roche). Ifng mRNA levels shown in figures were normalized to the housekeeping gene L32. The primer sequences were synthesized at the Integrated DNA Technologies.
• Quantification and Statistical Analysis
Statistics
Details concerning the statistical analysis methods are provided in each figure legend. Briefly, all data were analyzed using GraphPad Prism 7 software and were shown as mean and the standard error of the mean (SEM). Statistical significance was determined by Log-rank test for survival curves, 2 way ANOVA with Bonferroni’s multiple hypothesis correction for changes in body weight, or Mann-Whitney test for bacterial burdens and data on cytokines and cell numbers. Statistical significance is indicated by*, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. A p value < 0.05 was considered statistically significant. The exact value of n, representing the number of mice in the experiments depicted, is indicated in the figure legends.
Supplementary Material
Highlights.
-
–
ILC3 are required for early host defense during Y. enterocolitica infection.
-
–
IFNγ from CCR6−ILC3 is essential for protection of mice from Yersinia.
-
–
HVEM expression by ILC3 is important for IFNγ production following infection.
-
–
LIGHT is the ligand for HVEM signaling in regulating ILC3-derived IFNγ production.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (P01 DK46763, R01 AI61516 and MIST U01 AI125955 to M.K.; MIST U01 AI125957 to H.C; S10RR027366), the Crohn’s and Colitis Foundation of America (CCFA-254582 to Shui J-W), the Uehrara Foundation (to D.T.), and National Research Foundation (NRF) of Korea (2013R1A1A2057931 to GY.S.; 2016R1A4A1010115 to PH.K.). We thank the staff of the Microscopy & Histology Core (Angela Denn), Flow Cytometry Core and the Department of Laboratory Animal Care (DLAC) at the LJI for excellent technical assistance, SDx Histopathology Inc., (Carlsbad, CA) help with identifying YE and histology analysis, Zheng Fu for help with statistical analysis, and Nicolas Thiault for illustrating our graphical abstract.
Footnotes
• Data and Software Availability
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahn YO, Weeres MA, Neulen ML, Choi J, Kang SH, Heo DS, Bergerson R, Blazar BR, Miller JS, and Verneris MR (2015). Human group3 innate lymphoid cells express DR3 and respond to TL1A with enhanced IL-22 production and IL-2- dependent proliferation. European journal of immunology. 45(8), 2335–2342. DOI: 10.1002/eji.201445213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature. 517(7534), 293–301. DOI: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- Autenrieth IB, Kempf V, Sprinz T, Preger S, and Schnell A (1996). Defense mechanisms in Peyer's patches and mesenteric lymph nodes against Yersinia enterocolitica involve integrins and cytokines. Infection and immunity. 64(4), 1357–1368. Published online 1996/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, and Hardt WD (2003). Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infection and immunity. 71(5), 2839–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund AK, Forkel M, Picelli S, Konya V, Theorell J, Friberg D, Sandberg R, and Mjosberg J (2016). The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nature immunology. 17(4), 451–460. Published online 2016/02/16 DOI: 10.1038/ni.3368. [DOI] [PubMed] [Google Scholar]
- Bottone EJ (1997). Yersinia enterocolitica: the charisma continues. Clinical microbiology reviews. 10(2), 257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, and Powrie F (2010). Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. 464(7293),1371–1375. Published online 2010/04/16 DOI: 10.1038/nature08949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, and Freeman GJ (2009). The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev. 229(1), 244–258. Published online 2009/05/12 DOI: 10.1111/j.1600-065X.2009.00783.x. [DOI] [PubMed] [Google Scholar]
- Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, Shah K, Barbosa-Morais NL, Harris N, and Veiga-Fernandes H (2017). Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature. 549(7671), 277–281. Published online 2017/09/05 DOI: 10.1038/nature23469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, Lennerz JK, Doherty JM, Mills JC, and Colonna M (2009). A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 457(7230), 722–725. DOI: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan W, Costantino N, Li R, Lee SC, Su Q, Melvin D, Court DL, and Liu P (2007). A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res. 35(8), e64 DOI: 10.1093/nar/gkm163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohavy O, Zhou J, Granger SW, Ware CF, and Targan SR (2004). LIGHT expression by mucosal T cells may regulate IFN-gamma expression in the intestine. Journal of immunology. 173(1), 251–258. Published online 2004/06/24. [DOI] [PubMed] [Google Scholar]
- Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, and Hymowitz SG (2005). Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. The Journal of biological chemistry. 280(47), 39553–39561. Published online 2005/09/20 DOI: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- Cover TL, and Aber RC (1989). Yersinia enterocolitica. The New England journal of medicine. 321(1), 16–24. DOI: 10.1056/NEJM198907063210104. [DOI] [PubMed] [Google Scholar]
- DePaolo RW, Kamdar K, Khakpour S, Sugiura Y, Wang W, and Jabri B (2012). A specific role for TLR1 in protective T(H)17 immunity during mucosal infection. The Journal of experimental medicine. 209(8), 1437–1444. DOI: 10.1084/jem.20112339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G, and Littman DR (2004). Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science. 305(5681), 248–251. Published online 2004/07/13 DOI: 10.1126/science.1096472. [DOI] [PubMed] [Google Scholar]
- Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, and Littman DR (2004). An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nature immunology. 5(1), 64–73. DOI: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- Echeverry A, Saijo S, Schesser K, and Adkins B (2010). Yersinia enterocolitica promotes robust mucosal inflammatory T-cell immunity in murine neonates. Infection and immunity. 78(8), 3595–3608. DOI: 10.1128/IAI.01272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, Sun Y, and Fu YX (2006). NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. 107(4), 1342–1351. Published online 2005/10/15 DOI: 10.1182/blood-2005-08-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaus P, Honkela A, and Rattray M (2012). Identifying differentially expressed transcripts from RNA-seq data with biological variation. Bioinformatics. 28(13), 1721–1728. Published online 2012/05/09 DOI: 10.1093/bioinformatics/bts260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Obata T, Kunisawa J, Sato S, Ivanov II, Lamichhane A, Takeyama N, Kamioka M, Sakamoto M, Matsuki T, et al. (2014). Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. 345(6202), 1254009 DOI: 10.1126/science.1254009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronke K, Kofoed-Nielsen M, and Diefenbach A (2017). Isolation and Flow Cytometry Analysis of Innate Lymphoid Cells from the Intestinal Lamina Propria. Methods Mol Biol. 1559, 255–265. Published online 2017/01/08 DOI: 10.1007/978-1-4939-6786-5_17. [DOI] [PubMed] [Google Scholar]
- Guo X, Muite K, Wroblewska J, and Fu YX (2016). Purification and Adoptive Transfer of Group 3 Gut Innate Lymphoid Cells. Methods Mol Biol. 1422, 189–196. Published online 2016/06/02 DOI: 10.1007/978-1-4939-3603-8_18. [DOI] [PubMed] [Google Scholar]
- Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, Levy M, Salame TM, Weiner A, David E, et al. (2016). The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell. 166(5), 1231-1246 e1213. Published online 2016/08/23 DOI: 10.1016/j.cell.2016.07.043. [DOI] [PubMed] [Google Scholar]
- Herro R, Shui JW, Zahner S, Sidler D, Kawakami Y, Kawakami T, Tamada K, Kronenberg M, and Croft M (2018). LIGHT-HVEM signaling in keratinocytes controls development of dermatitis. The Journal of experimental medicine. 215(2), 415–422. Published online 2018/01/18 DOI: 10.1084/jem.20170536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibiza S, Garcia-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, Misic AM, Bartow-McKenney C, Larson DM, Pavan WJ, et al. (2016). Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature. 535(7612), 440–443. Published online 2016/07/15 DOI: 10.1038/nature18644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Toellner KM, White A, McConnell FM, Gaspal FM, Parnell SM, Jenkinson E, Anderson G, and Lane PJ. (2006). Neonatal and adult CD4+ CD3-cells share similar gene expression profile, and neonatal cells up-regulate OX40 ligand in response to TL1A (TNFSF15). Journal of immunology. 177(5), 3074–3081. [DOI] [PubMed] [Google Scholar]
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d'Hargues Y, Goppert N, Croxford AL, Waisman A, Tanriver Y, et al. (2013). A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. 494(7436), 261–265. DOI: 10.1038/nature11813. [DOI] [PubMed] [Google Scholar]
- Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, Monticelli LA, Moriyama S, Putzel GG, Rakhilin N, et al. (2017). The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature. 549(7671), 282–286. Published online 2017/09/05 DOI: 10.1038/nature23676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luci C, Reynders A, Ivanov II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, et al. (2009). Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nature immunology. 10(1), 75–82. DOI: 10.1038/ni.1681. [DOI] [PubMed] [Google Scholar]
- Marsters SA, Ayres TM, Skubatch M, Gray CL, Rothe M, and Ashkenazi A (1997). Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-kappaB and AP-1. The Journal of biological chemistry. 272(22), 14029–14032. [DOI] [PubMed] [Google Scholar]
- Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, Sciume G, Richard AC, Hayes ET, Gomez-Rodriguez J, et al. (2014). The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal immunology. 7(4), 958–968. DOI: 10.1038/mi.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, and Cheroutre H (2007). Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 317(5835), 256–260. DOI: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- Murphy TL, and Murphy KM (2010). Slow down and survive: Enigmatic immunoregulation by BTLA and HVEM. Annual review of immunology. 28, 389–411. DOI: 10.1146/annurev-immunol-030409-101202. [DOI] [PubMed] [Google Scholar]
- Narni-Mancinelli E, Chaix J, Fenis A, Kerdiles YM, Yessaad N, Reynders A, Gregoire C, Luche H, Ugolini S, Tomasello E, et al. (2011). Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proceedings of the National Academy of Sciences of the United States of America. 108(45), 18324–18329. Published online 2011/10/25 DOI: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, West N, Tung S, Seddon BP, Uhlig HH, et al. (2016). ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. 5, e10066 Published online 2016/01/19 DOI: 10.7554/eLife.10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramskold D, Wang ET, Burge CB, and Sandberg R (2009). An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol. 5(12), e1000598 Published online 2009/12/17 DOI: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadighi Akha AA, McDermott AJ, Theriot CM, Carlson PE Jr., Frank CR, McDonald RA, Falkowski NR, Bergin IL, Young VB, and Huffnagle GB (2015). Interleukin-22 and CD160 play additive roles in the host mucosal response to Clostridium difficile infection in mice. Immunology. 144(4), 587–597. DOI: 10.1111/imm.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samten B, Townsend JC, Weis SE, Bhoumik A, Klucar P, Shams H, and Barnes PF (2008). CREB, ATF, and AP-1 transcription factors regulate IFN-gamma secretion by human T cells in response to mycobacterial antigen. Journal of immunology. 181(3), 2056–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanos SL, Bui VL, Mortha A, Oberle K, Heners C, Johner C, and Diefenbach A (2009). RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nature immunology. 10(1), 83–91. DOI: 10.1038/ni.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh-Takayama N, Vosshenrich CA, Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam K, Cerf-Bensussan N, Mandelboim O, et al. (2008). Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity. 29(6), 958–970. DOI: 10.1016/j.immuni.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Shui JW, and Kronenberg M (2013). HVEM: An unusual TNF receptor family member important for mucosal innate immune responses to microbes. Gut microbes. 4(2), 146–151. DOI: 10.4161/gmic.23443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shui JW, Larange A, Kim G, Vela JL, Zahner S, Cheroutre H, and Kronenberg M (2012). HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature. 488(7410), 222–225. DOI: 10.1038/nature11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Dorman L, Viggiano V, Cippitelli M, Ghosh P, Rice N, and Young HA (1997). Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. The Journal of biological chemistry. 272(48), 30412–30420. [DOI] [PubMed] [Google Scholar]
- Simoni Y, Fehlings M, Kloverpris HN, McGovern N, Koo SL, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang CL, et al. (2017). Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity. 46(1), 148–161. Published online 2016/12/18 DOI: 10.1016/j.immuni.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, Mack M, Cella M, and Colonna M (2015). Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. The Journal of experimental medicine. 212(11), 1869–1882. DOI: 10.1084/jem.20151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, and Artis D (2011). CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 34(1), 122–134. DOI: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MW, Huang Y, Wang-Zhu Y, Ware CF, Cheroutre H, and Kronenberg M (2013). BTLA interaction with HVEM expressed on CD8(+) T cells promotes survival and memory generation in response to a bacterial infection. PloS one. 8(10), e77992 DOI: 10.1371/journal.pone.0077992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MW, Turovskaya O, Shaikh RB, Kim G, McCole DF, Pfeffer K, Murphy KM, Ware CF, and Kronenberg M (2008). A crucial role for HVEM and BTLA in preventing intestinal inflammation. The Journal of experimental medicine. 205(6), 1463–1476. DOI: 10.1084/jem.20071160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamada K, Shimozaki K, Chapoval AI, Zhai Y, Su J, Chen SF, Hsieh SL, Nagata S, Ni J, and Chen L (2000). LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. Journal of immunology. 164(8), 4105–4110. Published online 2001/02/07. [DOI] [PubMed] [Google Scholar]
- Trulzsch K, Oellerich MF, and Heesemann J (2007). Invasion and dissemination of Yersinia enterocolitica in the mouse infection model. Advances in experimental medicine and biology. 603, 279–285. DOI: 10.1007/978-0-387-72124-8_25. [DOI] [PubMed] [Google Scholar]
- Trulzsch K, Sporleder T, Igwe EI, Russmann H, and Heesemann J (2004). Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infection and immunity. 72(9), 5227–5234. DOI: 10.1128/IAI.72.9.5227-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu TC, Brown NK, Kim TJ, Wroblewska J, Yang X, Guo X, Lee SH, Kumar V, Lee KM, and Fu YX (2015). CD160 is essential for NK-mediated IFN-gamma production. The Journal of experimental medicine. 212(3), 415–429. Published online 2015/02/26 DOI: 10.1084/jem.20131601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, et al. (2010). Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 33(5), 736–751. Published online 2010/11/26 DOI: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walisser JA, Glover E, Pande K, Liss AL, and Bradfield CA (2005). Aryl hydrocarbon receptor-dependent liver development and hepatotoxicity are mediated by different cell types. Proceedings of the National Academy of Sciences of the United States of America. 102(49), 17858–17863. Published online 2005/11/23 DOI: 10.1073/pnas.0504757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, et al. (2017). The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 549(7672), 351–356. Published online 2017/09/14 DOI: 10.1038/nature24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, Yan D, Xu M, Lee WP, and Grogan JL (2014). TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal immunology. 7(3), 730–740. DOI: 10.1038/mi.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. (2008). Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine. 14(3), 282–289. DOI: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.