

ABSTRACT
Normal metabolism requires a controlled balance between anabolism and catabolism. It is not completely known how this balance can be retained when the level of nutrient supply changes in the long term. We found that in murine liver anabolism, as represented by the phosphorylation of RPS6KB (ribosomal protein S6 kinase), was soon elevated while catabolism, as represented by TFEB (transcription factor EB)-directed gene transcription and lysosomal activities, was downregulated after the administration of a high-fat diet (HFD). Surprisingly, neither the alteration in RPS6KB phosphorylation nor that in TFEB functions was static over the long course of HFD feeding. Instead, the 2 signals exhibited dynamic alterations in opposite directions, which could be explained by the dependence of MTORC1 (MTOR complex 1) activation on TFEB-supported lysosome function and the feedback suppression of TFEB by MTORC1. Disruption of the dynamics by enforced expression of TFEB in HFD-fed mice at the peaks of MTORC1 activation restored lysosome function. Consistently, interference of MTORC1 activation with rapamycin or with a constitutively activated RRAGA mutant at the peak or nadir of MTORC1 oscillation enhanced or reduced the lysosome function, respectively. These treatments also improved or exacerbated hepatic steatosis and liver injury, respectively. Finally, there was a significant inverse correlation between TFEB activation and steatosis severity in the livers of patients with non-alcohol fatty liver diseases, supporting the clinical relevance of TFEB-regulated events. Thus, maintaining catabolic function through feedback mechanisms during enhanced anabolism, which is caused by nutrient oversupply, is important for reducing liver pathology.
KEYWORDS: Autophagy, high-fat diet, lipophagy, lysosome, MTORC1, NAFLD, TFEB
Introduction
Coordinated anabolism and catabolism is an essential requirement of metabolic homeostasis. The lysosome is best known for its degradative functions and its catabolic roles in metabolism. However, its role in anabolism has recently been revealed by the studies on the activation of the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) [1–3]. MTORC1 has a central role in cell metabolism, proliferation and survival [2–5]. MTORC1 promotes anabolism and protein synthesis by phosphorylating its substrates RPS6KB and EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) [3,6]. In addition, activated MTORC1 promotes lipogenesis [7–9], suppresses ketogenesis [10], and prolongs blood glucose clearance [5,9–13].
Strikingly, activation of MTORC1 can be heavily dependent on the lysosome. In response to nutrients, a full activation of MTORC1 requires the presence of both amino acids and growth factors, which act through different pathways [2–4]. MTORC1 is recruited to the lysosome via the RRAG proteins, which are small guanosine triphosphatases (GTPases) and interact with MTORC1 in an amino acid-dependent manner [14,15]. Conversely, signals initiated by various growth factors lead to the suppression of the TSC1-TSC2 complex [16,17]. The TSC complex binds to and suppresses RHEB on the lysosome membrane, but dissociates from RHEB when TSC2 is phosphorylated in response to growth factor stimulation [18]. The GTP-bound form of RHEB can now stimulate the kinase activity of MTORC1 that has been recruited to the lysosome membrane via RRAG (Ras related GTP binding).
The lysosome not only provides the membrane platform for the interaction of MTORC1 with its activators, but is also the site where amino acids can be sensed and multiple regulatory mechanisms can be exerted. Mammals have 4 RRAG proteins, RRAGA to RRAGD, which form obligate heterodimers consisting of RRAGA or RRAGB with RRAGC or RRAGD. Amino acids can promote the loading of RRAGA or RRAGB with GTP, which enables the lysosome-associated heterodimer to recruit MTORC1 to the lysosomal membranes [15]. The RRAG GTPase is regulated by a lysosome-associated super-complex composed of the Ragulator complex, SLC38A9 and vacuolar-type H+-translocating ATPase (v-ATPase) [3,19,20]. The ATPase activity of the v-ATPase appears to be essential to deliver the amino acids signal from the lysosomal lumen to the Ragulator and RRAG GTPases [20,21]. The Ragulator complex is composed of 5 components, LAMTOR1/p18, LAMTOR2/p14, LAMTOR3/MP1, LAMTOR4/c7orf59 and LAMTOR5/HBXIP; and the pentamer has GEF activity toward the RRAG GTPase [22].
It is thus important that there is a sufficient supply of lysosomes, and that the lysosome maintains its functional integrity to support MTORC1 activation. A key regulator of lysosome biogenesis and function is TFEB, which is a bHLH-LZ transcription factor [23–25]. TFEB is critical in the regulation of a gene network known as coordinated lysosomal expression and regulation (CLEAR) [26,27]. The CLEAR network has identified hundreds of TFEB targets that regulate lysosomal biogenesis and function. In addition, TFEB-regulated genes can affect autophagosome biogenesis, the fusion between autophagosomes and lysosomes, and the degradation of the autophagic content in the lysosome [28]. TFEB is activated under nutrient-depleted condition and translocates from the cytosol to the nucleus to induce its target genes [1,28–30].
As a promoter for anabolism, MTORC1 inhibits catabolism by suppressing macroautophagy/autophagy activation via inactivating ULK1 [31], and by suppressing lysosomal degradation via inactivating TFEB. In cultured cells under nutrient-replete conditions, MTORC1 phosphorylates TFEB at several serine/threonine residues, including serine 211 (S211) [29,32], which traps TFEB in the cytosol via YWHA/14–3-3 [32]. MTORC1 may also inhibit lysosomal function by other means [33]. Thus MTORC1 has a dual role in promoting anabolism and suppressing catabolism to achieve a coordinated metabolic control.
However, a major issue with this model of metabolic control is that suppression of TFEB by activated MTORC1 can also reduce lysosome biogenesis and lysosome function, which would impair the activation of MTORC1. This may not be a particular concern in studies using cultured cells where alteration of nutrient conditions is transient. However, if nutrient-activated MTORC1 promotes anabolism while suppressing catabolism in the long term, this would cause metabolic imbalance and likely lead to pathological consequences. It is thus likely that MTORC1 activation will subside in the constant presence of nutrients. It is attractive to hypothesize that suppression of TFEB by MTORC1 in the long-term presence of superfluous nutrients may actually serve as a self-limiting feedback mechanism to reduce MTORC1 activation to the basal level.
The present study investigated these issues in murine livers. The in vivo model is suitable for examining long-term signaling dynamics in a pathophysiologically relevant context. As such, we subjected mice to a high-fat diet to stimulate anabolism and maintained the stimulation for up to 32 weeks. We found that MTORC1 signaling was not static or linearly maintained but exhibited a damping oscillation pattern over the long term after an initial elevation. Interestingly, the alteration of TFEB was in the opposite direction to that of MTORC1. Disruption of the temporal pattern of MTORC1 or TFEB activation changed lysosome functions and altered the progression of hepatic steatosis and liver injury. The findings collectively indicated the importance of restricting anabolism and restoring catabolic function in the long-term presence of a high level of nutrients. Furthermore, there was an inverse relationship of TFEB activation and hepatic steatosis in biopsied liver samples of patients with non-alcoholic fatty liver disease (NAFLD), suggesting the clinical relevance of the finding made in the mouse model and that the MTORC1-TFEB signaling axis could be a potential therapeutic target for managing NAFLD in patients.
Results
The activation of RPS6KB/p70S6K and the suppression of TFEB oscillated during long-term high-fat diet feeding
The activation of MTORC1 is commonly measured by the phosphorylation of one of its common targets, RPS6KB/pP70S6K, which promotes anabolism (Figure 1(a,b). The level of phosphorylated RPS6KB (p-RPS6KB) was low before the HFD started, but steadily increased in the first 3 weeks of HFD feeding. The level became reduced at 6 weeks and bottomed out at 10 weeks of feeding. However, the p-RPS6KB level was increased again at 16 week of feeding, although at a lower level, which was reduced by 32 weeks, approaching the basal level. We further confirmed that the level of phosphorylated RPS6 (ribosomal protein S6), a downstream target of RPS6KB, oscillated in the same pattern at key time points, although the level of total RPS6 was eventually downregulated at later time points (Figure 1(c,d)). Thus, the MTORC1 activity had a dynamic change during the long-term presence of superfluous nutrients with an overall pattern of early elevation followed by a damping oscillation.
Figure 1.
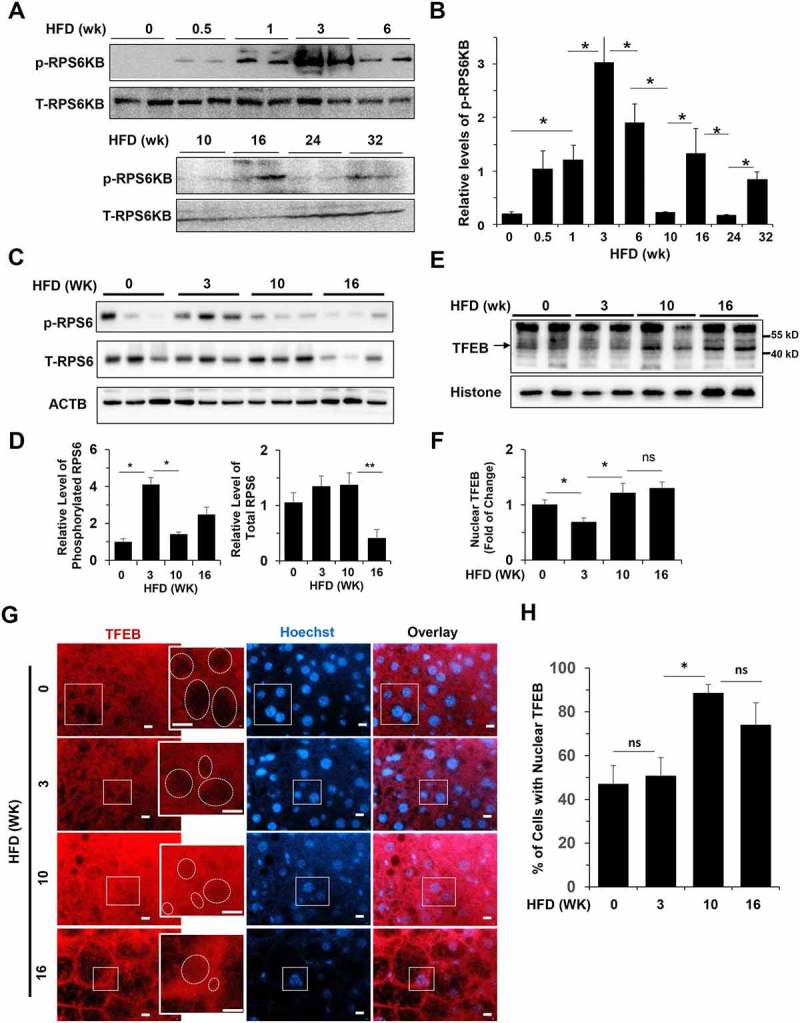
Activation of hepatic TFEB and RPS6KB oscillated in opposite directions in HFD-fed mice. (a-b) Levels of phosphorylated (p-RPS6KB) and total (T-RPS6KB) RPS6KB were determined in liver lysates prepared from mice fed with HFD for the indicated time. Each lane represents one sample (a). Densitometry analysis was performed for phosphorylated-RPS6KB, which was normalized to the level of total RPS6KB (b). (c-d) Levels of phosphorylated (p-RPS6) and total (T-RPS6) RP S6 were determined in liver lysates prepared from mice fed with HFD for the indicated time. Each lane represents one individual mouse (c). Densitometry analysis was performed for p-RPS6, which was normalized to the level of total RPS6, and total RPS6, which was normalized to the level of ACTB/β-actin (d). (e-f) The hepatic nuclear fraction prepared from mice fed with HFD for the indicated time was subjected to immunoblotting assay (e). Densitometry analysis was performed for TFEB, which was normalized to the level of histone (f). (g-h) Mice were fed with HFD for the indicated durations. Paraffin sections of liver tissue were stained with anti-TFEB and Hoechst 33328 (g). Boxed areas were enlarged in the insets, which illustrate the representative hepatocytes. Dotted circles indicate the nucleus, some of which were positive for TFEB signals. The percentage of cells with TFEB-positive nuclei was quantified (h). *, p < 0.05; **, p < 0.01; ns, not significant; n = 3–6 per group. Scale bar = 10 μm.
To understand the mechanism of this dynamic change of the anabolic RPS6KB activation, we examined another MTORC1 target, TFEB, which represents the catabolic activity, in the long-term presence of superfluous nutrients. Phosphorylation of TFEB by MTORC1 prevented nuclear translocation of TFEB and thus its catabolic function [29,32]. The nuclear fraction of the liver was isolated from mice fed with a HFD at 3, 10 and 16 weeks, at which time the phosphorylation of RPS6KB and RPS6 had dynamic changes. Compared to the control group fed with the regular diet, 3-wk of HFD feeding reduced the level of nuclear TFEB, which, however, bounced back at 10-weeks and 16-weeks (Figure 1(e,f)). The direction of the alteration of TFEB nuclear translocation was in general the opposite to that of RPS6KB activation, i.e. an initial decline followed by a later elevation, consistent with the opposite functions of the 2 molecules.
To confirm TFEB translocation in the hepatocyte we performed immunostaining (Figure 1(g)). The immunostaining analysis does not quantify the actual amount of TFEB in the nucleus vs in the cytosol, but allows the analysis on specific cell types. Based on the fluorescence intensity, it seemed that only a small amount of TFEB would be found in the nucleus. The percentage of hepatocytes with nuclear TFEB was thus quantified (Figure 1(h)). Although the percentage of hepatocytes with TFEB after 3 weeks of HFD feeding did not alter based on this assay, it was elevated after 10 and 16 weeks of HFD feeding, as seen in the immunoblotting assay.
The 2 immunological methods examining TFEB translocation had methodological variations. We thus evaluated the functional consequences of TFEB translocation using more quantitative methods. First we examined the transcriptional expression of 6 TFEB target genes, which encode lysosomal enzymes and autophagy-related molecules [28] and found that all of them showed a dynamic alteration with a decreased expression at the 3-week and 16-week HFD feeding time points, but a restored expression at the 10-week feeding point (Figure 2(a)). As a comparison, 2 other autophagy molecules, ATG7 and BECN1, which do not seem to be regulated by TFEB [28], did not show similar alterations (Figure S1). We then examined the activity of CTSB (cathepsin B), CTSD, CTSE, LIPA/LAL (lysosomal acid lipase A) and ACP2 (acid phosphatase 2, lysosomal), all reflecting the lysosomal function promoted by TFEB. The activity of all of these enzymes declined as HFD started, but rebounded by 10 weeks of feeding (Figure 2(b)). In addition, TFEB also promotes lysosome biogenesis, which, as measured by the abundance of a lysosomal marker, LAMP1 (lysosomal-associated membrane protein 1), also showed a similar pattern of alteration in response to high-fat diet feeding (Figure 2(c)). Taken together, these assays assessed the functional consequences of TFEB activation, and showed dynamic changes consistent with those of TFEB nuclear translocation following HFD administration. Furthermore, these changes in the catabolic function of TFEB were in the opposite direction as the changes in the anabolic RPS6KB activation, reflecting the coordinated MTORC1 function in response to superfluous nutrient stimulation.
Figure 2.
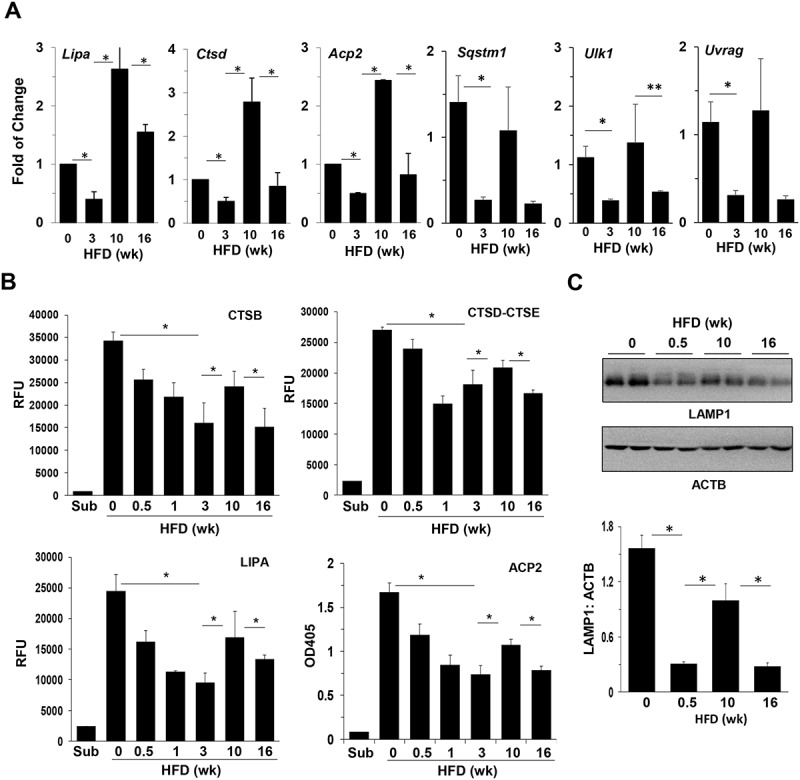
Hepatic lysosomal functions oscillated in HFD-fed mice. (a) The hepatic mRNA levels of the selected TFEB targets were analyzed by qRT-PCR in mice fed with HFD for the indicated times. (b) Liver lysates were analyzed for the activity of lysosomal enzymes. (c) Expression of LAMP1 in the livers of HFD-fed mice was determined by immunoblotting assay and quantified by densitometry. Each lane represents 1 individual mouse. *, p < 0.05; n = 4–6 per group. Acp2, acid phosphatase 2, lysosomal; Ctsd, cathepsin D; Lipa, lysosomal acid lipase A; RFU, relative fluorescence units; Sqstm1/p62, sequestosome 1; Ulk1, unc-51 like kinase 1; Uvrag, UV radiation resistance associated gene.
Autophagy degradation oscillated with the dynamics of TFEB function
Autophagy is one of the major downstream events regulated by MTORC1 [31]; autophagy activation can be suppressed by MTORC1. In addition, degradation of autophagosomes requires robust lysosome functions, which are promoted by TFEB, but could be inhibited by MTORC1 via its negative effect on TFEB translocation. We thus examined whether autophagy flux also demonstrated a dynamic change during the course of HFD feeding.
GFP-LC3 transgenic mice were fed with a regular diet or HFD for different times. The number of GFP-LC3 puncta per cell was used to assess the level of autophagosomes/autolysosomes as previously described [34]. The hepatic level of GFP-LC3 puncta was quite stable in mice fed with regular chow diet up to 38 weeks (Figure S2). Autophagy flux in the liver can be assessed with the injection of chloroquine (CQ) 16 h before sacrifice to suppress the lysosome function. CQ treatment thus results in a higher level of GFP-LC3 puncta due to the blockage of autophagic degradation. The level of degradation can be measured by the difference between the CQ and non-CQ groups and were quite stable under the chow diet regimen (Figure S2).
However, only 3 days into the HFD feeding regimen, the number of GFP-LC3 puncta was increased and remained higher until 16 weeks (Figure 3(a,b). The degree of degradation as measured by the difference between the CQ and non-CQ groups was variable at different time points (Figure 3(b)). An immunoblotting assay for the level of LC3-II, the membrane form of LC3, demonstrated a similar trend (Figure 3(c,d). The results indicated that the degradation of autophagosomes fluctuated with the troughs at 0.5, 3 and 16 weeks, and the peaks at 0, 1 and 10 weeks after HFD started, but showed an overall reduction (Figure 3(e)). Autophagy synthesis in the presence of HFD, as measured by the difference in GFP-LC3 puncta between the HFD-CQ and the regular chow-CQ groups, was also in a declining trend with a small degree of fluctuation that was in parallel with that of autophagy degradation (Figure 3(e)). The pattern of the degradation curve was in parallel with that of TFEB activation (Figure 1(b), Figure 2), but opposite to the phosphorylation of RPS6KB (Figure 3(f)), consistent with the function of TFEB in promoting autophagy-lysosome degradation capability and the function of MTORC1 in suppressing autophagy.
Figure 3.
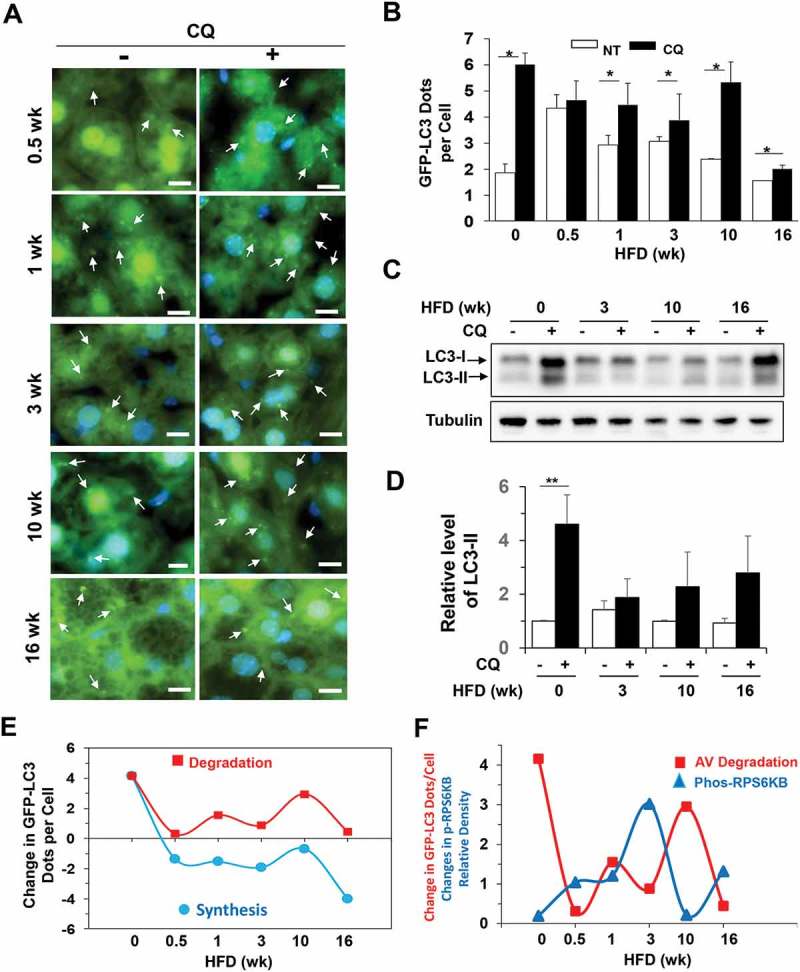
Autophagy degradation was dynamically regulated in HFD-fed mice. (a-b) GFP-LC3 transgenic mice at the age of 8 weeks were fed with HFD for the indicated time, and then treated with (CQ) or without (NT) CQ (60 mg/kg, i.p.) 16 h before sacrifice. Cryosections of the livers were stained with Hoechst 33328. GFP-LC3 puncta represented autophagosomes (a) and were quantified (b). The values at time zero were obtained from 8-week-old mice fed with regular diet. (c-d) Mice were fed with HFD for the indicated duration and then treated with or without CQ (60 mg/kg, i.p.) 16 h before sacrifice. Liver lysates were analyzed by immunoblotting for LC3. The level of LC3-II was normalized to that of tubulin and expressed as the fold of change over the control diet with no CQ treatment (0 week). (e) Autophagosome synthesis and degradation were calculated using the GFP-LC3 puncta data above and plotted against the duration of HFD feeding. (f) The changes in degradation of the GFP-LC3 puncta (autophagosomes or AV) and phosphorylated RPS6KB levels (from Figure 1F) were compared on the same time scale. The changes of the 2 parameters were in opposite directions at each time point. *, p < 0.05; **, p < 0.01; n = 3–6 per group. Scale bar: 10 μm. Arrows indicate GFP-LC3 puncta. For clarity and for the illustrative purpose, not all puncta are indicated.
We then assessed how autophagic degradation of lipid could be altered. Lipid degradation can be mediated by an autophagic mechanism in a process known as lipophagy [35–37]. Cryosections of the liver were stained with a lipophilic BODIPY dye, and intrahepatic lipid droplets were quantified (Figure 4(a,b)). In addition, the level of hepatic triglycerides was measured biochemically (Figure 4(c)). Results from the 2 assays were consistent and indicated that the hepatic lipid level was progressively increased over a 16-wk course of HFD feeding. However, autophagic degradation of lipids could still be detected based on the differential increments following CQ treatment. Notably, lipid degradation measured in this way was relatively larger in amount at 0.5–1 week and 10 weeks after HFD feeding, but relatively smaller at 3 weeks and 16 weeks after HFD feeding (Figure 4(d)). The pattern of fluctuation was in parallel with those of autophagy degradation (Figure 3(f)), TFEB translocation (Figure 1(f)) and lysosomal enzyme activities (Figure 2(b)). These findings indicated that autophagic degradation capability could affect the lipid level in hepatocytes.
Figure 4.
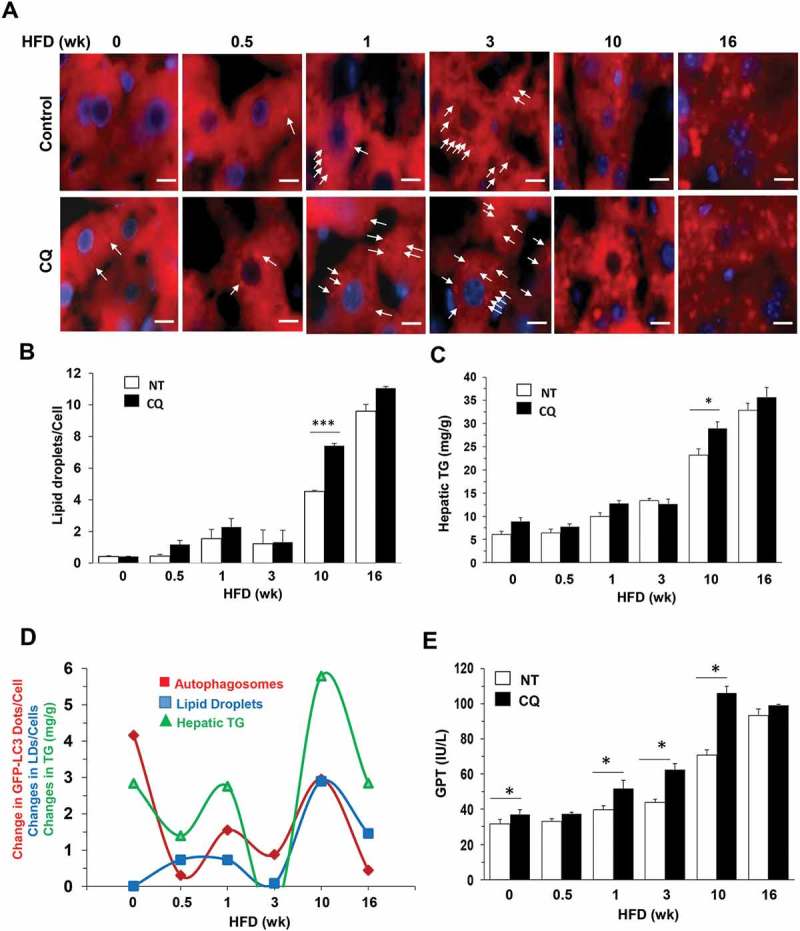
Alterations of lipophagy in the liver of HFD-fed mice. (a-b) Mice were fed with HFD for different times and given CQ or not treated (NT) 16 h before sacrifice. Cryosections of the liver were stained with BODIPY-581/591 for lipids droplets (a), which were quantified as the number per cell (b). (c) Hepatic triglycerides (TG) were measured in the same group of mice. (d) The difference between the CQ plus group and control group was what was degraded in the lysosome. The difference was noted as the ‘changes’ in the degradation of autophagosomes, the level of lipid droplets or the level of hepatic TG, which were plotted against the duration of HFD feeding. Data of the degraded autophagosomes (GFP-LC3 puncta) were from Figure 3D. The changes of these 3 parameters were in the same directions at almost all time points, implying that they are associated with the same process related to lipophagy. (e) GPT/ALT levels were measured in the same group of mice as in panel C. *: p < 0.05, n = 3–6 per group. Scale bar: 10 μm. Lipid droplets were small in both size and number in samples with shorter HFD duration (0–3 weeks) and are indicated by arrows. Lipid droplets in samples with longer HFD duration (10–16 wk) were large in size and number and are not indicated by arrows.
Lipophagy can affect the severity of steatosis, which in turn can affect liver injury. We found that GPT/ALT (glutamic pyruvic transaminase, soluble/alanine transaminase) levels were increased over the time with HFD feeding, but CQ co-treatment exacerbated the injury further (Figure 4(e)). Interestingly, the exacerbation of liver injury, and thus the promotion of GPT/ALT level, by CQ was more notable at the 10-week time point than at the 16-week time point (Figure 4(e)), consistent with the finding of a higher lipophagic activity at 10 weeks than that at 16 weeks (Figure 4(b,c)). Thus there was a more significant impact of CQ suppression on liver injury at 10 weeks than at 16 weeks. Overall, the lysosome function and autophagy degradation were downregulated in long-term HFD administration but in a non-linear way, which would be associated with the oscillating MTORC1 signaling.
The subcellular location of TFEB correlated with the severity of hepatic steatosis in NAFLD patients
The above findings made in a mouse model of fatty liver disease prompted us to examine whether a correlation of TFEB activation and hepatic steatosis could be also found in NAFLD patients. Liver biopsy samples of clinically diagnosed NAFLD patients (Table S1) were H&E stained for steatosis assessment and immunostained with an anti-TFEB antibody to determine its location. Indeed, TFEB was detected in hepatocyte cytoplasm in some cells, but in the nucleus in other cells (Figure 5(a)). The percentage of cells with nuclear TFEB was determined and correlated with the steatosis score for each sample (Figure 5(b)). A significant negative correlation (r = −0.841, p < 0.0001) was found in which more severe steatosis was associated with a lower percentage of cells with nuclear-positive TFEB (Figure 5(b)). Conversely, nuclear TFEB did not show a significant correlation with BMI (r = −0.389, p = 0.0734) (Figure 5(c)). However, nuclear TFEB location did not seem to be highly correlated with serum triglycerides, cholesterol or GPT/ALT levels in these patients (Table S1). Overall, the results supported the idea that TFEB activation could be an important factor in determining the severity of hepatic steatosis in mice or in NAFLD patients.
Figure 5.
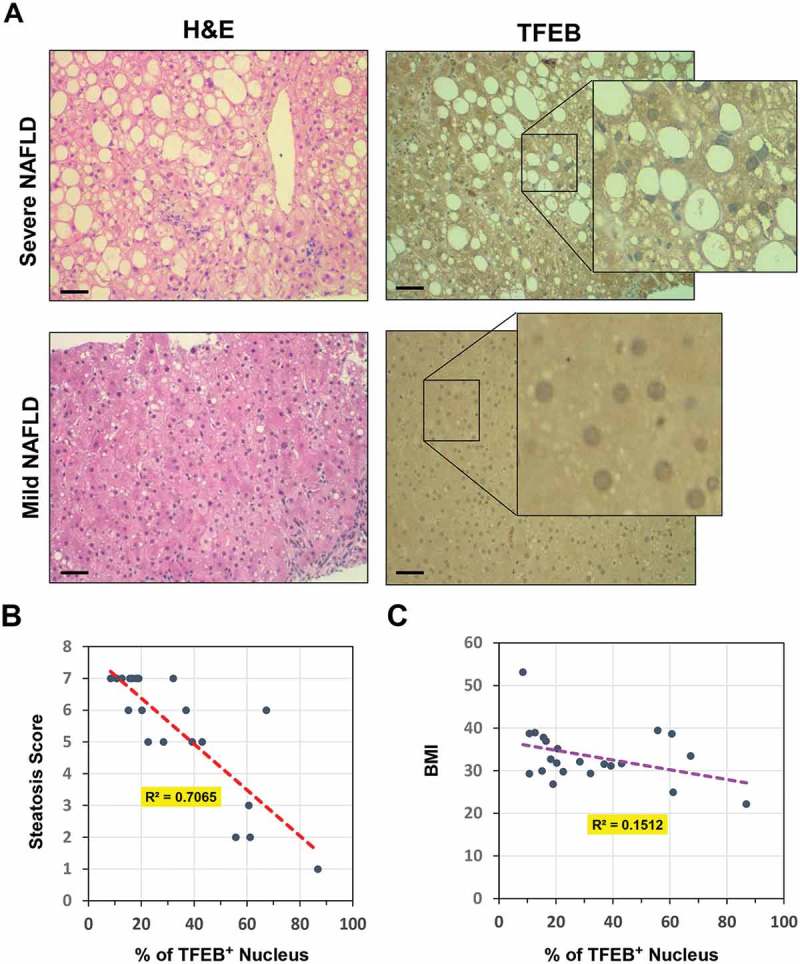
Nuclear location of hepatic TFEB was inversely correlated with steatosis in NAFLD patients. (a) The liver biopsy sample of NAFLD patients were stained with H&E or with an anti-TFEB antibody. Representative mild and severe cases of steatosis are shown. Scale bar: 50 μm. Insets show enlarged images. Minor steatosis was associated with a higher percentage of cells with nuclear TFEB. (b-c) Correlation of percentage of cells with TFEB-positive nucleus with hepatic steatosis score (b) or BMI (c). Number of patients (n) = 22.
Overexpression of TFEB resulted in a resumption of lysosome functions and reduced hepatic steatosis and liver injury
The above findings suggested that the dynamics of lysosome functions, lipophagic activities, hepatic steatosis and injury were likely regulated by the dynamics of the MTORC1-TFEB regulatory circuit. The oscillation reflected the dependence of MTORC1 on the lysosomal function that was supported by TFEB, but the activated MTORC1 in turn inhibited TFEB, thus dampening its own activity. Subsequently the de-repressed TFEB restored the lysosome function and reinstated MTORC1 activation, thus starting the next round of oscillation, although at a lower level. We thus examined whether disruption of the MTORC1-TFEB regulatory circuit could lead to changes in the outcomes. We hypothesized that if the level of TFEB could be restored from an exogenous source, the negative impact on lysosome functions by MTORC1 could be overcome, thus improving hepatic outcomes.
Toward that end, adenoviral vectors carrying wild type or S211A Tfeb were given to mice that had been fed with HFD for 12 weeks. S211 is the major site for MTORC1-mediated phosphorylation [29,32]. After the additional 4 weeks of HFD feeding, samples were collected (Figure 6(a)) and the expression of TFEB was confirmed (Figure 6(b)). There was a lower level of phosphorylated TFEB (Figure 6(b,c) and a higher level of non-phosphorylated TFEB (Figure 6(b, d) in the mice receiving Ad-Tfeb, particularly, mice receiving Ad-S211A-Tfeb. The period from 12 to 16 weeks was the period when MTORC1 activity was elevated and TFEB activation was reduced in response to HFD feeding. We found that overexpression of TFEB indeed enhanced hepatic lysosome enzyme activities (Figure 6(e)). In addition, several genes involved in lipid oxidation, lipolysis and cholesterol conversion to bile acids were also increased (Figure 6(f)) as reported previously [38]. These changes were correlated with a reduced progression of fatty liver and the associated symptoms, and resulted in an improvement in body weight, liver weight and the liver:body weight ratio (Figure 6(g)), reduced hepatic lipid content (Figure 6(h)) and liver injury (Figure 6(i)). It also reduced serum cholesterol level, although not serum triglyceride levels (Figure 6(i)). Expression of the TFEBS211A mutant had a more significant impact in several aspects than the expression of wild-type TFEB, consistent with its ability to resist MTORC1-mediated phosphorylation [29].
Figure 6.
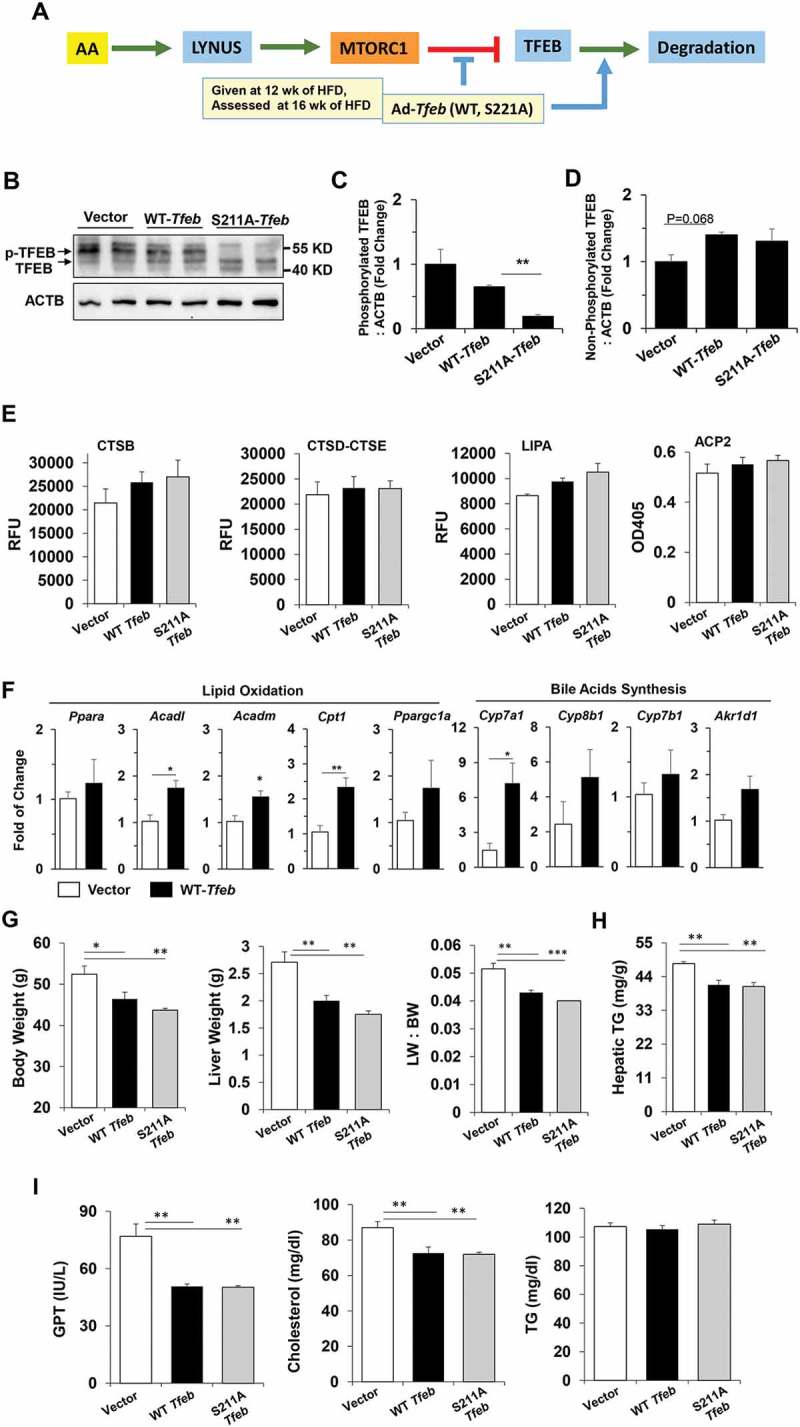
Overexpression of TFEB disrupted the oscillation and improved hepatic status in HFD-fed mice. (a) Scheme of experimentation. Mice fed with HFD for 12 weeks were given adenovirus expressing wild-type TFEB or the TFEBS211A mutant, or adenoviral vector, followed by 4 more weeks of HFD feeding before being sacrificed. (b-d) Expression of non-phosphorylated TFEB and phosphorylated TFEB (p-TFEB) in the liver was examined by immunoblotting assay (b). The relative levels of phosphorylated TFEB (c) and non-phosphorylated TFEB (d) were quantified, which were normalized to ACTB. (e) Hepatic lysosomal enzyme activities were determined in each group of the mice. (f) Hepatic expression of genes involved in lipid metabolism was assessed by qRT-PCR. (g-i) The following parameters were determined: body weight, liver weight, and liver:body weight ratio (g), hepatic TG (h), and serum levels (i) of GPT/ALT, cholesterol and TG. *, p < 0.05; **, p < 0.01; n = 3–6 per group. Acadl, acyl-Coenzyme A dehydrogenase, long-chain; Acadm, acyl-Coenzyme A dehydrogenase, medium chain; Akr1d1, aldo-keto reductase family 1, member D1; Cpt1, carnitine palmitoyltransferase 1a, liver; Cyp7a1, cytochrome P450, family 7, subfamily a, polypeptide 1; Cyp7b1: cytochrome P450, family 7, subfamily b, polypeptide 1; Cyp8b1, cytochrome P450, family 8, subfamily b, polypeptide 1; Ppargc1a/Pgc1-α, peroxisome proliferative activated receptor, gamma, coactivator 1 alpha; Ppara/Ppar-α, peroxisome proliferator activated receptor alpha.
To investigate the effect of Tfeb overexpression at the beginning of HFD treatment when steatosis was still minor, we gave Ad-Tfeb at the first day of HFD treatment and collected samples 3 weeks later (Figure S3A). This was also the period when MTORC1 activity was elevated and TFEB activation was reduced in response to HFD feeding (Figure 1(e)). As in the study with 16 weeks of feeding (Figure 6), overexpression of TFEB also promoted lysosomal functions with enhanced lysosomal enzyme activities in this short-term regimen (Figure S3B). Although the effect of TFEBS211A was more prominent, overexpression of both the wild-type and mutant TFEB resulted in reduced liver weight, hepatic triglycerides, liver injury, and serum cholesterol level, though not serum TG level (Figure S3C-E). These results suggested that elevation of TFEB at either an early or later stage of a hepatic steatosis condition could result in improved outcomes by overcoming the inhibition from MTORC1.
Modification of MTORC1 activity affected TFEB translocation, lysosome functions and hepatic pathology
Rapamycin, an MTORC1 inhibitor, is able to improve hepatic steatosis and insulin resistance in HFD-fed mice [37]. To determine whether such treatments could also rescue TFEB activation, we administrated rapamycin at the time when MTORC1 was highly activated in this model, i.e. the 3-week or 16-week time point (Figure 7(a)). As anticipated, rapamycin inhibited the phosphorylation of RPS6KB and EIF4EBP1 (Figure 7(b)), and promoted nuclear translocation of TFEB (Figure 7(c-e)). Corresponding to the de-suppression of TFEB, expression of representative TFEB target genes was elevated (Figure 7(f)) and lysosomal enzyme activities were enhanced (Figure 7(g)). Thus suppression of MTORC1 by rapamycin could result in a reactivation of TFEB, promoting lysosome degradation.
Figure 7.
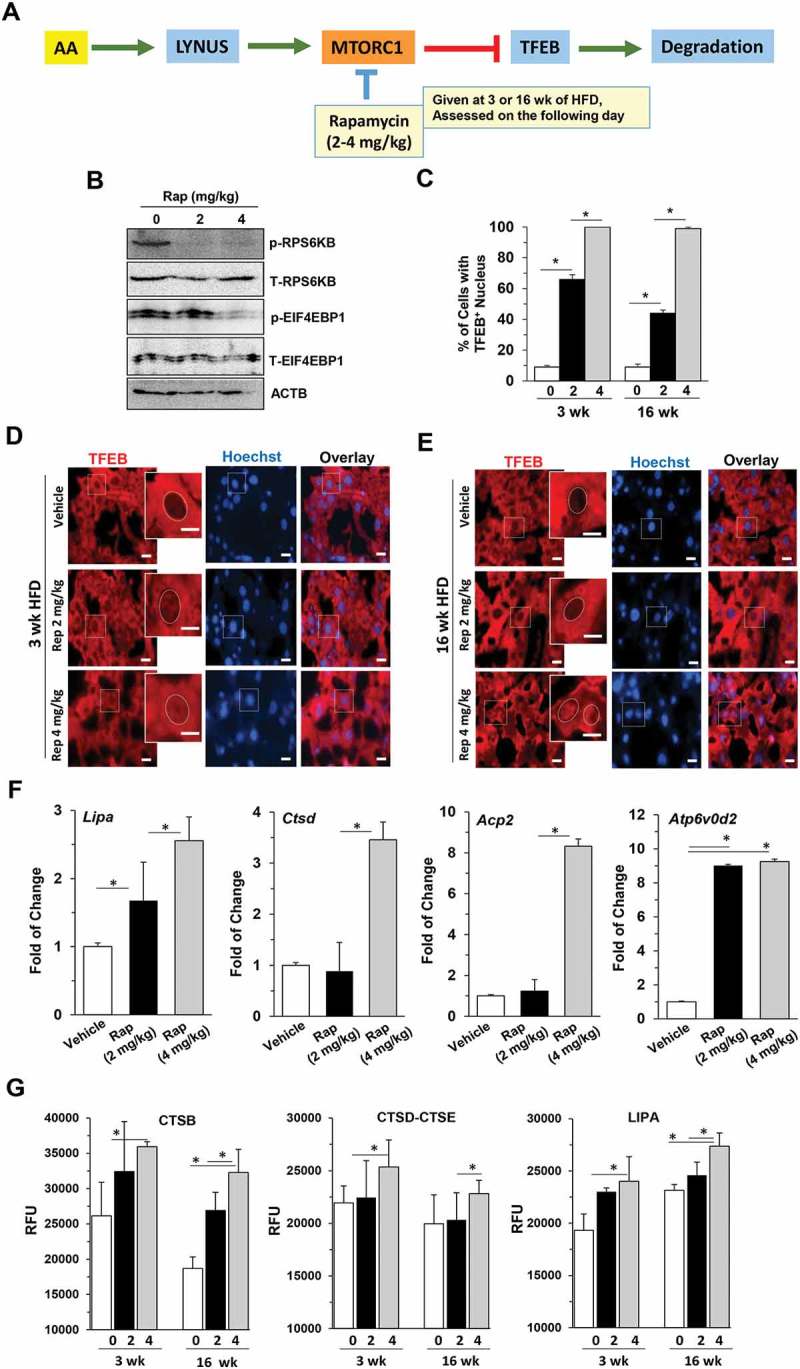
Suppression of MTORC1 disrupted the oscillation pattern of TFEB signaling and improved hepatic status in HFD-fed mice. (a) Experimental scheme. Mice fed with HFD for 3 or 16 weeks were given 2 mg/kg or 4 mg/kg rapamycin and sacrificed 16 h later. (b) Hepatic lysates were prepared from the 16-wk groups and analyzed by immunoblotting assay. The letter p indicates the phosphorylated form whereas T indicates all forms. (c-e) The percentage of cells with TFEB-positive nuclei (c) was quantified from livers stained with anti-TFEB antibody and Hoechst 33328 after 3-week (d) or 16-week (e) HFD feeding. Boxed areas were enlarged in the insets, which illustrate the representative hepatocytes. Dotted circles indicate the nucleus. (f) The mRNA levels of the selected TFEB targets were determined by qRT-PCR in mice fed with HFD for 16 weeks. (g) Hepatic lysosomal enzyme activities were determined. *, p < 0.05; **, p < 0.01; n = 3 per group. Scale bar: 10 μm. Acp2, acid phosphatase 2, lysosomal; Atp6V0d2, ATPase, H+ transporting, lysosomal V0 subunit D2; Ctsd, cathepsin D; Lipa, lysosomal acid lipase 2.
We then tested the hypothesis that persistent, instead of oscillating, activation of MTORC1 could lead to sustained lysosome suppression and exacerbated liver injury. An adenoviral vector carrying a constitutively activated HA-tagged Rraga mutant (RragaGTP/GTP) was administrated to mice fed with HFD for 6 weeks, which were analyzed after another 4 weeks of HFD feeding (Figure 8(a)). The total HFD feeding lasted for 10 weeks. This RRAGA mutant assumes the GTP-bound conformation and is able to support MTORC1 activation without amino acid stimulation through the lysosome [14,15]. We hypothesized that the overexpression of this mutant RRAGA would reverse the decreased MTORC1 activity that was noted in the period from 6 to 10 weeks (Figure 1(e)).
Figure 8.
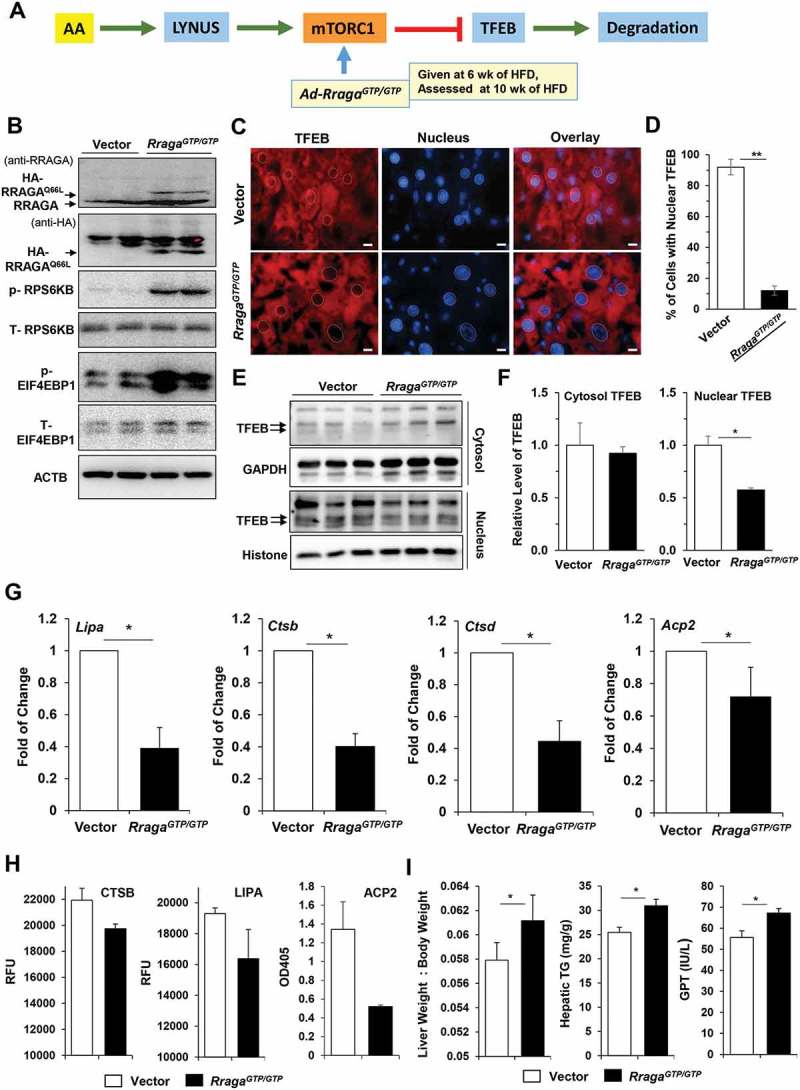
Overexpression of a constitutively activated RRAGA mutant disrupted the MTORC1 oscillation pattern and impaired liver function. (a) Scheme of experimentation. Mice fed with HFD for 6 weeks were given Ad-RragaGTP/GTP, or adenoviral vector, followed by 4 more weeks of HFD feeding before being sacrificed. (b) Liver lysates were analyzed by immunoblotting for RRAGA expression and MTORC1 activation. (c-d) Cryosections of the livers were stained with anti-TFEB and Hoechst 33328. The nuclei are indicated with dotted circles (c) and the percentage of cells with nuclear TFEB were quantified (d). (e-f) Liver lysates were fractionated to the cytosol and the nucleus for immunoblotting analysis (e). The levels of TFEB in Ad-RragaGTP/GTP groups relative to the vector groups were determined by densitometry in both fractions (f). (g) The mRNA levels of the selected TFEB targets were determined by qRT-PCR. (h) Hepatic lysosomal enzyme activities were determined. (i) The following parameters were determined: liver:body weight ratio, hepatic triglycerides, and serum GPT. *, p < 0.05; **, p < 0.01; n = 3 per group, Scale bar: 10 μm. Acp2, acid phosphatase 2, lysosomal; Ctsb, cathepsin B; Ctsd, cathepsin D; Lipa, lysosomal acidic lipase A.
The exogenous RragaGTP/GTP was successfully expressed in the livers as determined by immunoblotting analysis using anti-RRAGA and anti-HA antibodies. The MTORC1 activity was re-elevated in the RragaGTP/GTP group with a higher level of phosphorylation of RPS6KB and EIF4EBP1 than that in the control group (Figure 8(b)). This elevated MTORC1 activity in turn reversed the subcellular location of TFEB, resulting in a decreased nuclear location in the RragaGTP/GTP-administered mice as shown by both immunostaining and immunoblotting assays (Figure 8(c-f)). Consequently, the mRNA expression of TFEB targets was suppressed (Figure 8(g)), and the lysosomal enzyme activities were impaired (Figure 8(h)) in the RragaGTP/GTP group. Functionally, the liver:body weight ratio, the level of hepatic triglyceride, and the level of serum GPT/ALT were all increased (Figure 8(i)) in the RragaGTP/GTP group. Interestingly, overexpression of RragaGTP/GTP did not seem to upregulate the expression of many genes involved in de novo lipid synthesis that could be affected by MTORC1 (Figure S4). These findings suggested that the effect of RRAGAGTP/GTP could be more related to the effect of MTORC1 on the lysosome and TFEB, and that suppression of TFEB activity by sustaining MTORC1 function using RRAGAGTP/GTP exacerbated hepatic steatosis and injury.
Discussion
This study was performed to understand how the regulatory signaling of MTORC1 and TFEB responded to the persistent stimulation of a superfluous level of nutrients in vivo. The study indicates that 1) the catabolic function represented by TFEB and the anabolic function represented by RPS6KB oscillated in opposite directions in the long-term presence of superfluous nutrients through feedback regulation; 2) this antagonistic oscillation could sustain catabolic function while reducing anabolic response; 3) enhancing TFEB or inhibiting MTORC1 improved, whereas sustaining MTORC1 exacerbated, the hepatic pathology; 4) the beneficial TFEB activation was damped, correlated with the continuous development of hepatic steatosis and injury despite the fact that the MTORC1 activity was also damped; 5) there was an inverse correlation between TFEB activation and hepatic steatosis in the liver of NAFLD patients, suggesting the clinical relevance of this regulatory loop.
The dynamics of MTORC1 and TFEB signaling
MTORC1 is activated in response to nutrients. However, hepatic MTORC1 activity in normally fed mice is kept at a low basal level based on RPS6KB phosphorylation status and TFEB nuclear translocation. The low MTORC1 level can be important for preventing normal cells from uncontrolled growth. The MTORC1 activity rises quickly to a higher level when mice are exposed to a diet containing a larger amount of nutrient. Thus MTORC1 is activated not only during the transition from the fasting to fed status as shown in previous studies, but also during the transition from a normal diet to a high-fat diet as shown in the present study. Thus the activation of MTORC1 may be better defined as the response to the change in nutrient level, rather than to the presence of nutrients.
MTORC1 activation has opposite impacts on anabolism and catabolism but here we have observed that both demonstrate dynamic changes. Anabolic functions represented by the phosphorylation of RPS6KB is elevated following the increase in nutrients, but eventually subsides to the basal level. Conversely, catabolism, as represented by nuclear TFEB translocation, TFEB-directed gene transcription and lysosomal enzyme activities, is reduced following the administration of high-fat diet, but can bounce back later. The observed time points in this study at which the oscillation is peaked or bottomed may likely reflect the oscillation at the upward or the downward side of the cycle, rather than the exact points of maximal changes. The actual time points may vary with the diet scheme and may not be as important as the alterations per se. We have observed the returning of hepatic RPS6KB activity to the basal level in one cycle in the observation period after an initial rise in another model of superfluous nutrient feeding (Figure S5). Thus the key point is that MTORC1 activity is markedly elevated upon hyper-nutrient stimulation, which then subsides to the base line in the long-term diet regimen, perhaps reflecting an adaption process.
Based on current literature, the opposing dynamics of RPS6KB phosphorylation and TFEB-directed lysosomal functions likely resulted from the dependence of MTORC1-RPS6KB activation on TFEB-supported lysosome function, and the subsequent suppression of TFEB translocation by activated MTORC1. In turn, suppressed TFEB leads to downregulated lysosome biogenesis and function, which diminishes MTORC1 activation. The latter allows a degree of de-repression of TFEB, restoration of lysosome function and lysosome biogenesis, which in turn results in MTORC1 elevation once again. Thus, the activation of MTORC1 could be negatively regulated by its own action in inhibiting TFEB activity, while the activation of TFEB could be also negatively affected by its functional support of MTORC1 activation in the presence of nutrients (Figure S6).
Whereas MTORC1 activation is mainly regulated by amino acids and growth factors, the MTORC1-TFEB feedback loop is important so that the anabolic activation of MTORC1 will not be sustained at a high level, which can be pathogenic [3]. Indeed, deletion of Rptor, a regulatory associated protein of MTORC1, prevents hepatic steatosis in HFD-fed mice [10]. In addition, pharmacological suppression of MTORC1 with rapamycin can reduce hepatic steatosis and liver injury in a HFD model [37]. Thus, the feedback suppression of MTORC1 by the downregulated lysosome function seems to be a self-restraining response. Consistently, overexpression of the constitutively activated RRAGAGTP/GTP mutant protein sustains MTORC1 activation in a manner that is independent of lysosome function, thus avoiding the feed-back inhibition, which leads to enhanced hepatic steatosis and liver injury. The pathogenic effect of RRAGAGTP/GTP overexpression might be more related to the inhibition of TFEB by MTORC1 due to RRAG’s lysosomal location, but not much to the potential promotion of de novo lipid synthesis by MTORC1 (Figure S4).
The in vivo oscillating activation of the anabolic RPS6KB stimulated by high fat diet follows the same pattern that was first observed in the in vitro activation of NFKB complex by TNF [39], and also in the in vitro activation of TP53 by irradiation [40]. In these cases, the oscillation is caused by the feedback inhibition by the products of NFKB and TP53, i.e. NFKBIA/I-κBα and MDM2, respectively. Our study represents the first in vivo example of signaling oscillation on a much longer time scale, and validates the general principle derived from the in vitro observations. Moreover, the determination of the in vivo dynamics in this study may allow the exploration of the pathophysiological significance of such signaling dynamics in a disease model.
The pathological significance of TFEB suppression
It is critical to note that while MTORC1’s anabolic function is eventually dampened, hepatic steatosis continuously progresses, indicating that factors other than anabolic synthesis also play important roles. These factors include an increased uptake of, and a decreased catabolic removal of, fatty acids. The present study confirms earlier works indicating the suppression of autophagy [36,41,42] by high-fat diet, but mostly reveals the inhibition of TFEB and lysosome function and how MTORC1-TFEB dynamics could affect autophagy and liver pathogenesis.
The dynamic alterations of TFEB activation is reflected in the subcellular location of TFEB, the expression of TFEB downstream genes, the activities of lysosomal enzymes, and autophagic degradation. However, lysosome function improvement is followed by MTORC1 reactivation, and TFEB activation is subjected to MTORC1 suppression. Thus, TFEB function alters at a repressed level and its activity does not return to the pre-HFD level despite the fact that MTORC1 activation was gradually reduced to the pre-HFD basal level. If TFEB activation is enhanced, such as by overexpression, during the course of HFD, lysosome function is indeed improved with reduced steatosis and metabolic syndrome as shown here. Conversely, liver-specific deletion of Tfeb increased hepatic steatosis during high-fat diet feeding [38]. The beneficial function of TFEB is likely related to the enhanced degradative function of the lysosome, which is instrumental to lipophagy [35]. It is noted that TFEB can also have a role in lipid utilization independent of the lysosome, particularly during fasting [38]. Overexpression of TFEB in mouse liver leads to increased expression of genes for fatty acid oxidation and ketogenesis. We confirmed in our studies the elevation of genes for lipid oxidation and lipid utilization (cholesterol conversion to bile acids) in TFEB-expressing livers (Figure 6(c)). The impact of TFEB on metabolism could thus be multi-dimensional.
It thus seems that enhancing TFEB and lysosome functionality may be more critical than inhibiting MTORC1 in facilitating the resolution of fatty liver diseases. There is a clear correlation in TFEB activation in the liver of NAFLD patients, with a high level of activation being associated with a lower level of steatosis. This observation raises several potentially interesting issues. One is what determines that some patients had a higher TFEB activation than others. Understanding this may help to understand why some people are more prone to fatty liver diseases. The second issue is whether TFEB activation status could serve as a prognostic marker, although detection of hepatic TFEB involves invasive procedures. While the use of rapamycin is not ideal because of the conceived side effects, modulation of TFEB in those patients with low TFEB activation status may be an alternative choice for future therapeutic considerations.
Materials and methods
Animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee at Indiana University, and at the Institute for Nutritionals Sciences, Chinese Academy of Sciences. C57Bl/6J wild-type mice and GFP-LC3 transgenic mice [34] of both sex were used in the study. Mice were housed in a specific pathogen-free facility with a 12-h light-dark cycle. At 8 weeks of age, mice were provided ad libitum normal chow diet, or high-fat diet (Research diets, D12492), or high- fat, high-sucrose diet (Research Diets, D12327). At the designated time point, mice were sacrificed in the morning between 9 am-12 noon, and blood and livers were harvested for analysis. Chloroquine was given by intraperitoneal (i.p.) injection at the dose of 60 mg/kg. Rapamycin (Sigma-Aldrich, R8781) was given by i.p. injection at the dose of 2 and 4 mg/kg. Expression of the wild-type and S211A mutant of TFEB [29], or RragaGTP/GTP(RRAGAQ66L) [14] was mediated by adenoviral vectors (2x109 viral particle/mouse, intravenously) as previously described.
Analysis of autophagy flux in GFP-LC3 mice
GFP-LC3 transgenic mice were treated as described with or without CQ (60 mg/kg, i.p.) 16 h before sacrifice. Cryosections of livers were prepared as previously described [34]. Briefly, liver tissue was fixed in 4% paraformaldehyde for 4 h, followed by a gradual equilibrium with 5–20% sucrose (Sigma-Aldrich, S7903). Livers were then fixed in OCT compound (Fisher Scientific, 23–730-571) and subjected to cryosectioning. The cryosections were stained with Hoechst 33328 and images were obtained using fluorescence microscopy (Nikon Eclipse TE-200). GFP-LC3 puncta were quantified per cells. Autophagy flux was analyzed as previously described [43]. Autophagy synthesis under HFD treatment was defined as the difference between the value of the HFD + CQ group and the value of chow diet + CQ group. Because the levels of GFP-LC3 puncta in the chow diet groups were virtually unchanged from 8 weeks to 38 weeks in both non-CQ and CQ groups (Figure S1), we used the values from the 8-weeks groups to calculate the autophagy synthesis under HFD feeding. Autophagy degradation was defined as the difference between the value of the HFD + CQ group and the value of the HFD group.
Immunoblotting, immunofluorescence staining and immunohistochemistry assays
Liver tissue was homogenized in RIPA buffer with protease and phosphatase inhibitors. The post-nuclear supernatant was used for protein separation by SDS-PAGE, which is followed by transfer to PVDF membranes. Antibodies against the following molecules were used: total RPS6KB (Cell Signaling Technology, 9202; 1:1000 dilution), phosphorylated-RPS6KB (Thr389; Cell Signaling Technology, 9234; 1:1000 dilution), total RPS6 (Cell Signaling Technology, 2217; 1:1000 dilution), phosphorylated RPS6 (Ser235/236; Cell Signaling Technology, 2211; 1:1000 dilution), total EIF4EBP1 (Cell Signaling Technology, 9452; 1:1000 dilution), phosphorylated EIF4EBP1 (Thr37/46; Cell Signaling Technology, 9459; 1:1000 dilution), LC3B (Sigma-Aldrich, L7543; 1:1000), ATG7 (Cell Signaling Technology, 2631; 1:1000 dilution), BECN1/BECLIN 1 (Santa Cruz Biotechnology, S11427; 1:1000 dilution), SQSTM1/p62 (MBL International Corp., PM045; 1:1000 dilution), TFEB (Abcam, 113,372; 1:1000 dilution; or Santa Cruz Biotechnology, SC11009; 1:200 for immunostaining; BETHYL Lab, A303-673A-M for immunoblotting), LAMP1 (University of Iowa Hybridoma Center, 1D4B; 1:1000 dilution), ACTB/β-actin (Sigma-Aldrich, A5441; 1:5000 dilution), GAPDH (Novus Biologicals, NB 300–221; 1:1000) and H3F3/histone H3 (Cell Signaling Technology, 4499; 1:1000 dilution). Horseradish peroxidase-conjugated secondary antibodies (111–035-045, and 115–035-062; 1:5000 dilution) were from the Jackson Immuno-Research Laboratory. Blots were developed using Immobilon Chemiluminescent HRP substrate (EMD Millipore, WBKLS0500). Images were acquired and quantified with a Kodak 4000 image station and its companion software or with ImageJ.
For immunostaining, cryosections prepared from frozen liver tissue or paraffin sections were blocked with 5% serum in phosphate-buffered saline (10 mM phosphate, 2.7 mM KCl, 137 mM NaCl, pH 7.4)) containing 0.1% Triton X-100 (Sigma-Aldrich, X100) (PBS-Tx) for 1 h and then incubated with an anti-TFEB antibody in 5% serum in PBS-Tx. The incubation was 2 h at room temperature or overnight at 4°C. Sections were washed in PBS-Tx, following by incubation with Cy-3-conjugated secondary antibody (Jackson Immuno-Research Laboratory, 111–165-144) for 1 h at room temperature. The sections were finally stained with Hoechst 33342 (1 μg/ml) for 5 min. Images were acquired with a Nikon Eclipse TE 200 immunofluorescence microscope and the companion NIS-Elements AR3.2 software.
For immunohistochemistry, paraffin sections of liver biopsy were subjected to antigen retrieval in sodium citrate buffer (10 mM, pH 6.0). After wash, the sections were stained with an anti-TFEB antibody (Abcam, 113,372; 1:100 dilution), washed and then stained with HRP-conjugated goat anti-rabbit antibody. After thorough washes, the substrate was added.
Measurement of lysosome enzyme activity
Lysosomal enzyme activities were measured as previously described [37]. Briefly, livers were homogenized in lysis Buffer A (0.1 M sodium acetate, 0.1 M sodium chloride) for activities of CTSB (pH 5.5) and CTSD-CTSE (pH 4.0), in Lysis B (0.2 M sodium acetate [Sigma-Aldrich, S2889], 0.01% Tween-80 [Sigma-Aldrich, P1354], pH 5.5) for LIPA activity, or in a commercial assay buffer for ACP2 activity using a commercial assay kit (Sigma-Aldrich, CS0740). Samples with equal amounts of proteins (100 µg for ACP2, 5 µg for all other enzymes) were used in the assay. The substrates used are Z-FR-AMC (Enzo Life Sciences, BML-P139) for CTSB, Mca-GKPILFFRLK (Dnp)-D-R (Enzo Life Sciences, BML-P145-0001) for CTSD-CTSE, 4-methylumbelliferyl oleate (Sigma-Aldrich, 75,164) for LIPA, and 4-nitrophenyl phosphate (Sigma-Aldrich, N2765) for ACP2. The lysates and substrates were incubated at 37°C for 90 min (LIPA), 45 min (cathepsins) or 30 min (ACP2). The enzyme activity was determined using a multi-function spectrometer (Infinite M200Pro, Tecan, Morrisville, NC, USA) at OD405 for ACP2, and at the following fluorescence excitation/emission wavelength: 365 nm/440 nm for CTSB, 328 nm/393 nm for CTSD-CTSE, and 327 nm/449 nm for LIPA.
Measurement of hepatic steatosis and injury
GPT/ALT, triglyceride and cholesterol level were measured using corresponding commercial assay kits (Pointe Scientific, A7526, T7532, C7510) per the manufacturer’s instruction. Hepatic triglycerides were extracted as previously described [43]. The histological severity of steatosis was scored using the NAFLD scoring system published by the Non-alcoholic Steatohepatitis Clinical Research Network [44]. To quantify lipid droplets, hepatic cryosections were stained for 10 min with BODIPY-581/591-C11 (ThermoFisher Scientific, D3861) prepared in 0.4% Triton X-100 at the concentration of 0.1 mM, and were then examined under a fluorescence microscope (Nikon Eclipse TE-200) equipped with Nikon’s Elements AR3.2 software.
Analysis of the histology of human samples
Liver biopsy samples were from patients with clinically diagnosed NAFLD at the Indiana University Health Pathology Laboratory (Table S1). All samples were de-identified and the study was approved by the Institution Review Board of Indiana University. Only formalin-fixed and paraffin-embed samples were used in this study for immunohistochemistry analysis. The staining patterns of TFEB location were correlated with steatosis scores or body mass index (BMI) using the linear fit function of Microsoft Excel.
Gene expression analysis
Total RNA was extracted from 30 mg of frozen liver tissue using GeneJET RNA purification kit (Thermo Fisher Scientific, K0731). RNA quality and concentration was quantified using a Tecan NanoQuant Plate on Infinite M200Pro spectrometer (Tecan, Switzerland). cDNA was prepared using M-MLV reverse transcriptase (Thermo Fisher Scientific, N8080018) per the manufacturer’s instruction. Gene expression was quantified using SYBR Green master mix (Thermo Fisher Scientific, A25780) and a 7500 Fast Real-Time PCR System (Life Technologies-Applied Biosystems, Waltham, MA, USA). The 2−ΔΔCt method was used for quantification. Results were normalized using the housekeeping gene PPIA/cyclophilin A. Primer sequences used for the amplification are listed in Table S2.
Statistical analysis
Statistical analyses were performed with Sigma Stat 3.5. Data were expressed as mean± SE. Significance was determined using Student’s t test for two-group comparisons, or using one-way ANOVA with post-hoc analysis for multi-group comparisons. Correlation was analyzed using Pearson Product Moment Correlation. Results were considered statistically significant when p < 0.05.
Funding Statement
This work was supported by the HHS | NIH | National Institute on Alcohol Abuse and Alcoholism (NIAA) [R01AA021751];HHS | NIH | National Institute on Alcohol Abuse and Alcoholism (NIAA) [R21AA021450].
Abbreviations
- ACP2
acid phosphatase 2, lysosomal;
- EIF4EBP1
eukaryotic translation initiation factor 4E binding protein 1;
- GPT/ALT
glutamic pyruvic transaminase, soluble/alanine transaminase;
- HFD
high fat diet;
- LIPA/LAL
lysosomal acid lipase A;
- MTORC1
MTOR complex 1;
- NALFD
non-alcoholic fatty liver disease;
- RPS6
ribosomal protein S6;
- RPS6KB
ribosomal protein S6 kinase;
- RRAGA
Ras-related GTP binding Al TFEB: transcription factor EB
Acknowledgments
The authors are in debt to Dr. Kun-Liang Guan (University of California at San Diego) for providing RRAGAGTP/GTP construct. The work is in part supported by NIH grants (R21AA021450 and R01AA021751) to X-M Yin.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012. March 7;31(5):1095–1108. PubMed PMID: 22343943; PubMed Central PMCID: PMCPMC3298007. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jewell JL, Russell RC, Guan KL.. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013. March;14(3):133–139. PubMed PMID: 23361334; PubMed Central PMCID: PMCPMC3988467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Laplante M, Sabatini DM.. mTOR signaling in growth control and disease. Cell. 2012. April 13;149(2):274–293. PubMed PMID: 22500797; PubMed Central PMCID: PMCPMC3331679. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nature Cell Biology. 2013. June;15(6):555–564. PubMed PMID: 23728461; PubMed Central PMCID: PMCPMC3743096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. 2014. December 30;33C:55–66. [DOI] [PubMed] [Google Scholar]
- [6].Thoreen CC, Chantranupong L, Keys HR, et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012. May 3;485(7396):109–113. PubMed PMID: 22552098; PubMed Central PMCID: PMCPMC3347774. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ricoult SJ, Manning BD. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013. March 1;14(3):242–251. PubMed PMID: 23399656; PubMed Central PMCID: PMCPMC3589096. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002. May;109(9):1125–1131. PubMed PMID: 11994399; PubMed Central PMCID: PMCPMC150968. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Peterson TR, Sengupta SS, Harris TE, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011. August 5;146(3):408–420. PubMed PMID: 21816276; PubMed Central PMCID: PMCPMC3336367. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sengupta S, Peterson TR, Laplante M, et al. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature. 2010. December 23;468(7327):1100–1104. PubMed PMID: 21179166; eng. [DOI] [PubMed] [Google Scholar]
- [11].Yecies JL, Zhang HH, Menon S, et al. Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 2011. July 6;14(1):21–32. PubMed PMID: 21723501; PubMed Central PMCID: PMCPMC3652544. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kenerson HL, Yeh MM, Yeung RS. Tuberous sclerosis complex-1 deficiency attenuates diet-induced hepatic lipid accumulation. PubMed PMID: 21479224; PubMed Central PMCID: PMCPMC3066210. eng PloS One. 2011;63:e18075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Umemura A, Park EJ, Taniguchi K, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. Cell Metab. 2014. July 1;20(1):133–144. PubMed PMID: 24910242; PubMed Central PMCID: PMCPMC4079758. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by Rag GTPases in nutrient response. Nature Cell Biology. 2008. August;10(8):935–945. PubMed PMID: 18604198; PubMed Central PMCID: PMCPMC2711503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science (New York, NY). 2008. June 13;320(5882):1496–1501. PubMed PMID: 18497260; PubMed Central PMCID: PMCPMC2475333. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nature Cell Biology. 2002. September;4(9):648–657. PubMed PMID: 12172553; eng. [DOI] [PubMed] [Google Scholar]
- [17].Inoki K, Li Y, Xu T, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003. August 1;17(15):1829–1834. PubMed PMID: 12869586; PubMed Central PMCID: PMCPMC196227. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dibble CC, Elis W, Menon S, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012. August 24;47(4):535–546. PubMed PMID: 22795129; PubMed Central PMCID: PMCPMC3693578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010. April 16;141(2):290–303. PubMed PMID: 20381137; PubMed Central PMCID: PMCPMC3024592. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zoncu R, Bar-Peled L, Efeyan A, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science (New York, NY). 2011. November 4;334(6056):678–683. PubMed PMID: 22053050; PubMed Central PMCID: PMCPMC3211112. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jewell JL, Kim YC, Russell RC, et al. Differential regulation of mTORC1 by leucine and glutamine. Science (New York, NY). 2015. January 9;347(6218):194–198. PubMed PMID: 25567907; PubMed Central PMCID: PMCPMC4384888. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bar-Peled L, Schweitzer LD, Zoncu R, et al. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012. September 14;150(6):1196–1208. PubMed PMID: 22980980; PubMed Central PMCID: PMCPMC3517996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carr CS, Sharp PA. A helix-loop-helix protein related to the immunoglobulin E box-binding proteins. Mol Cell Biol. 1990. August;10(8):4384–4388. PubMed PMID: 2115126; PubMed Central PMCID: PMCPMC360994. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hodgkinson CA, Moore KJ, Nakayama A, et al. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. 1993. July 30;74(2):395–404. PubMed PMID: 8343963; eng. [DOI] [PubMed] [Google Scholar]
- [25].Hughes MJ, Lingrel JB, Krakowsky JM, et al. A helix-loop-helix transcription factor-like gene is located at the mi locus. J Biol Chem. 1993. October 5;268(28):20687–20690. PubMed PMID: 8407885; eng. [PubMed] [Google Scholar]
- [26].Sardiello M, Palmieri M, Di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science (New York, NY). 2009. July 24;325(5939):473–477. PubMed PMID: 19556463; eng. [DOI] [PubMed] [Google Scholar]
- [27].Palmieri M, Impey S, Kang H, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011. October 1;20(19):3852–3866. PubMed PMID: 21752829; eng. [DOI] [PubMed] [Google Scholar]
- [28].Settembre C, Di Malta C, Va P, et al. TFEB links autophagy to lysosomal biogenesis. Science (New York, NY). 2011. June 17;332(6036):1429–1433. PubMed PMID: 21617040; PubMed Central PMCID: PMCPMC3638014. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012. June;8(6):903–914. doi: 10.4161/auto.19653 Epub 2012 May 11. PubMed PMID: 22576015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011. January;12(1):21–35. PubMed PMID: 21157483; PubMed Central PMCID: PMCPMC3390257. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011. February;13(2):132–141. PubMed PMID: 21258367; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012. June 12;5(228):ra42 PubMed PMID: 22692423; PubMed Central PMCID: PMCPMC3437338. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou J, Tan SH, Nicolas V, et al. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013. April;23(4):508–523. PubMed PMID: 23337583; PubMed Central PMCID: PMCPMC3616426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004. March;15(3):1101–1111. PubMed PMID: 14699058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009. April 30;458(7242):1131–1135. PubMed PMID: 19339967; PubMed Central PMCID: PMCPMC2676208. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010. June 9;11(6):467–478. PubMed PMID: 20519119; PubMed Central PMCID: PMCPMC2881480. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013. May;58(5):993–999. PubMed PMID: 23339953; PubMed Central PMCID: PMCPMC3634371. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nature Cell Biology. 2013. June;15(6):647–658. PubMed PMID: 23604321; PubMed Central PMCID: PMCPMC3699877. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hoffmann A, Levchenko A, Ml S, et al. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science (New York, NY). 2002. November 8;298(5596):1241–1245. PubMed PMID: 12424381; eng. [DOI] [PubMed] [Google Scholar]
- [40].Lahav G, Rosenfeld N, Sigal A, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004. February;36(2):147–150. PubMed PMID: 14730303. [DOI] [PubMed] [Google Scholar]
- [41].Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2010. August;24(8):3052–3065. PubMed PMID: 20375270; PubMed Central PMCID: PMCPMC2909278. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Tanaka S, Hikita H, Tatsumi T, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology (Baltimore, Md). 2016. December;64(6):1994–2014. PubMed PMID: 27637015. [DOI] [PubMed] [Google Scholar]
- [43].Ding WX, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010. November;139(5):1740–1752. PubMed PMID: 20659474; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltimore, Md). 2005. June;41(6):1313–1321. PubMed PMID: 15915461; eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.