

ABSTRACT
The compound eye of the fruit fly Drosophila melanogaster is one of the most intensively studied and best understood model organs in the field of developmental genetics. Herein we demonstrate that autophagy, an evolutionarily conserved selfdegradation process of eukaryotic cells, is essential for eye development in this organism. Autophagic structures accumulate in a specific pattern in the developing eye disc, predominantly in the morphogenetic furrow (MF) and differentiation zone. Silencing of several autophagy genes (Atg) in the eye primordium severely affects the morphology of the adult eye through triggering ectopic cell death. In Atg mutant genetic backgrounds however genetic compensatory mechanisms largely rescue autophagic activity in, and thereby normal morphogenesis of, this organ. We also show that in the eye disc the expression of a key autophagy gene, Atg8a, is controlled in a complex manner by the anterior Hox paralog Lab (Labial), a master regulator of early development. Atg8a transcription is repressed in front of, while activated along, the MF by Lab. The amount of autophagic structures then remains elevated behind the moving MF. These results indicate that eye development in Drosophila depends on the cell death-suppressing and differentiating effects of the autophagic process. This novel, developmentally regulated function of autophagy in the morphogenesis of the compound eye may shed light on a more fundamental role for cellular self-digestion in differentiation and organ formation than previously thought.
Abbreviations: αTub84B, α-Tubulin at 84B; Act5C, Actin5C; AO, acridine orange; Atg, autophagy-related; Ato, Atonal; CASP3, caspase 3; Dcr-2; Dicer-2; Dfd, Deformed; DZ, differentiation zone; eGFP, enhanced green fluorescent protein; EM, electron microscopy; exd, extradenticle; ey, eyeless; FLP, flippase recombinase; FRT, FLP recognition target; Gal4, gene encoding the yeast transcription activator protein GAL4; GFP, green fluorescent protein; GMR, Glass multimer reporter; Hox, homeobox; hth, homothorax; lab, labial; L3F, L3 feeding larval stage; L3W, L3 wandering larval stage; lf, loss-of-function; MAP1LC3, microtubule-associated protein 1 light chain 3; MF, morphogenetic furrow; PE, phosphatidylethanolamine; PBS, phosphate-buffered saline; PI3K/PtdIns3K, class III phosphatidylinositol 3-kinase; PZ, proliferation zone; Ref(2)P, refractory to sigma P, RFP, red fluorescent protein; RNAi, RNA interference; RpL32, Ribosomal protein L32; RT-PCR, reverse transcription-coupled polymerase chain reaction; S.D., standard deviation; SQSTM1, Sequestosome-1, Tor, Target of rapamycin; TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labeling assay; UAS, upstream activation sequence; qPCR, quantitative real-time polymerase chain reaction; w, white
KEYWORDS: Autophagy, cell death, differentiation, Drosophila, eye development, genetic compensation, HOX, labial, pattern formation, transcriptional control
Introduction
Autophagy (cellular “self-eating”) is a lysosome-mediated self-degradation process of eukaryotic cells. As a main route of eliminating superfluous and damaged cytoplasmic constituents and ensuring macromolecule turnover, autophagy is required for maintaining cellular homeostasis. It also provides energy for the survival of cells under starvation. Although autophagy primarily functions as a prosurvival mechanism in terminally differentiated cells, under certain physiological and pathological settings it can also promote cell death [1–3]. In mammals, defects in autophagy can lead to accelerated aging and the development of various age-dependent pathologies including neurodegenerative diseases, cancer, diabetes, tissue atrophy and fibrosis, immune deficiency, compromised lipid metabolism, and infection by intracellular microbes [4–10].
During autophagy, parts of the cytoplasm are delivered into the lysosomal compartment for degradation by acidic hydrolases. Depending on the mechanism of delivery, 3 major types of autophagy can be distinguished: macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy (hereafter referred to as autophagy) involves the formation of a double membrane-bound compartment called the phagophore that sequesters the cytoplasmic material destined for degradation. The phagophore matures into an autophagosome, which then fuses with a lysosome, thereby generating a structure called autolysosome where degradation takes place [11–14].
The core mechanism of autophagy involves more than 30 autophagy-related (Atg) proteins, which are evolutionarily conserved from yeast to mammals [15]. Atg proteins are organized into functionally distinct complexes: i) the Atg1 kinase complex for inducing phagophore formation; ii) a class III phosphatidylinositol 3-kinase (PtdIns3K/Vps34) complex for vesicle nucleation; iii) a ubiquitin-like conjugation system for vesicle expansion; and iv) a recycling complex for recovering utility materials. The ubiquitin-like conjugation system mediates the transient conjugation of Atg8 (whose mammalian orthologs include the MAP1LC3/microtubule-associated protein 1 light chain 3 and the GABARAP/GABA type A receptor-associated protein families) to a phagophore membrane component, phosphatidylethanolamine (PE).
To date, 2 major developmental functions of autophagy have been uncovered [16,17]. First, it can lead to cell death via, or independently of, apoptosis, thereby removing, for example, larval tissues during metamorphosis in Drosophila [18]. Second, autophagy can selectively degrade specific proteins and organelles to mediate cellular differentiation [17]. However, exploring the function of autophagy in particular developmental events is still in the initial phase. For example, the process plays an important role in spore and fruiting body formation in fungi, and in the life cycle transition of pathogenic protozoans [19–22]. In the nematode Caenorhabditis elegans, autophagic degradation is required for the elimination of paternally distributed mitochondria from [23], and soma-germline separation in, early-stage embryos [24], elongation of the mid-stage embryo [25,26], as well as dauer larva formation [27]. In Drosophila, the process is critical for normal development by degrading larval tissues such as the fat body, salivary gland and midgut [18,28–30], and the removal of paternally delivered mitochondria from the zygote [31]. In chicken, autophagy is necessary for ear development [32]. In mammals, the elimination of maternally distributed gene products from early-stage embryos [33–35] and the embryo-to-neonate transition [36] are mediated by the autophagic process. It is also important in cellular differentiation, such as adipocyte, erythrocyte and lymphocyte maturation [37–39].
The compound eye of Drosophila, together with antenna, ocelli, head cuticle and palpus, develops from a larval primordium called eye-antennal imaginal disc (Figure 1) [40,41]. This organ is an epithelial bilayer; one layer is the disc proper, which is built up from columnar cells and gives rise to the retina, and the other layer called peripodial membrane that is involved in modulating columnar cell fates through emitting signaling cues [42]. Cells of the disc proper divide, grow, and then undergo differentiation into photoreceptors and accessory cells [43]. The border between the proliferating and differentiating cells is marked by the morphogenetic furrow (MF), which migrates from the posterior to anterior direction within the disc [44].
Figure 1.

Expression domains of eye selector genes and eye-specific drivers in the Drosophila eye disc. (A) Schematic representation of the Drosophila eye-antenna imaginal disc, which has 2 major parts: the antenna and eye fields (surface view). The main regions of the eye field are indicated (the proliferation and differentiation zones, PZ and DZ; the morphogenetic furrow, MF). Blue arrow shows the direction where the MF migrates. Expression domains of some eye disc-specific selector genes (grey) and different Gal4 drivers (blue) used in this study are shown. (B) Cross sectional view of the eye-antennal imaginal disc.
Tor (Target of rapamycin) kinase functions as a main upstream negative regulator of autophagy. Hyperactivation of Tor in the eye primordium leads to a massive reduction in the size of the adult organ and interferes with ommatidial patterning (ommatidia become fused or pitted) [45]. This intervention also delays the progression of MF, and causes disorganization or massive loss of photoreceptor cells [45,46]. Tor inactivation similarly compromises eye development by decreasing the rate of proliferation [47]. These data raise the possibility that autophagy is implicated in normal growth and morphogenesis of this organ. Indeed, silencing of Atg7 behind the MF by a GMR-Gal4 driver was reported to result in a rough eye phenotype with fused and enlarged ommatidia [48]. Conversely, Atg7 loss-of-function (lf) mutant animals are characterized by normal eye morphology [49], and, also using GMR-Gal4, knockdown of the Atg1, Atg4a, Atg5, Atg8a, Atg9, Atg12 or Atg18a genes has no effect on ommatidial structure [48,50]. Furthermore, depletion of Atg1, Atg7, Atg8a and Atg12 proteins, performed at 25°C and without coexpressing Dcr-2 (Dicer-2) that would make gene silencing more efficient, also does not interfere with eye development [51]. Eye morphology likewise remains unaffected by overexpressing a dominant-negative mutant allele of Atg1 [52]. Due to these contradictory data, the role of autophagy in Drosophila eye development remains to be elucidated.
In this study we examined the eye disc-specific accumulation of Atg5 and Atg8a proteins, as well as autophagic structures, and found that while the proteins are detectable nearly ubiquitously in each part of the organ, but most abundantly in the area of the anteriorly located prospective head cuticle, autophagic compartments display a specific distribution pattern, predominantly accumulating within and behind the MF (the latter corresponds to the differentiation zone; DZ). We further demonstrated that RNA interference (RNAi)-mediated depletion of Atg proteins in the developing eye disc by drivers being active in the MF can severely compromise eye formation. In the affected animals, eye development was completely or partially inhibited as a consequence of ectopic cell death. However, the effect of lf mutations in Atg genes on eye development was largely rescued by genetic compensatory mechanisms involving the action of alternative transcripts, paralogs or maternally deposited factors. We also found that the Hox gene lab (labial) is expressed in front of and along the MF, and that Atg8a expression is strongly influenced in these regions by Lab deficiency. These data reveal a novel, developmentally regulated role for autophagy; its cell death-suppressing function is essential for columnar cells in the Drosophila eye primordium to survive, thereby acting as a prerequisite for eye morphogenesis. Since this live-or-die cell fate decision is likely to occur in several cell types during development, autophagy may play a more fundamental role in tissue formation than previously thought.
Results
Autophagic structures accumulate in a specific pattern in the eye primordium
Under normal conditions, autophagy operates at basal levels in terminally differentiated cells to maintain normal macromolecule turnover. During differentiation however when cellular constituents are largely reorganized, the process may exhibit an increased activity in the affected cells. To investigate the potential role of autophagy in Drosophila eye development, we first examined the accumulation pattern of 2 key autophagy proteins, Atg5 and Atg8a, as well as Atg5- and Atg8a-positive autophagic structures in the eye primordium of wandering L3 stage (L3W) larvae. At this stage the eye disc is divided into 2 major regions by the MF, the anteriorly located proliferation zone (PZ) and the posteriorly located DZ (Figure 1) [40]. We used an Atg5-specific antibody (Figure. S1) to label Atg5 accumulation in this organ. Atg5 is known to localize to the growing phagophore and remain there until recycling eventually from the autophagosome [53]. Using conventional fluorescent microscopy, the antibody staining revealed abundant Atg5 accumulation in each part of the eye disc, but most obviously in the regions of the prospective head cuticle (indicated by yellow arrows in Figure 2A and S2). Semiconfocal and confocal microscopy resolutions however uncovered a relatively large amount of Atg5-positive foci labelling early autophagosomal structures in the MF and DZ, as compared with other areas of the organ (Figure 2B to B’’’, C to C’’’’ and S7A). Consistent with these data, anti-Atg8a antibody staining performed with confocal microscopy also revealed basal levels of autophagic activity in the antennal field and PZ, but much higher levels in the MF and DZ (Figure 2D to D’’’ and S3A to A’’’, S4, S7B). It is worth to note that anti-Atg8a antibody staining was also highly specific as the expression of Atg8a-specific double-stranded RNA (dsRNA) almost completely abolished protein accumulation in the eye disc (Figure. S4), and that Atg8a protein, similar to Atg5, was distributed nearly ubiquitously in the eye primordium, most evidently in the prospective head cuticle and MF (see later in this study). Thus, the intensity of Atg5 and Atg8a accumulation did not coincide with the distribution of autophagic structures; while the proteins accumulated nearly ubiquitously in the entire antennal-eye disc, the presence of Atg5- and Atg8a-positive foci (autophagic structures) was mainly concentrated to the regions of MF and DZ. A similar punctuated pattern was detected in these regions when the expression of an UAS-mCherry-Atg8a reporter, which marks phagophores, autophagosomes and autolysosomes, was driven by ey-Gal4(II) in the entire eye disc (Figure 2E to E’’’ and S3B to B’’’, S5B, S7C). Staining by LysoTracker Red, which marks acidic compartments including autolysosomes, lysosomes and multivesicular bodies, also revealed a punctuated pattern predominantly behind the MF (Figure. S6 and S7D). Together, these results point to an unequal distribution for autophagic activity in different parts of the developing eye tissue; autophagic structures predominantly accumulate in the MF and DZ (Figure 2, S3, and S6, S7). The other parts of the eye field, together with the antennal field, exhibit only basal levels of autophagy. These data suggest that the autophagic process is involved in the differentiation and/or survival of retinal precursor cells.
Figure 2.

Autophagic structures accumulate in a specific pattern in the Drosophila eye disc. (A to A’’’) Anti-Atg5 antibody staining shows a nearly uniform Atg5 accumulation in the eye disc, with highest levels in the areas of prospective head cuticle (yellow arrows). Green foci in the differentiation zone (DZ) correspond to Atg5-positive structures (early autophagosomal structures). Pictures were taken by conventional fluorescence microscopy. (B to B’’’) Optical sectioning by a semiconfocal microscopy reveals an unequal distribution of Atg5-positive autophagic structures in the eye disc, predominantly in the MF and DZ. (C to C’’’’) Confocal microscopy image showing anti-Atg5-positive autophagic structures. Ato (red) is specific marker for labeling the MF. (D to D’’’) Anti-Atg8a antibody staining indicates autophagic structures, using optical sectioning of a semiconfocal microscopy. Green foci indicate autophagosomal and autolysosomal membranes. Atg8a-positive structures accumulate most abundantly along and behind the MF (indicated by a white arrow). (E to E’’’) mCherry-Atg8a reporter gene driven by ey-Gal4(II) is expressed almost in the entire eye field. Red foci label autophagosomes and autolysosomes. mCherry-Atg8a-positive structures accumulate most evidently in the DZ. Images in panels A to E’’’ are positioned as antenna parts are up; bars: 100 µm; samples were prepared from L3W larvae. MF: morphogenetic furrow. Hoechst staining indicates nuclei. Animals were maintained at 25°C.
Downregulation of Atg genes in the eye disc impairs the development of the organ
Next, we monitored whether silencing of Atg genes in the eye primordium affects the development of this organ. In previous studies, GMR-Gal4 was used to control the expression of UAS-Atg RNAi constructs in the eye disc [48,50]. However, the activity of this driver was only detectable behind the MF (i.e. within the DZ; Figure 1A and S5A), and even its own expression disturbs eye development [54]. In addition, the expression of GMR-Gal4 is not restricted exclusively to the eye field [55]. Hence, we used 2 ey-Gal4 drivers, ey-Gal4(II) and ey-Gal4(III), to target gene silencing to a broader area of the eye primordium, including regions in front of, along, and behind the MF (Figure 1A and S5B, C). Importantly, these drivers per se did not affect eye formation (Table S1 and Figure 3A,D,F). ey-Gal4(II)- or ey-Gal4(III)-driven silencing of Atg genes led to aberrant eye disc and adult eye morphology ranging from small size through abnormal shape to the complete absence of the organ (Table S1 and Figure 3B,C,E,F and S8). Each of the major Atg protein complexes was represented in this set of silencing experiments (Figure 3F). For example, depletion of Atg101 (induction complex) and Atg14 (PtdIns3K complex) with the ey-Gal4(II) driver resulted in aberrant eye morphology with penetrance of 96.67% and 78.26%, respectively. In addition, we silenced Atg3 (conjugation system) by ey-Gal4(III) (note that Atg3 depletion with ey-Gal4(II) caused the lack of the entire eye disc and pupal lethality; Figure. S8D). Atg3 RNAi/ey-Gal4(III) animals displayed aberrant eye phenotype with penetrance of 93.1% in males and 82.4% in females. Downregulation of Atg genes by so7-Gal4 driver being active in almost the entire eye field (Figure. S5D) similarly affected eye formation (Figure. S9). These results suggest that the function of Atg genes in front of and/or within the MF is critical for normal eye development, while depletion of Atg proteins in the DZ alone is not sufficient to compromise the morphogenesis of this organ.
Figure 3.
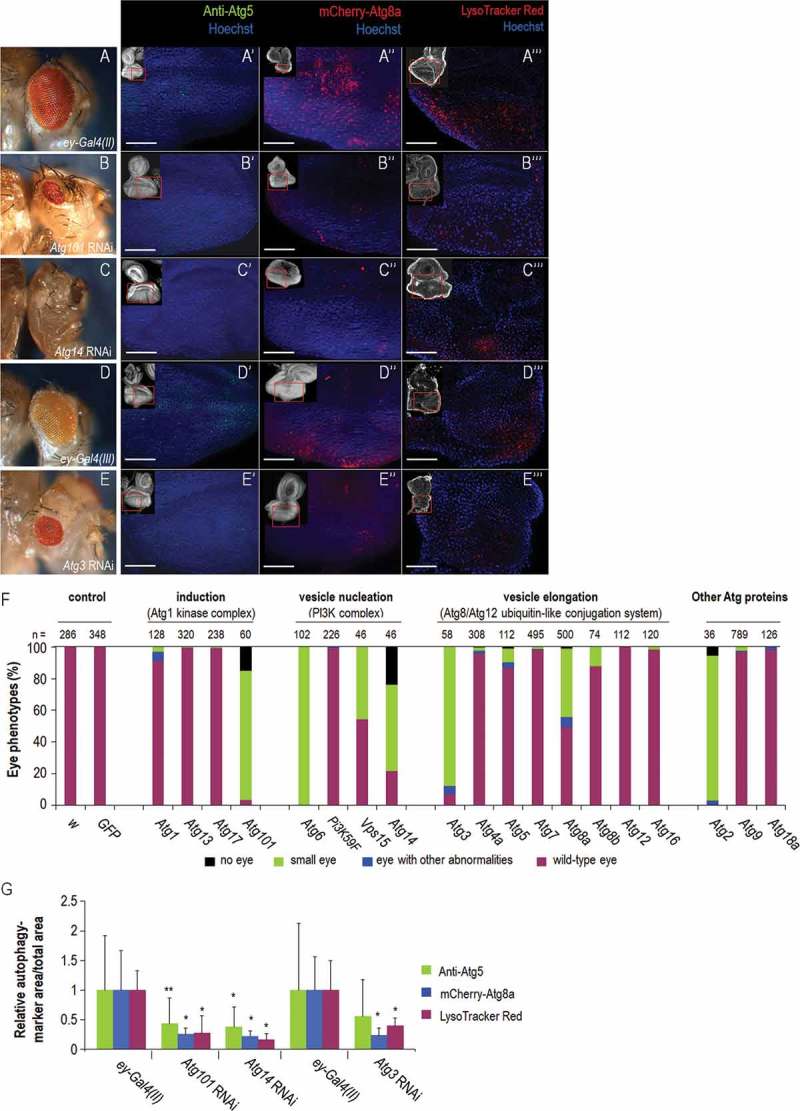
Silencing of Atg genes in the eye disc can severely compromise eye morphology in the affected adults. (A) Stereomicrograph of an ey-Gal4(II)/+ adult head, which served as a control for silencing of Atg101 and Atg14, showing wild-type eye morphology. (A’) Atg5 antibody staining indicates early autophagic structures (green dots) in the eye disc of an ey-Gal4(II)/+control animal. (A’’) mCherry-Atg8a accumulation in the eye disc of an ey-Gal4(II)/+ control animal. Fluorescent foci (red) indicate autophagosomal and autolysosomal structures. (A’’’) LysoTracker Red staining marks acidic structures in the eye disc of an ey-Gal4(II)/+ control animal. (B) Small eye phenotype of an ey-Gal4(II)/Atg101 RNAi adult. (B’ to B’’’) Silencing of Atg101 in the eye disc leads to reduced levels of Atg5- (B’) mCherry-Atg8a- (B’’) and LysoTracker Red- (B’’’) positive structures. (C) The eyeless phenotype of an ey-Gal4(II)/Atg14 RNAi adult. (C’ to C’’’) Depletion of Atg14 in the eye disc leads to reduced levels of Atg5- (C’) mCherry-Atg8a- (C’’) and LysoTracker Red- (C’’’) positive structures. (D) Stereomicrograph of an ey-Gal4(III)/+ adult head, which served as a control for silencing Atg3, showing wild-type eye. (D’) Atg5 accumulation, (D’’) mCherry-Atg8a expression and (D’’’) LysoTracker Red staining in the ey-Gal4(III)/+ genetic background. (E) Stereomicrograph of an ey-Gal4(III)/Atg3 RNAi adult head showing a small eye phenotype., (E’) Silencing of Atg3 in the eye disc leads to a reduced amount of Atg5-, (E’’) mCherry-Atg8a-, and (E’’’) LysoTracker Red-positive foci. In images A’ to A’’’, B’ to B’’’, C’ to C’’’, D’ to D’’’ and E’ to E’’’, the antenna part is up; bars: 50 µm. At the upper left corner of each image, the red rectangle indicates the area enlarged. Eye disc samples were dissected from L3W larvae. (F) Silencing of Atg genes in the eye primordium can severely compromise the development of the organ. The penetrance of eye phenotypes may depend on the efficiency of the RNAi constructs used (also see Figs. S9 and S10). In some cases, like Atg2, Atg3, Atg6, Atg14 and Atg101, the phenotype is manifested with a nearly full penetrance. (G) Effect of Atg3, Atg14 and Atg101 RNAi treatments on autophagic activity in the eye disc of L3W larvae. The ratio of anti-Atg5/mCherry-Atg8a/LysoTracker Red-positive structures and the area of entire eye disc in each image is shown as averages, the data represent relative values. Bars represent mean ±S.D., *: P < 0.05, **: P < 0.01, two-sample Student t test, t test for unequal variances or Mann-Whitney U test. Temperature: 29°C, with the exception of E’’: 18°C, and D to D’’’, E, E’, E’’’: 25°C. In fluorescence images, the background expression was highly reduced in order to strengthen the visibility of puncta.
Knockdown of certain Atg genes, eg Atg3, Atg14 and Atg101, was manifested as abnormal eye development with a relatively high (over 50%) penetrance while silencing of other Atg genes, such as Atg5 and Atg13, did not influence or only slightly affected normal eye formation (Table S1 and Figure 3F). This may have resulted from the different effectiveness of RNAi constructs we assayed. Indeed, assessing mRNA or protein levels in the eye disc of Atg RNAi animals showed a significant reduction in the level of a given mRNA in those cases where the majority of individuals expressed an aberrant eye phenotype, but no change in samples without an obvious phenotype (Figure. S10 and S11). For example, the corresponding Atg protein levels were not significantly changed in Atg5 RNAi and Atg13 RNAi female samples showing no phenotype in response to knockdown (Table S2 and Figure. S11). This phenomenon was particularly obvious in case of Atg8a, which was targeted by different RNAi constructs (Figure S12A). The construct without effect [Atg8a RNAi(V20)] was not capable of lowering the accumulation of Atg8a isoforms, whereas the constructs leading to phenotype [Atg8a RNAi(GD) and Atg8a RNAi(TRiP-1)] considerably reduced their amount, as compared with control (Figure. S12B, C). Consistent with these results, the number of autophagic structures was also significantly reduced in Atg RNAi animals with compromised eye morphology, but not altered in those RNAi samples displaying normal eye development, as compared with their corresponding ey-Gal4 controls (Figure. 3A’ to A’’’, B’ to B’’’, C’ to C’’’, D’ to D’’’, E’ to E’’’, G and S13).
To further demonstrate the specificity of eye phenotypes caused by Atg gene knockdowns, we could largely rescue normal eye development in Atg RNAi animals by introducing a wild-type copy of the corresponding Atg gene. First, the eye phenotype of Atg8a RNAi and Atg14 RNAi animals was considerably suppressed by an Atg8a reporter transgene (eGFP-Atg8a; see later in the manuscript) and a genomic fragment covering Atg14 (g-Atg14) [56], respectively (Figure S12D and S14). Then, an extra copy of a genomic fragment (DC352) that covers Atg101 was introduced into Atg101 RNAi animals. DC352 represents a transgenic duplication specific to Atg101 (http://flybase.org/reports/FBab0046578.html), and in a genetic background containing DC352, Atg101 RNAi animals characteristically had normal eye morphology (Figure. S15A). The presence of DC352 also restored autophagic activity to nearly normal levels in Atg101 RNAi eye samples (Figure. S15B).
Some eye selector genes including ey (eyeless), Optix and eya (eyes absent) are expressed in the peripodial membrane, yet with unknown function [57], and ey-Gal4(II) is also active in this part of the eye disc [42]. To examine the possible contribution of autophagy in the peripodial membrane to eye development, we inactivated Atg genes exclusively in this tissue by using c311-Gal4 driver [42], and found no alteration in the eye structure of animals tested (Table S2 and Figure. S16). Thus, autophagy influences eye development in the disc proper only. Together, we conclude that decreasing the activity of Atg genes in front of and within the MF severely interferes with Drosophila eye development.
Genetic compensatory mechanisms largely rescue autophagic activity and normal eye development in Atg loss-of-function mutant animals
We also assessed eye development in Atg lf mutant animals to further confirm the importance of the autophagic process in the formation of the organ. Since mutations in certain Atg genes are known to cause lethality during early development, we analyzed genetic mosaics to determine the size and morphology of adult eye clonally deficient in an Atg protein. Alternatively, homozygous mutant larvae resulted from the cross of heterozygous parents were monitored. Contrary to previous data reporting almost no effect for mutations in Atg genes on Drosophila eye formation [49], we found that mutational inactivation of Atg17 and Atg1 can seriously affect normal eye morphology. Atg17d130 mutant larvae for instance could exhibit even the complete absence of the eye field, i.e. a phenotype without eyes (Figure. S17A), while Atg17 and Atg1 lf mutant adults occasionally displayed a small eye phenotype (Figure S17B, F). However, defects in eye development were detectable at much lower penetrance in these autophagy-defective mutant systems – only in a few animals among many hundreds we examined – than in Atg RNAi animals, in some of which the manifestation of the eye phenotype was almost fully penetrant (Table S1 and Figure 3F). However, the specificity of eye phenotypes seen in Atg17d130 mutant larvae is supported by the fact that normal morphogenesis of the larval eye disc could be significantly rescued by introducing a transgene that contains the wild-type copy of the gene (Figure. S17C). The fully penetrant lethality of Atg17d130 mutant pupae was also highly suppressed by this transgene; almost half of the transgenic mutants remained alive (Figure. S17D). Furthermore, we observed that in Atg17d130 mutant larvae, unlike control, the htt (huntingtin) gene became strongly overexpressed (Figure. S17E). Because htt codes for a protein functioning as a scaffold for selective autophagy [58], its hyperactivation in the Atg17d130 mutant background may explain why mutant larvae exhibit defects in eye development with a low penetrance only (Atg17 also acts as a scaffold to recruit other Atg proteins to the phagophore assembly site).
It has been recently revealed that genetic compensation induced by deleterious mutations but not gene knockdowns results in a much milder phenotypic effect in mutant animals, as compared with the corresponding RNAi backgrounds [59]. This prompted us to investigate the mechanisms rescuing normal autophagic functions in Atg mutant systems. We first measured the level of the newly identified 3 Atg8a mRNA isoforms (splice variants), A, B and C, in the eye disc of L3W larvae by semi-quantitative RT-PCR, and found that A is expressed abundantly, B is present only at very low levels, while C is not detectable (Figure 4A and S18). We further showed that an Atg8a mutant allele, KG07569, interferes with isoform A only in this organ (Figure 4A,B), and in the Atg8aKG07569 mutant background, the expression of Atg8a-A ceased, while isoform B became highly activated, as compared with the control (w1118) genetic background (Figure 4B and S18). In addition, a weak induction of Atg8a-C transcription was also detectable (Figure 4B and S18). Next, we monitored transcript levels of Atg8b, the sole paralog of Atg8a [60,61], in control versus Atg8aKG07569 mutant samples. The analysis demonstrated the transcriptional activation of Atg8b in response to Atg8a-A deficiency (in control samples Atg8b was not expressed) (Figure 4C). Another Atg8a-A mutant allele, d4, represents a deletion covering the first exonic sequences, that is present only in splice variant A (Figure 4A) [62]. Using a primer pair, one member of which is specific to the region that overlaps with deletion d4 and hence expected to produce no amplification product, we could detect Atg8a-A transcript in Atg8ad4 mutant samples (Figure 4D). Together, these data imply that ectopic expression of splice variants (Atg8a-B and -C) and/or a paralog (Atg8b), as well as a trans-splicing-like mechanism (when 2 primary RNA transcripts are joined and ligated) may rescue some Atg8a-A activities in Atg8a-A lf mutant eye samples.
Figure 4.

Genetic compensatory mechanisms rescue autophagic activity in Atg loss-of-function mutant backgrounds. (A) The structure of Atg8a gene. Orange boxes represent coding sequences, connecting lines indicate intronic sequences, grey boxes show untranslated regulatory elements. The 3 isoforms, A, B and C, are indicated. (A’) Expression levels of the 3 Atg8a isoforms in the eye disc. Semi-quantitative PCR was performed with isoform-specific primers; the number of amplification cycles (NACs) was 40. Atg8a-A is expressed more abundantly than Atg8a-B (for quantification, see Fig. S14). (B) Expression levels of the Atg8a isoforms in an Atg8a lf mutant background affecting isoform A (allele KG07569). While Atg8a-A expression ceased, Atg8a-B became upregulated, as compared with the control (w1118) background (yellow arrow). NACs were 34, and under this setting Atg8a-B is not detectable. (C) Atg8b, a paralog of Atg8a, is upregulated in an Atg8a mutant background (arrow). In panels B and C, Act5C and RpL32 were used as internal controls. (D) Atg8a-A transcript can be detectable in mutant animals bearing a deletion allele of Atg8a, d4 (one of the primers was designed to the region covering the deletion). (E) The expression of Atg18b, a paralog of Atg18a, is activated in an Atg18a mutant background, but not in control (w1118). (F) The Atg4a paralog Atg4b became upregulated in Atg4a mutant animals, as compared with the control (w1118) background. In panels D to F, Act5C was used as an internal control, arrows show the increased transcript levels. (G to G’’’) Atg8a-specific antibody staining displays Atg8a-positive structures in the eye disc of control RNAi (G) and Atg8a RNAi (G’) animals, as well as of control (G’’) versus Atg8aKG07569 mutant (G’’’) animals. ey-Gal(II) driver was used with UAS-Dcr-2. (G’’’’) Quantification of Atg8a-positive structures in genotypes shown in panels G to G’’’. Bars represent mean ±S.D., **: P < 0.01; ***: P < 0.001; Mann-Whitney U test. (H) Atg5-specific structures (red) in cells clonally defective for Atg17 function (not green). (H’) The corresponding uncolored picture. (I) Atg5 accumulation in cells deficient in Atg1 (not green) and in control cells (green). (I’) The corresponding uncolored picture. In panels H’ and I’, the dotted lines indicate homozygous mutant cells without Atg17 and Atg1 activity, respectively. In images G’ to G’’’, H and I, the antenna part is up; bars: 50 µm. Eye disc samples were dissected from L3W larvae. Temperature was 25°C.
We observed similar compensatory mechanisms for mutations in Atg18a and Atg4a that also possess a well-defined paralog, Atg18b and Atg4b, respectively. Atg18b became activated in the Atg18aKG07569 mutant background (Figure 4E and S19), while Atg4b was upregulated in an Atg4a mutant background, as compared with control eye disc samples (w1118) (Figure 4F and S20). Consistent with results above, a significant amount of Atg8a-positive autophagic structures was detectable in Atg8aKG07569 mutant, but not in Atg8a RNAi(GD) samples (Figure 4G to G’’’’). These data indicate that Atg8aKG07569 mutant eye samples are not completely defective for autophagy (indeed, Atg8aKG07569 mutant adults had no defect in eye formation, but nearly half of the Atg8a RNAi(GD) animals exhibited obvious malformations in eye morphology; Table S1 and Figure F). We could also readily identify autophagic structures in eye disc cells clonally deficient in Atg17 or Atg1 function (Figure 4H to I’).
Knockdown of Atg13 and Atg17 had almost no effect on eye development (Table S1 and Figure 3F). Deletion alleles of Atg13 and Atg17 did also not change (Atg13) or only occasionally altered (Atg17) the morphogenesis of this organ (Figure. S17). This is particularly interesting, as these mutations effectively abolish the transcriptional activity of the corresponding genes in the fat body [63,64]. Analyzing homozygous mutant progeny of heterozygous parents however revealed the presence of both Atg13 transcript and protein in the eye disc of L3W larvae (Figure. S21A to A’’ and S22A, A’). Similar to these results, Atg17-specific mRNA could also be detected in eye disc samples dissected from homozygous Atg17 mutants (Figure. S21B, B’ and S22B). Since both mutations (Atg13∆81 and Atg17d130) represent large deletions covering a significant part of the corresponding coding region, the presence of transcripts (and proteins) could be the consequence of maternally contributed factors. Using a dominant female sterile technique (with the use of ovoD1 dominant negative mutation), we generated homozygous Atg13 mutants with no maternal Atg13 product, and found that animals die prior to the L3W stage (note that homozygous Atg13 mutants with maternal contribution die as pupae) (Figure. S21A’’’). Probably due to these mechanisms, specific transcripts and autophagic structures accumulated, although at lowered levels than in controls, in Atg13 and Atg17 mutant eye disc samples (Figure. S22 and S23). Together, these results raise the possibility that maternal effect can also rescue autophagic activity in the eye disc of larvae homozygous for certain Atg mutations and derived from heterozygous parents.
To further prove the specificity of genetic compensation eliminating the phenotypic manifestation of Atg lf mutations, we silenced Atg14 in the Atg14∆5.2 mutant genetic background (importantly, Atg14 encodes a single transcript and has no paralog). Atg14 RNAi animals exhibited a compromised eye phenotype with a relatively high penetrance (Figure 3F and Table S1), while the ∆5.2 mutation [56] did not influence eye morphology (Figure 5). If genetic compensatory mechanisms rescue normal eye morphology in Atg14∆5.2 mutants, one would expect the suppression of the eye phenotype caused by RNAi treatment in the mutant background (in the mutant, there is no transcript that the RNAi could degrade). Indeed, the presence of the ∆5.2 mutation highly rescued normal eye development in Atg14 RNAi samples (in females, the penetrance of wild-type eye morphology increased from 60% to 95%, in males, it was elevated from 50% to 80%) (Figure 5).
Figure 5.
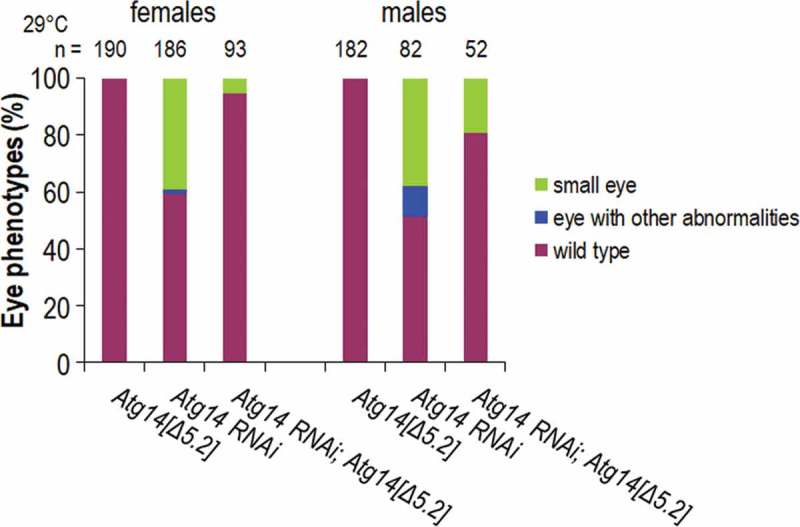
Genetic compensation rescues normal eye development in Atg14∆5.2 mutants. Atg14-specific RNAi treatment causes highly penetrant defects in eye development in both genders. A loss-of-function mutation in Atg14, ∆5.2, however, does not influence eye morphology. In Atg14∆5.2 mutants with no Atg14 transcript, the eye phenotype caused by Atg14 RNAi treatment is significantly suppressed (the mutation eliminates the transcript on which RNAi would act).
Based on genetic compensation discussed above we postulate that lf mutations in Atg genes do not completely eliminate autophagy functions in the affected tissues, thereby masking the phenotypic manifestation of mutant alleles. In good accordance with this assumption, the level of Ref(2)P/SQSTM1/p62 serving as a substrate for autophagy varied significantly among different Atg mutant animals (Figure. S24). The most significant Ref(2)P accumulation was evident in Atg13 and Atg17 mutant samples, the gross mutant phenotype of which appears to be the most severe (lethal) among those examined (the other mutants are viable). Thus, in the latter samples, autophagy still operates, although at decreased levels as compared with control.
Knockdown of Atg genes in the eye disc triggers apoptotic cell death
Reduced activity of Atg genes in the entire eye disc can retard eye development; the affected animals frequently displayed a small eye or eyeless phenotype (Table S1 and Figure 3). To address whether these morphological defects result from, at least in part, excessive cell death, we monitored the amount of cells with apoptotic features in normal (control) versus autophagy deficient eye disc samples. We found that samples from animals depleted for Atg3, Atg14 or Atg101 show a much higher number of TUNEL-positive (i.e., fragmented DNA-containing) cells than those derived from the corresponding control [ey-Gal4(II)/+ or ey-Gal4(III)/+] animals (Figure 6A to E,i). We also performed acridine orange (AO) staining on eye discs of L3W stage larvae to detect acidic compartments, whose accumulation is also a characteristic feature of cells undergoing apoptosis. Samples from Atg3, Atg14 and Atg101 RNAi animals showed increased levels of AO-positive cells, relative to the corresponding controls (ey-Gal4) (Fig. 6A’ to E’, I). The elevated number of TUNEL- and AO-positive cells in Atg RNAi samples was evident both in front of and behind the MF.
Figure 6.
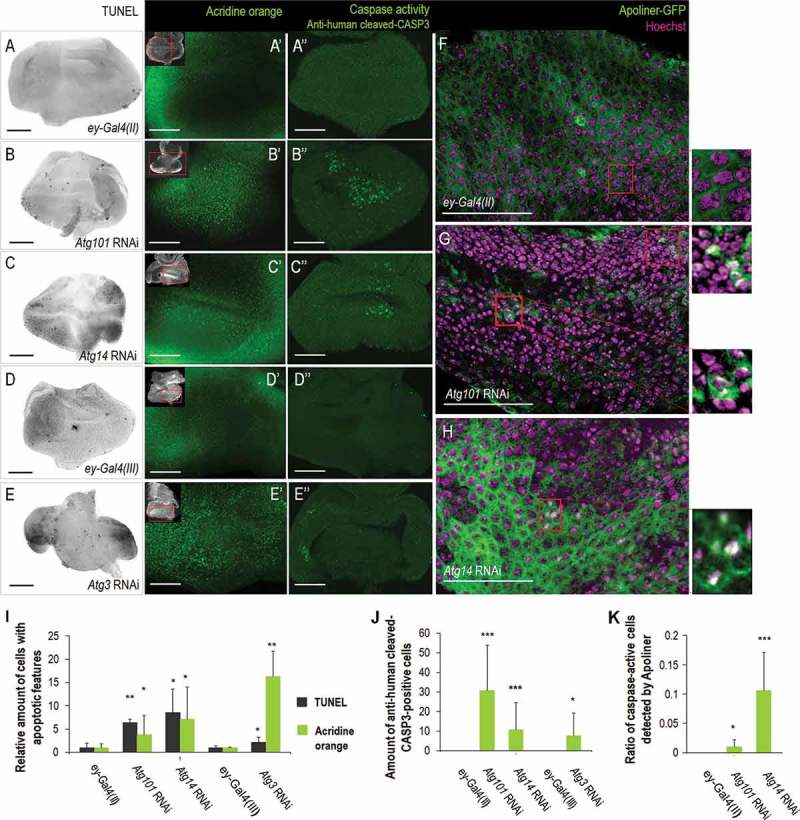
Downregulation of Atg genes in the eye disc triggers apoptosis. (A) TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) staining reveals only a few fragmented DNA-containing nuclei in the eye disc of an ey-Gal4(II)/+ larva (control). (B, C) TUNEL-positive cells in Atg101 RNAi and Atg14 RNAi samples. In the RNAi samples, higher numbers of TUNEL-positive nuclei are evident, as compared with controls. (D) ey-Gal4(III)/+ served as a control for (E) the Atg3 RNAi sample. (A’ to E’) Acridine orange (AO) staining identifies acidic (apoptotic) cell bodies (green foci). Control ey-Gal4/+ samples (A’, D’) contain much fewer AO-positive structures than the corresponding RNAi samples (B’, C’, E’). In images A to H, the antenna part is up; bars: 50 µm. At the upper left corner of AO-stained images, a small picture shows the entire eye-antenna imaginal disc and a red rectangle indicates the area enlarged. Eye discs were dissected from L3W larvae. (A’’ to E’’) Human cleaved-CASP3/Caspase-3-specific antibody staining reveal cells showing increased caspase activity and presumably undergoing apoptosis. Control samples (A’’ and D’’) contain no human cleaved CASP3 immunoreactive cell while the corresponding RNAi samples do (B’’, C’’, E’’). (F to H) The Apoliner-gfp reporter gene functions as a sensor for effector caspase activity in cells undergoing apoptosis. Apoliner-GFP normally binds the plasma membrane (green), but effector caspases (primarily Drice and Cp1) cleaves the nuclear localization signal-GFP tag from the membrane, thereby transferring GFP into the nucleus (white signal, as a result of GFP and Hoechst dye colocalization). (F) There is no detectable level of effector caspase activity in the eye disc of an ey-Gal4(II)/+ larva (control). Silencing of Atg101 (G) and Atg14 (H) in the eye primordium increases the number of nuclei with white signal, as compared with control samples. Enlarged boxes represent disc area in higher magnification, eye discs were dissected from L3W larvae. (I) Quantification of cells with apoptotic features in control (ey-Gal4) versus Atg RNAi genetic backgrounds. Average numbers of TUNEL-positive nuclei (grey) and the area of AO-positive structures (green) are indicated. (J) The amount of cells showing caspase-associated immunoreactivity in control (ey-Gal4) versus Atg RNAi animals. (K) Quantification of cells with effector caspase activity, detected by Apoliner (from panels F to H). In panel I, data are normalized to their own control, in panels I to K, bars represent mean ± S.D., *: P < 0.05, **: p < 0.01, ***: P < 0.005, two-sample Student t test, t test for unequal variances or Mann-Whitney U test. Temperature for silencing Atg14 and Atg101 was 29°C, for silencing Atg3 was 25°C.
Consistent with these data, human cleaved-CASP3/caspase-3-specific antibody staining also revealed elevated amounts of cells showing increased caspase activity in samples dissected from Atg3-, Atg14- and Atg101 RNAi animals, as compared with their corresponding controls (Figure. 6A’’ to E’’, J). This implies increased levels of cell death because this human cleaved-CASP3-specific antibody reveals CASP9-like Dronc activity in Drosophila, at least in part due to generating cleaved Drice and cleaved Dcp1 effector caspases [65]. A UAS-Apoliner reporter has previously been developed to effectively detect effector caspase activity in dying apoptotic cells [66]. Using this tool, we observed intense enzymatic activity in samples dissected from certain Atg RNAi animals (Figure 6F–H,K); in the enlarged part of panels G and H, intense white labeling – that marks cell death events – is visible). However, contrary to what we found by TUNEL and AO staining, caspase activation was predominantly detectable in front of the MF (in the PZ and prospective head cuticle). This implies that downregulation of Atg genes in the eye disc triggers at least 2 types of programmed cell death, a caspase-independent and caspase-dependent apoptosis. The former mainly occurs in the DZ, while the latter appears in front of the MF. Alternatively, the elimination of cell corpses is perturbed in the DZ, or the sign of human cleaved-CASP3-specific antibody may reflect apoptosis-independent caspase activity in the PZ [65].
In sum, we conclude that defects in autophagy in the developing eye disc promote apoptotic cell death, and this effect is likely to contribute to reduction in size of the affected adult eye. Inhibiting autophagy in the DZ alone (GMR-Gal4) does not impair eye development. Thus, autophagic activity in front of and/or within the MF is necessary for the survival of columnar cells in the entire eye disc.
The Hox gene lab (labial) is expressed in the disc proper where it modulates the transcription of Atg8a-a
As demonstrated above, the distribution of Atg8a-positive autophagic structures exhibited a specific pattern in the developing eye tissue, locating predominantly in the MF and DZ (Figure 2D to E’’’ and S3, S7). This observation prompted us to investigate whether autophagy in this tissue is regulated by developmental factors. Transcriptional control of certain Atg genes, including Atg8a, plays an important role in autophagy induction [52,67,68]. Atg8a encodes 3 isoforms, A, B and C, out of which Atg8a-A appears to function primarily in early phases of eye development (Fig. 4A’, B).
To gain insights into the possible mechanisms underlying Atg8a-A regulation during eye development, we searched for conserved binding sites of developmental regulatory factors in the Atg8a locus (including both regulatory and coding regions), and identified 2 putative conserved binding sites for Hox proteins (Homeobox-containing transcription factors, a subset of homeotic proteins), master regulators of early developmental events. One of these newly identified sites is located in the first intron of Atg8a-A, while the other is located within its 3ʹ untranslated region (3ʹ UTR) (Figure 7A). In close proximity to these Hox regulatory elements, putative binding sites for Hox cofactors including Exd (Extradenticle) and Hth (Homothorax) were also identified. The intronic binding site appears to be specific to Lab, whereas the 3ʹ UTR binding site seems to be specific to Dfd (Deformed), but other Hox proteins cannot be excluded (the putative Lab binding site is actually similar to an alternative Lab consensus binding sequence identified in the regulatory region of the Drosophila gene CG11339) [69,70]. Both lab and Dfd are expressed in the peripodial membrane of the eye-antennal disc [71,72]. To investigate the expression pattern of these Hox genes in more detail, in situ hybridization assays were performed by using antisense lab and Dfd RNA probes. Specificity of the probes was confirmed by in situ hybridizations which recapitulated the formerly established expression patterns at certain embryonic stages (FlyBase) (Figure 7B,C and S25A) [73,74]. According to these results, lab was mainly expressed in the MF and in the area from which the head cuticle develops, as well as weak staining was detectable in other parts of the PZ and in the peripodial membrane (Figure 7D,D’). It is worth to note that this newly identified expression pattern for lab is much wider in this organ than reported previously [71,72]. As strong accumulation of Atg8a-positive autophagic structures was also evident in the MF (Figure 2D to E’’’ and S3), we propose that Atg8a-A and its potential transcriptional regulator Lab share activity domains in the eye disc. We also examined Dfd expression, and found that it is only evident in the peripodial membrane (Figure. S25B, B’), as reported previously [71,72]. This expression domain was further confirmed by analyzing a Dfd-GFP reporter system (Figure. S25C to D’).
Figure 7.
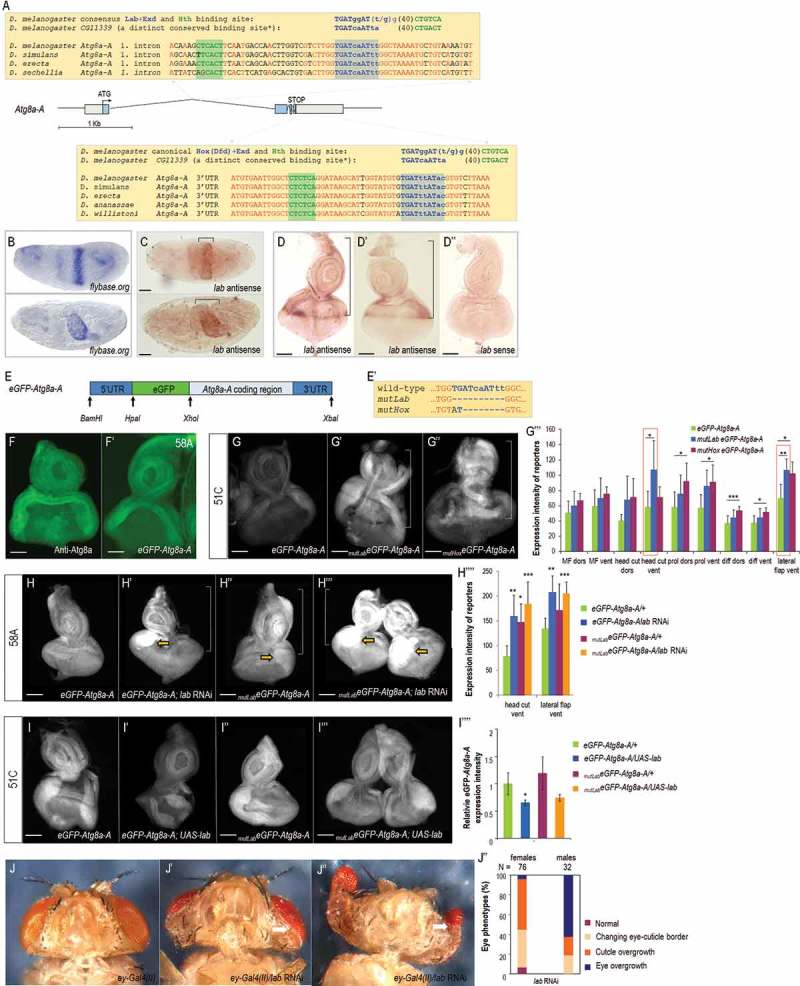
Lab represses Atg8a in the regions of prospective ventral head cuticle and ventral lateral flap. (A) Structure of the Atg8a-A coding region and the position of the 2 conserved Hox-Exd (blue letters) and Hth (green letters) binding sites. Blue boxes indicate coding sequences, connecting lines correspond to intronic sequences, and grey boxes represent 5ʹ and 3ʹ untranslated regions (UTRs). The ATG site and STOP codon are also indicated. Parts of Atg8a-A coding sequences from Drosophila species were aligned. Identical nucleotides nearby the Hox-Exd-Hth binding sites are represented by red letters. Nucleotides that belong to the Lab site are in uppercase, those belong to the Exd site are in lowercase. The canonical Hox-Exd binding site is indicated. *A distinct consensus Lab-Exd-Hth site that was identified in CG11339 gene [69]. (B) Localization of the lab transcript in the 13th embryonic stage (up) and in a late 16th embryonic stage (bottom), according to the FlyBase [73]. (C) In situ hybridization of lab RNA shows an expression pattern being identical to those found previously (in panel C). This shows the specificity of the probe (antisense lab RNA). (D, D’) In situ hybridization of antisense lab RNA in the eye disc. lab is mainly expressed in the morphogenetic furrow and in the region of prospective head cuticle. (D’’) In situ hybridization of sense lab RNA in the eye disc shows no specific staining (negative control). (E) Structure of an eGFP-Atg8a-A reporter gene driven by endogenous regulatory elements. Restriction enzymes used for cloning are indicated (arrows). (E’) Sequences deleted from the mutated versions of the reporter are indicated by dashes. (F) Anti-Atg8a antibody staining on an eye disc. Conventional (non-confocal) fluorescent picture displaying Atg8a protein distribution, rather than autophagic structures as it was shown in Figure. 2(d) to D’’’ and S3A to A’’’. (F’) Expression pattern of eGFP-Atg8a-A reporter in the eye disc. Conventional (non-confocal) image. Atg8a-specific antibody staining (F) and GFP reporter analysis (F’) reveal similar accumulation patterns. (G to G’’) eGFP-Atg8a-A expression is significantly enhanced in regions anterior to the MF when either of the potential Hox|Exd binding sites was mutated (in the first intron or 3ʹ UTR, shown in panel E’), as compared to the control reporter. 51C and 58A represent cytological regions. (G’’’) Quantification of expression (pixel) intensity of eGFP-Atg8a-A reporter with wild-type vs. mutant Hox binding sequences in 9 different regions of the eye disc (these regions are shown in Fig. S24). Red frames indicate regions where expression levels statistically differ between wild-type and potential Lab binding-site-mutated constructs. (H to H’’’) eGFP-Atg8a-A expression in eye discs from animals with lab deficiency. The area of excessive Atg8a expression is indicated by arrows. ey-Gal4(II) was used as a driver. (H’’’’) Quantification of Atg8a-A expression intensity in genetic background indicated. Only the 2 eye disc regions where significant differences had been observed (G’’’) were assayed. (I to I’’’) eGFP-Atg8a-A expression in eye discs from animals with a lab-hyperactive genetic background. Ectopic Lab represses Atg8a expression. ey-Gal4(III) was used as a driver. (I’’’’) Quantification of Atg8a-A expression intensity in genetic background indicated. In panels F, F’, H to H’’’ and I to I’’’, pictures were taken by conventional fluorescence microscopy, i.e. without (semi)confocal sectioning. (J to J’’) Eye morphology in lab RNAi adults. Control (J) and RNAi (J’, J’’) samples. Ventral view. In panels J’ and J’’, arrows indicate the region with cuticle overgrowth. ey-Gal4(II) was used a driver. (J’’’) Quantification of eye phenotypes in animals depleted for Lab. In panels G’’’, H’’’’, I’’’’ and J’’’, bars represent mean ±S.D., *: P < 0.05, **: P < 0.01, ***: P < 0.005; two-sample Student t test or t test for unequal variances. In panels D to D’’, F to G’’, H to H’’’ and I to I’’’ the antenna part is up; bars 50 µm. Eye discs were dissected from L3W larvae. Experiments were carried out at 25°C (A to G’’’, I to I’’’’) or 29°C (H to H’’’’, J to J’’’).
To test whether the 2 newly identified conserved Hox binding sites in the Atg8a-A locus are functional in vivo, we generated an eGFP-Atg8a-A reporter construct containing endogenous upstream and downstream regulatory sequences, together with the entire coding region fused with eGFP (Figure 7E). This construct involves both of the putative Hox binding sites identified in this study. Using site-directed mutagenesis, we further generated 2 mutant versions of the construct. One of them lacks the intronic (i.e., Lab-specific) Exd-Hox binding site (mutLabeGFP-Atg8a-A), while the other misses the 3ʹ UTR (i.e. Dfd-specific) Exd-Hox binding site (mutHoxeGFP-Atg8a-A) (Fig. 7E’). Importantly, the wild-type reporter was capable of recapitulating the accumulation pattern of Atg8a proteins, obtained by anti-Atg8a antibody staining and using conventional fluorescent microscopy (Figure 7F,F’). The expression intensity of the mutant reporters – integrated into different genomic environments, the 51C and 58A cytological regions – was significantly elevated in the anterior part of the eye disc, in front of the MF, as compared with the control (non-mutated) construct (Figure 7G to G’’). To determine precisely the area(s) where Lab may repress Atg8a-A expression, we divided the eye disc into 9 parts, and determined mutLabeGFP-Atg8a-A expression levels in these subregions (Figure. 7G’’’ and S26). Quantification of mutLabeGFP-Atg8a-A expression intensity showed that the absence of the putative Lab binding site leads to elevated expression in the ventral prospective head cuticle and lateral flap (bars highlighted by red frames in Figure. 7G’’’). mutHoxeGFP-Atg8a-A expression was also much stronger, mainly in the prospective head cuticle and ventral lateral flap, than the corresponding control (Figure. 7G’’’). Based on these data we propose that these potential Hox binding sites are functional in vivo, and that Lab, and perhaps (an)other Hox protein(s), directly regulates Atg8a-A in these regions. As Dfd accumulates in the peripodial membrane but not in the disc proper (Figure. S25B to D’), we focused on the putative Lab binding site only in further experiments.
To confirm the inhibitory effect that Lab exerts on Atg8a-A expression in front of the MF, we downregulated and overexpressed lab from drivers that are active in this area. Indeed, the former intervention strongly upregulated (Figure 7H to H’’’’) while the latter inhibited (Figure 7(i) to I’’’’) eGFP-Atg8a-A expression. Excessive expression of the reporters was most evident in the region of the ventral head cuticle and lateral flap (yellow arrows in Figure. 7H’ to H’’’). Thus, Lab inhibits Atg8a-A expression in front of the MF, especially in the region of the prospective head cuticle. The inhibitory effect of Lab hyperactivation on Atg8a-A expression was abolished when the Lab binding site mutant reporter version was examined (Figure 7I to I’’’), confirming the in vivo functionality of this particular Lab binding site.
The expression profile of these reporters highly coincided with the aberrant eye morphology of lab RNAi adult flies. Depletion of lab led to a shift in the ventral head-eye cuticle border in favor of the head cuticle (the white arrow in Figure. 7J’, J’’). This phenotype was often associated with the overgrowth of the head cuticle as well as with the lateral overgrowth of adult eyes (Figure. 7J’’’), morphological features that have been described previously for lab4 mutants [72]. We conclude that Lab inhibits the expression of Atg8a-A in the ventral prospective head cuticle and ventral lateral flap via the newly identified putative binding site.
Lab upregulates Atg8a-A in the MF
As shown above, autophagic structures abundantly accumulate in the MF and DZ (Figure 2 and S3, S6, S7). lab mRNA was also readily detectable in the MF, but not in the DZ (Figure 7D,D’). Upon these data, we investigated how Lab influences the transcription of Atg8a-A in the MF. To address this issue we silenced lab by ey-Gal4(III) driver that is active in the MF and DZ (Figure 1 and S5C). Semi-quantitative PCR experiments revealed highly reduced levels of Atg8a-A transcript in lab RNAi eye disc samples, as compared with controls (Figure 8A). Downregulating lab by GMR-Gal4 driver being active only in the DZ, however, did not alter Atg8a-A transcript levels (lab is not expressed in the DZ; Figure 7D, D’) (Fig. S27). Next, relative transcript levels of Atg8a-A were determined by quantitative real-time PCR in eye disc samples dissected from L3W larvae with control versus lab RNAi and lab overexpressing genetic backgrounds (Figure 8B). Data convincingly showed that samples defective for lab contain significantly fewer Atg8a-A transcripts while those hyperactive for lab display higher levels of Atg8a-A transcripts than control ones. Thus, Lab activates Atg8a-A expression in the MF. Taken together, we suggest that in the eye primordium, Lab has a dual role in the regulation of Atg8a-A activity. First, Lab inhibits Atg8a-A transcription in the prospective ventral head cuticle and lateral flap, presumably through a Lab-Exd-specific binding site we identified in the first intron (Figure 7G to I’’’’). It is likely that this regulatory interaction plays a role in the determination of the normal head-eye cuticle border (Figure 7J to J’’’). Second, Lab promotes the expression of Atg8a-A in the MF to activate autophagy (Figure 8A,B). These opposite effects of Lab on Atg8a-A activity may be mediated by different Hox cofactors.
Figure 8.
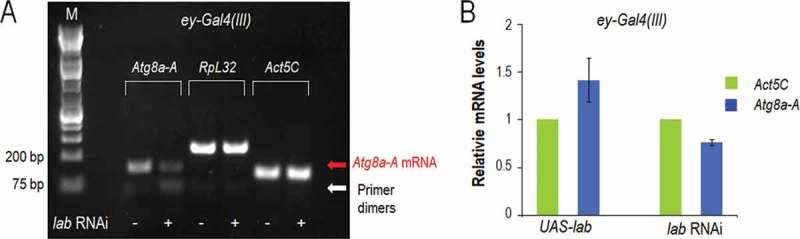
Lab activates Atg8a-A expression in the MF. (A) Semi-quantitative RT-PCR experiment displays reduced levels of Atg8a-A transcript (red arrow) in lab RNAi [driven by ey-Gal4(III)] eye discs, as compared with untreated control samples. RpL32 and Act5C serve as internal controls. M; molecular size marker. ey-Gal4(III) is expressed in the area of the MF and DZ. Note that lab RNAi driven by GMR-Gal4 that is active in the DZ only does not affect Atg8a transcript levels (Fig. S25). (B) qPCR showing relative levels of Atg8a-A mRNA in lab-hyperactive (UAS-lab) versus lab-depleted [driven by ey-Gal4(III)] genetic backgrounds, normalized to their own controls and mRNA levels of internal control genes. Act5C served as an internal control. Eye disc samples of L3W larvae were assayed. In panel B, bars represent mean ±S.D. Temperatures were 29°C (A and lab RNAi part of B) or 25°C (UAS-lab part of B).
Lab activates autophagy in the MF and DZ
As shown above, Lab increases Atg8a-A expression in the MF (Figure 8 and S27). This finding raised the intriguing possibility that Lab influences eye development, at least in part, through modulating autophagic activity. The overexpression of lab in the eye disc by ey-Gal4(II) driver led to the formation of headless adults. Thus, we overexpressed lab by an eye-specific driver with a weaker activity domain, ey-Gal4(III), and found that this intervention results in small eyes or a phenotype without eyes in the affected adults, with almost a full penetrance (Figure 9A,B) (in females: no eye, 10.20%; small eye, 89.80%, normal eye, 0%; and in males: no eye, 18.75%; small eye, 75.00%; normal eye, 6.25%). On the contrary, eye disc-specific silencing of lab resulted in eye overgrowth and also compromised eye development through altering the boundary of the head-eye cuticle (Figure 7J to J’’’ and 9C, D).
Figure 9.

Lab promotes autophagic activity in the differentiation zone in a cell non-autonomous way. (A to B’’’’’) Overexpression of lab enhances while its silencing (C to D’’’’’) reduces autophagic activity in the DZ. Both interventions can lead to excessive cell death revealed by TUNEL and acridine orange (AO) staining. (A) Stereomicrograph of an ey-Gal4(III)/+ adult head, serving as a control for lab overexpression. It shows normal eye morphology. (A’) Atg5 accumulation in the eye disc of an ey-Gal4(III)/+ control animal. (A’’) mCherry-Atg8a accumulation in the eye disc of an ey-Gal4(II)/+ animal (control). Red foci correspond to autophagosomes and autolysosomes. (A’’’) LysoTracker Red-positive structures in the eye disc of an ey-Gal4(III)/+ control animal. Red foci indicate lysosomes, autolysosomes and multivesicular bodies. (A’’’’) TUNEL staining reveals only a few fragmented DNA-containing nuclei (i.e. cells undergoing apoptosis) in the eye disc of an ey-Gal4(III)/+ control animal. (A’’’’’) AO staining identifies only a few apoptotic cell bodies in the eye disc of an ey-Gal4(III)/+ a control animal. (B) Stereomicrograph of an UAS-lab/+; ey-Gal4(III)/+ adult head with reduced eye morphology. (B’) Overexpression of lab during eye development leads to enhanced Atg5 accumulation, (B’’) mCherry-Atg8a expression, (B’’’) LysoTracker Red-positive staining, and (B’’’’) increased numbers of TUNEL-positive and (B’’’’’) AO-positive structures. (C to C’’’’’) Samples from ey-Gal4(II), UAS-Dcr-2/+ animals, serving as controls for lab RNAi background (D to D’’’’’). Controls exhibit normal eye morphology. (D) Stereomicrograph of an ey-Gal4(II), UAS-Dcr-2/lab RNAi adult head displaying obvious defects in eye morphology (see also Figure 6(j) to J’’). Silencing of lab during eye development leads to reduced amount of (D’) Atg5-, (D’’) mCherry-Atg8a- and (D’’’) LysoTracker Red-positive foci, as well as (D’’’’) increased amounts of TUNEL- and (D’’’’’) AO-positive nuclei. In panels A’ to A’’’’’, B’ to B’’’’’, C’ to C’’’’’ and D’ to D’’’’’, the antenna part is up; bars: 50 µm. At the upper left corner of each image, the red rectangle indicates the enlarged area. Eye disc samples were prepared from L3W larvae. (E) Quantification of the effect of lab overexpression and (F) the effect of lab silencing and lab4 mutation on autophagic activity in the MF and DZ. The ratio of areas of anti-Atg5-/mCherry-Atg8a-/anti-Atg8a-/LysoTracker Red-positive structures and the entire eye disc in each image (eye disc) is on average, data are normalized to the corresponding control. (G) Quantification of the effect of lab overexpression, silencing and lab4 mutation on apoptosis in the eye disc. The ratio of the number of TUNEL-positive nuclei/the area of the AO-positive structures and the entire eye disc in each image (eye disc) is on average; data are compared to their own control. On panels E to G, bars represent mean ± S.D., *: P < 0.05, **: P < 0.01; two-sample Student t test, t test for unequal variances or Mann-Whitney U test. (H) Transmission electron micrograph showing several cells with apoptotic features (arrows) in columnar cells from an animal overexpressing lab in the eye disc; bar 200 nm. Experiments were carried out at 25°C (UAS-lab and lab4) or at 29°C (lab RNAi and its control).
lab overexpression in the eye disc enhanced autophagic activity (Figure. 9A’ to A’’’, B’ to B’’’, E). Conversely, silencing or mutational inactivation of lab in this organ lowered the amount of autophagic structures (Figure. 9C’ to C’’’, D’ to D’’’, F and S28, S31A to A’’). Indeed, the amount of Atg5- and Atg8-positive structures was significantly increased in lab-hyperactive (Figure. 9B’, B’’, E) but decreased in lab-defective genetic backgrounds (Figure. 9D’, D’’, F and S28A, A’, B, B’, S31A to A’’). Similarly, the number of acidic compartments became higher when lab was overexpressed (Figure. 9B’’’, E), but became smaller when lab was silenced or inactivated (Figure. 9D’’’, F, and S28A’’, B’’). These results indicate that lab induces autophagic activity in the MF and DZ. We hypothesize that this effect of lab in the DZ is likely to be realized in a cell non-autonomous manner (as we could detect no lab transcript in this disc region), probably through stable products of target genes it regulates.
We also studied the complex regulatory relationship between lab and autophagy in the fat body of L3F larvae. In good agreement with data we obtained from the MF, fat body cells clonally defective for lab exhibited very low amounts of LysoTracker-Red-positive (acidic) structures, as compared with control cells (Figure. S29A to A’’). In addition, fat body cells clonally overexpressing lab contained much higher amounts of Atg8a-positive autophagic structures than non-overexpressing control ones (Figure. S29B, B’). Thus, in certain cell types, including columnar cells in the MF and larval fat body cells, Lab activates autophagy. The fact that Lab induces autophagy in the larval fat body was somehow unexpected since the other Hox proteins were reported to redundantly inhibit developmental autophagy in fat body cells [61]. Thus, Lab may be the sole Drosophila Hox paralog that activates the autophagic process in this tissue.
To further distinguish the role of lab in the peripodial membrane from its role in the disc proper (as ey-Gal4 drivers are active in both disc proper and peripodial membrane), we used c311-Gal4 driver [42] to silence lab specifically in the peripodial membrane. This intervention enhanced the amount of acidic compartments in columnar cells (Figure. S30). Since lab knockdown driven by ey-Gal4(II) inhibited autophagy in these cells, it is likely that lab is endogenously active in certain columnar cells where it modulates the autophagic process.
Both overexpression and inactivation of lab in the eye disc cause excessive cell death
As demonstrated above, Lab activates autophagy in the MF at least in part through enhancing Atg8a-A expression (Figure 8A, B, 9B to B’’’, D to D’’’, E, F and S28, S31A to A’’). Then, autophagic activity remains high in the DZ in a cell non-autonomous manner (Figure 7D, D’). Since defects in autophagy strongly induced ectopic cell death in this organ (Figure 6), we asked whether deregulation of lab similarly affects cell survival in the developing eye tissue. We found that depletion of lab leads to a massive elevation in the number of TUNEL-positive nuclei and acidic cell bodies, mainly in the DZ (by 2.84 and 1.53 times, respectively) (Figure. 9C’’’’ to D’’’’’, G). Lab deficiency also markedly increased the amount of human cleaved-CASP3-positive cells showing elevated caspase-associated immunoreactivity, but, unlike AO-positive cells, this change was predominantly evident in the PZ and prospective head cuticle (Figure. S31B to B’’, C, C’). lab overexpression similarly increased the number of TUNEL- (8.35 times) and AO-positive (3.49 times) structures (Figure. 9A’’’’, A’’’’’, B’’’’, B’’’’’, G), and the amount of cells with chromatin condensation (Figure 9H). We conclude that the Hox protein Lab, a master regulator of early development, promotes the survival of columnar cells in the eye primordium via, at least in part, fine tuning autophagy. This effect of Lab in the DZ may occur indirectly.
Discussion
Under normal cellular settings, autophagy operates at basal levels to maintain the homeostasis and long-term survival of terminally differentiated cells [75]. Cellular stress factors, however, can trigger autophagic activity at both transcriptional and posttranscriptional levels. This response involves various signaling cues and regulatory proteins [76–78]. The autophagic process also becomes activated during numerous developmental events [16,17,34,35]. For example, during Drosophila metamorphosis the degradation of larval tissues is primarily achieved by autophagy [18,30], or at very early stages of mammalian development the elimination of maternally-deposited factors occurs via autophagic degradation [17]. However, little is known about the key regulatory proteins that control the activity of Atg genes in developmental processes. Hox proteins, master regulators of early animal development, modulate autophagy in the Drosophila fat body [61]. This regulatory interaction between Hox factors and autophagy suggests a much stricter developmental control of the autophagic process than was previously assumed.
In this study we demonstrated that autophagic structures accumulate in a specific pattern in the Drosophila eye disc, predominantly in the MF and DZ (Figure 2, and S3, S6, S7), and that this pattern does not reflect the distribution of 2 key Atg proteins, Atg5 and Atg8a, which, using conventional fluorescent microscopy, were detected nearly ubiquitously in this organ, but most intensely in the area from which the head cuticle develops (Figures 2A, and 6F, F’). Other parts of the developing eye tissue displayed only basal levels of autophagic structures. Thus, autophagy displays a characteristic spatial activity pattern in the eye disc of L3W larvae, raising the possibility that lysosome-mediated cellular self-degradation contributes to the morphogenesis of this organ.
We further showed that silencing of several Atg genes can seriously compromise the development of the Drosophila compound eye (Table S1 and Figure 3A to F and S8, S9). In this set of experiments Atg RNAi constructs were driven by ey-Gal4(II), ey-Gal4(III) or so7-Gal4 that have a broad expression domain in the eye primordium (Figure. S5B to D). The effectiveness of RNAi constructs was increased by parallel-expressing Dcr-2 (making RNAi more efficient), and animals were maintained at 29°C, which is the optimum temperature for Gal4 proteins to bind the UAS sequence. The pleiotropic effect of Atg gene knockdowns included severe reduction in organ size (small eye phenotype), even the complete absence of the organ (eyeless phenotype), and alteration in organ shape (aberrant eye morphology). Some of the Atg RNAi constructs we assayed influenced eye growth and morphogenesis with high penetrance, while other constructs proved highly or completely ineffective (Table S1 and Figure 3F). The former constructs were capable of significantly reducing both the transcriptional and translational activity of the corresponding Atg genes as well as the amount of autophagic structures (Figure. S10, S12 and S13). Contrary to these functional RNAi samples, the latter failed to lower the corresponding protein levels, and were unable to modulate autophagic activity (Figure. S11 to S13). To provide an additional evidence for the specificity of eye phenotypes caused by Atg knockdowns, we rescued normal eye development in Atg8a-, Atg14- and Atg101 RNAi animals by a transgene carrying the wild-type copy of the corresponding Atg gene (Fig. S12D and S14) or a duplication bearing the wild-type copy of Atg101 (Figure S15). In addition, downregulation of Atg genes specifically in the peripodial membrane did not affect eye morphogenesis (Table S2 and Figure. S16).
Previous studies have detected no obvious defect in adult eye morphology when Atg genes are silenced by GMR-Gal4 driver [48,50]. It is possible that GMR-Gal4 is expressed in less excessive levels in the eye disc than ey-Gal4(II) and ey-Gal4(III) do. Alternatively, the function of autophagy is more significant in the MF and/or PZ where GMR-Gal4 is not active.
We also explored the effect of lf mutations in Atg genes on eye development in this organism. In the literature several studies have reported no influence of autophagy on this developmental paradigm [48–52]. Contrary to these data, we found that mutational inactivation of Atg17 and Atg1 can impede eye formation (Figure. S17). Some of the mutant animals failed to develop the organ. The percentage of eye phenotypes in these mutant backgrounds however was relatively low, appearing only in the minority of animals examined. Lf mutations in other Atg genes did not affect eye formation. It has been recently demonstrated in zebrafish that genetic compensatory mechanisms attenuate the phenotypic effect of deleterious mutations but not gene knockdowns [59]. In accordance with these findings, mutations in Atg4a, Atg8a, and Atg18a led to the activation or upregulation of the corresponding paralogous genes, Atg4b, Atg8b and Atg18b, respectively (Figure 4A to C and S12F, S19, S20). Moreover, splice variants of Atg8a, A, B and C, identified only recently were expressed in an orchestrated way to compensate their own deficiency; in the eye disc isoform A is active (and B in a lesser extent), and a mutation that specifically inhibits Atg8a-A resulted in the transcriptional activation or upregulation of isoforms B and C (Figure 4A–F) and S12, S18). We also showed the presence of maternally contributed transcripts in homozygous Atg13 and Atg17 mutant samples derived from heterozygous parents (Figure. S21 and S22). Thus, multiple compensatory mechanisms can abolish eye phenotypes in Atg mutant samples. As an evidence, the Atg14∆5.2 mutation, which alone did not affect eye development, strongly suppressed the highly penetrant eye phenotype of Atg14 RNAi animals (the mutation eliminates the transcripts the RNAi could act on) (Figure 5). The parallel expression of (a) paralog(s) and/or splice variant(s), as well as maternally contributed gene products, each have the potential to rescue autophagic activity in a certain Atg mutant background. In other words, many Atg mutant animals examined so far are not completely defective for autophagy. Indeed, we could readily detect autophagic structures in eye disc samples dissected from Atg8a, Atg13, Atg17 and Atg1 lf mutants (Figure 4G to I’ and S23). In the light of these results, the functional analysis of Atg mutant systems requires more attention in future genetic studies on Drosophila and on other models [79].
In Atg RNAi eye disc samples displaying an obvious phenotype, reduction in autophagic activity was accompanied with enhanced amounts of cells with apoptotic features (Figure 6). Although mutational inactivation of autophagy is known to trigger apoptosis in mammalian cell lines and nematodes [80,81], one can argue that the increased number of apoptotic cell corpses observed in these autophagy deficient systems is simply a consequence of failure in the heterophagic elimination of dying cells, a process that also requires Atg gene function [82,83]. However, an increase in caspase activity pointed to excessive levels of apoptosis rather than defects in the engulfment of dying cells (Figure. 6A’’ to E’’, F to H, J, K). Both methods (staining with human cleaved-CASP3-specific antibody and using the Apoliner caspase sensor) essentially led to the same observation, i.e. increased levels of apoptotic cell death. This is important because human cleaved-CASP3-specific antibody staining alone could mark positive cells independently of caspase activity [65]. Hence, our present data provide evidence for a role of Atg genes in preventing columnar cells from undergoing apoptosis in the Drosophila eye disc. In clonal analysis of Atg lf mutations with ref(2)P accumulation (also known as SQSTM1 and p62 in mammals) there was no apparent cell death effect. Although the lethal Atg13∆81 and Atg17d130 mutations significantly increased ref(2)P levels (Figure. S24), autophagic activity was still observable in these mutant samples (Figure. S23). Presumably, this was due to the presence of maternally contributed factors, explaining why the clonal cells contained autophagic structures (Figure 4H to I’). Alternatively, apoptotic cell death occurred in Atg mutant cell clones but an apoptosis-induced compensatory proliferation mechanism rescued a nearly-normal eye morphology [84].
In mammalian cell cultures, upregulation of the Atg8 ortholog MAP1LC3B by the transcription factor TFEB leads to elevated autophagy [67]. In Drosophila, expression levels of Atg1 and Atg8a are also proportional to autophagic activity [52,68]. Since Atg1 plays a role in brain development in an autophagy-independent manner [85], we focused on the Atg8a genomic region to found potential binding sites for transcription factors that may regulate autophagy during eye morphogenesis. We identified 2 conserved Hox binding sites within the Atg8a coding sequences (Figure 7A). Furthermore, conserved binding sites for 2 Hox co-factors, Exd and Hth, were also uncovered in the close vicinity of these Hox regulatory elements (Figure 7A). These sequence data are consistent with a recent finding that Hox proteins including Dfd, Ubx and Abd-B redundantly inhibit autophagy in the fat body to prevent a premature degradation of the organ [61].
By generating an endogenously regulated eGFP-Atg8a-A translational fusion reporter (Figure 7E) and its 2 mutant derivatives lacking either of the 2 newly identified conserved Hox-Exd binding sites (Figure. 7E’), we revealed that both of these sites are functional in vivo, i.e. they are responsive to regulatory cues (Figure 7F to I’’’’). In front of the MF, particularly in the prospective ventral head cuticle, Atg8a-A proteins accumulated at much higher levels in the Lab binding site mutant versions than in the corresponding control (Figure. 7G’’’). Thus, the intronic regulatory element may mediate Atg8a-A repression by a specific Hox protein, Lab, in the anterior part of the eye disc. In contrast, quantification of Atg8a-A transcript levels in the MF (Figure 8A,B), together with the analysis of autophagic activity (Figure 2 and S7), unambiguously showed that Lab activates Atg8a-A in this eye disc region. In accordance with these results, lab was also expressed at relatively high levels in the PZ, particularly the prospective head cuticle, and in the MF (Figure 7D and D’). Consistent with these observations, lab deficiency in the eye disc led to decreased activity of autophagy, while lab hyperactivity elevated the amount of autophagic structures in the MF and DZ (Figure 9Ato D’’’ and S28, S31). Thus, lab is required for establishing physiological levels of autophagy in the eye disc, most probably by upregulating Atg8a in the MF and downregulating this gene in front of the MF, especially in the regions from which the ventral head cuticle develops. In addition to modulating autophagic activity, dysregulation of lab in the eye disc caused an excess in the amount of columnar cells undergoing apoptosis (Figure. 9A’’’’ to D’’’’’, G, H and S31).
Based on these data we propose a model that Lab exerts a dual effect on Atg8a-A expression in the developing eye primordium (Figure 10). First, Lab represses Atg8a-A in the prospective ventral head cuticle. This regulatory interaction may depend on the Lab regulatory sequence we identified in the first intron of Atg8a-A (Figure 7A), and be required for the correct formation of the head-eye cuticle border (Figure 7J to J’’’). Second, Lab activates Atg8a-A expression within the MF (Figure 8). As a result, autophagic structures are generated abundantly in this eye region (Figure 2 and S3, S6, S7). As the MF moves anteriorly, autophagy activity remains elevated in the DZ. As lab transcripts were largely undetectable in the latter area (Figure 7D, D’), autophagic regulation is achieved by factors other than Lab. Nevertheless, in the MF and DZ, intense autophagy promotes the survival, and likely differentiation, of photoreceptor and accessory cells (Figure 10). We propose that Lab is critical for normal eye development in Drosophila through supporting survival and differentiation of columnar cells. Together, these data may shed light into a more prominent role of autophagy in tissue shaping and organ development than previously thought. As autophagy is implicated in several human developmental disorders, such as Vici syndrome and myopathies [5,8–10], findings presented by this study may also provide a better understanding of the mechanisms underlying such pathologies, thereby having significant medical relevance.
Figure 10.
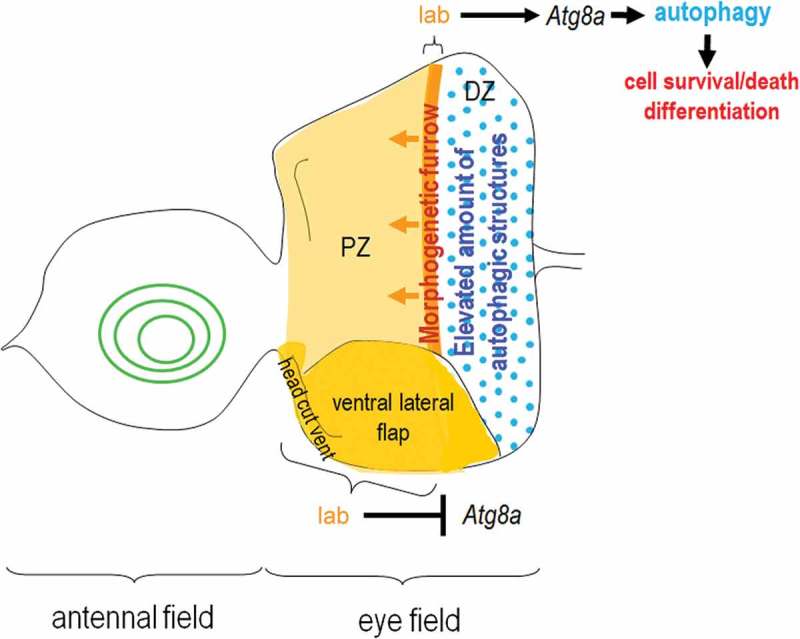
Model for how lab regulates Atg8a-A and influences autophagic activity in the eye disc. Lab may repress Atg8a expression in the regions of prospective ventral head cuticle and ventral lateral flap, while Atg8a expression and autophagic activity in the MF are induced. Levels of autophagic activity remain elevated behind the moving MF (i.e. in columnar cells), which presumably occurs in a cell non-autonomous way. The differentiated regulation of Atg8a expression and autophagy in the eye disc by Lab may involve distinct Hox cofactors. Brown coloring indicates areas where lab transcript is detectable; ochre shows the areas (prospective ventral head cuticle and ventral lateral flap) where Lab inhibits Atg8a expression; orange coloring indicates the region (MF) where lab activates Atg8a. Blue dots show high levels of autophagic structures. PZ, proliferation zone; DZ, differentiation zone; arrows indicate activation, and the bar represents inhibitory interaction.
Materials and methods
Fly stocks, genetics and conditions
Drosophila strains were maintained on standard cornmeal-sugar-agar medium. Stocks were obtained from the Bloomington Drosophila Stock Center (referred to as ‘BL’), the Vienna Drosophila RNAi Center (referred to as ‘v’) and the Drosophila Genetic Resource Center, Kyoto (referred to as ‘DGRC’). Other strains were gift from members of the Drosophila research community. We used the following RNAi lines to silence autophagic genes:
Atg1 RNAi (Atg1JF02273, BL26731 and Atg1HMS02750, BL44034)
Atg2 RNAi (Atg2HMS01198, BL34719 and Atg2JF02786, BL27706)
Atg3 RNAi (Atg3HMS01348, BL34359)
Atg4a RNAi (Atg4aJF03003, BL28367 and Atg4aHMS01482, BL35740)
Atg5 RNAi (Atg5JF02703; BL27551 and Atg5HMS01244, BL34899 and Atg5KK108904, v104461)
Atg6 RNAi (Atg6JF02897, BL28060 and Atg6HMS01483, BL35741)
Atg7 RNAi (Atg7JF02787, BL27707 and Atg7HMS01358, BL34369)
Atg8a RNAi (Atg8aGD4654, v43097, Atg8aJF02895, BL28989 and Atg8aHMS01328, BL34340)
Atg8b RNAi (Atg8bHMS01245, BL34900)
Atg9 RNAi (Atg9JF02891, BL28055 and Atg9HMS01246, BL34901)
Atg10 RNAi (Atg10HMS02026, BL40859)
Atg12 RNAi (Atg12KK111564, v102362 and Atg12HMS01153, BL34675)
Atg13 RNAi (Atg13KK100340, v103381 and Atg13HMS02028, BL40861)
Atg14 RNAi (Atg14KK100903, v108559)
Atg16 RNAi (Atg16HMS01347, BL34358)
Atg17 RNAi (Atg17KK101847, v104864)
Atg18a RNAi (Atg18aJF02898, BL28061 and Atg18aHMS01193, BL34714)
Atg101 RNAi (Atg101KK101226, v106176 and Atg101HMS01349, BL34360)
Vps15 RNAi (Vps15HMS00908, BL34092 and Vps15GL00085, BL35209)
Vps34/Pi3K59F RNAi (Vps34HMS00261, BL33384 and Vps34GL00175, BL36056)
In this study the following Gal4 drivers were used:
ey-Gal4(II) was also obtained from BDSC (w*; P{GAL4-ey.H}, 3–8, BL5534)
ey-Gal4(III) was kindly provided by Barry Dickson (Janelia Research Campus, Ashburn, Virginia, US) GMR-Ga4 (w*; P{GAL4-ninaE.GMR}12, BL1104)
so7-Gal4 (y1 w*; P{so7-GAL4}A/TM6B, BL26810)
c311-Gal4 (y1; P{GawB}c311, BL5937)
Ubi-Gal4 (w*; P{Ubi-GAL4.U}2/CyO, BL32551)
UAS-Dcr-2 (w1118; P{UAS-Dcr-2.D}2, BL24650 and w1118; P{UAS-Dcr-2.D}10, BL24651) was used to enhance the efficiency of long hairpin RNAi constructs.
The following mutant stocks were used:
Atg1 (Atg1KG07993 in yd2, w1118, ey-FLP, GMR-lacZ; Atg1KG07993, FRT80B/TM6B, 111645, DGRC and Atg125 in Atg125, FRT80B/TM6B – kindly provided by Tamas Maruzs, BRC, Szeged, Hungary)
Atg4a (w1118; Mi{ET1}Atg4MB03551, BL23542)
Atg18a (w*; P{SUPor-P}Atg18KG03090 ry506/TM6B, BL13945, modified)
Atg310 [86]
Atg7Δ77[49]
Atg8aKG07569 (outcrossed variant of BL14639)
Atg8ad4 [62]
Atg13Δ81 [63]
Atg14Δ5.2 (hs-FLP; FRT82B, Atg14Δ5.2/TM6C) [56]
Atg17d130 (w*; FRT2A, FRT82B, Atg17d130) [64]
DC352 (w1118; Dp(1;3) DC352, PBac{DC352}VK00033, BL30762) and g-Atg14 [56] were used as the genomic rescue of Atg101 and Atg14, respectively. We applied UAS-Atg17-GFP [64] for the rescue experiment of Atg17d130.
Atg3, Atg7, Atg8a, Atg13 and Atg17 mutant alleles and UAS-Atg17-GFP were gift from Gábor Juhász (Eötvös University, Budapest, Hungary), while Atg14 mutant allele and g-Atg14 were kindly provided by Tor Erik Rusten (Oslo University Hospital, Oslo, Norway).
Genotype w1118 (BL3605) was used as the control.
UAS-GFP-mCherry-Atg8a [87,88] and UAS-Apoliner (w*; P{UAS-Apoliner}5, BL32122), were both recombined with ey-Gal4(II) and were used to examine autophagy and effector caspase activity, respectively.
To analyze Hox gene functions, the following strains were used:
Dfd-GFP (y1 w*; PBacVK00037, BL30877)
UAS-lab (w1118; P{UAS-lab.M}X2, BL7300)
lab RNAi (labKK107959, v100311)
w*; FRT82B, lab4 [72]
We obtained the following strains to perform mosaic analysis:
w*; Ubi-GFP, FRT80B (BL1620)
yd2, w1118, ey-FLP GMR-lacZ; RpS174, w+, FRT80B/TM6B (BL5621)
yd2, w1118, ey-FLP, GMR-lacZ; FRT82B, w+, l(3)cl-R31/TM6B (BL5620)
w*; neoFRT82B, ry506 (BL2035 with the replacement of the first chromosome to w*)
w*, ey-FLP; FRT82B, Ubi-GFP/TM6B (a gift from Deborah Hursh)
w*, hs-FLP; FRT82B, Ubi-GFP (a gift from Deborah Hursh)
y*, w*, hs-FLP; Act< CD2< Gal4, UAS-GFP, nls, 4-mCherry-Atg8a [89]
w*; P{neoFRT}82B P{ovoD1-18}3R/st1 βTub85DD ss1 es/TM3, Sb1 (BL2149) was applied for the Dominant Female Sterile technique [90].
Immunohistochemistry
Fixation and immunostaining of imaginal discs were essentially carried out as described previously [91]. The following primary antibodies were used: rabbit anti-Atg5 (Sigma Aldrich, AV54267) at 1:1000; mouse anti-GFP (Merck Millipore, MAB3580) at 1:500; guinea pig anti-Atonal (gift from Daniel R. Marenda, Drexel University, Philadelphia) [92] at 1:200, rat anti-Atg8a (kindly provided by Gábor Juhász, Eötvös University, Budapest, Hungary) [64,88] at 1:200; rabbit anti-cleaved CASP3/caspase-3 (Cell Signaling Technology, 9661) at 1:400. For nuclear staining, Hoechst 33,342 (0.1 mg/ml, Molecular Probes, H-1399) dye was used. Alexa Fluor 488 and Texas Red (Life Technologies, A21210, A11088, T2767) at 1:500 were used as secondary antibodies.
Lysotracker red and acridine orange staining, TUNEL assay
L3W stage larvae were dissected in PBS (Sigma, P4417), and stained for the eye-antennal imaginal disc (together with the mouth hook and larval brain) using the fluorescent dye LysoTracker Red (Life Technologies, L7528) in 1:1000 dilution for 2 min. Samples were washed once with PBS, and incubated 2 times in PBS for 2.5 min. Eye-antennal discs were mounted into glycerol:PBS (4:1) containing Hoechst 33,342 (0.1 mg/ml; Molecular Probes, H-1399). Acridine orange staining was carried out as follows. L3W larvae were dissected in PBS, eye-antennal imaginal discs were stained with acridine orange (0.01 mg/ml in PBS) for 2 min. Samples were washed once in PBS, then incubated in PBS for 3 min. Discs were mounted into glycerol:PBS (4:1). TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end-labelling) assay was performed as described previously [93]. The following reagents were used: Equilibrium Buffer (Merck Millipore, S7106), Reaction Buffer (Merck Millipore, S7105), TdT enzyme (Merck Millipore, S7107) anti-digoxigenin-AP (Roche, 11,093,274,910), NBT-BCIP solution (Sigma, 72,091).
Analysis of autophagy in larval fat body samples
Preparation of fat bodies was carried out in PBS (Sigma, P4417) solution. LysoTracker Red staining was executed as described above. Covering was achieved in glycerol:PBS (4:1) solution containing 0.1 mg/ml Hoechst 33,342 (Life Technologies, H-1399). Starvation was achieved by transferring larvae onto 20% sucrose solution for 3 h, well-fed condition was provided by using a medium containing 0.825 g cornmeal, 0.405 g sugar, 0.585 g yeast, 2 ml water 3 h prior to dissection.
In situ hybridization
To detect labial mRNA in the eye disc, in situ hybridization was performed using anti-digoxigenin-AP (Roche, 11,093,274,910) and NBT-BCIP solution (Sigma, 72,091) [94]. mRNAs were isolated from 10 mg of wandering larvae lysate with Pure Link RNA Mini Kit (Thermo Fisher Scientific, 12183018A), cDNAs were generated by reverse transcription (RevertAid First Strand cDNA Synthesis Kit, Thermo Fisher Scientific, K1621). The probe for in situ hybridization was generated by labial specific primers (forward: 5ʹ-ACT ACC TGC CAG TGG AAT CG-3ʹ and reverse: 5ʹ-TTC AAC TTT GCT TGC TCG TG-3ʹ).
Western blotting
For anti-Atg5 (rabbit; Sigma Aldrich, AV54267) specificity test, fat body samples were dissected from well-fed L3 stage (76–90 h) Drosophila larvae. In other cases, proteins were isolated from eye-antennal imaginal discs (20 pairs/sample) of wandering L3 larvae. Membranes were probed with anti-Ref(P)2 (rabbit, 1:2500) [62], anti-Atg13 (rat, 1:5000) [64], anti-Atg8a (rabbit, 1:2500) [88] – all of these were kindly provided by Gábor Juhász, Eötvös University, Budapest, Hungary, alpha-Tub84B (mouse, 1:2500; Sigma, T6199), anti-rabbit IgG alkaline phosphatase (1:1000; Sigma, A3687), anti-mouse IgG alkaline phosphatase (1:1000; Sigma, A5153) and anti-rat IgG alkaline phosphatase (1:1000; Sigma, A8438), and developed by NBT-BCIP solution (Sigma, 72,091). Two technical parallels were carried out in each case.
Generation of eGFp-Atg8a-A reporter constructs
An endogenously regulated eGFP-Atg8a-A reporter construct was generated, containing a 268 base pair-long promoter element, the full length Atg8a-A coding sequence except from the stop codon, and eGFP inserted into the end of the upstream regulatory sequence. For PCR amplification, the following primers were used: Atg8a-A promoter element, forward 5ʹ-CGC GGA TCC GCG GCA GTG TGA CCG TAG GTG TG-3ʹ and reverse 5ʹ-ACA GTT AAC TGT GAT TGC AAT GAA GAG GTA ATT GG-3ʹ; eGFP, forward 5ʹ-ACA GTT AAC TGT CAT CCT GGT CGA GCT GGA-3ʹand reverse 5ʹ-CCG CTC GAG CGG CTT GTA CAG CTC GTC CAT GC-3ʹ; the translation initiation site, forward 5ʹ-CCG CTC GAG CGG ATG AAG TTC CAA TAC AAG GAG GAG-3ʹ and reverse 5ʹ-TGC TCT AGA GCA TCT TCC TGT CAC TTA TCG CTG A-3ʹ. PCR experiments were performed with High Fidelity PCR Enzyme Mix (Thermo Fisher Scientific, K0191). PCR fragments were ligated into the vector pattB (7418 base pair, Getentry accession number: KC896839). In vitro mutagenesis was performed by the QuikChange® XL II Site-Directed Mutagenesis Kit (Agilent Technologies, 200,521–5). Mutagenesis of the putative Hox|Exd binding site was performed with the following primers: forward 5ʹ-GGT CGT CTT GGG GCT AAA AT-3ʹ and reverse 5ʹ-CCA AGA CGA CCA TTT TAG CC-3ʹ. Using these primers, a ∆Hox-Exd (1st intron) deficient eGFP-Atg8a-A plasmid was generated, which lacks the TGATCAATTT sequence. Mutagenesis of the putative Hox|Exd binging site in the 3ʹ UTR was made by the following primers: forward 5ʹ-CAC GAT GCA ACA AAA TTC TGT GTG TGT ATG GTT ACG AAT AGG AC-3ʹ and reverse 5ʹ-CAC AGA ATT TTG CAT CGT GGT CCT ATT CGT AAC CAT ACA CA-3ʹ. Using these primers, a ∆Hox-Exd (3ʹ UTR)-defective eGFP-Atg8a-A construct was generated, lacking the sequence CATATTTAG. Drosophila transgenic lines were created by the ΦC31-based integration system [95], attP-51C, attP-58A and attP-68E were used as landing sites. After performing initial tests, attP-51C and attP-58A insertions were used for further experiments.
Microscopy
TUNEL- and eGFP-Atg8a images were captured with an Olympus BX51 microscope (Eötvös University, Budapest, Hungary) (with a UPlanApo 20x/.070 objective), equipped with a F-ViewII camera (Olympus, Eötvös University, Budapest, Hungary), and the AnalySIS software. Semi-confocal fluorescent images were captured with a Zeiss Axioimager Z1 upright microscope (Eötvös University, Budapest, Hungary) (with Plan-NeoFluar 10 × 0.3 NA, Plan-NeoFluar 40 × 0.75 NA and Plan-Apochromat 63x 1,4 NA objectives) equipped with an ApoTome; and AxioVision 4.82 and ImageJ 1.45s software were used to examine and evaluate the data obtained. Confocal images of fixed samples were acquired on a Zeiss LSM710 inverted confocal microscope with a Plan-Apochromat 20x/0.8 M27 objective (MTA TTK, Budapest, Hungary); line averaging: 8x; scanning mode: sequential unidirectional; excitation: 405 nm (Hoechst33,342), 488 nm (eGFP), and 543 nm (mCherry); main dichroic beam splitter: MBS-405 (Hoechst 33,342), MBS-488 (eGFP) and MBS-458/543 (mCherry); Hoechst 33342 was detected 410IF, eGFP was detected between 493 to 575 nm, and mCherry was detected 578IF. Transmission images were captured with the 405 nm laser line. Images were processed by using ZEN software. Photographs of adult eyes were taken with a Nikon SMZ1000 Stereomicroscope (with Nikon Plan APO 1x WD70 objective) equipped with a Media Cybernetics, Evolution MP 5.0 Mega-pixel camera (Eötvös University, Budapest, Hungary) using the QCapture Pro 5.0 software. Stereomicrographs were processed with CombineZ5.
Transmission electron microscopy
Eye-antenna discs were dissected from wandering L3 larvae in PBS, and were fixed with 2% formaldehyde, 0.5% glutaraldehyde, 3 mM CaCl2 and 1% sucrose in 0.1 M Na-cacodylate, pH 7.4 for 1 h at room temperature. After washing with 0.1 M cacodylate buffer, samples were incubated in 0.5% osmium tetroxide for 1 h and in half-saturated aqueous uranyl acetate (for 30 min, at RT), dehydrated in graded series of ethanol, embedded in LR White according to the manufacturer’s instructions, and cured for 24 h at 60°C. Ultrathin sections (70 to 80 µm) were stained with 4% uranyl acetate in 50% methanol (for 8 min) and lead citrate (for 3 min) and were examined on a Jeol JEM-1011 transmission electron microscope (Eötvös University, Budapest, Hungary) at 60 kV, and images were obtained with an Olympus/SIS Morada CCD camera (Eötvös University, Budapest, Hungary), using the Olympus/SIS iTEM software.
PCR experiments
30 pairs of eye-antenna disc were dissected from wandering L3 larvae in PBS, collected in TRI Reagent® solution (Zymo Research, R2050-1–50), and homogenized. RNA isolation was done according to the Direct-zol™ RNA MiniPrep kit (Zymo Research, R2050) protocol, which also includes a DNAse treatment. Reverse transcription was performed using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, K1621).
The following primers were used in semi-quantitative RT-PCR to amplify internal controls: Act5C/Actin5C, forward 5ʹ- GGA TAC TCC CGA CAC AA-3ʹ and reverse 5ʹ-GAG CAG CAA CTT CGT CA-3ʹ; Gapdh1, forward 5ʹ-AAA AAG CTC CGG GAA AAG G-3ʹ and reverse 5ʹ-AAT TCC GAT CTT CGA CAT GG-3ʹ; RpL32, forward 5ʹ-GCT AAG CTG TCG CAC AAA TGG-3ʹ and reverse 5ʹ-GTA GCC AAT GCC TAG CTT GTT C-3ʹ (for experiments shown in Figure 4) or 5ʹ-CTT GTT CGA TCC GTA ACC GAT G-3ʹ (for experiments shown in Figure 8). For detection of Atg4a, forward 5ʹ-TGG TCA GAT GGT TCT CGC C-3ʹ and reverse 5ʹ-TTC AAG GCA GCG CTT TAA GG-3ʹ; Atg4b, forward 5ʹ-TGG TCA GAT GGT TCT CGC C-3ʹ and reverse 5ʹ-AAG GCA CAT GGG GTT TTG G-3ʹ; Atg18a, forward 5ʹ-CAG AAA CCA TGA GCC TGC-3ʹ and reverse 5ʹ-AGA CGC TCG ATG AGG AAC AG-3ʹ; Atg18b, forward 5ʹ-CTT TAC TTC CCT GTC CGT GC-3ʹ and reverse 5ʹ-TAA AGT GCA TCT TGA GGC-3ʹ; Atg3, forward 5ʹ- CAA GTC AAT TGA GAG AGC CAT C-3ʹ and reverse 5ʹ-TGT CGC TAT CTG GAG TGT GC-3ʹ; Atg13, forward 5ʹ-GAG GAC TAC GAC AAG CTG GT-3ʹ and reverse 5ʹ-AGT TTG TCC CTG CCT CTC TC-3ʹ; Atg14, forward 5ʹ-CCA TCT GGA CGT GAA CAA TG-3ʹ and reverse 5ʹ-GCA GAG AGT TTT CGT CCT CTG-3ʹ; Atg17, forward 5ʹ-GCC ATG AGA AGC TCT GCC TA-3ʹ and reverse 5ʹ-TAC AAG GTG AGC GAG TCC TG-3ʹ; Atg101, forward 5ʹ-CAC CTG ACG ACC CTC CAT-3ʹ and reverse 5ʹ-GGG ATC CAA AGT CAC AAT ACT GA-3ʹ. Semi-quantitative RT-PCR for Atg8a: one common reverse primer was used to all isoforms 5ʹ-CGT GAT GTT CCT GGT ACA GGG A-3ʹ, the forward primers were the followings: Atg8a-A, 5ʹ-CAA TAC AAG GAG CAC GC-3ʹ; Atg8a-B, 5ʹ-AGT CAT AGA TGC GCT GA-3ʹ; Atg8a-C, 5ʹ-ATT CCA GAG CCA AGG AAA TG-3ʹ. For Atg8b, forward 5ʹ-ATC CGC AAG CGT ATC AAT CT-3ʹ and reverse 5ʹ-TGA CGA CGT TGT CTG CTT CT-3. For htt, forward 5ʹ-GGT GGT CAA TAG TGG AGT GC-3ʹ and reverse 5ʹ-GCG STT ATC TCC GGG TCA TC-3ʹ. 3 to 4 technical parallels were carried out.
For quantitative real-time PCR experiments, the following primers were used: Atg8a-A, forward 5ʹ-GGT CAG TTC TAC TTC CTC ATT CGC-3ʹ and reverse 5ʹ-ATA GTC CTC GTG ATG TTC CTG-3ʹ. Act5C/Actin5C was used as internal control: forward 5ʹ-CCA GAG ACA CCA AAC CGA AAG-3ʹ and reverse 5ʹ-GAG CAG CAA CTT CGT C-3ʹ. Quantitative real-time PCR was carried out with a Roche LightCycler 480 instrument (Eötvös University, Budapest, Hungary) using the LightCycler® 480 SYBR Green I Master kit (Roche, 03003230001). Three parallel measurements were done, and repeated once.
Image analysis
AxioVision 4.82 and ImageJ 1.45s software [96] were used to examine and evaluate data. Quantification of dot-like structures (foci) was carried out on cut views, which had been generated by maximum intensity projection of 1 μm thick optical sections in AxioVision 4.82. Image were opened in ImageJ, then channel splitting, background subtraction (rolling ball radii were 5 to 15 in case of eye discs, while for fat body samples were 1 to 5), using default threshold (with adjustment when it was necessary) and analyzing particles were done. TUNEL- and cleaved human CASP3-antibody-positive cells were counted manually in ImageJ, using cut views. Apoliner-GFP-positive nuclei were counted manually in ImageJ, using single optical sections. Regions of the eye filed were identified according to the nuclear staining. Quantification of eGFP-Atg8a-A-expression was performed on conventional fluorescent images of the columnar cells-ward side of the eye-antennal discs. Measurement of mean pixel intensity of the selected region of the eye field was done in ImageJ. Quantifications of gel bands from semi-quantitative RT-PCRs or western blots were also carried out in ImageJ, using densitometry analyses. These quantifications highly depend on the number of amplification cycles and/or the time of exposure.
Statistics
A Lilliefors test were used assess whether there was a normal distribution of samples examined. If it was normal, the F test was performed to compare 2 variances in the case of independent samples. If the variances were equal, a two-sample Student t test was used otherwise, a t test for unequal variances (also called the Welch’s t test) was applied. If the distribution of a sample is not normal, Mann-Whitney U test was performed. In the case of paired samples, paired t test was applied for normal distribution; else Wilcoxon signed-rank test was used.
Bioinformatics
The sequence of Atg8a-A genomic regions from 4 Drosophila species (D. melanogaster, D. simulans, D. erecta and D. sechellia) were obtained from FLYBASE (www.flybase.com) [97]. Conserved Hox|Exd and Hth binding sites [69,98,99] were identified by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) [100]. Potential binding sites were aligned with ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/) [101].
Funding Statement
This work was supported by the Hungarian Scientific Research Found [K109349];Hungarian Scientific Research Found [NK78012];Hungarian Scientific Research Found [K82039];Hungarian Brain Reserach Program [KTIA_NAP_13].
Acknowledgments
This work was supported by the Hungarian Scientific Research Found grants NK78012 and K109349 to T.V. and M.S., the Hungarian Brain Research Program (KTIA_NAP_13) and OTKA grant K82039 to J.M, and by the New National Excellence Program of the Ministry of Human Capacities. A part of Drosophila stocks was obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) and Vienna Drosophila Resource Center (VDRC, www.vdrc.at). Other Drosophila strains and reagents were kindly provided by Barry Dickson, Deborah Hursh, Gábor Juhász, Daniel R. Marenda, Tamás Maruzs and Máté Varga. qPCR experiments and the evaluation of results were assisted by Mónika Kosztelnik. The authors also thank Regina Preisinger, Beatrix Supauer, Tünde Pénzes, Gabriella Szabados, Judit Botond and Sára Simon for the excellent technical assistance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contribution
V.B., T.Kov., A.M., P.L., S.Sz., Á.R., P.I.K., T.L., G.H., M.E., J.M., K.T-V., T.V., and M.S. performed experiments; V.B., T.Kov., A.M., M.E., T.Kor., J.M., K.T-V., T.V., and M.S. designed experiments and analyzed data; V.B., T.Kov., A.M., J.M., K.T-V., T.V. and M.S. wrote the manuscript.
Supplementary Material
Supplemental data for this article can be accessed here.
References
- [1].Liu Y, Levine B.. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2014;22:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Das G, Shravage BV, Baehrecke EH.. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol. 2012;4:a008813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Takacs-Vellai K, Bayci A, Vellai T.. Autophagy in neuronal cell loss: a road to death. BioEssays. 2006;28:1126–1131. [DOI] [PubMed] [Google Scholar]
- [4].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vellai T, Takács-Vellai K. Regulation of protein turnover by longevity pathways. Adv Exp Med Biol. 2010;694:69–80. [DOI] [PubMed] [Google Scholar]
- [6].Vellai T. Autophagy genes and ageing. Cell Death Differ. 2009;16:94–102. [DOI] [PubMed] [Google Scholar]
- [7].Vellai T, Takacs-Vellai K, Sass M, et al. The regulation of aging: does autophagy underlie longevity? Trends Cell Biol. 2009;19:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sandri M, Coletto L, Grumati P, et al. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J Cell Sci. 2013;126:5325–5333. [DOI] [PubMed] [Google Scholar]
- [11].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sigmond T, Barna J, Tóth ML, et al. Autophagy in Caenorhabditis elegans. Methods Enzymol. 2008;451:521–540. [DOI] [PubMed] [Google Scholar]
- [13].Varga M, Fodor E, Vellai T. Autophagy in zebrafish. Methods. 2015;75:172–180. [DOI] [PubMed] [Google Scholar]
- [14].Fodor E, Sigmond T, Ari E, et al. Methods to study autophagy in zebrafish. Methods Enzymol. 2017;588:467–496. [DOI] [PubMed] [Google Scholar]
- [15].Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. [DOI] [PubMed] [Google Scholar]
- [17].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Otto GP, Wu MY, Kazgan N, et al. Macroautophagy is required for multicellular development of the social amoeba Dictyostelium discoideum. J Biol Chem. 2003;278:17636–17645. [DOI] [PubMed] [Google Scholar]
- [20].Voigt O, Poggeler S. Autophagy genes Smatg8 and Smatg4 are required for fruiting-body development, vegetative growth and ascospore germination in the filamentous ascomycete Sordaria macrospora. Autophagy. 2013;9:33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Besteiro S, Williams RA, Morrison LS, et al. Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J Biol Chem. 2006;281:11384–11396. [DOI] [PubMed] [Google Scholar]
- [22].Alvarez VE, Kosec G, Sant’Anna C, et al. Autophagy is involved in nutritional stress response and differentiation in Trypanosoma cruzi. J Biol Chem. 2008;283:3454–3464. [DOI] [PubMed] [Google Scholar]
- [23].Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science. 2011;334:1141–1144. [DOI] [PubMed] [Google Scholar]
- [24].Zhang Y, Yan L, Zhou Z, et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell. 2009;136:308–321. [DOI] [PubMed] [Google Scholar]
- [25].Erdelyi P, Borsos E, Takacs-Vellai K, et al. Shared developmental roles and transcriptional control of autophagy and apoptosis in Caenorhabditis elegans. J Cell Sci. 2011;124:1510–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Borsos E, Erdélyi P, Vellai T. Autophagy and apoptosis are redundantly required for C. elegans embryogenesis. Autophagy. 2011;7:557–559. [DOI] [PubMed] [Google Scholar]
- [27].Melendez A, Talloczy Z, Seaman M, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. [DOI] [PubMed] [Google Scholar]
- [28].Juhász G, Csikós G, Sinka R, et al. The Drosophila homolog of Aut1 is essential for autophagy and development. FEBS Lett. 2003;543:154–158. [DOI] [PubMed] [Google Scholar]
- [29].Denton D, Shravage B, Simin R, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tracy K, Baehrecke EH. The role of autophagy in Drosophila metamorphosis. Curr Top Dev Biol. 2013;103:101–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Politi Y, Gal L, Kalifa Y, et al. Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Dev Cell. 2014;29:305–320. [DOI] [PubMed] [Google Scholar]
- [32].Aburto MR, Sanchez-Calderon H, Hurle JM, et al. Early otic development depends on autophagy for apoptotic cell clearance and neural differentiation. Cell Death Dis. 2012;3:e394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tsukamoto S, Kuma A, Murakami M, et al. Autophagy is essential for preimplantation development of mouse embryos. Science. 2008;321:117–120. [DOI] [PubMed] [Google Scholar]
- [34].Hale AN, Ledbetter DJ, Gawriluk TR, et al. Autophagy: regulation and role in development. Autophagy. 2013;9:951–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Di Bartolomeo S, Nazio F, Cecconi F. The role of autophagy during development in higher eukaryotes. Traffic. 2010;11:1280–1289. [DOI] [PubMed] [Google Scholar]
- [36].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. [DOI] [PubMed] [Google Scholar]
- [37].Baerga R, Zhang Y, Chen PH, et al. Targeted deletion of autophagy-related 5 (atg5) impairs adipogenesis in a cellular model and in mice. Autophagy. 2009;5:1118–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kundu M, Lindsten T, Yang CY, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112:1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Miller BC, Zhao Z, Stephenson LM, et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy. 2008;4:309–314. [DOI] [PubMed] [Google Scholar]
- [40].Ready DF, Hanson TE, Benzer S. Development of the Drosophila retina, a neurocrystalline lattice. Dev Biol. 1976;53:217–240. [DOI] [PubMed] [Google Scholar]
- [41].Haynie JL, Bryant PJ. Development of the eye-antenna imaginal disc and morphogenesis of the adult head in Drosophila melanogaster. J Exp Zool. 1986;237:293–308. [DOI] [PubMed] [Google Scholar]
- [42].Gibson MC, Schubiger G. Peripodial cells regulate proliferation and patterning of Drosophila imaginal discs. Cell. 2000;103:343–350. [DOI] [PubMed] [Google Scholar]
- [43].Pappu KS, Mardon G. Genetic control of retinal specification and determination in Drosophila. Int J Dev Biol. 2004;48:913–924. [DOI] [PubMed] [Google Scholar]
- [44].Wolff T, Ready DF. The beginning of pattern formation in the Drosophila compound eye: the morphogenetic furrow and the second mitotic wave. Development. 1991;113:841–850. [DOI] [PubMed] [Google Scholar]
- [45].Hennig KM, Colombani J, Neufeld TP. TOR coordinates bulk and targeted endocytosis in the Drosophila melanogaster fat body to regulate cell growth. J Cell Biol. 2006;173:963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang Y-H, Huang M-L. Reduction of Lobe leads to TORC1 hypoactivation that induces ectopic Jak/STAT signaling to impair Drosophila eye development. Mech Dev. 2009;126:781–790. [DOI] [PubMed] [Google Scholar]
- [47].Juhasz G, Hill JH, Yan Y, et al. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen SF, Kang ML, Chen YC, et al. Autophagy-related gene 7 is downstream of heat shock protein 27 in the regulation of eye morphology, polyglutamine toxicity, and lifespan in Drosophila. J Biomed Sci. 2012;19:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Juhasz G, Érdi B, Sass M, et al. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Velentzas PD, Velentzas AD, Pantazi AD, et al. Proteasome, but not autophagy, disruption results in severe eye and wing dysmorphia: a subunit- and regulator-dependent process in Drosophila. PloS One. 2013;8:e80530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pérez E, Das G, Bergmann A, et al. Autophagy regulates tissue overgrowth in a context-dependent manner. Oncogene. 2015;34:3369–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kramer JM, Staveley BE. GAL4 causes developmental defects and apoptosis when expressed in the developing eye of Drosophila melanogaster. Genet Mol Res. 2003;2:43–47. [PubMed] [Google Scholar]
- [55].Li WZ, Li SL, Zheng HY, et al. A broad expression profile of the GMR-GAL4 driver in Drosophila melanogaster. Genet Mol Res. 2012;11:1997–2002. [DOI] [PubMed] [Google Scholar]
- [56].Katheder NS, Khezri R, O’Farrell F, et al. Microenvironmental autophagy promotes tumour growth. Nature. 2017;541:417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Atkins M, Mardon G. Signaling in the third dimension: the peripodial epithelium in eye disc development. Dev Dyn. 2009;238:2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ochaba J, Lukacsovich T, Csikos G, et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci USA. 2014;111:16889–16894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Rossi A, Kontarakis Z, Gerri C, et al. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature. 2015;524:230–233. [DOI] [PubMed] [Google Scholar]
- [60].Scott RC, Schuldiner O, Neufeld TP. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell. 2004;7:167–178. [DOI] [PubMed] [Google Scholar]
- [61].Banreti A, Hudry B, Sass M, et al. Hox proteins mediate developmental and environmental control of autophagy. Dev Cell. 2014;28:56–69. [DOI] [PubMed] [Google Scholar]
- [62].Pircs K, Nagy P, Varga A, et al. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila. PloS One. 2012;7:e44214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20:2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Nagy P, Karpati M, Varga A, et al. Atg17/FIP200 localizes to perilysosomal Ref(2)P aggregates and promotes autophagy by activation of Atg1 in Drosophila. Autophagy. 2014;10:453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Fan Y, Bergmann A. The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 2010;17:534–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Bardet PL, Kolahgar G, Mynett A, et al. A fluorescent reporter of caspase activity for live imaging. Proc Natl Acad Sci USA. 2008;105:13901–13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Simonsen A, Cumming RC, Brech A, et al. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–184. [DOI] [PubMed] [Google Scholar]
- [69].Ebner A, Cabernard C, Affolter M, et al. Recognition of distinct target sites by a unique Labial/Extradenticle/Homothorax complex. Development. 2005;132:1591–1600. [DOI] [PubMed] [Google Scholar]
- [70].Slattery M, Riley T, Liu P, et al. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147:1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Diederich RJ, Pattatucci AM, Kaufman TC. Developmental and evolutionary implications of labial, Deformed and engrailed expression in the Drosophila head. Development. 1991;113:273–281. [DOI] [PubMed] [Google Scholar]
- [72].Stultz BG, Park SY, Mortin MA, et al. Hox proteins coordinate peripodial decapentaplegic expression to direct adult head morphogenesis in Drosophila. Dev Biol. 2012;369:362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Kumar S, Konikoff C, Van Emden B, et al. FlyExpress: visual mining of spatiotemporal patterns for genes and publications in Drosophila embryogenesis. Bioinformatics. 2011;27:3319–3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bergson C, McGinnis W. An autoregulatory enhancer element of the Drosophila homeotic gene Deformed. EMBO J. 1990;9:4287–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. [DOI] [PubMed] [Google Scholar]
- [76].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Pietrocola F, Izzo V, Niso-Santano M, et al. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol. 2013;23:310–322. [DOI] [PubMed] [Google Scholar]
- [78].He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Mauvezin C, Ayala C, Braden CR, et al. Assays to monitor autophagy in Drosophila. Methods. 2014;68:134–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Boya P, Gonzalez-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Takacs-Vellai K, Vellai T, Puoti A, et al. Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol. 2005;15:1513–1517. [DOI] [PubMed] [Google Scholar]
- [82].Qu X, Zou Z, Sun Q, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. [DOI] [PubMed] [Google Scholar]
- [83].Huang S, Jia K, Wang Y, et al. Autophagy genes function in apoptotic cell corpse clearance during C. elegans embryonic development. Autophagy. 2013;9:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Fan Y, Bergmann A. Apoptosis-induced compensatory proliferation. The cell is dead. Long live the Cell! Trends Cell Biol. 2008;18:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Toda H, Mochizuki H, Flores R 3rd, et al. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nagy P, Varga A, Kovacs AL, et al. How and why to study autophagy in Drosophila: it’s more than just a garbage chute. Methods. 2014;75:151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nezis IP, Shravage BV, Sagona AP, et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol. 2010;190:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Takats S, Nagy P, Varga A, et al. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013;201:531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Arsham AM, Neufeld TP. A genetic screen in Drosophila reveals novel cytoprotective functions of the autophagy-lysosome pathway. PloS One. 2009;4:e6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chou TB, Perrimon N. The autosomal FLP-DFS technique for generating germline mosaics in Drosophila melanogaster. Genetics. 1996;144:1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Klein T. Immunolabeling of imaginal discs In: Dahmann C, editor. Drosophila: Methods in Molecular Biology New York (NY): Humana Press; 2008. p. 253–263. [DOI] [PubMed] [Google Scholar]
- [92].Melicharek D, Shah A, DiStefano G, et al. Identification of novel regulators of atonal expression in the developing Drosophila retina. Genetics. 2008;180:2095–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].McCall K, Baum J, Cullen K, et al. Visualizing Apoptosis In: Henderson D, editor. Drosophila cytogenetics protocols. New York (NY): Humana Press; 2004. p. 431–442. [Google Scholar]
- [94].Wilk R, Murthy SUM, Yan H, et al. In situ hybridization: fruit fly embryos and tissues In: Gallagher SR and Wiley EA, editors. Current protocols essential laboratory techniques. New Jersey (NJ): John Wiley & Sons, Inc; 2008. [Google Scholar]
- [95].Bischof J, Maeda RK, Hediger M, et al. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci USA. 2007;104:3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Schneider CA, Rasband WS, Eliceiri KW. NIH image to image J: 25 years of image analysis. Nat Methods. 2012;9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Attrill H, Falls K, Goodman JL, et al. FlyBase: establishing a gene group resource for drosophila melanogaster. Nucleic Acids Res. 2016;44:D786–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Takács-Vellai K, Vellai T, Chen EB, et al. Transcriptional control of Notch signaling by a HOX and a PBX/EXD protein during vulval development in C. elegans. Dev Biol. 2007;302:661–669. [DOI] [PubMed] [Google Scholar]
- [99].Regos A, Lengyel K, Takacs-Vellai K, et al. Identification of novel cis-regulatory regions from the Notch receptor genes lin-12 and glp-1 of Caenorhabditis elegans. Gene Expr Patterns. 2013;13:66–77. [DOI] [PubMed] [Google Scholar]
- [100].Madden T. The BLAST sequence analysis tool In: The NCBI handbook. 2nd ed. Bethesda (MD): National Center for Biotechnology Information (US); 2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK153387/ [Google Scholar]
- [101].Larkin MA, Blackshields G, Brown NP, et al. ClustalW and ClustalX version 2. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.