

Abstract
Objective
To characterize the etiologies and clinical features at diagnosis of patients with hemophagocytic lymphohistiocytosis (HLH) and correlate these baseline features with survival using an etiopathogenically guided multivariable model.
Patients and Methods
The Spanish Group of Autoimmune Diseases HLH Study Group, formed in 2013, is aimed at collecting adult patients with HLH diagnosed in internal medicine departments between January 3, 2013, and October 28, 2017.
Results
The cohort consisted of 151 patients (91 men; mean age, 51.4 years). After a mean follow-up of 17 months (range, 1-142 months), 80 patients died. Time-to-event analyses for death identified a worse survival curve for patients with neoplasia (P<.001), mixed microbiological infections (P=.02), and more than 1 infection (P=.01) and glucocorticoid monotherapy (P=.02). According to univariate analyses, platelets of less than 100,000/mm3 (hazard ratio [HR], 3.39; 95% CI, 1.37-8.40), leukopenia (HR, 1.81; 95% CI, 1.01-3.23), severe hyponatremia (HR, 1.61; 95% CI, 1.02-2.54), disseminated intravascular coagulation (HR, 1.87; 95% CI, 1.05-3.34), bacterial infection (HR, 1.99; 95% CI, 1.09-3.63), mixed microbiological infections (HR, 3.42; 95% CI, 1.38-8.46), and 2 or more infectious triggers (HR, 2.95; 95% CI, 1.43-6.08) were significantly associated with death. In contrast, peripheral adenopathies (HR, 0.63; 95% CI, 0.40-0.98) and the immunosuppressive drug/intravenous immunoglobulin/biological therapies (HR, 0.44; 95% CI, 0.20-0.96) were protective against all-cause mortality. Multivariable Cox proportional hazards regression analysis identified 2 or more infectious triggers (HR, 3.14; 95% CI, 1.28-7.68) as the only variable independently associated with death.
Conclusion
The mortality rate of adult patients diagnosed with HLH exceeds 50%. Infection with more than 1 microbiological agent was the only independent variable associated with mortality irrespective of the underlying disease, epidemiological profile, clinical presentation, and therapeutic management.
Abbreviations and Acronyms: GC, glucocorticoid; HLH, hemophagocytic lymphohistiocytosis; ICU, intensive care unit; ID, immunosuppressive drug; IVIG, intravenous immunoglobulin; NK, natural killer
Hemophagocytic lymphohistiocytosis (HLH, also called hemophagocytic syndrome) is an immune-mediated life-threatening disease caused by impaired natural killer (NK) and cytotoxic T-cell function.1 It was first described in 1939 by pediatricians, and therefore the disease has been characterized overwhelmingly in children, although the number of studies in adults is increasing. Etiopathogenically, genetic defects are linked to the development of HLH in children, whereas adults have a more complex scenario, with 2 main groups of etiological factors: underlying diseases and conditions that increase the risk of developing HLH, and external factors that initiate the pathogenic hyperinflammatory process (overwhelmingly infections).2 The heterogeneous pathogenic scenario in adults, in which different predisposing diseases and conditions are often mixed with different triggers, together with the high mortality rate, makes HLH one of the most complex and life-threatening clinical diseases.
Prognostic studies in large cohorts of adult patients with HLH are limited. Some studies have focused on patients with a specific underlying disease or trigger, including HLH-related lymphoma,3 Still disease,4 lupus,5 human immunodeficiency virus infection,6 and Epstein-Barr virus infection.7 However, large studies include etiologically unselected patients, the largest of which are recently reported French studies including 162 patients,8, 9 and a Chinese study including 205 patients.10 In these studies, mortality rates varied widely, and a large list of prognostic factors was identified. Several clinical factors contribute to this heterogeneity, including significant differences in the frequency of underlying diseases, the rate of patients recruited from intensive care units (ICUs), the definition of the main outcome studied (death), and the statistical approaches used. These factors make the identification of independent prognostic factors of survival that could be extrapolated and therefore generalized to other HLH adult populations difficult.
The aim of this study was to characterize the etiologies and clinical features at diagnosis in a large cohort of Spanish adult patients with HLH and correlate these baseline features with survival using an etiopathogenically guided multivariable statistical model.
Methods
Patients
The HLH Study Group of the Spanish Group of Autoimmune Diseases (Grupo de Enfermedades Autoinmunes Sistémicas) was formed in 2013 with the aim of collecting a large series of Spanish adult patients with HLH diagnosed in internal medicine departments with substantial experience in the management of patients with systemic diseases. Between January 3, 2013, and October 28, 2017, 151 consecutive patients who fulfilled at least 5 of the 8 criteria proposed by the Histiocytosis Society in 200411 were included. The study protocol was approved by the Clinical Research Ethics Committee of the Hospital Clinic of Barcelona (HCB/2016/0183) and complied with the ethical standards of the Declaration of Helsinki. The institutional review board waived the need for informed consent because of the retrospective design and the high rate of mortality.
Definition of Variables
The date of HLH diagnosis was defined as confirmation of fulfillment of HLH criteria by the attending physician. For patients with recurrent hemophagocytic syndrome, only the first episode was considered. The end of follow-up was defined according to the last recorded Spanish National Healthcare System visit, which was the principal source of information on the health status. The primary outcome was all-cause mortality.
Variables assessed as prognostic factors for survival were collected by retrospective review of individual medical charts and classified into 4 groups:
-
1.
Epidemiological features. Age at diagnosis, sex, country of birth, active immunosuppression (defined as the use of glucocorticoids [GCs], immunosuppressive drugs [IDs], and/or biological therapies ≥1 month before HLH diagnosis), and underlying diseases/conditions present at HLH diagnosis, classified as neoplasia, autoimmune/rheumatic diseases, and others (chronic viral infections, solid organ transplantation).
-
2.
HLH features. Clinical features and organ involvements directly related to HLH were defined according to standard definitions.1 Laboratory values were collected as the maximum or minimum abnormal value measured during the HLH diagnostic process. Hemophagocytosis was defined as histological evidence of activated macrophages engulfing erythrocytes, platelets, and/or nucleated cells or their precursors, in bone marrow smears and/or biopsy of bone marrow, liver, spleen, or lymph node.8 The HScore12 was calculated only in those patients in whom all the values for the items included in the score were available.
-
3.
Infectious trigger. Clinical/microbiological evidence of active bacterial, viral, parasitic, and/or fungal infection identified during the HLH diagnostic process, confirmed by standard diagnostic procedures used in the standard of care practice of internal medicine. As a key differentiating aspect from previous studies, we classified infectious diseases as underlying chronic infections (mainly chronic human immunodeficiency virus and hepatitis C virus infections) and acute infections (diagnosed during the HLH diagnostic process as playing an acute role in triggering the hyperinflammatory response).
-
4.
Therapeutic interventions. The HLH-related therapeutic regimens were classified as GC monotherapy, GC combined with other drugs (IDs, intravenous immunoglobulins [IVIGs], and/or biologics), and chemotherapy (including etoposide-based and other regimens).
Statistical Analyses
Descriptive data are presented as mean and SD or median and interquartile range for continuous outcomes and number and percentage (%) for categorical outcomes. Time-to-event analyses for death are presented as Kaplan-Meier curves. An etiopathogenically guided statistical approach was designed on the basis of the most-commonly accepted hypothesis of the etiopathogenesis of HLH in adults1: epidemiological features, underlying diseases, and active immunosuppression at HLH diagnosis were considered as HLH-independent variables, whereas HLH clinical features, infectious triggers, and therapeutic interventions were considered as HLH-dependent variables forming part of the disease presentation and management. Univariate Cox proportional hazards regression analyses were performed to study the crude measure of association between all variables and death. Statistically significant HLH-dependent variables were entered into a multivariable Cox proportional hazards regression analysis adjusted for HLH-independent variables, which permitted identification of variables independently associated with all-cause mortality. The hazard ratios (HRs) and their 95% CIs obtained in the Cox regression analysis were calculated. All significance tests were 2-tailed, and values of P<.05 were considered significant. All analyses were conducted using the R version 3.0.3 for Windows statistical software package.
Results
Baseline Characterization
Baseline characteristics are summarized in Table 1. The cohort consisted of 151 patients, including 91 (60%) males (male:female ratio, 1.5:1), with a mean age at diagnosis of 51.4±21.5 years (range, 14-92 years); 26 (17%) patients were born outside Spain. The main group of underlying diseases consisted of autoimmune/rheumatic diseases in 53 (35%) patients, neoplasia in 48 (32%), chronic viral infections in 32 (21%), and solid organ transplantation in 8 (5%); 29 (19%) patients had 1 or more underlying disease, whereas 42 (28%) had no identified underlying disease/condition. Active immunosuppressive treatment was reported in 42 (28%) patients at HLH diagnosis. The main clinical HLH features included fever in 140 (93%) patients, splenomegaly in 96 (64%), hepatomegaly in 76 (50%), peripheral adenopathies in 74 (49%), general symptomatology in 71 (47%), and pulmonary involvement in 70 (46%) patients. Other clinical manifestations are presented in Table 1. The most common hematological abnormalities were anemia in 140 of 149 (94%) patients, thrombocytopenia in 136 of 150 (91%), and leukopenia in 115 of 150 (77%). The most frequent laboratory abnormalities included raised ferritin levels in 135 of 140 (96%), raised triglycerides in 128 of 140 (91%), raised AST transaminase in 117 of 147 (80%), raised lactate dehydrogenase in 115 of 146 (79%), and hyponatremia in 92 of 148 (62%). Soluble IL-2 receptor (CD25) levels were raised in 20 of 25 (80%), and NK-cell activity was absent or lower in 15 of 18 (83%). Hemophagocytosis was confirmed in 138 of 148 (93%) cases (in 3 cases there was no histopathological study, due to sudden death, or autopsy results).
Table 1.
| Feature | N | Patients | % |
|---|---|---|---|
| Epidemiology | |||
| Age at diagnosis (y), mean ± SD | 150 | 51.4±21.5 | |
| Sex: male | 151 | 91 | 60.3 |
| Born outside Spain | 151 | 26 | 17.2 |
| Underlying diseasesc | |||
| Autoimmune/rheumatologic diseases | 151 | 53 | 35.1 |
| Systemic lupus erythematosus | 151 | 13 | 8.6 |
| Inflammatory bowel disease | 151 | 7 | 4.6 |
| Still disease | 151 | 7 | 4.6 |
| Rheumatoid arthritis | 151 | 6 | 4.0 |
| Inflammatory myopathies | 151 | 4 | 2.6 |
| Mixed connective tissue disease | 151 | 4 | 2.6 |
| Vasculitis | 151 | 3 | 2.0 |
| Other autoimmune diseases | 151 | 9 | 6.0 |
| Neoplasia | 151 | 48 | 31.8 |
| T/NK-cell lymphoma | 151 | 14 | 9.3 |
| B-cell lymphoma | 151 | 12 | 7.9 |
| Other hematological neoplasia | 151 | 11 | 7.3 |
| Solid neoplasia | 151 | 11 | 7.3 |
| Chronic viral infections | 151 | 32 | 21.2 |
| Transplantation | 151 | 8 | 5.3 |
| No underlying disease identified | 151 | 42 | 27.8 |
| Active immunosuppressive treatment | 151 | 42 | 27.8 |
| Clinical presentation | |||
| a. Symptoms/organs involved | |||
| Duration of symptoms (wk), mean ± SD | 151 | 3.7±4.5 | |
| Fever (>37.5°C) | 151 | 140 | 92.7 |
| Splenomegaly | 151 | 96 | 63.6 |
| General symptoms | 151 | 71 | 47.0 |
| Hepatomegaly | 151 | 76 | 50.3 |
| Peripheral adenopathies | 151 | 74 | 49.0 |
| Pulmonary involvement | 151 | 70 | 46.4 |
| Renal involvement | 151 | 51 | 33.8 |
| Gastrointestinal involvement | 151 | 46 | 30.5 |
| Skin lesions | 151 | 42 | 27.8 |
| CNS involvement | 151 | 35 | 23.2 |
| Arthralgias/myalgias | 151 | 29 | 19.2 |
| b. Hematological & coagulation | |||
| Anemia | |||
| Hemoglobin <12 g/dL | 149 | 140 | 94.0 |
| Hemoglobin <9 g/dL | 149 | 104 | 69.8 |
| Hemoglobin <7 g/dL | 149 | 34 | 22.8 |
| Thrombocytopenia | |||
| Platelets <150,000 cells per mm3 | 150 | 136 | 90.7 |
| Platelets <100,000 cells per mm3 | 150 | 128 | 85.3 |
| Platelets <10,000 cells per mm3 | 150 | 21 | 14.0 |
| Leukopenia (leukocytes <4000 cells per mm3) | 150 | 115 | 76.7 |
| Neutropenia | |||
| Neutrophils <1500 cells per mm3 | 148 | 89 | 60.1 |
| Neutrophils <1000 cells per mm3 | 148 | 69 | 46.6 |
| Neutrophils <500 cells per mm3 | 148 | 34 | 23.0 |
| Hypofibrinogenemia (fibrinogen <1.5 g/L) | 146 | 30 | 20.5 |
| Disseminated intravascular coagulation | 151 | 20 | 13.2 |
| c. Biochemical | |||
| Hyperferritinemia | |||
| Ferritin >500 ng/mL | 140 | 135 | 96.4 |
| Ferritin >1000 ng/mL | 140 | 127 | 90.7 |
| Ferritin >10,000 ng/mL | 140 | 48 | 34.3 |
| Hypertriglyceridemia | |||
| Triglycerides >150 mg/dL | 140 | 128 | 91.4 |
| Triglycerides >265 mg/dL | 140 | 93 | 66.4 |
| Hyponatremia | |||
| <135 mmol/L | 148 | 92 | 62.2 |
| <130 mmol/L | 148 | 42 | 28.4 |
| Raised ALT transaminase | |||
| ALT transaminase >40 IU/L | 148 | 114 | 77.0 |
| ALT transaminase >100 IU/L | 148 | 78 | 52.7 |
| Raised AST transaminase | |||
| AST transaminase >40 IU/L | 147 | 117 | 79.6 |
| AST transaminase >100 IU/L | 147 | 84 | 57.1 |
| Raised lactate dehydrogenase | |||
| Lactate dehydrogenase >500 IU/L | 146 | 115 | 78.8 |
| Lactate dehydrogenase >1000 IU/L | 146 | 74 | 50.7 |
| d. Hemophagocytosis | |||
| Histopathological confirmation | 148 | 138 | 93.2 |
| e. HScore,d median (IQR) | 126 | 230 (200-267) | |
ALT = alanine aminotransferase; AST = aspartate aminotransferas; CNS = central nervous system; IQR = interquartile range; NK = natural killer.
Unless otherwise specified, the values are number (%).
Patients may have more than 1 underlying disease.
Only calculated in patients with all ítems measured/carried out (n=126).
Infectious triggers (see Supplemental Table 1, available online at http://mcpiqojournal.org/) were diagnosed in 95 (63%) patients, including acute viral infections in 47 (49%) patients, bacterial infections in 33 (35%), parasitic infections in 9 (9.5%) (including 8 cases of Leishmaniasis, which is endemic in Spain), and fungal infections in 13 (14%); 14 patients presented infection by 2 different microorganisms; specific anti-infective agents were administered according to standard therapeutic recommendations followed in internal medicine departments.
Specific therapies for HLH included glucocorticoids in 129 (85%) patients, etoposide in 28 (19%), cyclosporine A in 28 (19%), IVIGs in 22 (15%), and rituximab in 12 (8%); 19 (13%) patients did not receive IDs. The list of drugs and the therapeutic regimens used are summarized in Supplemental Table 2 (available online at http://mcpiqojournal.org/). Admission in ICU was required in 59 (39%) patients.
Survival Analysis
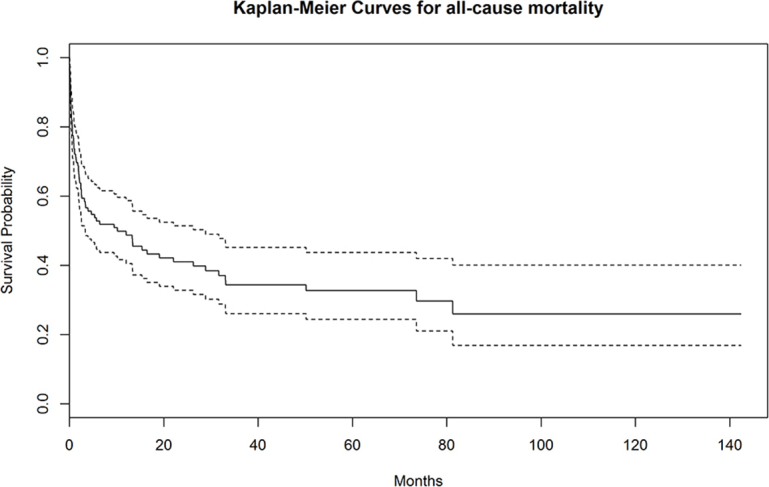
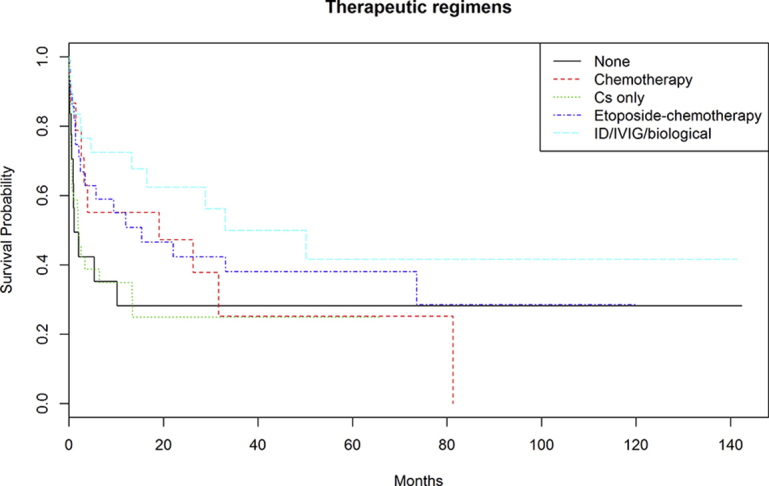
After a mean follow-up of 17 months (range, 1-142 months), 80 patients died. Supplemental Figure 1 (available online at http://mcpiqojournal.org/) shows the survival curve of the entire cohort; survival at 1, 2, 3, 6, and 12 months was 73.6%, 66.3%, 59.4%, 52.8%, and 48.8%, respectively. Time-to-event analyses for death (Kaplan-Meier curves) were made according to the main types of underlying diseases (autoimmune/rheumatic, neoplasia, others, multiple diseases, and none), microbiological classification of infectious triggers (bacteria, viruses, fungi/parasites, mixed infections, and none), number of infectious triggers (0, 1, >1), and HLH-specific therapeutic regimens (GC monotherapy, GC + other drugs, etoposide-based chemotherapy, other chemotherapies, and none). Figure 1 shows that the best survival curve was in patients with underlying autoimmune/rheumatic diseases and the worst was in those with neoplasia (log-rank P<.001). Figure 2 shows that the worst survival curves were for patients with mixed microbiological infections and those with bacterial infections (log-rank P=.02), whereas Figure 3 shows that the worst survival curve was in patients with more than 1 infectious trigger (log-rank P=.01). Supplemental Figure 2 (available online at http://mcpiqojournal.org/) shows that the best survival curves were in patients treated with IDs/IVIGs/biologics and etoposide-based therapies and the worst was in those treated with GC monotherapy (log-rank P=.02).
Figure 1.

Survival curves according to the main types of underlying diseases (autoimmune/rheumatic, neoplasia, others, multiple diseases, and none).
Figure 2.

Survival curves according to the microbiological classification of infectious triggers (bacteria, viruses, fungi/parasites, mixed infections, and none).
Figure 3.

Survival curves according to the number of infectious triggers (0, 1, >1).
Table 2 summarizes the HRs obtained in the univariate Cox proportional hazards regression analyses. The HLH-independent variables associated with death were age (HR, 1.03; 95% CI, 1.02-1.04), male sex (HR, 1.61; 95% CI, 1.01-2.56), and underlying neoplasia (HR, 1.84; 95% CI, 1.01-3.37). With respect to the HLH-dependent variables, platelets less than 100,000/mm3 (HR, 3.39; 95% CI, 1.37-8.40), leukopenia (HR, 1.81; 95% CI, 1.01-3.23), severe hyponatremia (HR, 1.61; 95% CI, 1.02-2.54), disseminated intravascular coagulation (HR, 1.87; 95% CI, 1.05-3.34), bacterial infection (HR, 1.99; 95% CI, 1.09-3.63), mixed microbiological infections (HR, 3.42; 95% CI, 1.38-8.46), and 2 or more infectious triggers (HR, 2.95; 95% CI, 1.43-6.08) were associated with death. In contrast, underlying autoimmune/rheumatic diseases (HR, 0.34; 95% CI, 0.16-0.74), peripheral adenopathies (HR, 0.63; 95% CI, 0.40-0.98), and ID/IVIG/biological therapies (HR, 0.44; 95% CI, 0.20-0.96) were protective against all-cause mortality.
Table 2.
| Variable | All-cause mortality (n=80) |
|
|---|---|---|
| Univariate HR (95% CIs) | P | |
| Epidemiological variables | ||
| Age at diagnosis (y) | 1.03 (1.02-1.04) | <.001 |
| Sex: male | 1.61 (1.01-2.56) | .04 |
| Underlying disease grouped | ||
| None | Reference | |
| Neoplasia | 1.84 (1.01-3.37) | .05 |
| Autoimmune/rheumatic disease | 0.34 (0.16-0.74) | .006 |
| Others | 0.98 (0.45-2.17) | .97 |
| Multiple (more than 1) | 1.24 (0.66-2.35) | .51 |
| Active ID therapies | 0.76 (0.45-1.28) | .30 |
| Clinical variables | ||
| Fever (>37.5°C) | 0.93 (0.43-2.03) | .86 |
| Splenomegaly | 0.88 (0.56-1.39) | .59 |
| Hepatomegaly | 1.24 (0.80-1.92) | .34 |
| Peripheral adenopathies | 0.63 (0.40-0.98) | .04 |
| Pulmonary involvement | 1.40 (0.91-2.18) | .13 |
| CNS involvement | 1.11 (0.64-1.91) | .71 |
| Skin lesions | 0.82 (0.50-1.37) | .45 |
| Gastrointestinal involvement | 1.03 (0.65-1.65) | .89 |
| Renal involvement | 1.33 (0.84-2.10) | .23 |
| Hemoglobin <9 g/dL | 1.71 (0.96-3.05) | .07 |
| Platelets <100,000 cells per mm3 | 3.39 (1.37-8.40) | .008 |
| WBC <4000 cells per mm3 | 1.81 (1.01-3.23) | .05 |
| Neutrophils <1000 cells per mm3 | 1.48 (0.90-2.42) | .12 |
| Triglycerides >150 mg/dL | 1.24 (0.54-2.86) | .61 |
| Natremia <130 mmol/L | 1.61 (1.02-2.54) | .04 |
| AST transaminase >100 IU/L | 0.89 (0.57-1.40) | .62 |
| ALT transaminase >100 IU/L | 0.81 (0.52-1.25) | .34 |
| Lactate dehydrogenase >450 IU/L | 0.84 (0.54-1.31) | .45 |
| Ferritin >500 ng/mL | 3.05 (0.42-21.97) | .27 |
| Fibrinogen <1.5 g/L | 1.29 (0.76-2.22) | .35 |
| Disseminated intravascular coagulation | 1.87 (1.05-3.34) | .03 |
| Histopathological hemophagocytosis | 1.71 (0.63-4.68) | .30 |
| HScore (≥169) | 1.29 (0.59-2.84) | .53 |
| Infectious trigger | ||
| Presence of infectious trigger | 1.53 (0.96-2.45) | .07 |
| Microbiological classification | ||
| No infection | Reference | |
| Bacteria | 1.99 (1.09-3.63) | .03 |
| Virus | 1.36 (0.79-2.34) | .28 |
| Fungi/parasites | 0.78 (0.27-2.24) | .65 |
| Mixed | 3.42 (1.38-8.46) | .008 |
| Number of infectious triggers | ||
| 0 | Reference | |
| 1 | 1.38 (0.85-2.24) | .20 |
| ≥2 | 2.95 (1.43-6.08) | .003 |
| Therapeutic interventions | ||
| No immunosuppressive drugs | Reference | |
| Glucocorticoid monotherapy | 1.21 (0.60-2.44) | .60 |
| ID/IVIG/biological therapies | 0.44 (0.20-0.96) | .04 |
| Chemotherapy | 0.69 (0.34-1.39) | .30 |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; CNS = central nervous system; HR = hazard ratio; ID = immunosuppressive drug; IVIG = intravenous immunoglobulin; WBC = white blood cell.
Values are represented as HRs and 95% CIs.
These variables were entered into a multivariable Cox proportional hazards regression analysis that identified 2 or more infectious triggers (HR, 3.14; 95% CI, 1.28-7.68) as the only variable independently associated with death (Table 3).
Table 3.
| Variable | All-cause mortality (n=80) |
|
|---|---|---|
| Multivariable HR (95% CIs)c | P | |
| Peripheral adenopathies | 0.76 (0.45-1.28) | .30 |
| Platelets <100,000 cells per mm3 | 1.92 (0.72-5.15) | .20 |
| WBC <4000 cells per mm3 | 1.54 (0.83-2.85) | .17 |
| Natremia <130 mmol/L | 1.41 (0.84-2.39) | .20 |
| Disseminated intravascular coagulation | 1.56 (0.81-3.00) | .19 |
| Number of infectious triggers | ||
| 0 | ||
| 1 | 0.95 (0.56-1.61) | .85 |
| ≥2 | 3.14 (1.28-7.68) | .01 |
| Microbiological classification | NCd | |
| No infection | ||
| Bacteria | ||
| Virus | ||
| Fungi/parasites | ||
| Mixed | ||
| Therapeutic interventions | ||
| No immunosuppressive drugs | Reference | |
| Glucocorticoid monotherapy | 1.37 (0.65-2.92) | .41 |
| ID/IVIG/biological therapies | 0.78 (0.33-1.85) | .57 |
| Chemotherapy | 0.71 (0.32-1.54) | .38 |
ID = immunosuppressive drug; IVIG = intravenous immunoglobulin; NC = not calculated; HR = hazard ratio; WBC = white blood cell.
Values are represented as HRs and 95% CIs.
Multivariable Cox proportional hazards regression analysis adjusted for age at diagnosis, sex, and underlying disease group.
To avoid multicollinearity with the number of triggers, the trigger group was not included in the model.
Discussion
One of the most critical clinical conditions in adults is HLH, with an all-cause mortality rate reported in large series ranging between 42%9 and 75%.13 Eight studies have analyzed prognostic factors in large cohorts of adults (>50 patients) in the last 10 years (see Supplemental Table 3, available online at http://mcpiqojournal.org/) and found that most factors identified preexist the HLH diagnosis (epidemiological features, underlying diseases). Epidemiological features were key prognostic markers, especially age,13, 14, 15, 16 but also male sex,13, 15 together with underlying diseases, with patients with underlying neoplasia9, 10, 14, 15, 17, 18 having a poor prognosis compared with other etiologies. With respect to HLH presentation, only splenomegaly13 was identified as a prognostic factor among HLH-related clinical features, while the number of prognostic laboratory markers was higher, including thrombocytopenia,10, 13, 14, 17 creatinine,15 prothrombin time,10, 16 ferritin,15 and fibrinogen.16 Other prognostic factors identified were closely related to ICU admission (sequential organ failure assessment [SOFA] score, shock, and requirement for life-sustaining therapies such as renal replacement).14, 16, 17 In total, these studies identified 13 different prognostic factors.
The wide list of prognostic factors identified until now for adult HLH may be due to several factors. First, the different frequencies of underlying diseases (which have per se a considerable influence on survival) included in each study; although all but 1 study15 identify neoplasia as the most frequent underlying disease, the frequency ranges widely from 30%14 to 77%17; in addition, it is unclear how the frequent overlap between different etiologies (especially between underlying diseases and infections) was defined and analyzed. Another source of heterogeneity is that some studies included only patients admitted to the ICU14, 16, 17 and different definitions of the primary outcome, including hospital mortality,16, 17 all-cause mortality,10, 13, 15, 18 and 1-month mortality.9, 14 The statistical approach was also heterogeneous, mainly including multivariable analysis using logistic regression models with different selection methods for variables at model entry, even though logistic regression is not recommended for the analysis of time-dependent outcomes.19 Few studies used more appropriate time-to-event analysis approaches, such as Kaplan-Meier9, 18 or Cox-regression10 models that allowed quantification of the measure of effect by means of HRs depending on the time of follow-up, whereas no study considered adjustment to control the confounding effects of overlapping or preexisting HLH variables.
Our results confirm, in the crude univariate analysis, the prognostic factors most frequently reported in previous studies, including age, male sex, underlying disease, thrombocytopenia, disseminated intravascular coagulation, infections, and therapeutic interventions. However, we also identified other, unreported factors, such as adenopathies, leukopenia, and severe hyponatremia. In the statistical adjustment, we used age and sex (usually used as variables of adjustment) but added underlying diseases. This pretest statistical design was based not only on the expected influence on patient survival of patient having a specific underlying disease, but also because some clinical and laboratory parameters identified in the crude analysis are closely linked to the clinical expression of the main underlying diseases (autoimmune diseases and neoplasia). According to the multivariable Cox proportional hazards regression analysis adjusted for HLH-independent variables, infection with more than 1 agent remained the only independent variable associated with survival in adult patients with HLH, an association that should be considered significant irrespective of the underlying disease, epidemiological profile, clinical presentation, and therapeutic management.
Therefore, we identified infection as the key independent factor for survival in adults with HLH. A few studies have focused on the role of acute infectious triggers in HLH survival, mainly in pediatric patients,20, 21 but also in adults treated with biological agents.22 However, some descriptive studies have reported higher mortality rates in patients presenting with bacterial infections13, 23, 24 or co-infections.21, 25 Our study reports, for the first time, that both the variety and number of infectious triggers implicated in HLH have a considerable influence on survival using time-to-event statistical models; the best survival curve was found for patients with parasitic/fungal infections, followed by those with viral infections, whereas the worst survival curves were for those with bacterial infections and multiple microbiological infections (of whom 71% also had bacterial infections in association with other microorganisms). Leishmaniasis, an endemic infection in Spain, was the most frequent parasitic infection related to HLH (8 cases, all born in Spain, of which 7 were reported from Madrid). These findings should encourage efforts to detect (and therefore treat) the infectious triggers of HLH (especially bacterial infections) as early as possible.
The time-to event analyses confirmed a worse prognosis of HLH in adult patients with underlying neoplasia,18 and the protective effect of having an autoimmune/rheumatic disease; descriptive studies have also suggested a better prognosis for HLH related to autoimmune diseases,13 with mortality rates of 13% in HLH-related autoimmune diseases,26 9.5% in HLH-related adult-onset Still disease,4 and 3% in HLH-related lupus.5 This compares with mortality rates of more than 80% in HLH-related lymphoma,13 and the figure may be even higher in patients with HLH-related NK/T-cell lymphoma.27 We also analyzed the role of therapeutic interventions in survival, and found better trends in patients treated with GCs plus other agents (IDs, IVIGs, or biologics) and those treated with etoposide. In contrast, patients treated with GC monotherapy and those who did not receive specific HLH therapy had the worst survival curves. Studies in children20, 28, 29 and adults5, 8, 30 have found similar results. These studies, together with our results, highlight the differentiated prognosis of HLH according to the underlying disease and the need for more-intensive immunosuppressive therapy than monotherapy with GCs. The survival rates reported in prognostic HLH studies including multiple etiologies should always be evaluated taking into account the frequency of each underlying disease in the corresponding cohort, and a lower survival rate should be expected in cohorts in which neoplasia predominates (clearly related to the high rate of high-grade associated hematological neoplasia) and a better rate in cohorts with a predominance of underlying autoimmune/rheumatic disease (as observed in our study).
Our results and those of other studies analyzing prognostic factors associated with survival in adult HLH should be evaluated with caution because of the retrospective design and the lack of an international consensus on the management and treatment of adult HLH. Because of the rarity of the disease and the often-life-threatening presentation, prospective studies or randomized controlled trials in patients with HLH are extremely difficult, and it may be anticipated that the level of evidence will remain limited to retrospective studies. An additional limitation of our study, as occurs in retrospective multicenter studies, is the lack of a central review of laboratory and histopathological data to ensure a homogeneous diagnostic approach. Therefore, we are now working on the development of a Spanish diagnostic and therapeutic protocol for adult patients with HLH, searching for a nationwide homogeneous approach to the disease. One additional future task could be the worldwide collection and analysis of larger real-life data sets involving the main specialties involved in the care of these patients (intensive care, internal medicine, rheumatology, pediatrics, and hematology) and specific etiologically driven substudies considering the variety and combinations of the underlying diseases and infectious triggers. The complexity of these patients, in whom different etiopathogenic scenarios coexist, together with a mortality rate of nearly 50%, poses a considerable clinical and therapeutic challenge.
Conclusion
This is the first study that characterizes the etiologies and clinical features at diagnosis of patients with HLH and correlates these baseline features with survival using an etiopathogenically guided multivariable model. The results suggest that the mortality rate of adult patients diagnosed with HLH exceeds 50%. There was worse survival for patients with neoplasia and mixed microbiological infections, with 1 or more infections being the only independent variable associated with mortality, irrespective of the underlying disease, epidemiological profile, clinical presentation, and therapeutic management.
Acknowledgement
We thank David Buss for technical assistance.
Footnotes
Potential Competing Interests: The authors report no competing interests.
Supplemental Online Material
Supplemental Figure 1.
Supplemental Figure 2.
Supplemental material can be found online at http://mcpiqojournal.org/. Supplemental material attached to journal articles has not been edited, and the authors take responsibility for the accuracy of all data.
References
- 1.Ramos-Casals M., Brito-Zerón P., López-Guillermo A., Khamashta M.A., Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503–1516. doi: 10.1016/S0140-6736(13)61048-X. Erratum in Lancet. 2014;383(9927):1464. [DOI] [PubMed] [Google Scholar]
- 2.Usmani G.N., Woda B.A., Newburger P.E. Advances in understanding the pathogenesis of HLH. Br J Haematol. 2013;161(5):609–622. doi: 10.1111/bjh.12293. [DOI] [PubMed] [Google Scholar]
- 3.Yu J.-T., Wang C.-Y., Yang Y., et al. Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol. 2013;92:1529–1536. doi: 10.1007/s00277-013-1784-3. [DOI] [PubMed] [Google Scholar]
- 4.Bae C.B., Jung J.Y., Kim H.A., Suh C.H. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features, predictive factors, and prognosis in 21 patients. Medicine (Baltimore) 2015;94(4):e451. doi: 10.1097/MD.0000000000000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lambotte O., Khellaf M., Harmouche H., et al. Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosus-associated hemophagocytic syndrome. Medicine (Baltimore) 2006;85(3):169–182. doi: 10.1097/01.md.0000224708.62510.d1. [DOI] [PubMed] [Google Scholar]
- 6.Fardet L., Lambotte O., Meynard J.L., et al. Reactive haemophagocytic syndrome in 58 HIV-1-infected patients: clinical features, underlying diseases and prognosis. AIDS. 2010;24(9):1299–1306. doi: 10.1097/QAD.0b013e328339e55b. [DOI] [PubMed] [Google Scholar]
- 7.Kogawa K., Sato H., Asano T., et al. Prognostic factors of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children: report of the Japan Histiocytosis Study Group. Pediatr Blood Cancer. 2014;61(7):1257–1262. doi: 10.1002/pbc.24980. [DOI] [PubMed] [Google Scholar]
- 8.Arca M., Fardet L., Galicier L., et al. Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol. 2015;168(1):63–68. doi: 10.1111/bjh.13102. [DOI] [PubMed] [Google Scholar]
- 9.Riviere S., Galicier L., Coppo P., et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127(11):1118–1125. doi: 10.1016/j.amjmed.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 10.Zhou M., Li L., Zhang Q., et al. Clinical features and outcomes in secondary adult hemophagocytic lymphohistiocytosis. QJM. 2018;111(1):23–31. doi: 10.1093/qjmed/hcx183. [DOI] [PubMed] [Google Scholar]
- 11.Henter J.I., Horne A., Arico M., et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 12.Fardet L., Galicier L., Lambotte O., et al. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. 2014;66(9):2613–2620. doi: 10.1002/art.38690. [DOI] [PubMed] [Google Scholar]
- 13.Li F., Yang Y., Jin F., et al. Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: a retrospective study of increasing awareness of a disease from a single-center in China. Orphanet J Rare Dis. 2015;10:20. doi: 10.1186/s13023-015-0224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barba T., Maucort-Boulch D., Iwaz J., et al. Hemophagocytic lymphohistiocytosis in intensive care unit: A 71-case strobe-compliant retrospective study. Medicine (Baltimore) 2015;94(51):e2318. doi: 10.1097/MD.0000000000002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Otrock Z.K., Eby C.S. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol. 2015;90(3):220–224. doi: 10.1002/ajh.23911. [DOI] [PubMed] [Google Scholar]
- 16.Valade S., Azoulay E., Galicier L., et al. Coagulation disorders and bleedings in critically ill patients with hemophagocytic lymphohistiocytosis. Medicine (Baltimore) 2015;94(40):e1692. doi: 10.1097/MD.0000000000001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buyse S., Teixeira L., Galicier L., et al. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med. 2010;36(10):1695–1702. doi: 10.1007/s00134-010-1936-z. [DOI] [PubMed] [Google Scholar]
- 18.Schram A.M., Comstock P., Campo M., et al. Haemophagocytic lymphohistiocytosis in adults: a multicentre case series over 7 years. Br J Haematol. 2016;172(3):412–419. doi: 10.1111/bjh.13837. [DOI] [PubMed] [Google Scholar]
- 19.Hosmer D.W., Lemeshow S. John Wiley; New York: 1999. Applied Survival Analysis: Regression Modeling of Time to Event Data. [Google Scholar]
- 20.Chen H.H., Kuo H.C., Wang L., et al. Childhood macrophage activation syndrome differs from infection-associated hemophagocytosis syndrome in etiology and outcome in Taiwan. J Microbiol Immunol Infect. 2007;40(3):265–271. [PubMed] [Google Scholar]
- 21.Qiang Q., Zhengde X., Shuang Y., Kunling S. Prevalence of coinfection in children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. 2012;34(2):e45–e48. doi: 10.1097/MPH.0b013e31822d4ea7. [DOI] [PubMed] [Google Scholar]
- 22.Brito-Zerón P., Bosch X., Pérez-de-Lis M., et al. BIOGEAS Study Group Infection is the major trigger of hemophagocytic syndrome in adult patients treated with biological therapies. Semin Arthritis Rheum. 2016;45(4):391–399. doi: 10.1016/j.semarthrit.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Nair V., Das S., Sharma A., et al. A clinicopathological analysis of 26 patients with infection-associated haemophagocytic lymphohistiocytosis and the importance of bone marrow phagocytosis for the early initiation of immunomodulatory treatment. Postgrad Med J. 2013;89(1050):185–192. doi: 10.1136/postgradmedj-2012-130955. [DOI] [PubMed] [Google Scholar]
- 24.Halacli B., Unver N., Halacli S.O., et al. Investigation of hemophagocytic lymphohistiocytosis in severe sepsis patients. J Crit Care. 2016;35:185–190. doi: 10.1016/j.jcrc.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 25.Lerolle N., Laanani M., Riviere S., et al. Diversity and combinations of infectious agents in 38 adults with an infection-triggered reactive haemophagocytic syndrome: a multicenter study. Clin Microbiol Infect. 2016;22(3):268.e1–268.e8. doi: 10.1016/j.cmi.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Kumakura S., Murakawa Y. Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheumatol. 2014;66(8):2297–2307. doi: 10.1002/art.38672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin Z., Wang Y., Wang J., et al. Multivariate analysis of prognosis for patients with natural killer/T cell lymphoma-associated hemophagocytic lymphohistiocytosis. Hematology. 2018;23(4):228–234. doi: 10.1080/10245332.2017.1385191. [DOI] [PubMed] [Google Scholar]
- 28.Bergsten E., Horne A., Arico M., et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long term results of the cooperative HLH-2004 study. Blood. 2017;130(25):2728–2738. doi: 10.1182/blood-2017-06-788349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin Y.K., Xie Z.D., Yang S., Lu G., Shen K.L. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis: a retrospective study of 78 pediatric cases in mainland of China. Chin Med J (Engl) 2010;123(11):1426–1430. [PubMed] [Google Scholar]
- 30.Kobayashi R., Tanaka J., Hashino S., et al. Etoposide-containing conditioning regimen reduces the occurrence of hemophagocytic lymphohistiocytosis after SCT. Bone Marrow Transplant. 2014;49(2):254–257. doi: 10.1038/bmt.2013.145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.