

Abstract
Doxorubicin is one of the most effective chemotherapy drugs used against solid tumors in the treatment of several cancer types. Two different mechanisms, (i) intercalation of doxorubicin into DNA and inhibition of topoisomerase II leading to changes in chromatin structure, (ii) generation of free radicals and oxidative damage to biomolecules, have been proposed to explain the mode of action of this drug in cancer cells. A genome-wide integrative systems biology approach used in the present study to investigate the long-term effect of doxorubicin in Saccharomyces cerevisiae cells indicated the up-regulation of genes involved in response to oxidative stress as well as in Rad53 checkpoint sensing and signaling pathway. Modular analysis of the active sub-network has also revealed the induction of the genes significantly associated with nucleosome assembly/disassembly and DNA repair in response to doxorubicin. Furthermore, an extensive re-wiring of the metabolism was observed. In addition to glycolysis, and sulfate assimilation, several pathways related to ribosome biogenesis/translation, amino acid biosynthesis, nucleotide biosynthesis, de novo IMP biosynthesis and one-carbon metabolism were significantly repressed. Pentose phosphate pathway, MAPK signaling pathway biological processes associated with meiosis and sporulation were found to be induced in response to long-term exposure to doxorubicin in yeast cells.
Introduction
Doxorubicin, a natural anthracycline antibiotic, is one of the most effective chemotherapy drugs used against solid tumors in the treatment of several cancer types. Like other anthracyclines, it is presumed to interact with DNA through intercalation, but the mechanism is not fully known1. One of the mechanisms proposed to explain the effect of doxorubicin on cancerous cells is the intercalation into DNA and thus leading to disruption of DNA repair, which would be mediated by topoisomerase II. Doxorubicin inhibits topoisomerase II, which overwinds DNA during transcription, thereby preventing the recombination of the DNA double strand, thus stopping DNA replication2. Another proposal is the generation of free radicals, which in turn might damage DNA and result in cell death3. There is also evidence on the enhancement of nucleosome turnover around promoters by doxorubicin, attributed due to its intercalation property4. These alterations in the nucleosome assembly are believed to affect mechanisms of cell killing during chemotherapy.
Research has been conducted on different types of cancer cell lines and cell models, with various concentrations of doxorubicin. Despite the fact that the responses of different cell types at transcriptional level show variations5,6, doxorubicin was found to play role in some additional cellular processes, including ceramide metabolism7 and cell cycle arrest8.
Resistance to chemotherapeutic agents is one of the major disadvantages of long-term anticancer treatment. Repeated doxorubicin administration leads to drug-resistant cancer cells and increased cytotoxicity9. In fact, cardiotoxicity is the most common doxorubicin-induced side effect. Therefore, most of the research conducted on doxorubicin has been focused on the elimination of the anti-therapeutic effects. Potential treatment options have been developed to reduce doxorubicin-mediated cardiotoxicity, such as lowering the dosage of doxorubicin, combined therapies with cardioprotective agents (e.g. dexrazoxane)10, through regulation of cardiac circular RNA expression11. The former approach of modulating the drug dosage was put forward, aiming to diminish the significant effects of oxidative stress, which is the major cause of cardiotoxicity, but subsequent studies revealed that minimizing or eliminating reactive oxygen species (ROS) did not solve the detrimental effects12. The underlying mechanisms of the cytotoxic side-effects are still not clear. Therefore, new approaches are required in order to prevent cardiotoxicity. Recent studies have already been started targeting the mechanisms causing apoptosis13,14.
Given the above-mentioned arguments, there has not been any unifying conclusion on the working mechanism of doxorubicin and its effects on cell metabolism are not fully established. Although several studies were designed to understand the mechanism of doxorubicin, most are focused on individual genes, whose expression has changed in the presence of doxorubicin, and ignored the system level genome-wide interactions in cells15–19. However, valuable information may be obtained through the integration of high-throughput -omics measurements at different levels with known metabolic/regulatory/interaction networks20, which may then be applied in studies for personalized medicine21.
Since the genes affected by chemotherapeutic drugs are well protected among eukaryotes, Saccharomyces cerevisiae, a eukaryotic model organism suitable to study the effects of chemicals on living cells at -omics level, has been an important tool for cancer research22,23. S. cerevisiae has been used to study the effects of doxorubicin on growth inhibition24, on cell cycle25, on DNA double-strand breaks26 and on environment-dependent protein complex dynamics27. Genome-wide screening with haploid and diploid S. cerevisiae deletion collections to identify the genes that induce doxorubicin resistance has also provided valuable information28,29. However, a systematic study using different approaches under similar conditions has been overlooked.
In the present study, an integrative systems biology approach was used to elucidate the long-term effect of doxorubicin in S. cerevisiae cells. Analysis and integration of the fluxomic data with transcriptomic response of yeast cells, which were grown and exposed to doxorubicin in well-controlled bioreactors, revealed an extensive rewiring of transcription and metabolism.
Results
Genome-wide transcriptomic response of yeast cells to the long-term presence of doxorubicin was analysed in the samples collected at the mid-exponential phase growth and integrated with the flux-balance analysis, followed by protein-protein interaction network in order to identify the long-term effect of the drug on living cells. The cells were grown in triplicates in batch cultures in the presence or absence of doxorubicin.
Fermentation characteristics of yeast cells
The preliminary shake flask experiments carried out in the presence of changing concentrations of doxorubicin (5 to 50 µM) indicated a decreasing growth rate with increasing concentrations of the drug. 20 µM doxorubicin, which was found to decrease the maximum growth rate from 0.3 h−1 to 0.1 h−1, was selected for further experiments (Supplementary Fig. S1). Yeast cells were shown to retain their >80% viability at this concentration28.
The cultures grown in the absence or presence of doxorubicin in bioreactors were characterized for their uptake/secretion rates during the mid-exponential phase. When the mass-balance-based biomass-specific rates for both conditions were compared, it was observed that the maximum growth rate was decreased from 0.27 ± 0.01 h−1 to 0.10 ± 0.01 h−1 in the presence of doxorubicin (Supplementary Table S1), as was the case in shake flask experiments. Since the growth rate was lower, glucose uptake, glycerol and ethanol production rates were also lower in the cultures grown in the presence of doxorubicin (Supplementary Table S1).
Intracellular Fluxes in the presence of doxorubicin
The fluxes through the metabolic network of S. cerevisiae were calculated for growth on glucose for the cultures grown in the absence and presence of doxorubicin. The rates presented in Table S1 were used as inputs, and model was solved by optimizing the maximization of the ATP production, to estimate the fluxes via flux balance analysis. Among 3498 reactions within the model, 278 reactions were different based on their magnitude of change (down: <0.8, up: >1.10). 260 fluxes were decreased, whereas 18 fluxes were increased in the presence of doxorubicin, compared to the control cultures (Supplementary Table S2).
The increased fluxes were mainly pentose phosphate (PP) pathway reactions and transport of O2. The decreased fluxes in the presence of doxorubicin were glycolytic reactions, transport reactions, and several reactions involved in amino acid biosynthesis (Supplementary Table S2). To find out the functional annotation biological process gene ontology (GO) terms of these reactions, the genes associated with the reactions of interest were identified. However, for the transport reactions the associations of gene-reaction were not provided. These gene sets (excluding transport) were used in GO-term enrichment analysis. The genes associated with the reactions, which showed increased flux values, were found to be mainly related to pentose phosphate shunt and oxidation-reduction process (Fig. 1). The genes associated with the reactions, which showed decreased fluxes, were found to be related to amino acid biosynthetic process, metabolic process, oxidation-reduction process, tRNA aminoacylation for protein translation among others (see Fig. 1 and Supplementary Table S3).
Figure 1.

GO biological process terms associated to differential fluxes, changed in response to doxorubicin plotted against negative log values (base10) of corrected p-values. Red and green colors indicate increased and decreased fluxes, respectively.
Transcriptional response to doxorubicin
The analysis of genome-wide transcriptional response of yeast cells to long-term exposure to doxorubicin revealed that 1279 genes were differentially and significantly (a fold change greater than 1.5 and p value < 0.05) expressed when compared to control (Supplementary Table S4).
A total of 976 genes were found to be up-regulated and GO biological process terms which were significantly (Benjamini-Hochberg corrected p value < 0.05) associated with 785 genes of this group excluding the genes with unknown biological function were identified (Supplementary Table S4). These genes significantly associated with biological processes such as sporulation resulting in formation of a cellular spore, response to stress, carbohydrate metabolic process, meiotic cell cycle, reciprocal meiotic recombination, fungal-type cell wall organization, and transmembrane transport displayed increased levels of expression when compared to control (Fig. 2, Supplementary Table S5). These genes were found to be significantly enriched in pathways related to starch, galactose, amino sugar and sucrose metabolisms, MAPK signaling pathway and meiosis.
Figure 2.
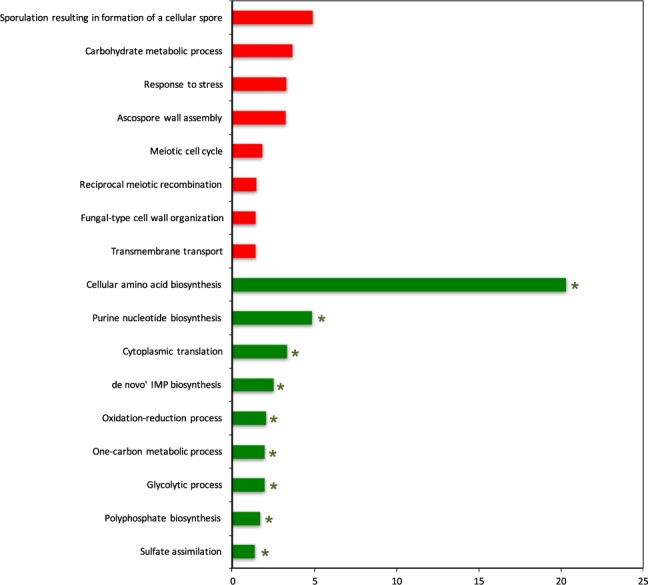
GO biological process terms significantly associated to genes differentially expressed in response to doxorubicin plotted against negative log values (base10) of corrected p-values. Red and green colors indicate up- and down-regulated biological processes, respectively. * indicates the terms that are also observed for differential fluxes (shown in Fig. 1).
A total of 35 transcription factors, which were mainly implicated in the regulation of cellular response to nutrient levels and general stress, were identified among the up-regulated genes. The major repressor of DNA damage regulated genes (RFX1), which is involved in DNA damage and replication checkpoint pathway, transcriptional factors involved in the regulation of metallothionein genes in response to DNA damage (CUP2) and in the pleiotropic drug resistance (PDR8, PDR1 and YRM1) were also observed to be induced in response to exposure to doxorubicin. The higher expression levels of RFX1, CUP2 and PDR8 were also confirmed by RT-qPCR analysis (Supplementary Figs S2 and S3).
Manual investigation of up-regulated genes indicated that several genes involved in cellular response to oxidative stress (TRR2, MHR1, ECM5, FRM2, HSP12, HSP31, GRX7, GPX1, SRX1, GCY1, MTL1, XBP1, TSA2, LOT6, MCR1, HBN1, VHR1, NTG1, NQM1, YJR096W, PRX1, GAD1), siderophore and iron ion transport and homeostasis (FIT3, FRE7, FIT2, HMX1, FRE6, FRE3, FRE4, FRE2, ARN1, ARN2) and copper ion import (FRE7, FRE6, FRE3, FRE4, FRE2) displayed significantly elevated levels of expression when compared to the transcriptomic response of the cells grown in the absence of doxorubicin. Furthermore, several genes involved in meiotic DNA double-strand break processing (EXO1, SGS1, SPO11, RAD50), double strand break formation (REC102, MEI4, REC104, SPO11, RAD50) and autophagy, CVT and mitophagy (ATG22, ATG23, ATG 29, ATG36, ATG39, ATG11, MON1) were up-regulated. The higher expression levels of SRX1, FRE7, SGS1, REC104 and ATG39 were further confirmed by RT-qPCR analysis (Supplementary Figs S2 and S3). Several structural and functional mitochondrial genes were also induced in yeast cells in the presence of doxorubicin.
A total of 303 genes were identified displaying significantly and differentially down-regulated expression levels when compared to control. The significantly associated GO biological process terms (Benjamini-Hochberg corrected p value < 0.05) with 292 down-regulated genes, excluding 11 genes encoding hypothetical proteins, were determined (Supplementary Table S4). The genes which were involved in amino acid biosynthesis, including the synthesis of lysine, methionine, arginine and isoleucine were observed to be down-regulated in response to the exposure of yeast cells to doxorubicin. Biological processes such as translation, glycolysis, sulfate assimilation, purine nucleotide biosynthesis, one-carbon metabolism and de novo IMP biosynthesis were also found to be affected in similar manner (Fig. 2). A total of 34 genes collected under a common translation term consist of the genes involved in ribosome biogenesis, ribosomal small subunit biogenesis and assembly and RNA processing. Analysis of the affected pathways indicated that biosynthesis of amino acids, glycolysis and gluconeogenesis, ribosome, sulfur metabolism were significantly repressed. A total of 10 transcription factors (TF) were identified as significantly down-regulated. Half of these TFs were involved in chromatin re-organization, restriction, silencing, nucleosome re-modeling and positioning (ABF1, TBF1, RAP1, CBF1, NHP6A).
Furthermore, the manual inspection of this group of genes indicated that the genes (HHT1, HTA2, HAT2, HTA1, HTB2, HTB1) involved in chromatin assembly and disassembly displayed significantly lower expression levels in response to doxorubicin exposure when compared to control. Several genes involved in the biosynthesis of cysteine, valine, leucine and arginine were also found to be significantly and differentially down-regulated. The genes involved in glutamine biosynthesis (HIS7, ADE6, ASN1, URA2, ADE4), NAD and de novo NAD biosynthesis from tryptophan (BNA7, BNA5, NMA1, BNA4), NADH oxidation (GPD1, ADH1, ADH5) and global nucleotide excision repair (HHT1, DOT1, ABF1) were repressed. The lower expression levels of HTB2, BNA4, and ADH5 were further confirmed by RT-qPCR analysis (Supplementary Figs S2, S3).
The independent measurements of gene expression and intracellular fluxes were consistent as implied by the common GO terms (Fig. 2, indicated by *). These two different types of -omics measurements revealed some common processes that had decreased flux and transcript levels in response to doxorubicin.
Modular analysis of differentially active sub-network
Differential expression analysis of transcriptomic data collected after long-term doxorubicin treatment of yeast cells in a batch environment revealed 785 significantly induced and 292 inhibited DEGs. A differential active gene network (DEGN) was constructed using differentially expressed genes and protein-protein interaction data. Modular topological analysis of DEGN, consisting of 896 proteins and 2291 interactions, by MCODE indicated the presence of 9 densely connected modules, which are significantly associated with distinct biological processes or having nodes with similar biological roles (Supplementary Table S6 and Fig. 3). Transcription factors of which target genes were over-represented in each module were identified to determine the plausible co-regulation using TF enrichment analysis.
Figure 3.

Modular organization of differentially active regulatory network (DEGRN). Nine modules identified by MCODE by using physical interaction network of DEGs (DEGN). Red nodes are up-regulated; green nodes are down-regulated while grey nodes are TFs that are not DEGs. Black connections represent physical interactions, blue arrows show positive regulation, red tapped arrows indicate inhibition, and grey arrows represent unspecified regulation. Rectangular nodes are genes/proteins while elliptic nodes are TFs. Regulatory interactions shown only for significant TFs while some TFs reside in modules were shown with only physical interactions. Inter-modular physical interactions are not shown.
Fkh2p, which is involved in chromatin remodeling and a negative role on chromatin silencing, was found to be significantly associated with M1, M5 and M3. The genes in M1 is mostly down-regulated (15 out of 17) in the presence of doxorubicin and significantly associated with chromatin assembly/disassembly. Four members of the modules also have roles in DNA repair mechanism. The second module with significantly enriched targets of Fkh2p, M5, consists of mostly down-regulated (8 out of 9) genes and was found not to be associated with any GO biological process term but was significantly associated with general metabolic pathways term. Manual investigation of the constituents indicated that most of the genes of this module is involved in amino acid biosynthesis. This module was also found to be significantly associated with Fhk1p, involved in chromatin remodeling, mitotic transcription regulation, transcription termination, mating-type switching, and pseudohyphal growth, Ndd1p, involved in the positive regulation of G2/M mitotic cell cycle and Msn1p, involved in the positive regulation of transcription in response to glucose limitation.
The third co-regulated module by Fhk2p, M3, has five out of seven genes up-regulated. Although no significant GO term or pathway association could be detected for this module, manual investigation indicated that the genes in this module were involved in stress response, signal transduction, trans-membrane transport activity and cell wall organization. YBL029C-A, which is a protein with unknown function, is known to be localized to cell peripherals and induced with DNA replication stress. Hot1p which has stress responsive regulatory role, the down-regulated transcription factor Hap3p which is a global regulator of respiratory gene expression, Tbf1p which is involved in the negative regulation of chromatin silencing and required for full snoRNA expression, Sok2p which plays a regulatory role in PKA signal transduction were found to have significantly enriched target genes in this module. M3 seems to be co-regulated with M4 by Sko1p, which is involved in stress response. TF enrichment analysis indicates that M3, M4 and M6 were co-regulated by the stress responsive factor Yap6p. The gene encoding Yap6p is also found to be significantly up-regulated in the presence of doxorubicin in the present study, also confirmed by RT-qPCR analysis (Supplementary Figs S2 and S3). TF enrichment analysis indicated that M4 is also co-regulated with M6 and M2 by Spt23p.
The smallest module M2 consists of 4 up-regulated DEGs and significant association with proteasome mediated ubiquitin dependent protein catabolism and gluconeogenesis biological process terms accordingly. Three proteins of this module are subunits of Gid complex and the forth node, MOH1, is a protein with unknown function and has interactions with all other three nodes. Three regulators of this module, Yap6p, Met32p and Nrg1p, are also up-regulated DEGs themselves. Yap6p and Nrg1p are stress response factors known to be Tup1-Ssn6 complex recruiters in response to changing environmental conditions30. Nrg1p is involved in glucose repression and glucose sensing and is a DNA binding cofactor that target Tup1-Ssn6 complex to starch degrading genes. Met32p is involved in the biosynthesis of methionine and Ert1p is involved in the regulation of gluconeogenesis and fermentable carbon utilization. The only significant TF with direct role in protein catabolism is Spt23p, which was shown to be activated by a mechanism that involves ubiquitin/proteasome-dependent processing31.
The module M6, with 12 genes up-regulated out of 13, was found to be significantly associated with protein folding/refolding, SRP-dependent co-translational protein targeting to membrane GO biological process terms and protein processing in ER pathway.
The largest module M9 with 34 nodes (23 out of 34 up-regulated) has a long and branched pathway-like topology (Fig. 3). This module was significantly associated with GO biological process terms, namely meiotic cell cycle, meiotic double strand break formation and reciprocal meiotic recombination. Manual investigation of the nodes revealed that the top part, which is formed by seven up-regulated nodes, is related to meiotic cell cycle. This part of the module is connected to the rest of the module via SSK22, which encodes a signaling protein. The following part (GSY1, GLY1, PCL10, and RGC1) has various roles in glycogen biosynthetic process. The next part has stress responsive nodes (PHO80, PHO81, PHO4, PCL1, NPL3, and MMS22). Especially MMS22 has an important role in double strand break repair and named after its MMS sensitivity. The module diverges into three branches on MMS22; the smallest branch includes down-regulated urea cycle related ARG3 and CAR1, the second branch has nodes with various biological roles and ends with two up-regulated nodes, which have roles in vitamin B6 biosynthetic process (SNO1 and SNZ3). The last and the longest branch has up-regulated autophagy related nodes, HSV2 and VAM7. This last part of the module also has nodes with no clear biological roles (TDA1, FRK1 and STB2). The only significant TF identified to be regulating this module is Hot1p, which targets nodes involved in glycogen and glycerol synthesis.
All five nodes of disconnected module M7 are up-regulated and all of them related to pheromone dependent MAPK signal transduction pathway leading to cell cycle arrest. Significantly up-regulated Rfx1p and Dal80p, which is a negative regulator of genes in multiple nitrogen degradation pathways, are significant TFs identified as co-regulators of M7.
There is no significant co-regulator for M8 which is significantly associated with phagosome pathway and establishment of mitotic spindle orientation and localization processes. Except from SWC3, which is a protein of unknown function, all nodes of the module are up-regulated. All up-regulated gene products have indirect roles in mitosis via dynein activity. Induction of these genes together with induced cell cycle arrest may imply that doxorubicin leads to organelle re-organization or nuclear instability as they are the additional roles of these nodes.
Discussion
Although the molecular mechanism of doxorubicin is not clear, two mechanisms (i) intercalation into DNA and inhibition of topoisomerase II leading to changes in chromatin structure and (ii) generation of free radicals and oxidative damage to biomolecules have been proposed as potential mode of action32. In order to shed a light on the underlying molecular mechanism of doxorubicin, we have used a systems based integrative approach in the present study. The genome-wide transcriptional response of yeast cells to long-term exposure to doxorubicin and the changes in fluxes identified using a genome scale model were comparatively analysed. Transcriptional re-wiring of the metabolism was also investigated by modular analysis of active sub-network via integrating transcriptome and interactome, and TF enrichment analysis.
Genes involved in response to oxidative stress were found to be significantly induced in the transcriptomic response of yeast cells to long-term exposure to doxorubicin, as expected. Although low levels of ROS is responsible for the survival and proliferation, induction of ROS levels is associated with tumorigenesis. However, a further increase may lead to apoptotic, autophagic, ferrotopic and necrotic cell death. Cancer cells have higher ROS levels than normal cells, which make them more susceptible to ROS inducing chemotherapeutic agents or radiation therapy than non-cancerous cells12,33.
PP pathway implicated in the synthesis of biological macromolecules and NADPH was found to be up-regulated (Fig. 4). NADPH creates a reducing environment, and reduces glutathione to prevent the oxidative stress34. Several genes involved in de novo NAD biosynthesis from tryptophan and NADH oxidation, were down-regulated. This observation is consistent with the work of Davies and Doroshow35, in which inhibition of the reduction of NAD+ to NADH by doxorubicin was reported. In another study, it was shown that NADH oxidase activity was inhibited by doxorubicin when exposed to mice36.
Figure 4.

Integrative analysis of central carbon metabolism in the presence of doxorubicin. Red and green arrows indicate increased and decreased fluxes, respectively. Red and green rectangles indicate up- and down-regulated genes, respectively.
Transcriptional level of the genes and fluxes throughout the glycolytic pathway were observed to be down-regulated (Fig. 4). An up-regulation of glycolysis and glucose uptake are the prominent features of cancer cells. Therefore, this pathway has been considered as a target for the development of new therapeutics37.
Sulfate assimilation pathway and expression levels of several genes involved in glutamine biosynthesis were repressed in response to the long-term presence of doxorubicin. Sulfate assimilation pathway is also closely linked to the glutathione production38. A decrease in the activity of this pathway might possibly result in a reduction at the level of glutathione, which is an electron donor in ROS detoxification and oxidative stress protection in ER.
The system-wide integrated approach used in this study indicated a pronounced re-wiring of the metabolism to cope with the stress caused by this chemical. In addition to glycolysis and sulphur assimilation, several pathways related to ribosome biogenesis/translation, nucleotide biosynthesis, and de novo IMP biosynthesis and one-carbon metabolism were observed to be repressed in response to doxorubicin (Fig. 5). One-carbon metabolism is involved in the synthesis of essential biomolecules such as nucleotides and amino acids as well as in epigenetic modifications. The up-regulation of this latter process in cancer has been reported due possibly to the increasing need of building blocks in proliferative cells39. Therapeutic agents that block one-carbon metabolism are found to be effective in cancer therapy39–41. However, the toxicity of these therapeutic agents to non-cancer cells created an obstacle for their widespread use.
Figure 5.

Integrative analysis of down-regulated pathways in response to doxorubicin. Green arrows indicate decreased fluxes. Red and green rectangles indicate up- and down-regulated genes, respectively.
The biosynthesis of several amino acids including the synthesis of lysine, methionine, arginine and isoleucine were observed to be significantly down-regulated in response to the exposure of yeast cells to doxorubicin. These results are in good correlation with the findings that the supplementation of single amino acid in minimal media protected yeast cells from doxorubicin toxicity42. The amino acids that showed highest protection were associated with TCA cycle.
Sterol/steroid biosynthesis pathways were found to be repressed in the presence of doxorubicin according to the analysis on differential fluxes, but not based on transcriptomic measurements. It has been shown that cancerous cells depict specific changes in various parts of lipid metabolism43, with some causing lipid metabolic reprogramming in cancer metastasis44,45, sterol molecules having roles in the regulation of cellular proliferation in cancer46.
Doxorubicin, as other anthracyclines, is considered to damage DNA through intercalation or direct alkylation and may result in the formation of DNA double-strand breaks47. DNA damage induces Rad53 checkpoint sensing and signaling pathway in yeast48. Several components of this pathway (MEC1, RAD24, HUG1) were observed to be significantly up-regulated upon long-term exposure of yeast cells to doxorubicin. The higher expression level of HUG1 was also confirmed by RT-qPCR analysis (Supplementary Figs S2 and S3). Detailed investigation of the transcriptional response indicated that the genes involved in cellular response to DNA damage and DNA repair were also induced. Several genes functioning in DNA repair were observed to be up-regulated upon doxorubicin exposure49. Modular analysis of DEGN also indicated that a module is significantly associated with nucleosome assembly/disassembly and several genes within this module was also found to be involved in DNA repair. Major transcriptional repressor of DNA damage regulated genes, RFX1, which recruits Tup1p/Cyc8p to the promoters of these genes, was found to be induced. The mammalian homolog of this gene, RFX-1, was shown to regulate several genes involved in immunity and cancer progression50,51. It has been reported that intercalation of doxorubicin causes torsional stress leading to the nucleosome destabilization results in anthracycline-mediated cell killing52.
Several copper import genes were also found to be induced within the framework of this study. Furthermore, CUP2, which is involved in the activation of the metallothionein genes in response to elevated copper levels and required for the regulation of copper genes in response to DNA damaging reagents, was observed to be significantly up-regulated in response to doxorubicin. A regulatory relationship between DNA damage and copper response in yeast was proposed through a genome-wide genetic screening and the up-regulation of copper import genes in response to DNA-damaging agents53. Although a detailed study is required to fully understand this relationship, it was proposed that Mac1p may exist in different redox forms in the cell53.
Meiosis and sporulation were observed to be significantly induced in response of yeast cells exposed to doxorubicin. Meiosis was shown to be up-regulated in the presence of doxorubicin in yeast cells49. A deficiency in glucose and nitrogen are known to induce meiosis followed G1 arrest of the mitotic cell cycle in diploid yeast cells54,55. When yeast cells starve for essential nutrients, sporulation and meiosis coupled with spore formation is induced56. However, absence of any nutritional deficiency in our experimental system indicates that G1 arrest and induction of meiosis are triggered by doxorubicin, which may cause a misperception in glucose sensing. Several transcription factors implicated in the regulation of cellular response to nutrient levels and general stress were also identified among the significantly up-regulated genes. The up-regulation of these biological processes in yeast cells subjected to long-term exposure to doxorubicin, indicates that the yeast cells experience a misperception of the nutritional environment in the presence of doxorubicin. Furthermore, several members of Snf3/Rgt2/Rgt1 (SSR) glucose sensing and signaling pathway and consequently hexose transporter genes were found to be significantly up-regulated to cope with this misperception.
Induction of carbohydrate metabolic process in the presence of doxorubicin reflects the up-regulation of several genes involved in the catabolism of galactose and maltose, as well as PP pathway and degradation of yeast storage carbohydrates glycogen and trehalose, which are known to be produced under glucose starvation conditions. This finding agrees well with a possible misperception in glucose availability.
The expression level of SNF1, the mammalian ortholog of the AMP-activated protein kinase (AMPK) involved in the regulation of cellular energy homeostasis and metabolism as well as in glucose sensing, was found to be significantly repressed in the present study. Inhibition of SNF1 in response to a DNA damaging agent, MMS, accompanied with a switch from respiration to fermentation is also reported in yeast cells57. Inhibition of AMPK in several non-carcinoma cells in response to doxorubicin have also been observed and a pre-activation of this pathway has been suggested to prevent doxorubicin-induced cell death and cardiotoxicity58–61. Both pro- and anti-tumorigenic roles of inhibitors and activators of AMPK in cancer therapy have been suggested62.
MAPK signaling pathway was found to be significantly induced in response to stress caused by the presence of doxorubicin. MAPK pathways are functional tools to adapt cells to stress by targeting a wide variety of downstream effectors including transcriptional factors63. The induction of MAPK pathway by anthracycline based anti-tumor antibiotics has been reported in neuroblastoma cells64, rat hepatoma cells65, human cervical carcinoma cells66, monoblasts67, glioblastoma cells68 and, in a time and dose dependent manner, in breast epithelial and carcinoma cells69,70. A dual role of MAPK/ERK pathway as a tumor suppressor and a pro-oncogenic signal has been well recognized71. Therefore, it should be noted that the understanding of the effect of doxorubicin on MAPK in each tumor type specific microenvironment is very important in designing single or combination therapies with doxorubicin.
Modular analysis of differentially active sub-networks revealed nine modules and investigation of transcriptional regulators of these modules indicates that Yap6p, Spt23p and Fkh2p are the most active transcriptional factors that each co-regulates more than two modules. Yap6p physically interacts with the Tup1-Cyc8 complex and recruits Tup1p, which is a general repressor of transcription, to its targets. Expression of this stress responsive TF is also up-regulated and its role in DNA damage was reported to be through chromatin remodeling72. Fkh2p has a major role in cell-cycle regulation and also has a role in chromatin remodeling. Its targets in the differentially active sub-network are mostly down-regulated in contrast to the targets of Yap6p and Spt23p. Targets of Spt23 in the network are related to degradation of enzymes required for gluconeogenesis. Down-regulation of gluconeogenesis is suggested as an attractive therapeutic strategy in recent cancer research73 and repression of gluconeogenesis through Spt23p can be considered as one of the anti-cancer effects of doxorubicin.
A genome-wide screen of S. cerevisiae diploid deletion collection resulted in identification of 376 resistance genes to doxorubicin29. Among those 376 genes, 43 (22 up, 21 down) were found to be differentially expressed in the presence of doxorubicin in the present study. 22 up-regulated genes were found to be enriched (p value < 0.05) in cell cycle, cellular response to DNA damage stimulus, DNA repair and DNA topological change terms. Among those, TOP3, RAD50 and RAD54 have roles in the homologous recombination pathway. Within the 21 down-regulated genes, genes involved in glycolysis, pentose phosphate pathway and purine biosynthesis were observed.
Unique mode of action of several ROS causing or DNA damaging agents were already reported. A comparative analysis indicated that exposure to doxorubicin triggers unique transcriptional response. Only less than 15% of DEGs that are sensitive to doxorubicin are common with DEGs identified as sensitive to DNA damaging agents; MMS and ionizing radiation74. The results of a study using cumene hydroperoxide was compared with a previous study where hydrogen peroxide was used as source of oxidative stress75,76. When those are compared with the results of the present study, it was observed that among the genes up-regulated in CHP and H2O2-induced stress, 56 out of 162 were found to be up-regulated in the present study. And among the genes down-regulated in CHP and H2O2-induced stress, 53 out of 481 were found to be down-regulated in the presence of doxorubicin. These variations in transcriptional response reveal that despite common expression patterns, larger part of the transcriptome reacts differently and production of ROS causing oxidative stress does not have a universal mechanism. Focusing on disparate results will guide us on understanding specific mechanisms of the drugs on cellular phenomena.
The intercalation of doxorubicin into DNA and inhibition of topoisomerase II leading to changes in chromatin structure has been proposed as one of the major effects of doxorubicin. We have compared the differentially expressed genes identified in the present study with genes specifically up- and down-regulated upon 10 min inactivation of TOP2, excluding the environmental stress response genes, in yeast cells77. 11 genes out of 97, identified to be selectively inhibited by the inactivation TOP2, were also observed to be down-regulated in the present study. Among the 173 selectively up-regulated genes by the inactivation of TOP2, 61 genes were also found to be up-regulated in the presence of doxorubicin in the present study. Despite requiring further investigation, the incidence of 61 common genes might be an indicator for the inactivation of TOP2, which is one of the important targets for anti-cancer drugs78, under doxorubicin exposure.
In addition to the generation of ROS and DNA damage, the present study indicates that doxorubicin induces an impressive re-wiring of metabolic and signaling pathways, which are already known or suggested as therapeutic targets in cancer. The changes, which are induced, are in the direction to alleviate the changes occurring in cancerous cells.
Further studies including time dependent response of yeast cells to different concentrations of doxorubicin and integration of transcriptomic response with metabolomics, proteomics and phosphoproteomics will be required to dissect the specific response of yeast cells to doxorubicin and its targets in the treatment of cancer. It should be noted that adaptation of yeast cells to the long-term presence of doxorubicin needs also to be considered. Future research on the genomic re-organizations and dynamic re-organization of the response at different -omics levels in the presence of different concentrations of doxorubicin would give further insight into the molecular mechanisms underlying the doxorubicin treatment and complement the present study. Although the essential central pathways were conserved among yeast and human, detailed studies in normal cells and tumor microenvironment will be required to elucidate the complex cellular mechanisms of this and other therapeutic agents.
Materials and Methods
Strain and preculture conditions
Homozygous ho∆/ho∆ strain of S. cerevisiae diploid BY4743 (MATa/MATΔ his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 lys2Δ0/+ met15Δ0/+ ura3Δ0/ura3Δ0) was kindly provided by Prof. Stephen G. Oliver. Cells were grown to stationary phase in shake flasks in YPD medium. Culture aliquots containing 50% (v/v) glycerol were kept at −80 °C until further use.
Precultures were grown in F1 minimal medium79 at 30 °C and 180 rpm overnight. F1 minimal medium had the following composition per liter: 20 g glucose, 3.13 g (NH4)2SO4, 2.0 g KH2PO4, 0.55 g MgSO4.7H2O, 0.1 g NaCl, 0.02 g uracil, 0.02 g L-histidine, 0.1 g L-lucine, 90 mg CaCl2.H2O, 70 µg ZnSO4.7H2O, 50 µg FeCl3.6H2O, 10 µg, CuSO4.5H2O, 10 µg H3BO3, 10 µg KI, 62 mg inositol, 14 mg thiamine.HCl, 4 mg pyridoxine, 4 mg Ca-pantothenate, and 0.3 mg biotin.
Shake flask experiments
To decide on the most appropriate drug concentration, S. cerevisiae cells were grown in F1 minimal media having different concentrations of doxorubicin (Sigma, Cat. No: D1515). Doxorubicin was added into the media before the inoculation of cells to have final concentration of 5, 10, 20, 50 µM. OD600 was followed.
Bioreactor experiments
Aerobic batch cultures were grown in F1 minimal medium (with 20 g/L glucose) in 2 L B-Braun Biostat B Plus bioreactors with a working volume of 1.5 L (Sartorius Stedim Systems GmbH, Melsungen, Germany). The cultivation temperature was controlled at 30 °C, and the pH was controlled at 5.5 with 1 M NaOH and 1 M HCl. The bioreactor was operated at a stirrer speed of 800 rpm and an aeration rate of 1.5 L/min. Under these conditions, oxygen transfer was sufficient because the dissolved oxygen never dropped below 85% of air saturation that was measured online with a DO sensor (VISIFERM DO 225, Hamilton Bonaduz AG, Switzerland). A silicone antifoam agent (Sigma-Aldrich, USA) was diluted 1:10 (v:v) and intermittently added to the medium. During the experiments, the dissolved oxygen tension, pH, temperature, and the carbon dioxide and oxygen in offgas were monitored online with a gas analyzer (BlueSens, Herten, Germany).
Control cultures were grown solely in F1 media, cultures with doxorubicin additionally contained 20 µM doxorubicin, added before the inoculation of cells. Cells were subjected to doxorubicin exposure approximately for 50 hours before the sample collection. Each fermentation experiment in bioreactor (control and with doxorubicin) was performed in triplicate.
Samples were collected at the mid-exponential phase at an OD600 of 0.6–0.7 for analysis of biomass, RNA and metabolites. The samples for RNA and metabolite analysis were immediately frozen in liquid nitrogen and stored at −80 °C until further processing.
Measurement of cell dry weight and analysis of extracellular metabolites
The cell dry weight was obtained gravimetrically: 4 × 1 mL broth was centrifuged (8000 rpm, 6 min) in previously weighed Eppendorf tubes and the cells were washed twice with demineralized water. The Eppendorf tubes containing cell pellets were dried in an oven at 70 °C for 48 h until constant weight.
The supernatants obtained by the centrifugation of the broth samples were used for analysis of extracellular metabolites. The concentrations of glucose, glycerol and ethanol in these samples were analyzed enzymatically according to the manufacturer’s instructions (Boehringer Mannheim/R-Biopharm, Darmstadt, Germany).
RNA extraction and microarray experiments
The control group and doxorubicin treated yeast cells were derived from three independent batch cultures grown in bioreactors as explained above. RNA extraction was carried out with QIAcube (Qiagen, USA) using the enzymatic lysis protocol (Qiagen RNeasy mini kit; Cat no: 74106). The quality and quantity were checked via UV–vis spectrophotometer (NanoDrop ND-1000, Thermo Fisher Scientific Inc., USA). RNA integrity number values were obtained using a microfluidics-based platform (Bioanalyzer 2100 Agilent Technologies, USA) and samples with RIN values 7–10 were processed for microarray analysis. All protocols were used according to their individual directions supplied by the manufacturer.
Microarray experiments were carried out on Yeast 2.0 Arrays, using GeneChip® 3′ IVT Express Kit for cDNA preparation and amplification (Affymetrix Inc., USA). Hybridization, wash and stain was carried out as described previously80. All the kit protocols were used as described by the manufacturer.
Microarray data acquisition and analysis
The data was processed using MATLAB Bioinformatics Toolbox. The datasets were normalized by Robust Multi-Array Average (RMA) via ‘affyrma’ command. The significance of gene expression was evaluated using unpaired t-test with a p-value threshold of 0.05 and fold change >1.5, and differentially expressed genes (DEGs) were identified. GO term enrichments were carried out in DAVID81 (Benjamini-Hochberg corrected p value < 0.05) after excluding the genes with unknown function from each up- and down-regulated gene sets.
RT-qPCR experiments
The expression level of RFX1, CUP2, PDR8, SRX1, FRE7, SGS1, REC104, ATG39, YAP6, HUG1, NHP6A, HTB2, BNA4, ADH5, ENO1, SHM2 and ARG4 were determined in the cells grown as control and in the presence of doxorubicin. FBA1 was used as the housekeeping gene82.
The primers were designed using Roche Array Design Centre with the default settings (see Supplementary Table S7). The amplicon sizes were around 60 bp. Initial concentration of RNA in all samples were set to be 50 ng/μl. Reverse transcription was performed using BioRad iScript cDNA Synthesis Kit, as described by the manufacturer, in instrument Applied Biosystems 2720 Thermal Cycler (USA). Real-time RT-qPCR with the cDNAs was then carried out with SensiFAST SYBR No-ROX Kit as described by the manufacturer (Bioline Reagents Ltd, UK, Cat no: BIO-98020). The PCR reactions were performed in a final reaction volume of 20 μl containing the final concentration of 0.5 µM of forward and reverse primers in Roche LightCycler 96 instrument (Roche Diagnostics GmbH, Mannheim, Germany). 96-well microplates and adhesive sealing films were manufactured by Axygen (USA).
Quantification cycle (Cq) values were called using the LightCycler 96 Application software yielding amplification plots. Relative gene expression values were calculated from Cq values using ΔΔCq method.
Metabolic flux analysis
Flux balance analysis (FBA), a common approach used to calculate the flow of metabolites through a network of biochemical reactions, was carried out using Yeast7.0 genome-scale metabolic model, developed for S. cerevisiae83. This model contains all the known biochemical reactions and the genes related to those. FBA is based on mass balance equations for each intracellular metabolite . In the stoichiometric matrix S (mxn) with m number of metabolites and n number of reactions, entries are the stoichiometric coefficients for the corresponding biochemical reaction and v (nx1) is the rate vector of the model. At (pseudo) steady-state an algebraic set of equations (S.v = 0) is obtained. FBA allows the calculation of intracellular fluxes of each reaction as a function of experimentally measured extracellular fluxes. In a typical case where n > m, the system is underdetermined, yet a unique solution can still be obtained under biologically relevant optimality criteria. Overall, when solved with a given a set of upper and lower bounds on v, FBA results in a distribution of metabolic fluxes, which minimize or maximize an objective function84. The biomass specific rates of glucose consumption, glycerol and ethanol productions were calculated from the respective mass balances for the liquid phase. These uptake/production rates were used as constraints. Alternate optima were eliminated to obtain a unique set of flux distributions, by applying flux variability analysis85. The resulting genome scale model being under-determined was optimized for maximum ATP hydrolysis. All calculations were performed using MATLAB and COBRA toolbox86, for each experiment individually (in total six cases).
Different fluxes between cultures grown in the absence and presence of doxorubicin were identified based on the fold change (down: <0.8, up: >1.10).
Integration of fluxome and transcriptome
To find out the functional annotation biological process GO terms of the reactions, which have different flux values between two conditions, the genes associated with these reactions were identified. These gene sets were then used in GO-term enrichment analysis, carried out in DAVID81 (Benjamini-Hochberg corrected p value < 0.05).
Modular analysis of active network
An active network, consisting of 915 proteins with 2301 connections, was constructed using DEGs and protein-protein interactions extracted from BIOGRID v.3.4.146. After removing the disconnected residues, the connected active protein-protein interaction network (DEGN) with 896 nodes and 2291 edges was identified.
DEGN was visualized by using Cytoscape v.3.5.1 and densely connected modules were extracted by using MCODE application of Cytoscape87,88. Default settings of MCODE were applied (haircut: on; fluff: off). Minimum number of nodes was set to be four.
In order to characterize biological significance of nine modules identified by MCODE, significantly associated (Benjamini-Hochberg corrected p value < 0.05) gene ontology terms and pathways to each module were identified by using DAVID 6.8 functional annotation tool89.
Transcription factors significantly associated to the modules (Bonferroni corrected p value < 0.01) were identified using YEASTRACT 201790. ‘Only DNA-binding evidence’ was used for regulatory interactions. Resulting regulatory network of densely connected modules of DEGs (DEGRN) was visualized by using Cytoscape v.3.5.1.
Electronic supplementary material
Acknowledgements
This research was financially supported by The Scientific and Technological Research Council of Turkey (TUBITAK) through Project No. 114C062, carried out at Bogazici University Department of Chemical Engineering. Authors would like to thank Yuk Yin Ng from Istanbul Bilgi University for his support of the RT-qPCR experiments.
Author Contributions
H.T.-N. designed and performed experiments, analysed data and wrote manuscript, M.E.K. analysed data and wrote manuscript, S.E. performed experiments, B.K. designed experiments, analysed data and wrote manuscript. All authors read and approved the manuscript.
Data Availability
The microarray data has been submitted to ArrayExpress at the European Bioinformatics Institute under accession number [E-MTAB-6634] in compliance with MIAME guidelines.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-31939-9.
References
- 1.Anders, C. et al. Pharmacokinetics and Efficacy of PEGylated Liposomal Doxorubicin in an Intracranial Model of Breast Cancer. Plos One8, 10.1371/journal.pone.0061359 (2013). [DOI] [PMC free article] [PubMed]
- 2.Pommier Y, Leo E, Zhang H, Marchand C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chemistry & Biology. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim S, et al. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Experimental and Molecular Medicine. 2006;38:535–545. doi: 10.1038/emm.2006.63. [DOI] [PubMed] [Google Scholar]
- 4.Yang F, Kemp C, Henikoff S. Doxorubicin Enhances Nucleosome Turnover around Promoters. Current Biology. 2013;23:782–787. doi: 10.1016/j.cub.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bar-On O, Shapira M, Hershko D. Differential effects of doxorubicin treatment on cell cycle arrest and Skp2 expression in breast cancer cells. Anti-Cancer Drugs. 2007;18:1113–1121. doi: 10.1097/CAD.0b013e3282ef4571. [DOI] [PubMed] [Google Scholar]
- 6.Ahn MJ, et al. The differential gene expression profiles between sensitive and resistant breast cancer cells to adriamycin by cDNA microarray. Cancer Res Treat. 2004;36:43–49. doi: 10.4143/crt.2004.36.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Hill R, Li Y, Norris J. Ceramide Glycosylation Catalyzed by Glucosylceramide Synthase and Cancer Drug Resistance. Role of Sphingolipids in Cancer Development and Therapy. 2013;117:59–89. doi: 10.1016/B978-0-12-394274-6.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen B, et al. Inhibition of mTORC2 Induces Cell-Cycle Arrest and Enhances the Cytotoxicity of Doxorubicin by Suppressing MDR1 Expression in HCC Cells. Molecular Cancer Therapeutics. 2015;14:1805–1815. doi: 10.1158/1535-7163.MCT-15-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira A, et al. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology. 2017;390:63–73. doi: 10.1016/j.tox.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Octavia Y, et al. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. Journal of Molecular and Cellular Cardiology. 2012;52:1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Gupta SK, et al. Quaking Inhibits Doxorubicin-Mediated Cardiotoxicity Through Regulation of Cardiac Circular RNA Expression. Circ Res. 2018;122:246–254. doi: 10.1161/CIRCRESAHA.117.311335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chio I, Tuveson D. ROS in Cancer: The Burning Question. Trends in Molecular Medicine. 2017;23:411–429. doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Z, et al. TAK1 inhibitor NG25 enhances doxorubicin-mediated apoptosis in breast cancer cells. Sci Rep. 2016;6:32737. doi: 10.1038/srep32737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao L, Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci Rep. 2017;7:44735. doi: 10.1038/srep44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao H, Dong YB, Bowling MT, Zhou HS, McMasters KM. Alteration of gene expression in melanoma cells following combined treatment with E2F-1 and doxorubicin. Anticancer Res. 2006;26:1947–1956. [PubMed] [Google Scholar]
- 16.Lehnhardt M, et al. Response rate of fibrosarcoma cells to cytotoxic drugs on the expression level correlates to the therapeutic response rate of fibrosarcomas and is mediated by regulation of apoptotic pathways. BMC Cancer. 2005;5:74. doi: 10.1186/1471-2407-5-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Troester MA, Hoadley KA, Parker JS, Perou CM. Prediction of toxicant-specific gene expression signatures after chemotherapeutic treatment of breast cell lines. Environ Health Perspect. 2004;112:1607–1613. doi: 10.1289/ehp.7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daigeler A, et al. Heterogeneous in vitro effects of doxorubicin on gene expression in primary human liposarcoma cultures. BMC Cancer. 2008;8:313. doi: 10.1186/1471-2407-8-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maillet A, et al. Modeling Doxorubicin-Induced Cardiotoxicity in Human Pluripotent Stem Cell Derived-Cardiomyocytes. Sci Rep. 2016;6:25333. doi: 10.1038/srep25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebrahim, A. et al. Multi-omic data integration enables discovery of hidden biological regularities. Nature Communications7, 10.1038/ncomms13091 (2016). [DOI] [PMC free article] [PubMed]
- 21.Nielsen J. Systems Biology of Metabolism: A Driver for Developing Personalized and Precision Medicine. Cell Metabolism. 2017;25:572–579. doi: 10.1016/j.cmet.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Guaragnella N, et al. The expanding role of yeast in cancer research and diagnosis: insights into the function of the oncosuppressors p53 and BRCA1/2. Fems Yeast Research. 2014;14:2–16. doi: 10.1111/1567-1364.12094. [DOI] [PubMed] [Google Scholar]
- 23.Boone C. Yeast systems biology: our best shot at modeling a cell. Genetics. 2014;198:435–437. doi: 10.1534/genetics.114.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buschini A, Poli P, Rossi C. Saccharomyces cerevisiae as an eukaryotic cell model to assess cytotoxicity and genotoxicity of three anticancer anthraquinones. Mutagenesis. 2003;18:25–36. doi: 10.1093/mutage/18.1.25. [DOI] [PubMed] [Google Scholar]
- 25.Saenko IV, Shutov AM, Rastorgueva EV. Doxorubicin and menadione reduce cell proliferation of Saccharomyces cerevisiae by different mechanisms. Tsitologiia. 2010;52:407–411. [PubMed] [Google Scholar]
- 26.Yang F, Kemp C, Henikoff S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutation Research-Fundamental and Molecular Mechanisms of Mutagenesis. 2015;773:9–15. doi: 10.1016/j.mrfmmm.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Celaj, A. et al. Quantitative analysis of protein interaction network dynamics in yeast. Molecular Systems Biology13, 10.15252/msb.20177532 (2017). [DOI] [PMC free article] [PubMed]
- 28.Xia L, Jaafar L, Cashikar A, Flores-Rozas H. Identification of genes required for protection from doxorubicin by a genome-wide screen in Saccharomyces cerevisiae. Cancer Res. 2007;67:11411–11418. doi: 10.1158/0008-5472.CAN-07-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westmoreland TJ, et al. Comparative genome-wide screening identifies a conserved doxorubicin repair network that is diploid specific in Saccharomyces cerevisiae. PLoS One. 2009;4:e5830. doi: 10.1371/journal.pone.0005830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanlon SE, Rizzo JM, Tatomer DC, Lieb JD, Buck MJ. The stress response factors Yap6, Cin5, Phd1, and Skn7 direct targeting of the conserved co-repressor Tup1-Ssn6 in S. cerevisiae. PLoS One. 2011;6:e19060. doi: 10.1371/journal.pone.0019060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoppe T, et al. Activation of a membrane-bound transcription factor by regulated ubiquitin/proteasome-dependent processing. Cell. 2000;102:577–586. doi: 10.1016/S0092-8674(00)00080-5. [DOI] [PubMed] [Google Scholar]
- 32.Thorn C, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenetics and Genomics. 2011;21:440–446. doi: 10.1097/FPC.0b013e32833ffb56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zou Z, Chang H, Li H, Wang S. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis. 2017;22:1321–1335. doi: 10.1007/s10495-017-1424-9. [DOI] [PubMed] [Google Scholar]
- 34.Traverso, N. et al. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Medicine and Cellular Longevity, 10.1155/2013/972913 (2013). [DOI] [PMC free article] [PubMed]
- 35.Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem. 1986;261:3060–3067. [PubMed] [Google Scholar]
- 36.Yao CX, Li WY, Zhang SF, Zhang HF, Zang MX. Effects of Doxorubicin and Fenofibrate on the activities of NADH oxidase and citrate synthase in mice. Basic Clin Pharmacol Toxicol. 2011;109:452–456. doi: 10.1111/j.1742-7843.2011.00748.x. [DOI] [PubMed] [Google Scholar]
- 37.Ganapathy-Kanniappan, S. & Geschwind, J. Tumor glycolysis as a target for cancer therapy: progress and prospects. Molecular Cancer12, 10.1186/1476-4598-12-152 (2013). [DOI] [PMC free article] [PubMed]
- 38.Pang, C. et al. Transcriptome and network analyses in Saccharomyces cerevisiae reveal that amphotericin B and lactoferrin synergy disrupt metal homeostasis and stress response. Scientific Reports7, 10.1038/srep40232 (2017). [DOI] [PMC free article] [PubMed]
- 39.Ducker GS, Rabinowitz JD. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017;25:27–42. doi: 10.1016/j.cmet.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farber S, Diamond LK. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N Engl J Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 41.Mehrmohamadi M, Liu X, Shestov AA, Locasale JW. Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 2014;9:1507–1519. doi: 10.1016/j.celrep.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dornfeld K, Madden M, Skildum A, Wallace KB. Aspartate facilitates mitochondrial function, growth arrest and survival during doxorubicin exposure. Cell Cycle. 2015;14:3282–3291. doi: 10.1080/15384101.2015.1087619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos C, Schulze A. Lipid metabolism in cancer. Febs Journal. 2012;279:2610–2623. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 44.Luo, X. et al. Emerging roles of lipid metabolism in cancer metastasis. Molecular Cancer16, 10.1186/s12943-017-0646-3 (2017). [DOI] [PMC free article] [PubMed]
- 45.Beloribi-Djefaflia, S., Vasseur, S. & Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis5, 10.1038/oncsis.2015.49 (2016). [DOI] [PMC free article] [PubMed]
- 46.Gabitova L, Gorin A, Astsaturov I. Molecular Pathways: Sterols and Receptor Signaling in Cancer. Clinical Cancer Research. 2014;20:28–34. doi: 10.1158/1078-0432.CCR-13-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Momparler RL, Karon M, Siegel SE, Avila F. Effect of Adriamycin on DNA, RNA, and Protein Synthesis in Cell-free Systems and lntact Cells. Cancer Research. 1976;36:2891–2895. [PubMed] [Google Scholar]
- 48.Branzei D, Foiani M. The Rad53 signal transduction pathway: Replication fork stabilization, DNA repair, and adaptation. Experimental Cell Research. 2006;312:2654–2659. doi: 10.1016/j.yexcr.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Demir AB, Koc A. High-Copy Overexpression Screening Reveals PDR5 as the Main Doxorubicin Resistance Gene in Yeast. PLoS One. 2015;10:e0145108. doi: 10.1371/journal.pone.0145108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng C, et al. Regulatory factor X1 is a new tumor suppressive transcription factor that acts via direct downregulation of CD44 in glioblastoma. Neuro-Oncology. 2014;16:1078–1085. doi: 10.1093/neuonc/nou010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su J, et al. RFX1-dependent activation of SHP-1 induces autophagy by a novel obatoclax derivative in hepatocellular carcinoma cells. Oncotarget. 2014;5:4909–4919. doi: 10.18632/oncotarget.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong K, Addinall SG, Lydall D, Rutherford JC. The yeast copper response is regulated by DNA damage. Mol Cell Biol. 2013;33:4041–4050. doi: 10.1128/MCB.00116-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neiman AM. Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics. 2011;189:737–765. doi: 10.1534/genetics.111.127126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Honigberg S, Purnapatre K. Signal pathway integration in the switch from the mitotic cell cycle to meiosis in yeast. Journal of Cell Science. 2003;116:2137–2147. doi: 10.1242/jcs.00460. [DOI] [PubMed] [Google Scholar]
- 56.Piekarska I, Rytka J, Rempola B. Regulation of sporulation in the yeast Saccharomyces cerevisiae. Acta Biochim Pol. 2010;57:241–250. [PubMed] [Google Scholar]
- 57.Kitanovic A, et al. Metabolic response to MMS-mediated DNA damage in Saccharomyces cerevisiae is dependent on the glucose concentration in the medium. FEMS Yeast Res. 2009;9:535–551. doi: 10.1111/j.1567-1364.2009.00505.x. [DOI] [PubMed] [Google Scholar]
- 58.Gratia S, et al. Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovascular Research. 2012;95:290–299. doi: 10.1093/cvr/cvs134. [DOI] [PubMed] [Google Scholar]
- 59.Wang S, Song P, Zou M. Inhibition of AMP-activated Protein Kinase alpha (AMPK alpha) by Doxorubicin Accentuates Genotoxic Stress and Cell Death in Mouse Embryonic Fibroblasts and Cardiomyocytes ROLE OF p53 AND SIRT1. Journal of Biological Chemistry. 2012;287:8001–8012. doi: 10.1074/jbc.M111.315812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu M, et al. Resveratrol protects cardiomyocytes from doxorubicin-induced apoptosis through the AMPK/P53 pathway. Molecular Medicine Reports. 2016;13:1281–1286. doi: 10.3892/mmr.2015.4665. [DOI] [PubMed] [Google Scholar]
- 61.Kobashigawa, L., Xu, Y., Padbury, J., Tseng, Y. & Yano, N. Metformin Protects Cardiomyocyte from Doxorubicin Induced Cytotoxicity through an AMP-Activated Protein Kinase Dependent Signaling Pathway: An In Vitro Study. Plos One9, 10.1371/journal.pone.0104888 (2014). [DOI] [PMC free article] [PubMed]
- 62.Faubert B, Vincent E, Poffenberger M, Jones R. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Letters. 2015;356:165–170. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 63.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 64.Guise, S., Braguer, D., Carles, G., Delacourte, A. & Briand, C. Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. Journal of Neuroscience Research63, 257–267, doi:10.1002/1097-4547(20010201)63:3<257::AID-JNR1019>3.0.CO;2-T (2001). [DOI] [PubMed]
- 65.Kim S, et al. DA-125, a novel anthracycline derivative showing high-affinity DNA binding and topoisomerase II inhibitory activities, exerts cytotoxicity via c-Jun N-terminal kinase pathway. Cancer Chemotherapy and Pharmacology. 2001;47:511–518. doi: 10.1007/s002800100276. [DOI] [PubMed] [Google Scholar]
- 66.Yeh P, et al. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochemical Pharmacology. 2002;63:1423–1430. doi: 10.1016/S0006-2952(02)00908-5. [DOI] [PubMed] [Google Scholar]
- 67.Mansat-De Mas V, et al. Protein kinase C zeta mediated Raf-1/extracellular-regulated kinase activation by daunorubicin. Blood. 2003;101:1543–1550. doi: 10.1182/blood-2002-05-1585. [DOI] [PubMed] [Google Scholar]
- 68.Yu H, Park J, Lee J, Choi K, Choi C. Constitutive Expression of MAP Kinase Phosphatase-1 Confers Multi-drug Resistance in Human Glioblastoma Cells. Cancer Res Treat. 2012;44:195–201. doi: 10.4143/crt.2012.44.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Small G, Somasundaram S, Moore D, Shi Y, Orlowski R. Repression of mitogen-activated protein kinase (MAPK) phosphatase-1 by anthracyclines contributes to their antiapoptotic activation of p44/42-MAPK. Journal of Pharmacology and Experimental Therapeutics. 2003;307:861–869. doi: 10.1124/jpet.103.055806. [DOI] [PubMed] [Google Scholar]
- 70.Kaltenmeier CT, et al. A Tumor Cell-Selective Inhibitor of Mitogen-Activated Protein Kinase Phosphatases Sensitizes Breast Cancer Cells to Lymphokine-Activated Killer Cell Activity. J Pharmacol Exp Ther. 2017;361:39–50. doi: 10.1124/jpet.116.239756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burotto M, Chiou V, Lee J, Kohn E. The MAPK Pathway Across Different Malignancies: A New Perspective. Cancer. 2014;120:3446–3456. doi: 10.1002/cncr.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tan K, et al. A systems approach to delineate functions of paralogous transcription factors: role of the Yap family in the DNA damage response. Proc Natl Acad Sci USA. 2008;105:2934–2939. doi: 10.1073/pnas.0708670105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khan MW, Chakrabarti P. Gluconeogenesis combats cancer: opening new doors in cancer biology. Cell Death Dis. 2015;6:e1872. doi: 10.1038/cddis.2015.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benton MG, Somasundaram S, Glasner JD, Palecek SP. Analyzing the dose-dependence of the Saccharomyces cerevisiae global transcriptional response to methyl methanesulfonate and ionizing radiation. BMC Genomics. 2006;7:305. doi: 10.1186/1471-2164-7-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sha W, Martins AM, Laubenbacher R, Mendes P, Shulaev V. The genome-wide early temporal response of Saccharomyces cerevisiae to oxidative stress induced by cumene hydroperoxide. PLoS One. 2013;8:e74939. doi: 10.1371/journal.pone.0074939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gasch AP, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nikolaou C, et al. Topoisomerase II regulates yeast genes with singular chromatin architectures. Nucleic Acids Research. 2013;41:9243–9256. doi: 10.1093/nar/gkt707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nitiss J, Targeting DNA. topoisomerase II in cancer chemotherapy. Nature Reviews Cancer. 2009;9:338–350. doi: 10.1038/nrc2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baganz, F., Hayes, A., Marren, D., Gardner, D. C. & Oliver, S. G. Suitability of replacement markers for functional analysis studies in Saccharomyces cerevisiae. Yeast13, 1563–1573, https://doi.org/10.1002/(SICI)1097-0061(199712)13:16<1563::AID-YEA240>3.0.CO;2-6 (1997). [DOI] [PubMed]
- 80.Cankorur-Cetinkaya A, Eraslan S, Kirdar B. Transcriptional remodelling in response to changing copper levels in the Wilson and Menkes disease model of Saccharomyces cerevisiae. Mol Biosyst. 2013;9:2889–2908. doi: 10.1039/c3mb70276f. [DOI] [PubMed] [Google Scholar]
- 81.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 82.Cankorur-Cetinkaya A, et al. A novel strategy for selection and validation of reference genes in dynamic multidimensional experimental design in yeast. PLoS One. 2012;7:e38351. doi: 10.1371/journal.pone.0038351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aung HW, Henry SA, Walker LP. Revising the Representation of Fatty Acid, Glycerolipid, and Glycerophospholipid Metabolism in the Consensus Model of Yeast Metabolism. Ind Biotechnol (New Rochelle N Y) 2013;9:215–228. doi: 10.1089/ind.2013.0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Orth JD, Thiele I, Palsson B. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mahadevan R, Schilling CH. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003;5:264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 86.Schellenberger J, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nature Protocols. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shannon P, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bader, G. & Hogue, C. An automated method for finding molecular complexes in large protein interaction networks. Bmc Bioinformatics4, 10.1186/1471-2105-4-2 (2003). [DOI] [PMC free article] [PubMed]
- 89.Huang D, Sherman B, Lempicki R. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teixeira MC, et al. The YEASTRACT database: an upgraded information system for the analysis of gene and genomic transcription regulation in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42:D161–166. doi: 10.1093/nar/gkt1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data has been submitted to ArrayExpress at the European Bioinformatics Institute under accession number [E-MTAB-6634] in compliance with MIAME guidelines.