

Abstract
Injectable or implantable PLGA devices for the sustained delivery of proteins have been widely studied and utilized to overcome the necessity of repeated administrations for therapeutic proteins due to poor pharmacokinetic profiles of macromolecular therapies. These devices can come in the form of microparticles, implants, or patches depending on the disease state and route of administration. Furthermore, the release rate can be tuned from weeks to months by controlling the polymer composition, geometry of the device, or introducing additives during device fabrication. Slow-release devices have become a very powerful tool for modern medicine. Production of these devices has initially focused on emulsion based methods, relying on phase separation to encapsulate proteins within polymeric microparticles. Process parameters and the effect of additives have been thoroughly researched to ensure protein stability during device manufacturing and to control the release profile. Continuous fluidic production methods have also been utilized to create protein laden PLGA devices through spray drying and electrospray production. Thermal processing of PLGA with solid proteins is an emerging production method that allows for continuous, high throughput manufacturing of PLGA/protein devices. Overall, polymeric materials for protein delivery remain an emerging field of research for the creation of single administration treatments for a wide variety of disease. This review describes, in detail, methods to make PLGA devices, comparing traditional emulsion based methods to emerging methods to fabricate protein-laden devices.
Graphical abstract
Manufacturing of PLGA based devices encapsulating proteins is dependent on the methodology, protein properties, and processing parameters.
Introduction
Peptide and protein drugs are some of the most effective therapies due to their highly specific interactions with biological targets to elicit a desired therapeutic effect.1 Systems ranging from low molecular weight growth factors and inhibitory agents to high molecular weight antibodies and viral nanoparticles have been utilized for regenerative medicine, disease treatment, and immunotherapy.2–5 Effective administration of these drugs requires repeated doses as the proteins are rapidly cleared and exhibit low half-lives in circulation.6 Protein therapeutics exhibit poor bioavailability when administered orally, limiting them to parenteral routes of administration. Intravenous, subcutaneous, and intramuscular injections are typically utilized for administration; however, repeated administration has low patient compliance, thus limiting the effectiveness of the protein therapeutics.7 Implantable sustained release devices are an alternative to repeat injections, allowing for a single administration followed by a controlled delivery of a protein therapeutic for an extended period. Lyophilized and solution formulations of protein therapeutics often require storage at 4 to −20°C to maintain stability before administration. This requirement can result in costly shipping conditions and limitations for utilization in developing countries where refrigeration may not be readily available. Encapsulation of proteins within solid-state implantable devices enhances the thermal stability of the protein, resulting in less stringent storage conditions.8,9
Polymeric materials for sustained drug delivery have been extensively studied for more than 50 years to create formulations for enteral, parenteral, and topical administration of therapeutic molecules. Peptides and proteins have been encapsulated within polymers to create a multitude of implantable or injectable hydrogels, microparticles, nanofibers, and monolithic devices.10,11 These devices have been produced through a number of different processing methods and consist of a wide variety of synthesized and commercially available polymers.12,13 The release of proteins from these devices is driven by the erosion and formation of pores in the polymer matrix by hydrolytic or triggered degradation of the polymer. The protein then diffuses out of the encapsulating polymer driven by chemical potential, resulting in a sustained release. Polyesters are ubiquitous in drug delivery systems owing to their biocompatibility, biodegradability, processability, and ability to tune release rate.14 In particular, poly(lactic-co-glycolic acid) (PLGA) is one of the most popular polyesters for drug delivery and is utilized clinically in several FDA approved medical devices.15–17
PLGA Material Properties
PLGA is a copolymer of lactic and glycolic acid that completely biodegrades under in vivo conditions and the resulting monomers are readily metabolized and cleared.18 The glass transition temperature (Tg) and crystallinity of PLGA is dependent on the ratio of lactic to glycolic acid and whether it is a random or block copolymer.19 PLGA can also be end-capped with hydrophobic, acidic, or basic groups based on the polymerization chemistry.20 These factors affect the release of drugs encapsulated within PLGA materials and allow for the tuning of release duration from time frames of 10 to over 45 weeks.21 PLGA is soluble in many organic solvents and exhibits relatively low melt temperatures, making it amenable to many solution and traditional plastic processing techniques.22,23 Owing to these favorable properties for in vivo sustained release of therapeutics, PLGA has been extensively studied to create a wide variety of PLGA materials laden with peptide and protein drugs.
The composition of lactic and glycolic acid in PLGA affects the release profile through the crystallinity of the polymer matrix. Poly(glycolic acid) (PGA) is highly crystalline as a homopolymer and poly(lactic acid) (PLA) can exhibit varying degrees of crystallinity depending on the stereochemistry of the polymer.24 The crystallinity of the PLGA copolymer depends both on the amount of glycolic acid units, the stereochemistry of the lactic acid units, and whether the PLGA is a block or random copolymer.25,26 PLGA with a higher crystallinity reduces the release rate of encapsulated proteins due to the lower chain flexibility and inability of water to penetrate the crystalline regions of the polymer.27 The size of the PLGA devices also has an impact on the release profile. In general, the smaller the device the faster the release owing to the higher surface area to volume ratio. The larger the surface area relative to the volume of the device, the more surface is in contact with the release medium allowing for more rapid penetration of the medium and release of the protein.28,29 However, in practice the relationship between size and release rate may be more complex as the size differences effect device stability during solvent based manufacturing steps of emulsification, solvent removal rate, and the resulting protein dispersion within the material.30
The release of proteins from PLGA devices is dependent on the physicochemical character of the encapsulated protein and different interaction types (i.e. ionic, hydrophobic, adsorption etc.) with the PLGA matrix. Protein release is also dependent on the molecular weight of the PLGA used, with lower molecular weight PLGA generally yielding more rapid release profiles.31,32 This is due to less physical entanglement between the polymer chains and more rapid pore formation during initial hydration. Lower molecular weight PLGA also exhibits more rapid generation of oligomeric species that are able to diffuse out of the bulk polymer matrix, further increasing the porosity of the system and allowing the encapsulated proteins to more rapidly diffuse through the resulting pores.33 Additives to the PLGA device formulation can be used to affect the release profile as well. Pore forming additives in the device are typically low molecular weight hydrophilic molecules or polymers that are able to easily diffuse out of the polymer matrix during hydration, leaving behind channels and pores that allow the encapsulated proteins to rapidly diffuse out.34–36 PEG and sugars have been shown to act as porogens, as well as stabilizing agents for protein encapsulation.37,38 Therefore, there is a balance in designing PLGA systems with these stabilizing additives and controlling the release profile of the encapsulated protein. Incorporation of basic additives, such as metal hydroxide salts, has been shown to slow the release profile and enhance the stability of proteins during release by neutralizing carboxylic acid groups generated during PLGA hydrolysis.39,40 Neutralizing these acidic groups reduces further hydrolysis of nearby PLGA chains and results in an overall diminishment in the polymer degradation rate.
Emulsion Based PLGA Microparticle Production
PLGA microparticles (5 – 100 μm in diameter) encapsulating peptides and proteins are one of the most prevalent devices researched for sustained protein delivery.21,41 Protein encapsulated microparticles can be readily prepared through a number of solvent evaporation methods and there have been a variety of emulsion methods developed for protein incorporation.42 Microparticle suspensions can be easily administered through typical intramuscular injection methods, obviating the need for surgery to administer the polymeric device.43,44 Additives in the formulation and the microparticle geometry allow for further control over the protein release rate and biodegradation.
Single Emulsion
Emulsion based methods for microparticle production are some of the most widely used in protein encapsulation research. Single emulsion, oil-in-water (O/W), is a microparticle production method where both the polymer and protein are co-dissolved in an organic solvent and emulsified in an aqueous solution.45 The organic solvent is evaporated, resulting in spherical polymer/protein microparticles. This method is facile, however this method is typically suited for hydrophobic drugs and not hydrophilic proteins. Hydrophilic molecules encapsulated via this method have a tendency to migrate to the outer aqueous phase.46,47
Double Emulsion
Double emulsion processes, water-in-oil-in-water (W/O/W), are more commonly used for microparticle production with proteins, as they more efficiently encapsulate water soluble peptides and proteins (Figure 1).31,48 The basic principle of double emulsion microparticle production involves dissolving the protein in an aqueous solution that is added to PLGA dissolved in an organic phase, typically dichloromethane (DCM) or ethyl acetate, and the emulsification of the two phases via stirring. The resulting emulsion is then added to a large volume of water typically containing a poly(vinyl alcohol) (PVA) stabilizer. The organic phase is removed under reduced pressure and the solid microspheres encapsulating the protein are separated from the solution and dried. Microparticles prepared via the double emulsion methodology laden with proteins have been used for delivery of vaccines, regenerative growth factors, and regulatory hormones.49–54 Lupron Depot®, Nutropin Depot®, and Sandostatin LAR® Depot are PLGA microparticle devices for peptide and protein delivery that have been approved by the FDA for treatment of prostate cancer, growth hormone deficiency, and acromegaly, respectively.55
Figure 1.
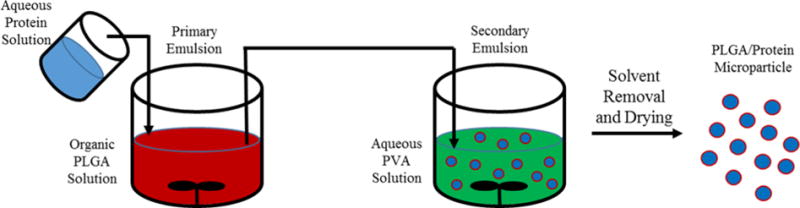
Schematic of protein encapsulation within PLGA microparticles via the double emulsion process (W/O/W).
The W/O/W methodology for microparticle production is highly dependent on the concentrations of PLGA and stabilizing polymer, emulsification volumes, emulsification energy applied, viscosities of the different phases, and additives.56–60 Controlling these different factors is essential in controlling the microparticle size, protein stability, and release kinetics. Additionally, the size, surface chemistry, and loading level of the encapsulated protein can have a dramatic effect on the release kinetics.61,62 The release of the protein through the polymer matrix involves diffusion through pores and water channels formed during swelling and degradation.63 Thus, the smaller hydrodynamic size of the encapsulated protein the more rapidly it is able to diffuse through small pores and channels. The surface charge and hydrophilic characteristic of the encapsulated protein can interact with carboxylic acid moieties present on the ends of PLGA, affecting the release through ionic and hydrogen bonding forces.64–66 Hydrophobic surfaces of a protein can also inhibit the release through Van der Waals interaction with hydrophobic portions of PLGA.67,68 These binding interactions control the diffusional rate of the protein through water channels within the polymer matrix, controlling release rate. Increased loading of the protein drug also results in a more rapid release, attributed to more protein present at the surface layer of the microparticle.69,70 The initial hydration and swelling of the microparticle allows for the protein to diffuse out, leaving behind more pores and channels allowing for more protein to diffuse out and accelerate release.
During W/O/W production of microparticles, the protein therapeutic is exposed to an aqueous/organic interface during emulsification with the polymer. Proteins in aqueous solution are folded to minimize hydrophobic interactions with the surrounding water, and the resulting active conformation of proteins typically has a hydrophobic core.71,72 Exposure to the aqueous/organic interface induces protein unfolding, adsorption, and aggregation.39,73,74 This is due to protein rearrangement to a more thermodynamically stable state in response to exposure to the hydrophobic organic solvent.75 Shear forces present during emulsification can also contribute to the denaturation process. The resulting unfolded and aggregated proteins can exhibit lower biological activity due to the misfolded conformations.76,77 This result can be reversible for small peptides and proteins that exhibit reversible unfolding events, however for complex and larger therapeutic proteins this effect can be highly detrimental.78,79 There have been many studies utilizing stabilizing additives to reduce the adsorption of proteins along the aqueous/organic interface.80–82 These additives are typically amphiphilic sugars or polymers that align along the interface and effectively “shield” the protein from interacting with the hydrophobic polymer solution. For example, the model protein α-chymotrypsin was encapsulated within PLGA microspheres with several different stabilizing sugars and poly(ethylene glycol) (PEG). The resulting aggregation and retained activity of α-chymotrypsin was analyzed and the addition of PEG during emulsion increased the retained activity from 70 to 96%. The increase in retained activity was attributed to PEG completely occupying the aqueous/organic interface, preventing the protein from aggregating.83 Covalent attachment of PEG to lysozyme has also been shown to improve protein stability during microsphere encapsulation instead of free PEG additive, owing to the same mechanism of shielding the protein from the interface.84,85
The amount of protein encapsulated within the PLGA microparticle versus the targeted amount is known as the encapsulation efficiency shown by the equation below.
Where:
massExtracted Protein = mass of protein recovered per mass of PLGA microparticles after removal of PLGA via organic solvent
massTheoretical Mass Loading = theoretical maximum mass of protein per mass of PLGA microparticles
Loading levels of proteins within PLGA microparticles are typically 1 – 10 wt% and the encapsulation efficiency using emulsion based production can vary from 30 – 95%.86–89 The encapsulation efficiency of peptides and proteins is dependent on the physicochemical characteristic of the protein and stabilization of the primary and secondary emulsion. If the microparticles formed during the primary emulsion are not properly stabilized before addition into the second aqueous phase, the encapsulated protein can leach into the aqueous phase and lower the encapsulation efficiency.90,91 Using higher molecular weight PLGA for microparticle production increases the encapsulation efficiency, likely due to lower polymer chain mobility limiting the ability of proteins to diffuse out of the microsphere during the second emulsion process.92 Protein leaching can be mitigated by tuning the pH of the aqueous phase to favor protein solubility and the pH of the secondary emulsion aqueous phase to decrease protein solubility.92,93 Leaching into the secondary emulsion aqueous phase can also be decreased by metal complexation of the protein to decrease aqueous solubility.94 PLGA has also been functionalized with small molecules that have resulted in increased encapsulation efficiency, potentially due to favorable interactions between residues on the encapsulated protein and the functional group conjugated to PLGA.95 While these approaches have resulted in improved encapsulation efficiency, effective loading of PLGA microparticles with proteins requires optimization of the emulsion process and conditions.
A recent example of double emulsion production for PLGA microparticles encapsulating a peptide for in vivo extended release depot application is microspheres containing the peptide liraglutide for diabetes treatment.96 The recombinantly produced peptide liraglutide is currently used as an FDA approved treatment for type 2 diabetes and binds the glucagon- like peptide-1 receptor, resulting in insulin secretion.97 Liraglutide was incorporated into PLGA (MW = 30 kDa, 50/50 lactide/glycolide) microspheres via the double emulsion method to create an injectable formulation for sustained release of the peptide and depression of blood glucose levels over 30 days. Twelve microsphere formulations were tested to determine the optimal conditions for liraglutide encapsulation and release, varying the volume of the polymer solution, the volume of the second emulsion aqueous phase, PVA stabilizer content, sonication time, and stirring speed (Figure 2 (A.)). Varying these parameters yielded encapsulation efficiencies from 47 – 74% and particle diameters from 6.8 to 16.2 μm. Based on the in vitro release profile, particles with a diameter of 9.4 μm were used for in vivo studies and a single injection of the liraglutide encapsulated microspheres was shown to be as effective as daily injections of liraglutide solution (Figure 2 (B.) and (C.)). While the liraglutide loaded PLGA microspheres were effective, this example highlights the many microparticle production parameters that must be considered when developing PLGA/protein microparticle systems produced via W/O/W double emulsion methods.
Figure 2.
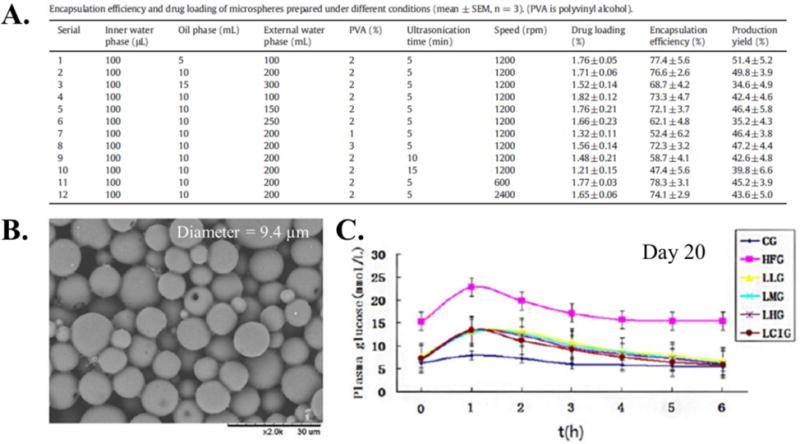
(A.) Table of parameters for production of liraglutide encapsulated PLGA (MW = 30 kDa, 50/50 lactide/glycolide) microspheres via the W/O/W emulsion process. (B.) SEM micrograph of microspheres used for in vivo studies produced using parameters from Serial 2. (C.) In vivo plasma glucose response of rat treatment groups after sugar gavage administration. The treatment groups were: CG = negative control, HFG = positive control, LLG = single injection of microspheres containing 0.9 mg liraglutide, LMG = single injection of microspheres containing 1.8 mg liraglutide, LHG = single injection of microspheres containing 3.6 mg of liraglutide, and LCIG = daily injection of 0.06 mg of liraglutide.96
Another recent example of extended release PLGA microparticles is the encapsulation of a Leishmania protein antigen NH36 for vaccination against the parasitic infection leishmaniasis.98 Vaccines require booster doses in order to elicit an effective humoral immune response, thus single administration depot formulations can obviate the need for repeat injections and increase patient compliance. The 36 kDa NH36 protein was effectively encapsulated at 83% encapsulation efficiency using the W/O/W double emulsion method. Vaccination also requires the stimulation of appropriate receptors, including toll-like receptors (TLRs), often accomplished through the co-administration of immunostimulatory adjuvants alongside the antigen.99 PLGA microspheres themselves enhance the uptake of the antigen by antigen presenting cells and adjuvants can also be encapsulated within microspheres to supply a sustained release of adjuvant with the antigen.100 In this study, the hydrophobic TLR 3M-052 was encapsulated within separate microspheres using the single emulsion W/O method that is more suitable for hydrophobic drug encapsulation. Co-administration of the NH36 and 3M-052 loaded microspheres elicited a higher response indicative of leishamaniasis immunity relative to multiple injections of NH36. This example highlights the utility of emulsion prepared PLGA microparticle formulations for single administration vaccine development and considerations in different emulsion methods for encapsulation of hydrophilic and hydrophobic components of a vaccine cocktail.
Coacervation
Coacervation is a production method for protein-laden PLGA microparticles involving phase separation of a PLGA solution around an aqueous protein solution (Figure 3).101 The microparticles are prepared by first emulsifying the aqueous protein solution in an organic solution of PLGA. An organic PLGA nonsolvent is then added to the emulsion, causing the polymer to phase separate due to the first organic solvent being extracted by the PLGA non-solvent.102 The PLGA microparticles encapsulating the protein are further solidified by the addition of a second non-solvent and separated via centrifugation, filtration, or sieving. Coacervation is more efficient at encapsulation of hydrophilic proteins than single emulsions processes, as the aqueous phase is completely encapsulated by the organic phase during polymer phase separation.103,104 The absence of the second aqueous phase that is present in double emulsion processes also typically yields higher encapsulation efficiencies during coacervation because the protein cannot diffuse out of the inner aqueous phase. Hydrophobic drugs can also be encapsulated via this process through co-dissolution with PLGA in the first organic phase and use of a second organic phase that is a nonsolvent for both the drug and the polymer.105,106
Figure 3.
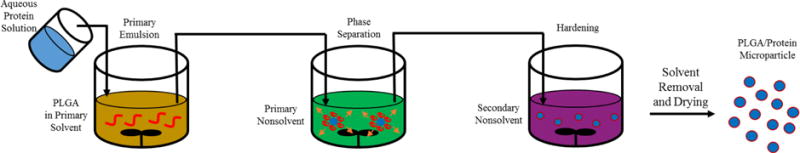
Schematic of microparticle production via coacervation. The protein solution (blue) is added to the polymer (red) dissolved in the primary organic solvent (orange) and emulsified. The emulsion is then added to the primary nonsolvent (green) to induce phase separation where the polymer coalesces around the aqueous droplet and the primary solvent mixes with the primary nonsolvent. The coalesced polymer/protein droplets is then added to a secondary non-solvent (purple) that induces further phase separation and hardening of the polymer around the inner protein rich phase.
The primary solvent containing PLGA is typically DCM or ethyl acetate, similar to other emulsion processes. The first nonsolvents used to induce phase separation included vegetable and silicone oils, poly(butadiene), poly(dimethylsiloxane), poly(methacrylics), and parrafins.107 These nonsolvents must be immiscible with both the polymer and the aqueous solution, while effectively extracting the primary organic solvent. The second nonsolvent must be miscible with the first nonsolvent and is used to wash away both the first nonsolvent and the primary solvent, which further hardens the microspheres by solvent extraction. Examples of the second nonsolvent include hexanes, heptanes, ether, and mineral oil.107 As with the single and double emulsion production methods, the protein being encapsulated is in contact with an aqueous/organic interface which can lead to protein destabilization during coacervation. Stabilization of the protein can be accomplished through addition of small molecule stabilizers, as previously described, provided they are insoluble in the first and second nonsolvents.108
The stirring rate and temperature used during phase separation are critical factors during coacervation due to a tendency for the particles to aggregate and coalesce during solvent extraction.102 Furthermore, changes in stirring rate control the size of the microparticles and insufficient agitation can entirely suppress microparticle formation. The polymer concentration in the primary emulsion and volumetric ratios of polymer solution to protein solution are critical in the controlling the successful formation of microspheres.109,110 The ratio of the aqueous phase to polymer phase is typically 0.02 to 0.12 for stable coacervation.102 This ratio window can limit the amount of protein that can be encapsulated within the microparticle via coacervation. This is due to the phase separation and encapsulation process relying on the interfacial energies during polymer microparticle hardening.111 The coacervation process is also highly dependent on the rate of addition of the first nonsolvent to extract the primary solvent, owing to the non-equilibrium nature of coacervation and dependency of the microparticle size and morphology on the kinetics of phase separation.112
An example of coacervation encapsulation of a protein antigen is the tetanus toxoid that was encapsulated within PLGA microparticles to elicit neutralizing antibodies in a murine model.113 The microparticles had tetanus toxoid with an aluminum adjuvant encapsulated via coacervation at 0.45 wt% loading and an encapsulation efficiency of 96% (Figure 4 (A.)). In vivo administration of the microparticles elicited enhanced antibody titers relative to administration with free tetanus toxoid with aluminium (Figure 4 (B.)). Another example of coacervation encapsulation is the production of PLGA microparticles with the luteinizing hormone releasing hormone analog triptorelin for contraceptive application.114 A series of microparticles were prepared using different volumetric ratios of dichloromethane, methanol additive amounts to the dichloromethane phase, and amount of silicone oil as the first nonsolvent. This parameter study was used to develop a phase diagram elucidating the stability window for successful microparticle formation (Figure 4 (D.)). These parameters were used to optimize the microparticle diameter, size dispersity, and release properties. The encapsulation efficiency of triptorelin for all samples was 70 – 73% regardless of coacervation conditions. Both of these examples highlight the consistent and high loading levels achieved using coacervation, but also demonstrate that the process parameters for coacervation must be thoroughly optimized to produce microparticles with the desired properties.
Figure 4.
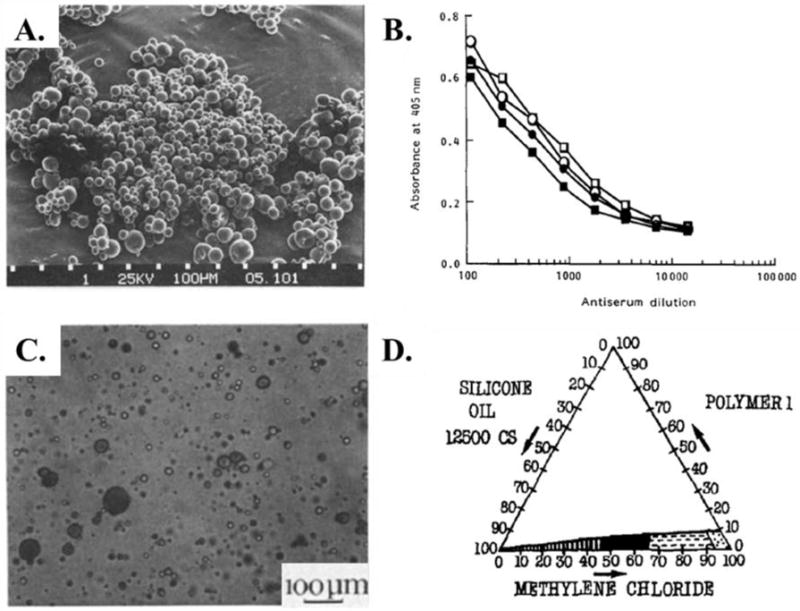
(A.) SEM micrograph of PLGA microparticles encapsulating tetanus toxoid prepared via coacervation. (B.) Antibody response of mice immunized with microparticles releasing tetanus toxoid (open symbols) and tetanus toxoid adsorbed to aluminium (closed symbols).113 (C.) Optical image of PLGA microparticles encapsulating triptorelin during phase separation. (D.) Phase diagram for PLGA microparticle formation via coacervation with the stability window shown in black.114
While coacervation has advantages in ease and efficiency of encapsulation of hydrophilic drugs, the complex variables controlling microparticle size and particle morphology have limited the widespread application in protein laden microparticle production. Recent studies with protein coacervates have been focused on forming complexes between polyelectrolyte polymers and proteins that become insoluble upon complexation.115,116 These coacervates do not require organic solvent for production have less dependence on mixing and volumetric ratio parameters. However, polyelectrolyte coacervation is limited to proteins bearing enough charged residues to effectively complex with the charged polymer.
Continuous Fluidic PLGA Material Production Methods
Emulsion based processes have been successful in creating clinically used peptide and protein microparticle depot systems, however there are inherent scalability issues with batch production methods.117 Fluid spraying methodologies have been developed to overcome the batch nature of typical emulsion production methods, owing to the continuous and high-throughput spraying production. The basic principle of spray based microparticle production is forcing an emulsion of PLGA and protein solutions through a nozzle and then rapidly drying to solidify the formed protein laden material.
Spray Drying
Spray drying was originally used in the pharmaceutical industry to create solid particles from small molecule solutions and has recently been expanded to drug laden polymeric particle production.118 The process first involves the creating of a O/W emulsion of an organic solution containing PLGA and an aqueous solution containing a peptide or protein therapeutic. Typical spray drying processes then use a two-fluid atomizing nozzle where the emulsion is flowed through the inner channel and a stream of hot gas is flowed through the outer channel. Both streams are then forced through a small opening and the air stream causes the fluid to be broken up into small particles. The atomized particles are then flowed into a drying chamber to remove the solvent and collected via inertial or filtration separation. This process can be run in a continuous manner, provided the particles can be effectively removed during production and the collection method is not prone to clogging.
While the two-fluid atomizing nozzle is one of the most widely used, there are several other systems that have been utilized for microparticle production (Figure 5). Rotary atomizers are systems comprising a rotating disk at the nozzle exit where droplets are formed through centrifugal force at the edge of the disk.119 A pressure nozzle involves simply forcing the fluid through a narrow barrel at high pressure, forcing the stream to break up into small particles.120 Ultrasonic atomization consists of using ultrasonic vibrations to form droplets during flow through the nozzle.121 Three-fluid atomizing nozzle have also been explored to eliminate the primary emulsion step in two-fluid atomization processes. Both the polymer solution and protein solution are flowed separately and are mixed in the nozzle immediately before atomization. The three-fluid method can also be utilized to mix an emulsion with another polymer solution to form more complex core-shell microparticle systems.122,123
Figure 5.
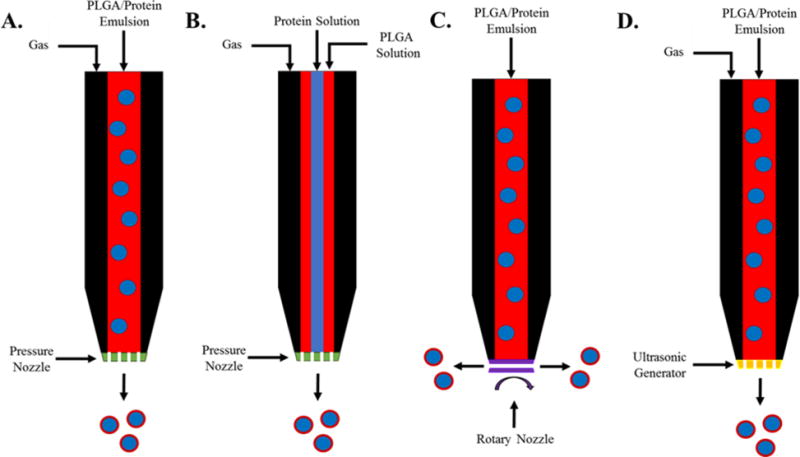
Schematics of inlet flows and nozzle designs for (A.) two-fluid pressure nozzle spray drying, (B.) three-fluid pressure nozzle spray drying, (C.) rotary nozzle spray drying, and (D.) ultrasonic nozzle spray drying.
While all of the atomization methods have been successful in creating microparticles laden with proteins, each has distinct advantages and disadvantages.124–127 The two-fluid atomization method produces particles with a narrow distribution and offers good control over the particle size, however it can suffer from scale-up limitations due to the drying residence time in the nozzle. The three-fluid atomization method eliminates the emulsion step and streamlines the production process, but is highly sensitive to the flow rates and viscosities of the three-fluids being fed into the nozzle. Pressure nozzles are simple in design and application, but control over particle size can be difficult and the narrow orifice can block during microparticle production. Rotary atomizers are more mechanically complex due to multiple moving parts, but do not suffer blockage issues and exhibit low particle dispersities. Particles prepared via ultrasonic atomization exhibit uniform and controlled particle sizes, however the ultrasonic energies imparted during production can destabilize the protein being encapsulated.
Regardless of the atomization method, successful formulation of microparticles via spray drying requires rapid solvent removal and drying to solidify the polymer around the encapsulated protein. The solvent removal primarily takes place when the solution comes into contact with the heated gas applied during atomization, thus the contact time with the heated air must be controlled to allow for adequate drying and hardening of the microparticle.128 The solvent system used for polymer dissolution must be volatile and the addition of azeotrope forming cosolvent systems have been utilized to enhance the drying rate.129,130 Control of the particle size, particle dispersity and drug distribution within the polymer is highly dependent on both the flow rate through the atomization nozzle and the drying rate, thus tuning these parameters for the desired microparticle property can be a complex process.131–133
Spray drying for protein laden microspheres utilizes emulsion, thus it can suffer similar protein destabilization along the aqueous/organic interface observed with the previously discussed emulsion based production methods. The atomization and drying process also involve exposure to hot gas, which can further induce destabilization due to protein exposure to a water/air interface. Proteins can exhibit adsorption and unfolding in response to a water/air interface due to the differences in polarity between the two phases.134 In addition, proteins exhibit further destabilization with applied shear forces at the water/air interface.135 Shear forces are applied during the atomization process, thus protein stability during spray drying can be highly dependent on the flow rates and atomization method utilized. The microparticle is also rapidly subjected to high temperatures during atomization and solvent removal inducing thermal denaturation and aggregation.136
A recent example of spray drying was a study utilizing three-fluid atomization to create PLGA microspheres laden with lysozyme via a one-step process (Figure 6 (A.)).137 The aim of the study was to optimize the process parameters during microparticle production to control microparticle morphology, microparticle size, protein stability, protein distribution within the particle, and in vitro release. The solvent system used for PLGA did not have a dramatic effect on the particle size and residual moisture, indicating the solvents were effectively removed during drying in the lab-scale spray dryer. While the solvent system did not affect the particle size, both the type of solvent used and the feed rates of the organic and aqueous phases dramatically effected the distribution of lysozyme within the microparticle. The use of both acetone and acetonitrile in place of dichloromethane for the organic phase, Figure 6 (B.), resulted in extensive lysozyme migration to the particle surface and high burst release values during in vitro release (75%, 100%, and 40% for acetone, acetonitrile, and dichloromethane respectively). This was attributed to the low surface tension gradient between the acetone or acetonitrile phase and the aqueous phase, reducing the ability of the PLGA to spread and cover the lysozyme droplets. Increasing the feed rate of the organic phase relative to the aqueous phase also reduced the burst release and surface content of lysozyme, owing to more PLGA phase available to fully encapsulate the inner aqueous phase. The encapsulated lysozyme had minimal secondary structure loss and a trehalose stabilizer was used to shield the protein from interface induced denaturation during spray drying. This example demonstrated the high variability of microparticle properties prepared via three-fluid spray drying in response to solvent systems and feed rates. Furthermore, the parameters developed in the study were for a lab-scale spray drying system that will vary during scale-up to larger systems with different drying residence times and throughput rates.
Figure 6.
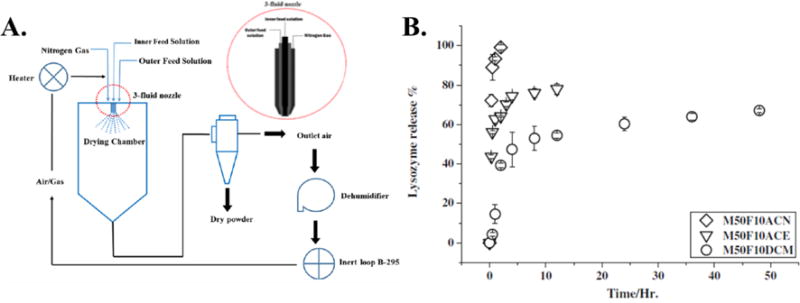
(A.) Schematic of the three-fluid atomizer and spray drying process. (B.) In vitro release profile of lysozyme from spray dried microspheres prepared using acetonitrile (M50F10ACN), acetone (M50F10ACE), and dichloromethane (M50F10DCM).137
Electrospinning and Electrospraying
Electrospinning and electrospraying are similar methods utilizing electric potential to generate polymer droplets, known as electrohydrodynamic (EHD) production techniques, containing proteins that can be subsequently dried for the formation of solid materials (Figure 7).138–140 A polymer/protein emulsion at the tip of a needle is subjected to a strong electric potential that causes the build-up of charge on the liquid droplet. When the charge reaches a critical level it overcomes the surface tension of the liquid and causes a jet or droplets of the solution to discharge from the tip. The discharged solution is collected onto a grounded plate or rotating drum and the solvent is either removed via evaporation during discharge or further drying depending on the volatility of the solvent. Both methods have the ability to create polymeric materials in the micro- to nano-scale laden with protein therapeutics.141,142
Figure 7.
Schematic of the electrospinning and electrospraying process with a rotating drum collector.
Electrospinning is utilized to create fibrous materials, while electrospraying is used to create particle systems. Both methods use similar needle and applied voltage set-ups, and the fabrication of fibers versus particles is controlled by polymer concentration and the viscoelastic properties of the solution.143,144 Electrospinning requires a continuous stream of polymer solution to form uniform fibers, therefore a higher polymer concentration and higher viscoelastic property of the solution prevents break-up of the stream during discharge from the needle. In contrast, effective particle formation during electrospraying requires a lower polymer concentration with low viscoelasticity to promote break-up of the droplets. The solvent used to dissolve the polymer will affect these properties, making solvent selection an important aspect of designing electrospinning or electrospraying processes.145 Beaded fibers will develop if the solution properties fall in the middle of those for particle or fiber formation. Electrospinning and electrospraying have also been utilized simultaneously to embed micro- or nanoparticles within the fibrous materials.146,147 The size and morphology of the fibers and particles formed via EHD is controlled by the applied voltage and flow rate of solution through the needle.148,149 In general, higher applied voltage and lower flow rates result in fibers or particles with smaller diameters. Controlling the solvent evaporation rate is an important aspect of electrospraying, as too rapid evaporation rates can result in collapse of the particles and formation of wrinkled and porous structures.148,150 Electrospray microparticle formation does not require the co-administration of hot gas to induce particle formation and drying, removing a potential source of thermal denaturation for proteins being encapsulated within the particles.
PLGA fibers and particles formed via EHD have been widely utilized as tissue engineering scaffolds for regenerative medicine and as implantable drug delivery devices.10,151,152 Protein therapeutics can be incorporated into the electrospun fibers via surface attachment or incorporation into the polymer matrix. Successful delivery of surface attached proteins requires that the attachment can be released over time via hydrolysis, degradation, or specific enzymatic activity.153–155 While surface attachment has been effective, it requires excess protein and extensive washing steps for attachment and the loading of proteins on the surface can suffer from batch-to-batch variability.156,157 PLGA microparticles and nanoparticles produced via electrospraying are not typically subjected to surface modification and the bioactive agent is encapsulated within the particle. Encapsulation of proteins within the PLGA matrix can be accomplished by creating an emulsion of the protein solution with the polymer prior to electrospinning.158,159 Utilizing an emulsion for EHD can have deleterious effects on fiber or particle integrity depending on the solvent volumetric ratios, limiting the amount of protein that can be incorporated into the polymeric material.160 Furthermore, there are still the inherent protein stability issues involved with creating an aqueous/organic emulsion containing proteins. Protein destabilization can be further induced via the strong shear forces induced during expulsion of the liquid droplet from the needle tip.161,162 The incorporation of emulsion and protein stabilizers can further alter the viscoelastic properties of the solution, complicating the integrity of fibers or particles formed during EHD.
While emulsion EHD methods have been successful in generating protein laden polymeric materials, co-axial EHD methodology has been developed to create more complex and stable formulations.163 Co-axial EHD involves the simultaneous flow of an inner aqueous phase containing the protein and an outer phase containing the polymer. The two fluids meet at the tip of the needle and are simultaneously discharged from the tip to form a core-shell structure encapsulating the protein in the interior of the polymeric structure.164 The resulting material avoids complications with protein dispersion throughout a polymer matrix by localizing protein within the interior of the structure.165 Controlling the flow rates of both fluids is essential in the successful formation of the core-shell structure and the thickness of the polymer shell can be tuned via the flow rate of the polymer phase.166,167 The polymer concentration and molecular weight are also important parameters due to the influence on the solution viscoelastic properties.168 These process parameters make the process more complex relative to emulsion EHD and effective core-shell structure formation may be difficult to achieve. While co-axial EHD with two fluids has been widely utilized in research, the process has been expanded to three fluid systems to create more complex systems for release of multiple drugs at different rates.169
Protein encapsulation via co-axial EHD avoids protein instability issues by eliminating the emulsion step and lowering the size of the interface between the organic and aqueous phase.170,171 Elimination of the emulsion step also increases the simplicity of the overall process and makes it one-step. Encapsulation efficiency is also typically increased during co-axial EHD due to total encapsulation of the inner aqueous phase.172 The protein must diffuse through the encapsulating PLGA layer in order be released and the polymer must reach a level of porosity to permit protein penetration. This limitation can result in long lag times before protein release, however the effect can be mitigated by incorporating porogens in the polymer layer to accelerate protein diffusion.173
A recent study compared co-axial and emulsion electrospray processes to encapsulate the model protein bovine serum albumin (BSA) within PLGA microparticles.174 Co-axial electrospraying resulted in particles having a size an order of magnitude lower than those prepared via emulsion electrospraying and exhibited narrower size distributions (Figure 8 (A.) and (B.)). Particles prepared via emulsion electrospraying exhibited a rougher, wrinkled surface morphology relative to those prepared via co-axial electrospraying (Figure 8 (D.) and (E.)). This difference was attributed to uneven solvent removal due to the heterogeneous dispersion of organic and aqueous phases in the emulsion. The co-axial technique localized the polymer phase on the outside of the particle, allowing for even solvent removal and PLGA coating of the inner, protein rich phase (Figure 8 (C.) and (F.). This effect also increased the encapsulation of BSA from 74% to 89% for particles prepared via emulsion or co-axial electrospraying respectively. Analysis of BSA distribution within the microparticles resulted in BSA localized in the interior of co-axial microparticles and on the exterior of emulsion microparticles, consistent with the expected location of the aqueous phase based on the production method. The burst release of BSA during in vitro release was decreased from 26% to 10% for emulsion and co-axial prepared particles, with both types reaching 50 – 55% cumulative release over 6 weeks (Figure 8 (G.) and (H.). The release study indicated that the co-axial electrospray prepared microparticles exhibited a more consistent release and protein was able to consistently diffuse out over the duration of the study. Overall, the study highlighted the control over release, protein dispersion, and particle morphology afforded by utilizing co-axial versus emulsion electrospray production of PLGA microparticles. However, both production methods had 7 formulations tested to determine the optimal conditions and compositions for particle production, demonstrating the sensitivity of electrospraying to the process parameters.
Figure 8.
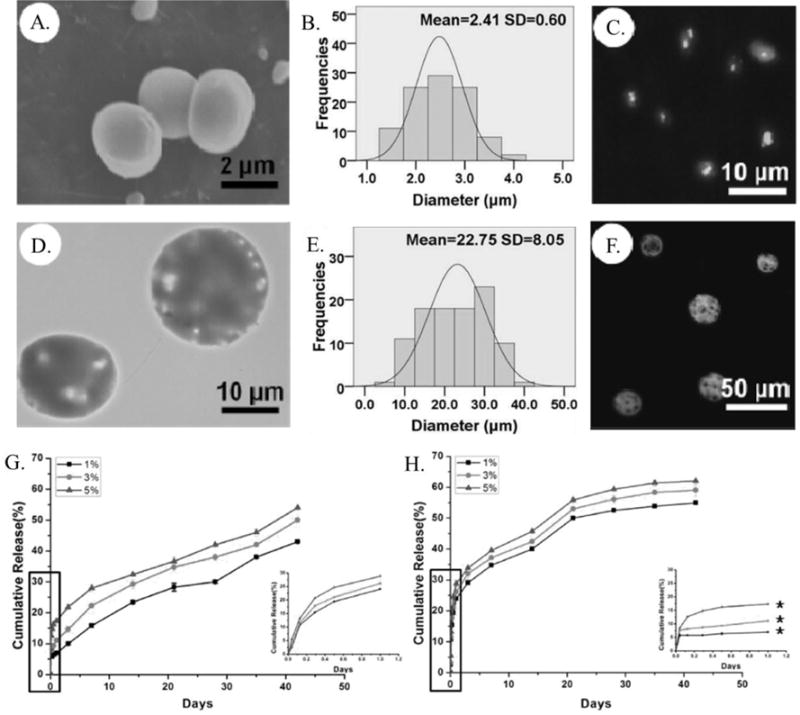
(A.) SEM micrograph and (B.) size distribution histogram of PLGA microparticles prepared via co-axial electrospray. (C.) Confocal microscopy image of the distribution of fluorescently labeled BSA within PLGA microparticles prepared via co-axial electrospray. (D.) SEM micrograph and (E.) size distribution histogram of PLGA microparticles prepared via co-axial electrospray. (F.) Confocal microscopy image of the distribution of fluorescently labeled BSA within PLGA microparticles prepared via co-axial electrospray. Release profile of BSA from microparticles prepared via (G.) emulsion and (H.) co-axial electrospray with the release over the first day shown by inset.174
Thermal Processing
Thermal processing of PLGA materials into solid, implantable materials in the solid and melt state is an alternative to the traditional solvent based production methods. In general, thermal processing involves applying heat and pressure to induce coalescence between PLGA particles to form a macroscopic assembly. The PLGA particles can be loaded with proteins before or during processing and the resulting assembly can be implanted for sustained release. PLGA particles have been developed into larger implantable devices through sintering, where particles are subjected to heat in a mold and allowed to fuse together.175–177 This methodology is advantageous, as it is high throughput and allows for continuous operation.178 The encapsulation efficiency of the therapeutic is also high, owing to the closed nature of the process, and allows for good control over the loading level. The melting temperature (Tm) and glass transition temperature (Tg) of PLGA are dependent on the stereochemistry of the lactic acid units and the percentage of glycolic acid in the copolymer. The Tm and Tg can range from 80 – 160°C and 33 – 60°C respectively based on the type of PLGA used.19,22 PLGA with a 50:50 ratio of lactic to glycolic acid exhibits the lowest Tm and Tg and has been the most widely used for thermal processing application. Additionally, viscosity modifiers such as PEG or sugars can be used as plasticizing agents to lower the required temperature for thermal processing.179,180 Temperatures typically used for successful thermal processing of proteins with PLGA are in the range of 80 – 105°C. These temperatures readily and fully denature proteins when in solution, however processing proteins in the solid state imparts further thermal stability due to the lower hydration state and resulting restriction of protein molecular motion.181,182 This allows for proteins to be processed at the elevated temperatures required for melt encapsulation, however the encapsulated proteins can still exhibit some denaturation and aggregation depending on the biochemical properties of the protein and additives used. Studies with synthesized block copolymers of poly(ε-caprolactone)-b-poly(ethylene glycol)-b-poly(ε-caprolactone) and poly(2-Isopropoxy-2-oxo-1,3,2-dioxaphospholane)-b-poly(L-lactic acid) have yielded biodegradable polymers with melt temperatures in the range of 35 – 55°C and were used to encapsulate proteins via melt processing.183–185 The proteins exhibited enhanced stability when thermally processed at lower temperatures, however these polymers are not readily available to most researchers.
Extrusion
Protein encapsulation via thermal processing has predominately been accomplished via melt extrusion, where solid PLGA and protein are mixed together and melted in a heated barrel with a conveying screw until it is forced through a die and solidified (Figure 9). Extrusion has been extensively utilized for encapsulation of small molecule drugs with excipients for conventional pharmaceutical formulations.186 However, melt extrusion normally requires high temperatures to melt the polymer matrix and high levels of shear imparted by the conveying screw, which can have detrimental effects on protein secondary structure and retained biological activity. As previously stated, melt extrusion with protein/PLGA blends requires temperatures of 80 - 105°C. Hydrated proteins typically exhibit Tm values of 50 - 70°C, indicating that above these temperatures the proteins denature and form misfolded oligomeric and aggregated species.187 Additionally, proteins in the aqueous state have Tg values in the range of −75 – 30°C, indicating that above these temperatures the polypeptide chain is able to move and rearrange in order to achieve different conformations.188,189 However, when water is removed, the resulting solid protein exhibits Tg values of 130 - 185°C due to the restricted molecular motion of the protein chains in response to the absence of the plasticizing water molecules.190–192 The shift in Tg is beneficial when designing melt extrusion and processing proteins in the solid state, as the restricted molecular motion of solid proteins can help retain the correct conformation after extrusion at elevated temperatures.
Figure 9.
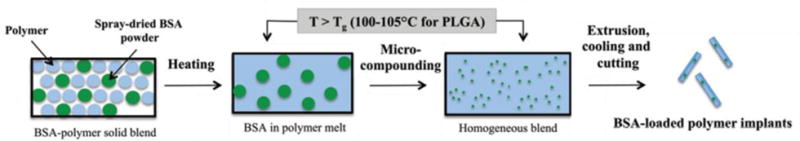
Schematic of the melt extrusion process for encapsulation of BSA with PLGA.193
Thermal processing of PLGA/protein blends has largely focused on the model proteins lysozyme and BSA due to their relative expense and ability to be obtained in high quantity. Both proteins were able to be successfully incorporated into rod shaped PLGA implants via twin-screw and small scale syringe ram-extrusion. The impact of processing temperature, stabilizing additives, and surface biochemistry on protein stability was studied during melt extrusion. Investigation of the secondary structure of both proteins exhibited varying degrees of denaturation in response to processing temperature and the use of stabilizing PEG, sugar, and salt additives.194,195 The secondary structure of the protein was denatured in response to increasing temperature, likely due to the applied energy overcoming the activation energy for protein unfolding in the solid state. The mechanism of stabilization via the PEG or sugar additive is theorized to be similar to stabilization during protein emulsification, where the additive aligns along the protein/polymer interface to shield the protein from the hydrophobic polymer melt.193 Covalent attachment of PEG to lysozyme has also been shown to stabilize the protein, likely owing to a similar shielding mechanism as PEGylated protein stabilization during emulsion microparticle production.196 The enzymatic activity of lysozyme was directly affected by the processing temperature and is related to the thermally induced denaturation and aggregation in response to melt processing.194 There is also the potential for thermally induced chemical reactions between the surface residues of the protein and PLGA during melt processing, such as acylation or thioester formation.197
The release properties and stability of proteins during in vitro release from rod shaped PLGA implants prepared via melt processing are dependent, similarly to microparticle systems, on additives, protein loading, protein dispersion, and protein surface properties. The polymer rods have a lower surface area to volume ratio relative to PLGA microparticles, therefore the polymer must undergo extensive water uptake and degradation to effectively form a porous network that allows the protein to diffuse out of the matrix. This limitation has been overcome via the incorporation of low molecular weight PEG and hydrophilic molecules as porogens to accelerate release.198 Incorporation of basic metal salts has also been shown to increase the release, however this diminished aggregation of proteins within the matrix rather than eliciting pore formation.40 Proteins have a tendency to form non-covalent aggregates during extrusion and during release as a consequence of the acidic microenvironment within PLGA. The basic metal salts neutralize the acid formed during release and result in less aggregation, which allows the smaller monomeric proteins to more readily diffuse through the polymeric implant. Increased loading of the protein additive also leads to more rapid release due to the more protein localized closer to the surface that can be released during the early hydration and degradation phases of the implant surface layer.194,198
The aggregation of proteins during melt processing can also lead to segregation within the polymer matrix, which results in increased burst release and decreased protein stability (Figure 10 (A.) and (B.)).195 Alteration of the protein surface properties with amphiphilic or hydrophobic moieties has been shown to increase the release rate relative to the unmodified protein.195,196 For example, the covalent attachment of the hydrophobic dye, fluorescein isothiocyanate (FITC) to BSA, exhibited an increase in release rate relative to native BSA (Figure 10 (C.) and (D.)). The enhanced release rate was attributed to decreased aggregation of the protein during extrusion due to the surface chemistry interacting more favorably with the hydrophobic PLGA matrix and more readily dispersing within the matrix. An advantage of proteins immobilized within polymeric matrices via thermal processing is increased thermal stability during storage, which can increase the ability to utilize protein therapeutics in developing countries where refrigeration is not readily available. Modification of BSA with FITC maintained the same release profile after prolonged storage, while the native BSA had an altered release profile. This indicated a change in protein aggregation or interaction with the PLGA matrix and demonstrated that the FITC modification prevented changes in the BSA/PLGA formulation during storage.
Figure 10.
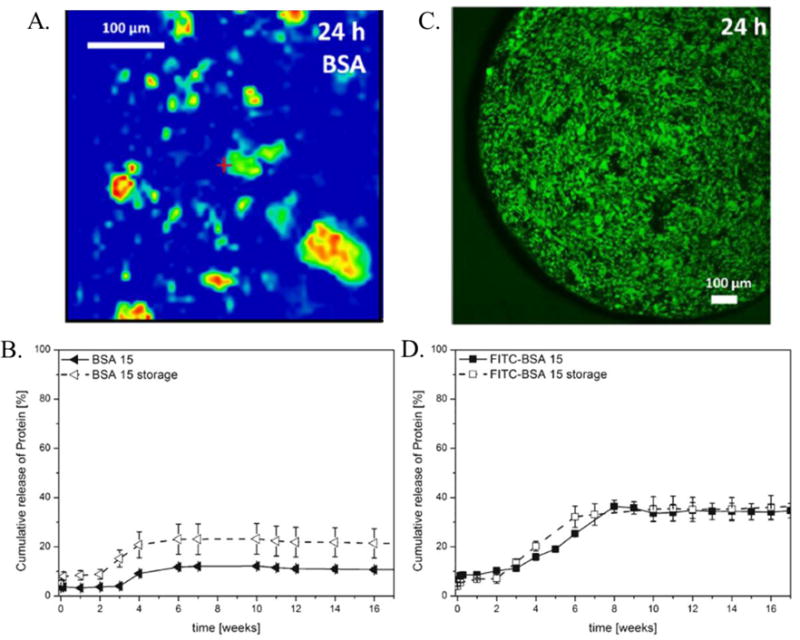
(A.) Dispersion of BSA within a PLGA cylinder prepared via twin-screw extrusion and (B.) in vitro release of BSA from cylinders before and after storage at 30°C for 8 weeks. (C.) Dispersion of BSA modified with hydrophobic FITC dye within a PLGA cylinder prepared via twin-screw extrusion and (D.) in vitro release of BSA modified with hydrophobic FITC dye from cylinders before and after storage at 30°C for 8 weeks.195
The aggregation of proteins within the PLGA matrix can also be mitigated by controlling the initial particle size of the solid protein before melt extrusion with PLGA. In general, a smaller particle size of the protein results in less aggregation and segregation of protein within the PLGA matrix. This phenomenon is thought to be a result of simple separation of adjacent protein particles, preventing proteins from unfolding to form more thermodynamically stable aggregated species with adjacent proteins. The initial protein particle size can be controlled through the drying process when the protein is taken from the aqueous to the solid state or through mechanical forces breaking up particles after drying. Spray drying has been utilized to control the particle size of BSA prior to melt extrusion with PLGA and yielded solid BSA particles exhibiting sizes of 1 – 5 μm.193 Spray drying also allowed for the incorporation of stabilizing additives during protein preparation. The spray dried BSA with excipients exhibited enhanced thermal stability and sustained release after melt extrusion with PLGA. Ball milling has been utilized to mechanically break-up protein solids into smaller particles prior to melt extrusion. BSA subjected to ball milling exhibited a reduction in particle size from 46.7 to 17.7 μm and a dramatic reduction in aggregated regions of BSA within PLGA cylinders after melt extrusion.197 Application of ball milling to reduce the particle size of lysozyme and glucose oxidase prior to melt processing also yielded materials that had decreased aggregated regions of protein within the polymer matrix.199 The recovered milled proteins exhibited enhanced retained secondary structure and enzymatic activity after melt extrusion compared to the unmilled proteins with larger particle sizes. Aggressive milling generates excess physical stress and thermal energy and milling at high frequencies resulted in protein denaturation and instability. Thus, there is an ideal milling window for controlling the solid protein particle size prior to extrusion with PLGA. These examples demonstrate how both the surface chemistry and the size of the solid protein being encapsulated with PLGA have a dramatic effect on the protein aggregation and stability during melt extrusion.
Studies with PLGA/protein blend extrusion have currently focused on model proteins with no therapeutic application to understand the impact of protein physicochemical properties and process parameters on protein stability and release. The expansion of melt extrusion to therapeutically relevant proteins has been largely limited by the amount of protein needed extrusion and the stability of the protein therapeutic. However, a recent study has utilized the virus-like particle (VLP) Qβ to create PLGA based single administration vaccine devices via melt extrusion.200 Qβ is a protein nanoparticle that has been widely studied as a vaccine platform, but also exhibits high thermal stability and can be produced in high yield through recombinant expression. These factors made it an attractive candidate for developing therapeutically relevant vaccination devices using melt extrusion with PLGA. Processing of Qβ with PLGA yielded intact VLPs with limited aggregation and application of the Qβ/PLGA material in vivo resulted in identical antibody titers relative to a standard soluble vaccination schedule (Figure 11). Furthermore, the IgG subtypes generated were identical between the PLGA device vaccination and standard vaccination, indicating the melt processed device did not elicit a different cellular immune response relative to standard vaccination. This study demonstrated the utility of melt extrusion to create therapeutically relevant materials for single administration vaccines. Studies are currently underway expanding the methodology to other viral nanoparticles to create implantable devices for vaccination and cancer immunotherapy applications.
Figure 11.
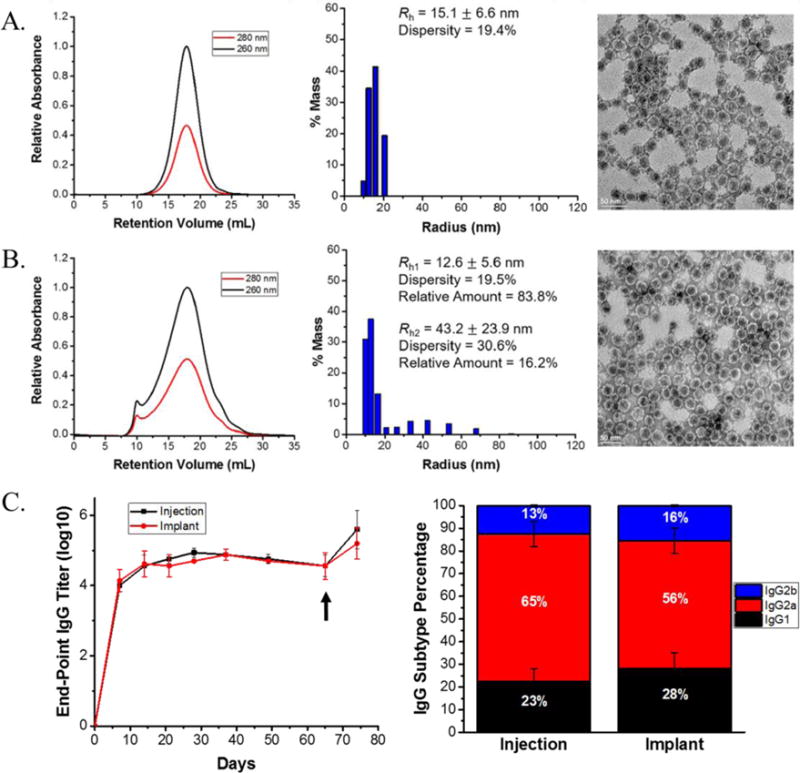
(A.) Size exclusion chromatography chromatogram, dynamic light scattering histogram, and transmission electron micrograph image of native Qβ VLPs showing the correct 30 nm size and icosahedral morphology. (B.) Size exclusion chromatography chromatogram, dynamic light scattering histogram, and transmission electron micrograph image of Qβ VLPs after melt extrusion with PLGA showing the appearance of a small aggregate peak in the chromatogram and histogram. (C.) anti-Qβ IgG end-point titers and subtypes generated of mice immunized via 3 biweekly injections of Qβ solution, denoted “Injection”, and mice implanted once with Qβ/PLGA material, denoted “Implant”. The black arrow indicates an injection of Qβ solution for all mice to challenge the immunological memory.200
Conclusion
Encapsulation of therapeutic peptides and proteins within polymeric materials for sustained release has been a widely studied field of research to decrease the need for repeated injections to elicit a therapeutic effect. Among the polymers studied, PLGA is one of the most widely utilized owing to the high degree of tunability and processability afforded by PLGA. These PLGA/protein materials have been most commonly prepared via emulsion techniques to create injectable microparticle formulations. While emulsion production methods have been effective and have led to several clinically used devices, they can suffer from protein instability and difficulty in scale-up due to batch-to-batch variation. There have been extensive studies that have shown the addition of stabilizing additives and fine-tuning of processing conditions can overcome protein instability. Continuous production methods, such as spray-drying and electrospraying, have been developed to overcome the batch nature of typical emulsion methods. These methodologies can be more readily scaled, but have limitations in optimization of process parameters and throughput.
Melt extrusion has been studied as an alternate continuous production method and protein laden cylindrical implants have been successfully prepared using this technique. There are high thermal and shear stresses during extrusion that can negatively influence protein stability and the impact of additives and process parameters has been researched to increase the stability of the encapsulated proteins. Melt extrusion has almost exclusively focused on model protein systems, however the methodology has been expanded to create materials laden with more complex and therapeutically relevant viral nanoparticles for vaccine applications. While these studies have shown initial success, the incorporation of therapeutically relevant into PLGA implants requires further research into how protein size, biochemical properties, and extrusion conditions affect protein integrity and release.
Production methodologies for protein laden PLGA devices requires in depth study of the factors controlling protein stability and release during protein encapsulation. Balancing of these parameters is imperative in successful device production, however the parameters developed during lab-scale production may not readily apply to scaled-up processing. Transitioning PLGA/protein device manufacturing toward industrial scales will require a focus on more readily scalable techniques in the laboratory setting. Extrusion and other continuous processing methods are advantageous from this stand-point as the parameters developed can be easily applied to larger manufacturing systems. However, the continuous processing methods apply harsher conditions requiring a balance between designing a high-throughput, scalable process that also maintains the structure and function of the encapsulated protein. The knowledge base on production methodologies for protein loaded PLGA materials is vast, however, new processes are being constantly introduced to improve upon therapeutic effect and production efficiency. The past provides a wealth of knowledge, while the future still remains bright for the therapeutic application of these materials and its safety.
Acknowledgments
The authors are financially supported in part by a grant from the Clinical and Translational Science Collaborative of Cleveland, 4UL1TR000439 from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health.
Footnotes
ORCID iD: 0000-0002-0491-3742
ORCID iD: 0000-0001-5869-6942
Contributor Information
Parker W. Lee, Email: parker.w.lee@case.edu, Department of Macromolecular Science and Engineering, School of Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
Jonathan K. Pokorski, Email: jon.pokorski@case.edu, Department of Macromolecular Science and Engineering, School of Engineering, Case Western Reserve University, Cleveland, Ohio 44106, United States.
References
- 1.Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov. 2005;4:298–306. doi: 10.1038/nrd1695. [DOI] [PubMed] [Google Scholar]
- 2.Leader B, Baca QJ, Golan DE. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 3.Lee K, Silva EA, Mooney DJ. Growth factor delivery-based tissue engineering: general approaches and a review of recent developments. J R Soc Interface. 2011;8:153–170. doi: 10.1098/rsif.2010.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinmetz NF. Viral nanoparticles as platforms for next-generation therapeutics and imaging devices. Nanomedicine Nanotechnol Biol Med. 2010;6:634–641. doi: 10.1016/j.nano.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steele JFC, Peyret H, Saunders K, Castells-Graells R, Marsian J, Meshcheriakova Y, et al. Synthetic plant virology for nanobiotechnology and nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9:e1447. doi: 10.1002/wnan.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655–672. doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moeller EH, Jorgensen L. Alternative routes of administration for systemic delivery of protein pharmaceuticals. Drug Discov Today Technol. 2008;5:e89–e94. doi: 10.1016/j.ddtec.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Morlock M, Koll H, Winter G, Kissel T. Microencapsulation of rh-erythropoietin, using biodegradable poly(d,l-lactide-co-glycolide): protein stability and the effects of stabilizing excipients. Eur J Pharm Biopharm. 1997;43:29–36. [Google Scholar]
- 9.Shao PG, Bailey LC. Porcine insulin biodegradable polyester microspheres: stability and in vitro release characteristics. Pharm Dev Technol. 2000;5:1–9. doi: 10.1081/pdt-100100513. [DOI] [PubMed] [Google Scholar]
- 10.Ramakrishna Seeram, Zamani M, Prabhakaran Molamma P. Advances in drug delivery via electrospun and electrosprayed nanomaterials. Int J Nanomedicine. 2013;8:2997. doi: 10.2147/IJN.S43575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagels RF, Prud’homme RK. Polymeric nanoparticles and microparticles for the delivery of peptides, biologics, and soluble therapeutics. J Controlled Release. 2015;219:519–535. doi: 10.1016/j.jconrel.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Langer R, Folkman J. Polymers for the sustained release of proteins and other macromolecules. Nature. 1976;263:797–800. doi: 10.1038/263797a0. [DOI] [PubMed] [Google Scholar]
- 13.Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem Rev. 2016;116:2602–2663. doi: 10.1021/acs.chemrev.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Washington KE, Kularatne RN, Karmegam V, Biewer MC, Stefan MC. Recent advances in aliphatic polyesters for drug delivery applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9:e1446. doi: 10.1002/wnan.1446. [DOI] [PubMed] [Google Scholar]
- 15.Brannon-Peppas L. Recent advances on the use of biodegradable microparticles and nanoparticles in controlled drug delivery. Int J Pharm. 1995;116:1–9. [Google Scholar]
- 16.Heller J. Controlled drug release from poly(ortho esters) — A surface eroding polymer. J Controlled Release. 1985;2:167–177. [Google Scholar]
- 17.Wang Y. FDA’s regulatory science program for generic PLA/PLGA-based drug products. Am Pharm Rev. 2016;20 [Google Scholar]
- 18.Vert M, Mauduit J, Li S. Biodegradation of PLA/GA polymers: increasing complexity. Biomaterials. 1994;15:1209–1213. doi: 10.1016/0142-9612(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 19.Park Peter In Pyo, Jonnalagadda Sriramakamal. Predictors of Glass Transition in the Biodegradable Polylactide and Poly-lactide-co-glycolide Polymers. J Appl Polym Sci. 2006;100:1983–1987. [Google Scholar]
- 20.Burkersroda F von, Schedl L, Göpferich A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials. 2002;23:4221–4231. doi: 10.1016/s0142-9612(02)00170-9. [DOI] [PubMed] [Google Scholar]
- 21.Shive, Anderson Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 22.Lan P, Zhang Y, Gao Q, Shao H, Hu X. Studies on the synthesis and thermal properties of copoly(L-lactic acid/glycolic acid) by direct melt polycondensation. J Appl Polym Sci. 2004;92:2163–2168. [Google Scholar]
- 23.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers. 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilding DK, Reed AM. Biodegradable polymers for use in surgery—polyglycolic/poly(actic acid) homo- and copolymers: 1. Polymer. 1979;20:1459–1464. [Google Scholar]
- 25.Hutchinson FG, Furr BJA. Biodegradable polymer systems for the sustained release of polypeptides. J Controlled Release. 1990;13:279–294. [Google Scholar]
- 26.O’Hagan DT, Jeffery H, Davis SS. The preparation and characterization of poly(lactide-co-glycolide) microparticles: III. Microparticle/polymer degradation rates and the in vitro release of a model protein. Int J Pharm. 1994;103:37–45. [Google Scholar]
- 27.Asano M, Fukuzaki H, Yoshida M, Kumakura M, Mashimo T, Yuasa H, et al. In vivo characteristics of low molecular weight copoly (D,L-lactic acid) formulations with controlled release of LH-RH agonist. Biomaterials. 1989;10:569–573. doi: 10.1016/0142-9612(89)90065-3. [DOI] [PubMed] [Google Scholar]
- 28.Batycky RP, Hanes J, Langer R, Edwards DA. A theoretical model of erosion and macromolecular drug release from biodegrading microspheres. J Pharm Sci. 1997;86:1464–1477. doi: 10.1021/js9604117. [DOI] [PubMed] [Google Scholar]
- 29.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berkland C, Pollauf E, Raman C, Silverman R, Kim K (Kevin), Pack DW. Macromolecule Release from Monodisperse PLG Microspheres: Control of Release Rates and Investigation of Release Mechanism. J Pharm Sci. 2007;96:1176–1191. doi: 10.1002/jps.20948. [DOI] [PubMed] [Google Scholar]
- 31.Cohen S, Yoshioka T, Lucarelli M, Hwang LH, Langer R. Controlled Delivery Systems for Proteins Based on Poly(Lactic/Glycolic Acid) Microspheres. Pharm Res. 1991;8:713–720. doi: 10.1023/a:1015841715384. [DOI] [PubMed] [Google Scholar]
- 32.Alonso MJ, Gupta RK, Min C, Siber GR, Langer R. Biodegradable microspheres as controlled-release tetanus toxoid delivery systems. Vaccine. 1994;12:299–306. doi: 10.1016/0264-410x(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 33.Bodmer D, Kissel T, Traechslin E. Factors influencing the release of peptides and proteins from biodegradable parenteral depot systems. J Controlled Release. 1992;21:129–137. [Google Scholar]
- 34.Cleek RL, Ting KC, G Eskin S, Mikos AG. Microparticles of poly(dl-lactic-co-glycolic acid)/poly(ethylene glycol) blends for controlled drug delivery. J Controlled Release. 1997;48:259–268. [Google Scholar]
- 35.Jiang W, Schwendeman SP. Stabilization and Controlled Release of Bovine Serum Albumin Encapsulated in Poly(D, L-lactide) and Poly(ethylene glycol) Microsphere Blends. Pharm Res. 2001;18:878–885. doi: 10.1023/a:1011009117586. [DOI] [PubMed] [Google Scholar]
- 36.Yeh M-K. The stability of insulin in biodegradable microparticles based on blends of lactide polymers and polyethylene glycol. J Microencapsul. 2000;17:743–756. doi: 10.1080/02652040050161738. [DOI] [PubMed] [Google Scholar]
- 37.Jaganathan KS, Rao YUB, Singh P, Prabakaran D, Gupta S, Jain A, et al. Development of a single dose tetanus toxoid formulation based on polymeric microspheres: a comparative study of poly(D,L-lactic-co-glycolic acid) versus chitosan microspheres. Int J Pharm. 2005;294:23–32. doi: 10.1016/j.ijpharm.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 38.Ruan G, Feng S-S, Li Q-T. Effects of material hydrophobicity on physical properties of polymeric microspheres formed by double emulsion process. J Control Release. 2002;84:151–160. doi: 10.1016/s0168-3659(02)00292-4. [DOI] [PubMed] [Google Scholar]
- 39.Zhu G, Mallery SR, Schwendeman SP. Stabilization of proteins encapsulated in injectable poly (lactide- co-glycolide) Nat Biotechnol. 2000;18:52–57. doi: 10.1038/71916. [DOI] [PubMed] [Google Scholar]
- 40.Zhu G, Schwendeman SP. Stabilization of Proteins Encapsulated in Cylindrical Poly(lactide-co-glycolide) Implants: Mechanism of Stabilization by Basic Additives. Pharm Res. 2000;17:351–357. doi: 10.1023/a:1007513425337. [DOI] [PubMed] [Google Scholar]
- 41.Han FY, Thurecht KJ, Whittaker AK, Smith MT. Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading. Front Pharmacol. 2016;7 doi: 10.3389/fphar.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teekamp N, Duque LF, Frijlink HW, Hinrichs WL, Olinga P. Production methods and stabilization strategies for polymer-based nanoparticles and microparticles for parenteral delivery of peptides and proteins. Expert Opin Drug Deliv. 2015;12:1311–1331. doi: 10.1517/17425247.2015.1003807. [DOI] [PubMed] [Google Scholar]
- 43.Chue P. Long-acting risperidone injection: efficacy, safety, and cost-effectiveness of the first long-acting atypical antipsychotic. Neuropsychiatr Dis Treat. 2007;3:13–39. doi: 10.2147/nedt.2007.3.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhee Y-S, Sohn M, Woo BH, Thanoo BC, DeLuca PP, Mansour HM. Sustained-Release Delivery of Octreotide from Biodegradable Polymeric Microspheres. AAPS PharmSciTech. 2011;12:1293–1301. doi: 10.1208/s12249-011-9693-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arshady R. Preparation of biodegradable microspheres and microcapsules: 2. Polylactides and related polyesters. J Controlled Release. 1991;17:1–21. [Google Scholar]
- 46.Ito F, Fujimori H, Makino K. Factors affecting the loading efficiency of water-soluble drugs in PLGA microspheres. Colloids Surf B Biointerfaces. 2008;61:25–29. doi: 10.1016/j.colsurfb.2007.06.029. [DOI] [PubMed] [Google Scholar]
- 47.Jain R, Shah NH, Malick AW, Rhodes CT. Controlled drug delivery by biodegradable poly(ester) devices: different preparative approaches. Drug Dev Ind Pharm. 1998;24:703–727. doi: 10.3109/03639049809082719. [DOI] [PubMed] [Google Scholar]
- 48.Lewis Danny H. Biodegradable polymers as drug delivery systems. Marcel Dekker; New York: 1990. Controlled release of bioactive agents from lactide/glycolide polymers; pp. 1–41. [Google Scholar]
- 49.Wang Q, Tan MT, Keegan BP, Barry MA, Heffernan MJ. Time course study of the antigen-specific immune response to a PLGA microparticle vaccine formulation. Biomaterials. 2014;35:8385–8393. doi: 10.1016/j.biomaterials.2014.05.067. [DOI] [PubMed] [Google Scholar]
- 50.Challacombe SJ, Rahman D, Jeffery H, Davis SS, O’Hagan DT. Enhanced secretory IgA and systemic IgG antibody responses after oral immunization with biodegradable microparticles containing antigen. Immunology. 1992;76:164–168. [PMC free article] [PubMed] [Google Scholar]
- 51.Wu J, Qi K, Xu Z, Wan J. Glucagon-like peptide-2-loaded microspheres as treatment for ulcerative colitis in the murine model. J Microencapsul. 2015;32:598–607. doi: 10.3109/02652048.2015.1065923. [DOI] [PubMed] [Google Scholar]
- 52.Sanders LM, Kent JS, McRae GI, Vickery BH, Tice TR, Lewis DH. Controlled release of a luteinizing hormone-releasing hormone analogue from poly(d,l-lactide-co-glycolide) microspheres. J Pharm Sci. 1984;73:1294–1297. doi: 10.1002/jps.2600730927. [DOI] [PubMed] [Google Scholar]
- 53.Cleland JL, Mac A, Boyd B, Yang J, Duenas ET, Yeung D, et al. The stability of recombinant human growth hormone in poly(lactic-co-glycolic acid) (PLGA) microspheres. Pharm Res. 1997;14:420–425. doi: 10.1023/a:1012031012367. [DOI] [PubMed] [Google Scholar]
- 54.Lochmann A, Nitzsche H, von Einem S, Schwarz E, Mäder K. The influence of covalently linked and free polyethylene glycol on the structural and release properties of rhBMP-2 loaded microspheres. J Control Release. 2010;147:92–100. doi: 10.1016/j.jconrel.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 55.Mundargi RC, Babu VR, Rangaswamy V, Patel P, Aminabhavi TM. Nano/micro technologies for delivering macromolecular therapeutics using poly(D,L-lactide-co-glycolide) and its derivatives. J Control Release. 2008;125:193–209. doi: 10.1016/j.jconrel.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 56.Mao S, Xu J, Cai C, Germershaus O, Schaper A, Kissel T. Effect of WOW process parameters on morphology and burst release of FITC-dextran loaded PLGA microspheres. Int J Pharm. 2007;334:137–148. doi: 10.1016/j.ijpharm.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 57.Chaisri W, Hennink WE, Okonogi S. Preparation and characterization of cephalexin loaded PLGA microspheres. Curr Drug Deliv. 2009;6:69–75. doi: 10.2174/156720109787048186. [DOI] [PubMed] [Google Scholar]
- 58.Florence AT, Whitehill D. The formulation and stability of multiple emulsions. Int J Pharm. 1982;11:277–308. [Google Scholar]
- 59.Yang Y-Y, Chung T-S, Ping Ng N. Morphology, drug distribution, and in vitro release profiles of biodegradable polymeric microspheres containing protein fabricated by double-emulsion solvent extraction/evaporation method. Biomaterials. 2001;22:231–241. doi: 10.1016/s0142-9612(00)00178-2. [DOI] [PubMed] [Google Scholar]
- 60.Jeffery H, Davis SS, O’Hagan DT. The preparation and characterization of poly(lactide-co-glycolide) microparticles. II. The entrapment of a model protein using a (water-in-oil)-in-water emulsion solvent evaporation technique. Pharm Res. 1993;10:362–368. doi: 10.1023/a:1018980020506. [DOI] [PubMed] [Google Scholar]
- 61.Park TG, Yong Lee H, Sung Nam Y. A new preparation method for protein loaded poly(d,l-lactic-co-glycolic acid) microspheres and protein release mechanism study. J Controlled Release. 1998;55:181–191. doi: 10.1016/s0168-3659(98)00050-9. [DOI] [PubMed] [Google Scholar]
- 62.Blanco D, Alonso MJ. Protein encapsulation and release from poly(lactide-co-glycolide) microspheres: effect of the protein and polymer properties and of the co-encapsulation of surfactants. Eur J Pharm Biopharm. 1998;45:285–294. doi: 10.1016/s0939-6411(98)00011-3. [DOI] [PubMed] [Google Scholar]
- 63.Bawa R, A Siegel R, Marasca B, Karel M, Langer R. An explanation for the controlled release of macromolecules from polymers. J Controlled Release. 1985;1:259–267. [Google Scholar]
- 64.Péan J-M, Venier-Julienne M-C, Boury F, Menei P, Denizot B, Benoit J-P. NGF release from poly(d,l-lactide-co-glycolide) microspheres. Effect of some formulation parameters on encapsulated NGF stability. J Controlled Release. 1998;56:175–187. doi: 10.1016/s0168-3659(98)00086-8. [DOI] [PubMed] [Google Scholar]
- 65.Lam XM, Duenas ET, Daugherty AL, Levin N, Cleland JL. Sustained release of recombinant human insulin-like growth factor-I for treatment of diabetes. J Controlled Release. 2000;67:281–292. doi: 10.1016/s0168-3659(00)00224-8. [DOI] [PubMed] [Google Scholar]
- 66.Sophocleous AM, Desai K-GH, Mazzara JM, Tong L, Cheng J-X, Olsen KF, et al. The nature of peptide interactions with acid end-group PLGAs and facile aqueous-based microencapsulation of therapeutic peptides. J Controlled Release. 2013;172:662–670. doi: 10.1016/j.jconrel.2013.08.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crotts G, Park TG. Stability and release of bovine serum albumin encapsulated within poly(d,l-lactide-co-glycolide) microparticles. J Controlled Release. 1997;44:123–134. [Google Scholar]
- 68.Paillard-Giteau A, Tran VT, Thomas O, Garric X, Coudane J, Marchal S, et al. Effect of various additives and polymers on lysozyme release from PLGA microspheres prepared by an s/o/w emulsion technique. Eur J Pharm Biopharm. 2010;75:128–136. doi: 10.1016/j.ejpb.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 69.Turner P, Coombes AGA, Al-Rubeai M. Measuring the heterogeneity of protein loading in PLG microspheres using flow cytometry. J Controlled Release. 2004;96:193–205. doi: 10.1016/j.jconrel.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 70.Wong HM, Wang JJ, Wang C-H. In Vitro Sustained Release of Human Immunoglobulin G from Biodegradable Microspheres. Ind Eng Chem Res. 2001;40:933–948. [Google Scholar]
- 71.Finkelstein AV, Galzitskaya OV. Physics of protein folding. Phys Life Rev. 2004;1:23–56. doi: 10.1016/j.plrev.2017.01.025. [DOI] [PubMed] [Google Scholar]
- 72.Dill KA. Dominant forces in protein folding. Biochemistry (Mosc) 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 73.Norde W, Favier JP. Structure of adsorbed and desorbed proteins. Colloids Surf. 1992;64:87–93. [Google Scholar]
- 74.Zhai J, Miles AJ, Pattenden LK, Lee T-H, Augustin MA, Wallace BA, et al. Changes in β-Lactoglobulin Conformation at the Oil/Water Interface of Emulsions Studied by Synchrotron Radiation Circular Dichroism Spectroscopy. Biomacromolecules. 2010;11:2136–2142. doi: 10.1021/bm100510j. [DOI] [PubMed] [Google Scholar]
- 75.Zhai J li, Day L, Aguilar M-I, Wooster TJ. Protein folding at emulsion oil/water interfaces. Curr Opin Colloid Interface Sci. 2013;18:257–271. [Google Scholar]
- 76.Ghaderi R, Carlfors J. Biological activity of lysozyme after entrapment in poly(d,l-lactide-co-glycolide)-microspheres. Pharm Res. 1997;14:1556–1562. doi: 10.1023/a:1012122200381. [DOI] [PubMed] [Google Scholar]
- 77.Van de Weert M, Hoechstetter J, Hennink WE, Crommelin DJA. The effect of a water/organic solvent interface on the structural stability of lysozyme. J Controlled Release. 2000;68:351–359. doi: 10.1016/s0168-3659(00)00277-7. [DOI] [PubMed] [Google Scholar]
- 78.Cleland JL, Jones AJ. Stable formulations of recombinant human growth hormone and interferon-gamma for microencapsulation in biodegradable microspheres. Pharm Res. 1996;13:1464–1475. doi: 10.1023/a:1016063109373. [DOI] [PubMed] [Google Scholar]
- 79.Kim HK, Park TG. Microencapsulation of human growth hormone within biodegradable polyester microspheres: Protein aggregation stability and incomplete release mechanism. Biotechnol Bioeng. 1999;65:659–667. doi: 10.1002/(sici)1097-0290(19991220)65:6<659::aid-bit6>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 80.Johansen P, Men Y, Audran R, Corradin G, Merkle HP, Gander B. Improving Stability and Release Kinetics of Microencapsulated Tetanus Toxoid by Co-Encapsulation of Additives. Pharm Res. 1998;15:1103–1110. doi: 10.1023/a:1011998615267. [DOI] [PubMed] [Google Scholar]
- 81.Sah H. Stabilization of proteins against methylene chloride/water interface-induced denaturation and aggregation. J Control Release. 1999;58:143–151. doi: 10.1016/s0168-3659(98)00148-5. [DOI] [PubMed] [Google Scholar]
- 82.Pérez C, Griebenow K. Improved activity and stability of lysozyme at the water/CH2Cl2 interface: enzyme unfolding and aggregation and its prevention by polyols. J Pharm Pharmacol. 2001;53:1217–1226. doi: 10.1211/0022357011776667. [DOI] [PubMed] [Google Scholar]
- 83.Pérez-Rodriguez C, Montano N, Gonzalez K, Griebenow K. Stabilization of alpha-chymotrypsin at the CH2Cl2/water interface and upon water-in-oil-in-water encapsulation in PLGA microspheres. J Control Release. 2003;89:71–85. doi: 10.1016/s0168-3659(03)00074-9. [DOI] [PubMed] [Google Scholar]
- 84.Diwan M, Park TG. Pegylation enhances protein stability during encapsulation in PLGA microspheres. J Controlled Release. 2001;73:233–244. doi: 10.1016/s0168-3659(01)00292-9. [DOI] [PubMed] [Google Scholar]
- 85.Pai SS, Tilton RD, Przybycien TM. Poly(ethylene glycol)-Modified Proteins: Implications for Poly(lactide-co-glycolide)-Based Microsphere Delivery. AAPS J. 2009;11:88–98. doi: 10.1208/s12248-009-9081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Montalvo-Ortiz BL, Sosa B, Griebenow K. Improved Enzyme Activity and Stability in Polymer Microspheres by Encapsulation of Protein Nanospheres. AAPS PharmSciTech. 2012;13:632. doi: 10.1208/s12249-012-9794-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lim MPA, Lee WL, Widjaja E, Loo SCJ. One-step fabrication of core–shell structured alginate–PLGA/PLLA microparticles as a novel drug delivery system for water soluble drugs. Biomater Sci. 2013;1:486–493. doi: 10.1039/c3bm00175j. [DOI] [PubMed] [Google Scholar]
- 88.Li W-I, Anderson KW, Mehta RC, Deluca PP. Prediction of solvent removal profile and effect on properties for peptide-loaded PLGA microspheres prepared by solvent extraction/evaporation method. J Controlled Release. 1995;37:199–214. [Google Scholar]
- 89.Emami J, Hamishehkar H, Najafabadi AR, Gilani K, Minaiyan M, Mahdavi H, et al. A Novel Approach to Prepare Insulin-Loaded Poly (Lactic-Co-Glycolic Acid) Microcapsules and the Protein Stability Study. J Pharm Sci. 2009;98:1712–1731. doi: 10.1002/jps.21544. [DOI] [PubMed] [Google Scholar]
- 90.Carrascosa C, Espejo L, Torrado S, Torrado JJ. Effect of γ-Sterilization Process on PLGA Microspheres Loaded with Insulin-Like Growth Factor - I (IGF-I) J Biomater Appl. 2016;18:95–108. doi: 10.1177/088532803038026. [DOI] [PubMed] [Google Scholar]
- 91.Shi M, Yang Y-Y, Chaw C-S, Goh S-H, Moochhala SM, Ng S, et al. Double walled POE/PLGA microspheres: encapsulation of water-soluble and water-insoluble proteins and their release properties. J Controlled Release. 2003;89:167–177. doi: 10.1016/s0168-3659(02)00493-5. [DOI] [PubMed] [Google Scholar]
- 92.Blanco-Príeto MJ, Fattal E, Gulik A, Dedieu JC, Roques BP, Couvreur P. Characterization and morphological analysis of a cholecystokinin derivative peptide-loaded poly(lactide-co-glycolide) microspheres prepared by a water-in-oil-in-water emulsion solvent evaporation method. J Controlled Release. 1997;43:81–87. [Google Scholar]
- 93.Silva AL, Rosalia RA, Sazak A, Carstens MG, Ossendorp F, Oostendorp J, et al. Optimization of encapsulation of a synthetic long peptide in PLGA nanoparticles: low-burst release is crucial for efficient CD8(+) T cell activation. Eur J Pharm Biopharm. 2013;83:338–345. doi: 10.1016/j.ejpb.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 94.Tracy MA. Development and Scale-up of a Microsphere Protein Delivery System. Biotechnol Prog. 1998;14:108–115. doi: 10.1021/bp9701271. [DOI] [PubMed] [Google Scholar]
- 95.Selmin F, Puoci F, Parisi OI, Franzé S, Musazzi UM, Cilurzo F. Caffeic Acid-PLGA Conjugate to Design Protein Drug Delivery Systems Stable to Irradiation. J Funct Biomater. 2015;6:1–13. doi: 10.3390/jfb6010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu J, Williams GR, Branford-White C, Li H, Li Y, Zhu L-M. Liraglutide-loaded poly(lactic-co-glycolic acid) microspheres: Preparation and in vivo evaluation. Eur J Pharm Sci. 2016;92:28–38. doi: 10.1016/j.ejps.2016.06.018. [DOI] [PubMed] [Google Scholar]
- 97.Lorenz M, Evers A, Wagner M. Recent progress and future options in the development of GLP-1 receptor agonists for the treatment of diabesity. Bioorg Med Chem Lett. 2013;23:4011–4018. doi: 10.1016/j.bmcl.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 98.Wang Q, Barry MA, Seid CA, Hudspeth EM, McAtee CP, Heffernan MJ. 3M052 as an adjuvant for a PLGA microparticle-based Leishmania donovani recombinant protein vaccine. J Biomed Mater Res B Appl Biomater. 2017 doi: 10.1002/jbm.b.33965. [DOI] [PubMed] [Google Scholar]
- 99.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 100.Rice-Ficht AC, Arenas-Gamboa AM, Kahl-McDonagh MM, Ficht TA. Polymeric particles in vaccine delivery. Curr Opin Microbiol. 2010;13:106–112. doi: 10.1016/j.mib.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 101.Jalil R, Nixon JR. Biodegradable poly(lactic acid) and poly(lactide-co-glycolide) microcapsules: problems associated with preparative techniques and release properties. J Microencapsul. 1990;7:297–325. doi: 10.3109/02652049009021842. [DOI] [PubMed] [Google Scholar]
- 102.Nihant N, Grandfils C, Jérôme R, Teyssié P. Microencapsulation by coacervation of poly(lactide-co-glycolide) IV. Effect of the processing parameters on coacervation and encapsulation. J Controlled Release. 1995;35:117–125. [Google Scholar]
- 103.Nihant N, Stassen S, Grandfils C, Jérome R, Teyssié P, Goffinet G. Microencapsulation by coacervation of poly(lactide-co-glycolide). III. Characterization of the final microspheres. Polym Int. 1994;34:289–299. [Google Scholar]
- 104.Sanders LM, McRae GI, Vitale KM, Kell BA. Controlled delivery of an LHRH analogue from biodegradable injectable microspheres. J Controlled Release. 1985;2:187–195. [Google Scholar]
- 105.Leelarasamee N, Howard SA, Malanga CJ, Luzzi LA, Hogan TF, Kandzari SJ, et al. Kinetics of drug release from polylactic acid—hydrocortisone microcapsules. J Microencapsul. 1986;3:171–179. doi: 10.3109/02652048609031571. [DOI] [PubMed] [Google Scholar]
- 106.Lapka GG, Mason NS, Thies C. Process for preparation of microcapsules. 1986 [Google Scholar]
- 107.Thomasin C, Hô NT, Merkle HP, Gander B. Drug microencapsulation by PLA/PLGA coacervation in the light of thermodynamics. 1. Overview and theoretical considerations. J Pharm Sci. 1998;87:259–268. doi: 10.1021/js970047r. [DOI] [PubMed] [Google Scholar]
- 108.Pérez C, Griebenow K. Effect of salts on lysozyme stability at the water-oil interface and upon encapsulation in poly(lactic-co-glycolic) acid microspheres. Biotechnol Bioeng. 2003;82:825–832. doi: 10.1002/bit.10632. [DOI] [PubMed] [Google Scholar]
- 109.Stassen S, Nihant N, Martin V, Grandfils C, Jérôme R, Teyssié P. Microencapsulation by coacervation of poly(lactide-co-glycolide): 1. Physicochemical characteristics of the phase separation process. Polymer. 1994;35:777–785. [Google Scholar]
- 110.Wu XS. Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers: Part III. Drug delivery application. Artif Cells Blood Substit Immobil Biotechnol. 2004;32:575–591. doi: 10.1081/bio-200039635. [DOI] [PubMed] [Google Scholar]
- 111.Heinrich M, Wolf BA. Kinetics of phase separation: trapping of molecules in nonequilibrium phases. Macromolecules. 1990;23:590–596. [Google Scholar]
- 112.Wu Xue Shen. Encyclopedic Handbook of Biomaterials and Bioengineering. Marcel Dekker; New York: 1995. Preparation, characterization, and drug delivery applications of microspheres based on biodegradable lactic/glycolic acid polymers; pp. 1151–2000. [Google Scholar]
- 113.Esparza I, Kissel T. Parameters affecting the immunogenicity of microencapsulated tetanus toxoid. Vaccine. 1992;10:714–720. doi: 10.1016/0264-410x(92)90094-z. [DOI] [PubMed] [Google Scholar]
- 114.Ruiz JM, Tissier B, Benoit JP. Microencapsulation of peptide: a study of the phase separation of poly(d,l-lactic acid-co-glycolic acid) copolymers 50/50 by silicone oil. Int J Pharm. 1989;49:69–77. [Google Scholar]
- 115.Black KA, Priftis D, Perry SL, Yip J, Byun WY, Tirrell M. Protein Encapsulation via Polypeptide Complex Coacervation. ACS Macro Lett. 2014;3:1088–1091. doi: 10.1021/mz500529v. [DOI] [PubMed] [Google Scholar]
- 116.Chu H, Gao J, Chen C-W, Huard J, Wang Y. Injectable fibroblast growth factor-2 coacervate for persistent angiogenesis. Proc Natl Acad Sci. 2011;108:13444–13449. doi: 10.1073/pnas.1110121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schwendeman SP, Shah RB, Bailey BA, Schwendeman AS. Injectable controlled release depots for large molecules. J Control Release. 2014;190:240–253. doi: 10.1016/j.jconrel.2014.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Synder HE. Pharmaceutical spray drying: solid-dose process technology platform for the 21st century. Ther Deliv. 2012;3:901–912. doi: 10.4155/tde.12.64. [DOI] [PubMed] [Google Scholar]
- 119.Huang LX, Kumar K, Mujumdar AS. A comparative study of a spray dryer with rotary disc atomizer and pressure nozzle using computational fluid dynamic simulations. Chem Eng Process Process Intensif. 2006;45:461–470. [Google Scholar]
- 120.Panda A, Meena J, Katara R, Majumdar DK. Formulation and characterization of clozapine and risperidone co-entrapped spray-dried PLGA nanoparticles. Pharm Dev Technol. 2016;21:43–53. doi: 10.3109/10837450.2014.965324. [DOI] [PubMed] [Google Scholar]
- 121.Bittner B, Kissel T. Ultrasonic atomization for spray drying: a versatile technique for the preparation of protein loaded biodegradable microspheres. J Microencapsul. 1999;16:325–341. doi: 10.1080/026520499289059. [DOI] [PubMed] [Google Scholar]
- 122.Wan F, Maltesen MJ, Andersen SK, Bjerregaard S, Baldursdottir SG, Foged C, et al. Modulating Protein Release Profiles by Incorporating Hyaluronic Acid into PLGA Microparticles Via a Spray Dryer Equipped with a 3-Fluid Nozzle. Pharm Res. 2014;31:2940–2951. doi: 10.1007/s11095-014-1387-2. [DOI] [PubMed] [Google Scholar]
- 123.Pabari RM, Sunderland T, Ramtoola Z. Investigation of a novel 3-fluid nozzle spray drying technology for the engineering of multifunctional layered microparticles. Expert Opin Drug Deliv. 2012;9:1463–1474. doi: 10.1517/17425247.2012.734295. [DOI] [PubMed] [Google Scholar]
- 124.Wan F, Yang M. Design of PLGA-based depot delivery systems for biopharmaceuticals prepared by spray drying. Int J Pharm. 2016;498:82–95. doi: 10.1016/j.ijpharm.2015.12.025. [DOI] [PubMed] [Google Scholar]
- 125.Filková I, Huang L, Mujumdar A. Handbook of Industrial Drying. Fourth. CRC Press; 2014. Industrial Spray Drying Systems; pp. 191–226. [Google Scholar]
- 126.Van Oort M, Sacchetti M. Inhalation Aerosols. CRC Press; 2006. Spray-Drying and Supercritical Fluid Particle Generation Techniques; pp. 307–346. [Google Scholar]
- 127.Cal K, Sollohub K. Spray Drying Technique. I: Hardware and Process Parameters. J Pharm Sci. 2010;99:575–586. doi: 10.1002/jps.21886. [DOI] [PubMed] [Google Scholar]
- 128.Vehring R, Foss WR, Lechuga-Ballesteros D. Particle formation in spray drying. J Aerosol Sci. 2007;38:728–746. [Google Scholar]
- 129.Wan F, Bohr A, Maltesen MJ, Bjerregaard S, Foged C, Rantanen J, et al. Critical Solvent Properties Affecting the Particle Formation Process and Characteristics of Celecoxib-Loaded PLGA Microparticles via Spray-Drying. Pharm Res. 2013;30:1065–1076. doi: 10.1007/s11095-012-0943-x. [DOI] [PubMed] [Google Scholar]
- 130.Takada S, Uda Y, Toguchi H, Ogawa Y. Application of a spray drying technique in the production of TRH-containing injectable sustained-release microparticles of biodegradable polymers. PDA J Pharm Sci Technol. 1995;49:180–184. [PubMed] [Google Scholar]
- 131.Wan F, Wu JX, Bohr A, Baldursdottir SG, Maltesen MJ, Bjerregaard S, et al. Impact of PLGA molecular behavior in the feed solution on the drug release kinetics of spray dried microparticles. Polymer. 2013;54:5920–5927. [Google Scholar]
- 132.Lebrun P, Krier F, Mantanus J, Grohganz H, Yang M, Rozet E, et al. Design space approach in the optimization of the spray-drying process. Eur J Pharm Biopharm. 2012;80:226–234. doi: 10.1016/j.ejpb.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 133.Meeus J, Lenaerts M, Scurr DJ, Amssoms K, Davies MC, Roberts CJ, et al. The Influence of Spray-Drying Parameters on Phase Behavior, Drug Distribution, and In Vitro Release of Injectable Microspheres for Sustained Release. J Pharm Sci. 2015;104:1451–1460. doi: 10.1002/jps.24361. [DOI] [PubMed] [Google Scholar]
- 134.Leiske DL, Shieh IC, Tse ML. A Method To Measure Protein Unfolding at an Air-Liquid Interface. Langmuir ACS J Surf Colloids. 2016;32:9930–9937. doi: 10.1021/acs.langmuir.6b02267. [DOI] [PubMed] [Google Scholar]
- 135.Maa YF, Hsu CC. Protein denaturation by combined effect of shear and air-liquid interface. Biotechnol Bioeng. 1997;54:503–512. doi: 10.1002/(SICI)1097-0290(19970620)54:6<503::AID-BIT1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 136.Maa YF, Prestrelski SJ. Biopharmaceutical powders: particle formation and formulation considerations. Curr Pharm Biotechnol. 2000;1:283–302. doi: 10.2174/1389201003378898. [DOI] [PubMed] [Google Scholar]
- 137.Wan F, Maltesen MJ, Andersen SK, Bjerregaard S, Foged C, Rantanen J, et al. One-step production of protein-loaded PLGA microparticles via spray drying using 3-fluid nozzle. Pharm Res. 2014;31:1967–1977. doi: 10.1007/s11095-014-1299-1. [DOI] [PubMed] [Google Scholar]
- 138.Zeng J, Chen X, Xu X, Liang Q, Bian X, Yang L, et al. Ultrafine fibers electrospun from biodegradable polymers. J Appl Polym Sci. 2003;89:1085–1092. [Google Scholar]
- 139.Subbiah T, Bhat GS, Tock RW, Parameswaran S, Ramkumar SS. Electrospinning of nanofibers. J Appl Polym Sci. 2005;96:557–569. [Google Scholar]
- 140.Almería B, Deng W, Fahmy TM, Gomez A. Controlling the morphology of electrospray-generated PLGA microparticles for drug delivery. J Colloid Interface Sci. 2010;343:125–133. doi: 10.1016/j.jcis.2009.10.002. [DOI] [PubMed] [Google Scholar]
- 141.Huang Z-M, Zhang Y-Z, Kotaki M, Ramakrishna S. A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Compos Sci Technol. 2003;63:2223–2253. [Google Scholar]
- 142.Sridhar R, Ramakrishna S. Electrosprayed nanoparticles for drug delivery and pharmaceutical applications. Biomatter. 2013;3:e24281. doi: 10.4161/biom.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zong X, Kim K, Fang D, Ran S, Hsiao BS, Chu B. Structure and process relationship of electrospun bioabsorbable nanofiber membranes. Polymer. 2002;43:4403–4412. [Google Scholar]
- 144.Shenoy SL, Bates WD, Frisch HL, Wnek GE. Role of chain entanglements on fiber formation during electrospinning of polymer solutions: good solvent, non-specific polymer–polymer interaction limit. Polymer. 2005;46:3372–3384. [Google Scholar]
- 145.Luo CJ, Stride E, Edirisinghe M. Mapping the Influence of Solubility and Dielectric Constant on Electrospinning Polycaprolactone Solutions. Macromolecules. 2012;45:4669–4680. [Google Scholar]
- 146.Lavielle N, Hébraud A, Schlatter G, Thöny-Meyer L, Rossi RM, Popa A-M. Simultaneous Electrospinning and Electrospraying: A Straightforward Approach for Fabricating Hierarchically Structured Composite Membranes. ACS Appl Mater Interfaces. 2013;5:10090–10097. doi: 10.1021/am402676m. [DOI] [PubMed] [Google Scholar]
- 147.Ramier J, Bouderlique T, Stoilova O, Manolova N, Rashkov I, Langlois V, et al. Biocomposite scaffolds based on electrospun poly(3-hydroxybutyrate) nanofibers and electrosprayed hydroxyapatite nanoparticles for bone tissue engineering applications. Mater Sci Eng C Mater Biol Appl. 2014;38:161–169. doi: 10.1016/j.msec.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 148.Malik SA, Ng WH, Bowen J, Tang J, Gomez A, Kenyon AJ, et al. Electrospray synthesis and properties of hierarchically structured PLGA TIPS microspheres for use as controlled release technologies. J Colloid Interface Sci. 2016;467:220–229. doi: 10.1016/j.jcis.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 149.Cui W, Li X, Zhou S, Weng J. Investigation on process parameters of electrospinning system through orthogonal experimental design. J Appl Polym Sci. 2007;103:3105–3112. [Google Scholar]
- 150.Bohr A, Yang M, Baldursdóttir S, Kristensen J, Dyas M, Stride E, et al. Particle formation and characteristics of Celecoxib-loaded poly(lactic-co-glycolic acid) microparticles prepared in different solvents using electrospraying. Polymer. 2012;53:3220–3229. [Google Scholar]
- 151.Jahangiri A, Adibkia K. Applications of electrospinning/electrospraying in drug delivery. BioImpacts BI. 2016;6:1–2. doi: 10.15171/bi.2016.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li M, Mondrinos MJ, Gandhi MR, Ko FK, Weiss AS, Lelkes PI. Electrospun protein fibers as matrices for tissue engineering. Biomaterials. 2005;26:5999–6008. doi: 10.1016/j.biomaterials.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 153.Kim S-E, Lee PW, Pokorski JK. Biologically Triggered Delivery of EGF from Polymer Fiber Patches. ACS Macro Lett. 2017;6:593–597. doi: 10.1021/acsmacrolett.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zomer Volpato F, Almodóvar J, Erickson K, Popat KC, Migliaresi C, Kipper MJ. Preservation of FGF-2 bioactivity using heparin-based nanoparticles, and their delivery from electrospun chitosan fibers. Acta Biomater. 2012;8:1551–1559. doi: 10.1016/j.actbio.2011.12.023. [DOI] [PubMed] [Google Scholar]
- 155.Zou J, Yang Y, Liu Y, Chen F, Li X. Release kinetics and cellular profiles for bFGF-loaded electrospun fibers: Effect of the conjugation density and molecular weight of heparin. Polymer. 2011;52:3357–3367. [Google Scholar]
- 156.Luo Y, Nartker S, Miller H, Hochhalter D, Wiederoder M, Wiederoder S, et al. Surface functionalization of electrospun nanofibers for detecting E. coli O157:H7 and BVDV cells in a direct-charge transfer biosensor. Biosens Bioelectron. 2010;26:1612–1617. doi: 10.1016/j.bios.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 157.Kim S-E, Wallat JD, Harker EC, Advincula AA, Pokorski JK. Multifunctional and spatially controlled bioconjugation to melt coextruded nanofibers. Polym Chem. 2015;6:5683–5692. doi: 10.1039/C5PY00282F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang Y, Xia T, Zhi W, Wei L, Weng J, Zhang C, et al. Promotion of skin regeneration in diabetic rats by electrospun core-sheath fibers loaded with basic fibroblast growth factor. Biomaterials. 2011;32:4243–4254. doi: 10.1016/j.biomaterials.2011.02.042. [DOI] [PubMed] [Google Scholar]
- 159.Chen X, Wang J, An Q, Li D, Liu P, Zhu W, et al. Electrospun poly(l-lactic acid-co-ɛ-caprolactone) fibers loaded with heparin and vascular endothelial growth factor to improve blood compatibility and endothelial progenitor cell proliferation. Colloids Surf B Biointerfaces. 2015;128:106–114. doi: 10.1016/j.colsurfb.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 160.Yao S, Liu H, Yu S, Li Y, Wang X, Wang L. Drug-nanoencapsulated PLGA microspheres prepared by emulsion electrospray with controlled release behavior. Regen Biomater. 2016;3:309–317. doi: 10.1093/rb/rbw033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yang Y, Li X, Qi M, Zhou S, Weng J. Release pattern and structural integrity of lysozyme encapsulated in core-sheath structured poly(DL-lactide) ultrafine fibers prepared by emulsion electrospinning. Eur J Pharm Biopharm. 2008;69:106–116. doi: 10.1016/j.ejpb.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 162.Yaghoobi N, Faridi Majidi R, Faramarzi M ali, Baharifar H, Amani A. Preparation, Optimization and Activity Evaluation of PLGA/Streptokinase Nanoparticles Using Electrospray. Adv Pharm Bull. 2017;7:131–139. doi: 10.15171/apb.2017.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhang L, Huang J, Si T, Xu RX. Coaxial electrospray of microparticles and nanoparticles for biomedical applications. Expert Rev Med Devices. 2012;9:595–612. doi: 10.1586/erd.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Enayati M, Chang M-W, Bragman F, Edirisinghe M, Stride E. Electrohydrodynamic preparation of particles, capsules and bubbles for biomedical engineering applications. Colloids Surf Physicochem Eng Asp. 2011;382:154–164. [Google Scholar]
- 165.Zamani M, Prabhakaran MP, Thian ES, Ramakrishna S. Protein encapsulated core-shell structured particles prepared by coaxial electrospraying: investigation on material and processing variables. Int J Pharm. 2014;473:134–143. doi: 10.1016/j.ijpharm.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 166.Jiang H, Hu Y, Li Y, Zhao P, Zhu K, Chen W. A facile technique to prepare biodegradable coaxial electrospun nanofibers for controlled release of bioactive agents. J Controlled Release. 2005;108:237–243. doi: 10.1016/j.jconrel.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 167.Yuan S, Lei F, Liu Z, Tong Q, Si T, Xu RX. Coaxial Electrospray of Curcumin-Loaded Microparticles for Sustained Drug Release. PLOS ONE. 2015;10:e0132609. doi: 10.1371/journal.pone.0132609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ramachandran R, Junnuthula VR, Gowd GS, Ashokan A, Thomas J, Peethambaran R, et al. Theranostic 3-Dimensional nano brain-implant for prolonged and localized treatment of recurrent glioma. Sci Rep. 2017;7:srep43271. doi: 10.1038/srep43271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lee Y-H, Bai M-Y, Chen D-R. Multidrug encapsulation by coaxial tri-capillary electrospray. Colloids Surf B Biointerfaces. 2011;82:104–110. doi: 10.1016/j.colsurfb.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 170.Liao I-C, Leong KW. Efficacy of engineered FVIII-producing skeletal muscle enhanced by growth factor-releasing co-axial electrospun fibers. Biomaterials. 2011;32:1669–1677. doi: 10.1016/j.biomaterials.2010.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Xie J, Ng WJ, Lee LY, Wang C-H. Encapsulation of protein drugs in biodegradable microparticles by co-axial electrospray. J Colloid Interface Sci. 2008;317:469–476. doi: 10.1016/j.jcis.2007.09.082. [DOI] [PubMed] [Google Scholar]
- 172.Xie J, Wang C-H. Encapsulation of proteins in biodegradable polymeric microparticles using electrospray in the Taylor cone-jet mode. Biotechnol Bioeng. 2007;97:1278–1290. doi: 10.1002/bit.21334. [DOI] [PubMed] [Google Scholar]
- 173.Liao I-C, Chen S, Liu JB, Leong KW. Sustained viral gene delivery through core-shell fibers. J Control Release Off J Control Release Soc. 2009;139:48–55. doi: 10.1016/j.jconrel.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang Y, Yang X, Liu W, Zhang F, Cai Q, Deng X. Controlled release behaviour of protein-loaded microparticles prepared via coaxial or emulsion electrospray. J Microencapsul. 2013;30:490–497. doi: 10.3109/02652048.2012.752537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rahman CV, Ben-David D, Dhillon A, Kuhn G, Gould TWA, Müller R, et al. Controlled release of BMP-2 from a sintered polymer scaffold enhances bone repair in a mouse calvarial defect model. J Tissue Eng Regen Med. 2014;8:59–66. doi: 10.1002/term.1497. [DOI] [PubMed] [Google Scholar]
- 176.Rabin C, Liang Y, Ehrlichman RS, Budhian A, Metzger KL, Majewski-Tiedeken C, et al. In vitro and in vivo demonstration of risperidone implants in mice. Schizophr Res. 2008;98:66–78. doi: 10.1016/j.schres.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Qian F, Stowe N, Liu EH, Saidel GM, Gao J. Quantification of in vivo doxorubicin transport from PLGA millirods in thermoablated rat livers. J Control Release. 2003;91:157–166. doi: 10.1016/s0168-3659(03)00237-2. [DOI] [PubMed] [Google Scholar]
- 178.Wang C-K, Wang W-Y, Meyer RF, Liang Y, Winey KI, Siegel SJ. A rapid method for creating drug implants: translating laboratory-based methods into a scalable manufacturing process. J Biomed Mater Res B Appl Biomater. 2010;93:562–572. doi: 10.1002/jbm.b.31617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kranz H, Ubrich N, Maincent P, Bodmeier R. Physicomechanical properties of biodegradable poly(D,L-lactide) and poly(D,L-lactide-co-glycolide) films in the dry and wet states. J Pharm Sci. 2000;89:1558–1566. doi: 10.1002/1520-6017(200012)89:12<1558::aid-jps6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 180.Owen GR, Jackson JK, Chehroudi B, Brunette DM, Burt HM. An in vitro study of plasticized poly(lactic-co-glycolic acid) films as possible guided tissue regeneration membranes: material properties and drug release kinetics. J Biomed Mater Res A. 2010;95:857–869. doi: 10.1002/jbm.a.32865. [DOI] [PubMed] [Google Scholar]
- 181.Huson MG, Strounina EV, Kealley CS, Rout MK, Church JS, Appelqvist IAM, et al. Effects of Thermal Denaturation on the Solid-State Structure and Molecular Mobility of Glycinin. Biomacromolecules. 2011;12:2092–2102. doi: 10.1021/bm200080h. [DOI] [PubMed] [Google Scholar]
- 182.Pikal MJ, Rigsbee D, Roy ML. Solid state stability of proteins III: Calorimetric (DSC) and spectroscopic (FTIR) characterization of thermal denaturation in freeze dried human growth hormone (hGH) J Pharm Sci. 2008;97:5122–5131. doi: 10.1002/jps.21386. [DOI] [PubMed] [Google Scholar]
- 183.Stanković M, de Waard H, Steendam R, Hiemstra C, Zuidema J, Frijlink HW, et al. Low temperature extruded implants based on novel hydrophilic multiblock copolymer for long-term protein delivery. Eur J Pharm Sci. 2013;49:578–587. doi: 10.1016/j.ejps.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 184.Stanković M, Tomar J, Hiemstra C, Steendam R, Frijlink HW, Hinrichs WLJ. Tailored protein release from biodegradable poly(ε-caprolactone-PEG)-b-poly(ε-caprolactone) multiblock-copolymer implants. Eur J Pharm Biopharm. 2014;87:329–337. doi: 10.1016/j.ejpb.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 185.Iwasaki Y, Takemoto K, Tanaka S, Taniguchi I. Low-Temperature Processable Block Copolymers That Preserve the Function of Blended Proteins. Biomacromolecules. 2016;17:2466–2471. doi: 10.1021/acs.biomac.6b00641. [DOI] [PubMed] [Google Scholar]
- 186.Ghebre-Selassie I, Martin C. Pharmaceutical Extrusion Technology. CRC Press; 2003. [Google Scholar]
- 187.Michnik A. Thermal stability of bovine serum albumin DSC study. J Therm Anal Calorim. 2003;71:509–519. [Google Scholar]
- 188.Ringe D, Petsko GA. The ‘glass transition’ in protein dynamics: what it is, why it occurs, and how to exploit it. Biophys Chem. 2003;105:667–680. doi: 10.1016/s0301-4622(03)00096-6. [DOI] [PubMed] [Google Scholar]
- 189.Shinyashiki N, Yamamoto W, Yokoyama A, Yoshinari T, Yagihara S, Kita R, et al. Glass Transitions in Aqueous Solutions of Protein (Bovine Serum Albumin) J Phys Chem B. 2009;113:14448–14456. doi: 10.1021/jp905511w. [DOI] [PubMed] [Google Scholar]
- 190.Pikal MJ, Rigsbee Dr, Roy Ml. Solid state chemistry of proteins: I. glass transition behavior in freeze dried disaccharide formulations of human growth hormone (hGH) J Pharm Sci. 2007;96:2765–2776. doi: 10.1002/jps.20960. [DOI] [PubMed] [Google Scholar]
- 191.Van Donkelaar LHG, Martinez JT, Frijters H, Noordman TR, Boom RM, van der Goot A-J. Glass transitions of barley starch and protein in the endosperm and isolated from. Food Res Int. 2015;72:241–246. [Google Scholar]
- 192.Matveev YI, Grinberg VY, Sochava IV, Tolstoguzov VB. Glass transition temperature of proteins. Calculation based on the additive contribution method and experimental data. Food Hydrocoll. 1997;11:125–133. [Google Scholar]
- 193.Rajagopal K, Wood J, Tran B, Patapoff TW, Nivaggioli T. Trehalose Limits BSA Aggregation in Spray-Dried Formulations at High Temperatures: Implications in Preparing Polymer Implants for Long-Term Protein Delivery. J Pharm Sci. 2013;102:2655–2666. doi: 10.1002/jps.23634. [DOI] [PubMed] [Google Scholar]
- 194.Ghalanbor Z, Körber M, Bodmeier R. Improved lysozyme stability and release properties of poly(lactide-co-glycolide) implants prepared by hot-melt extrusion. Pharm Res. 2010;27:371–379. doi: 10.1007/s11095-009-0033-x. [DOI] [PubMed] [Google Scholar]
- 195.Cossé A, König C, Lamprecht A, Wagner KG. Hot Melt Extrusion for Sustained Protein Release: Matrix Erosion and In Vitro Release of PLGA-Based Implants. AAPS PharmSciTech. 2017;18:15–26. doi: 10.1208/s12249-016-0548-5. [DOI] [PubMed] [Google Scholar]
- 196.Lee P, Towslee J, Maia J, Pokorski J. PEGylation to Improve Protein Stability During Melt Processing. Macromol Biosci. 2015;15:1332–1337. doi: 10.1002/mabi.201500143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ghalanbor Z, Körber M, Bodmeier R. Protein release from poly(lactide-co-glycolide) implants prepared by hot-melt extrusion: Thioester formation as a reason for incomplete release. Int J Pharm. 2012;438:302–306. doi: 10.1016/j.ijpharm.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 198.Ghalanbor Z, Körber M, Bodmeier R. Interdependency of protein-release completeness and polymer degradation in PLGA-based implants. Eur J Pharm Biopharm. 2013;85:624–630. doi: 10.1016/j.ejpb.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 199.Lee PW, Maia J, Pokorski JK. Milling Solid Proteins to Enhance Activity After Melt-Encapsulation. Int J Pharm. 2017;533:254–265. doi: 10.1016/j.ijpharm.2017.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lee PW, Shukla S, Wallat JD, Danda C, Steinmetz NF, Maia J, et al. Biodegradable Viral Nanoparticle/Polymer Implants Prepared via Melt-Processing. ACS Nano. 2017;11:8777–8789. doi: 10.1021/acsnano.7b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]