

Supplemental Digital Content is available in the text
Keywords: cleidocranial dysplasia, phenotype–genotype correlation, RUNX2, supernumerary teeth
Abstract
Rationale:
Supernumerary teeth are those that teeth in excess number than the normal count. It is usually associated with genetic syndromes when present in more numbers. Several causal genes, such as APC, NHS, TRPS1, EVC and RUNX2, have been identified. However, etiology of supernumerary teeth remains largely unclear.
Patient concerns:
A family with the clinical diagnosis of supernumerary teeth, short stature and craniofacial dysplasia was examined.
Diagnoses:
Molecular genetic analysis found that mutation occurred in the RUNX2 gene. On the basis of this finding and clinical manifestations, the final diagnosis of cleidocranial dysplasia was made.
Interventions:
Whole exome sequencing (WES) of DNA samples was performed to identify the disease-causing mutation, including the affected child and mother as well as the healthy father.
Outcomes:
A novel mutation of RUNX2 (c.473C>A; p.A158E) was identified in both patients, but not in normal family member and in-house database containing 3,000 Chinese Han individuals WES. This mutation was further confirmed by Sanger sequencing and predicted to be deleterious by several commonly used algorithms, including SIFT, PPT-2, MutationTaster and Proven. Furthermore, phenotype-genotype correlation analyses of all published 239 cases with different mutations in RUNX2 revealed significant association of supernumerary teeth and facial dysplasia with the Runt domain of the encoded protein.
Lessons:
This is the first WES study to identify genetic cause in Chinese patients with a novel RUNX2 mutation. Our findings expanded the mutation spectrum and clinical features of the disease and facilitated clinic diagnosis and genetic counseling.
1. Introduction
Supernumerary teeth are those that teeth in excess number than the normal count. The prevalence of supernumerary teeth is 1.5% to 3.5% in permanent dentition.[1] Supernumerary teeth are usually associated with genetic syndromes, such as Gardner's syndrome, Nance–Horan syndrome, Trichorhinophalangeal syndrome, cleidocranial dysplasia (CCD), and Opitz BBB/G syndrome. Several causative genes, such as APC, NHS, TRPS1, Runt-related transcription factor 2 (RUNX2), and MID1, have been identified to be responsible for the formation of supernumerary teeth.
Considering the genetical heterogeneities in affected individuals with supernumerary teeth and craniofacial dysplasia, we applied whole-exome sequencing (WES) and biomedical informatics analysis to identify potential causes for molecular diagnosis at clinic.[2] Here we report the identification of a novel mutation within the Runt domain of RUNX2 in a familial case with supernumerary teeth and craniofacial dysplasia (part of phenotypes in the disorder of cleidocranial dysplasia) and also present mini-meta analysis of phenotype–genotype correlations of the disorder.
Cleidocranial dysplasia (CCD; OMIM 119600) is an autosomal dominant inheritable skeletal disorder with an estimated incidence of 1/1000,000 with no gender or ethnic predilection.[3] The main phenotypes of CCD are mainly related to clavicular dysplasia, delayed closure or non-closure of the fontanels, multiple wormian bones (also known as intra sutural bones), short stature, midface hypoplasia, and supernumerary teeth.[4,5] Other phenotypes, including brachydactyly with hypoplastic distal phalanges or pubic bones, scoliosis, brachycephaly, depressed nasal bridge, are also described in literatures.[6,7] This disease is often caused by loss function or haploinsufficiency of the RUNX2 gene, which plays an important role in the differentiation of osteoblasts and the maturation of chondrocytes.[5,8–10] RUNX2 protein contains a Runt domain, an N-terminal stretch of glutamine/alanine repeats (Q/A domain), and a C-terminal proline/serine/threonine-rich (PST) domain.[11,12] It has been reported that chromosomal deletions, translocations, missense, nonsense, splice site mutations and frame-shift are the common mutations in RUNX2.[13] To date, more than 182 heterozygous mutations in RUNX2 have been identified in affected individuals with CCD (HGMD database). Majority of RUNX2 mutations happened in the Runt domain, and the most common mutations are missense mutations.[13]
2. Methods
2.1. Clinical manifestations
A family with the clinical diagnosis of supernumerary teeth and craniofacial dysplasia at School of Stomatology in Shandong University was examined. The proband (III-1, indicated by an arrow in the pedigree, Fig. 1A) was a 16-year-old girl; the affected mother was 48 years old. The affected individuals underwent clinical evaluation, such as craniofacial examination, oral/dental evaluation, and radiological imaging. Endocrinologic laboratory tests, electrocardiogram, and color Doppler echocardiography were also performed for the mother due to her chest dull pain. Oral examination was conducted to record characteristics, such as tooth eruption and retention, enamel development state and periodontal and occlusal conditions. Photographs of the intra-oral view, both shoulder joints, head and face were obtained in both patients.
Figure 1.
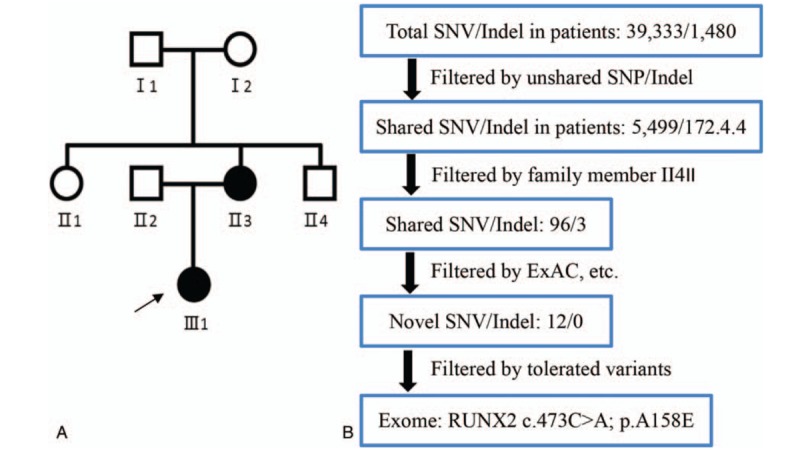
Pedigree and whole-exome sequencing. (A) Genetic pedigree of a Chinese family with supernumerary teeth and craniofacial dysplasia. Proband, indicated with an arrow. (B) Whole-exome sequencing (WES) and bioinformatics analysis. The schematic illustrates the main steps of WES analysis.WES = whole-exome sequencing.
2.2. Whole-exome sequencing and bioinformatics analysis
This study was approved by the ethics committee of School of Stomatology, Shandong University. Informed consent was obtained from the parents. Two milliliters of peripheral blood samples were collected from each member of the trio. Genomic DNAs were extracted using Universal Genomic DNA Kit (ComWin Biotech, Beijing, China). Whole-exome sequencing was performed (Angen Gene Medicine Tech, Beijing, China). Whole-exome capture was carried out using the Agilent SureSelect Human All Exon Kit (Agilent Technologies, Santa Clara, CA) and high-throughput sequencing by the Illumina HiSeq 2000 sequencer instrument (Illumina, San Diego, CA). The steps of WES in silico analysis of potential genetic causes are summarized (Fig. 1B). All variants were identified through Genome Analysis Toolkit (GATK). The results were filtered to exclude synonymous variants. Deleterious single-nucleotide variants (SNVs) were predicted by SIFT, PolyPhen-2, MutationTaster and Proven programs. Identified causal mutation in RUNX2 (GenBank acc. no.: NM_001024630.3, NP_001019801.3) was further confirmed by Sanger Sequencing (ABI 3500 Genetic Analyzer, Applied Biosystems, Foster City, CA) using specific primers (forward: 5′-CATTCCTGTCGGCCATTACT-3′; reverse: 5′-GAAAAACACTCAACTTCATCTGGA-3′).
3. Results
3.1. Clinical findings
Both the proband and the mother had the chief complaint of delayed eruption of permanent anterior teeth. The proband showed a short stature (156 cm) and bodyweight at 50 kg. In physical examination, the proband face appeared to be broad forehead, frontal bossing, orbital hypertelorism, mid-face hypoplasia, and protruding mandible (Fig. 2A). Her nasal bridge appears to be low. Intraoral examination revealed persistence of the primary teeth, delayed eruption of the permanent teeth, supernumerary teeth, an Angle class III malocclusion, negative overjet, bilateral posterior crossbite, and high arched palate (Fig. 2A). The Panoramic radiograph showed at least one unerupted supernumerary tooth in the maxilla, and nine supernumerary teeth in the mandible (Fig. 2C, III-1). Both shoulders of the proband could not be approximated to midline either actively or passively. However, chest and posteroanterior cephalometric radiograph examinations were declined by the family members. The color Doppler echocardiography showed mitral and tricuspid valve regurgitation in patient II-3 and tricuspid valve regurgitation in patient III-1 (Supplementary Fig. 1).
Figure 2.
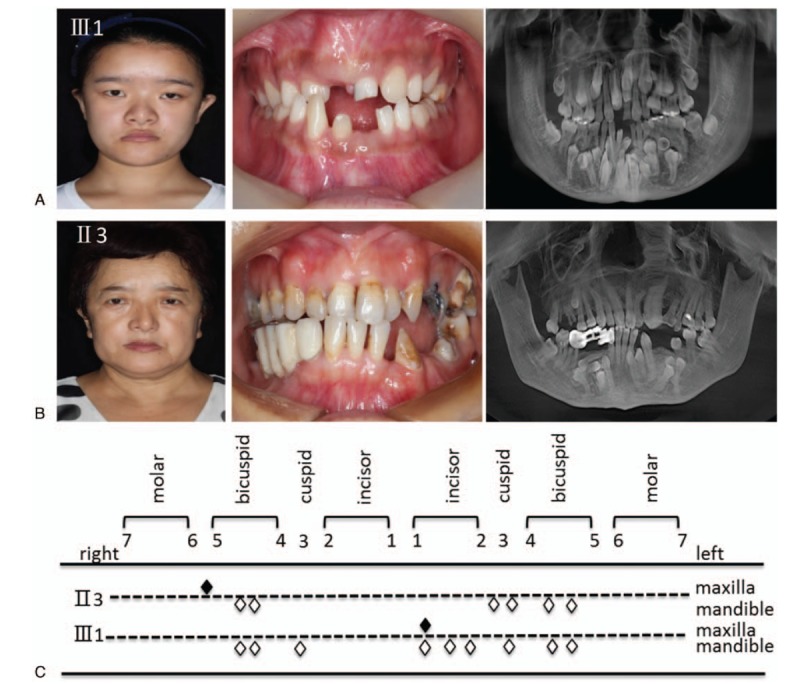
Clinical manifestations. (A) Photographs show phenotypic characteristics of the proband. (B) Photographs show phenotypic characteristics of the affected mother. (C) The appearance of supernumerary teeth in the patients. White and black rhombuses denote supernumerary teeth in the mandible and maxilla, respectively.
The proband's mother (i.e., III-3) was also short (154 cm). Her facial appearance was similar to her daughter, with a prominent forehead and large fontanelles, hypertelorism and a depressed nasal bridge, mid-face hypoplasia and mandibular hyperplasia (Fig. 2B). No abnormal hypermobility of the shoulders was found. Oral manifestations include an edge-to-edge occlusion of the anterior teeth, negative overjet, retention of deciduous teeth, and a high-arched palate. Panoramic radiograph showed one supernumerary tooth in the maxilla and 6 supernumerary teeth in the mandible, failure of secondary dentition eruption (Fig. 2B). She had a fixed denture for her unerupted supernumerary teeth (Fig. 2C). The color Doppler echocardiography showed mitral regurgitation and tricuspid regurgitation (Supplementary Fig. 1, II-3).
3.2. Genetic analysis
Mutational analysis of RUNX2 revealed a novel missense mutation (c.C473A; p.A158E) in both affected subjects, which was further confirmed by Sanger sequencing (Fig. 3A). This mutation has not been previously reported (HGMD). This substitution of Alanine 158 with Glutamic acid is within Runt domain (Fig. 3B, indicated by a red triangle). Alanine 158 in RUNX2 is evolutionarily conserved in all examined vertebrates (Fig. 3C), suggesting that this residue is functionally important. To understand the pathogenetic effect and provide insight for a better understanding of phenotype determining mechanisms, we examined the crystal structure of RUNX1 that shares a high homology of Runt domain with RUNX2 sequence. In the ternary complex formed with CBFβ and DNA (Protein Data Bank: 1H9D), Arg-134 of RUNX1 is found to correspond to Ala-158 of RUNX2, thereby interacting with the DNA molecule (Fig. 4). Considering the conservation of the Runt domains of RUNX1-3 for a similar DNA binding mode, we infer that the pathogenic effects of the p.A158E mutation of RUNX2 might result in an impairment of its DNA binding capability.
Figure 3.
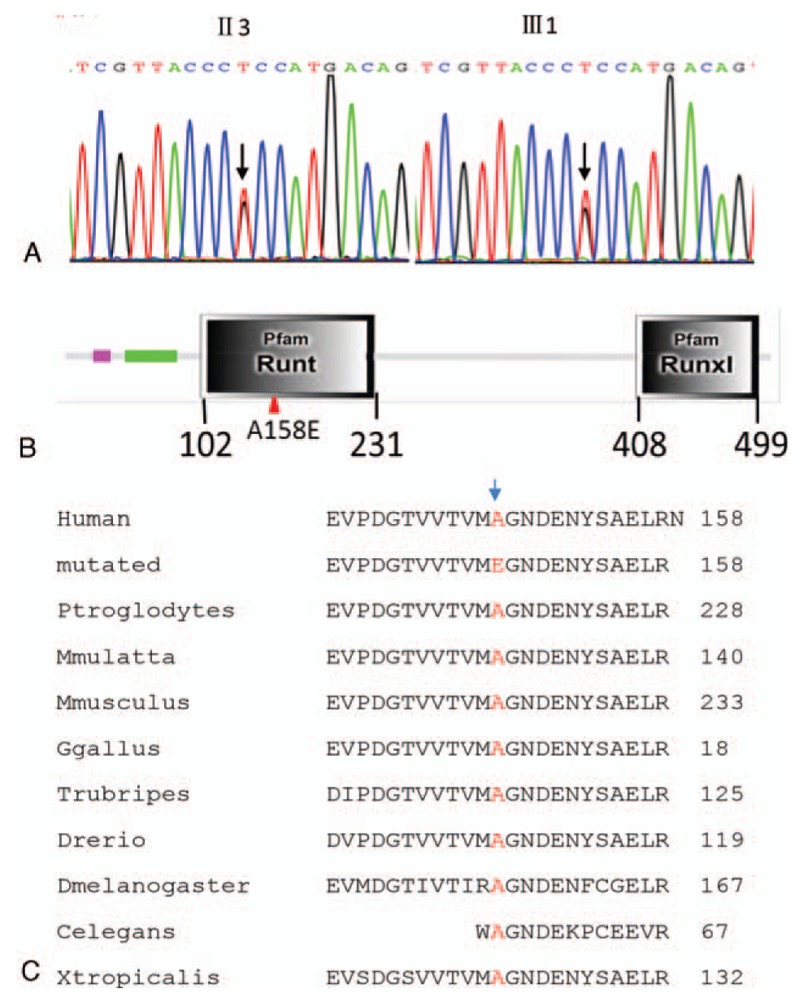
Mutation identification and characterization. (A) Sequencing of exon 3 of RUNX2 shows the heterozygous mutant allele with C>A missense mutation (c.473C>A), which causes a protein change at amino acid 158 (p.A158E). (B) The protein structure of RUNX2 (altered position is indicated by a red triangle). (C) Comparison of sequences of the p.A158-containing regions of RUNX2 in 11 different species.
Figure 4.
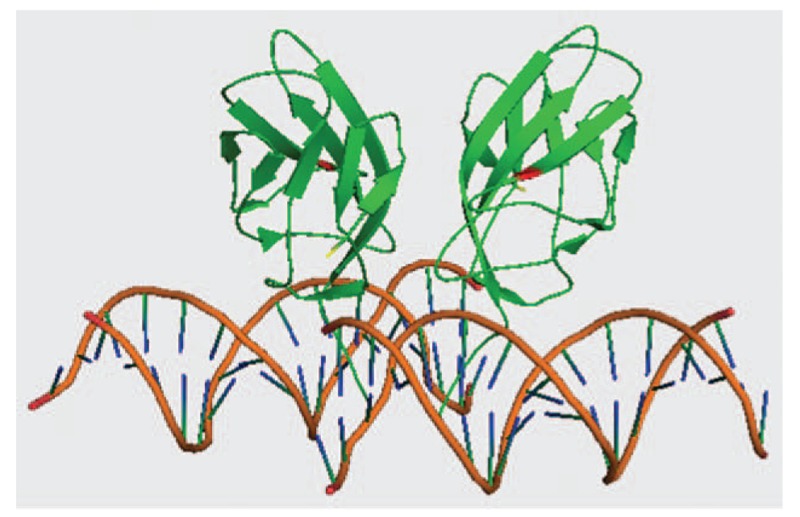
Ribbon diagram of the RD:CBFβ:DNA ternary complex and mutated residues. The RD and CBFβ are shown in green, while DNA is in orange. The amino acid mutated at 158 (p.A158E) is shown in red. The 3D structural model is established using PyMOL Molecular Graphics System.
3.3. Genotype–phenotype correlation
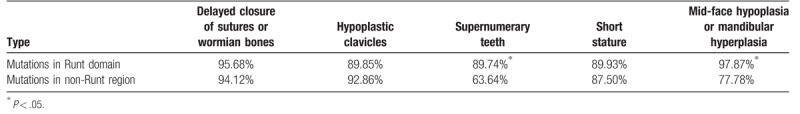
By extensive literature analysis, we compared various phenotypes between 183 patients with missense and 25 patients with nonsense mutations (details are provided in Supplementary Table 1) that were clustered in the Runt domain of RUNX2 (Table 1). The phenotypes are presented in 5 categories, including delayed closure of sutures or wormian bones, hypoplastic clavicles, supernumerary teeth, short stature, and mid-face hypoplasia or mandibular hyperplasia (Table 1). However, we found no significant difference, in terms of these phenotypes, between the groups with missense and nonsense, suggesting a crucial effect of missense mutations within the Runt domain on the encoded whole protein. Next, we analyzed the effects of mutations locations, either in Runt domain or non-Runt domain region, on clinical manifestations (Table 2). In a total of 239 patients, selected clinical manifestations presented in 208 of them with nonsense or missense mutations clustered in Runt domain were compared with that in the remaining 31 cases with nonsense or missense mutations in non-Runt domain regions. As results, mutation rates in Runt domain are significantly higher in patients with supernumerary teeth and maxillary hypoplasia as well as in patients with mid-face hypoplasia and mandibular hyperplasia than that in non-Runt domain regions (P < .05, Table 2), suggesting the importance of Runt domain in dental and craniofacial development.
Table 1.
Comparison of effects of mutation types in the Runt domain on phenotypic spectrum.

Table 2.
Comparison of effects of mutation locations on clinical manifestations.
4. Discussion
Supernumerary teeth, as an important diagnostic clue, have been demonstrated to be associated multiple syndromes, such as Gardner's syndrome (OMIM 175100) caused by haploinsufficiency of the APC gene (OMIM 611731), Nance–Horan syndrome (OMIM 302350) by mutations in NHS (OMIM 302350), Trichorhinophalangeal syndrome (OMIM 190350) by TRPS1 (OMIM 604386), cleidocranial dysostosis (OMIM 119600) by RUNX2 (OMIM 600211), and Opitz BBB/G syndrome (OMIM 300000) by MID1 (OMIM 300552). Careful physical examination is fundamental for diagnosis of patients with supernumerary teeth whether to be a genetical syndrome or not. Given clinical diagnosis remains unclear, genetic analysis using WES is recommended to uncover potential genomic defects underlying supernumerary teeth.
In the present study, we report 2 cases of a rare autosomal dominant syndrome associated with multiple supernumerary teeth and craniofacial dysplasia. To identify the underlying genetic defect, we employed WES to explore possible causative gene and identified a novel missense mutation in codon 473 of RUNX2. This method can identify all sequence variations in coding regions, which makes results more reliable. Furthermore, several commonly used algorithms, such as PolyPhen-2 and MutationTaster, revealed that mutation p.A158E in RUNX2 with deleterious effects and altered protein structure. This appears the first study to identify the genetic cause in patients with CCD-like phenotypes in China utilizing the WES approach.
Affected individuals with CCD-associated phenotypes usually have characteristic clinical features. They tend to show prominent forehead, widely spaced eyes, flat nasal bridge, maxillary hypoplasia, relative mandibular prognathism contribute to skeletal Class III tendency, prolonged retention of the primary dentition, and presence of multiple supernumerary teeth, with consequent delayed and impacted eruption of permanent teeth.[14] These facial features, multiple supernumerary teeth and altered eruption patterns are of significance at clinic. But the most characteristic clinical features are delayed closure of fontanels or open fontanels, hypoplasia or aplasia one or both clavicles or completely absent in 10% patients permitting inability to approximate the shoulders anteriorly, and short stature.[5,15,16] Other conditions share some characteristics with CCD. For instance, 16q22 deletion syndrome shows the same manifestations of fontanel and clavicle as CCD. Mandibuloacral dysplasia syndrome also shows short stature, delayed closure of cranial sutures and dysplastic clavicles. Yunis–Varon syndrome exhibits similar manifestations in delayed closure of cranial sutures and dysplastic clavicles. Based on clinical and radiographic findings, diagnosis of typical CCD generally is not a challenge since it can be differentiated from symptoms of cranium, clavicles and teeth. However, clinical and radiographic manifestations of the affected family members in the present cases only showed part of manifestations, such as craniofacial dysplasia and dental abnormalities.
The etiology of CCD is usually caused by hypomorphic or haploinsufficiency of RUNX2 (runt-related transcription factor 2, OMIM 600211) on chromosome 6p21.[5]RUNX2 is essential for differentiation of osteoblast and dental stem cells, and thus involved in bone and tooth formation. Further evidence for the correlation of RUNX2 with CCD is similar phenotypes in hypoplastic clavicles and defective skull formation in heterozygous (Runx2+/-) mice to that in patients with CCD.[10] Around 60% to 70% of cases with CCD are found to carry pathogenic variants in RUNX2, while remaining cases with CCD appear spontaneously with no apparent genetic causes.[17,18]
RUNX2 spans approximately 200-kb in genome with eight coding exons.[19] The RUNX2 protein is a transcription factor[20] with multiple functional domains as described below.[21] The QA domain regulates transactivation activity of RUNX2 target genes.[21,22] The Runt domain consisting of 128 amino acids is responsible for heterodimerizations with CBFβ and DNA binding.[23–25] The nuclear localization signal (NLS) regulates the accumulation of the RUNX2 protein into nuclei.[26,27] The PST domain is necessary for interactions with other transcription factors and RUNX2-mediated transcriptional regulation.[28] To date, more than 182 different mutations in RNNX2 have been identified (HGMD database). While nonsense and frame-shift mutations occur throughout the RUNX2 gene, the majority missense mutations are found to be clustered in Runt domain [29,30], like in our cases.
Multiple studies attempted to describe genotype–phenotype correlations in CCD, but the results were inconclusive. For instance, short statures were often seen in the patients with impairment of Runt domain; correlation was also noted between the height scores and the number of supernumerary teeth.[31] However, later studies failed to confirm these observations.[11,32,33] Here, we analyzed all the missense/nonsense mutations with or without the Runt domain involvement, and found no significant association between short stature and different locations of mutations. We also found no evidence of a correlation between short stature and supernumerary teeth. Additional study showed that mutations in Runt domain were associated with severe dental anomalies, such as multiple impacted and supernumerary teeth, while mutations outside of Runt domain showed mild dental anomalies.[34] Through a mini-meta analysis, we found many more cases with supernumerary teeth and maxillary hypoplasia or mandibular protrusion with missense mutations in Runt domain.
5. Conclusion
In summary, we identified a novel missense mutation of RUNX2 in 2 patients by WES and showed potential correlations between facial phenotypes and missense mutations in Runt domain through a mini-meta analysis. These findings expanded our understanding of the correlation between the spectrum of CCD mutations and phenotypes. Further studies are required to establish the genotype–phenotype correlations in more details.
Acknowledgments
The authors are thankful for the participation of the family in this study. The authors gratefully acknowledge for Dr Richard Leapman's supports and guidance on this research at NIBIB, NIH.
Author contributions
Data curation: Dan Ma, Jun Zhang.
Formal analysis: Dan Ma, Jun Zhang.
Funding acquisition: Tao Cai, Jun Zhang.
Investigation: Dan Ma, Jun Zhang.
Project administration: Xuxia Wang, Jun Guo, Tao Cai.
Resources: Dan Ma, Xuxia Wang, Jun Zhang.
Supervision: Tao Cai, Jun Zhang.
Writing – original draft: Dan Ma.
Writing – review & editing: Tao Cai.
Supplementary Material
Footnotes
Abbreviations: CCD = cleidocranial dysplasia, RUNX2 = Runt-related transcription factor 2, WES = whole exome sequencing.
This study was supported by the National Natural Science Foundation of China (No. 81371180).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Mahabob MN, Anbuselvan GJ, Kumar BS, et al. Prevalence rate of supernumerary teeth among non-syndromic South Indian population: an analysis. J Pharm Bioallied Sci 2012;4:S373–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yu P, Yang W, Han D, et al. Mutations in WNT10B are identified in individuals with oligodontia. Am J Hum Genet 2016;99:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wu LZ, Su WQ, Liu YF, et al. Role of the RUNX2 p.R225Q mutation in cleidocranial dysplasia: a rare presentation and an analysis of the RUNX2 protein structure. Genet Mol Res 2014;13:1187–94. [DOI] [PubMed] [Google Scholar]
- [4].Jarvis JL, Keats TE. Cleidocranial dysostosis. A review of 40 new cases. Am J Roentgenol Radium Ther Nucl Med 1974;121:5–16. [DOI] [PubMed] [Google Scholar]
- [5].Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999;36:177–82. [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang C, Zheng S, Wang Y, et al. Mutational analysis of RUNX2 gene in Chinese patients with cleidocranial dysplasia. Mutagenesis 2010;25:589–94. [DOI] [PubMed] [Google Scholar]
- [7].Callea M, Bellacchio E, Di Stazio M, et al. A case of cleidocranial dysplasia with peculiar dental features: pathogenetic role of the RUNX2 mutation and long term follow-up. Oral Health Dent Manag 2014;13:548–51. [PubMed] [Google Scholar]
- [8].Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997;89:755–64. [DOI] [PubMed] [Google Scholar]
- [9].Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 1997;89:747–54. [DOI] [PubMed] [Google Scholar]
- [10].Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997;89:765–71. [DOI] [PubMed] [Google Scholar]
- [11].Baumert U, Golan I, Redlich M, et al. Cleidocranial dysplasia: molecular genetic analysis and phenotypic-based description of a Middle European patient group. Am J Med Genet A 2005;139A:78–85. [DOI] [PubMed] [Google Scholar]
- [12].Jaruga A, Hordyjewska E, Kandzierski G, et al. Cleidocranial dysplasia and RUNX2-clinical phenotype–genotype correlation. Clin Genet 2016;90:393–402. [DOI] [PubMed] [Google Scholar]
- [13].Becker A, Bimstein E, Shteyer A. Interdisciplinary treatment of multiple unerupted supernumerary teeth. Report of a case. Am J Orthod 1982;81:417–22. [DOI] [PubMed] [Google Scholar]
- [14].Dore DD, MacEwen GD, Boulos MI. Cleidocranial dysostosis and syringomyelia. Review of the literature and case report. Clin Orthop Relat Res 1987. 229–34. [PubMed] [Google Scholar]
- [15].Nebgen D, Wood RS, Shapiro RD. Management of a mandibular fracture in a patient with cleidocranial dysplasia: report of a case and review of the literature. J Oral Maxillofac Surg 1991;49:405–9. [DOI] [PubMed] [Google Scholar]
- [16].Levanon D, Negreanu V, Bernstein Y, et al. AML1, AML2, and AML3, the human members of the Runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics 1994;23:425–32. [DOI] [PubMed] [Google Scholar]
- [17].Machol K, Mendoza-Londono R, Lee B. Cleidocranial dysplasia spectrum disorder. University of Washington, Seattle, Gene Reviews®. Seattle, WA:1993. [PubMed] [Google Scholar]
- [18].Sapp JP, Eversole LR, Wysocki GP. Contemporary Oral and Maxillofacial Pathology. St. Louis: Mosby; 1997. [Google Scholar]
- [19].Bruderer M, Richards RG, Alini M, et al. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater 2014;28:269–86. [DOI] [PubMed] [Google Scholar]
- [20].Callea M, Fattori F, Yavuz I, et al. A new phenotypic variant in cleidocranial dysplasia (CCD) associated with mutation c.391C>T of the RUNX2 gene. BMJ Case Rep 2012;2012:bcr1220115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thirunavukkarasu K, Mahajan M, McLarren KW, et al. Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfbeta. Mol Cell Biol 1998;18:4197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kauffenstein G, Tamareille S, Prunier F, et al. Central role of P2Y6 UDP receptor in arteriolar myogenic tone. Arterioscler Thromb Vasc Biol 2016;36:1598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ogawa E, Maruyama M, Kagoshima H, et al. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila Runt gene and the human AML1 gene. Proc Natl Acad Sci U S A 1993;90:6859–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kagoshima H, Shigesada K, Satake M, et al. The Runt domain identifies a new family of heteromeric transcriptional regulators. Trends Genet 1993;9:338–41. [DOI] [PubMed] [Google Scholar]
- [25].Kamamoto M, Machida J, Miyachi H, et al. A novel mutation in the C-terminal region of RUNX2/CBFA1 distal to the DNA-binding Runt domain in a Japanese patient with cleidocranial dysplasia. Int J Oral Maxillofac Surg 2011;40:434–7. [DOI] [PubMed] [Google Scholar]
- [26].Quack I, Vonderstrass B, Stock M, et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet 1999;65:1268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kanno T, Kanno Y, Chen LF, et al. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol Cell Biol 1998;18:2444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zaidi SK, Javed A, Choi JY, et al. A specific targeting signal directs Runx2/Cbfa1 to subnuclear domains and contributes to transactivation of the osteocalcin gene. J Cell Sci 2001;114:3093–102. [DOI] [PubMed] [Google Scholar]
- [29].Lo Muzio L, Tete S, Mastrangelo F, et al. A novel mutation of gene CBFA1/RUNX2 in cleidocranial dysplasia. Ann Clin Lab Sci 2007;37:115–20. [PubMed] [Google Scholar]
- [30].Kim HJ, Nam SH, Kim HJ, et al. Four novel RUNX2 mutations including a splice donor site result in the cleidocranial dysplasia phenotype. J Cell Physiol 2006;207:114–22. [DOI] [PubMed] [Google Scholar]
- [31].Yoshida T, Kanegane H, Osato M, et al. Functional analysis of RUNX2 mutations in cleidocranial dysplasia: novel insights into genotype–phenotype correlations. Blood Cells Mol Dis 2003;30:184–93. [DOI] [PubMed] [Google Scholar]
- [32].Suda N, Hattori M, Kosaki K, et al. Correlation between genotype and supernumerary tooth formation in cleidocranial dysplasia. Orthod Craniofac Res 2010;13:197–202. [DOI] [PubMed] [Google Scholar]
- [33].Tessa A, Salvi S, Casali C, et al. Six novel mutations of the RUNX2 gene in Italian patients with cleidocranial dysplasia. Hum Mutat 2003;22:104. [DOI] [PubMed] [Google Scholar]
- [34].Bufalino A, Paranaiba LM, Gouvea AF, et al. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Dis 2012;18:184–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.