

ABSTRACT
Mitochondrial homeostasis is maintained by removing dysfunctional, ubiquitinated mitochondria from the network via PRKN-dependent mitophagy. MYO6, a unique myosin that moves towards the minus ends of actin filaments, forms a complex with PRKN and is selectively recruited to damaged mitochondria by binding to ubiquitin. On the mitochondrial surface, this myosin motor initiates the assembly of F-actin cages, which serve as a quality control mechanism to isolate dysfunctional mitochondria thereby preventing their refusion with neighboring populations. MYO6 also plays a role in the later stages of the mitophagy pathway by tethering endosomes to actin filaments facilitating mitophagosome maturation and autophagosome-lysosome fusion.
KEYWORDS: Actin, mitochondrial quality control, mitophagy, MYO6, myosin VI, PRKN, Parkin
In eukaryotic cells, a healthy mitochondrial network is maintained through several quality control mechanisms including fission and fusion events and delivery of damaged mitochondrial proteins to lysosomes in mitochondria-derived vesicles. As a last resort, entire dysfunctional organelles are isolated and cleared by a specialized selective autophagy pathway termed mitophagy; one of the best-studied mechanisms involves PRKN/Parkin that, when mutated, causes Parkinson disease. After mitochondrial damage, such as loss of membrane potential, the E3 ubiquitin ligase PRKN is recruited to the outer mitochondrial membrane (OMM) where it extensively ubiquitinates OMM proteins. Autophagy receptors, including OPTN, CALCOCO2/NDP52, and TAX1BP1 then recognize and capture ubiquitinated mitochondria targeting them for engulfment in phagophores, precursors to autophagosomes, which ultimately fuse with lysosomes for degradation.
MYO6 (myosin VI), a highly specialized myosin motor protein, can directly bind to the autophagy receptors and ubiquitin. This motor plays an important role in starvation-induced as well as selective autophagy including xenophagy and associates with PRKN in response to mitochondrial damage in a proteomics-based screen. Recently, we investigated the role of MYO6 and actin filaments in PRKN-dependent mitochondrial quality control (Figure 1) [1]. We observe the selective recruitment of MYO6 from intracellular vesicles, plasma membrane and cytosol to PRKN-positive damaged mitochondria after 2-h treatment with the protonophore CCCP. Surprisingly, siRNA knockdown experiments reveal that MYO6 recruitment to mitochondria does not involve the mitophagy receptors. In contrast, MYO6 is directly targeted to mitochondria by binding to ubiquitin because mutating the MYO6 ubiquitin-binding (MyUb) domain strongly impairs recruitment. Furthermore, MYO6 translocation is dependent on PRKN E3 ligase activity and inhibited in cells with the PRKNC431S mutation. Interestingly, MYO6 forms a complex with PRKN independent of CCCP treatment suggesting that this complex may not only exist on damaged mitochondria as previously described but also, for example, in the endocytic pathway. MYO6 is present on RAB5-positive endosomes, which play a role in PRKN-mediated mitochondrial elimination through the endosomal degradation pathway.
Figure 1.
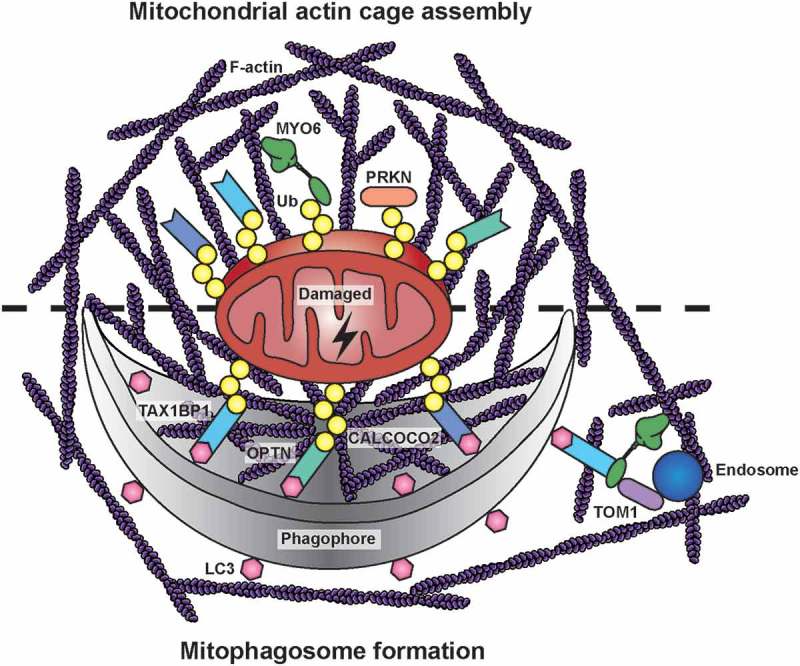
Mitochondrial actin structures during PRKN-dependent mitophagy. After a mitochondrial insult, PRKN is recruited to damaged mitochondria, where it extensively ubiquitinates outer mitochondrial membrane proteins. MYO6 is directly targeted to dysfunctional mitochondria by binding to ubiquitin (Ub) chains and, together with the actin regulators CDC42, ARP2/3, FMN/formins, and WASL/N-WASP, it initiates the assembly of F-actin cages. These actin filaments isolate damaged organelles destined for mitophagy, thereby preventing their refusion with neighboring populations. Autophagy receptors, such as OPTN, CALCOCO2, and TAX1BP1, are recruited independently to ubiquitinated mitochondria targeting them for engulfment in phagophores by binding to LC3. The mitochondrial actin cage may persist for several hours and thus could facilitate phagophore formation and mitophagosome maturation. The latter process involves TOM1-positive endosomes tethered to actin filaments in the vicinity of autophagosomes by docking of MYO6 to the autophagy receptors on the outer leaflet of the autophagosome.
Myosin motors utilize actin filaments for their specific cellular activity, such as transporting cargo or tethering organelles. However, they can also regulate the actin cytoskeleton and modulate actin network organization depending on cellular demands. After CCCP treatment to induce mitochondrial damage, we detect 2 distinct F-actin assembly events on mitochondria. We observe a first burst (within minutes) of transient mitochondrial actin filament polymerization known to mediate mitochondrial fission. This process involves non-muscle myosin II, which can assemble into bipolar filaments to provide the force for constriction. After a drop in mitochondrial actin levels, we observe a second wave (after 2 h) of actin filament assembly leading to stable actin cage formation around fragmented mitochondria; this encaging process requires MYO6 as it is inhibited by ectopic expression of the MYO6 tail lacking the motor domain and actin binding ability. The assembly of these actin cages is regulated by signalling downstream of the Rho-family GTPase CDC42, but not RHO or RAC1, and actin nucleators including the ARP2/3 complex, FMN/formins and WASL/N-WASP. Although the precise mechanism of MYO6-dependent actin regulation is still an open question, we recently identified two GEFs for Rho-family GTPases, ARHGEF12/LARG and DOCK7, associated with MYO6.
These MYO6-induced actin cages restrict mitochondrial fragment size to potentially create more ‘bite-size’ mitochondria for engulfment by the autophagy machinery. We demonstrate that the absence of mitochondrial actin cages through inhibition of actin nucleation accelerates refusion of fragmented mitochondria and network reformation after CCCP washout. Thus, MYO6-dependent actin cages sequester and isolate fragmented mitochondria destined for mitophagy thereby restricting their refusion with neighboring populations and reintegration into the mitochondrial network. This ‘caging’ role of actin serves as an additional quality control step at the onset of mitophagy that is distinct from the actin burst during mitochondrial fission and the dynamic actin cycling through mitochondrial subpopulations thought to regulate steady-state network morphology.
We postulate that the MYO6-dependent actin cages persist over several hours and therefore might have additional roles during mitophagy. For example, the actin meshwork could serve as a structural scaffold for the growing phagophore and could support fusion of individual phagophores into a single mitophagosome. At a later stage, MYO6 could anchor TOM1-positive endosomes to actin filaments surrounding the mitophagosome to promote fusion of endosomes with autophagosomes. This process is required for autophagosome maturation to enable fusion with lysosomes for subsequent degradation. Indeed, we observe a kinetic delay of mitophagy in myo6 knockout (KO) MEFs suggesting a role for MYO6 in the efficient clearance of mitochondria-loaded autophagosomes in line with our previous findings that MYO6 delivers endosomes to autophagosomes in the final stages of autophagy.
The mitophagy defect in cells lacking MYO6 leads to downstream mitochondrial dysfunction. In myo6 KO MEFs, total mitochondrial mass increases but mitochondrial respiration is impaired suggesting that loss of MYO6 results in an accumulation of functionally impaired mitochondria. MYO6-depleted HEK293 cells display further mitochondrial deficiencies, as they do not grow in medium containing galactose as the sole carbon source, which forces cells to rely on mitochondrial oxidative phosphorylation for energy production.
Taken together, this work underscores the importance of MYO6 in the assembly of actin filament cages as a critical quality control step in damage-induced PRKN-mediated mitophagy. Although considerable progress has been made in elucidating the involvement of the actin cytoskeleton in regulating mitochondrial network dynamics and fission/fusion events, future research is needed to determine the importance of the actin cytoskeleton and associated machinery in the spatial and temporal regulation of mitophagy and mitochondrial quality control.
Funding Statement
This work was supported by the Medical Research Council under Grant MR/N000048/1 and British Heart Foundation under Grant PG/15/12/31280.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Kruppa AJ, Kishi-Itakura C, Masters TA, et al. Myosin VI-dependent actin cages encapsulate parkin-positive damaged mitochondria. Dev Cell. 2018;44:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]