

Abstract
Molecular signals are the guiding force of development, imparting direction upon cells to divide, migrate, differentiate, etc. The mechanisms by which a cell can receive and transduce these signals into measurable actions remains a ‘black box’ in developmental biology. Primary cilia are ubiquitous, microtubule-based organelles that dynamically extend from a cell to receive and process molecular and mechanical signaling cues. In the last decade, this organelle has become increasingly intriguing to the research community due to its ability to act as a cellular antenna, receive and transduce molecular stimuli, and initiate a cellular response. In this review, we discuss the structure of primary cilia, emphasizing how the ciliary components contribute to the transduction of signaling pathways. Furthermore, we address how the cilium integrates these signals and conveys them into cellular processes such as proliferation, migration and tissue patterning. Gaining a deeper understanding of the mechanisms used by primary cilia to receive and integrate molecular signals is essential, as it opens the door for the identification of therapeutic targets within the cilium that could alleviate pathological conditions brought on by aberrant molecular signaling.
Keywords: primary cilia, Hedgehog, Wnt, Pdgf, proliferation, migration
I. Introduction
In the late 1800’s the Swiss anatomist Karl Zimmermann identified a thin, singular, hair-like structure on the surface of several different types of mammalian cells. At that time, without any knowledge of how the field of molecular biology would unfold, Zimmermann hypothesized that these structures played a sensory role for the cell (Zimmermann, 1898). After this intriguing discovery, it was over a hundred years before a resurgence of interest occurred in the organelle, now called a primary cilium (Sorokin, 1962; Sorokin, 1968) (Fig. 1). It was not until the discovery of intraflagellar transport (IFT) in the unicellular flagellate Chlamydomonas (Kozminski et al., 1993) that the role of primary cilia during development and disease began to be appreciated (Huangfu et al., 2003; Pazour et al., 2000).
Figure 1. Cilia and their structure.

Heterogeneity in ciliary structure. (A) Nodal cilia with either a 9+0 or 9+2 microtubule arrangement (inset), (B) primary cilia with a 9+0 microtubule arrangement (inset) and (C) airway cilia with a 9+2 microtubule structure (inset). (D) The detailed structure of a primary cilia, schematizing four main components: the basal body, transition zone, axoneme, and ciliary membrane.
Since the initial studies linking defects in the cilium to the onset of disease, it has become widely accepted that vertebrate species use primary cilia to transduce a number of signaling pathways (reviewed in (Singla and Reiter, 2006). The mechanisms by which cilia participate in signal transduction; however, are diverse and the precise details remain nebulous. With current estimates suggesting that well over 1000 proteins contribute to the structure and/or function the primary cilium (Boldt et al., 2016) and rapidly improving genome editing technologies becoming readily accessible, there is tremendous potential to manipulate ciliary composition to modulate molecular signals. While the possibilities of harnessing the power of the cilium are becoming more realistic, there is still much to be learned about the intricacies of cilia-dependent signal transduction.
Herein, we discuss the specialized components of primary cilia and how they participate in the signal transduction of several different molecular pathways. We highlight certain ciliary proteins that both localize to the cilium and function to modulate transduction of molecular signals. Furthermore, we attempt to link the role for primary cilia in signal transduction with their participation in guiding cell behaviors while examining how loss of cilia can result in congenital birth defects. Undoubtedly, the continued study of primary cilia in these contexts holds great promise for future therapeutic options for diseases in which signal transduction and cell behaviors have run amok.
II. Constructing the primary cilia
Cilia have been identified on almost every cell type in the developing embryo (Wheatley, 1982; Wheatley et al., 1996). There is some diversity in the structure of cilia, which is exemplified by the presence of a number of specialized types of cilia including nodal, primary, and olfactory cilia (Fig. 1A–C). Despite the variation between different types of specialized cilia, the fundamental structure and several components of cilium are highly conserved. Cilia of all types are made up of four key components: the basal body, the transition zone, the axoneme, and the ciliary membrane (Fig. 1). Although some structural and molecular variations in these components give the organelle unique functions in different cell types, for the most part, these components are identifiable across several tissues. Below we summarize each of the components that make up the primary cilium and briefly discuss key proteins that can alter the function of the organelle.
Basal Body
Centrioles are cylindrical cellular structures that are composed of nine sets of microtubule triplets. In cells that are actively dividing, the two centrioles play an essential role in formation of the mitotic spindle. Each centriole replicates during S-phase, ensuring that the newly generated cell will also have two centrioles. Thus, through this mechanism, each cell contains an older ‘mother’ centriole and a newly generated ‘daughter’ centriole. The mother centriole is distinct from the daughter centriole in that it has two sets of appendages, the distal and subdistal appendages (Paintrand et al., 1992). During times of quiescence, the distal and subdistal appendages on the mother centriole act as sites to assemble a ciliary vesicle and assist with plasma membrane docking, respectively (Fig. 1D) (Anderson, 1972; Sorokin, 1962). Upon the completion of these events, the mother centriole is transformed into the basal body of the cilium. The basal body remains connected to the daughter centriole by filaments at its proximal end, such that the daughter centriole is oriented roughly perpendicular to the basal body (Bahe et al., 2005). Once docked to the plasma membrane the basal body functions as the microtubule organizing center for the primary cilium.
To initiate and regulate ciliogenesis, several proteins must either be removed or recruited to the basal body. Distal appendages are critical for the recruitment of Tau tubulin kinase 2 (Ttbk2), which is required for the removal of centrosomal protein of 110kD (Cp110), an inhibitor of ciliogenesis, from the distal end of the mother centriole (Goetz et al., 2012; Spektor et al., 2007). It is thought that Cp110 functions structurally to prevent microtubule growth and centriole length, as the protein does not have any enzymatic activity (Chen et al., 2002; Specktor et al. 2007). This hypothesis is supported by ultrastructural studies reporting that Cp110 localizes to the distal ends of centrioles, forming a cap above the growing microtubules (Kleylein-Sohn et al., 2007; reviewed in Kobayashi and Dynlacht, 2011). The Cp110-mediated ‘capping’ of the basal body acts as a negative regulator of primary cilia extension and a checkpoint against inappropriate formation of primary cilia. Ciliogenesis requires the removal of the Cp110 cap from the basal body. Several proteins that localize to the distal tip of the basal body, including C2-calcium domain containing 3 (C2cd3) and Oral-facial-digital 1 (Ofd1) (Thauvin-Robinet et al., 2014), are necessary for coordinating the removal of Cp110. Disruptions to this process often result in ciliopathies.
Oral-facial-digital (OFD) syndromes (OFD1-14) are rare genetic disorders characterized by the co-presentation of abnormalities in the face (hypertelorism, low-set ears, cleft lip/palate), oral cavity (lingual hamartoma, abnormal frenulae, lobulated tongue) and digits (brachydactyly, polydactyly) (reviewed in Bruel et al., 2017). OFD14 is caused by mutations in C2cd3, a protein localized to the distal aspect of the basal body. C2cd3 is required for the formation of the distal appendages and for ciliary vesicle docking to the mother centriole (Thauvin-Robinet et al., 2014; Ye et al., 2014). Studies in chick, mouse and humans have together suggested a conserved role for C2cd3 in basal body function and ciliogenesis (Chang et al., 2014; Hoover et al., 2008; Schock et al., 2015; Thauvin-Robinet et al., 2009; Thauvin-Robinet et al., 2014). Loss of C2cd3 results in short centrioles without subdistal appendages, distal appendages that are incapable of elongation, and impaired ciliogenesis due to the failure to remove Cp110 from the distal tip of the basal body (Tsang and Dynlacht, 2013; Ye et al., 2014). Interestingly, diseases can also arise when hyperelongation of the centriole occurs. Mutations in Ofd1, a protein that co-localizes with C2cd3 to the distal basal body, result in a hyperelongated basal body with unstable microtubules and result in a subtype of OFD, OFD1 (Thauvin-Robinet et al., 2014). Thus, Ofd1 and C2cd3 antagonize each other to establish and regulate centriole length. Taken together, these studies suggest a model whereby centriole length is carefully regulated and essential for proper ciliogenesis. Further identification of the cadre of proteins that localize and function at the basal body will surely be essential for understanding the mechanisms of pathology causal to ciliopathies.
Transition Zone
The transition zone begins at the distal end of the basal body and forms a ‘gate’ between the ciliary and cellular compartments. This gate is established by two structures within the transition zone which anchor the most proximal region of the growing cilium to the adjacent membrane. First, the transition fibers connect the basal body to the ciliary membrane via nine centriolar appendages (Anderson, 1972) (Fig. 1D). Transition fibers are required for ciliogenesis and function in the extended primary cilium to regulate entry and exit of proteins into the mature cilium. Second, Y-shaped fibers or ‘Y-linkers’ connect the projected microtubules of the axoneme to the overlying ciliary membrane, with one end attached to microtubules and the other two ends attached to the ciliary membrane (Fig. 1D). Cargo destined for the primary cilium uses the transition zone as a docking point and must move through the transition zone’s narrow passageway into the ciliary compartment. In vertebrates, it has been suggested that transition zone components may also be involved in loading cargo onto the intraflagellar transport system (Garcia-Gonzalo et al., 2011; Gilula and Satir, 1972; Williams et al., 2011). Ciliary localization sequences built into membrane-associated proteins can interact with trafficking complexes, like the BBSome, to cross through the transition zone. The combined efforts of ciliary localization sequences and trafficking complexes allows larger proteins from the cytoplasm to enter and exit the primary cilium (Dishinger et al., 2010).
Many proteins localize to, and are required for, transition zone function. These proteins can be separated into two interacting and sometimes overlapping protein complexes: the Nephronophthisis (NPHP) complex, and the Meckel Syndrome (MKS) complex. Named for its association with the human ciliopathy nephronophthisis (medullary cystic kidney disease), the NPHP complex includes Nphp4 and Rpgr-interacting protein 11 (Rpgrip11) (Arts et al., 2007; Mollet et al., 2005). The MKS complex, named for its association with the human ciliopathy Meckel Syndrome (characterized by renal cystic dysplasia, occipital encephalocele, polydactyly, hepatic developmental defects and pulmonary hypoplasia), includes Tectonic (Tctn) proteins, B9 domain proteins (B9d, Mks), Coiled-coil proteins (Ccd2a) and Transmembrane proteins (Tmem) (Barker et al., 2014; Chih et al., 2011; Garcia-Gonzalo et al., 2011; Huang et al., 2011). Loss of function studies of proteins within the transition zone reveal that the transition zone is important for ciliogenesis and establishing ciliary composition. Mutations in proteins of the MKS complex cause ciliogenesis defects which often result in embryonic lethality (Chih et al., 2011; Garcia-Gonzalo et al., 2011; Sang et al., 2011; Weatherbee et al., 2009). Mutations in proteins of the NPHP complex result in only mild ciliogenic defects affecting only photoreceptor cilia and sperm flagella (Jiang et al., 2009; Jiang et al., 2008; Won et al., 2011). Despite the severity of some embryological defects associated with mutations in transition zone proteins, there are also a host of transition zone mutations that do not impair ciliogenesis. Understanding the complex interactions between these protein complexes and the mechanisms of transition zone function will be necessary for guiding future research directions.
Axoneme
The distinguishing feature of the cilium is the axoneme. The axoneme is a microtubule-based extension that protrudes from the apical surface of the cell (Fig. 1D). The axoneme is made up of nine sets of microtubule doublets extending from the basal body. The organization of axonemal microtubules confers function upon the cilium as either a motile cilium or non-motile (primary) cilium (Fig. 1A–C). While the axonemes of both motile and primary cilia are composed of a ring of nine pairs of microtubules, they differ in their central microtubule components. The central portion of motile cilia possess two extra microtubules; thus, they are referred to as having a ‘9+2’ microtubule arrangement. The presence of the central microtubule pair allows for large motor protein complexes called dyneins to connect to the outer ring of microtubules (reviewed in Smith and Yang, 2004). Dyneins are able to generate the force necessary for ciliary movement. While the presence of the central microtubule pair and the 9+2 conformation typically translates into motility, it is more accurate to use the presence or absence of dynein arms to determine motility of the cilium (Afzelius, 1976; Blum et al., 1977). In contrast, non-motile primary cilia traditionally lack these central microtubules, and therefore their structure is referred to as ‘9+0’ (Sorokin, 1968; Wheatley, 1969).
Extension of the ciliary axoneme requires intraflagellar transport (IFT). IFT describes the bi-directional transport of particles along microtubules (Kozminski et al., 1993). Building upon the microtubule template established by the basal body, IFT utilizes two microtubule-based motors (kinesin-II and dynein) to carry tubulins and other axonemal subunits to the ciliary tip and carry bi-products back to the ciliary base. In IFT, anterograde transport (from cell body towards ciliary tip) is carried out by IFT-B complexes and kinesin II motors to build the microtubule scaffold and carry essential ciliary proteins to the distal tip of the axoneme. Disruptions in components of the IFT-B complex and kinesin-2 anterograde motor complexes prevent ciliogenesis, as the tubulin subunits cannot be transported to the growing end of the axoneme (Takeda et al., 1999; Taulman et al., 2001). Conversely, retrograde transport (from ciliary tip towards cell body) is carried out by IFT-A complexes and dynein motors to return molecular cargo back to the cell body. Disruptions to the IFT-A complex or dynein motors result in a functionally impaired, dysmorphic cilium. Most frequently, cilia with impaired retrograde transport will have a bulbous tip due to cargo remaining trapped at the ciliary tip, unable to be carried back down to the cell body (Cortellino et al., 2009; Qin et al., 2011; Tran et al., 2008). Successful IFT allows the axoneme to extend via microtubule lengthening. The axonemal extension is stabilized via post-translational modifications of tubulin subunits including acetylation (L’Hernault and Rosenbaum, 1985), detyrosination (Hallak et al., 1977), polyglutamylation (Edde et al., 1990), and polyglycylation (Redeker et al., 1994). Once stabilized, numerous receptors and ion channels localize to the specialized membrane surrounding the fully extended axoneme.
The ciliary membrane
The ciliary membrane is a specialized membrane covering the axoneme, connected to peripheral microtubules by the Y-linkers of the transition zone (Gilula and Satir, 1972; Williams et al., 2011). While the ciliary membrane is continuous with the plasma membrane of the cell, it originates from the membrane of the ciliary vesicle and is structurally and physiologically different from the cell membrane due to a unique lipid composition and the presence of ion channels and osmotic capabilities (Garcia-Gonzalo et al., 2015; Nauli et al., 2003; Satir and Gilula, 1970). Variation in lipid composition can underlie changes in channel activity and electrical excitability in response to receptors on the ciliary membrane. Phosphoinositides serve as signature motifs for different cellular membranes and often are required for the function of membrane proteins. For example, phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2), is a known modulator of many ion channels. In the cilium, the ratio of phosphatidylinositol 4-phosphate (PI4P) predominates over PI(4,5)P2. This variation in phosphoinositides supports the localization of certain G protein coupled receptors or channels to the cilium (Chavez et al., 2015; Garcia-Gonzalo et al., 2015). The distinct ciliary membrane composition supports the preferential localization of receptors from many signaling pathways (Rohatgi et al., 2007; Schneider et al., 2005). Thus, the unique make-up of the ciliary membrane confers the ability of the cilium to function as a ‘cellular antenna’.
III. Signaling pathways that are modulated by ciliary function
Perhaps the most intriguing aspect of the primary cilium is its association with the transduction of several signaling pathways. Components of many major developmental pathways localize to the cilium. Furthermore, trafficking through the cilium is required for the processing, in the way of acetylation, sumoylation, phosphorylation and proteolytic cleavages, of several members of these pathways. Below we summarize what is known regarding the role of the cilium in transduction of some key developmental pathways and introduce more recently discovered ciliary proteins that have been shown to impact cilia-dependent transduction of these molecular signals.
Hedgehog signaling pathway
The Hedgehog (Hh) signaling pathway has been studied for decades in a plethora of contexts from organogenesis to disease (Nüsslein-Volhard and Wieschaus, 1980; Chang et al., 1994; Chiang et al., 1996; Hebrok et al., 2000; St-Jacques et al., 1999; Yao et al., 2002; Zhang et al., 2006). In brief, the pathway functions via a Hh ligand binding to the Patched (Ptch) receptor (Ingham et al., 1991). Hh-Ptch binding alleviates Ptch-mediated repression of the G protein-coupled receptor Smoothened (Smo) (Stone et al., 1996). Smo then transduces signals downstream via the subsequent activation of the Gli transcription factors, which translocate into the nucleus and upregulate the expression of Hh target genes. Although simple in principle, the pathway has several levels of complexity built-in, including three unique ligands (Sonic Hedgehog (Shh), Indian Hedgehog (Ihh) and Desert Hedgehog (Dhh)) that can be post-translationally processed to modify their activity and ability to signal in an autocrine or paracrine fashion, and a number of co-receptors that serve to alter Hh-binding affinity (Growth arrest specific 1 (Gas1), Cell adhesion associated, oncogene regulated (Cdo), and Brother of Cdo (Boc)) (Allen et al., 2011; Izzi et al., 2011; Martinelli and Fan, 2007). Furthermore, Hh signals are transduced through three Gli transcription factors, two of which (Gli2 and Gli3) are bimodal, functioning as both activators and repressors (Bai et al., 2004; Dai et al., 1999; McDermott et al., 2005; Sasaki et al., 1999).
In addition to the variability in ligands, receptors, and transcription factor activity, there are also several regulator proteins known to impact the transduction of the pathway whose roles are not fully understood. For example, Suppressor of Fused (Sufu) is a Gli-binding protein essential for normal embryonic development (Pearse et al., 1999; Pham et al., 1995). Sufu has long been regarded as a negative regulator of the pathway, mostly due to the close phenotypic resemblance of Sufu−/− and Ptch1−/− murine embryos, which both have a ventralized neural tube (Cooper et al., 2005; Svard et al., 2006). Although multiple mechanisms for how Sufu mediates pathway repression have been proposed (Chen et al., 2009; Cheng and Bishop, 2002; Humke et al., 2010; Kogerman et al., 1999; Lin et al., 2014; Methot and Basler, 2000; Shi et al., 2014), the prevailing view in the field suggests it acts as a cytoplasmic constraint for the nuclear translocation of the Gli transcription factors (Ding et al., 1999; Humke et al., 2010; Tukachinsky et al., 2010). Recent studies; however, have suggested that rather than solely acting as a repressor of the pathway, Sufu functions as a chaperone to shuttle Gli activators into, and Gli repressor out of, the nucleus. These studies further suggest that Sufu binds to chromatin with both Gli isoforms to generate a transcriptional response (Zhang et al., 2017). Based on continued studies such as these, it is clear that the mechanisms behind a single signaling event are tremendously complicated and we still have much to learn regarding Hh signal transduction. Some of this complexity has recently been explained with the discovery that the pathway utilizes the primary cilium to coordinate signaling.
The link between primary cilia and the Hh pathway was established when ENU generated ciliary mutants were found to have disrupted Hh signaling (Huangfu et al., 2003) and when several components of the pathway, including Ptch, Smo, and Gli were found to localize to the cilium itself (Corbit et al., 2005; Haycraft et al., 2005; Rohatgi et al., 2007). Identification of the cilium in this capacity allowed for its superimposition onto the known steps of Hh pathway transduction. With this in mind, the current generalized hypothesis is that Hh ligand binds to the Ptch receptor at the cilium, this binding alleviates Ptch-mediated repression of Smo, and allows for Smo to traffic into the cilium (Corbit et al., 2005; Rohatgi et al., 2007). Via mechanisms that remain unclear, activated ciliary Smo then works through kinesin family member 7 (Kif7) to generate a competent Gli activator that is imported into the nucleus to induce target gene expression (Liem et al., 2007). It is unclear as to if Sufu must dissociate from Gli (Law et al., 2012; Tukachinsky et al., 2010), if Sufu accompanies Gli activator into the nucleus (Zhang et al., 2017) (Fig. 2A). In the absence of Hh ligand, Ptch remains localized to the ciliary membrane, preventing the entrance of Smo (Rohatgi et al., 2007) (Fig. 2B). The Sufu-Gli complex moves to the basal body of the cilium where Gli undergoes a multi-site phosphorylation by Protein kinase A (PKA) (Hammerschmidt et al., 1996; Jiang and Struhl, 1995; Li, 1995), Gylcogen synthase kinase 3 (Gsk3) and Casein kinase 1 (Ck1). Phosphorylated Gli is recognized and ubiquitinated by E3 ubiquitin ligase and finally processed by the proteasome into the truncated Gli repressor (Pan et al., 2006; Pan and Wang, 2007). The Sufu-Gli repressor complex then moves to the nucleus where it recruits the Sap18-Sin3 co-repressor complex to repress Gli target genes (Cheng and Bishop, 2002; Paces-Fessy et al., 2004) (Fig. 2B).
Figure 2. Cilia-dependent Hh signaling.

Activity of Hh pathway components in the (A) presence and (B) absence of Shh. (C) Loss of Inpp5e dampens the ability of the cilium to transduce a Shh signal, thus rendering the cilia “desensitized”. (D) Loss of EvC/EvC2 prevents Gli2/3 dissociation from Sufu, thus preventing transcription of downstream targets and generating a cilium “desensitized” to a Shh signal.
Several ciliopathies are due to impaired cilia-dependent Hh signaling. Inositol polyphosphate 5-phosphatase E (Inpp5e) is a lipid 5-phosphatase which localizes to the axoneme and stabilizes the structure of the primary cilium (Jacoby et al., 2009). Loss of Inpp5e in mice causes many ciliopathy-related phenotypes, including anophthalmia (loss of one or both eyes), polydactyly, cystic kidneys, bifid sternum, cleft palate, and exencephaly (Chavez et al., 2015). Mutations in Inpp5e have been linked to two human ciliopathies, Joubert and MORM (mental retardation, truncal obesity, retinal dystrophy, and micropenis) syndromes (Chavez et al., 2015). Mutations in Inpp5e cause a dampening of the intracellular response to a Shh signal (Chavez et al., 2015; Garcia-Gonzalo et al., 2015). When Inpp5e activity is lost, G-protein-coupled receptor 161 (Gpr161) accumulates in primary cilia (Fig. 2C). Gpr161 negatively regulates the Hh signal response by increasing the cAMP response to promote PKA-dependent phosphorylation of Gli3 and its subsequent processing into the Gli repressor (Mukhopadhyay et al., 2013). Thus, increased Gpr161 activity leads to an over-production of Gli repressor, a bulbous ciliary axoneme and reduced Hh signaling (Chavez et al., 2015) (Fig. 2C).
EvC ciliary complex subunit 1 and EvC ciliary complex subunit 2 (EvC and EvC2) are two additional proteins that localize to the cilium, impact Hh signaling, and when mutated result in a human ciliopathy (Ellis van-Creveld syndrome). EvC and EvC2 co-localize in primary cilia (Blair et al., 2011; Ruiz-Perez et al., 2000; Ruiz-Perez et al., 2003). These proteins are believed to function as mediators between the activation of Smo and the processing of Gli into functional activator isoforms (Fig. 2D). Upon activation of the pathway, Smo and EvC2 form a complex restricted to a distinct ciliary compartment called the EvC zone. Impeding EvC2 function blocks Hh signaling at a specific step between Smo and the downstream regulators PKA and Sufu, preventing activation of Gli transcription factors (Dorn et al., 2012). One hypothesis suggests that EvC and EvC2 interact together with Smo to modulate the interaction between Sufu and Gli within the primary cilium (Yang et al., 2012). Additional insights into the role of these proteins and Hh signal transduction will need to be gained in order to better understand disease causing mechanisms.
Wnt signaling pathway
Similar to Hh, Wnt is a lipid-modified secreted ligand, of which many isoforms exist (reviewed in Nusse, 2003). Wnt binds to the 7-pass transmembrane receptor Frizzled (Fzd), and co-receptor Lipoprotein-receptor related protein (Lrp) (Bhanot et al., 1996; Pinson et al., 2000; Tamai et al., 2000; Wehrli et al., 2000). Upon binding to Fzd, Lrp is phosphorylated, recruiting a protein called Dishevelled (Dvl) to become activated (Fig. 3A) (Klingensmith et al., 1994; Noordermeer et al., 1994). Dvl interacts with a destruction complex containing Adenomatous polyposis coli (Apc) and Axin2 (Cliffe et al., 2003; Jonkers et al., 1997; Salic et al., 2000; Yost et al., 1998). With the inactivation of this destruction complex, β-catenin can accumulate in the cytoplasm and translocate to the nucleus to promote transcriptional activation with T-cell factor/Lymphoid enhancer factor (Tcf/Lef) and histone modification complexes, which leads to expression of Wnt target genes (Fig. 3A) (Behrens et al., 1996; Molenaar et al., 1996). In the absence of Wnt, Dvl does not inhibit the destruction complex, which then allows for β-catenin to be phosphorylated by Gsk3-β, ubiquitinated, and degraded by the proteasome (Fig. 3B). Without β-catenin accumulation and translocation to the nucleus, the Tcf/Lef complex recruits histone deacetylase complexes (HDACs) to repress target gene expression (Fig. 3B) (Brannon et al., 1997; Cavallo et al., 1998; Chen et al., 1999). Wnt can also signal through the non-canonical, planar cell polarity (PCP) pathway. PCP is independent of β-catenin and is predominantly coordinated by Dvl proteins that elicit microtubule and actin cytoskeletal remodeling via calcium-ion signaling or through several Dvl interaction partners and downstream mediators (reviewed in Yang and Mlodzik, 2015).
Figure 3. Hypothesized cilia-dependent Wnt signaling.

(A) Hypothesized (and controversial) activity of Wnt pathway components in the (A) presence and (B) absence of Wnt. (C) Loss of Kif3a causes increased translocation of β-catenin to the nucleus and increased target gene expression, thus “sensitizing” the cell to Wnt signaling.
The supposition that the cilium was necessary for Wnt signal transduction began with reports that several components of the Wnt pathway, including Inversin, Vangl-2, Gsk3-β, and Apc, localize near the primary cilium or basal body (Morgan et al., 2002; Ross et al., 2005; Simons et al., 2005; Veland et al., 2009). This hypothesis was bolstered by the demonstration that Inversin regulated Dvl and acted as a switch between canonical and non-canonical Wnt pathways (Gerdes et al., 2007; Ross et al., 2005; Simons et al., 2005). Subsequent studies hypothesized that the cilium functioned to restrain Wnt signaling, based on the observation that the loss of the ciliary protein Kinesin family member 3A (Kif3a) correlated with an accumulation of cytoplasmic β-catenin and increased expression of Wnt-target genes (Corbit et al., 2008). This study hypothesized that Kif3a affected Wnt/β-catenin signaling by controlling the phosphorylation of Dvl. In support of this mechanism, a second study reported that there was increased Wnt activity, via repression of nuclear β-catenin accumulation, in deciliated cells (Lancaster et al., 2011).
Despite these findings in support of a role for cilia in Wnt signal transduction, concurrent studies reported several convincing pieces of data suggesting primary cilia do not regulate Wnt signaling (Huang and Schier, 2009; Ocbina et al., 2009). First, loss of numerous ciliary genes, including Ift88, Ift72, Dync2h1 and Kif3a, did not impact Wnt-responsiveness, as determined by LacZ expression in the BAT-gal reporter line (Ocbina et al., 2009). Second, loss of these ciliary proteins did not affect Axin2 expression, as determined by in situ hybridization (Ocbina et al., 2009). Third, mouse embryonic fibroblasts (MEFs) derived from Ift88−/− and Ift172−/− mutant embryos responded to Wnt3a in a fashion similar to wild-type MEFs subject to the same treatment. Finally, these studies reported that MEFs from numerous ciliary mutants had no problem shifting between canonical and non-canonical Wnt signaling (Ocbina et al., 2009). These findings were validated in a zebrafish model. Maternal-zygotic oval (MZ ovl) mutants, which lack Ift88, retained normal canonical and non-canonical Wnt signaling and cellular processes guided by non-canonical Wnt activity (e.g., convergent extension) were not disrupted (Huang and Schier, 2009). Together, these results suggested the cilium was not necessary for either canonical or non-canonical Wnt signal transduction. Additional support for rejecting a role for cilia in Wnt signal transduction came with the failure of ciliopathic mouse models to present with phenotypes indicative of disrupted Wnt signaling (Ocbina et al., 2009).
There are several possibilities for the existence of these conflicting results. It is possible that several ciliary proteins have tissue- and species-specific roles, such that differing results can be obtained depending upon which tissue and which model system are used. In addition, many ciliary proteins have proposed cilia-independent functions, and these could be affecting Wnt signaling outside of primary cilia. A recent study found that Kif3a, a protein known to be essential for ciliogenesis, plays a role in Wnt signaling in a ciliary-independent fashion (Kim et al., 2016). Specifically, it was reported that Kif3a plays a role in both maintaining Wnt secretion, activating the pathway in an autocrine manner, and intracellular signal transduction of canonical signaling; however, these results were not recapitulated when ciliogenesis was prevented using pharmacological agents (Kim et al., 2016). While it is indeed tempting to speculate that the cilium is necessary for Wnt signaling, especially due to the numerous similarities to the Hh pathway (Kalderon, 2002), it is clear that the primary cilium is not utilized by each pathway in the same way.
Platelet-derived growth factor pathway
The Platelet-derived growth factor (Pdgf) signaling pathway is necessary for numerous cellular processes during development, including the proliferation, survival, and migration of several different cell types (reviewed in Heldin and Westermark, 1999). The ligands of the pathway form homo or heterodimers of PdgfA-D. These ligand dimers bind to the extracellular domains of the appropriate Pdgf receptors (Pdgfr), causing dimerization and trans auto-phosphorylation of the receptors. As tyrosine-kinase receptors, the activity of the receptors creates a phosphorylation mark which can be recognized by adapter molecules of two different signaling cascades: p85 of the Akt/phosphatidylinositol 3-kinase (PI3K) cascade, and Growth factor receptor bound protein 2 (Grb2) of the Mitogen-activated protein kinase/Extracellular signal-regulated kinase (Mek/Erk) cascade. Signaling through these cascades can eventually lead to cell proliferation (Davis and Stroobant, 1990; Fantauzzo and Soriano, 2014; Pierce et al., 1994; Yi et al., 1996), differentiation (Li et al., 2014; Pierce et al., 1994; Richardson et al., 1988), and survival (Artus et al., 2013; Fantauzzo and Soriano, 2014; Othberg et al., 1995).
While not nearly as heavily studied as the Hh or Wnt pathways in the context of their relationship to the primary cilium, a number of studies have reported convincing data to support an interaction between the Pdgf pathway and the cilium. Pdgfrα expression is up-regulated during ciliogenesis, and the expression and ligand-mediated activation of the receptor are blocked in Ift88−/− and Ift172−/− MEFs (Schneider et al., 2005; Umberger and Caspary, 2015). In addition, Pdgfrα localizes to the ciliary membrane along the axoneme, binds its ligand, dimerizes and trans-autophosphorylates while located along the primary cilium. In Ift20-depleted cells, Pdgfrα localizes aberrantly to the plasma membrane and is hyperactivated upon ligand stimulation (Schmid et al., 2017). Thus, these data strongly support the hypothesis that Pdgf signaling requires the cilium to function normally.
While the link between the expression and localization of Pdgfrα is well supported, the detailed mechanism of how the cilium functions in the transduction of a Pdgf signal is less clear. Studies using fibroblasts report that ciliary signaling through Pdgfrα involves the activation of Akt and Erk1/2 at the ciliary base (Clement et al., 2013), and loss of Ift20 in cranial neural crest cells causes suppression of Pdgf-Akt signaling, resulting in decreased osteogenic proliferation, mineralization, and increased cell death (Noda et al., 2016). While not much else is known regarding the detailed mechanism of PDGF signal transduced through the cilium, the phenotypic overlap between ciliary mutants and PDGF mutants suggests this field of study will continue to grow in the future.
Calcium signaling
Calcium (Ca2+) is the most common signal transduction element in cells across all species and plays a number of physiological roles in cellular processes including fertilization, muscle contraction, neuronal transmission and cell motility (reviewed in Clapham, 2007). The link between Ca2+ signaling and the cilium arose for several reasons. First, Ca2+ permeant ion channels (Polycystins), were reported to localize to the ciliary membrane (Bai et al., 2008; Kottgen et al., 2008; Pazour et al., 2002). Second, when primary cilia are bent by the mechanical force (e.g., fluid-flow), Ca2+ is conducted through polycystin complexes (Nauli et al., 2003; Praetorius and Spring, 2001). Finally, a variety of cells and tissues during development, including bone (Malone et al., 2007; Temiyasathit et al., 2012; Xiao et al., 2011), kidney epithelia (Leyssac, 1986; Praetorius and Spring, 2001, 2003), the embryonic node (Yoshiba et al., 2012; Yuan et al., 2015) and cartilage (Phan et al., 2009; Wann et al., 2012) require primary cilia-dependent, fluid-force induced calcium signaling to undergo proper developmental processes.
In the kidney, it is hypothesized that Ca2+ signaling is mediated via the mechanical stimulation of Polycystin1/2 (Pc1/2, encoded by Pkd1 or Pkd2, respectively) on the primary cilium (Nauli et al., 2003; Pazour et al., 2002). When the extracellular domain of Pc1 is activated in response to fluid flow, the Pc2 ion channel opens, allowing for Ca2+ influx. This intraciliary Ca2+ influx is proposed to lead to the release of intracellular calcium stores from the ER (Nauli et al., 2003). Ablation or mislocalization of Pc1/2 to the cellular membrane results in a dampening of Ca2+ signaling (Nauli et al., 2003). Loss of function mutations affecting Pc1 or Pc2 cause autosomal dominant polycystic kidney disease (ADPKD) (Hughes et al., 1995; Mochizuki et al., 1996). ADPKD manifests in patients by causing cyst formation in the kidney, pancreas, and liver, and is the most common kidney disease diagnosed in the US. While mutations in either Pkd1 or Pkd2 during development cause cyst formation, evidence supports that cyst formation in the human disease likely occurs due to both disrupted flow-induced Ca2+ signaling (Boehlke et al., 2010; Nauli et al., 2003), and undiscovered alternative roles for these proteins during adulthood (Piontek et al., 2007).
At the embryonic node, cilia-dependent, calcium signaling is hypothesized to be necessary for left-right patterning (Nonaka et al., 2002; Yoshiba et al., 2012; Yuan et al., 2015). The mouse node contains both motile and non-motile cilia (Sulik et al., 1994; McGrath et al., 2003; Tabin and Vogan, 2003). Loss of cilia, upon individual knockout of Kif3a or Kif3b, caused laterality defects such as situs inversus (Marszalek et al., 1999; Nonaka et al., 1998; Takeda et al., 1999). These studies investigated monocilia present on ventral nodal cells, and reported some of these 9+0 monocilia partook in rotational motion strong enough and synchronized enough to generate a leftward flow (Nonaka et al., 1998; Takeda et al., 1999). The fluid flow generated by that movement stimulates the adjacent primary cilia (McGrath et al., 2003; Tabin and Vogan, 2003). In support of the hypothesis that influx of Ca2+ is the signaling event which triggers asymmetric gene expression and determination of the future ‘left’, several studies have shown that Ca2+ oscillations occur at the node (Takao et al., 2013; Yuan et al., 2015). In 2015, Yuan et al., utilized a Ca2+-fluorescent reporter fused to a ciliary-localized protein necessary for ciliogenesis (ADP ribosylation factor like GTPase 13B; Arl13b) to conclude that Ca2+ influx in the cilium preceded cytosolic Ca2+ waves, and that this event is the earliest molecular signal of asymmetric left-right organization at the node. These studies support the hypothesis that fluid flow in the vertebrate node stimulates the adjacent primary cilia to release intracellular Ca2+, which can lead to induction of ‘left’ side gene expression. This hypothesis has relied upon the idea that increases in intraciliary Ca2+ precede Ca2+ influx in the cytoplasm; however, recent reports have challenged this idea.
Despite firm support within the field for a cilia-dependent, Ca2+ influx mechanism for establishing the L-R axis, recent studies argue that flux in intraciliary Ca2+ levels do not affect intracellular Ca2+ levels and that fluid flow at the node does not change intraciliary Ca2+ levels at all (Delling et al., 2013, 2016). This group did not detect mechanically-induced Ca2+ increases in primary cilia in response to physiological or even highly supraphysiological levels of fluid flow, and suggested flow-induced fluorescent Ca2+ reporter changes in the cilium were an artifact of experimental conditions. Taken together, the study concluded that mechanosensation, if it originates in primary cilia, is not via Ca2+ signaling. These new data propose the possibility that something entirely different from ciliary-dependent Ca2+ signaling is required for left-right polarity establishment in the vertebrate embryonic node.
While this controversy has yet to be solved, there are several possible reasons for these contradictory results. First, since the Ca2+ influx is transient, it is possible these studies were examining different temporal windows. Second, cell culture conditions are known to alter biological readouts. It’s possible that variation in experimental design, including selection of Ca2+ indicators and their associated Kd are responsible for differing data. With technological advances, and as additional knowledge about ciliary function improve, discrepancies such as these are likely to be worked out in the near future.
IV. Cellular processes that require primary cilia
The molecular signals processed through the primary cilium are used to guide the cell through several developmental processes. Thus, the cilium sits at the intersection of molecular cues and cellular processes including proliferation, migration, patterning, and mechanosensation throughout embryonic development, organogenesis, and into adulthood. The extent of ciliary participation in these vital cellular processes explains why ciliopathies have pleiotropic phenotypes, affecting several different organ systems (Fig. 5).
Figure 5. Primary cilia mutations have pleiotropic effects during development.

(A) Wild type primary cilia extension from a vertebrate cell during development. (B) Disrupted ciliogenesis can result in shortened and bulbous cilium (top), or complete loss of the primary cilium (bottom). (C) Primary cilia are required for proper patterning of motor neurons within the neural tube. (D) Mutations in anterograde Ift proteins (i.e. Ift88 or Kif3a) result in a dorsalization of the neural tube (left). Mutations in retrograde Ift proteins (i.e. Ift139) result in a ventralization of the neural tube (right). (E) Primary cilia are required for patterning of the limbs and digit identity. (F) Loss of primary cilia can cause polydactyly. (G) Patterning and development of the mandibular prominence of the developing craniofacial complex requires primary cilia for proper tongue and submandibular gland development. (H) Loss of primary cilia on neural crest cells in the developing mandible results in aglossia (loss of the tongue) and hypoplastic submandibular glands. (I) Patterning and development of the frontonasal prominence requires primary cilia. (J) Loss of primary cilia on neural crest cells leads to mid-facial widening, bilateral cleft palate, and a duplicated nasal septum. (K) Wild type kidney development requires functional primary cilia. (L) Mutations in primary cilia are known to cause Polycystic Kidney Disease, resulting in cysts forming within kidney.
Cell division and the primary cilium
Throughout development, cells must constantly make the decision whether to divide or become post-mitotic and differentiate. Primary cilia are closely linked to cellular proliferation as they must be retracted prior to entrance into the cell cycle. Furthermore, proper cell cycle progression requires continuous suppression of primary cilia formation in proliferating cells (Rieder et al., 1979; Tucker et al., 1979). At the crux of this mutually exclusive relationship between the primary cilium and cell division is the fact that the centrioles used for assembling the mitotic spindle during cell division are the same centrioles which are modified to become the basal body. Ciliogenesis initiates as the cell enters the G1/G0 phase, whereas ciliary disassembly occurs at S/G2 as the cell re-enters the cell cycle (Tucker et al., 1979). The process starts again upon exit from the cell cycle, as the mother centriole forms the basal body to initiate ciliogenesis (Sorokin, 1962; Sorokin, 1968).
Whereas the rationale for the mutually exclusive relationship between the ciliogenesis and cell division is clear, we are now beginning to identify several proteins that function to induce or repress these two cellular processes. First, in addition to their roles in intraflagellar transport, two IFT proteins have been identified to have roles in the cell cycle. Partial knockdown of the Ift27 results in defects in cytokinesis and elongation of the cell cycle (Qin et al., 2007). Depletion of Ift88 promotes cell-cycle progression to S, G2, and M-phases, whereas overexpression of Ift88 prevents G1-S transition and induces apoptotic cell death (Robert et al., 2007). Second, two proteins important in cell division, the scaffold protein Trichoplein (Inoko et al., 2012; Nishizawa et al., 2005) and Aurora A kinase (AurkA) (Pan et al., 2004; Pugacheva et al., 2007) are also important for resorption of the cilium. During proliferation, Trichoplein localizes to both mother and daughter centrioles, yet is lost during quiescence. Exogenous expression of Trichoplein inhibits primary cilia assembly, whereas depletion induces primary cilia assembly (Inoko et al., 2012). Activation of AurkA induces ciliary resorption in quiescent cells, whereas knockout of AurkA induces primary cilia formation, resulting in cell cycle arrest at the G0/G1 phase. Furthermore, this cell cycle arrest can be rescued via removal of IFT proteins, essential for ciliogenesis (Inoko et al., 2012). Trichoplein binds and activates AurkA directly and Trichoplein-dependent repression of ciliogenesis requires its ability to bind and activate AurkA. These data suggest a mechanism by which the trichoplein-AurkA pathway promotes G1 progression via the continuous suppression of primary cilia assembly. Interestingly, AurkA also interacts with Inpp5e (Plotnikova et al., 2015). AurkA phosphorylates Inpp5e, leading to the eventual transcriptional downregulation of AurkA. This relationship brings forth a possible mechanism as to how Inpp5e promotes ciliary stability and how the switch between ciliogenesis and cell proliferation is controlled.
Ciliary length has also been correlated to cell division. Primary cilia are typically between 1 and 10um long but can extend to over 20um. Various factors including injury/hypoxia (Verghese et al., 2008; Verghese et al., 2011), cytoskeletal dynamics (Miyoshi et al., 2011) and autophagy (Tang et al., 2013; Tang et al., 2014) can influence the length of cilia. It is hypothesized that increased ciliary length would allow for the ciliary membrane to be more densely decorated with receptors and channels, and thus increase sensitivity of the organelle. Several proteins are known to influence ciliary length. Nuclear distribution gene E homologue 1 (Nde1) is a centriolar protein which negatively regulates ciliary length (Kim et al., 2011b). Nde1 expression is robust during mitosis and extremely weak during quiescence. Loss of Nde1 generates longer cilia and causes a delay in cell cycle re-entry (Kim et al., 2011b). Furthermore, additional studies have reported that cell cycle-dependent mechanisms can control ciliary length through an Nde1 associated pathway (Maskey et al., 2015). Anaphase-promoting complex (Apc) is another protein associated with both cell division and ciliary length. Apc is an E3 ligase which is localized to the basal body. Apc regulates the onset of anaphase and destabilizes axonemal microtubules in the primary cilium. Inhibition of Apc and its co-activator Cdc20 increases ciliary length, while overexpression of Cdc20 suppresses cilium formation (Wang et al., 2014). Taken together, these data strongly suggest that there is highly regulated molecular crosstalk between ciliary and cell cycle components.
Since the presence or absence of cilia is tightly coupled to cell growth and proliferation, an obvious question arises: could intervention at the level of deciliation machinery serve as a possible therapeutic option for cancer? This option does appear to have some potential as proteins associated with cilium disassembly, including AurkA and the NIMA-related kinase (Nek2), are upregulated in numerous human cancers (Bischoff et al., 1998; Hayward et al., 2004; Sen et al., 1997; Wai et al., 2002). Although inhibitors of the ciliary disassembly machinery are being explored as possible therapeutic options, there are caveats to this approach. Interestingly, despite what is known about the mutually exclusive relationship between cell proliferation and ciliogenesis, some types of cancers, including basal cell carcinoma and medulloblastoma, are frequently ciliated (Han et al., 2009; Wong et al., 2009). Other cancers, including melanoma, breast, pancreatic, renal, and prostate cancer, are frequently unciliated (Basten et al., 2013; Hassounah et al., 2013; Kim et al., 2011a; Menzl et al., 2014; Seeley et al., 2009). Adding to this complexity are additional studies that demonstrate that cilia differentially regulate Hh signaling in tumors dependent upon how the pathway is activated. For example, tumors caused by excessive activation of Smo occur only in the presence of cilia, whereas tumors caused by over-expression of activated Gli2 occur when cilia are removed (Han et al., 2009; Wong et al., 2009). Thus, in tumors, cilia have both positive and negative effects on the Hh pathway. These studies emphasize the complexity of the relationship between cilia, molecular signal transduction, and cancer, and suggest that before inhibition of the deciliation machinery could be considered as a therapeutic option, a deeper understanding of the basic mechanisms of cilia-dependent signal transduction must be reached.
Cell migration and primary cilia
Coordinated, directionally-persistent migration is crucial for normal embryonic development. Cell migration occurs via the regulation and manipulation of the cytoskeleton, as well as a repositioning of cellular organelles like the centrosome in polarized cells (reviewed in Maninova et al., 2014). Defects in the formation, organization, or function of primary cilia impair the cellular processes associated with cell migration (Higginbotham et al., 2012; Jones et al., 2012; Schneider et al., 2010; Tobin et al., 2008), and are associated with migratory disorders in mice (Higginbotham et al., 2012; Tobin et al., 2008) and chickens (Schock et al., 2015). Furthermore, during migration, primary cilia have been reported to orient in parallel to the migration plane of traveling cells (Albrecht-Buehler, 1977; Katsumoto et al., 1994; Schneider et al., 2010).
Several molecular connections between primary cilia and cell migration machineries have been established. RhoGTPases are key regulators of cell dynamics including cell-cell interactions, polarity, and contraction, and have been implicated in cellular processes such as differentiation and migration. RhoGTPases have been specifically implicated in aberrant cell migration and many ciliopathies have been linked to disruptions in Rho activity (Coon et al., 2012; Valente et al., 2010). Recently it was determined that RhoGTPases localize to the basal body (Hernandez-Hernandez et al., 2013; Valente et al., 2010; Veland et al., 2013), thus potentially serving as a bridge between the cilium and cell migration.
Cilia-dependent Pdgfrα signaling has also been proposed as an important mechanism during polarization/cell migration. Na+/H+ exchange protein 1 (Nhe1) localizes to the leading edge of the plasma membrane during migration and connects the reorganizing actin cytoskeleton to the plasma membrane via integrins (Clement et al., 2013). Primary cilia-dependent Pdgfrα signaling contributes to Nhe1 localization at the leading edge through both Akt and Mek/Erk signaling (Clement et al., 2013). While the Mek/Erk signaling cascade seems to control where along the plasma membrane Nhe1 translocates to, therefore defining where the cell views the ‘leading edge’ during migration; Akt signaling is required to initiate Nhe1 translocation (Clement et al., 2013). Without primary cilia, cells cannot adequately signal through Pdgfrα to define their leading edge during migration (Schneider et al., 2010).
A more controversial link between primary and cell migration is via the Wnt/PCP pathway. Inversin acts at the decisive branch point between canonical and noncanonical Wnt signaling by targeting cytoplasmic Dvl for degradation, thus promoting β-catenin turnover and PCP (Simons et al., 2005). Inversin localizes to the primary cilium and cells that lack Inversin exhibit decreased polarization and cell migration associated with a dispersed accumulation of Gsk3-β and phosphorylated β-catenin at the ciliary base, elevated canonical Wnt signaling, and aberrant activation and leading edge localization of RhoGTPases (Otto et al., 2003; Simons et al., 2005). Although these data suggest a link between cilia, Wnt/PCP signaling and migration, Inversin and other ciliary components hypothesized to affect Wnt signaling also localize to extraciliary domains, such as the leading edge of migrating cells. Thus, some aspect of PCP-mediated regulation of cell polarization and migration may occur independent of the cilium (Boehlke et al., 2013; Cui et al., 2013; Werner et al., 2013).
Additional roles for primary cilia during development
In addition to being involved with proliferation and migration, primary cilia are also required for patterning during development. The most classic example for the requirement of primary cilia in patterning is in the neural tube (Fig. 5A–D). The neural tube is patterned on a dorso-ventral axis to generate distinct zones of neural progenitor cells. Hh, Wnt, and Bone morphogenetic protein (Bmp) signals are all necessary for proper patterning of the neural tube. Whereas, Wnt and Bmp are necessary for specifying the dorsal fates, a Shh morphogen gradient is essential for establishing ventral fates (Chiang et al., 1996; Yamada et al., 1993). Shh emanates from the notochord and floor plate of the neural tube establishing the highest levels of Shh at the most ventral aspect of the neural tube and the lowest levels of Shh at the most dorsal aspect of the neural tube. Along this gradient six different domains of neural progenitor sub-types are established, including motor neurons and interneurons (Ericson et al., 1997). Loss of Shh signaling impairs the specification of the most ventral neural cell fates (Chiang et al., 1996), whereas hyperactivation of the pathway causes an expansion of ventral cell fates (Roelink et al., 1995).
Loss of IFT-B proteins, including Ift88 or Ift172, results in dorsalization of the neural tube. This phenotype is similar to that caused by a loss of Shh signaling; however, cell types that require low levels of Hh signaling are specified. The dampened severity of the ciliopathic phenotype is due to cilia also being required for processing the Gli repressor (Haycraft et al., 2005; Huangfu and Anderson, 2005; Liu et al., 2005; May et al., 2005). IFT-A mutants, including Ift139−/−, which allow for formation of cilia with abnormal morphology, exhibit phenotypes similar to those observed with excessive Hh signaling and ventralization of the neural tube (Tran et al., 2008). Primary cilia are also required for anterior-posterior patterning of the limb (Haycraft et al., 2005; Haycraft et al., 2007; Norman et al., 2009) (Fig. 5E, F) and patterning of the facial prominences (Brugmann et al., 2010; Chang et al., 2016; Haycraft et al., 2007; Millington et al., 2017) (Fig. 5G–J). Interestingly, as in the neural tube, ciliopathic phenotypes present in the limb and the face are very reminiscent of Hh signaling mutations. Finally, as discussed earlier, primary cilia can also function as mechanosensors, capable of detecting fluid flow or mechanical force. In this capacity, ciliary function is important for both kidney (Fig. 5K, L) and skeletal development (Koyama, et al., 2007; Muhammad et al., 2012; Zhang et al., 2003).
V. Conclusions
Primary cilia are undoubtedly important for signal transduction. Although much has been gained from our study of the cilium, many questions still exist. For example, does the cilium function to integrate molecular signals from several signaling pathways? Could manipulation of the primary ciliome be used as a therapeutic option for disease that have aberrant Hh, Wnt or Pdgf signaling? One confounding issue for addressing questions such as these are the remaining controversies in the field (e.g., the exact mechanisms of ciliary-dependent Hh transduction, Wnt signal transduction, Ca2+ signaling at the node). Continued study of this organelle will undoubtedly expand our understanding of molecular signal transduction and could potentially improve the way we treat diseases; however, with several remaining controversies in the field, is it possible that even within primary cilia there is a yet to be appreciated degree of heterogeneity?
Although cilia have a degree of heterogeneity based upon which organ system they are associated with (e.g., nodal, versus olfactory, versus airway cilia); primary cilia have historically been considered homogeneous based on their conserved structure and components (basal body, transition zone, 9+0 microtubule axoneme arrangement). Several observations and experimental findings suggest we have been overlooking the prospect that there is a larger degree of heterogeneity among primary cilia, even those extending from a single tissue, than previously thought. First, not all ciliopathies present with identical phenotypes. For example, Bardet Biedl Syndrome (BBS) Type 2 (Bbs2), Oral-Facial Digital syndrome (OFD) Type 1 (Ofd1), and Ellis-Van Crevald (EVC) syndrome (EvC or EvC2) all present with distinct phenotypes. While intellectual disability can occur in both BBS and OFD1; BBS patients frequently present with obesity and hypogonadism, whereas OFD1 patients can experience lobulated tongues, cleft palate, hypertelorism and micrognathia. EVC patients can experience tooth defects also common to OFD and cardiac defects also common in BBS, but they more commonly experience abnormally short arms and legs, and hypoplastic nails (Hennekam et al., 2010). One possible explanation for this observation is that the expression of ciliary genes responsible for these ciliopathies are not identical. For example, analysis of Bbs2 and Ofd1, and EvC2 expression via in situ hybridization, reveals that each gene has areas of robust expression that coincide with the area predominantly affected in the associated ciliopathy. Thus, the ciliopathic phenotype will vary depending upon which tissue the ciliary gene is robustly expressed in. Although this seems an obvious correlation, a comprehensive comparison of the primary ciliome between tissues affected in ciliopathies has yet to be performed.
In addition to ciliary genes having non-ubiquitous expression patterns, primary cilia heterogeneity has also been suggested to occur via ciliary proteins having tissue specific function. For example, mutations in the Mks1 gene affect cilia formation in a tissue and cell-type dependent manner (Weatherbee et al., 2009). While cilia formation is impaired at the node, forelimb mesenchyme, lung mesenchyme, and brain of Mks1 mutants, cilia are still present on the multiciliated cells of the lung and bile ducts of the liver (Weatherbee et al., 2009). Additionally, although some Mks1 mutant phenotypes (fusion and forking of the ribs, reduced mineralization in the skull, and pulmonary hypoplasia) can easily be explained by a loss of Hh signaling, limb abnormalities reflect an expansion of Hh signaling, and the neural tube presents with phenotypes associated with both an expansion and reduction in Hh signaling (Weatherbee et al., 2009). This evidence supports the hypothesis that not only may cilia be heterogeneous, but that the function ciliary genes play may also be tissue-dependent. Further analysis of the ciliome will need to be done to address questions such as these.
If there is indeed a high level of heterogeneity among primary cilia, it is easy to see how this could be extrapolated to propose that primary cilia may be a “tunable” organelle, which ultimately may make primary cilia more sensitive to some signaling pathways in a tissue or cell-type dependent manner. For example, could ectopic overexpression of a ciliary component necessary for Hh signal transduction make a cell more sensitive to the Hh signaling pathway? In this manner, ciliary heterogeneity may contribute to different sensitivities tissues have to signaling pathways and change how a primary cilium responds to or transduces a particular signal. Temporal and spatial analysis of the ciliome, coupled with a better understanding of how the cilium in different cell types transduce molecular signals could make a significant impact on how we understand signal transduction in the future.
Figure 4. Cilia-dependent Pdgfrα signaling.
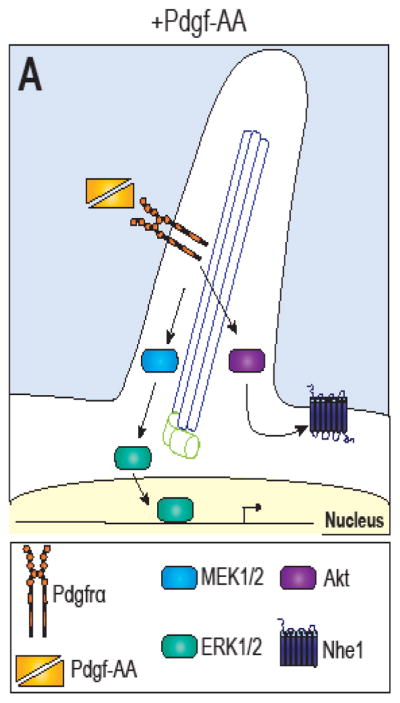
(A) Pdgfrα localizes to the membrane of the primary cilia. In the presence of its ligand, Pdgf-AA, Mer/Erk and Akt pathways are activated.
Highlights.
Primary cilia are necessary for transduction of several signaling pathways
Primary cilia are required for Hh signal transduction
The role of primary cilia in Wnt and Ca2+ signaling is controversial
Loss of primary cilia results in a class of diseases called ciliopathies
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- Albrecht-Buehler G. Phagokinetic tracks of 3T3 cells: parallels between the orientation of track segments and of cellular structures which contain actin or tubulin. Cell. 1977;12:333–339. doi: 10.1016/0092-8674(77)90109-x. [DOI] [PubMed] [Google Scholar]
- Allen BL, Song JY, Izzi L, Althaus IW, Kang JS, Charron F, Krauss RS, McMahon AP. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev Cell. 2011;20:775–787. doi: 10.1016/j.devcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RG. The three-dimensional structure of the basal body from the rhesus monkey oviduct. The Journal of cell biology. 1972;54:246–265. doi: 10.1083/jcb.54.2.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nature genetics. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Artus J, Kang M, Cohen-Tannoudji M, Hadjantonakis AK. PDGF signaling is required for primitive endoderm cell survival in the inner cell mass of the mouse blastocyst. Stem cells (Dayton, Ohio) 2013;31:1932–1941. doi: 10.1002/stem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahe S, Stierhof YD, Wilkinson CJ, Leiss F, Nigg EA. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. The Journal of cell biology. 2005;171:27–33. doi: 10.1083/jcb.200504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is Gli dependent and involves an activator function of Gli3. Dev Cell. 2004;6:103–115. doi: 10.1016/s1534-5807(03)00394-0. [DOI] [PubMed] [Google Scholar]
- Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO reports. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker AR, Renzaglia KS, Fry K, Dawe HR. Bioinformatic analysis of ciliary transition zone proteins reveals insights into the evolution of ciliopathy networks. BMC Genomics. 2014;15:531. doi: 10.1186/1471-2164-15-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basten SG, Willekers S, Vermaat JS, Slaats GG, Voest EE, van Diest PJ, Giles RH. Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia. 2013;2:2. doi: 10.1186/2046-2530-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C, Chan CS, Novotny M, Slamon DJ, Plowman GD. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. The EMBO journal. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair HJ, Tompson S, Liu YN, Campbell J, MacArthur K, Ponting CP, Ruiz-Perez VL, Goodship JA. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biology. 2011;9:14–14. doi: 10.1186/1741-7007-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum JJ, Hayes A, Whisnant CC, Rosen G. Effect of spin-labeled maleimide on 14S and 30S dyneins in solution and on demembranated ciliary axonemes. Biochemistry. 1977;16:1937–1943. doi: 10.1021/bi00628a028. [DOI] [PubMed] [Google Scholar]
- Boehlke C, Kotsis F, Buchholz B, Powelske C, Eckardt KU, Walz G, Nitschke R, Kuehn EW. Kif3a guides microtubular dynamics, migration and lumen formation of MDCK cells. PloS one. 2013;8:e62165. doi: 10.1371/journal.pone.0062165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Godel M, Muller K, Herbst M, Hornung M, Doerken M, Kottgen M, Nitschke R, Igarashi P, Walz G, Kuehn EW. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldt K, van Reeuwijk J, Lu Q, Koutroumpas K, Nguyen TM, Texier Y, van Beersum SE, Horn N, Willer JR, Mans DA, Dougherty G, Lamers IJ, Coene KL, Arts HH, Betts MJ, Beyer T, Bolat E, Gloeckner CJ, Haidari K, Hetterschijt L, Iaconis D, Jenkins D, Klose F, Knapp B, Latour B, Letteboer SJ, Marcelis CL, Mitic D, Morleo M, Oud MM, Riemersma M, Rix S, Terhal PA, Toedt G, van Dam TJ, de Vrieze E, Wissinger Y, Wu KM, Apic G, Beales PL, Blacque OE, Gibson TJ, Huynen MA, Katsanis N, Kremer H, Omran H, van Wijk E, Wolfrum U, Kepes F, Davis EE, Franco B, Giles RH, Ueffing M, Russell RB, Roepman R, Group UKRD. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun. 2016;7:11491. doi: 10.1038/ncomms11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannon M, Gomperts M, Sumoy L, Moon RT, Kimelman D. A beta-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes Dev. 1997;11:2359–2370. doi: 10.1101/gad.11.18.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel AL, Franco B, Duffourd Y, Thevenon J, Jego L, Lopez E, Deleuze JF, Doummar D, Giles RH, Johnson CA, Huynen MA, Chevrier V, Burglen L, Morleo M, Desguerres I, Pierquin G, Doray B, Gilbert-Dussardier B, Reversade B, Steichen-Gersdorf E, Baumann C, Panigrahi I, Fargeot-Espaliat A, Dieux A, David A, Goldenberg A, Bongers E, Gaillard D, Argente J, Aral B, Gigot N, St-Onge J, Birnbaum D, Phadke SR, Cormier-Daire V, Eguether T, Pazour GJ, Herranz-Perez V, Goldstein JS, Pasquier L, Loget P, Saunier S, Megarbane A, Rosnet O, Leroux MR, Wallingford JB, Blacque OE, Nachury MV, Attie-Bitach T, Riviere JB, Faivre L, Thauvin-Robinet C. Fifteen years of research on oral-facial-digital syndromes: from 1 to 16 causal genes. Journal of medical genetics. 2017;54:371–380. doi: 10.1136/jmedgenet-2016-104436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA. A primary cilia-dependent etiology for midline facial disorders. Hum Mol Genet. 2010;19:1577–1592. doi: 10.1093/hmg/ddq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallo RA, Cox RT, Moline MM, Roose J, Polevoy GA, Clevers H, Peifer M, Bejsovec A. Drosophila Tcf and Groucho interact to repress Wingless signalling activity. Nature. 1998;395:604–608. doi: 10.1038/26982. [DOI] [PubMed] [Google Scholar]
- Chang CF, Chang YT, Millington G, Brugmann SA. Craniofacial Ciliopathies Reveal Specific Requirements for GLI Proteins during Development of the Facial Midline. PLoS genetics. 2016;12:e1006351. doi: 10.1371/journal.pgen.1006351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CF, Schock EN, O’Hare EA, Dodgson J, Cheng HH, Muir WM, Edelmann RE, Delany ME, Brugmann SA. The cellular and molecular etiology of the craniofacial defects in the avian ciliopathic mutant talpid2. Development. 2014;141:3003–3012. doi: 10.1242/dev.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Lopez A, von Kessler DP, Chiang C, Simandl BK, Zhao R, Seldin MF, Fallon JF, Beachy PA. Products, genetic linkage and limb patterning activity of a murine hedgehog gene. Development (Cambridge, England) 1994;120:3339–3353. doi: 10.1242/dev.120.11.3339. [DOI] [PubMed] [Google Scholar]
- Chavez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of Ciliary Phosphoinositide Content Regulates Trafficking and Sonic Hedgehog Signaling Output. Developmental cell. 2015;34:338–350. doi: 10.1016/j.devcel.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Chen G, Fernandez J, Mische S, Courey AJ. A functional interaction between the histone deacetylase Rpd3 and the corepressor groucho in Drosophila development. Genes Dev. 1999;13:2218–2230. doi: 10.1101/gad.13.17.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, Zhang X, Hui CC, Chuang PT. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009;23:1910–1928. doi: 10.1101/gad.1794109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Indjeian VB, McManus M, Wang L, Dynlacht BD. CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Developmental cell. 2002;3:339–350. doi: 10.1016/s1534-5807(02)00258-7. [DOI] [PubMed] [Google Scholar]
- Cheng SY, Bishop JM. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc Natl Acad Sci U S A. 2002;99:5442–5447. doi: 10.1073/pnas.082096999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2011;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Clement DL, Mally S, Stock C, Lethan M, Satir P, Schwab A, Pedersen SF, Christensen ST. PDGFRalpha signaling in the primary cilium regulates NHE1-dependent fibroblast migration via coordinated differential activity of MEK1/2-ERK1/2-p90RSK and AKT signaling pathways. Journal of cell science. 2013;126:953–965. doi: 10.1242/jcs.116426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe A, Hamada F, Bienz M. A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr Biol. 2003;13:960–966. doi: 10.1016/s0960-9822(03)00370-1. [DOI] [PubMed] [Google Scholar]
- Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I, Lowe M, Beales PL, Aguilar RC. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Human molecular genetics. 2012;21:1835–1847. doi: 10.1093/hmg/ddr615. [DOI] [PubMed] [Google Scholar]
- Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, Bale AE. Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development. 2005;132:4407–4417. doi: 10.1242/dev.02021. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, Calmont A, Jarnik M, Burch J, Zaret KS, Larue L, Bellacosa A. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Developmental biology. 2009;325:225–237. doi: 10.1016/j.ydbio.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Chatterjee B, Lozito TP, Zhang Z, Francis RJ, Yagi H, Swanhart LM, Sanker S, Francis D, Yu Q, San Agustin JT, Puligilla C, Chatterjee T, Tansey T, Liu X, Kelley MW, Spiliotis ET, Kwiatkowski AV, Tuan R, Pazour GJ, Hukriede NA, Lo CW. Wdpcp, a PCP protein required for ciliogenesis, regulates directional cell migration and cell polarity by direct modulation of the actin cytoskeleton. PLoS biology. 2013;11:e1001720. doi: 10.1371/journal.pbio.1001720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Akimaru H, Tanaka Y, Maekawa T, Nakafuku M, Ishii S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J Biol Chem. 1999;274:8143–8152. doi: 10.1074/jbc.274.12.8143. [DOI] [PubMed] [Google Scholar]
- Davis JB, Stroobant P. Platelet-derived growth factors and fibroblast growth factors are mitogens for rat Schwann cells. The Journal of cell biology. 1990;110:1353–1360. doi: 10.1083/jcb.110.4.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504:311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delling M, Indzhykulian AA, Liu X, Li Y, Xie T, Corey DP, Clapham DE. Primary cilia are not calcium-responsive mechanosensors. Nature. 2016;531:656–660. doi: 10.1038/nature17426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Fukami S, Meng X, Nishizaki Y, Zhang X, Sasaki H, Dlugosz A, Nakafuku M, Hui C. Mouse suppressor of fused is a negative regulator of sonic hedgehog signaling and alters the subcellular distribution of Gli1. Curr Biol. 1999;9:1119–1122. doi: 10.1016/s0960-9822(99)80482-5. [DOI] [PubMed] [Google Scholar]
- Dishinger JF, Kee HL, Jenkins PM, Fan S, Hurd TW, Hammond JW, Truong YN, Margolis B, Martens JR, Verhey KJ. Ciliary entry of the kinesin-2 motor KIF17 is regulated by importin-beta2 and RanGTP. Nat Cell Biol. 2010;12:703–710. doi: 10.1038/ncb2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn KV, Hughes CE, Rohatgi R. A Smoothened-Evc2 Complex Transduces the Hedgehog Signal at Primary Cilia. Dev Cell. 2012 doi: 10.1016/j.devcel.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edde B, Rossier J, Le Caer JP, Desbruyeres E, Gros F, Denoulet P. Posttranslational glutamylation of alpha-tubulin. Science. 1990;247:83–85. doi: 10.1126/science.1967194. [DOI] [PubMed] [Google Scholar]
- Ericson J, Briscoe J, Rashbass P, van Heyningen V, Jessell TM. Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harbor Symposia on Quantitative Biology. 1997;62:451–466. [PubMed] [Google Scholar]
- Fantauzzo KA, Soriano P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes & development. 2014;28:1005–1017. doi: 10.1101/gad.238709.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, Garcia-Verdugo JM, Katsanis N, Hildebrandt F, Reiter JF. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nature genetics. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G, 3rd, Abedin M, Schurmans S, Inoue T, Reiter JF. Phosphoinositides Regulate Ciliary Protein Trafficking to Modulate Hedgehog Signaling. Dev Cell. 2015;34:400–409. doi: 10.1016/j.devcel.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, Badano JL, Katsanis N. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nature genetics. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- Gilula NB, Satir P. The ciliary necklace. A ciliary membrane specialization. The Journal of cell biology. 1972;53:494–509. doi: 10.1083/jcb.53.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Liem KF, Jr, Anderson KV. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell. 2012;151:847–58. doi: 10.1016/j.cell.2012.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallak ME, Rodriguez JA, Barra HS, Caputto R. Release of tyrosine from tyrosinated tubulin. Some common factors that affect this process and the assembly of tubulin. FEBS Lett. 1977;73:147–150. doi: 10.1016/0014-5793(77)80968-x. [DOI] [PubMed] [Google Scholar]
- Hammerschmidt M, Bitgood MJ, McMahon AP. Protein kinase A is a common negative regulator of Hedgehog signaling in the vertebrate embryo. Genes and Development. 1996;10:647–658. doi: 10.1101/gad.10.6.647. [DOI] [PubMed] [Google Scholar]
- Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nature medicine. 2009;15:1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassounah NB, Nagle R, Saboda K, Roe DJ, Dalkin BL, McDermott KM. Primary cilia are lost in preinvasive and invasive prostate cancer. PloS one. 2013;8:e68521. doi: 10.1371/journal.pone.0068521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS genetics. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64:7370–7376. doi: 10.1158/0008-5472.CAN-04-0960. [DOI] [PubMed] [Google Scholar]
- Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development (Cambridge, England) 2000;127:4905–4913. doi: 10.1242/dev.127.22.4905. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Hennekam RCM, Krantz ID, Allanson JE. Gorlin’s Syndromes of the Head and Neck. 5. Oxford University Press; New York: 2010. [Google Scholar]
- Hernandez-Hernandez V, Pravincumar P, Diaz-Font A, May-Simera H, Jenkins D, Knight M, Beales PL. Bardet-Biedl syndrome proteins control the cilia length through regulation of actin polymerization. Hum Mol Genet. 2013;22:3858–3868. doi: 10.1093/hmg/ddt241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Eom TY, Mariani LE, Bachleda A, Hirt J, Gukassyan V, Cusack CL, Lai C, Caspary T, Anton ES. Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev Cell. 2012;23:925–938. doi: 10.1016/j.devcel.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover AN, Wynkoop A, Zeng H, Jia J, Niswander LA, Liu A. C2cd3 is required for cilia formation and Hedgehog signaling in mouse. Development. 2008;135:4049–4058. doi: 10.1242/dev.029835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN, Greenberg CR, McLeod DR, Bernier FP, Chudley AE, Muller T, Shboul M, Logan CV, Loucks CM, Beaulieu CL, Bowie RV, Bell SM, Adkins J, Zuniga FI, Ross KD, Wang J, Ban MR, Becker C, Nurnberg P, Douglas S, Craft CM, Akimenko MA, Hegele RA, Ober C, Utermann G, Bolz HJ, Bulman DE, Katsanis N, Blacque OE, Doherty D, Parboosingh JS, Leroux MR, Johnson CA, Boycott KM. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. American journal of human genetics. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Schier AF. Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development. 2009;136:3089–3098. doi: 10.1242/dev.041343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nature genetics. 1995;10:151–160. doi: 10.1038/ng0695-151. [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24:670–682. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, Taylor AM, Nakano Y. Role of the Drosophila patched gene in positional signalling. Nature. 1991;353:184–7. doi: 10.1038/353184a0. [DOI] [PubMed] [Google Scholar]
- Inoko A, Matsuyama M, Goto H, Ohmuro-Matsuyama Y, Hayashi Y, Enomoto M, Ibi M, Urano T, Yonemura S, Kiyono T, Izawa I, Inagaki M. Trichoplein and Aurora A block aberrant primary cilia assembly in proliferating cells. The Journal of cell biology. 2012;197:391–405. doi: 10.1083/jcb.201106101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzi L, Levesque M, Morin S, Laniel D, Wilkes BC, Mille F, Krauss RS, McMahon AP, Allen BL, Charron F. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev Cell. 2011;20:788–801. doi: 10.1016/j.devcel.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compere P, Schiffmann SN, Gergely F, Riley JH, Perez-Morga D, Woods CG, Schurmans S. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nature genetics. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- Jiang J, Struhl G. Protein kinase A and hedgehog signaling in Drosophila limb development. Cell. 1995;80:563–572. doi: 10.1016/0092-8674(95)90510-3. [DOI] [PubMed] [Google Scholar]
- Jiang ST, Chiou YY, Wang E, Chien YL, Ho HH, Tsai FJ, Lin CY, Tsai SP, Li H. Essential role of nephrocystin in photoreceptor intraflagellar transport in mouse. Hum Mol Genet. 2009;18:1566–1577. doi: 10.1093/hmg/ddp068. [DOI] [PubMed] [Google Scholar]
- Jiang ST, Chiou YY, Wang E, Lin HK, Lee SP, Lu HY, Wang CK, Tang MJ, Li H. Targeted disruption of Nphp1 causes male infertility due to defects in the later steps of sperm morphogenesis in mice. Hum Mol Genet. 2008;17:3368–3379. doi: 10.1093/hmg/ddn231. [DOI] [PubMed] [Google Scholar]
- Jones TJ, Adapala RK, Geldenhuys WJ, Bursley C, AbouAlaiwi WA, Nauli SM, Thodeti CK. Primary cilia regulates the directional migration and barrier integrity of endothelial cells through the modulation of hsp27 dependent actin cytoskeletal organization. Journal of cellular physiology. 2012;227:70–76. doi: 10.1002/jcp.22704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers J, Korswagen HC, Acton D, Breuer M, Berns A. Activation of a novel proto-oncogene, Frat1, contributes to progression of mouse T-cell lymphomas. The EMBO journal. 1997;16:441–450. doi: 10.1093/emboj/16.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalderon D. Similarities between the Hedgehog and Wnt signaling pathways. Trends in cell biology. 2002;12:523–531. doi: 10.1016/s0962-8924(02)02388-7. [DOI] [PubMed] [Google Scholar]
- Katsumoto T, Higaki K, Ohno K, Onodera K. The orientation of primary cilia during the wound response in 3Y1 cells. Biology of the cell/under the auspices of the European Cell Biology Organization. 1994;81:17–21. doi: 10.1016/0248-4900(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Kim J, Dabiri S, Seeley ES. Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma. PloS one. 2011a;6:e27410. doi: 10.1371/journal.pone.0027410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Suh YA, Oh JH, Lee BR, Kim J, Jang SJ. KIF3A binds to β-arrestin for suppressing Wnt/β-catenin signalling independently of primary cilia in lung cancer. Scientific Reports. 2016;6:32770. doi: 10.1038/srep32770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol. 2011b;13:351–360. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleylein-Sohn J, Westendorf J, Le Clech M, Habedanck R, Stierhof YD, Nigg EA. Plk4-induced centriole biogenesis in human cells. Developmental cell. 2007;13:190–202. doi: 10.1016/j.devcel.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Klingensmith J, Nusse R, Perrimon N. The Drosophila segment polarity gene dishevelled encodes a novel protein required for response to the wingless signal. Genes Dev. 1994;8:118–130. doi: 10.1101/gad.8.1.118. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Dynlacht BD. Regulating the transition from centriole to basal body. The Journal of cell biology. 2011;193:435–444. doi: 10.1083/jcb.201101005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, Toftgard R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- Kottgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G. TRPP2 and TRPV4 form a polymodal sensory channel complex. The Journal of cell biology. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama E, Young B, Nagayama M, Shibukawa Y, Enomoto-Iwamoto M, Iwamoto M, Maeda Y, Lanske B, Song B, Serra R, Pacifici M. Conditional Kif3a ablation causes abnormal hedgehog signaling topography, growth plate dysfunction, and excessive bone and cartilage formation during mouse skeletogenesis. Development. 2007;134:2159–2169. doi: 10.1242/dev.001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A. 1993;90:5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Hernault SW, Rosenbaum JL. Chlamydomonas alpha-tubulin is posttranslationally modified by acetylation on the epsilon-amino group of a lysine. Biochemistry. 1985;24:473–478. doi: 10.1021/bi00323a034. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Schroth J, Gleeson JG. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nature cell biology. 2011;13:700–707. doi: 10.1038/ncb2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law KK, Makino S, Mo R, Zhang X, Puviindran V, Hui CC. Antagonistic and cooperative actions of Kif7 and Sufu define graded intracellular Gli activities in Hedgehog signaling. PloS one. 2012;7:e50193. doi: 10.1371/journal.pone.0050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyssac PP. Changes in single nephron renin release are mediated by tubular fluid flow rate. Kidney international. 1986;30:332–339. doi: 10.1038/ki.1986.189. [DOI] [PubMed] [Google Scholar]
- Li A, Xia X, Yeh J, Kua H, Liu H, Mishina Y, Hao A, Li B. PDGF-AA promotes osteogenic differentiation and migration of mesenchymal stem cell by down-regulating PDGFRalpha and derepressing BMP-Smad1/5/8 signaling. PloS one. 2014;9:e113785. doi: 10.1371/journal.pone.0113785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Function of protein Kinase A in hedgehog signal transduction and drosophilaImagianl Disc Development. Cell. 1995;80:553–562. doi: 10.1016/0092-8674(95)90509-x. [DOI] [PubMed] [Google Scholar]
- Liem KF, Jr, He M, Ocbina PJ, Anderson KV. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc Natl Acad Sci U S A. 2009;106:13377–82. doi: 10.1073/pnas.0906944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yao E, Wang K, Nozawa Y, Shimizu H, Johnson JR, Chen JN, Krogan NJ, Chuang PT. Regulation of Sufu activity by p66beta and Mycbp provides new insight into vertebrate Hedgehog signaling. Genes Dev. 2014;28:2547–2563. doi: 10.1101/gad.249425.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Malone AM, Anderson CT, Tummala P, Kwon RY, Johnston TR, Stearns T, Jacobs CR. Primary cilia mediate mechanosensing in bone cells by a calcium-independent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13325–13330. doi: 10.1073/pnas.0700636104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maninova M, Iwanicki MP, Vomastek T. Emerging role for nuclear rotation and orientation in cell migration. Cell adhesion & migration. 2014;8:42–48. doi: 10.4161/cam.27761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci U S A. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli DC, Fan CM. Gas1 extends the range of Hedgehog action by facilitating its signaling. Genes Dev. 2007;21:1231–1243. doi: 10.1101/gad.1546307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey D, Marlin MC, Kim S, Kim S, Ong EC, Li G, Tsiokas L. Cell cycle-dependent ubiquitylation and destruction of NDE1 by CDK5-FBW7 regulates ciliary length. Embo j. 2015;34:2424–2440. doi: 10.15252/embj.201490831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Developmental biology. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- McDermott A, Gustafsson M, Elsam T, Hui CC, Emerson CP, Jr, Borycki AG. Gli2 and Gli3 have redundant and context-dependent function in skeletal muscle formation. Development. 2005;132:345–357. doi: 10.1242/dev.01537. [DOI] [PubMed] [Google Scholar]
- McGrath J, Somlo S, Makova S, Tian X, Brueckner M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- Menzl I, Lebeau L, Pandey R, Hassounah NB, Li FW, Nagle R, Weihs K, McDermott KM. Loss of primary cilia occurs early in breast cancer development. Cilia. 2014;3:7. doi: 10.1186/2046-2530-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot N, Basler K. Suppressor of fused opposes hedgehog signal transduction by impeding nuclear accumulation of the activator form of Cubitus interruptus. Development. 2000;127:4001–4010. doi: 10.1242/dev.127.18.4001. [DOI] [PubMed] [Google Scholar]
- Millington G, Elliott KH, Chang YT, Chang CF, Dlugosz A, Brugmann SA. Cilia-dependent GLI processing in neural crest cells is required for tongue development. Developmental biology. 2017;424:124–137. doi: 10.1016/j.ydbio.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Kasahara K, Miyazaki I, Asanuma M. Factors that influence primary cilium length. Acta medica Okayama. 2011;65:279–285. doi: 10.18926/AMO/47009. [DOI] [PubMed] [Google Scholar]
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. doi: 10.1126/science.272.5266.1339. [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S. Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Human molecular genetics. 2005;14:645–656. doi: 10.1093/hmg/ddi061. [DOI] [PubMed] [Google Scholar]
- Morgan D, Eley L, Sayer J, Strachan T, Yates LM, Craighead AS, Goodship JA. Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Human molecular genetics. 2002;11:3345–3350. doi: 10.1093/hmg/11.26.3345. [DOI] [PubMed] [Google Scholar]
- Muhammad H, Rais Y, Miosge N, Ornan EM. The primary cilium as a dual sensor of mechanochemical signals in chondrocytes. Cellular and Molecular Life Sciences. 2012;69:2101–2107. doi: 10.1007/s00018-011-0911-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Wen X, Ratti N, Loktev A, Rangell L, Scales SJ, Jackson PK. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013;152:210–223. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nature genetics. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nishizawa M, Izawa I, Inoko A, Hayashi Y, Nagata K, Yokoyama T, Usukura J, Inagaki M. Identification of trichoplein, a novel keratin filament-binding protein. J Cell Sci. 2005;118:1081–1090. doi: 10.1242/jcs.01667. [DOI] [PubMed] [Google Scholar]
- Noda K, Kitami M, Kitami K, Kaku M, Komatsu Y. Canonical and noncanonical intraflagellar transport regulates craniofacial skeletal development. Proc Natl Acad Sci U S A. 2016;113:E2589–2597. doi: 10.1073/pnas.1519458113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka S, Shiratori H, Saijoh Y, Hamada H. Determination of left-right patterning of the mouse embryo by artificial nodal flow. Nature. 2002;418:96–99. doi: 10.1038/nature00849. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Noordermeer J, Klingensmith J, Perrimon N, Nusse R. dishevelled and armadillo act in the wingless signalling pathway in Drosophila. Nature. 1994;367:80–83. doi: 10.1038/367080a0. [DOI] [PubMed] [Google Scholar]
- Norman RX, Ko HW, Huang V, Eun CM, Abler LL, Zhang Z, Sun X, Eggenschwiler JT. Tubby-like protein 3 (TULP3) regulates patterning in the mouse embryo through inhibition of Hedgehog signaling. Hum Mol Genet. 2009;18:1740–1754. doi: 10.1093/hmg/ddp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287:795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Nusse R. Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development. 2003;130:5297–5305. doi: 10.1242/dev.00821. [DOI] [PubMed] [Google Scholar]
- Ocbina PJ, Tuson M, Anderson KV. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PloS one. 2009;4:e6839. doi: 10.1371/journal.pone.0006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othberg A, Odin P, Ballagi A, Ahgren A, Funa K, Lindvall O. Specific effects of platelet derived growth factor (PDGF) on fetal rat and human dopaminergic neurons in vitro. Experimental brain research. 1995;105:111–122. doi: 10.1007/BF00242187. [DOI] [PubMed] [Google Scholar]
- Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nature genetics. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paces-Fessy M, Boucher D, Petit E, Paute-Briand S, Blanchet-Tournier MF. The negative regulator of Gli, Suppressor of fused (Sufu), interacts with SAP18, Galectin3 and other nuclear proteins. Biochem J. 2004;378:353–362. doi: 10.1042/BJ20030786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintrand M, Moudjou M, Delacroix H, Bornens M. Centrosome organization and centriole architecture: their sensitivity to divalent cations. J Struct Biol. 1992;108:107–128. doi: 10.1016/1047-8477(92)90011-x. [DOI] [PubMed] [Google Scholar]
- Pan J, Wang Q, Snell WJ. An aurora kinase is essential for flagellar disassembly in Chlamydomonas. Dev Cell. 2004;6:445–51. doi: 10.1016/s1534-5807(04)00064-4. [DOI] [PubMed] [Google Scholar]
- Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Wang B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J Biol Chem. 2007;282:10846–10852. doi: 10.1074/jbc.M608599200. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. The Journal of cell biology. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol. 2002;12:R378–380. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- Pearse RV, 2nd, Collier LS, Scott MP, Tabin CJ. Vertebrate homologs of Drosophila suppressor of fused interact with the gli family of transcriptional regulators. Developmental biology. 1999;212:323–336. doi: 10.1006/dbio.1999.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham A, Therond P, Alves G, Tournier FB, Busson D, Lamour-Isnard C, Bouchon BL, Preat T, Tricoire H. The Suppressor of fused gene encodes a novel PEST protein involved in Drosophila segment polarity establishment. Genetics. 1995;140:587–598. doi: 10.1093/genetics/140.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan MN, Leddy HA, Votta BJ, Kumar S, Levy DS, Lipshutz DB, Lee SH, Liedtke W, Guilak F. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis and rheumatism. 2009;60:3028–3037. doi: 10.1002/art.24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce GF, Tarpley JE, Allman RM, Goode PS, Serdar CM, Morris B, Mustoe TA, Vande Berg J. Tissue repair processes in healing chronic pressure ulcers treated with recombinant platelet-derived growth factor BB. Am J Pathol. 1994;145:1399–1410. [PMC free article] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nature medicine. 2007;13:1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikova OV, Seo S, Cottle DL, Conduit S, Hakim S, Dyson JM, Mitchell CA, Smyth IM. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci. 2015;128:364–372. doi: 10.1242/jcs.161323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. The Journal of membrane biology. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. The Journal of membrane biology. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–1363. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1456–1461. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Wang Z, Diener D, Rosenbaum J. Intraflagellar transport protein 27 is a small G protein involved in cell-cycle control. Curr Biol. 2007;17:193–202. doi: 10.1016/j.cub.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redeker V, Levilliers N, Schmitter JM, Le Caer JP, Rossier J, Adoutte A, Bre MH. Polyglycylation of tubulin: a posttranslational modification in axonemal microtubules. Science. 1994;266:1688–1691. doi: 10.1126/science.7992051. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Jensen CG, Jensen LC. The resorption of primary cilia during mitosis in a vertebrate (PtK1) cell line. J Ultrastruct Res. 1979;68:173–85. doi: 10.1016/s0022-5320(79)90152-7. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Pringle N, Mosley MJ, Westermark B, Dubois-Dalcq M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell. 1988;53:309–319. doi: 10.1016/0092-8674(88)90392-3. [DOI] [PubMed] [Google Scholar]
- Robert A, Margall-Ducos G, Guidotti JE, Brégerie O, Celati C, Bréchot C, Desdouets C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J Cell Sci. 2007;120:628–37. doi: 10.1242/jcs.03366. [DOI] [PubMed] [Google Scholar]
- Roelink H, Porter JA, Chiang C, Tanabe Y, Chang DT, Beachy PA, Jessell TM. Floor plate and motor neuron induction by different concentrations of the amino-terminal cleavage product of sonic hedgehog autoproteolysis. Cell. 1995;81:445–455. doi: 10.1016/0092-8674(95)90397-6. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science (New York, NY) 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, Tan PL, Phillips HM, Leroux MR, Henderson DJ, Murdoch JN, Copp AJ, Eliot MM, Lupski JR, Kemp DT, Dollfus H, Tada M, Katsanis N, Forge A, Beales PL. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nature genetics. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, Marino B, Dallapiccola B, Wright M, Meitinger T, Polymeropoulos MH, Goodship J. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nature genetics. 2000;24:283–286. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Tompson SW, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, Hamel B, Gibbs JL, Young ID, Wright MJ, Goodship JA. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. American journal of human genetics. 2003;72:728–732. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, Lee E, Mayer L, Kirschner MW. Control of beta-catenin stability: reconstitution of the cytoplasmic steps of the wnt pathway in Xenopus egg extracts. Mol Cell. 2000;5:523–532. doi: 10.1016/s1097-2765(00)80446-3. [DOI] [PubMed] [Google Scholar]
- Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M, Huntzicker EG, Sfakianos MK, Sandoval W, Bazan JF, Kulkarni P, Garcia-Gonzalo FR, Seol AD, O’Toole JF, Held S, Reutter HM, Lane WS, Rafiq MA, Noor A, Ansar M, Devi AR, Sheffield VC, Slusarski DC, Vincent JB, Doherty DA, Hildebrandt F, Reiter JF, Jackson PK. Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell. 2011;145:513–528. doi: 10.1016/j.cell.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- Satir P, Gilula NB. THE CELL JUNCTION IN A LAMELLIBRANCH GILL CILIATED EPITHELIUM: Localization of Pyroantimonate Precipitate. The Journal of cell biology. 1970;47:468–487. doi: 10.1083/jcb.47.2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid FM, Schou KB, Vilhelm MJ, Holm MS, Breslin L, Farinelli P, Larsen LA, Andersen JS, Pedersen LB, Christensen ST. IFT20 modulates ciliary PDGFRalpha signaling by regulating the stability of Cbl E3 ubiquitin ligases. The Journal of cell biology. 2017 doi: 10.1083/jcb.201611050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Cammer M, Lehman J, Nielsen SK, Guerra CF, Veland IR, Stock C, Hoffmann EK, Yoder BK, Schwab A, Satir P, Christensen ST. Directional cell migration and chemotaxis in wound healing response to PDGF-AA are coordinated by the primary cilium in fibroblasts. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2010;25:279–292. doi: 10.1159/000276562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Schock EN, Chang CF, Struve JN, Chang YT, Chang J, Delany ME, Brugmann SA. Using the avian mutant talpid2 as a disease model for understanding the oral-facial phenotypes of oral-facial-digital syndrome. Dis Model Mech. 2015;8:855–866. doi: 10.1242/dmm.020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M. Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res. 2009;69:422–430. doi: 10.1158/0008-5472.CAN-08-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–2200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- Shi Q, Han Y, Jiang J. Suppressor of fused impedes Ci/Gli nuclear import by opposing Trn/Kapbeta2 in Hedgehog signaling. Journal of cell science. 2014;127:1092–1103. doi: 10.1242/jcs.142828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nature genetics. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006;313:629–633. doi: 10.1126/science.1124534. [DOI] [PubMed] [Google Scholar]
- Smith EF, Yang P. The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton. 2004;57:8–17. doi: 10.1002/cm.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. The Journal of cell biology. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin SP. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J Cell Sci. 1968;3:207–230. doi: 10.1242/jcs.3.2.207. [DOI] [PubMed] [Google Scholar]
- Spektor A, Tsang WY, Khoo D, Dynlacht BD. Cep97 and CP110 suppress a cilia assembly program. Cell. 2007;130:678–690. doi: 10.1016/j.cell.2007.06.027. [DOI] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes and Development. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, Noll M, Hooper JE, de Sauvage F, Rosenthal A. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- Sulik K, Dehart DB, Iangaki T, Carson JL, Vrablic T, Gesteland K, Schoenwolf GC. Morphogenesis of the murine node and notochordal plate. Developmental Dynamics. 1994;201:260–278. doi: 10.1002/aja.1002010309. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Tabin CJ, Vogan KJ. A two-cilia model for vertebrate left-right axis specification. Genes Dev. 2003;17:1–6. doi: 10.1101/gad.1053803. [DOI] [PubMed] [Google Scholar]
- Takao D, Nemoto T, Abe T, Kiyonari H, Kajiura-Kobayashi H, Shiratori H, Nonaka S. Asymmetric distribution of dynamic calcium signals in the node of mouse embryo during left-right axis formation. Developmental biology. 2013;376:23–30. doi: 10.1016/j.ydbio.2013.01.018. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A−/− mice analysis. The Journal of cell biology. 1999;145:825–836. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, Zhong Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature. 2013;502:254–257. doi: 10.1038/nature12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Zhu M, Zhong Q. Self-eating to remove cilia roadblock. Autophagy. 2014;10:379–381. doi: 10.4161/auto.27346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12:589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temiyasathit S, Tang WJ, Leucht P, Anderson CT, Monica SD, Castillo AB, Helms JA, Stearns T, Jacobs CR. Mechanosensing by the primary cilium: deletion of Kif3A reduces bone formation due to loading. PloS one. 2012;7:e33368. doi: 10.1371/journal.pone.0033368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Franco B, Saugier-Veber P, Aral B, Gigot N, Donzel A, Van Maldergem L, Bieth E, Layet V, Mathieu M, Teebi A, Lespinasse J, Callier P, Mugneret F, Masurel-Paulet A, Gautier E, Huet F, Teyssier JR, Tosi M, Frebourg T, Faivre L. Genomic deletions of OFD1 account for 23% of oral-facial-digital type 1 syndrome after negative DNA sequencing. Human mutation. 2009;30:E320–329. doi: 10.1002/humu.20888. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Lee JS, Lopez E, Herranz-Perez V, Shida T, Franco B, Jego L, Ye F, Pasquier L, Loget P, Gigot N, Aral B, Lopes CA, St-Onge J, Bruel AL, Thevenon J, Gonzalez-Granero S, Alby C, Munnich A, Vekemans M, Huet F, Fry AM, Saunier S, Riviere JB, Attie-Bitach T, Garcia-Verdugo JM, Faivre L, Megarbane A, Nachury MV. The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nature genetics. 2014 doi: 10.1038/ng.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin JL, Di Franco M, Eichers E, May-Simera H, Garcia M, Yan J, Quinlan R, Justice MJ, Hennekam RC, Briscoe J, Tada M, Mayor R, Burns AJ, Lupski JR, Hammond P, Beales PL. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet-Biedl syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6714–6719. doi: 10.1073/pnas.0707057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nature genetics. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang WY, Dynlacht BD. CP110 and its network of partners coordinately regulate cilia assembly. Cilia. 2013;2:9. doi: 10.1186/2046-2530-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979;17:527–535. doi: 10.1016/0092-8674(79)90261-7. [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umberger NL, Caspary T. Ciliary transport regulates PDGF-AA/alphaalpha signaling via elevated mammalian target of rapamycin signaling and diminished PP2A activity. Mol Biol Cell. 2015;26:350–358. doi: 10.1091/mbc.E14-05-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Logan CV, Mougou-Zerelli S, Lee JH, Silhavy JL, Brancati F, Iannicelli M, Travaglini L, Romani S, Illi B, Adams M, Szymanska K, Mazzotta A, Lee JE, Tolentino JC, Swistun D, Salpietro CD, Fede C, Gabriel S, Russ C, Cibulskis K, Sougnez C, Hildebrandt F, Otto EA, Held S, Diplas BH, Davis EE, Mikula M, Strom CM, Ben-Zeev B, Lev D, Sagie TL, Michelson M, Yaron Y, Krause A, Boltshauser E, Elkhartoufi N, Roume J, Shalev S, Munnich A, Saunier S, Inglehearn C, Saad A, Alkindy A, Thomas S, Vekemans M, Dallapiccola B, Katsanis N, Johnson CA, Attie-Bitach T, Gleeson JG. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nature genetics. 2010;42:619–625. doi: 10.1038/ng.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland IR, Awan A, Pedersen LB, Yoder BK, Christensen ST. Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiology. 2009;111:p39–p53. doi: 10.1159/000208212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veland IR, Montjean R, Eley L, Pedersen LB, Schwab A, Goodship J, Kristiansen K, Pedersen SF, Saunier S, Christensen ST. Inversin/Nephrocystin-2 is required for fibroblast polarity and directional cell migration. PloS one. 2013;8:e60193. doi: 10.1371/journal.pone.0060193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verghese E, Weidenfeld R, Bertram JF, Ricardo SD, Deane JA. Renal cilia display length alterations following tubular injury and are present early in epithelial repair. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2008;23:834–841. doi: 10.1093/ndt/gfm743. [DOI] [PubMed] [Google Scholar]
- Verghese E, Zhuang J, Saiti D, Ricardo SD, Deane JA. In vitro investigation of renal epithelial injury suggests that primary cilium length is regulated by hypoxia-inducible mechanisms. Cell biology international. 2011;35:909–913. doi: 10.1042/CBI20090154. [DOI] [PubMed] [Google Scholar]
- Wai DH, Schaefer KL, Schramm A, Korsching E, Van Valen F, Ozaki T, Boecker W, Schweigerer L, Dockhorn-Dworniczak B, Poremba C. Expression analysis of pediatric solid tumor cell lines using oligonucleotide microarrays. Int J Oncol. 2002;20:441–451. [PubMed] [Google Scholar]
- Wang W, Wu T, Kirschner MW. The master cell cycle regulator APC-Cdc20 regulates ciliary length and disassembly of the primary cilium. Elife. 2014;3:e03083. doi: 10.7554/eLife.03083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wann AK, Zuo N, Haycraft CJ, Jensen CG, Poole CA, McGlashan SR, Knight MM. Primary cilia mediate mechanotransduction through control of ATP-induced Ca2+ signaling in compressed chondrocytes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:1663–1671. doi: 10.1096/fj.11-193649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Hum Mol Genet. 2009;18:4565–4575. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O’Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- Werner ME, Ward HH, Phillips CL, Miller C, Gattone VH, Bacallao RL. Inversin modulates the cortical actin network during mitosis. American journal of physiology Cell physiology. 2013;305:C36–47. doi: 10.1152/ajpcell.00279.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley DN. Cilia in cell-cultured fibroblasts. I. On their occurrence and relative frequencies in primary cultures and established cell lines. Journal of anatomy. 1969;105:351–362. [PMC free article] [PubMed] [Google Scholar]
- Wheatley DN. The centriole, a central enigma of cell biology. Elsevier Biomedical Press; New York, N.Y: 1982. [Google Scholar]
- Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell biology international. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, Yoder BK, Leroux MR. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. The Journal of cell biology. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J, Marin de Evsikova C, Smith RS, Hicks WL, Edwards MM, Longo-Guess C, Li T, Naggert JK, Nishina PM. NPHP4 is necessary for normal photoreceptor ribbon synapse maintenance and outer segment formation, and for sperm development. Hum Mol Genet. 2011;20:482–496. doi: 10.1093/hmg/ddq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH, Jr, Dlugosz AA, Reiter JF. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nature medicine. 2009;15:1055–1061. doi: 10.1038/nm.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Dallas M, Qiu N, Nicolella D, Cao L, Johnson M, Bonewald L, Quarles LD. Conditional deletion of Pkd1 in osteocytes disrupts skeletal mechanosensing in mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:2418–2432. doi: 10.1096/fj.10-180299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Pfaff SL, Edlund T, Jessell TM. Control of cell pattern in the neural tube: motor neuron induction by diffusible factors from notochord and floor plate. Cell. 1993;73:673–686. doi: 10.1016/0092-8674(93)90248-o. [DOI] [PubMed] [Google Scholar]
- Yang C, Chen W, Chen Y, Jiang J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Research. 2012;22:1593–1604. doi: 10.1038/cr.2012.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mlodzik M. Wnt-Frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt) Annual review of cell and developmental biology. 2015;31:623–646. doi: 10.1146/annurev-cellbio-100814-125315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HH, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002;16:1433–1440. doi: 10.1101/gad.981202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Zeng H, Ning G, Reiter JF, Liu A. C2cd3 is critical for centriolar distal appendage assembly and ciliary vesicle docking in mammals. Proc Natl Acad Sci U S A. 2014;111:2164–2169. doi: 10.1073/pnas.1318737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi ES, Lee H, Yin S, Piguet P, Sarosi I, Kaufmann S, Tarpley J, Wang NS, Ulich TR. Platelet-derived growth factor causes pulmonary cell proliferation and collagen deposition in vivo. Am J Pathol. 1996;149:539–548. [PMC free article] [PubMed] [Google Scholar]
- Yoshiba S, Shiratori H, Kuo IY, Kawasumi A, Shinohara K, Nonaka S, Asai Y, Sasaki G, Belo JA, Sasaki H, Nakai J, Dworniczak B, Ehrlich BE, Pennekamp P, Hamada H. Cilia at the node of mouse embryos sense fluid flow for left-right determination via Pkd2. Science. 2012;338:226–231. doi: 10.1126/science.1222538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost C, Farr GH, 3rd, Pierce SB, Ferkey DM, Chen MM, Kimelman D. GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell. 1998;93:1031–1041. doi: 10.1016/s0092-8674(00)81208-8. [DOI] [PubMed] [Google Scholar]
- Yuan S, Zhao L, Brueckner M, Sun Z. Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr Biol. 2015;25:556–567. doi: 10.1016/j.cub.2014.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chen XM, Sun ZJ, Bian Z, Fan MW, Chen Z. Epithelial expression of SHH signaling pathway in odontogenic tumors. Oral Oncol. 2006;42:398–408. doi: 10.1016/j.oraloncology.2005.09.008. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Murcia NS, Chittenden LR, Richards WG, Michaud EJ, Woychik RP, Yoder BK. Loss of the Tg737 protein results in skeletal patterning defects. Dev Dyn. 2003;227:78–90. doi: 10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Shen L, Law K, Zhang Z, Liu X, Hua H, Li S, Huang H, Yue S, Hui CC, Cheng SY. Suppressor of Fused Chaperones Gli Proteins To Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol Cell Biol. 2017:37. doi: 10.1128/MCB.00421-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann KW. Beitrage zur Kenntnis einiger Drusen und Epithelien. Archmikr Anat u Entwick. 1898;52:552. [Google Scholar]