

ABSTRACT
The signal adapter MyD88, an essential component of Toll-like receptor (TLR) signaling, is important for gut-microbiome interactions. However, its contribution to cancer and its cell-type specific functions are controversially discussed. Therefore, we generated new tissue-specific mouse models and analyzed the clinical importance in human colorectal cancer. A gene-trap was inserted into the murine Myd88 gene (Myd88LSL), yielding MyD88-deficient background with Cre-mediated re-expression in myeloid (MYEL) or intestinal epithelial cells (IECs). These lines were bred with the Apc1638N model that develops invasive adenocarcinoma and analyzed at 12 months. Further, two patient collectives of colorectal cancer (n = 61, and n = 633) were analyzed for expression of Myd88 and TLRs. MyD88 expression was significantly increased in carcinomas, and increased intratumoral levels of MyD88 and TLR pathway components were associated with significantly shorter disease-free (P = .011), and overall survival (P < .0001). In accordance, fully MyD88-deficient mice showed highly significantly decreased tumor incidence, tumor numbers, increased survival, and, importantly, fully lacked malignant lesions. Thus, MyD88 is essential for tumorigenesis and especially progression to malignancy. Tissue-specific re-expression of MyD88 highly significantly increased tumor initiation by differing mechanisms. In intestinal epithelia, MyD88 enhanced epithelial turnover, whereas in myeloid cells, it led to increased production of tumor- and stemness-enhancing cytokines, significantly associated with altered expression of adaptive immune genes. However, neither re-expression of MyD88 in IECs or myeloid cells was sufficient for malignant progression to carcinoma. Thus, MyD88 crucially contributes to colorectal cancer initiation and progression with non-redundant and cell-type specific functions, constituting an attractive therapeutic target.
KEYWORDS: APC, colorectal cancer, mouse model, Toll-like receptors, Tumor immunology
Introduction
Chronic inflammation is a pivotal risk factor for the development of solid tumors, such as colorectal cancer,1 and the pro-inflammatory NF-κB (nuclear factor κB) signaling pathway is a key promoter of inflammation-associated carcinogenesis.2,3 In mouse models for inflammation-induced colorectal carcinogenesis, inhibition of the NF-κB pathway lead to decreased lesion formation.2 Moreover, also sporadic colorectal carcinomas often feature a pro-inflammatory microenvironment, even in the absence of inflammatory bowel disease. Among the upstream signaling pathways that induce NF-κB and promote carcinogenesis feature prominently the Toll-like receptors (TLRs).4 Polymorphisms in TLR genes are associated with solid tumors,5,6 and increased expression of TLR4 and the intracellular TLR adaptor myeloid differentiation response gene 88 (MyD88) is associated with poor prognosis in colorectal cancer.7 TLRs represent a highly conserved group of receptors triggering rapid initiation of inflammatory immune responses.8 Located at the plasma membrane and in endosomes, TLRs bind to microbial pathogen-associated molecular patterns (PAMPs) as well as host-derived danger-associated molecular patterns (DAMPs).9 Therefore, TLR/MyD88-signaling constitutes a major functional hub between the gut and the microbiome. TLRs as well as interleukin-1 family receptors recruit the intracellular adaptor proteins MyD88 and TRIF, respectively, and induce distinct signaling pathways. The MyD88-dependent pathway activates the NF-κB- and MAPK-pathways, inducing the production of pro-inflammatory cytokines.10 Moreover, NF-κB protects intestinal epithelial cells from apoptosis in a cell-autonomous fashion.2 Since MyD88 is a central protein linking TLRs and potentially oncogenic signaling pathways, many previous studies analyzed its role in tumorigenesis. MyD88-dependent signaling was found to prevent lesion formation upon chemical induced inflammation and carcinogenesis (AOM/DSS treatment).11 Moreover, MyD88 has been described to dampen the development of Helicobacter-induced gastric malignancies.12 However, global Myd88-deficiency in the genetic mouse model ApcMin/+ strongly reduced benign polyp formation,13 and the tumor promoting role of MyD88 was further attributed to activation of the kinases ERK1/2 (extracellular signal–regulated kinase 1 and 2) in intestinal epithelia.14 However, a more detailed analysis of the signaling pathway is crucial, as Myd88 is known to be expressed in several cell types within the intestine.15 Therefore, our goal was to determine the cell-type specific role of MyD88 in vivo. Moreover, the preclinical models used here not only recapitulates the early steps of colorectal carcinogenesis, i.e., aberrant crypts and benign adenoma, but rather allows the analysis of the contribution of MyD88 to the clinically important adenoma-carcinoma transition without chemical-induced chronic inflammation. MyD88 expression was crucially required for progression to malignancy, with non-redundant, tumor-enhancing functions both in intestinal epithelia and in myeloid cells. Further, we found an association of MyD88 signaling with expression of intratumoral T-cell markers and epithelial-mesenchymal transition, hitherto not reported for intestinal cancer.
Results
MyD88/TLR-signaling components are overexpressed and associated with poor prognosis in human colorectal cancer
Expression of MyD88 and TLR2 was significantly upregulated in human colorectal carcinoma (CRC) on mRNA level (n = 51), compared to normal colon (n = 25, Fig. 1A). In good accordance, intratumoral expression of Tlr2 and Tlr4 was increased in the Apc1638N/+ cancer mouse model (Fig. 1A). Prognostic association was assessed with the “The Cancer Genome Atlas (TCGA)” data set in 629 CRC patients. The analysis comprised the signal adaptors MYD88 and TRAF6, TLR4-coreceptors CD14 and LY96, and MyD88-mediated TLRs expressed in the large intestine (TLR1, TLR2, TLR4, TLR5, TLR6, TLR7, TLR8, and TLR9). Increased expression was found in 228 (37%) of all samples, most frequently upregulation of CD14 (n = 104, 16%), or MYD88 (n = 80, 13%)(not shown). Upregulation was highly significantly associated with poor overall survival in Kaplan-Meier survival analysis (log-rank test: P = .0009), and with decreased disease-free survival (log-rank test: P = .0013, Fig. 1B).
Figure 1.
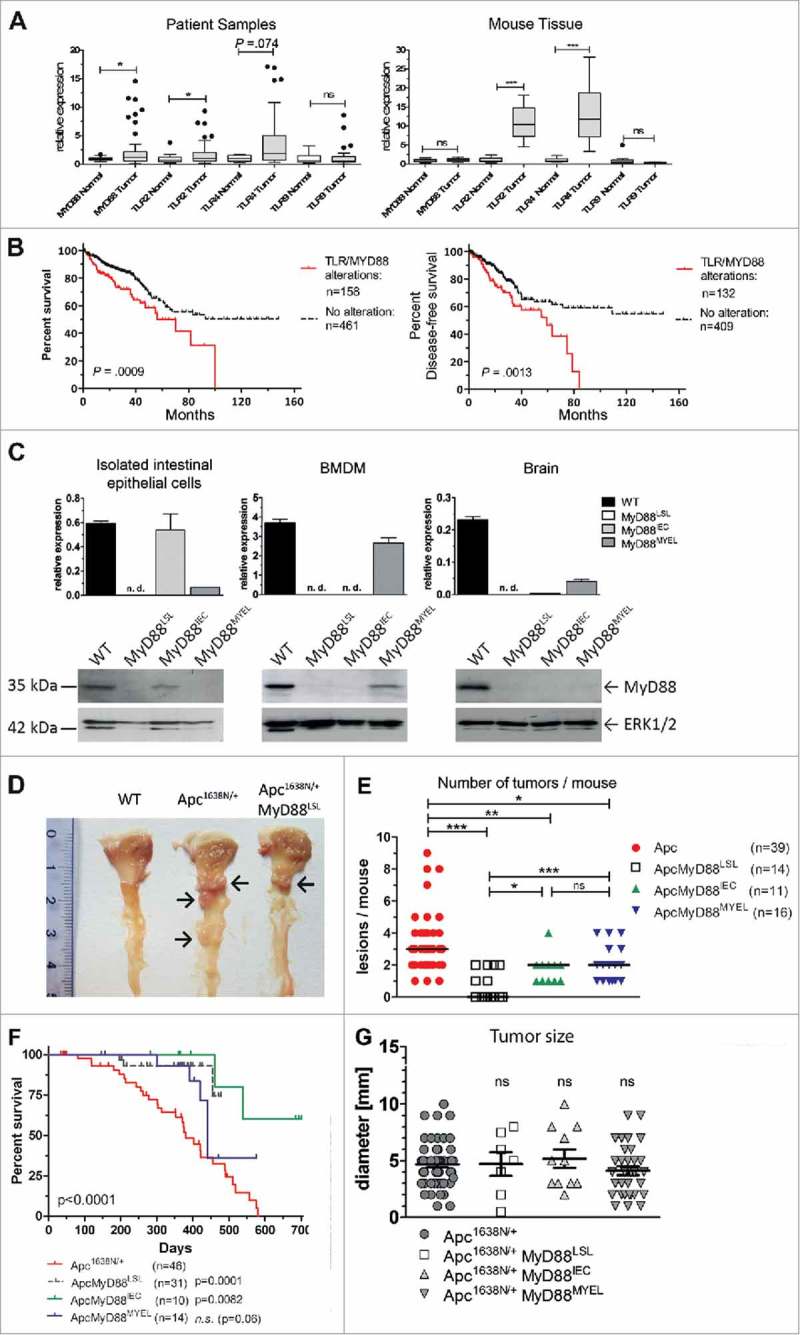
Myd88/TLR signaling is frequently overexpressed in colorectal cancer and associated with prognosis. A, TLR signaling components are upregulated in human (left) and murine (right) intestinal tumors compared to normal mucosa, as verified by qRT-PCR for Myd88, TLR2, TLR4 and TLR9. Human colorectal cancer (n = 51 patients) shows significant upregulation of Myd88 and TLR2 transcripts. Right panel: colon cancer model Apc1638N (n = 15 mice per group) shows a highly significant intratumoral upregulation of TLR2 and TLR4. *P < .05; ***P < .01; ns: not significant. B, Alterations in the TLR pathway are highly significantly associated with poor overall survival (log-rank test: P = .0009), as well as with poor disease-free survival (log-rank test: P = .0013). (C-H) Genetic “switch on” mouse models demonstrate that intestinal carcinogenesis depends on MyD88 expression in both epithelial and myeloid cells. C, Tissue-specific re-expression of Myd88 in intestinal epithelial cells (IEC) was achieved in MyD88IEC mice, or in bone marrow derived macrophages in the MyD88MYEL strain. Expression was analyzed on mRNA level by qRT-PCR (n = 4 mice/group; top panel). No expression was detected in control tissue (brain). Bottom panel: representative example for successful and tissue-specific “switch on” of MyD88 expression on protein level (immunoblot). Loading control: total ERK1/2. D, Macroscopic analysis of representative tissue samples from mice at 12 months of age: wildtype control is tumor-free, Apc1638N/+-model shows several tumors in proximal duodenum (arrows), Apc1638N/+ MyD88LSL mice have strongly reduced tumor formation (arrow). E, Median tumor numbers per animal. Compared to parental line (Apc1638N/+), tumors per animal are significantly reduced in MyD88-deficient mice (Apc1638N/+ MyD88LSL; P = .00018), as well as in mice with re-expression in IECs (Apc1638N/+ MyD88IEC; P = .0123), or in myeloid cells (Apc1638N/+ MyD88LSL; P = .0245). Re-expression of MyD88 in IECs, as well as in myeloid cells is sufficient for a significant, but partial restoration of the tumor phenotype, (Apc1638N/+ MyD88IEC: P = .0256; Apc1638N/+ MyD88MYEL: P = .0037). F, Kaplan-Meier survival analysis show significantly enhanced tumor-specific survival for mice with global MyD88-deficiency, or re-expression of MyD88 in intestinal epithelia, as compared to the parental Apc1638N/+ strain. G, No differences in tumor size were observed. Macroscopically visible lesions were measured along the largest diameter.
MyD88 is required for tumor initiation and tumor progression in vivo
To assess the contribution of MyD88 in vivo, we generated a new “switch-on” mouse model that allows cell-type specific expression of Myd88, by insertion of a loxp-flanked transcriptional termination element (“LSL”) between exon 1 and 2 of the Myd88 gene.16 Crossing Myd88LSL/LSL mice with pvillin-Cre17 mice and LysM-Cre mice18 lead to excision of the intron-gene-trap and therefore re-expression of Myd88 on mRNA and protein level (Fig. 1C) in intestinal epithelial (Myd88IEC) or myeloid cells (bone marrow derived macrophages, Myd88MYEL; for a schematic representation of the mouse strains refer to Supplementary Fig. 1). Control tissue (e.g., brain) showed essentially no re-expression of Myd88 (Fig. 1C). Interbreeding of Myd88LSL/LSL, Myd88IEC and Myd88MYEL animals with the genetic model Apc1638N/+ for colorectal cancer19 enabled the analysis of MyD88-mediated signaling during tumor formation. At 12 months of age, all examined Apc1638N/+ mice developed intestinal tumors with an average of 3.4 ± 1.9 tumors per animal, whereas wildtype controls remained tumor-free. MyD88-deficient mice had a striking and highly significant reduction in tumorigenesis (Fig. 1D and E). The number of lesions per animal was highly significantly reduced compared to parental Apc1638N/+ mice (Fig. 1E), as well as tumor incidence, with 57% of MyD88-deficient mice protected from tumor formation (P = .0001; Table 1). Survival was highly significantly increased in MyD88-deficient mice compared to MyD88-proficient littermates (P = .0005, Fig. 1F). This was accompanied by decreased tumor-induced morbidity and splenomegaly (Supplementary Fig. 2B). Next, we dissected the tissue-specific contributions of MyD88 in epithelial versus myeloid cells. Importantly, Myd88 re-expression in intestinal epithelia, as well as in myeloid cells, resulted in tumor incidence of 100%, highly significantly increased over globally MyD88-deficient mice (Table 1). In accordance, the mean number of lesions was significantly increased compared to global MyD88-deficiency, but still significantly reduced compared to Apc1638N/+ animals (Fig. 1E). No significant difference in tumor number was seen between Apc1638N/+ MyD88IEC and Apc1638N/+ MyD88MYEL models (Fig. 1E). Survival was increased over the parental Apc1638N/+-model, even though the Apc1638N/+ MyD88MYEL line did not attain significantly better survival (Fig. 1F). Anatomical tumor distribution along the intestinal tract did not change compared to parental mice (Supplementary Table 1, Supplementary Fig. 2). To distinguish between tumor initiation and progression, macroscopically visible lesions were measured along the largest diameter. No significant differences in tumor size were observed between the four models, indicating negligible effects on tumor growth (Fig. 1G). Upon blinded histopathological analysis of tumors, major differences in tumor malignancy were observed. In accordance with earlier findings, Apc1638N/+ tumors were classified as low to high grade neoplasia and as invasive carcinoma, frequently featuring de-differentiated areas with dysplastic epithelial cells and prominent nuclear atypia (Fig. 2A). In contrast, lesions from the Apc1638N/+ MyD88LSL model had benign hyperplastic features and were mainly staged as adenomas, rarely as low grade intraepithelial neoplasia (Table 2, Fig. 2B). Both “switch-on” animal models (Apc1638N/+ MyD88IEC and Apc1638N/+ MyD88MYEL) presented tumors of low-grade intraepithelial neoplastic type, and in one case, a high-grade intraepithelial neoplasm was observed upon re-expression of MyD88 in IECs. However, carcinoma in situ or invasive carcinoma was never observed in the “switch-on” models (Fig. 2B, Table 2).
Table 1.
Tumor incidence and morbidity in the different mouse strains.
| Genotype | Mice (N) | Spleen weight (Mean ± SD) | Tumor Multiplicity (Mean ± SD) | Tumor Incidence | Incidence vs. Apc1638N/+ | Incidence vs. Apc1638N/+ MyD88LSL |
|---|---|---|---|---|---|---|
| Apc1638N/+ | 39 | 417 ± 294 mg | 3.4 ± 1.9 | 39 / 39 (100%) | – | P < .0001 |
| Apc1638N/+ MyD88LSL | 14 | 128 ± 83 mg | 0.7 ± 0.9 | 6 / 14 (43%) | P < .0001 | – |
| Apc1638N/+ MyD88IEC | 11 | 197 ± 114 mg | 1.7 ± 0,9 | 11 / 11 (100%) | n.s. | P = .0029 |
| Apc1638N/+ MyD88MYEL | 16 | 172 ± 133 mg | 2.1 ± 1.1 | 16 / 16 (100%) | n.s. | P = .0005 |
Tumor incidence was compared with two-sided Fisher's exact t test, data were compared to parental Apc1638N/+ mice, and Apc1638N/+ MyD88LSL animals, respectively.
Figure 2.
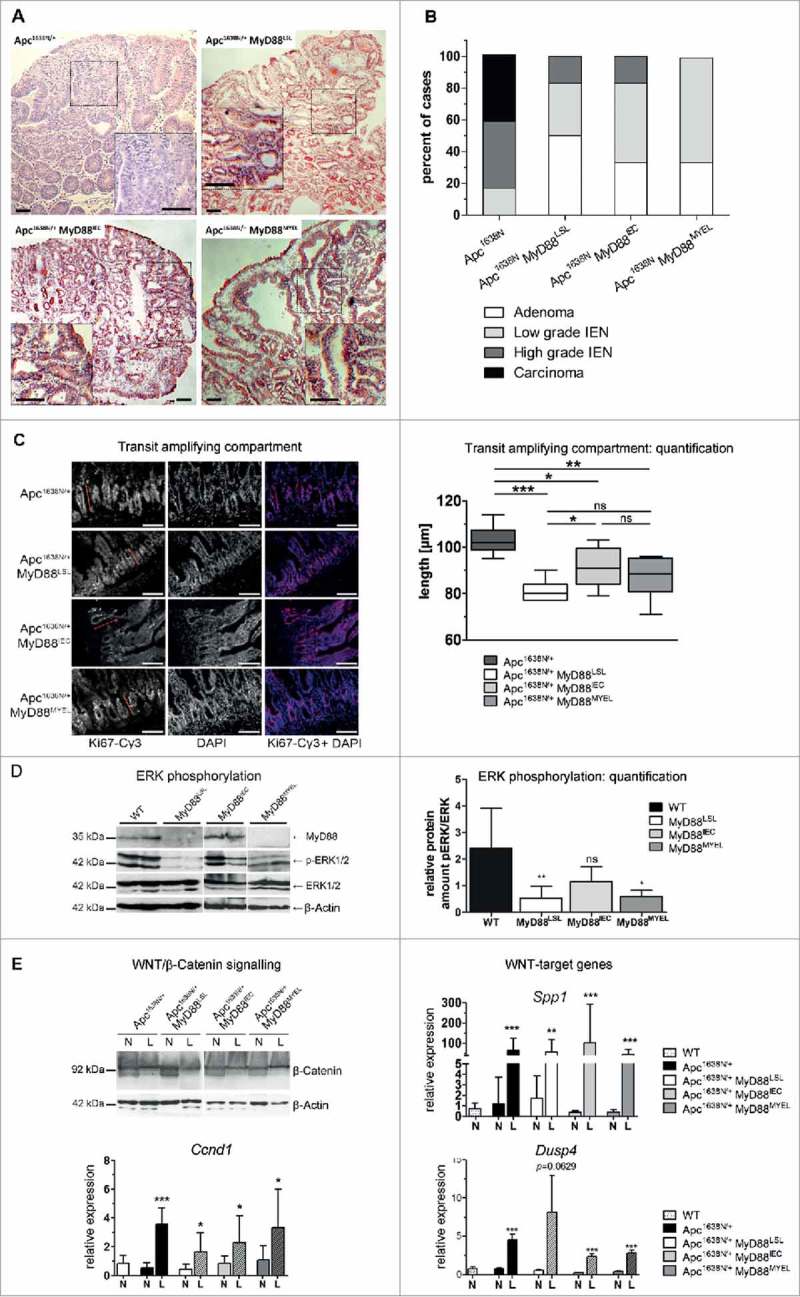
MyD88 is required for progression to malignancy and turnover of the intestinal epithelium. A, Typical histology of lesions from the different mouse strains, HE staining of tissue sections. Carcinoma in situ (Apc1638N/+), low to intermediate benign dysplasia (Apc1638N/+ MyD88LSL, Apc1638N/+ MyD88IEC, and Apc1638N/+ MyD88MYEL). Size bars: 100 µm. B, Tumor grading as performed by an experienced pathologist. Lesions were staged as adenomas, low to intermediate intraepithelial neoplasia, as well as invasive carcinoma. C, Extent of the transit amplifying compartment (TA) in jejunum, marked by Ki67-staining, depends on epithelial MyD88-expression. Nuclear counterstaining by DAPI, size bars: 40 µm. Quantification of TA according to published protocols showed a highly significant decrease in MyD88-deficient mice, rescued by re-expression in IECs (n≥5 per group). D, Immunoblot analysis of small intestinal tissue lysates from all four animal models revealed a-dependency of phospho-ERK1/2 levels from MyD88 expression in IECs (tissue from n = 6 mice analysed per group, shown here are two representative mice per genotype). The density of immunoblot signals was measured and is shown in relation to total ERK1/2 protein level (n = 6 mice). The relative amount of phospho-ERK1/2 was significantly reduced in Apc1638N/+ MyD88LSL (P = .0058), this decrease in pERK was rescued upon re-expression of MyD88 in IECs (not significantly differing from MyD88-proficient mice), but not by re-expression in myeloid cells in the line Apc1638N/+ MyD88MYEL (P = .0487), compared to the parental Apc1638N/+ strain. E, MyD88 deficiency does not alter aberrant activation of the canonical Wnt-pathway in lesions (L) compared to normal tissue (N), verified by detection of β-catenin in tumor lysates by immunoblot analysis, and by expression of the surrogate markers osteopontin (Spp1), CyclinD1 (Ccnd1) and dual specificity phosphatase 4 (Dusp4).
Table 2.
Tumor malignancy.
| Genotype | Scored Lesions (n) | Adenoma | Low-grade IEN | High-grade IEN | Invasive Carcinoma |
|---|---|---|---|---|---|
| Apc1638N/+ | 12 | 0 / 12 | 2 / 12 | 5 / 12 | 5 /12 |
| 0% | 17% | 42% | 42% | ||
| Apc1638N/+ | 6 | 3 / 6 | 2 / 6 | 1 / 6 | 0 /6 |
| MyD88LSL | 50% | 33% | 17% | 0% | |
| Apc1638N/+ | 6 | 2 / 6 | 3 / 6 | 1 / 6 | 0 /6 |
| MyD88IEC | 33% | 50% | 17% | 0% | |
| Apc1638N/+ | 6 | 2 / 6 | 4 / 6 | 0 /6 | 0 /6 |
| MyD88MYEL | 33% | 66% | 0% | 0% |
Tumor tissue sections were stained with hematoxylin/eosin and analyzed by a trained pathologist. Depending on the histological report, tumors were categorized as hyperproliferative adenoma, low-grade and high-grade intra-epithelial neoplasia (IEN), and invasive carcinoma.
MyD88 signaling is required for turnover of intestinal epithelia, but does not interfere with canonical WNT-signaling
Next, we assessed the role of MyD88 for proliferation and self-renewal in premalignant epithelia. Ki67-staining of intestinal tissue revealed a significant reduction of the transit-amplifying compartment in MyD88LSL mice, compared to the parental Apc1638N/+ strain. This reduced proliferation was rescued by re-expression of MyD88 in intestinal epithelia, but not in myeloid cells (Fig. 2C). Next, we investigated the signaling pathways associated with IEC proliferation, notably the MAP-kinase cascade, and analyzed phosphorylation of ERK1/2 in intestinal tissue lysates. Phospho-ERK1/2 levels in normal jejunum were significantly reduced in globally MyD88-deficient animals, clearly rescued by re-expression of Myd88 in IECs, but not in myeloid cells (Fig. 2D). In contrast, canonical WNT-signaling, which is aberrantly activated in Apc1638N/+ tumors, was essentially unaffected by absence of MyD88. Expression of Ccnd1 (CyclinD1), Spp1 (osteopontin), and Dusp4, targets of the canonical WNT-pathway,20 was elevated in tumors, but independent of MyD88, similar to intratumoral β-Catenin protein levels (Fig. 2E). As expected, intratumoral activation of the NF-κB pathway was decreased upon MyD88-deficiency, rescued by re-expression of MyD88 both in intestinal epithelia, as well as in myeloid cells (Supplementary Fig. 2C).
MyD88 is essential for production of tumor-enhancing cytokines in myeloid cells, and is associated with altered adaptive immune gene expression
Next, we investigated tumor-promoting cytokines for their tissue-specific dependence on MyD88. We observed significantly increased transcripts of the pro-inflammatory cytokines IL1β and especially high levels of IL6 in lesions from Apc1638N/+ mice, compared to normal tissue (Fig. 3A). This intratumoral increase depended on Myd88 expression and was rescued by MyD88 re-expression in myeloid cells, with only minor effects upon re-expression in intestinal epithelia. Of note, TNFα was independent of MyD88 and significantly upregulated in all tumors. Since microbial signals trigger MyD88-dependent signaling, we next incubated bone-marrow derived macrophages (MYEL), as well as isolated primary intestinal epithelial cells (IEC) from the different genetic mouse lines, with microbial-derived TLR-ligands and analyzed for secretion of IL6 by ELISA. In accordance with the mRNA expression analysis, macrophages robustly produced IL6 in a fully MyD88-dependent fashion, most efficiently after stimulation with ligands for Tlr2 and Tlr4. This confirms the specificity of Cre-mediated recombination. Further, IECs showed only minor IL6 secretion. Therefore, IL1β and IL6 provide pro-tumorigenic effects, their production being induced by MyD88-mediated signaling in tumor-associated myeloid cells.
Figure 3.
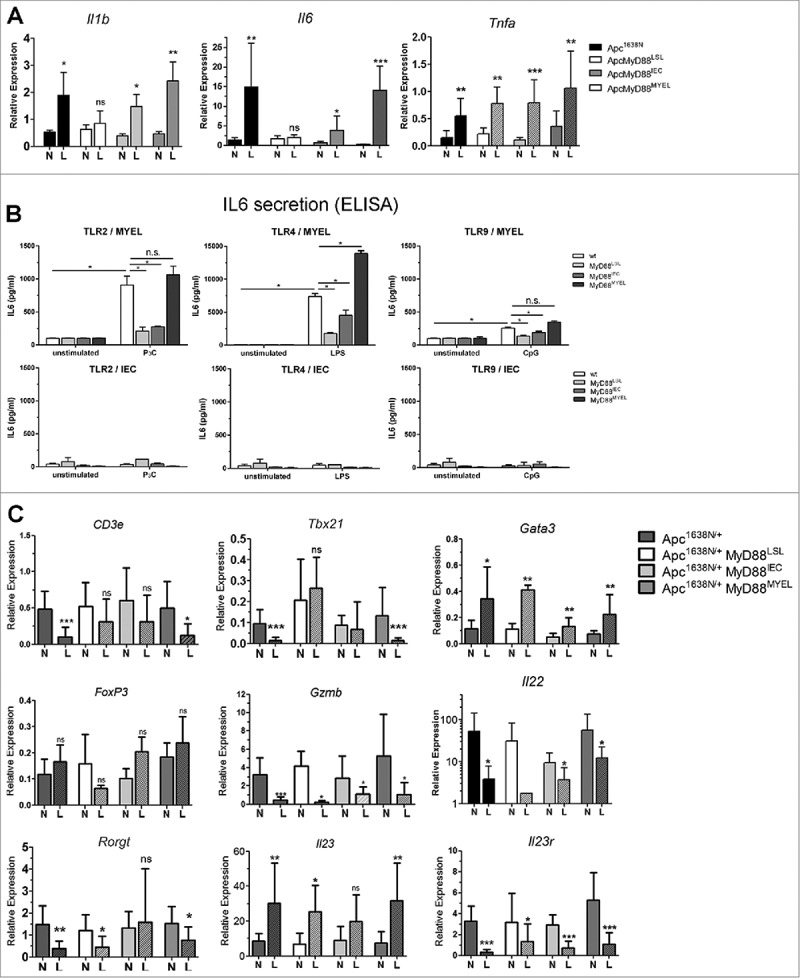
MyD88 signaling in macrophages shapes expression of cytokines and intratumoral T-cell marker transcripts. A, Expression of pro-inflammatory cytokines IL1β and IL6 is increased in tumors (L) compared to normal tissue in parental mice, crucially depending on Myd88 expression in myeloid cells, and only to a minor extent in IECs (n≥6 per group, error bars: s.d.), whereas TNFα is increased in tumors independent of MyD88, as verified by qRT-PCR. B, The tumor-promoting cytokine IL6 is derived from myeloid cells after stimulation with microbial-derived TLR ligands, but not from primary intestinal epithelial cells. Bone marrow derived macrophages were stimulated in vitro with Pam3Cys (TLR2 ligand), LPS (TLR4 ligand), or CpG DNA (TLR9 ligand), and IL6 secretion was tested by ELISA (n = 4), demonstrating exclusive dependency on MyD88 expression in the myeloid compartment. In contrast, isolated primary intestinal epithelial cells showed basically no detectable IL6 secretion. C, Intratumoral immune-regulatory transcripts of T-cell populations and T-cell produced cytokines were quantified by qRT-PCR. Of note, general T-cell transcripts (T-cell receptor subunit CD3e) were significantly reduced in parental tumors compared to normal tissue, similar to TH1-type cell transcripts (Tbx21). This intratumoral decrease in expression depends on MyD88-signalling in macrophages. Gata3 (Gata3, TH2 signature marker) was increased in tumors, essentially independent of MyD88. In contrast, Foxp3 (FoxP3, Treg signature marker) was significantly reduced in tumors upon global MyD88-deficiency. None of the other markers, including TH17-population markers, was found to depend on MyD88 expression (Gzmb (Granzyme B, effector CTLs and NK-cells), Il22 (Interleukin 22), Rorgt (RorgT, TH17 signature marker), Il23 (IL-23, ligand for IL23r), Il23r (IL23-receptor, TH17 signature marker).
Since MyD88-signalling contributes to innate as well as adaptive immunity, immune cell transcripts and densities of infiltrating T-cells and macrophages were analyzed in tissue sections. Tumor infiltration by macrophages (Mac1+), as well as of T-cells (CD3+/CD4+) was not significantly altered between the different mouse strains (Supplementary Fig. 3). In accordance, intratumoral expression of the macrophage hallmark transcript lysozyme M (Lyz2) was independent of MyD88 (Supplementary Fig. 3). However, MyD88 signaling was significantly associated with altered immune transcripts from intratumoral T-cell populations. The T-cell receptor subunit CD3e was significantly reduced in tumors in the parental Apc1638N/+ model compared to normal tissue, this was also observed by flow cytometry analysis for CD3-positive cells (not shown), indicating an intratumoral T-cell deprivation. Upon global Myd88-deficiency, however, CD3e was not reduced in tumors anymore. MyD88 re-expression in myeloid cells was sufficient to induce downregulation of CD3e expression in tumors (Fig. 3C). MyD88 signaling, especially in myeloid cells, was negatively associated with intratumoral expression of Tbx21 (Tbet), a hallmark transcript of anti-tumorigenic TH1 type cells. In contrast, signature markers of pro-tumorigenic TH2 cells (Gata3), anti-inflammatory Tregs (FoxP3), cytotoxic effector T-cells and NK-cells (Gzmb, Granzyme B), were essentially independent of MyD88 expression. Since TH17-type cells were shown to have both pro-tumorigenic and anti-tumorigenic capabilities, we analyzed several TH17–type markers. Of note, neither the transcription factor Ror-gT (Rorgt), the cytokines IL22 and IL23, nor the receptor Il23r, showed any association with MyD88-expression, even though they were all largely down-regulated in tumors, indicating an anti-tumorigenic role in the model used. The cytokine IL17 was not found to be differentially expressed in tumors or normal tissues, compared to wildtype controls (not shown).
MyD88 contributes to induction of EMT and stemness
Since tumors did not progress to malignancy upon MyD88-deficiency, we examined the role of MyD88 on epithelial-to-mesenchymal transition (EMT). Expression of EMT transcription factor Slug (Snai2) was significantly increased in tumors from Apc1638N/+ mice over normal tissue (Fig. 4A). Slug was also increased on the protein level in de-differentiated regions at the invasive front, clearly accumulating in the nucleus (Fig. 4B). Tumors from MyD88LSL or MyD88IEC mice showed no significant increase in Slug expression compared to normal intestine (Fig. 4A and B). Of note, re-expression of MyD88 in myeloid cells rescued the intratumoral expression of Slug (Fig. 4A and B). In addition, intratumoral expression of intestinal stem cell markers Lgr5 and Sox9 depended on MyD88 (Fig. 4A). The number of long-term label retaining cells, a functional marker for stem-cell-like properties, was tested by ten days of consecutive in vivo labeling and a 55 day chase period. Tissue cryosections from n = 4 mice per group were analyzed, and the number of intratumoral BrdU label-retaining cells strongly depended on MyD88. MyD88-deficient mice showed a significantly decreased number of label-retaining cells (Fig. 4C). However, neither expression in IECs nor in myeloid compartment fully restored the number of label-retaining cells.
Figure 4.
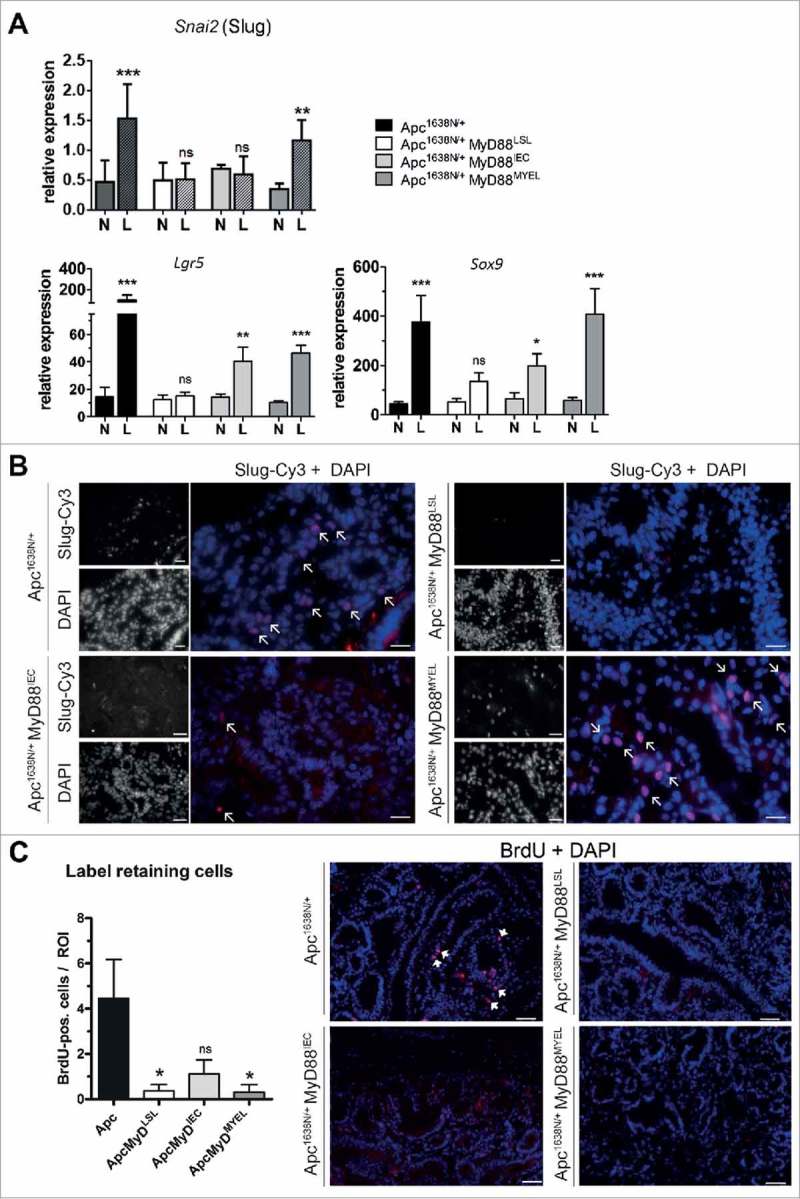
MyD88 signaling critically contributes to epithelial-mesenchymal transition (EMT) and stemness. A, MyD88 signaling is required for increased intratumoral expression of the EMT-inducing transcription factor Slug (Snai2), and the stem cell markers Lgr5 and Sox9. Re-expression of MyD88 in myeloid cells rescues the increase of Slug, whereas Lgr5 and SoX9 are partially rescued by expression in epithelial cells as well, as judged by qRT-PCR (n = 10 per group). B, Immunofluorescence staining on tumor cryosections (n = 5 per group). Slug is markedly expressed in the nuclei of tumor cells at the invasive front in the parental Apc-model, but is completely absent in MyD88-deficient mice. Partial and full rescue of Slug-expression can be seen upon re-expression in IECs, and in myeloid cells, respectively. Anti-Slug staining in red (fluorophore Cy3), nuclear counterstaining with DAPI in blue (size bars: 40 µm). C, Quantification of long-term label retaining cells in tumor tissue sections. Mice (n = 4 from each group) were subjected to BrdU injections for 10 days, sacrificed after 55 days of chase period. The number of BrdU-positive cells per low-power field (ROI) was significantly reduced in MyD88 deficient mice compared to parental Apc mice, and was essentially not rescued by tissue-specific re-expression. Right side: representative tumor tissue cryosections, stained with anti-BrdU antibody (red, positive cells marked with arrows), nuclear counterstaining with DAPI in blue (size bars: 50 µm). *P < .05; ns: not significant.
Discussion
The adapter protein MyD88 is a crucial mediator of pro-inflammatory pathways that are triggered by microbial-derived and endogenous danger signals, downstream of TLRs and IL1-receptors. However, the contribution of MyD88-mediated signaling to carcinogenesis is still under debate. Due to its broad expression, cell-type specific differences of MyD88 functions are presumed, but still little understood.13 NF-κB as a central downstream target of TLR signaling clearly contributes to cell survival of intestinal epithelia,21 and MyD88 increases the proliferation of intestinal epithelia.14,22 The results of our study show that MyD88 is required for intestinal tumor initiation, whereas effects of MyD88 on tumor growth were negligible. These results are in contrast to earlier findings obtained with a different genetic mouse model, where ApcMin-Myd88−/− mice highlighted a role of MyD88 in tumor growth rather than tumor formation or initiation.13 Of note, 57% of MyD88-deficient were protected from tumor formation in our study, with essentially no adverse side effects of the genetic deficiency. This makes MyD88 an interesting therapeutic target for tumor prevention and therapy. Our data show that MyD88 expression in either intestinal epithelia, or myeloid immune cells, is sufficient to induce development of benign tumors, but not for progression to the malignant carcinoma stage. Our results further highlight that MyD88-signaling may not interfere in the WNT-pathway, as anticipated before.14 In contrast, we observed an activation of the MAP-kinase and NF-κB pathways as main downstream targets of MyD88-dependent signaling. These results are opposed to earlier results reporting no effect on intestinal tumorigenesis for MyD88 in hematopoietic cells, based on reconstitution of ApcMin/+ Myd88−/− mice with bone marrow from wildtype mice.14 However, bone marrow replacement was performed at a time point when precursor lesions are already well established in the ApcMin/+ model. Therefore, it may be difficult to draw conclusions on the role of MyD88 in bone marrow derived cells in this particular model. In contrast, the genetic mouse model utilized here demonstrates a promoting role for MyD88-signaling in intestinal epithelia, as well as in myeloid cells. Moreover, we propose distinct and tissue-specific impacts for MyD88: in intestinal epithelia, MyD88 is required for cell homeostasis and renewal. MyD88-deficiency led to a strongly decreased proliferation with a decreased transit amplifying compartment, thus reducing the stochastic likelihood of a loss of heterozygosity (LOH) event for the tumor suppressor Apc (Suppl. Fig. 4). Therefore, tumor initiation is controlled by MyD88 in a cell-autonomous fashion in intestinal epithelial cells. However, re-expression of MyD88, neither in IECs nor myeloid cells, was sufficient to achieve the parental tumor phenotype of Apc1638N/+ mice, indicating a decisive contribution of stromal cells, such as macrophages and other myeloid immune cells. The production of tumor-promoting cytokines like IL1β and IL6 was strongly hampered upon MyD88-deficiency in myeloid cells, in line with other reports.13,22 In contrast, intratumoral production of the cytokines IL23 and TNFα did not depend on MyD88 expression in our hands, and TH17 transcripts were also essentially independent of Myd88, compared to earlier studies.13,23 These discrepancies may be at least in part explained by the different genetic mouse models used, and by the differences in tumor differentiation stages varying between carcinoma and adenoma. However, differences in gut microbiota and their derived TLR-ligands, which obviously occur between different animal facilities, cannot be excluded.
Importantly, however, MyD88 in myeloid cells was significantly associated with expression of regulatory transcripts associated with adaptive immune cell infiltrates. Transcripts from pro-tumorigenic TH2 type helper T-cell or cytotoxic effector cells were independent of MyD88. MyD88-expression in general, and specifcally in myeloid cells, was negatively associated with anti-tumorigenic TH1 type transcripts. In accordance, Apc1638N/+ mice show significantly enhanced tumor formation and aggressiveness in a T-/B-cell deficient background (not shown). Further, we have shown recently that specific microbiota, as likely initiators of TLR/MyD88-signalling, are associated with T-cell populations and prognosis in human colorectal cancer.24 Thus, blocking MyD88 in a therapeutic context might engage anti-tumoral effects of the adaptive immunity. Previously, TLR signaling was reported to induce EMT and tumor malignancy in hepatocellular cancer cell lines, however not for colorectal cancer.25 According to the current models, tumor progression and metastasis formation is attributable to cancer cells with prominent stemness properties.26 We found that expression of two master regulators of EMT and dedifferentiation, Slug and Snail, as well as of intestinal stem cells markers Lgr5 and Sox9, and the number of intratumoral BrdU label-retaining cells27 depended on MyD88 expression. In accordance, Tlr2 and Tlr4 were found to regulate the proliferation of intestinal stem cells and of Lgr5 expression, even though the effects were reported to be mediated either by the adapter Trif28 or MyD88.29 Of note, expression of MyD88 in myeloid cells was sufficient to induce expression of the transcription factor Slug, a master-induced of epithelia-mesenchymal transition, indicating that signaling factors released by macrophages induce EMT.
Taken together, our study reveals new roles for MyD88 in intestinal cancer, underscoring its suitability as potential therapeutic target,22 which could engage several parallel beneficial effects, like enhancing anti-tumoral adaptive immunity and blocking EMT and metastasis.
Patients and methods
Patient samples
The Ethics Committee of the Klinikum rechts der Isar approved the use of patient tissue samples (1926/7, and 5428/12). Informed, written consent of the patients had been obtained prior to the study. The publicly available Cancer Genome Atlas (TCGA) colorectal adenocarcinoma data set consisted of 633 CRC samples from 629 patients.
Animal experiments
Mice were kept at the animal facility of the Klinikum Rechts der Isar (Munich, Germany) under specific and opportunistic pathogen-free conditions (for details see supplementary methods). Approval has been obtained by the local authorities (District Government of Upper Bavaria; No. 55.2-1-54-2532-158-2015).
Mouse tissue sampling and ex vivo cell culture
Samples were obtained at 12 months, frozen in liquid nitrogen and stored at -80°C. For immunohistological analysis, tissue was embedded in Tissue Tek OCT (Sakura). For isolation of intestinal epithelial cells and bone marrow derived macrophages, see supplementary methods. Incubation with TLR-ligands in vitro, as well as ELISA assays for cytokine production were carried out as described in detail earlier.16
HE and immunofluorescence staining
6-µm-thick sections were cut from tissue blocks and processed by HE staining. All lesions were reviewed by a single, experienced pathologist. Immunofluorescence staining was performed as described before,30 for details see supplementary methods.
Quantitative analysis of mRNA and transcript levels
RNA was prepared from fresh-frozen patient or mouse tissue using RNeasy mini kit (QIAGEN). First-strand cDNA was synthesized from 1 mg total RNA using Reverse Transcription Kit (Fermentas, Thermo Fisher Scientific, Waltham, MS, USA). Quantitative RT-PCR analyses were performed using the Universal ProbeLibrary (Roche Diagnostics, Rotkreuz, Switzerland). mRNA expression levels were normalized to those of TFIID and were displayed as fold change relative to small intestine of WT mice. Oligonucleotide primers for quantitative RT-PCR were synthesized by Metabion (Martinsried, Germany). Accumulation of PCR amplification products was quantified on a LightCycler 480 Real-Time PCR system (Roche Diagnostics, Rotkreuz, Switzerland).
Western blot analysis
Equal amounts of protein were separated on 10% polyacrylamide gels and further subjected to immunoblotting following standard procedures [20]. Immunoreactive bands were detected using the following antibodies: anti-MyD88 (Abcam, Cambridge, UK); anti-phospho p44/42 (phospho-ERK1/2), anti-p44/42 (ERK1/2) and anti-Slug (Cell Signalling, Cambridge, UK); anti-β Aktin, anti-Ki67 and DAPI (2-(4-Carbamimidoylphenyll)-1H-indol-6-carboximidamide) (Sigma-Aldrich, Deisenhofen, Germany). All secondary antibodies were obtained from Jackson Immunoresearch (West Grove, PA, USA). Visualization was performed with an enhanced chemiluminescence substrate detection kit (Pierce, Rockford, IL, USA).
Statistical analysis
Statistical evaluation of differences among experimental groups was performed with GraphPad Prism® version 5.0 (GraphPad Software Inc., La Jolla, CA, USA) and SPSS software (version 16.0, SPSS Inc., Chicago, IL, USA). Normally distributed data were evaluated with Fisher's exact t-test, not normally distributed data were evaluated with Mann-Whitney test. The effect of altered MyD88/TLR-pathway component expression on patient survival, as well as survival data of murine models were evaluated by Kaplan-Meier survival analysis and log-rank test. P values < 0.05 were considered significant (*), p < 0.01 very significant (**) and p < 0.001 strongly significant (***).
Supplementary Material
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) under Grant JA 1024/3-1 to KPJ.
Disclosure of potential conflict of interest
No potential conflicts of interest were disclosed.
Competing interest
The authors declare no conflicts of interest.
Acknowledgments
The authors wish to thank Widya Johannes for excellent technical assistance, and Dr. Nicolas Gros for help with experiments, Dr. M. Laschinger and Dr. U. Nitsche for critical discussion, and Dr. Julia Slotta-Huspenina for histopathological evaluation.
Author contributions
All authors cooperated and contributed to the manuscript, and critically reviewed and approved it. Conception and design: KPJ, BH; Collection and assembly of data: AH, AC, KPJ; Data analysis and interpretation: AH, KPJ, BH; Manuscript writing: AH, KPJ.
References
- 1.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nature Rev Cancer. 2013;13:759–71. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 2.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. PMID:15294155. [DOI] [PubMed] [Google Scholar]
- 3.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappa B functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. PMID:15329734. [DOI] [PubMed] [Google Scholar]
- 4.Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, Visintin I, Rutherford T, Mor G. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–68. doi: 10.1158/0008-5472.CAN-05-3948. PMID:16585214. [DOI] [PubMed] [Google Scholar]
- 5.Achyut BR, Ghoshal UC, Moorchung N, Mittal B. Association of Toll-like receptor-4 (Asp299Gly and Thr399Ileu) gene polymorphisms with gastritis and precancerous lesions. Hum Immunol. 2007;68:901–7. doi: 10.1016/j.humimm.2007.10.006. PMID:18082569. [DOI] [PubMed] [Google Scholar]
- 6.Tahara T, Arisawa T, Wang F, Shibata T, Nakamura M, Sakata M, Hirata I, Nakano H. Toll-like receptor 2 -196 to 174del polymorphism influences the susceptibility of Japanese people to gastric cancer. Cancer Sci. 2007;98:1790–4. doi: 10.1111/j.1349-7006.2007.00590.x. PMID:17711514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang EL, Qian ZR, Nakasono M, Tanahashi T, Yoshimoto K, Bando Y, Kudo E, Shimada M, Sano T. High expression of Toll-like receptor 4/myeloid differentiation factor 88 signals correlates with poor prognosis in colorectal cancer. Br J Cancer. 2010;102:908–15. doi: 10.1038/sj.bjc.6605558. PMID:20145615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. PMID:20404851. [DOI] [PubMed] [Google Scholar]
- 9.Akira S, Takeda K. Toll-like receptor signalling. Nature Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 10.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. PMID:21235323. [DOI] [PubMed] [Google Scholar]
- 11.Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O'hUigin C, et al.. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med. 2010;207:1625–36. doi: 10.1084/jem.20100199. PMID:20624890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Banerjee A, Thamphiwatana S, Carmona EM, Rickman B, Doran KS, Obonyo M. Deficiency of the myeloid differentiation primary response molecule MyD88 leads to an early and rapid development of Helicobacter-induced gastric malignancy. Infect Immun. 2014;82:356–63. doi: 10.1128/IAI.01344-13. PMID:24166959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–7. doi: 10.1126/science.1140488. PMID:17615359. [DOI] [PubMed] [Google Scholar]
- 14.Lee SH, Hu LL, Gonzalez-Navajas J, Seo GS, Shen C, Brick J, Herdman S, Varki N, Corr M, Lee J, et al.. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat Med. 2010;16:665–70. doi: 10.1038/nm.2143. PMID:20473309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moresco EVY, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2013;21:R488–R93. doi: 10.1016/j.cub.2011.05.039. [DOI] [PubMed] [Google Scholar]
- 16.Gais P, Reim D, Jusek G, Rossmann-Bloeck T, Weighardt H, Pfeffer K, Altmayr F, Janssen KP, Holzmann B. Cutting edge: divergent cell-specific functions of MyD88 for inflammatory responses and organ injury in septic peritonitis. J Immunol. 2012;188:5833–7. doi: 10.4049/jimmunol.1200038. PMID:22586041. [DOI] [PubMed] [Google Scholar]
- 17.el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–93. doi: 10.1002/gene.20042. PMID:15282745. [DOI] [PubMed] [Google Scholar]
- 18.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrohages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–77. doi: 10.1023/A:1008942828960. PMID:10621974. [DOI] [PubMed] [Google Scholar]
- 19.Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, Renault B, Breukel C, Alt E, Lipkin M, Khan PM, et al.. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Nat Acad Sci U S A. 1994;91:8969–73. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohde F, Rimkus C, Friederichs J, Rosenberg R, Marthen C, Doll D, et al.. Expression of osteopontin, a target gene of de-regulated Wnt signaling, predicts survival in colon cancer. Int J Cancer J Int Du Cancer. 2007;121:1717–23. doi: 10.1002/ijc.22868. PMID:8090754. [DOI] [PubMed] [Google Scholar]
- 21.Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, Yost EA, Gruber AD, May MJ, Greten FR, et al.. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–6. doi: 10.1038/nature05590. PMID:17322906. [DOI] [PubMed] [Google Scholar]
- 22.Xie L, Jiang FC, Zhang LM, He WT, Liu JH, Li MQ, Zhang X, Xing S, Guo H, Zhou P. Targeting of MyD88 homodimerization by novel synthetic inhibitor TJ-M2010-5 in preventing colitis-associated colorectal cancer. J Natl Cancer Inst. 2015;108(4):djv364. doi: 10.1093/jnci/djv364. PMID:26712311. [DOI] [PubMed] [Google Scholar]
- 23.Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al.. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–8. doi: 10.1038/nature11465. PMID:23034650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cremonesi E, Governa V, Garzon JFG, Mele V, Amicarella F, Muraro MG, Trella E, Galati-Fournier V, Oertli D, Däster SR, et al.. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut. 2018;gutjnl-2016-313498. doi: 10.1136/gutjnl-2016-313498. PMID:29437871. [DOI] [PubMed] [Google Scholar]
- 25.Jia RJ, Cao L, Zhang L, Jing W, Chen R, Zhu MH, Zhu MH, Guo SW, Wu GB, Fan XY, et al.. Enhanced myeloid differentiation factor 88 promotes tumor metastasis via induction of epithelial-mesenchymal transition in human hepatocellular carcinoma. Cell Death Disease. 2014;5:e1103. doi: 10.1038/cddis.2014.71. PMID:24603331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. PMID:21376230. [DOI] [PubMed] [Google Scholar]
- 27.Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–84. doi: 10.1016/j.cell.2013.07.004. PMID:23870119. [DOI] [PubMed] [Google Scholar]
- 28.Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, Good M, Afrazi A, Marino R, Slagle D, et al.. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem. 2012;287:37296–308. doi: 10.1074/jbc.M112.375881. PMID:22955282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheeren FA, Kuo AH, van Weele LJ, Cai S, Glykofridis I, Sikandar SS, Zabala M, Qian D, Lam JS, Johnston D, et al.. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat Cell Biol. 2014;16:1238–48. doi: 10.1038/ncb3058. PMID:25362351. [DOI] [PubMed] [Google Scholar]
- 30.Janssen KP, el-Marjou F, Pinto D, Sastre X, Rouillard D, Fouquet C, Soussi T, Louvard D, Robine S. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. PMID:12145803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.