

Abstract
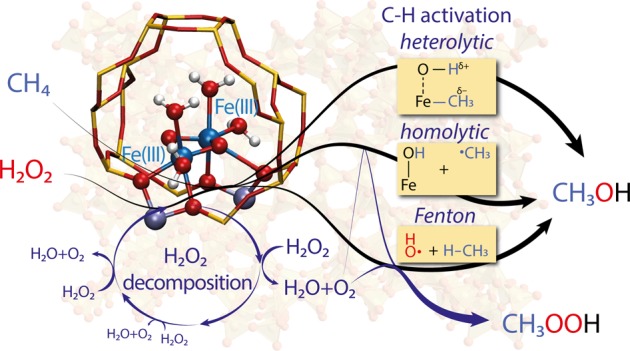
Periodic density functional theory (DFT) calculations were carried out to investigate the mechanism of methane oxidation with H2O2 over the defined Fe sites in Fe/ZSM-5 zeolite. The initial Fe site is modeled as a [(H2O)2–Fe(III)–(μO)2–Fe(III)–(H2O)2]2+ extraframework cluster deposited in the zeolite pore and charge-compensated by two anionic lattice sites. The activation of this cluster with H2O2 gives rise to the formation of a variety of Fe(III)-oxo and Fe(IV)-oxo complexes potentially reactive toward methane dissociation. These sites are all able to promote the first C–H bond cleavage in methane by following three possible reaction mechanisms: namely, (a) heterolytic and (b) homolytic methane dissociation as well as (c) Fenton-type reaction involving free OH radicals as the catalytic species. The C–H activation step is followed by formation of MeOH and MeOOH and regeneration of the active site. The Fenton-type path is found to proceed with the lowest activation barrier. Although the barriers for the alternative heterolytic and homolytic pathways are found to be somewhat higher, they are still quite favorable and are expected to be feasible under reaction conditions, resulting ultimately in MeOH and MeOOH products. H2O2 oxidant competes with CH4 substrate for the same sites. Since the oxidation of H2O2 to O2 and two [H+] is energetically more favorable than the C–H oxofunctionalization, the overall efficiency of the latter target process remains low.
Keywords: selective oxidation, catalytic reaction networks, DFT calculations, zeolites, heterogeneous catalysis, computational chemistry, selectivity control
1. Introduction
The conversion of methane to methanol is considered one of the greatest challenges in catalysis today.1−3 Currently, a two-step process via synthesis gas is used in industry. For economic reasons, a one-pot production of methanol at near-ambient conditions is desirable.4,5 The main challenge of this reaction is the targeted dissociation of the first C–H bond followed by formation of methanol, which is not the thermodynamically most stable oxidation product. It is easily converted further to other oxygenated hydrocarbons and carbon oxides. In the past hundred years, a tremendous amount of experimental and theoretical work on this process has been conducted.3,6−8 Although the field had witnessed significant achievements, no direct methane oxofunctionalization process suitable for industrial implementation has been delivered so far.
The simplest alternative to the current two-step process is the thermal oxidation of methane.9 Relatively high yields of oxygenated products can be achieved: 60% methanol selectivity at 12–13% methane conversion has been reported. Interestingly, it was proposed that the metal wall of the actual reactor is key to the low selectivity of the process, as it provides the catalytic sites for the overoxidation of methanol to carbon oxides. Although the methanol yields achieved by the thermal oxidation are quite impressive, it was concluded that no further improvement of the process could be delivered by optimization of operational conditions. The introduction of a catalyst capable of shifting selectivity away from the overoxidation path is necessary.5
Among the lower-temperature catalytic paths for methane oxofunctionalization, the catalytic system of Periana et al.10,11 stands out as the most successful. It utilizes (bpym)Pt(II)Cl2 homogeneous catalyst in concentrated sulfuric acid solvent at 200 °C, providing up to 81% selectivity to a methyl sulfate oxofunctionalization product at about 90% methane conversion. Despite its outstanding performance, this system is industrially not applicable due to the harsh reaction conditions and relatively low productivity.
In the past two decades, transition and nontransition metal clusters, such as Co, Fe, Cu, or Zn deposited in zeolites,12−20 came to the forefront of catalytic oxidation research. Mainly three oxidizing agents were applied in combination with these catalysts: (1) O2,12,14 (2) N2O18,19 in gas–solid-phase reactions, and (3) H2O216,17 in liquid–gas-phase reactions. The greatest problem with gas–solid-phase reactions is that they are semicatalytic. Catalyst oxidation, methane activation, and methanol formation by hydrolysis of the strongly adsorbed methoxo species are carried out in separate process stages. Additionally, the product is obtained as a very low-concentration solution, which entails further costs of separation. Recently, promising improvements were achieved by the cofeeding of water in the reaction mixture.21 An alternative solution is the liquid-phase reaction, where methane oxidation with liquid oxidants can be carried out in a single catalytic process. Among different systems considered, promising results were demonstrated by Hutchings and Hammond and their co-workers16,17,22−24, who investigated the oxidation of methane to methanol over an iron-modified (Fe/ZSM-5) zeolite-based catalyst with H2O2 as the oxidant.
Understanding the nature of the catalytic species and reaction mechanism is key to rational optimization and improvement of heterogeneous catalyst. Extraframework clusters present in metal-exchanged zeolites can have different chemical composition and be located at different positions. Distinct configurations and clusters might have different catalytic activity and contribute differently to the overall selectivity of the catalytic process.25−30 That is why a great deal of research has so far been devoted to discriminating the active site from the spectator species and identifying the reaction mechanism in zeolite catalysis. Panov and co-workers18,19 have proposed that when N2O is used as the oxidant, the catalytic species providing a high reactivity of Fe/ZSM-5 toward C–H activation is Fe(IV)=O, denoted as the α-oxygen. Later studies by Schoonheydt, Sels, Solomon, and co-workers31,32 provided additional experimental evidence to this assignment and identified such mononuclear species as the active site for gas-phase methane oxofunctionalization.
These experimental findings are in line with earlier computational studies on the nature of catalytic sites for benzene oxidation with N2O by Fe/ZSM-5.33 Li et al.34 carried out a comprehensive study on the stability and reactivity of different Fe-containing species in ZSM-5 zeolite. It was found that isolated Fe2+ species capable of forming the reactive α-oxygen sites can only be formed within a small fraction of cationic sites featuring a symmetric arrangement of lattice Al sites. Most of the extraframework positions were proposed to be occupied by the alternative oxygen-bridged iron clusters. Importantly, only the isolated sites were shown to contribute to the catalytic benzene oxidation process, as C–H activation over the other species yields highly stable surface structures that effectively deactivate the respective Fe site.
Very recently, Nørskov and co-workers35 reported a computational analysis of a wide range of materials for their ability to active C–H bonds in methane via a homolytic reaction mechanism (Scheme 1, pathway 2). In another example, researchers found that, in a [Cu3O3]2+ cluster deposited in the zeolite framework, the O atom with higher spin density is more reactive.36 In other cases, the authors compared clusters containing different metal ions. One study concluded that Ni is the best-performing among Cu, Zn, and Ni in homogeneous catalysts;37 another study found that Cu is the best among Ni, Co, Fe, Ag, and Au when these metals are deposited in a ZSM-5 zeolite framework.38
Scheme 1. Potential Mechanistic Pathways for C–H Bond Cleavage: (1) Heterolytic, (2) Homolytic, and (3) Fenton-type Activation Investigated in This Paper.
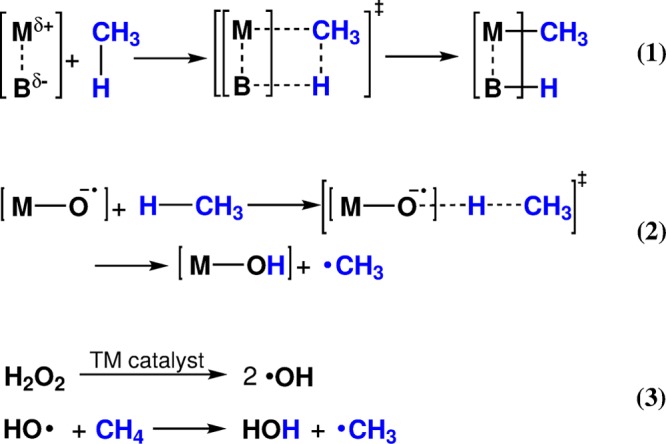
Two principal C–H activation mechanisms providing a path toward selective methane activation are usually distinguished: (1) heterolytic and (2) homolytic reaction mechanisms (Scheme 1).39,40 In the former case, the C–H bond is activated over an acid–base pair to form an anionic alkyl group stabilized by the acid (often a metal cation) and a proton is accepted by the base part of the active site. Note that the heterolytic C–H cleavage is not accompanied by any redox processes within the active site. The actual oxidation of the alkyl moiety should then take place in subsequent steps of the overall catalytic process. In the homolytic mechanism, the C–H bond breaks with the formation of two radical species: the alkyl radical and a formally H radical. The H radical represents a transient species that is readily accepted by the basic moiety of the active site, which is at the same time reduced with 1 e–. The subsequent rebound of the CH3 radical completes the two-electron reduction process and yields the oxidized organic product or surface-bound intermediate. Given the fact that homolytic C–H activation commonly yield a free alkyl radical while the energy losses due to cleavage of a strong C–H bond come predominantly from single-electron reduction of the active complex accompanied by its protonation, the basicity of the proton-accepting site (B) (that is, basically, the strength of the resulting B–H bond) has been recognized as one of the critical parameters defining the overall reactivity of a catalyst toward homolytic C–H activation.35,41 The direct relationships between this parameter and the computed barriers for C-H activation have been demonstrated in recent study by Nørskov and co-workers42 for a wide class of potential heterogeneous catalysts.
In the chemistry of iron (see Plietker),43 the preference for a particular C–H activation mechanism is usually defined by the oxidation state of the Fe site, its coordination environment, and the nature of the ligands.39 Heterolytic C–H cleavage (Scheme 1, pathway 1) is commonly observed over the lower-valent Fe sites (e.g., 3+ and 2+) conjugated with a strong base site. The homolytic C–H dissociation (Scheme 1, pathway 2) is more typical for higher-valent Fe sites that feature more covalent-type bonding and facilitate the oxidative activation of the substrate coupled with the reduction of Fe. In addition to these two very generic reaction channels, scientists distinguish one more mechanism specific to Fe chemistry: that is, the Fenton-type mechanism of C–H activation (Scheme 1, pathway 3). Although formally this mechanism can be regarded as a subtype of the homolytic C–H bond dissociation (pathway 2), it is commonly considered separately in view of the secondary role the Fe site plays in it. In Fenton chemistry, iron ions initiate the radical oxidation paths by producing free OH radical species, which activate C–H bonds.44,45 In the context of selective methane oxofunctionalization, Fenton-type reactions have been explored in works by Shulpin and co-workers46−48 who demonstrated that, in the presence of Fe(II) ions, methane can be oxidized with H2O2 and O2 to produce MeOOH as the main product. Similar chemistry has recently been proposed by Hutchings and co-workers49 to govern methane oxidation by Pd nanoparticles.
In many cases, only one type of activation is investigated. For example, the structure–activity relationship derived by Nørskov and co-workers35 is valid only under the assumption of homolytic C–H bond activation. However, there are density functional theory (DFT) studies that propose heterolytic methane dissociation over Cu and Fe clusters deposited in zeolites.50−52 In a complex system such as transition-metal-exchanged zeolites, different clusters can be found,34 which might promote diverse types of C–H bond activation. In our work, we aim to investigate how different Fe clusters influence the type and energetics of C–H bond cleavage. For this, we analyzed this reaction step over numerous clusters created from binuclear Fe(III)-oxo with H2O2 based on the catalytic system of Hutchings and co-workers.16 Mostly theoretical work focuses on the rate-determining C–H bond activation step, but here we also present the whole catalytic cycle, including formation of the active site, C–H bond activation, and product formation followed by regeneration of the initial site. All three previously introduced potential reaction mechanisms were considered for methane activation. Comprehensive description of the reaction network provides us with atomic-level insights into this immensely complicated heterogeneous catalytic process.
2. Model and Simulation Details
The spin-polarized periodic DFT calculations were carried out with the Vienna ab initio simulation package (VASP).53−56 The Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional57,58 was used together with a plane-wave basis set with a cutoff energy of 450 eV and the projector augmented wave (PAW) method.59,60 To account for the van der Waals interactions, the semiempirical Grimme’s dispersion correction with Becke–Jonson damping [DFT-D3(BJ)] method61 was used. A Gaussian smearing of the population of partial occupancies with a width of 0.05 eV was used during iterative diagonalization of the Kohn–Sham Hamiltonian. Brillouin zone sampling was restricted to the Γ point.62 Convergence was assumed to be reached when the force on each atom was below 0.04 eV·Å–1.
To locate transition states, the nudged elastic band method (NEB)63 was applied. The maximum energy geometry along the reaction path generated by the NEB method was further optimized by use of a quasi-Newton algorithm. In this procedure, only the extraframework atoms, and relevant framework atoms were relaxed. Vibrational frequencies were calculated by the finite difference method (0.02 Å atomic displacements) as implemented in VASP. Transition state showed a single imaginary frequency corresponding to the reaction path.
As an initial active-site model, a binuclear Fe cluster coordinated by framework oxygen atoms and extraframework μ-oxo and water molecules at its first coordination shell with a total coordination number of 6 for each iron center (octahedral environment), [(H2O)2–Fe(III)–(μO)2–Fe(III)–(H2O)2]2+, is taken into consideration. The cluster is placed over the eight-membered ring of the zeolite. Two Si atoms of the ring are substituted with Al in the T7 and T12 sites (Figure 1). The unit cell lattice parameters are optimized and are fixed throughout the calculations (a = 20.1, b = 19.8, c = 13.2, α = β = γ = 90°). We based our assumption or the initial site on the DFT study of Li et al.,34 which indicates this cluster as the most stable one among mono-, bi-, and tetranuclear species containing Fe(II) and Fe(III) ions in an oxidative aqueous environment. This is in agreement with the experimental and computational results of Hutchings and co-workers,16 who concluded with high uncertainty an [(H2O)(HO)–Fe–(μOH)2–Fe–(OH)(H2O)]2+ cluster as the representative species of their system. This cluster can be obtained from the previously described one by two H+ transfers from the H2O molecules to the bridging O atoms, which is a very facile reaction, and does not change the stability of the cluster significantly, as we demonstrate in our calculations.
Figure 1.
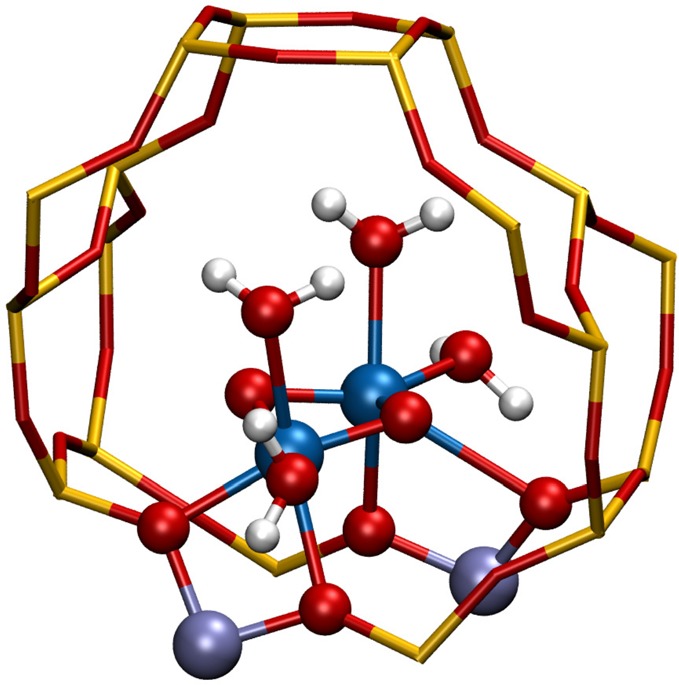
Initial [(H2O)2–Fe(III)–(μO)2–Fe(III)–(H2O)2]2+ model placed in the ZSM5 zeolite pore. The rest of the atoms in the periodic unit cell are omitted from the picture for the sake of visual clarity. Color code: Si, yellow; Al, purple; Fe, blue; O, red; H, white.
The octahedral environment may change during the course of the reaction; for example, one or both Fe atoms decoordinate from one of the framework O atoms. Due to the high number of structures, this is not elaborated in the text. Interested readers are invited to look at the files containing the geometry of each structure in Supporting Information.
All possible spin states (S = 0–5) were considered for the initial Fe cluster. DFT calculations point to the S = 3 state as the most stable one. However, in this state one H2O molecule decoordinates from the Fe and forms a H bond with another H2O molecule, thus providing an artificial stabilization to the overall system. The release of H2O is triggered by the change of spin state, which results in different orbital energies, a change of ligand field, and therefore a change in the preferred geometry of the Fe complex. A similar effect is observed for the S = 4 state. The second most stable configuration is the antiferromagnetically coupled S = 0 state (broken symmetry singlet). Spin density analysis shows that the absolute value of spin assigned to each atom is similar to those in the S = 5 state. As it was shown previously that antiferromagnetic coupling does not significantly influence the reactivity in the case of O-bridged Fe dimers,64 for the reactivity analysis we focused on the ferromagnetically coupled high-spin electronic configurations rather than the antiferromagnetically coupled S = 0 state. Extensive justification for this simplification has been provided by Baerends and co-workers.65 The preferred high- spin state changes over the course of the reaction with the oxidation state of the Fe cluster and the formation of radicals.
In this study, we base our mechanistic analysis on the discussion of relative electronic energies only, while the entropic effects are considered to have only a minor effect on the reaction profiles. This conclusion is supported by a series of test calculations, with the results summarized in Supporting Information.
Over the course of the reaction, H2O molecules leave the Fe sites and new H2O molecules are formed (e.g., upon decomposition of H2O2) that are not connected to the Fe atoms. These molecules form a hydrogen-bond network connecting the O- and H-sites associated with the cluster and the zeolite lattice. These networks can very easily rearrange upon methane adsorption and/or transformation without affecting the electronic properties of the reactive Fe species but influencing the overall energy of the system. As a result, we observed that the barrier heights and reaction energies for elementary steps can be quite substantially affected by the positions of these uncoordinated H2O [E(H-bond in water) = ∼20 kJ/mol]. Extensive analysis of the different isomeric structures involved in similar reaction steps shows that this is an artifact of the model. In the actual aqueous medium, these effects would have been counterbalanced by the presence of bulk H2O molecules. However, the comprehensive sampling required for such an extensive model is in conflict with the goals of the present study. Therefore, we neglected the respective effects and always removed noncoordinated physically adsorbed H2O molecules from the unit cell after their formation. The resulting desorption energy of H2O molecules is not discussed in this article, but it is shown in the reaction energy diagrams (e.g., reaction step 2×H2O/5 → 5 in Figure 8).
Figure 8.

Reaction energy diagram of methane to methanol formation, including formation of the active site, heterolytic C–H bond activation, formation of CH3OH, and active-site regeneration.
The numbering of structures in the paper is not presented in sequential order but follows individual reaction paths from the beginning to the formation of CH3OH or CH3OOH (as summarized in Figures S1 and S2). Conformational isomers and CH4 adsorbed in different positions of the same active site are marked with v after structure number. H2O in front of the structure number indicates an uncoordinated H2O molecule, which will be removed in a later step of the reaction.
Images of structures were created with Visual Molecular Dynamics (VMD) software.66,67
3. Results and Discussion
A schematic representation of the favorable reaction mechanisms identified in our study is shown in Figure 2. Detailed structures can be found in Figures S1 and S2. The active Fe(III) and Fe(IV) sites are formed by adsorption and dissociation of H2O2. Reorganization of the H ions results in different active sites. Our calculations show that Fe(III) species catalyze heterolytic and Fenton-type activation, while Fe(IV) species facilitate homolytic activation of methane. The Fe clusters are grouped on the basis of average formal oxidation state. For example, if one Fe atom is formally +2 while the other is in +4 oxidation state, the average is +3 and the cluster is considered an Fe(III) cluster. The structures of all clusters can be found in Figures S1 and S2.
Figure 2.

Schematic representation of the reaction network and most important steps underlying the oxidation of methane with H2O2 over Fe/ZSM-5 zeolite. Detailed descriptions of the structures can be found in Supporting Information.
From Fe(III) species, methanol can be formed via combination of the methyl group and an OH group before or after oxidation of the cluster by the peroxo ligand. In the cases of homolytic and Fenton-type mechanisms, a methyl radical is formed, which can further transform to CH3OH via a rebound mechanism or to CH3OOH via reaction with O2 (inevitably formed by a parallel H2O2 decomposition process) and abstraction of a H atom from the active site. Fe(IV) sites are the active species for both homolytic dissociation of methane and decomposition of H2O2 to O2 and [H+] ions. In the following sections, we provide detailed descriptions of each reaction step.
3.1. Active-Site Formation
The first step of the reaction is formation of the active site. All investigated pathways (Figure 3) start with adsorption of H2O2 in the zeolite pore (H2O2/1) and its subsequent coordination to the active site, where it substitutes one H2O molecule (2). These steps are exothermic by −44 kJ/mol. The conformational isomers 2v and 2vv, shown in Figure 3, are formed by rotation of the noncoordinated H2O molecule and H2O2. The difference in energy between these structures is an artifact of the model, resulting from the rearrangement of H bonds as described in the Model and Simulation Details section. This structural rearrangement is necessary to adequately probe alternative reaction pathways for the subsequent steps involving the H2O molecule as a proton mediator. From this stage, two alternative reaction channels can be distinguished that result in an active site: namely, (1) direct oxidation of both Fe(III) centers to Fe(IV), via homolytic cleavage of the peroxide moiety, and (2) deprotonation of H2O2 to form an Fe-bound peroxo ligand, leaving the formal oxidation state of the iron ions unchanged.
Figure 3.

Potential reaction pathways for active-site formation via reaction of the Fe cluster with H2O2 and schematic representations of key intermediates.
The first path proceeds with a barrier of only 56 kJ/mol and yields a transient OH radical (14) that readily subtracts a H atom from a neighboring coordinated H2O (E# = 10 kJ/mol), resulting in 2×H2O/15. The isomerization of this species by proton reshuffling gives 2×H2O/22 (ΔE = −3 kJ/mol) and 2×H2O/26 (ΔE = −73 kJ/mol). Such a water-assisted H transfer is a very facile reaction that, in the case of the 2×H2O/15 → 2×H2O/22 step, shows a barrier of 1 kJ/mol. Since we expect the activation barrier for similar H-bonding rearrangements to be on the same order of magnitude, the transition states were not located in other cases. This type of reaction for ferryl ion formation was considered earlier by Baerends and co-workers.68,69 An alternative path of 2×H2O/15 → 2×H2O/16 leads to the cleavage of one Fe-μO bond (ΔE = −33 kJ/mol). The diamond shape of the Fe-oxo cluster transforms to a near-linear Fe–O–Fe species and a terminal O is formed. Transition-state energy was found to be low for this type of reaction, as described in the next paragraph for the reaction 3 → H2O/4.
The second path starts with deprotonation of H2O2 by the bridging O site to form a bridging OH group and a terminal OOH ligand (3). The noncoordinating H2O molecule facilitates this reaction via a proton shuttling mechanism. Next, the Fe−μOH bond breaks with an activation barrier of only 16 kJ/mol to form an activated intermediate H2O/4, which is a near-linear Fe–O–Fe cluster with a terminal OH ligand. This reaction is similar to the previously described 2×H2O/15 → 2×H2O/16 transformation. The activation barrier of other Fe−μO bond cleavage reactions is expected to be in the same range as other similar reactions, and it is significantly lower than that of the rate-determining step; therefore, the activation barriers of similar steps were not calculated for other cases.
In the next reaction step, H2O is decoordinated from the Fe center. This is necessary because, during the heterolytic dissociation, formation of an Fe–C bond occurs. Since the complex is originally in octahedral coordination, this would not be possible without the removal of one ligand. We decided to decouple H2O decoordination from CH4 activation because otherwise the energy change resulting from forming H bonds and Fe–O bond breaking would artificially be included in the reaction and the transition-state energies of C–H bond dissociation. In the reaction H2O/4 → 2×H2O/5 (ΔE = 9 kJ/mol), one H2O molecule leaves the Fe atom. Removal of one H2O molecule from 2×H2O/5 results in H2O/5 (shown in Figure 3), and by removal of the second uncoordinated H2O molecule, 5 is obtained. In this case the Fe atom where the H2O was removed from has only five ligands. This will be the Fe atom that will form the Fe–C bond. We are going to focus on this Fe atom further in this paragraph. The removal of two H2O molecules, one coordinated and one uncoordinated, from 3 results in 11, which also has a five-coordinated Fe atom. The difference between 11 and 5 is that the Fe in 11 has one ligand that moves freely while the other four, the framework, and the bridging O atoms are in fixed positions. The Fe atom of 5 has two ligands that are rather flexible: an H2O molecule and an OH ligand. This difference leads to different structures. The Fe atom of 11 is in a square pyramid geometry, while the Fe of 5 is closer to a trigonal bipyramidal geometry.
The first pathway is thermodynamically more favorable than the second, while the second has lower reaction barriers and thus is kinetically more favorable. However, if we inspect the structures more closely, we realize that by cleaving the peroxo bond of 3(→8) (ΔE = −53 kJ/mol; E# = 78 kJ/mol) and reshuffling the protons of 8, the structures of the first pathway can be obtained. This indicates that the occurrence of all previously described structures is feasible.
To decrease the influence of fluctuating H bonds, the H2O molecules not coordinated to the Fe were removed (vide supra). This way the structures shown in Figure 3—4, 5, 11, 16, 22, and 26—were obtained. These are the active sites applied in the subsequent C–H bond activation steps. After removal of the extra water molecules, the difference in energy between 15, 16, 22, and 26 decreases from 127 to 23 kJ/mol. Their order of stability also changes: 16 becomes the most stable species among them. This energy difference is rather small. In the next section, we consider the active site and the gas-phase methane for all reactions as reference points to enable direct assessment of their reactivity.
3.2. C–H Bond Activation
The next step of the reaction is the C–H bond cleavage of methane. The previously described Fe(III) and Fe(IV) complexes were selected to act as active sites in the reaction. As can be seen in Figure 2, Fe(III) complexes catalyze the heterolytic and Fenton-type reactions while Fe(IV) complexes promote the homolytic oxidation of methane. Figure 4 shows a sample transition state for each case. The isosurface represents the spin density. Figure 4 A is a representative of the heterolytic dissociation (5 → 9). The bridging O subtracts H from CH4, and the CH3 ligand is already connected to Fe in the transition state, which means that the Fe orbitals participate in the reaction. The lack of spin density around C indicates that there are no unpaired electrons on the C orbitals, and this is indeed a heterolytic dissociation. Figure 4 B shows the transition state of reaction 26 → 27, a homolytic C–H bond dissociation. In this case the terminal O accepts the H atom and CH3 radical is formed. The C–H bond distance is smaller than the H–O bond distance, implying a late transition state. This is typical for the terminal O; however, in other type of O atoms (bridging O or OH ligand) this is not necessarily the situation. The spin density isosurface shows that C has more α- than β-electrons, indicative of a radical reaction. Figure 4C shows the transition state of a Fenton-type C–H bond dissociation (2 → 13). Spin density accumulates around the forming OH radical, which cleaves the C–H bond. This is also a radical reaction.
Figure 4.
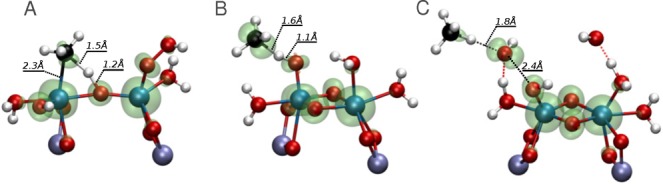
Transition states typical for (A) heterolytic dissociation, reaction 5 → 9; (B) homolytic dissociation, reaction 26 → 27; and (C) Fenton-type dissociation, reaction 2 → 13. The isosurface represents the spin density. Color coding: Al, purple; Fe, blue; O, red; C, black; H, white. The clusters shown here are part of a periodic model containing the unit cell of ZSM-5 zeolite, the Fe cluster, and the reactants.
In the following subsections we present the detailed cases of each type of CH4 activation.
3.2.1. Heterolytic Activation
The reaction energy diagram of heterolytic activation is shown in Figure 5. The reference point is the active site and gas-phase methane. At first methane is adsorbed in the zeolite pore, which is followed by heterolytic dissociation of the first C–H bond of CH4 and formation of a CH3 group and an OH ligand or H2O. For this reaction to occur, Fe needs to have an empty coordination site to accommodate the CH3 group. Hence one H2O ligand was decoordinated from one of the Fe atoms during formation of the active site followed by the removal of one or two physically adsorbed H2O molecules as described in the previous section (2×H2O/4 → 5 and H2O/5; 3 → 11).
Figure 5.
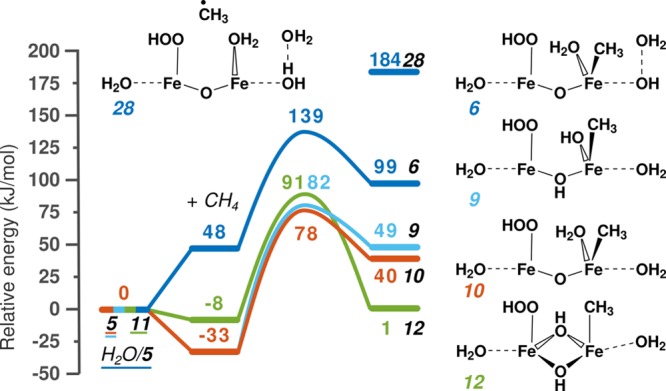
Reaction energy diagram of heterolytic C–H bond cleavage of methane. The reference point is the active site and gas-phase methane. The first step is methane adsorption in the zeolite pore, followed by heterolytic dissociation of the first C–H bond of methane and the formation of an Fe–CH3 moiety and an OH ligand or water, depending on the nature of the proton-accepting site.
The first possibility is the reaction H2O/5 → 6 (dark blue). It starts with CH4 adsorption in the zeolite pore. Since the Fe site is not located in the main channel, there is not enough space for the methane to coordinate favorably to the active site. The confinement of the framework and the cluster results in repulsive forces around the methane. This entails a positive adsorption energy and a relatively high reaction energy (99 kJ/mol) and reaction barrier (139 kJ/mol).
Reactions 5 → 9 (light blue) and 5 → 10 (orange) show the reaction over the same cluster as H2O/5 but with one fewer H2O molecule around the cluster. Methane is adsorbed at a different position, resulting in a negative adsorption energy. The difference between the two reactions is the activating O. In reaction 5 → 9, methane is activated by a bridging O (ΔE = 49 kJ/mol, E# = 82 kJ/mol), while in reaction 5 → 10, methane is activated by the terminal OH group (ΔE = 40 kJ/mol, E# = 78 kJ/mol). Since both the reaction barrier and energy are very similar, this indicates that the type of O does not influence the reactivity significantly.
Next, we compare the performance of two active sites, 5 and 11. As described previously, these two structures have an Fe atom with five ligands in different geometries. The comparison of paths 5 → 9 and 11 → 12 shows that this difference in geometry does not have an effect on the reaction barrier (91 vs 82 kJ/mol, respectively); however, formation of the octahedral environment greatly stabilizes 12 (reaction energy of 49 vs 1 kJ/mol).
It is interesting to take the adsorbed methane as the reference point for our comparison. The qualitative picture for 5 → 9, 5 → 10, and 11 → 12 does not change remarkably. However, reaction H2O/5 → 6 will have much lower reaction barrier and energy, so that it becomes the most favorable path. When confinement plays a role, the adsorption has to be taken into account.
As mentioned earlier, heterolytic activation occurs on Fe(III) sites. In Figure 5, structure 28 is the result of homolytic dissociation over H2O/5. The OH group of 5 accepts a H atom and a CH3 radical is formed. The reaction energy is 85 kJ/mol higher compared to the formation of intermediate 6, where instead of CH3 radical a CH3 ligand is formed. This is most likely due to the fact that homolytic dissociation entails the reduction of the Fe site, in this case to the Fe(II) state, while there is no formal oxidation state change for heterolytic dissociation.
3.2.2. Homolytic Activation
The reaction energy diagrams of homolytic C–H cleavage are shown in Figure 6. Methane is adsorbed in the zeolite, followed by its homolytic dissociation and formation of a CH3 radical and an OH ligand or H2O connected to Fe. The formal oxidation state of Fe in the active cluster is +4, which means that the high-spin state is 8/2. During the reaction, one Fe is reduced to +3 while C is formally oxidized. The highest possible spin state becomes 10/2 [Fe(III)–Fe(IV) and CH3 radical]; however, it can happen that the CH3 radical is antiferromagnetically coupled with the Fe cluster, and the more stable spin state remains 8/2 at the end of this reaction step. Figure 6 summarizes the results obtained for the most stable configurations.
Figure 6.

Reaction energy diagram of homolytic C–H bond cleavage of methane. The reference point is the active site and gas-phase methane. The first step is methane adsorption in the zeolite pore, followed by homolytic dissociation of the first C–H bond of methane and formation of a methyl radical and an OH ligand or water. The same-colored lines represent reaction pathways where the cluster and the H-accepting oxygen are the same but the position of adsorbed methane is different.
In Figure 6 the reactions are grouped on the basis of the type of O ligand that abstracts the H from CH4: (A) terminal, (B) bridging, or (C) OH group. The lowest reaction barriers over terminal O and OH are similar (for 22 → 27 and 16 → 21, E# = 64 and 80 kJ/mol, respectively); however, the reaction barrier over the bridging O is relatively high (E# = 139 kJ/mol). This is a result of the position of the Fe cluster, which is not located in the main channel but in a somewhat confined position. The bridging O is not easily accessible. The same reaction modeled in a different position in the main channel resulted in lower adsorption (−43 kJ/mol), reaction barrier (69 kJ/mol), and reaction energies (48 kJ/mol), which are comparable to the best values obtained for Fe=O and Fe–OH (transition-state structure shown in Figure S3). In general, this means that Fe=O, Fe–OH, and Fe−μO–Fe oxygens are all able to activate methane.
The same trend can be observed here as in the case of the heterolytic dissociation. The positive adsorption energy decreases the intrinsic barriers and reaction energies (reference point is the active site with adsorbed methane); however, compared to the original reference point (gas-phase methane and zeolite), the reactions with positive adsorption energy are less exothermic and have higher barriers. This is emphasized by the same-color reaction pathways, which represent routes where the activating O and the Fe cluster are the same but the methane is adsorbed in a different position in the zeolite. An example is the case of 16 → 19 and 16 → 19v (light blue in Figure 6 A). In the first case methane adsorbs above the cluster with a positive adsorption energy, while in the second case methane adsorbs in the main channel next to the cluster and accesses the terminal O from there. In the first case the intrinsic activation energy is a mere 65 kJ/mol, while in the second case it is 118 kJ/mol; however, if we take the gas-phase methane as reference, the second reaction becomes more favorable with a reaction barrier of 71 kJ/mol (vs 94 kJ/mol).
3.2.3. Fenton-type Activation
Fenton-type activation, as described earlier, is a type of homolytic C–H bond dissociation. The important distinction from the previously presented homolytic dissociation is that the role of the transition metal catalyst is to produce the active OH radicals. Here we present two possibilities for this type of reaction, for which reaction energy diagrams shown in Figure 7. The active sites are 2 and 4, already introduced in the previous section; 2 is formed by coordination of H2O2 to Fe, while 4 is formed by deprotonation of 2 and consecutive cleavage of the Fe−μOH group (Figure 3). In the transition states, the CH4 is effectively intact and an OH radical is formed. The reaction can be viewed as a redox reaction, in which the O atoms of H2O2 oxidize C in methane and Fe of the active site. The reaction yields CH3 radical, H2O molecule, and Fe(IV)Fe(III) complex. The Fenton-type methane activation is a highly favorable process (ΔE = −74 and −87 kJ/mol), proceeding with barriers of 25 and −5 kJ/mol (Figure 7).
Figure 7.
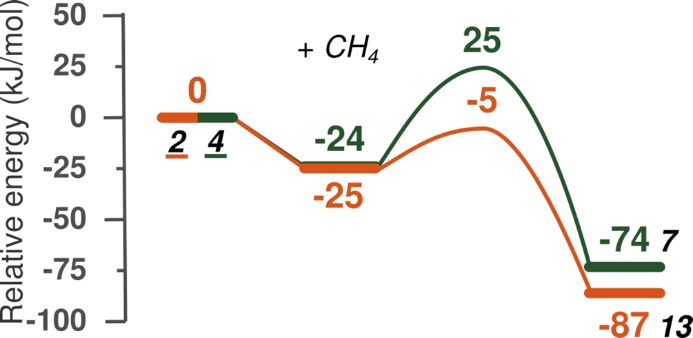
Reaction energy diagram of Fenton-type C–H bond cleavage of methane. The reference point is the active site and gas-phase methane. The first step is methane adsorption in the zeolite pore, followed by homolytic dissociation of the peroxo bond, formation of OH radical, and dissociation of the first C–H bond of methane, and then formation of a methyl radical and water molecule in the same step.
The lowest activation energy and thermodynamic favorability belongs to the Fenton-type activation. In general, Fenton-type reaction is undesired, because it is very difficult to control and this path is thought to decrease the selectivity of the overall process. The lowest reaction and activation energies of the heterolytic and homolytic dissociation pathways are comparable, which implies that both mechanisms are effectively possible, and all type of O ligands can potentially act as H acceptors, providing a favorable reaction channel for CH4 activation.
3.3. Closing the Reaction Cycle
3.3.1. Methanol Formation after Heterolytic Methane Activation
The heterolytic mechanism of C–H activation is seldom considered for the selective oxidation process, because the subsequent CH3OH formation would require a dual-site reductive elimination process. Figure 8 shows the full catalytic cycle through intermediates 10 and 9. It starts with activation of the binuclear Fe(III) site by H2O2 to obtain the 2×H2O/5 active site. The physically adsorbed water is then removed, as explained in the Model and Simulation Details section. This step results in a 148 kJ/mol energy loss of the model system. This is an artificial increase in the overall energy; however, it is necessary to show it here to keep the reference point constant throughout the reaction energy diagram. The same is true for the reaction step 2×H2O/22 → 22 in Figure 9. The desorption step is followed by adsorption and heterolytic dissociation of CH4. To form methanol, recombination of CH3 and an OH ligand is needed. This decreases the formal oxidation state of Fe(III)Fe(III) by 2, resulting in an Fe(II)Fe(II) structure. This is shown by the orange line in Figure 8, where 9 is transformed to 30 with a barrier of 35 kJ/mol and −96 kJ/mol reaction energy. To regenerate the catalyst, dissociation of the peroxo bond and rearrangement of H ions are needed. These steps are not calculated here; previous calculations indicate that the peroxo bond dissociation proceeds with a barrier of ∼40–70 kJ/mol, and the H atom transfer is almost barrierless.
Figure 9.

Reaction energy diagram of methane to methanol formation, including formation of the active site, homolytic C–H bond activation, formation of CH3OH, and active-site regeneration.
The other possibility is oxidation of the cluster prior to formation of MeOH. This route is indicated by a blue line in Figure 8. The activation barrier for peroxo bond (10 → 31) cleavage is 44 kJ/mol. H atom transfer to form 32 provides a 58 kJ/mol energy gain, which can be explained by the formation of an Fe(IV)Fe(IV) cluster from a formally Fe(V)Fe(III) intermediate. In the next step, MeOH is formed (33) with a barrier of 74 kJ/mol that is needed to reduce the Fe atoms. After desorption of MeOH, rearrangement of H atoms, and formation of a bridging O, regeneration of the initial Fe(III)–oxo cluster takes place. As described earlier, all these steps proceed with a low reaction barrier.
These data suggest that there is no energetic preference in the order of MeOH formation and oxidation of the Fe cluster defined.
3.3.2. Methanol Formation after Homolytic and Fenton-type Activation
The structures obtained after both homolytic and Fenton-type pathways are similar in nature: they contain a CH3 radical and an Fe(III)Fe(IV) cluster. Figure 9 shows the formation of methanol from 25, which is the result of combining the CH3 radical with an OH group of the Fe cluster. The reaction step requires 57 kJ/mol activation energy, and it has a reaction energy of −195 kJ/mol. With the substitution of methanol by one water molecule, we obtain the initial structure 1.
3.3.3. Methyl Hydroperoxide Formation and H2O2 Decomposition
If CH3 radicals are present in the liquid phase, formation of MeOOH is also possible. A potential pathway is illustrated in Figure 10. CH3OOH formation starts with 13, where a CH3 radical is formed via Fenton-type homolytic dissociation of methane. The first step of the reaction is adsorption of O2 in the zeolite. O2 is present in water, and it is also formed during the side reaction of H2O2 decomposition to H2O and O2. O2 and CH3 radical favorably combine to form an MeOO radical (34) with an energy gain of −201 kJ/mol. In the next step, there is H transfer between the Fe–OH and Fe–OH2 ligands (35) for the H atoms to get into more favorable position for the water-assisted H transfer to MeOO to yield MeOOH (CH3OOH + 2×H2O/36). As expected, this reaction has an activation barrier of only a few kilojoules per mole and is thermodynamically neutral. Compared to MeOH, the formation of MeOOH is thermodynamically similar: ΔE(O2/13 → CH3OOH + 2×H2O/36) = −202 kJ/mol vs ΔE(25 → 29) = −195 kJ/mol. The limiting factor is the O2 concentration in solution compared to the Fe–OH groups or framework O atoms close to the sites of formation of CH3 radicals.
Figure 10.

Reaction energy diagram of methane to CH3OOH formation, including formation of the active site, Fenton-type C–H bond activation, formation of CH3OOH, and active-site regeneration by H2O2 decomposition to O2.
To regenerate the initial site, complex 36 needs to be reduced. There are two reducing agents in the system: CH4 and H2O2. This means that the active site might cleave a second C–H bond or oxidize H2O2 to two [H+] ions and an O2 molecule. The latter option is illustrated in Figure 10. Starting from CH3OOH + 2×H2O/36, MeOOH and two uncoordinated H2O molecules are desorbed, and H2O2 is adsorbed. The first step is abstraction of one H atom from the H2O2 molecule and formation of an OOH radical (37). The second H atom is then abstracted after the rearrangement of the OOH radical, which comes with −39 kJ/mol energy gain due to the formation of a H-bonding interaction (37v). The reaction yields the initial site (O2/1). The total reaction energy of O2 formation from H2O2 is −52 kJ/mol, and the activation barrier of both H atom abstractions is less than 10 kJ/mol.
Both H2O2 →O2 + 2[H+] and CH4 → CH3 + [H] reactions occur on the same Fe(IV)–oxo species. This means that H2O2 and CH4 are competing for the same sites. Since the activation barrier of H2O2 oxidation is significantly lower than the barrier of C–H bond dissociation, and usually H2O2 concentration is significantly higher than that of methane, the dissociation of H2O2 will be favored over methane. This renders the usage of H2O2 impractical for methane activation in combination with high-valent Fe–oxo catalysts.
The total reaction energies of methanol and methyl hydroperoxide formation in Figures 8–10 are not directly comparable, due to removal of the physically adsorbed H2O molecules in Figures 8 and 9 and the adsorption of O2 in Figure 10. We can, however, make an estimation, for example, from Figure 9 by subtracting the desorption energy of the previously removed H2O molecules (ΔE = 103 kJ/mol) and adding the adsorption energy of O2 (ΔE = −20 kJ/mol). This gives ΔE(1 → 2×H2O + O2/29) ≈ −233 – 123 = −336 kJ/mol, which is comparable to the reaction energy for the alternative reaction channel ΔE(1 → CH3OOH + 2×H2O/36) = −353 kJ/mol. However, the calculations indicate that the formation of MeOOH is kinetically more favorable than the formation of MeOH (following Fenton-type or homolytic C–H bond dissociation), as the direct rebound of CH3 radical with an OH group proceeds with a barrier of E# ∼ 60 kJ/mol, while recombination of the CH3 radical with an O2 molecule is a barrierless reaction and the only barrier on the path to methyl hydroperoxide is associated with H atom abstraction, which our calculations predict to be very small (E# = 5 kJ/mol). Comparison with experimental data is difficult, due to the complexity of the reaction network and the potential decomposition of MeOOH to MeOH16 and to H2CO,47 and in situ analysis of the reaction mixture is not available. A batch reaction study performed by Hutchings and co-workers16 shows that, at the beginning of the reaction, selectivity toward MeOOH is high (∼60%), and it gradually decreases during the course of the reaction. This supports the kinetic preference for MeOOH formation over MeOH.
4. Conclusions
Different reaction paths for selective methane oxidation over a binuclear Fe site deposited in ZSM-5 zeolite were studied by periodic DFT calculations. The whole reaction cycle was investigated, including formation of the active site, CH4 activation, product formation, and regeneration of the initial site. Special attention was given to the rate-determining C–H bond dissociation step, the possibility of multiple mechanisms, and the influence of different active sites on them. These mechanisms were (1) heterolytic, (2) homolytic and (3) Fenton-type activation.
This study demonstrates that the system cannot be reduced to a single-site single-cycle concept. Even with the simplification to a single type of Fe cluster at a given position of the zeolite framework, the formation of multiple types of active sites is possible, catalyzing three mechanistically different C–H bond activations.
The possibility of formation of different Fe(III) and Fe(IV) clusters upon reaction with H2O2 was demonstrated. These sites were proven to be catalytically active. Fe(III) was established to promote heterolytic and Fenton-type reactions, while Fe(IV) was shown to promote the homolytic reaction.
We found that the geometry of the Fe complex significantly influences the reaction energy but not the barrier of heterolytic C–H bond activation. The calculations indicate that the type of activating O (Fe=O, Fe−μO–Fe, or Fe–OH) is unimportant from an energetic point of view for both types of activation. We also found that confinement of the zeolite has a significant effect on the reaction step.
Methanol can be formed following all types of C–H bond activation via recombination of CH3 and an OH ligand reducing the active site. If the C–H bond was previously heterolytically cleaved, this step can either precede or follow oxidation of the active site by peroxo bond cleavage. After homolytic and Fenton-type dissociation, this reduction step restores the initial oxidation state of the active site. MeOOH can be obtained from the reaction of CH3 radical and O2 molecule, followed by abstraction of a H atom from the active site. After this step, the Fe site is in the oxidized form available for further CH4 oxidation.
Since the reaction is accompanied by excessive consumption of H2O2, its decomposition was also investigated. We found that the same Fe(IV)–oxo sites promote the oxidation of both H2O2 and CH4. The first reaction will be dominant, as the activation barrier for O–H bond cleavage is significantly lower than for C–H bond cleavage. This is why we propose H2O2 to be an unsuitable oxidant in combination with high-valent Fe clusters.
Acknowledgments
We thank Elena Khramenkova (ITMO University) for carrying out some of the calculations. The Dutch Science Foundation (NWO) is gratefully acknowledged for financial support through the VIDI personal grant MetMOFCat. G.L. acknowledges financial support from NWO for her personal VENI grant (016.Veni.172.034). E.A.P. acknowledges partial support from the Ministry of Education and Science of the Russian Federation (Project 11.1706.2017/4.6). SurfSARA and NWO (The Netherlands Organisation for Scientific Research) are acknowledged for providing access to supercomputer resources.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b01672.
Seven figures showing schematic representations of reaction pathways, TS structure, and reaction energy diagrams; additional text describing free energy calculation; and coordinates of all calculated stable point and transition-state structures in the most stable spin state (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Arndtsen B. A.; Bergman R. G.; Mobley T. A.; Peterson T. H. Selective Intermolecular Carbon-Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res. 1995, 28, 154–162. 10.1021/ar00051a009. [DOI] [Google Scholar]
- Bergman R. G. Organometallic Chemistry: C-H Activation. Nature 2007, 446, 391–393. 10.1038/446391a. [DOI] [PubMed] [Google Scholar]
- Olivos-Suarez A. I.; Szécsényi A.; Hensen E. J. M.; Ruiz-Martinez J.; Pidko E. A.; Gascon J. Strategies for the Direct Catalytic Valorization of Methane Using Heterogeneous Catalysis: Challenges and Opportunities. ACS Catal. 2016, 6, 2965–2981. 10.1021/acscatal.6b00428. [DOI] [Google Scholar]
- Edwards J. H.; Foster N. R. The Potential for Methanol Production from Natural Gas by Direct Catalytic Partial Oxidation. Fuel Sci. Technol. Int. 1986, 4, 365–390. 10.1080/08843758608915816. [DOI] [Google Scholar]
- Zhang Q.; He D.; Zhu Q. Recent Progress in Direct Partial Oxidation of Methane to Methanol. J. Nat. Gas Chem. 2003, 12, 81–89. [Google Scholar]
- Ravi M.; Ranocchiari M.; van Bokhoven J. A. The Direct Catalytic Oxidation of Methane to Methanol-A Critical Assessment. Angew. Chem., Int. Ed. 2017, 56, 16464–16483. 10.1002/anie.201702550. [DOI] [PubMed] [Google Scholar]
- Parkyns N. D.; Warburton C. I.; Wilson J. D. Natural Gas Conversion to Liquid Fuels and Chemicals: Where Does It Stand?. Catal. Today 1993, 18, 385–442. 10.1016/0920-5861(93)80062-6. [DOI] [Google Scholar]
- Hammond C.; Conrad S.; Hermans I. Oxidative Methane Upgrading. ChemSusChem 2012, 5, 1668–1686. 10.1002/cssc.201200299. [DOI] [PubMed] [Google Scholar]
- Bone W. A.; Wheeler R. V. LVIII. - The Slow Oxidation of Methane at Low Temperatures. J. Chem. Soc., Trans. 1902, 81, 535–549. 10.1039/CT9028100535. [DOI] [Google Scholar]
- Periana R.; Taube D.; Evitt E.; Löffler D.; Wentrcek P.; Voss G.; Masuda T. A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol. Science 1993, 259, 340–343. 10.1126/science.259.5093.340. [DOI] [PubMed] [Google Scholar]
- Periana R.; Taube D.; Gamble S.; Taube H.; Satoh T.; Fujii H. Platinum Catalysts for The High-Yield Oxidation of Methane to a Methanol Derivative. Science 1998, 280, 560–564. 10.1126/science.280.5363.560. [DOI] [PubMed] [Google Scholar]
- Groothaert M. H.; Smeets P. J.; Sels B. F.; Jacobs P. A.; Schoonheydt R. A. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J. Am. Chem. Soc. 2005, 127, 1394–1395. 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- Beznis N. V.; Weckhuysen B. M.; Bitter J. H. Partial Oxidation of Methane over Co-ZSM-5: Tuning the Oxygenate Selectivity by Altering the Preparation Route. Catal. Lett. 2010, 136, 52–56. 10.1007/s10562-009-0206-6. [DOI] [Google Scholar]
- Shan J.; Huang W.; Nguyen L.; Yu Y.; Zhang S.; Li Y.; Frenkel A. I.; Tao F. Conversion of Methane to Methanol with a Bent Mono(μ-oxo)Dinickel Anchored on the Internal Surfaces of Micropores. Langmuir 2014, 30, 8558–8569. 10.1021/la501184b. [DOI] [PubMed] [Google Scholar]
- Xu J.; Zheng A.; Wang X.; Qi G.; Su J.; Du J.; Gan Z.; Wu J.; Wang W.; Deng F. Room Temperature activation of Methane over Zn Modified H-ZSM-5 Zeolites: Insight from Solid-State NMR and Theoretical Calculations. Chem. Sci. 2012, 3, 2932–2940. 10.1039/c2sc20434g. [DOI] [Google Scholar]
- Hammond C.; Forde M. M.; Ab Rahim M. H.; Thetford A.; He Q.; Jenkins R. L.; Dimitratos N.; Lopez-Sanchez J. A.; Dummer N. F.; Murphy D. M.; Carley A. F.; Taylor S. H.; Willock D. J.; Stangland E. E.; Kang J.; Hagen H.; Kiely C. J.; Hutchings G. J. Direct Catalytic Conversion of Methane to Methanol in an Aqueous Medium by Using Copper-Promoted Fe-ZSM-5. Angew. Chem., Int. Ed. 2012, 51, 5129–5133. 10.1002/anie.201108706. [DOI] [PubMed] [Google Scholar]
- Hammond C.; Dimitratos N.; Lopez-Sanchez J. A.; Jenkins R. L.; Whiting G.; Kondrat S. A.; Ab Rahim M. H.; Forde M. M.; Thetford A.; Hagen H.; Stangland E. E.; Moulijn J. M.; Taylor S. H.; Willock D. J.; Hutchings G. J. Aqueous-Phase Methane Oxidation over Fe-MFI Zeolites; Promotion Through Isomorphous Framework Substitution. ACS Catal. 2013, 3, 1835–1844. 10.1021/cs400288b. [DOI] [Google Scholar]
- Sobolev V. I.; Dubkov K. A.; Panna O. V.; Panov G. I. Selective Oxidation of Methane to Methanol on a FeZSM-5 Surface. Catal. Today 1995, 24, 251–252. 10.1016/0920-5861(95)00035-E. [DOI] [Google Scholar]
- Parfenov M. V.; Starokon E. V.; Pirutko L. V.; Panov G. I. Quasicatalytic and Catalytic Oxidation of Methane to Methanol by Nitrous Oxide over FeZSM-5 Zeolite. J. Catal. 2014, 318, 14–21. 10.1016/j.jcat.2014.07.009. [DOI] [Google Scholar]
- Kulkarni A. R.; Zhao Z.-J.; Siahrostami S.; Nørskov J. K.; Studt F. Cation-Exchanged Zeolites for the Selective Oxidation of Methane to Methanol. Catal. Sci. Technol. 2018, 8, 114–123. 10.1039/C7CY01229B. [DOI] [Google Scholar]
- Narsimhan K.; Iyoki K.; Dinh K.; Román-Leshkov Y. Catalytic Oxidation of Methane into Methanol over Copper-Exchanged Zeolites with Oxygen at Low Temperature. ACS Cent. Sci. 2016, 2, 424–429. 10.1021/acscentsci.6b00139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C.; Jenkins R. L.; Dimitratos N.; Lopez-Sanchez J. A.; Ab Rahim M. H.; Forde M. M.; Thetford A.; Murphy D. M.; Hagen H.; Stangland E. E.; Moulijn J. M.; Taylor S. H.; Willock D. J.; Hutchings G. J. Catalytic and Mechanistic Insights of the Low-Temperature Selective Oxidation of Methane over Cu-Promoted Fe-ZSM-5. Chem. - Eur. J. 2012, 18, 15735–15745. 10.1002/chem.201202802. [DOI] [PubMed] [Google Scholar]
- Hammond C.; Dimitratos N.; Jenkins R. L.; Lopez-Sanchez J. A.; Kondrat S. A.; Hasbi Ab Rahim M.; Forde M. M.; Thetford A.; Taylor S. H.; Hagen H.; Stangland E. E.; Kang J. H.; Moulijn J. M.; Willock D. J.; Hutchings G. J. Elucidation and Evolution of the Active Component within Cu/Fe/ZSM-5 for Catalytic Methane Oxidation: From Synthesis to Catalysis. ACS Catal. 2013, 3, 689–699. 10.1021/cs3007999. [DOI] [Google Scholar]
- Hammond C.; Hermans I.; Dimitratos N. Biomimetic Oxidation with Fe-ZSM-5 and H2O2 - Identification of an Active, Extra-Framework Binuclear Core and an FeIII-OOH Intermediate with Resonance-Enhanced Raman Spectroscopy. ChemCatChem 2015, 7, 434–440. 10.1002/cctc.201402642. [DOI] [Google Scholar]
- Boudart M. Catalysis by Supported Metals. Adv. Catal. 1969, 20, 153–166. 10.1016/S0360-0564(08)60271-0. [DOI] [Google Scholar]
- Boudart M.; McDonald M. A. Structure Sensitivity of Hydrocarbon Synthesis from CO and H2. J. Phys. Chem. 1984, 88, 2185–2195. 10.1021/j150655a004. [DOI] [Google Scholar]
- Hargreaves J. S. J.; Hutchings G. J.; Joyner R. W.; Kiely C. J. The Relationship Between Catalyst Morphology and Performance in the Oxidative Coupling of Methane. J. Catal. 1992, 135, 576–595. 10.1016/0021-9517(92)90055-M. [DOI] [Google Scholar]
- Meher S. K.; Ranga Rao G. Tuning, via Counter Anions, the Morphology and Catalytic Activity of CeO2 Prepared under Mild Conditions. J. Colloid Interface Sci. 2012, 373, 46–56. 10.1016/j.jcis.2011.09.050. [DOI] [PubMed] [Google Scholar]
- Lv P.; Lu Z.; Li S.; Ma D.; Zhang W.; Zhang Y.; Yang Z. Tuning Metal Cluster Catalytic Activity with Morphology and Composition: A DFT Study of O2 Dissociation at the Global Minimum of PtmPdn (m + n = 5) Clusters. RSC Adv. 2016, 6, 104388–104397. 10.1039/C6RA23266C. [DOI] [Google Scholar]
- Li G.; Vassilev P.; Sanchez-Sanchez M.; Lercher J.; Hensen E. J. M.; Pidko E. A. Stability and Reactivity of Copper Oxo-Clusters in ZSM-5 Zeolite for Selective Methane Oxidation to Methanol. J. Catal. 2016, 338, 305–312. 10.1016/j.jcat.2016.03.014. [DOI] [Google Scholar]
- Snyder B. E. R.; Vanelderen P.; Bols M. L.; Hallaert S. D.; Böttger L. H.; Ungur L.; Pierloot K.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. The Active Site of Low-Temperature Methane Hydroxylation in Iron-Containing Zeolites. Nature 2016, 536, 317–321. 10.1038/nature19059. [DOI] [PubMed] [Google Scholar]
- Snyder B. E. R.; Böttger L. H.; Bols M. L.; Yan J. J.; Rhoda H. M.; Jacobs A. B.; Hu M. Y.; Zhao J.; Alp E. E.; Hedman B.; Hodgson K. O.; Schoonheydt R. A.; Sels B. F.; Solomon E. I. Structural Characterization of a Non-Heme Iron Active Site in Zeolites That Hydroxylates Methane. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4565–4570. 10.1073/pnas.1721717115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Pidko E. A.; Van Santen R. A.; Feng Z.; Li C.; Hensen E. J. M. Stability and Reactivity of Active Sites for Direct Benzene Oxidation to Phenol in Fe/ZSM-5: A Comprehensive Periodic DFT Study. J. Catal. 2011, 284, 194–206. 10.1016/j.jcat.2011.07.008. [DOI] [Google Scholar]
- Li G.; Pidko E. A.; Van Santen R. A.; Li C.; Hensen E. J. M. Stability of Extraframework Iron-Containing Complexes in ZSM-5 Zeolite. J. Phys. Chem. C 2013, 117, 413–426. 10.1021/jp310374k. [DOI] [Google Scholar]
- Latimer A. A.; Kulkarni A. R.; Aljama H.; Montoya J. H.; Yoo J. S.; Tsai C.; Abild-Pedersen F.; Studt F.; Nørskov J. K. Understanding Trends in C-H Bond Activation in Heterogeneous Catalysis. Nat. Mater. 2017, 16, 225–229. 10.1038/nmat4760. [DOI] [PubMed] [Google Scholar]
- Mahyuddin M. H.; Tanaka T.; Shiota Y.; Staykov A.; Yoshizawa K. Methane Partial Oxidation over [Cu2(μ-O)]2+ and [Cu3(μ-O)3]2+ Active Species in Large-Pore Zeolites. ACS Catal. 2018, 8, 1500–1509. 10.1021/acscatal.7b03389. [DOI] [Google Scholar]
- McMullin C. L.; Pierpont A. W.; Cundari T. R. Complete Methane-to-Methanol Catalytic Cycle: A DFT Study of Oxygen Atom Transfer from N2O to Late-Row (MNi, Cu, Zn) β-diketiminate CH Activation Catalysts. Polyhedron 2013, 52, 945–956. 10.1016/j.poly.2012.07.019. [DOI] [Google Scholar]
- Arvidsson A. A.; Zhdanov V. P.; Carlsson P. A.; Grönbeck H.; Hellman A. Metal Dimer Sites in ZSM-5 Zeolite for Methane-to-Methanol Conversion from First-Principles Kinetic Modelling: Is the [Cu-O-Cu]2+ Motif Relevant for Ni, Co, Fe, Ag, and Au?. Catal. Sci. Technol. 2017, 7, 1470–1477. 10.1039/C6CY02521H. [DOI] [Google Scholar]
- Shilov A. E.; Shul’pin G. B. Activation of C-H Bonds by Metal Complexes. Chem. Rev. 1997, 97, 2879–2932. 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
- Copéret C. C-H Bond Activation and Organometallic Intermediates on Isolated Metal Centers on Oxide Surfaces. Chem. Rev. 2010, 110, 656–680. 10.1021/cr900122p. [DOI] [PubMed] [Google Scholar]
- Mayer J. M. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljama H.; Nørskov J. K.; Abild-Pedersen F. Theoretical Insights into Methane C-H Bond Activation on Alkaline Metal Oxides. J. Phys. Chem. C 2017, 121, 16440–16446. 10.1021/acs.jpcc.7b05838. [DOI] [Google Scholar]
- Plietker B., Ed. Iron Catalysis: Fundamentals and Applications; Wiley–VCH: Weinheim, Germany, 2008; 10.1007/978-3-642-14670-1. [DOI] [Google Scholar]
- Pignatello J. J.; Oliveros E.; MacKay A. Crit. Rev. Environ. Sci. Technol. 2006, 36, 1–84. 10.1080/10643380500326564. [DOI] [Google Scholar]
- Pestovsky O.; Stoian S.; Bominaar E. L.; Shan X.; Münck E.; Que L. Jr.; Bakac A. Aqueous FeIV = O: Spectroscopic Identification and Oxo-Group Exchange. Angew. Chem., Int. Ed. 2005, 44, 6871–6874. 10.1002/anie.200502686. [DOI] [PubMed] [Google Scholar]
- Shulpin G. B.; Nizova G. V. Formation of Alkyl Peroxides in Oxidation of Alkanes by H2O2 Catalyzed by Transition Metal Complexes. React. Kinet. Catal. Lett. 1992, 48, 333–338. 10.1007/BF02070104. [DOI] [Google Scholar]
- Nizova G. V.; Süss-Fink G.; Shul’pin G. B. Catalytic Oxidation of Methane to Methyl Hydroperoxide and Other Oxygenates under Mild Conditions. Chem. Commun. 1997, 397–398. 10.1039/a607765j. [DOI] [Google Scholar]
- Süss-Fink G.; Nizova G. V.; Stanislas S.; Shul’Pin G. B. Oxidations by the Reagent ’O2-H2O2 - Vanadate Anion - Pyrazine- 2-Carboxylic Acid’. Part 10. Oxygenation of Methane in Acetonitrile and Water. J. Mol. Catal. A: Chem. 1998, 130, 163–170. 10.1016/S1381-1169(97)00210-0. [DOI] [Google Scholar]
- Agarwal N.; Freakley S. J.; McVicker R. U.; Althahban S. M.; Dimitratos N.; He Q.; Morgan D. J.; Jenkins R. L.; Willock D. J.; Taylor S. H.; Kiely C. J.; Hutchings G. J. Aqueous Au-Pd Colloids Catalyze Selective CH4 Oxidation to CH3OH with O2 under Mild Conditions. Science 2017, 358, 223–227. 10.1126/science.aan6515. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K.; Shiota Y.; Yumura T.; Yamabe T. Direct Methane-Methanol and Benzene-Phenol Conversions on Fe-ZSM-5 Zeolite: Theoretical Predictions on the Reaction Pathways and Energetics. J. Phys. Chem. B 2000, 104, 734–740. 10.1021/jp991844b. [DOI] [Google Scholar]
- Fellah M. F.; Onal I. Direct Methane Oxidation to Methanol by N2O on Fe- and Co-ZSM-5 Clusters with and without Water: A Density Functional Theory Study. J. Phys. Chem. C 2010, 114, 3042–3051. 10.1021/jp9097292. [DOI] [Google Scholar]
- Montejo-Valencia B. D.; Pagán-Torres Y. J.; Martínez-Iñesta M. M.; Curet-Arana M. C. Density Functional Theory (DFT) Study to Unravel the Catalytic Properties of M-Exchanged MFI, (M = Be, Co, Cu, Mg, Mn, Zn) for the Conversion of Methane and Carbon Dioxide to Acetic Acid. ACS Catal. 2017, 7, 6719–6728. 10.1021/acscatal.7b00844. [DOI] [Google Scholar]
- Kresse G.; Hafner J. Ab initio Molecular Dynamics for Liquid Metals. Phys. Rev. B: Condens. Matter Mater. Phys. 1993, 47, 558–561. 10.1103/PhysRevB.47.558. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Hafner J. Ab initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous- Semiconductor Transition in Germanium. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 49, 14251–14269. 10.1103/PhysRevB.49.14251. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficient Iterative Schemes for ab initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Erratum: Generalized Gradient Approximation Made Simple (Physical Review Letters (1996) 77 (3865)). Phys. Rev. Lett. 1997, 78, 1396. 10.1103/PhysRevLett.78.1396. [DOI] [PubMed] [Google Scholar]
- Blöchl P. Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1994, 50, 17953–17979. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Monkhorst H. J.; Pack J. D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- Mills G.; Jónsson H.; Schenter G. K. Reversible Work Transition State Theory: Application to Dissociative Adsorption of Hydrogen. Surf. Sci. 1995, 324, 305–337. 10.1016/0039-6028(94)00731-4. [DOI] [Google Scholar]
- Hansen N.; Heyden A.; Bell A. T.; Keil F. J. A Reaction Mechanism for the Nitrous Oxide Decomposition on Binuclear Oxygen Bridged Iron Sites in Fe-ZSM-5. J. Phys. Chem. C 2007, 111, 2092–2101. 10.1021/jp065574q. [DOI] [Google Scholar]
- Belanzoni P.; Bernasconi L.; Baerends E. J. O2 Activation in a Dinuclear Fe(II)/EDTA Complex: Spin Surface Crossing as a Route to Highly Reactive Fe(IV)oxo Species. J. Phys. Chem. A 2009, 113, 11926–11937. 10.1021/jp9033672. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD - Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Stone J.An Efficient Library for Parallel Ray Tracing and Animation. M.Sc. thesis, Computer Science Department, University of Missouri—Rolla, 1998. [Google Scholar]
- Ensing B. Jr.; Buda F.; Blöchl P.; Baerends E. J. Chemical Involvement of Solvent Water Molecules in Elementary Steps of the Fenton Oxidation Reaction. Angew. Chem., Int. Ed. 2001, 40, 2893–2895. 10.1002/1521-3773(20010803)40:15<2893::AID-ANIE2893>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Buda F.; Ensing B.; Gribnau M. C. M.; Baerends E. J. DFT Study of the Active Intermediate in the Fenton Reaction. Chem. - Eur. J. 2001, 7, 2775–2783. 10.1002/1521-3765(20010702)7:13<2775::AID-CHEM2775>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.