

ABSTRACT
Immune checkpoint blockade is less efficient in patients bearing immunologically ‘cold’ tumors. Oncolytic viruses, which were originally discovered for their ability to preferentially kill malignant cells, can recondition the tumor microenvironment. Supporting this hypothesis, two new studies published in Science Translational Medicine show that adjuvant-like activities of oncolytic viruses make brain and breast tumors ‘hot’ and sensitize them for subsequent immune checkpoint blockade.
KEYWORDS: antibodies, antitumor response, antiviral response, CTLA-4, immunosurveillance, immunotherapy, immune checkpoint, Immunosurveillance, Inflammation and cancer, models of immunostimulation, oncolytic virus, PD-1, PD-L1, therapeutic antibodies
Cancer immunotherapies focus on (re)-educating the immune system of patients against their malignancies. Once appropriately activated, antitumor immune responses culminate in the elimination of cancer cells present in the initial tumor and in distant metastases. Such functionally active antitumor immune responses may eradicate macroscopic lesions and also establish active protection against relapse from micro-metastases, thus holding the key to long-term disease-free survival.1 Comprehensive preclinical and clinical analyses thus far have conclusively established that cancer-bearing hosts with appropriately programmed antitumor immune responses, mediated by the cells of the innate (NK cells, NKT cells, dendritic cells and macrophages) and adaptive (T and B cells) systems, demonstrate favorable outcome from cancers.2 Unfortunately, it is difficult to achieve the induction of fully functional antitumor immunity, since cancers often harbor an immunosuppressive microenvironment. Indeed, beyond passive evasion from immune recognition (‘immunoediting’), active immunosuppression constitutes a common strategy of malignant cells to avoid immunosurveillance and to form progressive cancers.3 As a result, many of the currently pursued immunotherapies aim at correcting immunological defects within the tumor-associated microenvironment (TME) with the objective to remove the brakes on antitumor immunity. Immune suppression within the TME is mediated by multifactorial, often interdependent, mechanisms, and thus requires multipronged immunotherapeutic approaches for its correction. Thus, the future of cancer therapies, including immunotherapies, lies in ‘strategic’ combinations of two or more complementary anticancer interventions.4,5
Of course, combination regimens require careful optimization of the timing of administration of each therapeutic agent so that such compounds accentuate the antitumor benefits of each other. This tenet of combination therapy was recently proven by two simultaneously published articles in Science Translational Medicine, by Samson et al.6 and Bourgeois-Daigneault et al.7 These studies demonstrate that oncolytic viruses (OVs),8 mainly known for their cancer-killing abilities, can be used as an initial priming agent to overcome TME-associated immunosuppression and generate a milieu conducive to favor the efficacy of subsequent checkpoint inhibitor immunotherapies in brain and breast cancers. These findings emphasize the importance of time-dependent repercussions of the combinatorial partners in promoting the therapeutic utility of combination immunotherapies. Most importantly, they support the emerging hypothesis that adjuvant-like properties of OVs, imbedded within the antiviral immunological events driven by its therapeutic administration, can be exploited to enhance the efficacy of cancer immunotherapies.9,10
Checkpoint molecules, which are expressed on cancer cells, antigen-presenting cells (APC, e.g., PD-L1, PD-L2, VISTA) or on lymphocytes (e.g., PD-1, CTLA-4), represent one of the major mechanisms through which cancers enforce immunosuppression (Fig. 1). When PD-1-expressing T and NK cells interact with PD-L1 expressed on cancer cells or APC, they become functionally impaired. Similarly, CTLA-4-expressing CD8+ T cells often display immunological tolerance towards tumors, and CTLA-4+ T regulatory (Tregs) contribute towards the TME-associated immunosuppression by inhibiting the functions of other immune cells. Such checkpoint molecule-mediated suppression of functionally active antitumor immunity facilitates the persistence of cancers. Additionally, checkpoint molecules directly promote the process of tumorigenesis. Thus, therapies that target checkpoint molecules promise to promote antitumor immunity and impair tumorigenesis. In the context of cancers, PD-1/PD-L1 and CTLA-4 checkpoints remain the most studied, and thus are right now the major therapeutic targets in the immuno-oncological pipeline.
Figure 1.
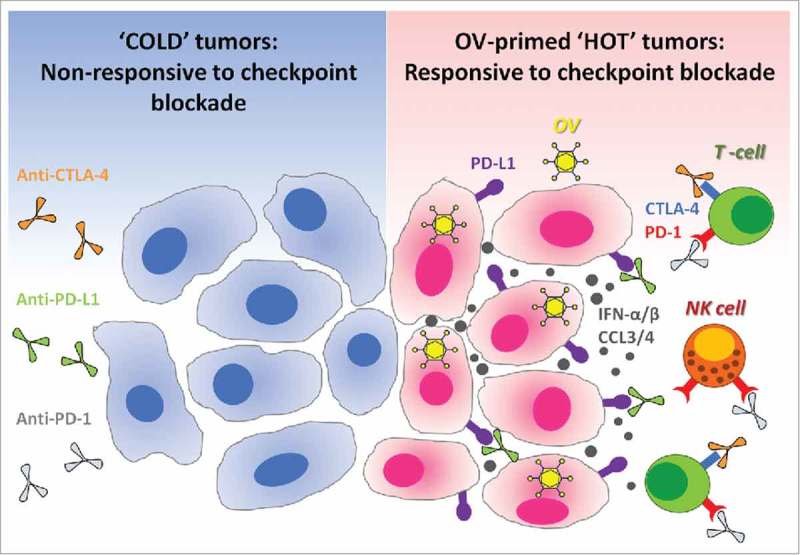
Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade cancer immunotherapies. Immune checkpoint blockade is inefficient in ‘cold’ tumors, which are poorly infiltrated by immune cells and also have low expression of PD-L1 on their surface. In the absence of available targets, immune checkpoint blockers like anti-PD-L1 (targeting PD-L1 expressed at the surface of cancer cells or on antigen-presenting cells), alone or in combination with anti-CTLA-4, remain therapeutically inefficient (left panel). Therapeutic administration of oncolytic viruses (OV) into tumors promotes strong antiviral immune response accompanied by the production of cytokines such as type-1 interferons and chemokines.17,26-28 Type-1 interferons promote the expression of PD-L1 on the surface of cancer cells, while chemokines like CCL3 and CCL4 attract immune cells which often express PD-1 or CTLA-4.29-32 Thus, antiviral immunological events inflame the tumor and make it ‘hot’. When checkpoint inhibitors are administered subsequently, they can bind to their respective targets on either cancer or immune cells. As a final result, oncolytic viruses sensitize tumors to the therapeutic effects of immune checkpoint inhibitors (right panel). OV: Oncolytic virus; NK cell: Natural killer cell; PD-1: Programmed death-1; PD-L1: Programmed death ligand-1; CTLA-4: Cytotoxic T-lymphocyte-associated protein 4; IFN-α/β: Interferon-alpha or beta; CCL3/4: Chemokine (C-C motif) ligand 3 or 4.
Therapeutic antibodies against CTLA-4, PD-1 or PD-L1 target the immunosuppressive interaction among checkpoint molecules on malignant and immune cells. Therapeutic checkpoint blockade hence promotes the clinically desired antitumor functionalities within immune cells. Overwhelming evidence thus far has documented the ability of checkpoint inhibitors, alone or in combination with complimentary immunotherapeutic interventions, to re-instate beneficial antitumor immunity and to improve the prognosis of patients bearing cancers of diverse origins and types. Not surprisingly, checkpoint blockade therapies represent one of the most promising cancer management modalities of the 21st century, and have reinvigorated our fight against cancers in clinics. That said, only 20% of cancer patients respond to immune checkpoint blockers targeting the PD-1/PD-L1 interaction. The efficacy of checkpoint blockade therapy is at least in part dictated by the immune landscapes within the TME.11 Tumors with higher density of infiltrating immune cells, termed as ‘hot’ tumors, are more responsive to checkpoint blockade therapies than ‘cold’ tumors with a scarce or null local immune infiltrate. Similarly, tumors that express PD-L1 on their surface, are more susceptible to anti-PD-1/PD-L1 antibody-based therapies. Thus, therapeutic modalities that can make tumors ‘hot’ or induce intratumoral PD-L1 expression are considered to be the ideal combinatorial partners for checkpoint blockade therapies.
One novel anticancer therapy consists in the administration of OVs, the first of which, Talimogene laherparepvec (commonly called ‘T-VEC’), was recently approved by the Food and Drug Administration (FDA) for the treatment of melanoma.12 OVs, primarily known for their cancer killing abilities (through a mechanism known as oncolysis), are now being acknowledged for their capacity to re-educate the host's immune system to induce clinically relevant antitumor immunities. Recent work from our groups, as well as from other laboratories, reveals that the antiviral innate responses induced following the administration of OVs to cancer-bearing hosts overturn multiple TME-associated immune evasion mechanisms and promote strong, multiclonal and protective antitumor immunity. Recently, we proposed that the antiviral immunological events induced following the administration of OVs turn tumor ‘hot’,9 and establish a TME that is conducive for enhancing the efficacy of checkpoint inhibitors.10 In congruence, the articles by Samson et al. and Bourgeois-Daigneault et al. provide the experimental proof that OVs may function as ‘neoadjuvants’ to prime the TME for checkpoint blockade.
For the treatment of brain tumors, OVs have been traditionally administered through intracranial routes, as it was believed that the delivery of OVs via any other route would not result in their proper delivery. However, as shown by Samson et al., oncolytic reovirus could successfully reach brain tissues in patients following intravenous administration.6 Most importantly, such systemically delivered reovirus promoted intratumoral accumulation of CD8+ T cells in eight out of nine patients, wherein CD3+ T cells were observed in and around blood vessels within tumors. In addition, higher expression of PD-L1 on high grade gliomas as well as on a brain-metastasized melanoma was observed in reovirus-treated patients, and it is reasonable to believe that PD-L1 was induced as a consequence of OV-induced type-1 interferons. When tested in a preclinical mouse model, mice implanted with GL261 tumors and primed with intravenous GM-CSF/reovirus, demonstrated significantly longer survival following subsequent treatment with anti-PD-L1 antibody, as compared to either treatment alone.6
Similarly, using patient-derived xenografts as well as preclinical models of triple-negative breast cancer (TNBC), Bourgeois-Daigneault et al. showed that the oncolytic rhabdovirus Maraba could be used to sensitize this otherwise refractory cancer to treatment with anti-PD-L1 and anti-CTLA-4 antibodies.7 Once again, these authors focused on addressing the issue of therapeutic timings around OV and checkpoint inhibitor administration. They found that administration of Maraba virus before breast cancer surgery created a ‘window of opportunity’ for the administration of checkpoint inhibitors immediately post-surgery. Using three different murine breast cancer models on two different genetic backgrounds (4T1 and EMT6 in BALB/c mice and E0771 in the C57 BL/6 strain), they showed that Maraba virus kills local TNBC cells, minimizes metastatic burden in lungs, promotes inflammation within TME, and induces tumor-specific antitumor immunity. Most importantly, Maraba also promoted the intratumoral infiltration of immune cells making the TNBC ‘hot’ and induced expression of PD-L1 on TNBC cells, eventually leading to the enhanced efficacy of anti-PD-L1 and anti-CTLA-4 antibody-based cancer immunotherapies when administered post-surgery. In line with our recently proposed hypothesis around the antitumor benefits of antiviral immunity elicited by OVs, this report suggests that OVs may be used to condition the TME and to sensitize it to the subsequent administration of immune checkpoint blockers.
The aforementioned preclinical studies echo the recent findings by Ribas et al.,13 who reported a 33% complete response rate amongst 21 patients with advanced melanoma, treated with T-VEC followed by anti-PD-1 antibody, in a phase 1b clinical trial. Together, these studies are forming a growing body of evidence that advocates for the use of OVs as adjuvants and priming agents. Of note, other mechanistically similar strategies using adjuvant-like immunostimulators (such as STING pathway agonists) have been reported to sensitize tumors to subsequent immune checkpoint blockade, further strengthening the argument for such a combinatorial approach.14,15 Similarly, it has been shown that immunogenic cell death (ICD) inducers such as oxaliplatin and cyclophosphamide sensitize KRAS-induced lung cancers to subsequent immune checkpoint blockade.16 It is not unreasonable to postulate that viruses, in particular T-VEC, can induce ICD because T-VEC has been genetically modified to remove genes that suppress the immune response and accentuate the premortem stress responses, including the type-1 interferon response, that characterize ICD.17-22 These emerging studies emphasize the need to rationally design for combination immunotherapies with respect to timing and dosing. Thus, preclinical experiments should compare various time-dependent dosing regimens of OVs and checkpoint inhibitors in a systematic fashion to streamline subsequent optimization of the combination regimen in clinical trials.
From the patient perspective, future preclinical and clinical exploration of OVs should consider the adverse effects of checkpoint inhibitors.23 By design, checkpoint inhibitors remove the inhibitory brakes that control effector immune cells. Unfortunately, this functional modulation applies to all immune cells, including those that are autoreactive. Thus far, almost all checkpoint inhibitors have found to prompt some degree of autoimmune side effects. These considerations are even more important in the context of OVs and checkpoint inhibitor combinations, as OVs themselves are criticized for their potential to evoke autoimmune responses.24 Thus, any potential OV-induced self-reactivities,25 promoted during the priming phase of this combination therapy, could be exacerbated by the subsequent administration of checkpoint inhibitors, further augmenting the risk of adverse events.
In summary, the combination of OVs with immune checkpoint inhibitors may result in therapeutic synergy. We hypothesize that a deeper understanding of the immunobiology of this combination will set the ground for further optimization of this therapeutic combination. As compared to other agents that are currently combined with checkpoint inhibitors, OVs provoke a particular pattern of changes in the TME. As each type of OV itself gets recognized by the host immune system, it first ignites an antiviral immune response to counter OV infection, replication and spread. In this sense, antiviral immunity has been considered to negatively affect OV therapy, as it ultimately purges the organism from OVs before the viruses can mediate complete oncolysis. Nonetheless, in reality this antiviral response facilitates the subsequent recognition of tumor antigens and the initiation or reactivation of tumor-specific T cell response. Importantly, some of the antiviral response elements, such as NK cells and virus-specific T cells, also contribute towards tumor destruction in their attempt to eradicate virally infected cells, knowing that malignant cells are preferentially infected by OVs. Currently however, the exact contribution of OV-induced antiviral and antitumor immunities towards antitumor therapeutic benefits remains unknown. As checkpoint inhibitors can affect the functionalities of these two immunities, the detailed understanding of these two OV-induced immune responses could hold the key to unlock the untapped translational potential of OV+checkpoint inhibitor combination therapies. Notwithstanding the biotherapeutic complexities to be unraveled in the future, strategically implemented sequential use of OV and checkpoint inhibitors are set to provide urgently needed novel therapeutic options for cancer patients.
Funding Statement
SG is supported by grants from the Canadian Institutes of Health Research (CIHR) and Beatrice Hunter Cancer Research Institute (BHCRI). GK's team is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancellerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; the European Commission (ArtForce); European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
References
- 1.Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell 2016;164:1233–47. doi: 10.1016/j.cell.2016.01.049. PMID:26967289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fridman WH, Zitvogel L, Sautes-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14:717–34. doi: 10.1038/nrclinonc.2017.101. PMID:28741618. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011;331:1565–70. doi: 10.1126/science.1203486. PMID:21436444. [DOI] [PubMed] [Google Scholar]
- 4.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 2015;161:205–14. doi: 10.1016/j.cell.2015.03.030. PMID:25860605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56–61. doi: 10.1126/science.aaa8172. PMID:25838373. [DOI] [PubMed] [Google Scholar]
- 6.Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, Thomson S, Corns R, Mathew RK, Fuller MJ, et al.. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10(422):pii:eaam7577. doi: 10.1126/scitranslmed.aam7577. PMID:29298869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bourgeois-Daigneault MC, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, Falls T3, St-Germain LE, Pelin A, Lichty BD, et al.. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10(422):pii:eaao1641. doi: 10.1126/scitranslmed.aao1641. PMID:29298865.27057469 [DOI] [PubMed] [Google Scholar]
- 8.Pol J, Buque A, Aranda F, Bloy N, Cremer I, Eggermont A, Erbs P, Fucikova J, Galon J, Limacher JM, et al.. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology 2016;5:e1117740. doi: 10.1080/2162402X.2015.1117740. PMID:27057469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gujar S, Pol JG, Kim Y, Lee PW, Kroemer G. Antitumor Benefits of Antiviral Immunity: an underappreciated aspect of oncolytic virotherapies. Trends Immunol. 2018;39(3):209–221. doi: 10.1016/j.it.2017.11.006. PMID:29275092. [DOI] [PubMed] [Google Scholar]
- 10.Lee PWK, Gujar S. Potentiating prostate cancer immunotherapy with oncolytic viruses. Nat Rev Urol. 2018. doi: 10.1038/nrurol.2018.10. PMID:29434366. [DOI] [PubMed] [Google Scholar]
- 11.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. doi: 10.1016/j.cell.2017.01.017. PMID:28187290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016;5:e1115641. doi: 10.1080/2162402X.2015.1115641. PMID:26942095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al.. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017;170:1109–19e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al.. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7:283ra52. doi: 10.1126/scitranslmed.aaa4306. PMID:25877890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gadkaree SK, Fu J, Sen R, Korrer MJ, Allen C, Kim YJ. Induction of tumor regression by intratumoral STING agonists combined with anti-programmed death-L1 blocking antibody in a preclinical squamous cell carcinoma model. Head Neck. 2017;39:1086–94. doi: 10.1002/hed.24704. PMID:28323387. [DOI] [PubMed] [Google Scholar]
- 16.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al.. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade Therapy. Immunity 2016;44:343–54. doi: 10.1016/j.immuni.2015.11.024. PMID:26872698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. PMID:27748397. [DOI] [PubMed] [Google Scholar]
- 18.Musella M, Manic G, De Maria R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017;6:e1314424. doi: 10.1080/2162402X.2017.1314424. PMID:28638743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for talimogene laherparepvec, a New Oncolytic virus immunotherapy. Clin Cancer Res. 2016;22:1048–54. doi: 10.1158/1078-0432.CCR-15-2667. PMID:26719429. [DOI] [PubMed] [Google Scholar]
- 20.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405–14. doi: 10.1038/nri3845. PMID:26027717. [DOI] [PubMed] [Google Scholar]
- 21.Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014;22:251–6. doi: 10.1038/mt.2013.220. PMID:24048442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Workenhe ST, Mossman KL. Rewiring cancer cell death to enhance oncolytic viro-immunotherapy. Oncoimmunology 2013;2:e27138. doi: 10.4161/onci.27138. PMID:24498567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, et al.. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–48. doi: 10.1016/j.ejca.2015.11.016. PMID:26765102. [DOI] [PubMed] [Google Scholar]
- 24.Bridle BW, Hanson S, Lichty BD. Combining oncolytic virotherapy and tumour vaccination. Cytokine Growth Factor Rev. 2010;21:143–8. doi: 10.1016/j.cytogfr.2010.02.009. PMID:20226716. [DOI] [PubMed] [Google Scholar]
- 25.Bridle BW, Li J, Jiang S, Chang R, Lichty BD, Bramson JL, Wan Y. Immunotherapy can reject intracranial tumor cells without damaging the brain despite sharing the target antigen. J Immunol. 2010;184:4269–75. doi: 10.4049/jimmunol.0901447. PMID:20237288. [DOI] [PubMed] [Google Scholar]
- 26.Chen A, Zhang Y, Meng G, Jiang D, Zhang H, Zheng M, Xia M, Jiang A, Wu J, Beltinger C, et al.. Oncolytic measles virus enhances antitumour responses of adoptive CD8(+)NKG2D(+) cells in hepatocellular carcinoma treatment. Sci Rep. 2017;7:5170. doi: 10.1038/s41598-017-05500-z. PMID:28701757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu X, Rivera A, Tao L, Zhang X. An HSV-2 based oncolytic virus can function as an attractant to guide migration of adoptively transferred T cells to tumor sites. Oncotarget 2015;6:902–14. doi: 10.18632/oncotarget.2817. PMID:25460506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleijn A, Kloezeman J, Treffers-Westerlaken E, Fulci G, Leenstra S, Dirven C, Debets R, Lamfers M. The therapeutic efficacy of the oncolytic virus Delta24-RGD in a murine glioma model depends primarily on antitumor immunity. Oncoimmunology 2014;3:e955697. doi: 10.4161/21624011.2014.955697. PMID:25941622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen F, Bobanga ID, Rauhe P, Barkauskas D, Teich N, Tong C, Myers J, Huang AY. CCL3 augments tumor rejection and enhances CD8(+) T cell infiltration through NK and CD103(+) dendritic cell recruitment via IFNgamma. Oncoimmunology 2018;7:e1393598. doi: 10.1080/2162402X.2017.1393598. PMID:29399390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer. 2017;117:1–7. doi: 10.1038/bjc.2017.136. PMID:28524159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He J, Hu Y, Hu M, Li B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci Rep. 2015;5:13110. doi: 10.1038/srep13110. PMID:26279307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, Honjo T. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–9. doi: 10.4049/jimmunol.1003208. PMID:21263073. [DOI] [PubMed] [Google Scholar]