

Summary
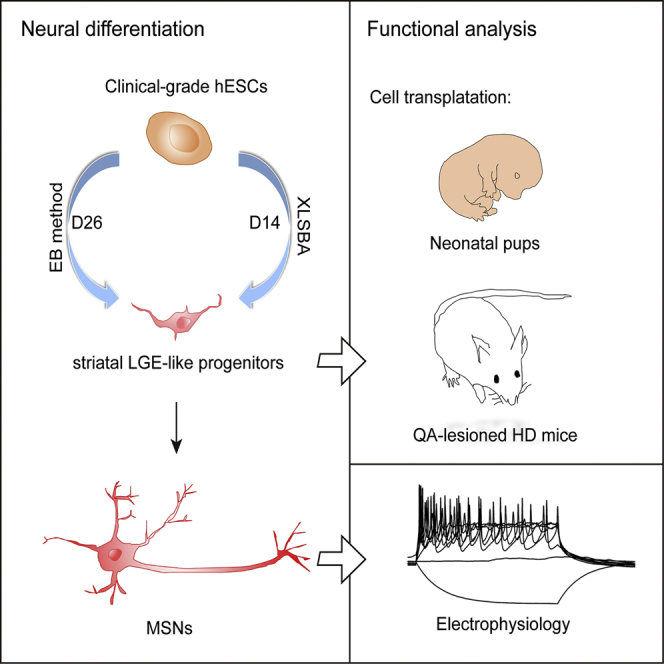
Differentiation of human pluripotent stem cells (hPSCs) into striatal medium spiny neurons (MSNs) promises a cell-based therapy for Huntington's disease. However, clinical-grade MSNs remain unavailable. Here, we developed a chemical recipe named XLSBA to generate clinical-grade MSNs from embryonic stem cells (ESCs). We introduced the γ-secretase inhibitor DAPT into the recipe to accelerate neural differentiation, and replaced protein components with small molecules. Using this optimized protocol we could efficiently direct regular human ESCs (hESCs) as well as clinical-grade hESCs to lateral ganglionic eminence (LGE)-like progenitors and striatal MSNs within less than half of the time than previous protocols (within 14 days and 21 days, respectively). These striatal cells expressed appropriate MSN markers and electrophysiologically acted like authentic MSNs. Upon transplantation into brains of neonatal mice or mouse model of Huntington's disease, they exhibited sufficient safety and reasonable efficacy. Therefore, this quick and highly efficient derivation of MSNs offers unprecedented access to clinical application.
Keywords: clinical-grade hESCs, differentiation, striatal medium spiny neurons, small molecules
Graphical Abstract
Highlights
-
•
DAPT expedites the differentiation of striatal projection neurons from hESCs
-
•
Small molecules can replace recombinant proteins to pattern LGE-like progenitors
-
•
The protocol allows clinical-grade hESCs to generate xeno-free neural progeny
In this article, Wu and colleagues showed that a cocktail of small molecules could induce clinical-grade hESCs into clinically compatible striatal LGE-like progenitors and GABAergic MSNs rapidly and efficiently.
Introduction
Neurological diseases such as Huntington's disease (HD) are progressive neurodegenerative disorders that threaten humans in the modern age. HD is caused by CAG trinucleotide expansion in exon 1 of the Huntington gene (HTT) (MacDonald et al., 1993), and exemplifies diseases that affect specific brain regions and subtypes of neurons in the brain. The cardinal neuropathological manifestation of HD includes the atrophy of basal ganglia, particularly the caudate nucleus and putamen (Lange et al., 1976, Vonsattel and DiFiglia, 1998), and the progressive loss of the major type of striatal GABAergic projection neurons named medium spiny neurons (MSNs). Although some symptoms of HD can be alleviated by medication, no effective treatment is currently available except the intrastriatal transplantation of fetus-derived ganglionic eminence cells reported in a trial (Rosser and Bachoud-Lévi, 2012).
Human pluripotent stem cells (hPSCs) can differentiate into desired neuronal subtypes including MSNs, providing an unlimited and readily accessible source of MSNs for neural replacement therapy of HD. Protocols including those employing dual-SMAD inhibition have been exploited to produce striatal MSNs (Arber et al., 2015, Aubry et al., 2008, Delli Carri et al., 2013, Ma et al., 2012, Nicoleau et al., 2013). Such endeavors advocate significantly for the understanding of neural differentiation and have shed light on the potential of human embryonic stem cell (hESC)-based cell replacement therapy. These protocols, however, are not designed for generation of clinical-grade cells and most of them are time-consuming, which increases the variability, yielding variable production of MSNs. In either protocol, recombinant proteins such as SHH, DKK1, or ACTIVIN A are used (Arber et al., 2015, Delli Carri et al., 2013, Nicoleau et al., 2013). Protein components in recipe increase the difficulties of quality control, and in addition increase the cost. Chemical compounds are cost-effective and easily quality controlled, exhibiting significant advantages over proteins. They can replace proteins of classic signaling to produce functional cortical neurons and nociceptors from hESCs (Chambers et al., 2012, Qi et al., 2017), suggesting that a chemical recipe is also possible for generating MSNs from hESCs.
Recipes free of proteins are particularly preferred for the manufacture of clinical-grade cells for therapy. We and colleagues have developed xeno-free hESCs with defined quality characteristics fitting for clinical therapy (Gu et al., 2017). To generate MSNs from these clinical-grade hESCs, we invented a small-molecule-based recipe and a streamlined protocol for efficient induction of lateral ganglionic eminence (LGE)-like cells and MSNs. This protocol is further adapted to the clinical-compatible conditions to generate clinical-grade MSNs, which facilitate the industrial manufacture and clinical use.
Results
DAPT Expedites the Differentiation of Striatal Projection Neurons from hESCs
The ability to produce specific subtypes of neurons such as MSNs from hPSCs has made considerable progress. However, existing hPSC differentiation protocols with extrinsic patterning factors require extended culture periods of as long as 80 days (Arber et al., 2015, Aubry et al., 2008, Delli Carri et al., 2013, Ma et al., 2012). For example, the well-established embryoid body (EB)-based differentiation strategy needs 40 days to generate striatal progenitors for cell transplantation (Figure 1A) (Ma et al., 2012). The protracted in vitro differentiation of hPSCs increases variabilities and costs for cell products in clinical application. Therefore, accelerating the differentiation is important for the generation of readily accessible clinical-grade MSNs used for therapy.
Figure 1.

DAPT Accelerates the Differentiation of Striatal Projection Neurons from hESCs
(A) Schematic overview of the differentiation strategies using either the EB or NSBS protocol to obtain striatal MSNs. BGIC represents BDNF, GDNF, IGF-1, and dibutyryl-cAMP. NSBS represents Noggin, SB431542, and SHH. BGAD represents BDNF, GDNF, AA, and DAPT.
(B) Representative images of immunofluorescence for particular stage markers during EB and NSBS differentiation. PAX6, SOX1, hNESTIN, and KI67 for neuroepithelia; FOXG1 and DLX2 for LGE-like progenitors; DARPP32, GABA, and TUJ1 for MSN GABA neurons. Ho, Hoechst 33258. Scale bars, 50 μm.
(C) Quantification for particular differentiation stage markers from (B). Efficiencies are presented as the percentage of positive cells ±SEM of all fields counted. ns, not significant; Student's t test.
(D) Relative mRNA expression for neuroblasts (DCX, TUJ1), mature neurons (MAP2), forebrain markers (FOXG1, SIX3), neural progenitors (NESTIN, SOX1, SOX2, PAX6), and LGE markers (DLX5) in H9-hESC derivatives generated with or without DAPT (day 18). Data are presented as mean ± SEM (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ns, not significant.
See also Figures S1 and S2.
N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) was proposed to block NOTCH signaling as a γ-secretase inhibitor (Dovey et al., 2001) and accelerate the neural differentiation (Chambers et al., 2012, Qi et al., 2017). To expedite the differentiation process for generating striatal MSNs from hESCs, we set up an EB and monolayer combination culture system, and introduced DAPT to the medium from day 11, based on the dual-SMAD inhibition approach (termed the NSBS protocol) (Chambers et al., 2009, Kirkeby et al., 2012a, Kirkeby et al., 2012b) (Figure 1A). Using the NSBS protocol, typical neural rosette structures appeared as early as day 5 of differentiation, as compared with day 15 if using the EB protocol (Figure 1B). Neuroepithelia (NE) expressing SOX1 and PAX6 were readily detected at day 5 of differentiation in the NSBS protocol, 10 days earlier than that in the control (Figures 1B and 1C). LGE-like progenitors expressing telencephalic marker FOXG1 and subpallial telencephalic marker DLX2 were detected on day-14 cultures, with a similar efficiency as that in the EB protocol on day 26 (Figures 1B and 1C). A similar amount of MSNs expressing TUJ1, GABA, and DARPP32 were generated at day 21, less than half of that used in the EB protocol (47 days) (Figures 1B and 1C).
DAPT significantly accelerated neural specification and neuronal maturation, as was also shown with qPCR. Administration of DAPT significantly increased levels of the neuroblast gene DCX, pan-neuronal gene TUJ1, mature neurons marker MAP2, and forebrain markers FOXG1 and SIX3, but not neural progenitor markers NESTIN, SOX1, and SOX2 (Figure 1D). It also increased cells expressing post-mitotic neuronal marker DLX5 (Eisenstat et al., 1999, Liu et al., 1997, Panganiban and Rubenstein, 2002) (Figure 1D).
Therefore, using a simple yet efficient NSBS protocol, we successfully expedited the generation of NE, LGE-like cells, and MSNs within 5 days, 14 days, and 21 days as compared with 15 days, 26 days, and 47 days for the EB protocol.
Optimizing SHH Pathway Activity for Robust Generation of Striatal MSNs
Appropriate activity of Sonic hedgehog (SHH) signaling is critical to induce LGE and can ventralize the hPSC derivatives (Campbell, 2003, Ma et al., 2012). Since DAPT significantly accelerates the MSN specification, fine-tuning the SHH activity is crucial in obtaining authentic MSNs in the optimized protocol. To determine an optimal level of SHH, we treated hESCs with 0, 100, 200, and 500 ng/mL SHH from day 0 to day 9 (Figure S1A). On day 14, while SHH concentration did not affect the expression of telencephalic marker FOXG1, 100 ng/mL SHH was sufficient to increase the expression of telencephalic marker OTX2, early developing striatal markers DLX2, and post-mitotic MSNs markers DLX5, FOXP2, and MEIS2 (Figure S2B). Immunostaining at day 17 indicated that cells expressing FOXP1, CALBINDIN, and DARPP32 were significantly increased upon treatment with 100 ng/mL SHH (Figures S2C and S2D). Among the TUJ1+ neurons, 66% were FOXP1+, 91% CALBINDIN+, and 77% DARPP32+ neurons (Figure S2D). However, SHH did not significantly alter the efficiency of neural differentiation as each group yielded ∼92% MAP2+ cells among TUJ1+ cells (Figures S2C and S2D).
The NSBS protocol favored the generation of ventral telencephalic cells and accelerated the neural differentiation, as was demonstrated by qPCR at three typical stages of MSN differentiation, namely NE, LGE, and MSN. Except for a decreasing gene expression of PAX6 and DLX2 at the NE and LGE stage, respectively, NESTIN, FOXG1, and SIX3 at the NE stage, GSH2, MASH1, DLX5, and DLX6 at the LGE stage, and FOXP1, MEIS2, CALBINDIN, DRD2, and ARPP21 at the MSN stage were all increased (Figures S2A–S2C).
Overall, these results indicated that 100 ng/mL SHH could enrich neuronal subtype MSN without compromising the efficiency of neural specification and neuronal maturation.
Generation of GABAergic MSNs from hESCs by Chemical Cocktails
A protein-free recipe is critical to obtain subtype-specific neurons from hPSCs for clinical applications. Small molecules such as the ALK2/ALK3 inhibitor LDN-193189 (L) and BMP inhibitor dorsomorphin (D) can replace NOGGIN for neuralization of hPSCs (Chambers et al., 2012, Kim et al., 2010, Morizane et al., 2011, Qi et al., 2017, Yu et al., 2008, Zhou et al., 2010). We thus chose L and D together with WNT signaling inhibitor XAV939(X), and SHH signaling agonists SAG or purmorphamine (PP), to coordinate the anterior-posterior and dorsal-ventral patterning, respectively, and direct hESCs into LGE-like progenitors. A combination of four small molecules (XAV939, LDN-193189, SB431542, and SAG, termed the XLSBA protocol) facilitated the induction of LGE-like precursors that expressed striatal and neuronal markers such as GSH2, MEIS2, FOXP1, FOXP2, NOLZ1, ARPP21, CTIP2, and DARPP32 (Figures 2A′ and 2B′). We further adjusted the duration and concentration of small molecules and optimized the protocol for efficient production of LGE-like cells (Figures 2A″–2A‴ and 2B″–B‴). Under such a condition, the transcript levels of GSH2, DLX5, FOXP2, and APRR21 were significantly increased (Figures 2C and 2D). In addition, abundant NESTIN+ (93.1% ± 1.0%), KI67+ (73.7% ± 0.7%), FOXG1+ (82.7% ± 0.7%), DLX2+ (76.3% ± 0.4%), and GSH2+ (87.2% ± 0.4%) cells were detected (Figures 2E and 2F). Thus, the XLSBA protocol can efficiently and quickly induce hESCs into LGE-like striatal progenitors.
Figure 2.

Generation of LGE-like Progenitors from hESCs by Chemical Cocktails
(A) Relative mRNA expression of striatal lineage genes to identify optimal small-molecule combination to replace noggin. (A′) Striatal lineage gene expression in H9-hESCs cultures differentiated in the presence of LDN-193193 (100 nM), dorsomorphin (100 nM), XAV939 (1 μM), or their combination from days 1–9. All expression levels are normalized to levels detected in hESCs. (A″) Anterior-posterior axis gene expression in H9-hESC cultures differentiated after treatment with different concentrations of XAV939 (0, 0.25, 1, and 4 μM). (A‴) Anterior-posterior axis gene expression in H9-hESCs cultures differentiated after treatment with 4 μM XAV939 for different time periods. X1 represents cell cultures treated with XAV939 for 1 day, X2 represents cell cultures treated for 2 days, and so on. Data are presented as mean ± SEM (n = 3). ns, not significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; one-way ANOVA, followed by Tukey's multiple comparisons tests.
(B) Relative mRNA expression of striatal lineage genes to identify optimal small molecule to replace SHH. (B′) Striatal lineage gene expression in H9-hESC cultures differentiated in the presence of purmorphamine (PP, 1 μM) or SAG (100 nM). All expression levels are normalized to levels detected in hESCs. (B″) Dorsal-ventral axis gene expression in H9-hESCs cultures differentiated after treatment with different concentrations of SAG (0, 10, 50, and 100 nM). (B‴) Dorsal-ventral axis gene expression in H9-hESCs cultures differentiated after treatment with 100 nM SAG for different time periods. A1 represents cell cultures treated with SAG for 1 day. A2 represents cell cultures treated for 2 days, and so on. Data are presented as mean ± SEM (n = 3). ns, not significant. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; one-way ANOVA followed by Tukey's multiple comparisons tests.
(C) Schematic overview of the optimized differentiation protocol for hESCs.
(D) Relative mRNA expression of striatal lineage genes treated with small molecules from days 0–5 and days 0–9. All expression levels are normalized to levels detected in hESCs. Data are presented as mean ± SEM. ns, no significant. ∗∗∗∗p < 0.0001; Student's t test.
(E) Immunostaining images for neural progenitor marker NESTIN, proliferative marker KI67, forebrain marker FOXG1, LGE progenitor markers GSH2 and DLX2, and post-mitotic MSN marker CTIP2 on day 11. Scale bar, 50 μm.
(F) Quantification for immunostaining of markers in (E). Error bar represents SEM.
The hESC-induced LGE-like progenitors efficiently converted into MSNs on day 21 of differentiation, as was shown by immunofluorescence of subtype-specific neuronal markers (Figures 3A–3D). Over 87% of the MAP2+ neurons expressed DARPP32 and more than 89% of the TUJ1+ neurons were GABA+ (Figures 3A and 3B), and a proportion of the TUJ1+ cells co-expressed other genes with enriched expression in the striatum such as CALBINDIN, demonstrating their striatal GABAergic MSN identity (Figures 3C and 3D). More mature neurons possessing dendritic spines also expressed PSD95 and SYNAPSIN 1 (Figure 3E). Except for the GABAergic neurons (89.5% ± 5.0%), 8.2% ± 1.2% of cells expressed tyrosine hydroxylase (TH; a marker for dopaminergic neurons). None of the differentiated cells expressed other neuronal subtype markers, such as choline acetyltransferase (CHAT; a marker of cholinergic neurons), 5-hydroxytryptamine (5-HT; a marker for serotonergic neurons), and vesicular glutamate transporter 1 (VGlut1; a marker for excitatory glutamatergic neurons) (Figures 3F and 3G). Thus, the XLSBA protocol primarily induces DARPP32+ GABAergic striatal projection neurons.
Figure 3.

Efficient Generation of DARPP32+ GABAergic Neurons from Clinical-Grade hESCs by Small-Molecule Cocktails
(A) Immunostaining images for TUJ1, MAP2, DARPP32, and GABA of day 21–26 cultures derived from hESC lines H9, H1, and clinical-grade Q-CTS-hESC-2 cell line. Scale bar, 50 μm.
(B) Quantification for the percentage of DARPP32 positive cells among MAP2+ and GABA+ cells among TUJ1+ neurons presented in (A). Data are presented as means ± SEM. ns, not significant, ∗p < 0.05, Student's t test.
(C) Representative confocal images of GABAergic MSNs markers DARPP32, GABA and CALBINDIN. Scale bar, 50 μm.
(D) Quantification for the percentage of DARPP32+ and CALBINDIN+ cells among TUJ1+ neurons, and DARPP32+ cells among GABA+ neurons presented in (C). Data are presented as means ± SEM.
(E) Immunostaining images of day-21 cultures for mature neuron markers SYNAPSIN1 and post-synaptic protein PSD95 in hESC-derived MSNs. Scale bar, 50 μm. The right panel shows accumulation of PSD95 on spines and dendrites, enlarged from the left image. Scale bar, 10 μm.
(F) Representative images of H9-hESCs cultures immunolabeled with TUJ1 and markers of neuronal subtypes (GABA, TH, CHAT, 5-HT, VGlut1). Scale bar, 50 μm.
(G) Quantification for the percentage of GABA+, TH+, CHAT+, 5-HT+, and VGlut1+ cells among TUJ1+ cells presented in (F). Efficiencies are presented as the percentage of positive cells ±SEM of all fields counted. ∗∗∗∗p < 0.0001, Student's t test.
XLSBA worked efficiently in additional hESC lines, particularly biosafe clinical-grade hESC line (Gu et al., 2017). Using the xeno-free Clinical Therapy Systems (CTS) reagents, the clinical-grade hESC line was efficiently differentiated into DARPP32-expressing MSNs, and the Q-CTS-hESC-2 line generated even more DARPP32+ cells (∼94.9% of MAP2+ neurons) than the regular hESC lines (Figures 3A and 3B). Therefore, the chemical XLSBA protocol is superior in rapidly and robustly inducing clinical-grade LGE/striatal progenitors and a homogeneous population of MSNs from hESCs.
hESC-Derived MSNs Progressively Mature and Act as Authentic GABAergic Neurons
To examine the functional properties of hESC-derived MSNs, we performed whole-cell patch-clamp recordings on hESC-derived MSNs at 20–24 days after differentiation. Current-clamp recording showed that all cells elicited single or multiple action potentials upon the injection of depolarizing currents (Figure 4A). As depolarizing voltage steps in voltage-clamp mode it elicited fast inward currents followed by slow outward currents, indicating opening of voltage-activated sodium and potassium channels, respectively (Figure 4B). Spontaneous action potentials were frequently detected (Figure 4C). Upon local administration of tetrodotoxin (TTX), traces of membrane action potentials were not detected in representative neurons and the inward currents were ablated (Figures 4D and 4E), indicating the involvement of TTX-sensitive voltage-gated sodium channels. These neurons became functionally mature over time. Whereas they initially displayed modest action potentials at 20 days, approximately 46% and 34% of the recorded cells fired at 22 and 24 days of differentiation, respectively (Figure 4F). The passive membrane properties of differentiating neurons (Table S1) exhibited negative resting membrane potential (RMP; −40 to −50 mV, Figure 4G). The input resistance values (∼2–4 GΩ, Figure 4H) decreased while capacitance values (20–25 pF, Figure 4I) were stable during their maturation in culture (days 20–24). Addition of GABA in culture elicited inward currents when cells were held at −70 mV (n = 19, Figure 4J), whereas addition of H2O could not (n = 12, Figure 4J). The currents were blocked by the GABA type A (GABA-A) receptor antagonist bicuculline (Figure 4J). Therefore, these results confirmed that hESC-derived neurons exhibited GABAergic identity.
Figure 4.

Electrophysiological Properties of GABAergic MSNs Differentiated from hESCs
(A) Representative traces of membrane potential responding to step depolarization by current injection steps from −10 pA to +60 pA in 10-pA increments. Membrane potential was current-clamped at around −65 mV.
(B) Representative traces of whole-cell currents in voltage-clamp mode; cells were held at −70 mV; step depolarization from −80 mV to +60 mV at 10-mV intervals was delivered. The inset shows Na+ currents.
(C) Spontaneous action potentials (APs) recorded from neurons of 24 days differentiation. No current injection was applied (n = 21).
(D) TTX blocked the membrane APs (n = 47).
(E) The Na+ currents of neurons were blocked by TTX. When the cells were treated with 1 μM TTX, the channel of Na+ currents was blocked completely (n = 47).
(F) Percentages of different AP spikes at 20, 22, and 24 days after differentiation. Increased complexity of AP spikes of differentiated cells over the maturation process. The majority of cells showed single spikes at 24 days differentiation, whereas a larger percentage of cells generated repetitive APs upon co-culture with human glia.
(G–I) Quantification of resting membrane potential (RMP, G), membrane resistance (Rin, H) and membrane capacitance (Cm, I) in neurons at 20, 22, and 24 days after differentiation. Error bars indicate ±SEM. ns, not significant. ∗∗p < 0.01; one-way ANOVA followed by Tukey's multiple comparisons tests.
(J) Focal application of GABA elicited inward membrane currents (n = 19), which was almost completely eliminated by bicuculline. Focal application of ddH2O could not induce inward membrane currents (n = 11).
(K–N) Averaged (means ±SEM) current-voltage relationship (I-V curves) for Na+ and K+ currents, recorded from hESC-derived neurons. ns, not significant. ∗p < 0.05; one-way ANOVA followed by Tukey's multiple comparisons tests.
Since astrocytes could promote neuron maturation (Johnson et al., 2007), we investigated whether co-culturing differentiated MSNs with human astrocytes would lead to more active electrophysiological characteristics. As anticipated, ∼77% of recorded MSNs (17 of 22 total cells) were capable of firing repetitively (Figure 4F) and displayed a more hyperpolarized RMP in the presence of astrocytes compared with those in the absence of astrocytes (Figure 4G), indicating more maturity. However, no significant change in membrane resistance (Rin) and membrane capacitance (Cm) was observed (Figures 4H and 4I) and peaks of the voltage-gated Na+ and K+ currents also stayed unchanged upon co-culture with human astrocytes (Figures 4K–4N). These results indicated that astrocytes promoted the maturation of hESC-derived striatal MSNs in our culture system to some extent.
hESC-Derived MSNs Become Mature and Functional in Brains of Neonatal and HD Mice
To confirm whether these LGE-like progenitors and MSNs are safe and functional, we transplanted 100,000 LGE-like progenitors into the striatum of P1 neonatal SCID mice (Figure 5A). Immunostaining for human nuclei (hN) indicated that a significant proportion of graft cells expressed DARPP32, FOXP1, FOXG1, and CALBINDIN 12 weeks after transplantation (Figures 5B and 5C). NESTIN+ or KI67+ proliferative cells were rare within the grafts (Figures 5B and 5C). The transplanted LGE-like progenitors differentiated into both the direct and indirect subtypes of MSNs, as evidenced by the presence of substance P+ (SP+) and enkephalin+ (ENK+) cells within the grafts (Figures 5D and 5E). While considerably grafted cells became TUJ1+ neurons (87.4% ± 1.9%), none expressed GFAP (a marker for astrocytes), OLIG2 (a marker for oligodendrocytes), or IBA1 (a marker for microglia) (Figures 5F and 5G). Except for a small population of CHAT+ neurons (9.61% ± 5.21%), GABA neurons (81.7% ± 8.1%) predominated in the population of transplanted cells (Figures 5H and 5I). Taken together, GABAergic MSN neuronal identity was successfully established from grafted progenitors.
Figure 5.

Survival, Differentiation, and Maturation of hESC-Derived LGE-like Progenitors in the Neonatal Mouse Brain
(A) Schematic overview of the transplantation: LGE-like progenitors from hESCs were differentiated for 12–14 days in vitro, and the cell cultures were injected into newborn mouse brain.
(B) Immunostaining of 12-week-old grafts in neonatal mice for human nuclei (hN) and DARPP32, FOXP1, FOXG1, CALBINDIN, KI67, or hNESTIN. Scale bar, 50 μm.
(C) Quantification of immunostaining of markers in (B), showing the percentage of indicated markers among hN+ cells. Data are presented as mean ± SEM.
(D) Immunostaining of 12-week-old grafts for hN, SP, and ENK. Scale bars, 50 μm. The right panels show enlarged images from the inset. Scale bars, 2 μm.
(E) Quantification of immunostaining of markers in (D), shown as a percentage of indicated markers among hN+ cells. Data are presented as mean ± SEM.
(F) Immunostaining images of 12-week-old grafts for hN and TUJ1, GFAP, OLIG2, and IBA1. Scale bar, 50 μm.
(G) Quantification of immunostaining of markers in (F). The majority of the grafted cell were TUJ1+ neurons with few being GFAP-expressing astrocytes and no OLIG2+ oligodendrocytes and IBA1+ microglia. Data are presented as mean ± SEM. ∗∗∗∗p < 0.0001.
(H) Representative images of 12-week-old grafts immunolabeled with hN and markers of neuronal subtypes GABA, TH, CHAT, 5-HT, and VGlut1. Scale bar, 50 μm.
(I) Quantification for the proportion of neural subtypes, determined by counting GABA-, TH-, CHAT-, 5-HT-, and VGlut1-positive cells among hN+ cells presented in (H). Data are presented as mean ± SEM. ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To further examine hESC-derived MSNs in an adult and diseased environment, we stereotactically transplanted the same amount of LGE-like progenitors into the striatum of quinolinic acid (QA)-lesioned mice, which mimic some motor deficits of human HD (Sanberg et al., 1989) (Figure 6A). These transplanted cells also gradually matured. Four weeks after transplantation, a vast majority of transplanted hN+ cells remained hNESTIN+ and KI67+ in the grafts (Figure 6B). KI67+ proliferative cells were reduced at week 8 post transplantation and eventually neither were detectable 16 weeks after transplantation (Figure 6B). Whereas in sham control animals no NEUN+ and DARPP32+ cells were left (Figure 6C), in those that received transplantation hN+ cells repopulated the lesions (Figure 6D) and 48.7% ± 2.8% of them were DARPP32+ (Figure 6E). These graft cells expressed GABA, TUJ1, CTIP2, and FOXP1. A small proportion of them were also SP+ and ENK+ cells (Figures 6E–6G). Unsurprisingly, the grafted cells expressed similar proportions of TUJ1, GABA, DARPP32, MEIS2, and KI67 among hN+ cells between mice transplanted cells by the EB and XLSBA protocols (Figures S3A and S3B). These results demonstrated that hESC-derived MSNs from XLSBA protocol could survive and mature in QA-lesioned mice just as for those from the EB method.
Figure 6.

hESC-Derived Progenitors Survive and Produce MSNs in QA-Lesioned Mice
(A) Experimental scheme for the transplantation of QA-lesioned mice and behavior tests.
(B) Microtome sections of QA-lesioned adult mouse brains at 4, 8, and 16 weeks post transplantation were analyzed by immunohistochemistry for hN and NESTIN or KI67. Scale bar, 50 μm.
(C) Adult SCID mice injected with QA and without cell transplantation showed a loss of neurons marked by NEUN staining and MSNs marked by DARPP32 staining on the lesion side (ctrl-ipsil), while normal on contralateral side of the striatum (ctrl-contral). Scale bar, 50 μm.
(D) Low magnification of a coronal brain section showing transplanted human cells labeled with hN residing in the striatum. Scale bar, 1,000 μm.
(E) Immunostaining images of 16-week-old grafts for GABA, DARPP32, TUJ1, FOXP1, substance P (SP), and enkephalin (Enk). Scale bar, 50 μm.
(F) Triple staining for DARPP32, CTIP2, and hN on a section of a 16-week-old grafts. Scale bar, 50 μm.
(G) Quantification of immunostaining of striatal markers, shown as a percentage of hN+ cells. Data are presented as means ± SEM.
(H) Representative tracing images of animals in open field tests.
(I) Open field tests indicated increased total distances in animals receiving cell grafts compared with sham control. Open field behaviors were analyzed by two-way ANOVA (∗∗∗p < 0.001; control group, n = 13; cell group, n = 13).
(J) Rotarod tests showed increased latency in animals transplanted with hESC-derived progenitors compared with sham control. The tests were analyzed by two-way ANOVA (∗∗p < 0.01, ∗∗∗∗p < 0.001; control group, n = 13; cell group, n = 13).
See also Figures S3–S5.
Monthly behavior tests (Figure 6A) showed that compared with the sham transplantation, animals receiving MSN transplantation exhibited significant improvement of behavior rating in open field tests (Figures 6H, 6I, and S4) and rotarod tests (Figure 6J). In addition, a similar degree of motor recovery was observed in mice receiving MSN progenitors generated by the XLSBA protocol and EB method (Figures S5A and S5B). These results indicated that hESC-derived MSNs were functional upon transplantation and could be used for therapy.
Discussion
Cell-based therapy requires readily available cell sources that meet the stringent criteria of human cellular and tissue-based product (HCT/P), namely clinically compliant hESC derivatives in this case. We developed a chemical recipe named XLSBA for rapid and efficient generation of committed striatal LGE-like progenitors and MSNs from hESCs. We also systematically evaluated the safety and efficacy of the XLSBA protocol in neonatal mouse and QA-lesioned mouse model of HD, and found that similar functional improvement after striatal transplantation with cells generated by XLSBA or EB methods. The XLSBA protocol can be used to generate clinical-grade MSNs, and is valuable for standardization and quality control in manufacturing and clinical cell transplantation.
The distinct advantages of small molecules over recombinant proteins include better cell permeability, no immunogenicity, lower cost, and rapid, reversible biological effects. As for the chemical recipe in this study, in addition to the dual-SMAD inhibition by small molecules (Arber et al., 2015, Chambers et al., 2012, Delli Carri et al., 2013, Nicoleau et al., 2013, Qi et al., 2017), all patterning factors were replaced by small molecules such as SAG for SHH signaling (Chen et al., 2002, Danjo et al., 2011), and tankyrase inhibitor XAV939 for inhibition of canonical WNT signaling (Huang et al., 2009). Through carefully titrating the dosage of each small molecule, we obtained an optimal combination of XAV939, SAG, and dual-SMAD inhibition, together with well-designed episodes of administration to efficiently produce committed striatal LGE-like progenitors. Compared with the less efficient and variable proportion of TUJ1+ (22%–93% of total cells) and DARPP32+ (20%–53% of TUJ1+ cells) cells using other protocols (Arber et al., 2015, Aubry et al., 2008, Delli Carri et al., 2013, Golas and Sander, 2016, Ma et al., 2012, Nicoleau et al., 2013), the XLSBA protocol generated ∼90% of DARPP32+ GABAergic MSNs and nearly pure TUJ1+ neurons. Such a homogeneous population of defined neurons inevitably advocates the therapeutic potential of cell transplantation.
Accelerating the production of MSNs from hPSCs has considerable potential in modeling development in vitro. First, refining the generation process of MSNs from hESCs will provide insight into the signaling programs and decipher key processes of MSN development. Various signalings coordinate temporally and spatially to fine-tune the cellular identity during development. Moderate SHH signaling, for example, is necessary to induce LGE fate (Aubry et al., 2008, Ma et al., 2012). A high dose, however, suppresses the telencephalic fate (Fasano et al., 2010). It is interesting that although a certain level of SHH signaling favors an optimal production of MSNs, extra SHH appears to be dispensable for GABAergic gene expression, as we revealed that LGE-like progenitors are indeed induced even in the absence of SHH. In addition, we found that 100 ng/mL SHH is sufficient for the maximal induction of LGE-like cells compared with that in previous studies, whereas 200 ng/mL SHH is needed to induce LGE identity (Aubry et al., 2008, Ma et al., 2012). This is also supported by a recent study, which reported that hPSCs produce both dorsal cortical glutamatergic and ventral GABAergic neurons by default (Floruta et al., 2017). Differences in the culture methods, such as monolayer versus aggregates, might induce variable intrinsic signaling, and DAPT, which drastically alters the temporal induction, might also reshuffle the orchestration of the cell signaling. Second, the recapitulation of several key developmental stages of MSN with significantly shortened time span would allow us to obtain samples for research more easily and quickly. A combination of hESC differentiation with single-cell transcriptome sequencing technology will encourage us further to identify new markers and probe their function in specific stages of developing MSNs. As human striatum development is a complex process, while neuronal monolayers may lack complexities in vivo, it will also be possible in the near future to establish three-dimensional organoids to mirror the process of striatal development more accurately based on the spatial relationships and interplay of multiple cell types during development.
Since human induced PSCs (hiPSCs) possess the same developmental potential as hESCs and carry related pathogenic gene components, they make it possible to model human diseases in vitro. Currently many disease-specific hiPSCs such as Parkinson’s disease, HD, and Alzheimer’s disease have been established (Park et al., 2008). The use of these patient-specific hiPSC lines can help determine the mechanisms of disease initiation, identify the pathogenesis of the disease, and promote the development of early intervention therapies. However, modeling HD with hiPSC-derived neurons often recapitulated partial relevant phenotypes (An et al., 2012, HD iPSC Consortium, 2012, Jeon et al., 2012), and additional cellular stressors are required to trigger essential HD-associated phenotypes such as the formation of mHTT aggregates (Jeon et al., 2012, Nekrasov et al., 2016). Recently, a paper pointed out that the manifestation of HD phenotypes is dependent on cellular age, because age signature retention through direct neuronal conversion could help to exhibit the mHTT aggregates, which are absent in iPSC-derived neurons with erased age marks (Victor et al., 2018). Therefore, it has been challenging to model disease progression using hiPSC-derived MSNs in vitro for neurodegenerative diseases with long latency such as HD. However, it becomes possible to capture the phenotypes of late-onset disease when we accelerate the appearance of pathological phenotypes with our short-term culture method in vitro. Meanwhile, by establishing a superior method for differentiated cells with minimal variability, HD hiPSCs can also be used for high-throughput drug screening. These may have profound implications for the development of personalized therapeutic interventions for neurodegenerative diseases such as HD.
Cell-based therapy has raised the hope of curing hitherto intractable human neurodegenerative diseases in clinical trials, since targeted differentiation of hPSCs into neuronal subtypes have begun to demonstrate their therapeutic potential in epilepsy, Parkinson's disease, and HD (Cunningham et al., 2014, Kikuchi et al., 2017, Ma et al., 2012). However, most hESC lines worldwide are research grade, as they were produced in the presence of animal components and without biosafety evaluation. Therefore they are not ideal for use in clinical trials. Currently xeno-free hESCs are available, such as the biosafe clinical-grade hESC lines accredited at the Chinese National Institutes for Food and Drug Control (Gu et al., 2017). In the context of clinical applications, in addition to hESCs their differentiated progeny cells must also be developed in adherence to Good Manufacture Practices, which will guarantee clinical-grade level for all procedures and reagents used for the generation of the cells to be grafted. To date, clinical-grade MSNs remain unavailable for HD therapy. With this goal in mind, we used commercially available CTS reagents that are free of xenogeneic components and obtained MSN precursor cells from clinical-grade hESCs. These would provide therapeutically safe cell sources for HD. For fetal grafts, the limited time for tests of quality control raised a major challenge (Nicoleau et al., 2011). Thus, differentiating hPSCs into neural progenies in an accelerating manner would not only provide timely, reliable, and scalable supply of cell sources but also save enough time to perform rigorous tests to ensure the biosafety of the preparation. The faster time frame and validated quality would enhance the reproducible production and facilitate the routine application of hPSC-derived striatal cells in clinical implementation. Therefore, this would take stem cell-based neural replacement therapy for HD patients one step closer to the clinic.
Experimental Procedures
hESC Culture and Neural Differentiation
hESCs (H9 and H1) were maintained on Matrigel; clinical-grade Q-CTS-hESC-2 line was cultured on vitronectin, and were dissociated with EDTA (Gibco) for passaging or dispase (Gibco) for differentiation (Beers et al., 2012). Differentiation of striatal MSNs by EB protocol is based on the procedures as previously reported (Ma et al., 2012). In brief, hESCs were detached to form EBs with dispase. After adhering to a plastic surface on day 7, definitive neuroepithelial cells with neural-tube-like rosettes were mechanically picked and transferred to dishes to form neurospheres on days 15–17. For immunostaining, neural progenitor clusters were dissociated with Accutase and plated on the Matrigel-coated coverslips. SHH, VPA, brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), insulin-like growth factor 1 (IGF-1), and cyclic AMP (cAMP) were added at appropriate times. For differentiation of MSNs by small molecules, differentiation media were described as previously (Kirkeby et al., 2012a, Kirkeby et al., 2012b). On day 0, hESCs were detached with dispase to form EBs in NIM. The EBs were then plated in NPM onto plastic plates coated with Matrigel on day 4. Within days 11–16 of differentiation, the cells were dissociated to small clusters with Accutase and replated onto Matrigel-coated plates in NDM. BDNF, GDNF, ascorbic acid (AA), and DAPT were added for terminal differentiation. For clinical-grade hESC differentiation, all CTS reagents were purchased from Life Technologies. Cells were reseeded on polyornithine/fibronectin/laminin-coated coverslips. Small-molecule compounds were as follows: XAV939 (0–4 μM, Selleck), dorsomorphin (100 nM, Selleck), LDN-193189 (100 nM, Selleck), SB431542 (10 μM, Merck), SAG (0–100 nM, Stemgent), purmorphamine (0–1 μM, Stemgent), AA (0.2 mM, Sigma), and DAPT (1 μM, Tocris). Recombinant growth factors were as follows: SHH (C25II; 0–500 ng/mL), NOGGIN (200 ng/mL), BDNF (20 ng/mL), GDNF (10 ng/mL), all from Peprotech. For detailed differentiation procedures, see Supplemental Experimental Procedures.
hESC-Derived Neural Progenitors and Human Astrocyte Co-cultures
Human astrocytes were purchased from ScienCell. Astrocyte culture medium consisted of DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco) and non-essential amino acids (Gibco). For electrophysiology, human astrocytes were passaged onto coverslips coated with poly-D-lysine (Sigma) in 24-well plates (5 × 104 per well) and incubated overnight at 37°C in the CO2 incubator. On the second day, ∼2 × 104 GABA neural progenitors differentiated from hESCs between 11 and 16 days were dissociated with Accutase and replated on top of human astrocytes in NDM supplemented with BDNF, GDNF, AA, and DAPT. Half of the medium was changed every other day. The cells were maintained in this medium for 8 days or more to detect their electrophysiological characteristics.
Immunocytochemical Analyses
Cultures were fixed in 4% paraformaldehyde, washed with PBS, and incubated in a blocking buffer (10% donkey serum and 0.3% Triton X-100 in PBS) for 60 min at room temperature before being incubated in primary antibodies (see Table S2) overnight at 4°C. Secondary antibodies were species-specific Alexa-dye conjugates (Invitrogen) and nuclei were stained with Hoechst 33258 (Ho in figures). Images were collected with a Zeiss LSM710 or Zeiss LSM780 (Carl Zeiss, Germany).
RNA Isolation and qRT-PCR
Total RNA was collected from cultured cells with TRIzol (Life Technologies). cDNA was generated from 500 ng of total RNA with the PrimeScript RT Reagent Kit with gDNA Eraser (Takara), and used as a template for the qPCR. qPCR was performed with the QuantStudio 6 Flex instrument (Applied Biosystems) by SYBR Green detection. The use of primer sequences and details are summarized in Table S3.
Neural Transplantation and Quinolinic Acid Striatal Lesions
All animal experiments were carried out in accordance with the instructions for the Care and Use of Animals in Research published by the Institute of Zoology of the Chinese Academy of Sciences. Transplantation into the neonatal NOD-SCID mice was conducted as described previously (Danjo et al., 2011). The establishment of QA-lesioned mouse model of HD and cell transplantation were performed as described previously (Ma et al., 2012) and as detailed in Supplemental Experimental Procedures.
Electrophysiology
Whole-cell current and clamp recordings of hESC-derived neurons were performed as described in detail in Supplemental Experimental Procedures.
Behavioral Tests
Behavioral tests were conducted before and after transplantation monthly until the animals were sacrificed.
Open Field Test
Mice were placed in the center of activity chambers (Clever Sys; 48 × 48 × 42 cm3). Activities were recorded for 10 min under normal conditions of lighting. Quantitative analysis was done on total distance.
Rotarod Test
An accelerating rotarod (YLS-4C, China) was used to test motor coordination. The mouse was placed on a rotating rod that accelerated from 4 to 40 rotations per minute in a period of 300 s. The period of time the mouse stayed on the rod was monitored and the three of a total of five runs in which the mouse performed best were recorded.
Statistical Analysis
For all experiments, analysis was derived from at least three independent experiments. GraphPad Prism 6 software was used for statistical analysis. In all studies, comparison of mean values was conducted with unpaired t test, one-way ANOVA, or two-way ANOVA. In all analyses, statistical significance was determined at the 0.01%, 0.1%, 1%, or 5% level (p < 0.0001, p < 0.001, p < 0.01, p < 0.05, respectively).
Author Contributions
M.W. contributed to data collection, analysis, and interpretation, and manuscript writing. D.Z. and C.B. established the HD model, and performed cell transplantation and behavior tests. T.M. performed whole-cell current and clamp recordings. W.Z. and L.X. performed qPCR analysis. Z.T. revised the manuscript. B.H. and Y.W. designed and supervised the research and revised the manuscript.
Acknowledgments
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16020604, XDA16030401), Key Research Program of the Chinese Academy of Sciences (ZDRW-ZS-2017-5), Program of Beijing Municipal Science and Technology Commission (Z181100001818002), and the National Key Research and Development Program of China (2016YFA0101402).
Published: August 30, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.08.005.
Contributor Information
Baoyang Hu, Email: byhu@ioz.ac.cn.
Yihui Wu, Email: yihuiwu@ioz.ac.cn.
Supplemental Information
References
- An M.C., Zhang N., Scott G., Montoro D., Wittkop T., Mooney S., Melov S., Ellerby L.M. Genetic correction of Huntington's disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012;11:253–263. doi: 10.1016/j.stem.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber C., Precious S.V., Cambray S., Risner-Janiczek J.R., Kelly C., Noakes Z., Fjodorova M., Heuer A., Ungless M.A., Rodriguez T.A. Activin A directs striatal projection neuron differentiation of human pluripotent stem cells. Development. 2015;142:1375–1386. doi: 10.1242/dev.117093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubry L., Bugi A., Lefort N., Rousseau F., Peschanski M., Perrier A.L. Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc. Natl. Acad. Sci. USA. 2008;105:16707–16712. doi: 10.1073/pnas.0808488105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers J., Gulbranson D.R., George N., Siniscalchi L.I., Jones J., Thomson J.A., Chen G. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012;7:2029–2040. doi: 10.1038/nprot.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. Dorsal-ventral patterning in the mammalian telencephalon. Curr. Opin. Neurobiol. 2003;13:50–56. doi: 10.1016/s0959-4388(03)00009-6. [DOI] [PubMed] [Google Scholar]
- Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Qi Y., Mica Y., Lee G., Zhang X.J., Niu L., Bilsland J., Cao L., Stevens E., Whiting P. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat. Biotechnol. 2012;30:715–720. doi: 10.1038/nbt.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.K., Taipale J., Young K.E., Maiti T., Beachy P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M., Cho J.H., Leung A., Savvidis G., Ahn S., Moon M., Lee P.K., Han J.J., Azimi N., Kim K.S. hPSC-derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell. 2014;15:559–573. doi: 10.1016/j.stem.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danjo T., Eiraku M., Muguruma K., Watanabe K., Kawada M., Yanagawa Y., Rubenstein J.L., Sasai Y. Subregional specification of embryonic stem cell-derived ventral telencephalic tissues by timed and combinatory treatment with extrinsic signals. J. Neurosci. 2011;31:1919–1933. doi: 10.1523/JNEUROSCI.5128-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delli Carri A., Onorati M., Lelos M.J., Castiglioni V., Faedo A., Menon R., Camnasio S., Vuono R., Spaiardi P., Talpo F. Developmentally coordinated extrinsic signals drive human pluripotent stem cell differentiation toward authentic DARPP-32+ medium-sized spiny neurons. Development. 2013;140:301–312. doi: 10.1242/dev.084608. [DOI] [PubMed] [Google Scholar]
- Dovey H.F., John V., Anderson J.P., Chen L.Z., de Saint Andrieu P., Fang L.Y., Freedman S.B., Folmer B., Goldbach E., Holsztynska E.J. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J. Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- Eisenstat D.D., Liu J.K., Mione M., Zhong W., Yu G., Anderson S.A., Ghattas I., Puelles L., Rubenstein J.L.R. DLX-1, DLX-2, and DLX-5 expression define distinct stages of basal forebrain differentiation. J. Comp. Neurol. 1999;414:217–237. doi: 10.1002/(sici)1096-9861(19991115)414:2<217::aid-cne6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Fasano C.A., Chambers S.M., Lee G., Tomishima M.J., Studer L. Efficient derivation of functional floor plate tissue from human embryonic stem cells. Cell Stem Cell. 2010;6:336–347. doi: 10.1016/j.stem.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Floruta C.M., Du R., Kang H., Stein J.L., Weick J.P. Default patterning produces pan-cortical Glutamatergic and CGE/LGE-like GABAergic neurons from human pluripotent stem cells. Stem Cell Rep. 2017;9:1463–1476. doi: 10.1016/j.stemcr.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golas M.M., Sander B. Use of human stem cells in Huntington disease modeling and translational research. Exp. Neurol. 2016;278:76–90. doi: 10.1016/j.expneurol.2016.01.021. [DOI] [PubMed] [Google Scholar]
- Gu Q., Wang J., Wang L., Liu Z.X., Zhu W.W., Tan Y.Q., Han W.F., Wu J., Feng C.J., Fang J.H. Accreditation of biosafe clinical-grade human embryonic stem cells according to Chinese regulations. Stem Cell Rep. 2017;9:366–380. doi: 10.1016/j.stemcr.2017.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HD iPSC Consortium Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012;11:264–278. doi: 10.1016/j.stem.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S.M., Mishina Y.M., Liu S., Cheung A., Stegmeier F., Michaud G.A., Charlat O., Wiellette E., Zhang Y., Wiessner S. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- Jeon I., Lee N., Li J.Y., Park I.H., Park K.S., Moon J., Shim S.H., Choi C., Chang D.J., Kwon J. Neuronal properties, in vivo effects, and pathology of a Huntington's disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30:2054–2062. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- Johnson M.A., Weick J.P., Pearce R.A., Zhang S.C. Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J. Neurosci. 2007;27:3069–3077. doi: 10.1523/JNEUROSCI.4562-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi T., Morizane A., Doi D., Magotani H., Onoe H., Hayashi T., Mizuma H., Takara S., Takahashi R., Inoue H. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson's disease model. Nature. 2017;548:592–596. doi: 10.1038/nature23664. [DOI] [PubMed] [Google Scholar]
- Kim D.S., Lee J.S., Leem J.W., Huh Y.J., Kim J.Y., Kim H.S., Park I.H., Daley G.Q., Hwang D.Y., Kim D.W. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 2010;6:270–281. doi: 10.1007/s12015-010-9138-1. [DOI] [PubMed] [Google Scholar]
- Kirkeby A., Grealish S., Wolf D.A., Nelander J., Wood J., Lundblad M., Lindvall O., Parmar M. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012;1:703–714. doi: 10.1016/j.celrep.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Kirkeby A., Nelander J., Parmar M. Generating regionalized neuronal cells from pluripotency, a step-by-step protocol. Front. Cell. Neurosci. 2012;6:64. doi: 10.3389/fncel.2012.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange H., Thorner G., Hopf A., Schroder K.F. Morphometric studies of the neuropathological changes in choreatic diseases. J. Neurol. Sci. 1976;28:401–425. doi: 10.1016/0022-510x(76)90114-3. [DOI] [PubMed] [Google Scholar]
- Liu J.K., Ghattas I., Liu S., Chen S., Rubenstein J.L.R. Dlx genes encode DNA-binding proteins that are expressed in an overlapping and sequential pattern during basal ganglia differentiation. Dev. Dyn. 1997;210:498–512. doi: 10.1002/(SICI)1097-0177(199712)210:4<498::AID-AJA12>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ma L., Hu B., Liu Y., Vermilyea S.C., Liu H., Gao L., Sun Y., Zhang X., Zhang S.C. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10:455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald M.E., Ambrose C.M., Duyao M.P., Myers R.H., Lin C., Srinidhi L., Barnes G., Taylor S.A., James M., Groot N. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Morizane A., Doi D., Kikuchi T., Nishimura K., Takahashi J. Small-molecule inhibitors of bone morphogenic protein and activin/nodal signals promote highly efficient neural induction from human pluripotent stem cells. J. Neurosci. Res. 2011;89:117–126. doi: 10.1002/jnr.22547. [DOI] [PubMed] [Google Scholar]
- Nekrasov E.D., Vigont V.A., Klyushnikov S.A., Lebedeva O.S., Vassina E.M., Bogomazova A.N., Chestkov I.V., Semashko T.A., Kiseleva E., Suldina L.A. Manifestation of Huntington's disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016;11:27. doi: 10.1186/s13024-016-0092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoleau C., Viegas P., Peschanski M., Perrier A.L. Human pluripotent stem cell therapy for Huntington's disease: technical, immunological, and safety challenges. Neurotherapeutics. 2011;8:562–576. doi: 10.1007/s13311-011-0079-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoleau C., Varela C., Bonnefond C., Maury Y., Bugi A., Aubry L., Viegas P., Bourgois-Rocha F., Peschanski M., Perrier A.L. Embryonic stem cells neural differentiation qualifies the role of Wnt/beta-Catenin signals in human telencephalic specification and regionalization. Stem Cells. 2013;31:1763–1774. doi: 10.1002/stem.1462. [DOI] [PubMed] [Google Scholar]
- Panganiban G., Rubenstein J.L. Developmental functions of the Distal-less/Dlx homeobox genes. Development. 2002;129:4371–4386. doi: 10.1242/dev.129.19.4371. [DOI] [PubMed] [Google Scholar]
- Park I.H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Zhang X.J., Renier N., Wu Z., Atkin T., Sun Z., Ozair M.Z., Tchieu J., Zimmer B., Fattahi F. Combined small-molecule inhibition accelerates the derivation of functional cortical neurons from human pluripotent stem cells. Nat. Biotechnol. 2017;35:154–163. doi: 10.1038/nbt.3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosser A.E., Bachoud-Lévi A.-C. Clinical trials of neural transplantation in Huntington's disease. Prog. Brain Res. 2012;200:345–371. doi: 10.1016/B978-0-444-59575-1.00016-8. [DOI] [PubMed] [Google Scholar]
- Sanberg P.R., Giordano M., Henault M.A., Nash D.R., Ragozzino M.E., Hagenmeyer-Houser S.H. Intraparenchymal striatal transplants required for maintenance of behavioral recovery in an animal model of Huntington's disease. J. Neural Transplant. 1989;1:23–31. doi: 10.1155/NP.1989.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor M.B., Richner M., Olsen H.E., Lee S.W., Monteys A.M., Ma C., Huh C.J., Zhang B., Davidson B.L., Yang X.W. Striatal neurons directly converted from Huntington's disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci. 2018;21:341–352. doi: 10.1038/s41593-018-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Yu P.B., Hong C.C., Sachidanandan C., Babitt J.L., Deng D.Y., Hoyng S.A., Lin H.Y., Bloch K.D., Peterson R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Su P., Li D., Tsang S., Duan E., Wang F. High-efficiency induction of neural conversion in human ESCs and human induced pluripotent stem cells with a single chemical inhibitor of transforming growth factor beta superfamily receptors. Stem Cells. 2010;28:1741–1750. doi: 10.1002/stem.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.