

Summary
The chromatin regulator FACT (facilitates chromatin transcription) is essential for ensuring stable gene expression by promoting transcription. In a genetic screen using Caenorhabditis elegans, we identified that FACT maintains cell identities and acts as a barrier for transcription factor-mediated cell fate reprogramming. Strikingly, FACT’s role as a barrier to cell fate conversion is conserved in humans as we show that FACT depletion enhances reprogramming of fibroblasts. Such activity is unexpected because FACT is known as a positive regulator of gene expression, and previously described reprogramming barriers typically repress gene expression. While FACT depletion in human fibroblasts results in decreased expression of many genes, a number of FACT-occupied genes, including reprogramming-promoting factors, show increased expression upon FACT depletion, suggesting a repressive function of FACT. Our findings identify FACT as a cellular reprogramming barrier in C. elegans and humans, revealing an evolutionarily conserved mechanism for cell fate protection.
Keywords: cell fate maintenance, cell fate safeguarding, reprogramming, chromatin remodeler, facilitates chromatin transcription, FACT, C. elegans, human fibroblasts
Graphical Abstract
Highlights
-
•
Chromatin regulator FACT blocks cellular reprogramming in C. elegans and humans
-
•
FACT maintains cell fates and antagonizes induction of ectopic fates in C. elegans
-
•
FACT depletion in human cells primes the transcriptome for reprogramming
Known barriers to cell fate reprogramming repress gene expression to prevent ectopic fates. Kolundzic et al. now show that the histone chaperone FACT, a positive regulator of gene expression, safeguards cell identities and acts as an evolutionarily conserved barrier for cell fate reprogramming in both C. elegans and human cells.
Introduction
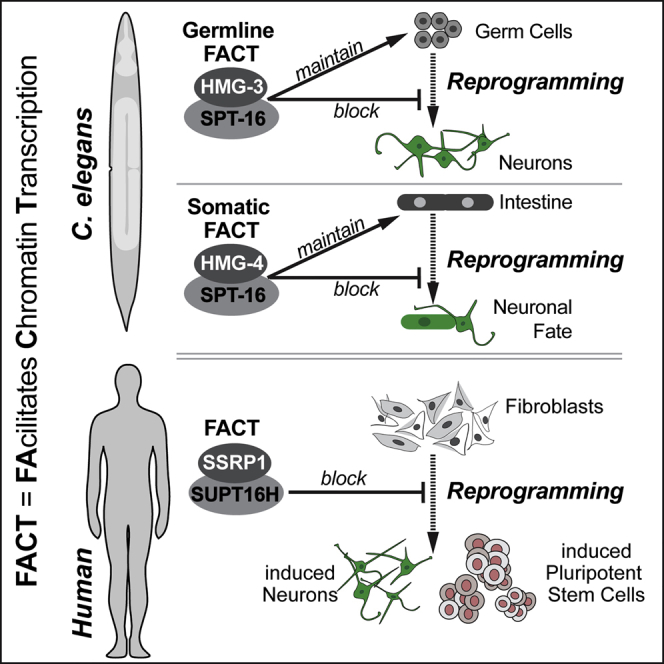
During development of metazoans, cells progressively lose plasticity and acquire specific fates. Mechanisms that restrict cell plasticity and safeguard the differentiated state remain incompletely understood (reviewed by Guo and Morris, 2017). Both positive and negative gene regulation are required during specification to maintain cell identities (Sánchez Alvarado and Yamanaka, 2014, Blau and Baltimore, 1991, Guo and Morris, 2017). Positive regulation by transcription factors (TFs) activates genes that define and maintain cell identities, while negative regulation restricts expression of genes belonging to other cell fates (Sánchez Alvarado and Yamanaka, 2014, Blau and Baltimore, 1991, Guo and Morris, 2017). For instance, the human zinc finger protein RE1-Silencing Transcription factor prevents expression of neural genes in non-neuronal cells (Chong et al., 1995) by recruiting epigenetic regulators that silence chromatin (Ballas et al., 2001, Roopra et al., 2004). Notably, such repressive chromatin regulators gained importance for cellular reprogramming because they can act as barriers for TF-mediated cell conversion (Becker et al., 2016, Cheloufi and Hochedlinger, 2017). Recent examples are the histone chaperones LIN-53 in Caenorhabditis elegans (RBBP4/CAF-1P48 in mammals) and Chromatin Assembly Factor 1 (CAF-1) in mouse which promote the formation of repressive chromatin and thereby block cell fate reprogramming (Cheloufi et al., 2015, Patel et al., 2012, Tursun et al., 2011). In contrast, chromatin regulators that are known to be non-repressive and promote gene expression have not been recognized as reprogramming barriers.
In a genetic screen for factors that safeguard cell fates in C. elegans, we identified that the histone chaperone FACT (facilitates chromatin transcription), which is essential for maintaining gene expression (Orphanides et al., 1998, Orphanides et al., 1999), blocks TF-mediated reprogramming of non-neuronal cells into neurons. FACT is a conserved heterodimeric complex consisting of SSRP1 (structure-specific recognition protein 1) and SUPT16H (suppressor of Ty 16 homolog) in mammals (Orphanides et al., 1998, Orphanides et al., 1999) but has not been studied in C. elegans. We discovered a germline-specific FACT isoform and show that FACT is required to maintain the intestinal and germline fate in C. elegans. Remarkably, the role of FACT as a reprogramming barrier is conserved, as its depletion in human fibroblasts enhances reprogramming to induced pluripotent stem cells (iPSCs) or neurons. Interestingly, transcriptome analysis (RNA-seq [RNA sequencing]) of human fibroblasts revealed that reprogramming-promoting factors, including Sal-like protein 4 (SALL4) (Parchem et al., 2014, Yang et al., 2008), have increased expression upon FACT depletion. Our findings suggest a more dichotomous role for FACT in gene regulation and imply that positive gene expression regulators contribute to impeding the induction of alternative cell fates. Overall, our study points to an evolutionarily conserved mechanism for safeguarding cell identities and identifies an unknown role for FACT in C. elegans and human cells.
Results
Whole-Genome RNAi Screen for Cell Fate-Safeguarding Factors in C. elegans
To reveal factors that safeguard cell identities in C. elegans, we challenged all tissues by overexpressing CHE-1, a neuron fate-inducing Zn-finger TF. CHE-1 normally specifies the glutamatergic ASE neuron fate, which is characterized by expression of the GCY-5 chemoreceptor (Figures 1A and S1A) (Yu et al., 1997). Using previously described transgenic animals that express the ASE neuron fate reporter gcy-5::GFP and allow ubiquitous CHE-1 expression upon heat shock (Patel et al., 2012, Tursun et al., 2011), we performed a whole-genome RNA interference (RNAi) screen in hermaphrodite adult worms to examine their F1 progeny for ectopic gcy-5::GFP induction upon broad che-1 overexpression (che-1oe) at the young adult stage. We identified 171 RNAi target genes (Table S1) that allow ectopic gcy-5::GFP induction upon depletion and represent different tissues: germline, epidermis, or intestine (Figures 1B and 1C). These genes are implicated in a variety of biological processes such as proteostasis, mitochondria function, and gene regulation by nuclear factors (Figures 1D–1F). Interestingly, we identified HMG-3, HMG-4, and SPT-16 that are orthologous to subunits of the essential chromatin remodeler FACT (Guindon et al., 2010, Ruan et al., 2008), as well as other genes known to functionally interact with FACT in other species such as SPT-5, EMB-5 (Spt6), and ISW-1 (Duina, 2011, McCullough et al., 2015) (Figure 1F). HMG-3 and HMG-4 are orthologs of human SSRP1, while SPT-16 is orthologous to SUPT16H (Figure 1G). Overall, we did not anticipate that depletion of FACT might promote cell fate conversion since it is primarily known for facilitating transcription rather than repressing ectopic gene expression. We therefore focused on characterizing FACT and the cell fate conversion effects upon its depletion.
Figure 1.

Whole-Genome RNAi Screening Strategy to Identify Cell-Fate-Safeguarding Factors in C. elegans
(A) Schematic representation: the ASER neuron is labeled by gcy-5::GFP. Overexpression of the Zn-Finger TF CHE-1 (che-1oe) combined with genome-wide RNAi led to discovery of genes preventing ectopic gcy-5::GFP induction in adults.
(B) Representative images of control animals, GFP induction in germline (lin-53, hmg-3, isw-1, and hecd-1 RNAi), intestine (spt-16, hmg-4, emb-5, and spt-5 RNAi), epidermis (hsp-1 RNAi), or germline and gut simultaneously (cco-1). Dashed lines indicate outline of animals. Scale bars represent 20 μm.
(C) Percentage of 171 genes whose knockdown allowed ectopic GFP induction in specific tissues.
(D) Gene ontology (GO) categories of genes from the screen.
(E and F) Proteostasis and FACT protein network based on genes identified in the screen. Network plots are based on known and predicted interactions from STRING (string-db.org) with minimal confidence score of 0.4. FACT complex proteins are highlighted in red.
(G) Models of FACT subunits in human and C. elegans. Conserved protein domains according to Pfam and InterPro are indicated: Nlob, N-terminal lobe domain; M24, metallopeptidase family M24; SPT16, FACT complex subunit Spt16p/Cdc68p; Rtt, histone chaperone Rttp106-like; SSRP1,structure-specific recognition protein; HMG, high mobility group box domain.
See also Table S1.
Depletion of HMG-3 Allows Germ Cell Reprogramming to Neurons
RNAi against hmg-3 allows CHE-1-dependent gcy-5::GFP induction in germ cells (Figure 2A). To exclude the possibility that depletion of HMG-3 causes non-specific de-silencing of transgenic reporters, we performed hmg-3RNAi in the absence of che-1oe. No changes in expression were detected for either gcy-5::GFP (Figures S1A and S1B) or two other reporters previously used to detect transgene de-silencing (Figures S1C and S1D) (Gaydos et al., 2014, Kelly et al., 2002, Patel and Hobert, 2017), suggesting that hmg-3RNAi creates permissiveness for CHE-1 to activate its target genes in germ cells. Induction of gcy-5::GFP expression by che-1oe upon hmg-3RNAi is accompanied by morphological changes of germ cells showing axo-dendritic-like projections (Figure 2A), indicating that germ cells converted into neuron-like cells. To assess the extent of conversion, we examined the nuclear morphology of converted germ cells and expression of neuronal genes. The gcy-5::GFP-positive cells display a nuclear morphology resembling neuronal nuclei (Figure 2A), and expression of pan-neuronal reporter genes, rab-3::NLS::RFP and unc-119::GFP (Stefanakis et al., 2015), further demonstrates a true conversion of germ cells into neuron-like cells (Figures 2A and S1E). Moreover, reprogrammed germ cells also express ift-20::NLS::RFP, a marker for ciliated neurons such as ASE, and the ASE and AWC-specific reporter ceh-36::GFP (Hobert, 2010, 2013) (Figures 2A and S1E). Importantly, transgene reporter expression reflects the endogenous expression of neuronal genes as shown by smFISH (single molecule fluorescence in situ hybridization). Transcripts from gcy-5, ceh-36, rab-3, unc-119, as well as from the conserved synaptic protein-encoding gene unc-10 (RIM), become expressed in the reprogrammed germ cells and are comparable in level to endogenous neurons (Figures 2B, 2C, S1F, and S1G). Furthermore, the acquisition of neuronal characteristics is accompanied by the loss of germline marker pie-1 and germ cell morphology (Figure S1H), corroborating the notion that germ cells convert into ASE neuron-like cells in hmg-3RNAi animals upon induction of CHE-1 expression.
Figure 2.

HMG-3 Inhibits Reprogramming of Germ Cells to ASE Neurons in C. elegans
(A) Depletion of hmg-3 leads to gcy-5::GFP induction in germ cells after che-1oe: white arrowheads indicate protrusions resembling neuron-like projections in gcy-5::GFP-positive cells. Dashed lines indicate outline of animals. Scale bars represent 20 μm and 5 μm in the magnification. Differential interference contrast (DIC) illustrates germ cell nuclei of hmg-3RNAi + che-1oe animals with changed nuclear morphology (stippled boxes mark magnification). Expression of ASE/AWC (ceh-36), sensory (ift-20), and pan-neuronal fate markers (rab-3, unc-119) can be detected in animals with gcy-5::GFP in the germline (outlined by dashed lines). Asterisk labels the germline distal tip. Scale bars represent 10 μm. For quantification, see Figure S1E.
(B) smFISH to detect transcripts derived from endogenous neuronal genes gcy-5, ceh-36, rab-3, unc-10, and unc119 in hmg-3RNAi germ cells. mRNA molecules are visualized as red dots. Controls were treated with mock hybridizations. Dashed boxes indicate magnified area. smFISH probes are described in STAR Methods. Scale bars represent 2 μm.
(C) Quantification of smFISH hybridization signals (red dots) per GFP-positive cells. For each condition, 20 GFP-positive cells were counted. Ordinary one-way ANOVA was used for statistical analysis. ∗∗∗∗, p < 0.0001. Error bars represent SD.
(D and E) Assessment of non-ASE neuron markers after che-1oe-induced germ cell reprogramming in hmg-3RNAi animals. (D) Representative images and (E) quantification of non-ASE markers glr-1::mCherry (interneurons), ttr-39::mCherry (GABAergic neurons), and ttx-3::mCherry (cholinergic interneuron) in reprogrammed germ cells (magnifications) expressing gcy-5::GFP. Dashed white lines outline the worm, and yellow lines outline the germline. Scale bars represent 20 μm and 5 μm in magnifications. Error bars represent SEM.
See also Figures S1 and S2.
Specificity of Germ Cell to Neuron Reprogramming in HMG-3-Depleted Animals
To examine whether CHE-1 reprograms germ cells in the hmg-3RNAi germline to properly specified ASE neurons, we tested the expression of markers for other neuron subtypes. CHE-1 does not induce GABAergic or cholinergic neuron reporters in hmg-3RNAi animals (Figures 2D and 2E), arguing that reprogrammed germ cells are not mis-specified but acquire a specific glutamatergic ASE fate. We next asked whether hmg-3 plays a widespread role in preventing germ cell conversion under ectopic expression of TFs. Mis-expression of the GABAergic neuron fate-inducing homeodomain TF UNC-30 (Jin et al., 1994) resulted in germ cell to GABAergic neuron conversion in hmg-3RNAi animals (Figures S2A–S2C). However, mis-expression of the myogenic TF HLH-1 (MyoD homolog) (Harfe et al., 1998) or the intestinal fate-inducing GATA-type TF ELT-7 (Riddle et al., 2013) failed to convert germ cells into muscle or gut-like cells, respectively (Figures S2B and S2D). This suggests that hmg-3RNAi specifically creates permissiveness for germ cell to neuron reprogramming. However, the negative results for HLH-1 and ELT-7 need cautious interpretation, as we cannot exclude the possibility of germline-mediated silencing of the respective transgenes. We tested whether mitotic or meiotic germ cells are susceptible to reprogramming using animals that carry a temperature-sensitive gain-of-function mutation of the Notch receptor GLP-1 (Germ Line Proliferation) that causes loss of meiotic germ cells (Pepper et al., 2003). Growth at a non-permissive temperature would lead to a loss of reprogramming if the converting germ cells belong to the meiotic pool. Germ cell conversion is not lost in glp-1(gf) mutants, suggesting that mitotic germ cells are the source for reprogramming into neuron-like cells (Figure S2E). This reprogramming is also independent of the cell-cycle activity of germ cells, as blocking cell-cycle progression by hydroxyurea (HU) (Gartner et al., 2004) did not inhibit germ cell conversion in hmg-3RNAi animals (Figure S2F). Taken together, depletion of the SSRP1 ortholog HMG-3 in C. elegans allows reprogramming of mitotic germ cells into specific neurons upon expression of neuron fate-inducing TFs.
Other FACT Subunits Prevent Neuron Fate Induction in the Intestine
While depletion of hmg-3 creates permissiveness for germ cell reprogramming, RNAi against hmg-4 and spt-16 allows induction of gcy-5::GFP in intestinal cells after che-1oe (Figures 3A and 3B). Intestinal cells showing gcy-5::GFP expression maintain their original morphological features, indicating an incomplete cell conversion (Figure 3A). However, smFISH revealed that the gcy-5::GFP-positive gut cells show expression of neuronal genes as seen in reprogrammed germ cells (Figures 3C–3E). The lack of morphological changes toward a neuron-like appearance might be due to the structural constraints of the intestine. Nevertheless, intestinal cells switch to a neuronal gene expression profile, which is stably maintained even 2 days after che-1oe similar to HMG-3 depletion-mediated germ cell to neuron reprogramming (Figures S2G and S2H). Overall, HMG-4 and SPT-16 prevent the induction of neuronal genes in the intestine, indicating that FACT plays a role in safeguarding cell identities of different tissues in C. elegans.
Figure 3.

FACT Subunits HMG-4 and SPT-16 Prevent Neuron Fate Induction in the Intestine
(A) RNAi against hmg-4 and spt-16 allows gcy-5::GFP induction in the intestine. Dashed lines indicate the outline of animals. Scale bars represent 20 μm.
(B) Quantification of animals showing gcy-5::GFP in the intestine in che-1oe + hmg-4RNAi or spt-16RNAi. n indicates the number of animals counted. Error bars represent SEM.
(C) Detection of transcripts derived from neuronal genes in hmg-4RNAi and spt-16RNAi intestinal cells by smFISH. Individual mRNA molecules are visualized as red dots. Controls were treated with mock hybridizations. Dashed boxes indicate the magnified area. Scale bars represent 2 μm.
(D and E) Quantification of smFISH hybridization signals (red dots) per GFP-positive cells. Quantification of neuronal transcripts in the intestine upon (D) hmg-4 RNAi and (E) spt-16 RNAi. For each condition, 20 GFP-positive cells were counted. Ordinary one-way ANOVA was used for statistical analysis. ∗∗, p < 0.01, ∗∗∗, p < 0.001, ∗∗∗∗, p < 0.0001. Error bars represent SD.
(F and G) Immunostaining of CRISPR-tagged (F) hmg-3 with HA and (G) hmg-4 with HA. Dashed lines indicate the outline of the animals. Scale bars represent 20 μm.
(H) Anti-SPT-16 immunostaining shows expression in somatic tissues and germline. Dashed lines indicate the outline of the animals. Scale bars represent 20 μm.
(I) Tissue-specific FACT isoforms prevent induction of neuronal genes in the germline and intestine.
See also Figure S2.
C. elegans Has a Germline-Specific FACT Isoform
The tissue-specific effects on ectopic gcy-5::GFP induction suggested distinct expression patterns of FACT genes. HMG-3 and HMG-4 share more than 90% amino acid homology with SSRP1 (Guindon et al., 2010, Ruan et al., 2008) and each other. To discriminate between HMG-3 and HMG-4, we tagged both with an HA (Human influenza hemagglutinin) epitope using CRISPR/Cas9 editing. Anti-HA immunostaining revealed that HMG-3 is exclusively expressed in the germline, explaining the distinct effect of hmg-3RNAi on permissiveness for reprogramming germ cells (Figure 3F). In contrast, HMG-4 is expressed predominantly in the soma with high intensity in the intestine and has only weak expression in the germline (Figure 3G). An antibody against SPT-16 revealed expression in all tissues with predominance in the intestine (Figure 3H). Because spt-16 RNAi during embryonic development caused early lethality, we could not assay for conversion of germ cells, which requires F1 RNAi as shown for the depletion of hmg-3 or other previously identified factors (Patel et al., 2012, Tursun et al., 2011).
Since HMG-4 can be detected with low levels in the germline, we assessed whether the germline-exclusive HMG-3 functions as a bona fide FACT subunit. We immunoprecipitated HMG-3::HA and HMG-4::HA and analyzed co-precipitated proteins by mass spectrometry (immunoprecipitation-mass spectrometry [IP-MS]) (Table S2). The most significant interacting protein for both is SPT-16 (Figures S2I and S2J), indicating that they predominantly associate with SPT-16. Notably, hmg-3 mutant animals (tm2539) are sterile and are not rescued by endogenous hmg-4 and, vice versa, the larval lethality of hmg-4 mutants (tm1873) is not rescued by endogenous hmg-3, suggesting non-redundant functions of both proteins.
Collectively, the specific RNAi effects together with the germline-exclusive expression of hmg-3 suggest that HMG-3 and SPT-16 form a germline-specific FACT that safeguards germ cells, while HMG-4 and SPT-16 form a FACT isoform that safeguards the intestinal fate (Figure 3I) in C. elegans.
FACT Depletion Affects Chromatin Accessibility for Transcription Factors
FACT promotes gene expression by nucleosome reorganization in yeast and human cells (reviewed in Hammond et al., 2017, Reddy et al., 2017); however, in C. elegans FACT has not been studied. Therefore, we profiled HMG-3 DNA-binding patterns by ChIP-seq (chromatin immunoprecipitation sequencing), as well as gene expression by RNA-seq and chromatin accessibility using ATAC-seq (Assay for Transposase-Accessible Chromatin sequencing) (Buenrostro et al., 2013) upon FACT depletion. Data derived from this global characterization of FACT is provided in Figures S3 and S4 and Tables S2 and S3.
To clarify why depletion of FACT leads to permissiveness for ectopic fate induction, we focused on chromatin accessibility changes detected by ATAC-seq without inducing che-1oe (see STAR Methods) (Figure 4A). Upon RNAi against hmg-4 or spt-16, average positional patterns of ATAC-seq around promoter-region peaks do not show drastic, uniform changes (Figure S3K), yet similar numbers of total peaks show small but significant increases and decreases (2,770 and 2,768 sites increased and 1,845 and 2,472 sites decreased for hmg-4RNAi and spt-16RNAi, respectively, FDR [false discovery rate], 0.01; see STAR Methods Table S3). A Spearman correlation of 0,809 of log2-fold changes between sites significantly (FDR 0.01) changed upon knockdown of hmg-4RNAi or spt-16RNAi suggests that these factors act together, confirming the specificity of the effects (Figure 4A).
Figure 4.

FACT-Depletion Causes Changes in Chromatin Accessibility and Gene Expression
(A) ATAC-seq using isolated nuclei of animals treated with RNAi against FACT subunits. Hexbin density scatterplot of hmg-4RNAi-normalized ATAC-seq log2-fold changes plotted against spt-16RNAi-normalized ATAC-seq log2-fold changes for ATAC-seq peaks that were significantly differential (FDR 0.01) in at least one of the conditions. Scale shows number of ATAC-seq peaks plotted per hexbin.
(B) Key de novo generated motifs with matching TFs and enrichment p values from ATAC-seq peaks closing (bottom) or opening (top) upon hmg-4RNAi or spt-16RNAi.
(C) Motif scanning of previously published JUN-1 (top) and ELT-1/-7 (bottom) motifs within sets of ATAC-seq peaks that either decrease (red), increase (green), or do not change (gray; adjusted p value > 0.5) upon FACT knockdown. Plotted is the maximum motif match score per ATAC-seq peak.
(D) RNAi against hmg-4 or spt-16 decreases expression of intestinal fate reporters. Dashed lines indicate the outline of the animals. Scale bars represent 20 μm.
(E) Quantification of intestinal fate reporter expression from (A). Two-way ANOVA test was used for statistical comparison, ∗∗∗ p < 0.001. At least 200 animals were counted for each condition. Error bars represent SEM.
(F) Immunostained intestines from animals treated with RNAi against hmg-4 and spt-16 to detect gut-specific ELT-2 and intermediate filament protein IFB-2. Scale bars represent 5 μm.
(G) In jun-1 (gk557) mutants, gcy-5::GFP induction in gut cells is suppressed. One-tailed Student’s t test was used for statistical comparison, ∗∗∗ p < 0.001. At least 150 animals were counted for each condition. Error bars represent SEM.
(H) FACT maintains intestinal fate identity and antagonizes JUN-1.
(I) Germline-specific P granules are lost upon hmg-3RNAi. Dashed lines outline the germline, and white asterisk indicates the distal tip end of the gonad. Scale bars represent 5 μm.
(J) Germ cell fate marker pie-1::mCherry::his-58 is lost upon hmg-3RNAi. Dashed lines outline the germline, white asterisk indicates distal end of the gonad. Scale bars represent 5 μm. At least 150 animals were counted for each condition. Error bars represent SEM. Paired-end Student’s t test was used for statistical comparison, ∗∗∗ p < 0.001.
(K) FACT maintains germ cell fate identity and antagonizes CHE-1-mediated reprogramming.
See also Figures S3 and S4 and Tables S3 and S6.
While decreased accessibility of genomic sites upon FACT depletion is in agreement with the complex’s known function as a gene expression activator (Hammond et al., 2017, Orphanides et al., 1998), the detection of many increased accessibility sites indicates that FACT may also be required to prevent ectopic gene expression induction, directly or indirectly. This notion is further supported by transcriptome analysis as depletion of hmg-4, or spt-16, leads to down- but also up-regulation of several genes (1,679 down- and 1,948 up-regulated for spt-16RNAi) (Figures S4A–S4F). Furthermore, genes with changed expression are enriched for changed ATAC-seq peaks (Figure S4H; see STAR Methods), suggesting the chromatin landscape and transcriptome concordantly alter, both positively and negatively, upon FACT depletion.
To gain insight into whether chromatin sites that change accessibility upon FACT RNAi are enriched for binding of TF families, we performed de novo motif analysis in ATAC-seq peaks, followed by scanning for matching known TF binding preferences (see STAR Methods) (Figures 4B, 4C, S4L, and S4M). In spt-16RNAi and hmg-4RNAi animals, motifs matching binding preferences for GATA TFs are enriched at closing sites (Figures 4B, 4C, and S4L). Interestingly, the GATA TFs ELT-2 and ELT-7 are the master regulators of the intestinal fate (Sommermann et al., 2010), and these closing sites are indeed highly enriched for ELT-2 binding based on available ChIP-seq data (Kudron et al., 2018) with a −log10 p value of 233.02 (spt-16RNAi) and 32.87 (hmg-4RNAi). Furthermore, a common TF-binding motif at opening chromatin sites matches that for JUN-1. Based on published ChIP-seq data (Kudron et al., 2018), JUN-1 binding is enriched in spt-16RNAi opening ATAC-seq peaks with a −log10 p value of 1.33 (Figures 4B, 4C, and S4M). Interestingly, the C. elegans bZIP TF JUN-1 is orthologous to the AP-1 subunit JUN and supports transcriptional reprogramming in the intestine upon fasting (Uno et al., 2013).
Decreased binding accessibility for ELT-2/7 TFs that are required for intestinal gene expression might reflect a loss of intestinal cell fate maintenance upon RNAi against hmg-4 or spt-16. Concomitantly, increased binding accessibility for JUN-1 might poise for gene expression changes in the intestine.
FACT Maintains the Intestinal and Germline Fate and Prevents Induction of Neuronal Genes by Antagonizing JUN-1
To test the physiological relevance of predicted changes in chromatin accessibility for different TFs in vivo, we performed RNAi against hmg-4 or spt-16 and examined the expression of intestinal genes with four different reporters (Figures 4D and 4E) and immunostained for ELT-2 TF and the gut-specific intermediate filament protein IFB-2 (Figure 4F). Both hmg-4 and spt-16 RNAi depleted intestinal gene expression (Figures 4D–4F), suggesting that gut fate maintenance is impaired upon loss of FACT. To test whether FACT antagonizes factors that promote gene expression changes (Figure 5D), we tested whether JUN-1 is required for gut reprogramming because hmg-4/spt-16 RNAi caused increased chromatin accessibility for JUN-1 binding sites. We found that the jun-1 (gk557) mutant background significantly decreases neuronal gene induction upon che-1oe in hmg-4RNAi and spt-16RNAi animals as compared to controls (Figure 4G), suggesting that JUN-1 promotes transcriptional reprogramming in the intestine upon FACT depletion (Figures 4G and 4H).
Figure 5.

FACT Depletion Increases Efficiency of Reprogramming Human Fibroblasts
(A) Reprogramming with human secondary fibroblasts carrying a doxycycline (DOX)-inducible OCT4/SOX2/KLF4/c-MYC (OSKM) cassette (Cacchiarelli et al., 2015) that were transfected with siRNAs against human SSRP1 or SUPTH16 before DOX induction.
(B) Alkaline phosphatase (AP) staining of stem cell colonies 21 days after DOX treatment. Control experiment is scrambled siRNA (see STAR Methods).
(C) Quantification of iPSC colonies (6 replicates) based on SSEA4 immunostaining for each knockdown condition. Paired Student’s t test was used for statistical comparison, ∗p < 0.05, ∗∗∗ p < 0.001. Error bars represent SD.
(D) Histological characterization of teratomas derived from grafting SSRP1 or SUPT16H depletion-derived iPSCs in mice. Teratomas reached 1.5 cm2 in size after 51–70 days and showed tissues of all three germ layers (ectoderm, endoderm, mesoderm). Scale bars represent 50 μm.
(E) Forced expression of Ascl1, Brn2, and Mytl1 in human fibroblasts (Normal Human Dermal Fibroblats = NHDF cells) and SSRP1 or SUPT16H depletion enhances reprogramming of fibroblasts to neurons. One-way ANOVA test was used for statistical comparison, ∗∗ p < 0.01. Error bars represent SEM.
See also Figure S5.
Next, we asked whether FACT plays a similar role in germline fate maintenance. Similar to the observed loss of intestinal gene expression upon hmg-4 and spt-16 RNAi, hmg-3 RNAi caused loss of germline fate markers. Levels of germline P granules, as well as expression of the germline-specific pie-1 reporter, significantly decreased (Figures 4I and 4J), indicating that FACT is required to maintain the germline fate (Figure 4K).
FACT Is a Reprogramming Barrier in Human Fibroblasts
As a chromatin regulator FACT is highly conserved, suggesting that its role in reprogramming may also be conserved. We tested whether FACT depletion in human fibroblasts enhances reprogramming using an hiF-T cell line that shows reprogramming to iPSCs with very low efficiency (Cacchiarelli et al., 2015). The hiF-T cells allow doxycycline (DOX)-inducible expression of OCT4, SOX2, KLF4, and C-MYC (OSKM) derived from a stable transgene ensuring comparable OSKM levels in repeat experiments (Cacchiarelli et al., 2015) (Figure 5A). The human FACT subunits SSRP1 and SUPT16H were depleted using small interfering RNA (siRNA) transfections for up to 4 days while transcript levels started recovering after 7 days (Figure S5A). OSKM induction 48 hr after FACT knockdown considerably increased the numbers of iPSC colonies—around 2-fold upon SUPTH16 depletion (Figures 5B and 5C). FACT depletion does not increase OSKM expression (Figure S5B), excluding the possibility of reprogramming enhancement due to the OSKM cassette being affected. These colonies show strong expression of several pluripotency markers, including NANOG, SSEA4 (Stage-specific embryonic antigen 4), and Tra-1-60 (Park et al., 2008) (Figures S5C and S5D). Pluripotency was also confirmed in a physiological context by transplanting iPSCs into mice, which then formed teratomas containing tissues from all three germ layers (Brivanlou et al., 2003, Hentze et al., 2009, Kooreman and Wu, 2010) (Figure 5D). Next, we asked whether FACT depletion in human fibroblast also enhances lineage reprogramming to neurons (Figure 5E). Our results suggest that FACT knockdown and forced expression of Ascl1, Brn2, and Mytl1 TFs that were previously shown to induce conversion of fibroblasts into neurons (Vierbuchen et al., 2010) increased the conversion up to 1.5-fold compared to the control (Figure 5E), although transduction efficiency was relatively low (15%). Interestingly, beta-III tubulin-positive neurons derived from FACT-depleted fibroblasts show a higher degree of projection complexity than control cells (Figure 5E), indicating that the reprogrammed cells are generated earlier or mature faster. Taken together, FACT depletion in human fibroblasts enhances reprogramming to iPSCs and neurons, demonstrating that FACT’s role as a safeguard of cellular identities is conserved in human cells.
FACT Depletion Directly and Indirectly Primes the Transcriptome for Pluripotency
To understand how FACT functions to enhance human fibroblast reprogramming, we performed ChIP-seq for SSRP1 and SUPT16H, as well as ATAC-seq and RNA-seq upon FACT knockdown (Figure 6A). We found that FACT is largely enriched around annotated transcription start sites (TSS) (Figure S6A). SSRP1 and SUPT16H show a high degree of correlation for promoter proximal ChIP-seq signal, gene expression changes upon knockdown, and altered accessibility within ATAC-seq peaks significantly changing (FDR 0.01) upon knockdown (Figures 6B and 6D), suggesting that, indeed, the two factors function together. FACT knockdown results in similar numbers of genes with increased (n = 2,447) and decreased (n = 2,183) expression (Figure 6C, FDR 0.1), as well as similar numbers of opening (n = 7,841) and closing (n = 8,117) transposase accessible regions (Figure 6D, FDR 0.01). Higher FACT ChIP-seq level for genes with significant promoter signal (FDR 0.1) does not translate to high or low expression levels (Figures 6E and S6B) or expression changes (up or down) upon FACT knockdown (Figures 6F and S6C). To further probe the relationship between FACT with chromatin structure and gene expression, we defined two classes of genes based on levels of promoter FACT ChIP-seq signal (see STAR Methods; Figures 6G and S6D). Genes with insignificant FACT levels and detected in the RNA-seq (FACT low; adjusted p value > 0.1) show a slightly lower expression level distribution than genes with significant FACT and detected in the RNA-seq (FACT high; adjusted p value < 0.1) (Figure S6E), yet the distributions of expression changes upon FACT knockdown were highly similar for both groups (Figure 6H and Table S6). Similarly, while the levels of overall promoter-region ATAC-seq counts were slightly lower for the FACT low group (Figure S6F), no obvious changes were seen in the distribution of promoter ATAC-seq fragment lengths (a measure of nucleosome positioning [Buenrostro et al., 2015]) (Figure 6I), nor were there drastic, uniform changes in mean positional patterns of ATAC-seq signal around annotated TSS for either group upon FACT knockdown (Figure 6J). Together, these observations suggest that FACT influences gene expression both positively and negatively and its presence or absence does not drastically alter chromatin accessibility genome wide. Interestingly, genes with high promoter FACT tend to be shorter than those with low promoter FACT (Figure S6G), which is consistent with similar observations for HMG-3 in worms (Figures S3 and S4).
Figure 6.

FACT Depletion Directly and Indirectly Primes the Transcriptome for Pluripotency
(A) Human fibroblasts were used for ChIP-seq analysis or transfected with siRNAs against human FACT subunits and used for ATAC-seq and RNA-seq analysis 48 hr after knockdown without OSKM induction.
(B) Correlation of SSRP1 and SUPT16H log2-ChIP versus input ratios for TSS windows classified as high for at least one of the two FACT components (see STAR Methods). Density scale reflects number of TSS windows plotted per hexbin. The Spearman correlation Rho value is given.
(C) Correlation of SSRP1 and SUPT16H knockdown RNA-seq log2-fold changes for genes that were significantly changed in at least one of the conditions (see STAR Methods; FDR 0.1). Density scale reflects number of genes plotted per hexbin. The Spearman correlation Rho value is given.
(D) Hexbin density scatterplot of SUPT16H knockdown normalized ATAC-seq log2-fold changes plotted against SSRP1 knockdown normalized ATAC-seq log2-fold changes for ATAC-seq peaks that were significantly differential (FDR 0.01) in at least one of the conditions. Scale shows number of ATAC-seq peaks plotted per hexbin. The Spearman correlation Rho value is given.
(E) Log2 ratio of SUPT16H ChIP versus Input plotted against Fragments Per Kilobase of transcript per Million mapped reads (FPKM) expression values from control RNA-seq for genes classified as FACT high and detected in the RNA-seq. Density scatterplot scale shows number of genes plotted per hexbin.
(F) Log2-fold changes in expression levels from RNA-seq after SUPT16H knockdown plotted against log2 ratio of SUPT16H ChIP versus Input for genes classified as FACT high and detected in the RNA-seq. Density scatterplot scale shows number of genes plotted per hexbin.
(G) Average positional profiles and heatmaps of library- and input-normalized SUPT16H ChIP-seq signal for genes whose TSS windows classified as FACT high or FACT low (see STAR Methods).
(H) Violin plots of RNA-seq log2-fold change distributions following FACT knockdown for genes classified as being FACT high or FACT low. Red jitter dots reflect genes whose expression changed significantly upon FACT knockdown (FDR < 0.1).
(I) Density distributions of control or FACT-knockdown ATAC-seq fragment lengths intersecting TSS windows for genes classified as FACT high or FACT low.
(J) Mean positional profiles and heatmaps of control or FACT-knockdown ATAC-seq signal for genes classified as FACT high or FACT low.
(K) −Log10 enrichment p values of genes assigned ATAC-seq peaks changing, left, or genes showing expression changes upon FACT-knockdown, right, intersecting with clusters of genes previously reported to display specific expression profile changes during OSKM-induced reprogramming (Cacchiarelli et al., 2015). I, early somatic; II, late somatic; III, metabolic processes; IV, late embryogenesis; V, early embryogenesis; VI, pre-implantation; VII, shared soma versus pluripotency; VIII, early pluripotency; IX, late pluripotency; X, neuro-ectoderm.
(L) RNA-seq fold changes upon FACT-knockdown for key genes previously implicated in reprogramming or pluripotency. Stars reflect adjusted p values.
(M) Browser shot of ATAC-seq signal and library-normalized RNA-seq signal upon SSRP1 (SSRNAi) or SUPT16H RNAi (SURNAi), and FACT ChIP-seq signals for the PRRX1 gene. ATAC-seq peaks and RNA-seq genes called as significantly changed are noted below the respective signal tracks.
(N) De novo-generated motifs from ATAC-seq peaks differentially changing upon FACT-knockdown and TF family-representatives matching the given motif.
(O and P) Browser shots as described in (M) for the (O) CEBPB and (P) SALL4 genes.
(Q) Zoomed-in browser shot of upstream enhancer-candidate for SALL4. Shown are control and FACT-knockdown signals for ATAC-seq and publicly available CEBPB ChIP-seq in human embryonic stem cells (ESCs). Two CEBPB biding sites were found within the 387-bp accessible region by matching the consensus RTTKCAYMAY and allowing one mismatch.
(R) Model for how FACT depletion primes for reprogramming: direct changes in gene expression for TFs such as CEBPB lead to promoter distal, indirect chromatin accessibility changes at genomic loci of key regulators such as SALL4.
FACT Depletion Increases Expression of Pluripotency-Promoting Factors while Decreasing Expression of Reprogramming-Inhibitors
To connect FACT to the observed reprogramming phenotype, we intersected our genome-wide datasets with gene clusters previously defined for their behavior over a reprogramming time course following induction of OSKM (Cacchiarelli et al., 2015). Genes with decreased expression or assigned ATAC-seq peaks that change upon FACT knockdown are significantly enriched in somatic clusters that display decreased expression upon OSKM induction (Figure 6K, clusters I and II). Furthermore, genes whose expression increases upon FACT knockdown are enriched in pluripotency clusters that have increased expression upon OSKM induction (Figure 6K, clusters VIII and X). Expression changes for several genes known to positively and negatively influence pluripotency (Figure 6L) provide strong molecular support for increased reprogramming efficiency upon FACT knockdown. For instance, FACT depletion leads to decreased expression of previously described reprogramming inhibitors such as the histone chaperone CAF1 subunits CHAF1B and RBBP7 (Cheloufi and Hochedlinger, 2017), repressive chromatin regulators including SUV39H1/2 and NR2F1 (Onder et al., 2012), PRRX1 (Polo et al., 2012, Yang et al., 2011), the phosphatase PTPN11 (Qin et al., 2014), as well as SUMO2 (Borkent et al., 2016) (Figure 6L). Concomitantly, FACT depletion increases expression of reprogramming-promoting factors such as ESRRB, LIN28, and SALL4 (Buganim et al., 2012), JMJD2C (Soufi et al., 2012), CEBPB (Chronis et al., 2017), FGF2 (Jiao et al., 2013), and BMP2 (Chen et al., 2011, Samavarchi-Tehrani et al., 2010) (Figure 6L). Visually inspecting these key reprogramming-associated genes (Figures 6M–6P and S6L) suggested that changes in gene expression upon FACT knockdown are likely to be a combination of direct effects on genes with substantial FACT occupancy and indirect effects through distal binding of TFs whose expression themselves changes. Indeed, 95% of ATAC-seq peaks that change upon FACT knockdown are distal to annotated TSS, and these changed regions were similarly assigned to FACT ChIP-seq high or low genes (Table S6).
FACT Depletion Increases Chromatin Accessibility for CEBP Family TFs at the Enhancer Region of the Reprogramming-Promoting Factor SALL4
To find candidates of distal-binding regulators, we performed de novo motif generation on sequences from distally annotating ATAC-seq peaks either opening or closing upon FACT depletion. A highly enriched motif at closing chromatin sites matches that for the Runt-related TF 1 (RUNX1) (Figures 6N and S6I). Interestingly, RUNX1 depletion has been shown to enhance reprogramming (Chronis et al., 2017), suggesting that decreased chromatin accessibility for RUNX1 binding sites upon FACT depletion could have analogous effects. Likewise, decreased accessibility for sequences matching the TEAD family TF motif (Figures 6N and S6I) could affect the TEAD/HIPPO pathway, another previously reported reprogramming barrier (Qin et al., 2012). Interestingly, motifs matching the CEBP family of TFs, which can enhance Mouse Embryonic Fibroblast (MEF) reprogramming (Chronis et al., 2017, Di Stefano et al., 2016), are enriched in ATAC-seq peaks opening upon FACT depletion. The CEBPB gene itself is highly occupied by FACT and up-regulated upon FACT depletion, yet has no obviously changing regulatory regions, and is therefore a candidate for direct regulation by FACT (Figure 6O). Meanwhile, the SALL4 gene, which has been strongly implicated in pluripotency (Buganim et al., 2012, Parchem et al., 2014, Yang et al., 2008), also shows high FACT occupancy and increased expression upon FACT knockdown (Figure 6P), yet a strong enhancer candidate ∼9 kb upstream of the SALL4 TSS is clearly opening upon FACT depletion and has two binding sites matching the CEBP TF family within a 387-bp sequence (Figure 6Q). Overall, we see a clear enrichment between genes showing reduced expression and genes having distal regulatory regions that close upon FACT knockdown, as well as between genes showing increased expression and genes having distal regulatory regions that open upon FACT knockdown, but not vice versa (Figure S6K). This leads us to propose a model where FACT safeguards somatic gene programs via proper control (both positive and negative) of transcriptional regulators and hinders the pluripotent state. Disruption of FACT leads to mis-regulation of such factors and, consequently, to indirect changes in the distal regulome, as exemplified by our mini CEBPB-SALL4 network (Figure 6R).
Discussion
The identification of FACT as a barrier for cell conversion supports a recent notion by Alvarado and Yamanaka (Sánchez Alvarado and Yamanaka, 2014) that cell fate maintenance factors have not been widely explored but might provide new avenues for reprogramming. Overall, the discovery of FACT as a reprogramming barrier was unexpected since FACT is predominantly known as a positive regulator of gene expression (Orphanides et al., 1998) (reviewed by Hammond et al., 2017, Reddy et al., 2017). It is anticipated that loss of FACT has both consequences: repression of genes that require FACT activity and increased gene expression due to indirect effects as a consequence of diminished factors that act as repressors. However, it is surprising that genes, such as CEBPB in fibroblasts, which are directly bound by FACT, show increased expression upon FACT depletion, suggesting a repressive function for FACT. While the increased accessibility of the SALL4 enhancer for CEBP binding illustrates a combined mode of direct and indirect effects that result in increased gene expression, it remains to be determined how chromatin sites, such as the SALL4 enhancer, become more accessible upon FACT depletion. Since 95% of changing chromatin sites in human cells are distal to annotated TSS, it is conceivable that TFs encoded by genes that change expression directly upon FACT depletion are apt to alter chromatin at distal enhancer sites. Also, loss of FACT might lead to more accessible chromatin due to persistence of nucleosome-depleted sites since FACT re-establishes the nucleosome signature after RNA polymerase II passage (Belotserkovskaya et al., 2003, Jamai et al., 2009). Additionally, decreased gene expression upon FACT depletion might obliterate the insulation of active and repressed chromatin regions as previously implied for cell identity genes (Dowen et al., 2014).
Overall, the preponderance of effects that favor cell fate conversion, such as decreased expression of reprogramming inhibitors and increased levels of promoting factors, might result in the observed reprogramming enhancement. This is specifically reflected by our finding that expression of SALL4 (Buganim et al., 2012, Parchem et al., 2014, Yang et al., 2008) together with the TFs ESRRB and LIN28B, which are all members of the deterministic pluripotency circuitry (Buganim et al., 2013), are increased upon FACT depletion. Generally, we speculate that depletion of global gene expression regulators such as FACT, or other previously identified reprogramming barriers including LIN-53, CAF-1, or SUMO2 (Borkent et al., 2016, Cheloufi et al., 2015, Tursun et al., 2011), lead to a disordered gene expression program of the host cell. This “gene expression chaos” is the window of opportunity for ectopically expressed TFs to re-establish a new ordered gene expression program resulting in cell identity conversion. While this proposed concept suggests a more generic permissiveness for reprogramming, further investigation will be required to determine the range of different target cell fates that can be generated by reprogramming upon FACT depletion. For application aspects, transient FACT depletion in human cells is sufficient to enhance reprogramming, thereby providing new avenues for clinical approaches. It has been shown that short-term OSKM expression is sufficient for in vivo reprogramming in mice, thereby preventing formation of tumors (Ocampo et al., 2016). In general, the combination of transient depletion of reprogramming barriers and short-term forced-expression of TFs will have fewer deleterious effects while ensuring efficient reprogramming.
Loss of FACT in C. elegans leads to reduced maintenance of the intestinal and germ cell fates. Strikingly, motifs for JUN-1 are enriched in both human fibroblasts and C. elegans at opening chromatin sites upon FACT depletion, indicating a remarkable conservation of gene regulation networks. Increased permissiveness for JUN-1 activity could promote changes in cell identities in C. elegans, which is in line with a previous study showing that JUN-1 promotes transcriptional reprogramming in the intestine together with the FOXO TF DAF-16 upon starvation (Uno et al., 2013). It is therefore conceivable that high levels of overexpressed CHE-1 co-opts JUN-1 to reprogram intestinal gene expression upon FACT depletion. Our observation that FACT depletion in C. elegans appears to allow mainly reprogramming to neurons needs to be interpreted with caution. Specifically, the transgenes to overexpress the intestine or muscle fate TFs ELT-7 or HLH-1, respectively, might suffer from germline-mediated silencing, which is a well-known phenomenon in C. elegans (Kelly and Fire, 1998). Furthermore, expression of ELT-7 and its downstream factor ELT-2 are suppressed upon FACT depletion. Hence, it is plausible that FACT depletion causes a rather unfavorable context for TFs such as ELT-7 to induce the intestinal fate in other tissues.
Generally, FACT’s combined role in maintaining gene expression and preventing chromatin binding accessibility for TFs such as CEBPB or Jun/JUN-1 in human fibroblasts and C. elegans seems conserved and might be essential to prevent deviant expression of genes belonging to other cell identities in both species. Seen in a broader context, FACT being an impediment for converting cell fates in C. elegans and human cells exemplifies that reprogramming barriers are evolutionarily conserved. This phenomenon is also reflected by the previously identified barrier for germ cell reprogramming LIN-53 in C. elegans (CAF-1p48/RBBP7 in mammals) (Tursun et al., 2011) and the LIN-53-containing histone chaperone CAF-1, which is a barrier during reprogramming of mouse fibroblasts (Cheloufi et al., 2015). Such evolutionary conservation is likely in metazoan organisms, which need to prevent the induction of ectopic gene expression programs. Challenges including global signaling events during development and tissue regeneration (e.g., wound healing), environmental stress, as well as aging could initiate ectopic gene expression profiles, resulting in cellular transformations if safeguarding mechanisms are not in place. In this context, our study demonstrates the versatility of C. elegans to serve as a powerful gene discovery tool for identifying unanticipated reprogramming barriers.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-SPT-16 rabbit polyclonal peptide antibody | This paper | N/A |
| anti-HA mono mouse antibody (12CA5) | Roche | Cat#11583816001 |
| ELT-2 mono mouse antibody | Developmental Studies Hybridoma Bank | Cat#455-2A4 |
| P-Granule mono mouse antibody | Developmental Studies Hybridoma Bank | Cat#OIC1D4 |
| intermediate filament subunit mono mouse antibody | Developmental Studies Hybridoma Bank | Cat#MH33 |
| SSEA-4, DyLight 488 conjugate mono mouse antibody | Thermo Scientific | Cat#MA1-021-D488 RRID: AB_2536688 |
| NANOG rabbit polyclonal antibody | Thermo Scientific | Cat#PA1-097 RRID: AB_2539867 |
| Anti-Beta III Tubulin Antibody | Sigma-Aldrich | Cat#AB9354 |
| Go-ChIP-Grade Purified anti-SSRP1 Antibody | BioLegend | Cat#609709 RRID: AB_2650946 |
| SPT16 (D7I2K) Rabbit mAb | Cell Signaling | Cat#12191 |
| Anti-HA tag antibody - ChIP Grade | Abcam | Cat#ab9110 RRID: AB_307019 |
| Bacterial and Virus Strains | ||
| Escherichia coli, OP50 | CAENORHABDITIS GENETICS CENTER (CGC) | WormBase ID: OP50 |
| Escherichia coli: HT115(DE3) | CAENORHABDITIS GENETICS CENTER (CGC) | WormBase ID: HT115(DE3) |
| C. elegans total RNAi Collection (Ahringer) | Source Bioscience | Cat#3318_Cel_RNAi_complete |
| C. elegans supplemental RNAi Collection (Ahringer) | Source Bioscience | Cat#3317_Cel_RNAi_supplement_whole |
| RTTA, tetO-Ascl1, tetO-Brn2 and tetO-Myt1l (BAM) | Vierbuchen et al. (2010) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Y-27632 2HCl ROCK1 inhibitor | Biozol Diagnostica | Cat#SEL-S1049-10MM |
| Critical Commercial Assays | ||
| Pluripotent Stem Cell 4-Marker Immunocytochemistry Kit | Thermo Fisher | Cat#A24881 |
| Alkaline Phosphatase Staining Kit II | Stemgent | Cat#00-0055 |
| Nextera DNA Library Preparation Kit | Illumina | Cat#FC-121-1030 |
| TruSeq RNA Library Prep Kit v2 | Illumina | Cat#RS-122-2001 |
| NEXTflex Rapid Directional qRNA-Seq Kit | Bioo Scientific | Cat#NOVA-5130-02D |
| Ribo-Zero Gold rRNA Removal Kit (H/M/R) | Illumina | Cat#MRZG12324 |
| iDeal ChIP-seq Kit for Transcription Factors | Daigenode | Cat#C01010055 |
| NEXTflex qRNA-Seq Kit v2 | Bioo Scientific | Cat#NOVA-5130-12 |
| NextSeq 500/550 High Output v2 kit, 150 cycles | Illumina | Cat#FC-404-2002 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE98758 |
| Human CEBP ChIP-seq | ENCODE | ENCFF377MTQ.bam |
| Human H2AFZ ChIP-seq | ENCODE | ENCFF255USS.bigWig |
| Human H3K27ac ChIP-seq | ENCODE | ENCFF858MGD.bigWig |
| Human H3K4me3 ChIP-seq | ENCODE | ENCFF837XME.bigWig |
| Worm ChIP-seq b0310.2_embryo | modENCODE/modERN | ENCFF230GNZ.bed |
| Worm ChIP-seq elt-1_l3 | modENCODE/modERN | ENCFF217KEA.bed |
| Worm ChIP-seq elt-2_l3 | modENCODE/modERN | ENCFF707HUO.bed |
| Worm ChIP-seq fkh-6_embryo | modENCODE/modERN | ENCFF671ZAU.bed |
| Worm ChIP-seq jun-1_l4 | modENCODE/modERN | spp.optimal.JUN-1_OP234_WA_L4_IP_Rep0.tagAlign_VS_JUN-1_OP234_WA_L4_Input_Rep0.tagAlign.regionPeak |
| Worm ChIP-seq pha-4_l4 | modENCODE/modERN | ENCFF209ONL.bed |
| Worm ChIP-seq skn-1_l4 | modENCODE/modERN | ENCFF798LMX.bed |
| Worm ChIP-seq unc-55_l2 | modENCODE/modERN | ENCFF346FAK.bed |
| Worm ChIP-seq ztf-16_embryo | modENCODE/modERN | ENCFF341MDZ.bed |
| Experimental Models: Cell Lines | ||
| Mouse: CF-1 MEF 2M Mito-C | Tebu-Bio | Cat#222GSC-6201M |
| Human: polycistronic human OCT4/KLF4/c-MYC/SOX2 (OKMS) cassette (hiF-Tcells) |
Cacchiarelli et al. (2015) Laboratory of Dr. Mikkelsen |
N/A |
| Human: Human Dermal Fibroblasts (NHDF) | Lonza | Cat#CC-2511 |
| Experimental Models: Organisms/Strains | ||
| N2 C. elegans wild isolate | Caenorhabditis Genetics Center | WormBase ID: N2 |
| JR3373 wIs125[hsp-16-2::elt-7 hsp-16-41::elt-7]; rrIs1 [elt-2::GFP + unc-119(+)] | Laboratory of Dr. Joel Rothman | WormBase ID: JR3373 |
| LW697 ccIs4810[pJKL380.4; lmn-1p::lmn-1::GFP::lmn-1 3'utr + pMH86; dpy-20(+)] I. | Caenorhabditis Genetics Center | WormBase ID: LW697 |
| OD56 ltIs 37[pie-1p::mCherry::his-58 unc-119(+)];unc-119(ed3) III. | Caenorhabditis Genetics Center | WormBase ID: OD56 |
| SS104 glp-4(bn2) I. | Caenorhabditis Genetics Center | WormBase ID: SS104 |
| NL2507 pkIs1582[let-858::GFP + rol-6(su1006)] | Caenorhabditis Genetics Center | WormBase ID: NL2507 |
| BAT28/OH9846 otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V. | Caenorhabditis Genetics Center | WormBase ID: OH9846 |
| BAT012 barIs12[elt-2prom::gfp; myo-3p::NmBirAo] | This paper | N/A |
| BAT026 otIs284 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V.; hdIs30 [glr-1::dsRED] | This paper | N/A |
| BAT032 glp-1(ar202) III.; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V. | This paper | N/A |
| BAT044 juIs244 [ttr-39prom::mCherry, ttx-3prom::gfp]; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V. | This paper | N/A |
| BAT046 otIs133 [ttx-3prom::mCherry]; otIs284 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V.; hdIs30 [glr-1::dsRED] | This paper | N/A |
| BAT068 otEX4945 [hs:hlh-1, rol-6(su1006)]; mgIs25 [unc-97prom::gfp] | This paper | N/A |
| BAT109 otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)] V. | This paper | N/A |
| BAT139 stIs10086 [ges-1::H1-Wcherry + unc-119(+)] | This paper | N/A |
| BAT160 itIs37 [pie-1p::mCherry::his-58(pAA64), unc-119(+)]; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)] ntIs1 [gcy-5p::GFP, lin-15(+)] V. | This paper | N/A |
| BAT282 barIs40 [vit-5::2xNLS::TagRFP] | This paper | N/A |
| BAT284 stIs10131 [elt-7::H1-wCherry + unc-119(+)] | This paper | N/A |
| BAT287 ntIs1 [gcy-5::gfp; lin-15b(+)] V. | This paper | N/A |
| BAT326 otIs263 [ceh-36prom::tagRFP]; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V. | This paper | N/A |
| BAT453 barEx147 [hsp-16.4prom::unc-30; hsp-16.2prom::unc-30; rol-6(su1006)]; juIs244 [ttr-39prom::mCherry, ttx-3prom::gfp] | This paper | N/A |
| BAT522 otis393 [ift-20prom::NLS::tagRFP]; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V. | This paper | N/A |
| BAT525 hmg-3 (tm2539) / dpy-5(e61) unc-13(e1091) I. | This paper | N/A |
| BAT527 otIs355 [rab-3prom::NLS::TagRFP]; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)] V. | This paper | N/A |
| BAT606 edIs6 [unc-119::gfp + pRF4[rol-6(su1006)]] IV.; otIs305 [hsp-16.48prom::che-1::3XHA::BLRP; rol-6(su1006)] V. | This paper | N/A |
| BAT1560 hmg-3(bar24[hmg-3::3xHA]) I. protein tag CRISPR engineered | This paper | N/A |
| BAT1753 hmg-3(bar24[hmg-3::3xHA]) I. 2x outcrossed | This paper | N/A |
| BAT1945 jun-1(gk557) II; otIs305[hsp::che-1::3xHA, rol-6] ntIs1[gcy-5::GFP] | This paper | N/A |
| BAT1967 hmg-4(bar32[hmg-4::3xHA]) III. protein tag CRISPR engineered | This paper | N/A |
| Oligonucleotides | ||
|
hmg-3(bar24) generation: hmg-3 sgRNA F TCTTatccgattcaccagaagact hmg-3 sgRNA R AAACagtcttctggtgaatcggat hmg-3::3xHA F1 cgatgagccgctaaaggcgaagaaagacgaatccgatgct gcttctgagtcttctggtgaatcggatTACCCATACGA CGTTCCAGA hmg-3::3xHA R1 gtaagaaggaaggcgaataaaaagcaacaataaaatatttag tcagaaaaTTAAGCGTAATCTGGGACGTCA hmg-3::3xHA F2 cgatgagccgctaaaggc hmg-3::3xHA R2 gtaagaaggaaggcgaataaaaagc hmg-3::3xHA Fs gatacggatgattccgatgacg hmg-3::3xHA Rs gaaggaaggcgaataaaaagcaac |
This paper | N/A |
|
dpy-10(cn64) sgRNA F TCTTgctaccataggcaccacgag dpy-10(cn64) sgRNA R AAACctcgtggtgcctatggtagc dpy-10(cn64) ssODN cacttgaacttcaatacggcaagatgagaatgactggaaaccgt accgcATgCggtgcctatggtagcggagcttcacatggcttc agaccaacagcct |
Paix et al. (2014) Arribere et al. (2016) |
N/A |
|
hmg-4(bar32) generation: hmg-4 sgRNA1 atccgattcatcagatccat hmg-4 sgRNA2 gtccgatggatctgatgaat hmg-4 ssODN_3xHA CCAAAAGAAGAATCAGAAGAGAGTAATAATGGCTC TGATGGATCTGATGAATCAGATGATTCAGATTAC CCATACGACGTTCCAGACTATGCCGGCTACCCCTA TGATGTCCCGGACTATGCAGGATCTTATCCATATG ACGTCCCAGATTACGCTTAAATTATTAATTTTGTTTC TTTTAAACTCGTGTACTATC hmg-4::3xHA F gattcggacgatgaggagc hmg-4::3xHA R cagaatgagatattcagacaacttgag |
This paper | N/A |
| smFISH probes, see Table S7 | This paper | N/A |
| Oligonucleotides used for qRT-PCR, see Table S7 | This paper | N/A |
| siRNA targeting sequences, see Table S7 | Dharmacon | N/A |
| Recombinant DNA | ||
| Peft-3::cas9-SV40_NLS::tbb-2 3'UTR | Addgene | Cat#46168 |
| pJJR50 | Adgene | Cat#75026 |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, (2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al. (2009) | http://samtools.sourceforge.net/ |
| JAMM | Ibrahim et al. (2015) | https://github.com/mahmoudibrahim/JAMM |
| Speakerscan | Megraw et al. (2009) | https://ohlerlab.mdc-berlin.de/software/A_Transcription_Factor_Affinity_Based_Code_for_Mammalian_Transcription_Initiation_78/ |
| cERMIT | Georgiev et al. (2010) | https://ohlerlab.mdc-berlin.de/software/cERMIT_82/ |
| UMI-tools | Smith et al. (2017) | https://github.com/CGATOxford/UMI-tools |
| Bedtools 2.23 | Quinlan, and Hall, (2010) | https://github.com/arq5x/bedtools2/releases |
| STAR | Dobin et al. (2013) | https://github.com/alexdobin/STAR |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Deep Tools | Ramírez et al. (2016) | https://github.com/deeptools/deepTools |
| EdgeR | Robinson et al. (2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| ggplot2 | Wickham (2016) | https://github.com/tidyverse/ggplot2 |
| Flexbar | Roehr et al. (2017) | https://github.com/seqan/flexbar |
| Picard | https://broadinstitute.github.io/picard/ | |
| FASTX-toolkit | http://hannonlab.cshl.edu/fastx_toolkit/ | |
| RSEM | Li and Dewey, (2011) | https://github.com/deweylab/RSEM |
| Tximport | Soneson et al. (2015) | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| Trimmomatic | Bolger et al. (2014) | https://github.com/timflutre/trimmomatic |
| PANTHER | Mi et al. (2013) | http://www.pantherdb.org |
| Tomtom | Gupta et al. (2007) | http://meme-suite.org/doc/tomtom.html?man_type=web |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Baris Tursun (baris.tursun@mdc-berlin.de).
Experimental Model and Subject Details
C. elegans Strains Used in the Study
Information on strains including genotypes are provided in Table S7. Hermaphrodites were used for phenotype scoring such as reprogramming effects, smFISH, reporter assays, and immunohistochemistry.
Nematode Culture
C. elegans strains were maintained using standard condition on OP50 bacteria at 20°C (Brenner, 1974). All heat-shock and temperature-sensitive strains were kept at 15°C (See Table S7).
Cell Culture
Human secondary fibroblasts carrying a doxycycline (DOX)-inducible, polycistronic human OCT4/KLF4/c-MYC/SOX2 (OKMS) cassette (hiF-Tcells) (Cacchiarelli et al., 2015) were cultured in hiF medium (DMEM/F12 Gltuamax supplemented with 10% FBS, 1% NEAA, 0,1% beta-mercaptoethanol 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 16 ng/ml FGFbasic). hiF-T cells were passaged every 3 days, using a splitting ratio of 1:3 as described before (Cacchiarelli et al., 2015). For experiments, 80.000 hiF-T cells/ well of a 12-well plate treated with attachment factor were seeded and incubated overnight at 37°C. According to the previously published study (Cacchiarelli et al., 2015), hiF-T cells were derived from BJ human foreskin fibroblasts (male gender).
Normal Human Dermal Fibroblasts (NHDF) were obtained from the company Lonza and were used for direct reprogramming experiments as described below. According to the manufacturer NHDF are primary human neonatal dermal fibroblasts derived from neonatal foreskins (male gender). Cells were first grown in Fibroblast Growth Medium (FGM, Invitrogen) before transferring them to neuronal medium (DMEM/F2, Invitrogen), apotransferrin (100mg/ml), insulin (5mg/ml), sodium selenite (30 nM), progesterone (20 nM), putrescine (100 nM), penicillin/streptomycin) supplemented with neurotrophic factors) as described before (Vierbuchen et al., 2010).
Method Details
RNAi in C. elegans
For RNAi, worms were grown on plates seeded with RNAi bacteria from Ahringer library (Source Bioscience). RNAi against Renilla luciferase (Rluc) was used as control. Whole-genome RNAi screen was designed as an F1 screen using a standard RNAi feeding protocol (Kamath and Ahringer, 2003). RNAi bacteria for the screen were seeded on 6-well plates and the screen was carried out in duplicates. In total, 19791 genes on 6 chromosomes were screened. Reprogramming experiments were carried out either as P0 for hmg-4 and spt-16, or F1 for hmg-3 RNAi using a standard feeding protocol (Kamath et al., 2003). For P0 experiments, worms were synchronized by bleaching and L1 larvae were put on RNAi plates; for F1 RNAi, synchronized L1s were grown at 15°C on normal food until they reached L4 stage when they were transferred on RNAi plates. Worms on RNAi plates were grown at 15°C until most of the P0 or F1 progeny reached L4 stage. The plates were heat-shocked at 37°C for 30 min followed by an overnight incubation at 25°C (Kolundzic et al., 2018, Tursun et al., 2011). RNAi in embryos against a number of genes causes lethality during early development which was also previously observed (Kamath et al., 2003). Therefore, we additionally performed RNAi only after birth of the animals for such RNAi targets. Plates were screened for presence of ectopic GFP the following day under a dissecting scope. To induce the Glp phenotype in glp-1(ar202), the animals were shifted to room temperature 8 hrs before the heat-shock. It is important to note that F1 RNAi against spt-16 caused embryonic lethality thereby not allowing to test for gcy-5::gfp induction in the germline as seen upon hmg-3 F1 RNAi. For double RNAi, bacteria were grown as saturated culture. The OD600 was measured to ensure that the bacteria were mixed in an appropriate 1:1 ratio and subsequently seeded on RNAi 6-well plates. The library screened for suppression in hmg-3-depleted background and entire results are listed in Table S4. For time course experiments, screening for the presence of ectopic GFP was done 0, 4, 8, 12, 24, 36 and 48 hrs after heats-hock. The C. elegans lines used in this study are listed and described in detail in STAR Methods.
Generation of CRISPR Alleles
CRISPR engineering was performed by microinjection using a PCR repair template and the dpy-10 co-CRISPR approach The injection mix contained a plasmid that drives expression of Cas9 (50ng/μl, a gift from John Calarco, Addgene #46168), one that drives expression of dpy-10(cn64) sgRNA (50ng/μl), a dpy-10(cn64) PAGE-purified 99mer single-stranded oligodeoxynucleotide (ssODN) HR template (50ng/μL; IDT), a plasmid expressing the sgRNA targeting the hmg-3 locus (dBT620, 50ng/μl) and a PCR repair template to introduce the 3xHA knock-in at the 3’ end of hmg-3 (90ng/μl). To generate the sgRNA plasmids, annealed oligo pairs were ligated into BbsI-digested pJJR50 (a gift from Mike Boxem, Addgene#75026). To generate the PCR repair template, 3xHA tag was flanked with approximately 50bp homology arms on both 3’ and 5’ side for insertion at the 3’ end of the hmg-3 or hmg-4 locus. Screening for successful knock-in events was done by pooling several injectants on the same plate and picking off independent F1 animals to generate transgenic lines(Arribere et al., 2016). Positive hits were homozygoused for the knock-in by singling and genotyping worms and the knock-in was confirmed by Sanger sequencing.
Sequences of the oligonucleotides used for the generation of hmg-3(bar24) and hmg-4(bar32) are reported in STAR Methods.
Antibody Staining
For staining anti-SPT16 (rabbit polyclonal peptide antiboy, 1:200) antibodies, worms were resuspended 0,025 % glutaraldehyde and frozen using freeze-crack protocol (Duerr, 2006). Acetone/methanol fixation was used to prevent gonad extrusion. For anti-HA staining (anti-HA mono mouse antibody, Roche, at 1:1000 dilution) whole worms were fixed and permeabilized following a previously described method (Bettinger et al., 1996). In brief, after washing, worms were resuspended in RFB (160 mM KCl; 40 mM NaCl; 20 mM EGTA; 10 mM Spermidine) + 2% formaldehyde followed by three freeze-thaw cycles. After incubation for 30 min at 25°C, the sample was washed with TTE (100 mM Tris pH 7,4; 1 % Triton; 1mM EDTA) and incubated for 4 h at 37°C with shaking in TTE + 1% beta-Mercaptoethanol. The sample was washed in BO3 buffer (10 mM H3BO3; 10 mM NaOH; 2 % Triton) and further incubated for 15 min at 37°C with shaking in BO3 buffer + 10 mM DTT. After another wash with BO3, BO3 buffer + 0,3 % H2O2 was added and incubated for 15 min at 25°C. The sample was washed once more with BO3, blocked with 0,2 % gelatin + 0,25 % Triton in PBS and stained. Primary antibodies were diluted in PBS with 0,25 % Triton + 0,2 % gelatin, added to the fixed worms and incubated overnight at 4°C. After washing in PBS + 0,25 % Triton, secondary antibodies (Alexa Fluor dyes at 1:1500 dilution) were applied and incubated overnight at 4°C. Samples were washed in PBS + 0,25 % Triton and mounted on glass slides with DAPI-containing mounting medium (Dianove, #CR-3448). For anti-P granule, ant-ELT-2 and anti-IFB-2 staining (anti-OIC1D4-s - P-granules, anti-455-2A4-s (ELT-2), anti-MH33-s - intermediate filament mono mouse antibody, repectively, Hybridoma bank, at 1:150 dilution) worms were dissected and processed as described before (Jones et al., 1996). In brief, worms were cut below the pharynx so that the gonads and intestines protrude, fixed for 15 min in 4% paraformaldehyde, followed by washing 3 times with PBS with 0,25 % Triton. Worms were then blocked for 1 hour in PBS with 0,25 % Triton + 0,2 % gelatin, followed by incubation with primary antibodies incubated overnight at 4°C. After washing in PBS + 0,25 % Triton, secondary antibodies (Alexa Fluor dyes at 1:1500 dilution) were applied and incubated overnight at 4°C. Samples were then washed in PBS + 0,25 % Triton and mounted on glass slides with DAPI-containing mounting medium as described above. The following antibodies were used: anti-OIC1D4-s (P-granules), anti-455-2A4-s (ELT-2), anti-MH33-s - intermediate filament (IFB-2), mono mouse antibodies, Hybridoma bank, at 1:150 dilution.
Single Molecule Fluorescent In Situ Hybridization (smFISH)
smFISH was performed using Custom Stellaris FISH probes, purchased from Biosearch Technologies and the staining was done according to the manufacturer’s protocol. Sequences of all the smFISH probes used are listed in STAR Methods.
Cell-Cycle Arrest by HU Treatment
Hydroxyurea (HU) treatment was carried out as previously described (Fox et al., 2011, Patel et al., 2012, Seelk et al., 2016). In brief, HU was added to seeded RNAi plates at a final concentration of 250 μM. L4 worms grown on RNAi plates were transferred to HU plates and incubated at room temperature for 5 hrs prior to heat-shock in order to induce CHE-1 expression. After overnight incubation, worms were assessed for GFP induction in the germline as described above.
siRNA Knockdown in Human Cells
hiF-T cells were cultured as described above and passaged every 3 days, using a splitting ratio of 1:3 as described before (Cacchiarelli et al., 2015). For siRNA experiments, 80.000 hiF-T cells/ well of a 12-well plate treated with attachment factor were seeded and incubated overnight at 37°C. siRNA knockdown was performed the following day by a reverse transfection method using DharmaFECT 1 and 40 nM siRNA pool reagents purchased from Dharmacon according to the manufacturer’s instructions. Sequences of all the siRNA reagents used are described in Table S8. To monitor knockdown efficiency, RNA was isolated using Qiagen RNeasy Plus Mini Kit 24h, 48h, 72h or 7 days after siRNA transfection. To control for OSKM expression after SSRP1 and SUPT16H depletion, RNA was isolated from siRNA transfected cells 48h after knockdown without prior Doxycycline treatment. cDNA was synthesized with oligo dT primers using the GoScript Reverse Transcriptase (Promega). cDNA was used for qPCR with the Maxima SYBR Green qPCR Master Mix (2X) (Thermo Scientific) according to the manufacturer’s instructions on an ABI PRISM 7700 system (Applied Biosystems). The real-time PCR data analysis was done by using comparative CT method (2008). Gene expression levels were calibrated to the housekeeping gene GAPDH and normalized to Rluc. Sequences of qRT-PCR primers used are described in STAR Methods.
Reprogramming Experiments with hiF-T Cells
Reprogramming experiments were performed by seeding hiF-T cells on irradiated mouse embryonic fibroblasts (MEF, GlobalStem) 24 h after siRNA transfection. Induction of OSKM by DOX supplementation (2μg/ml) was started the next day in hiF medium for the first 2 days and then in KSR medium (DMEM/F12 Gltuamax supplemented with 20% KSR, 1% NEAA, 0,1% beta-mercaptoethanol 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 8 ng/ml FGFbasic and ROCK1 inhibitor (Y-27632-2HCl, Biozol Diagnostica, final concentration 1μM)) as described previously (Cacchiarelli et al., 2015).
Reprogramming Experiments with NHDF Cells
Direct reprogramming experiments were performed by transducing Normal Human Dermal Fibroblasts (NHDF cells, Lonza) with RTTA, tetO-Ascl1, tetO-Brn2 and tetO-Myt1l (BAM) in a 1.2:1:1.5:2.5 ratio and supplemented with polybrene (c.f. 8 μg/ml) 24 h before siRNA transfection to deplete FACT. Induction of BAM by DOX supplementation (2 μg/ml) was started the next day in neuronal medium ((DMEM/F2 (Invitrogen), apotransferrin (100mg/ml), insulin (5mg/ml), sodium selenite (30 nM), progesterone (20 nM), putrescine (100 nM), penicillin/streptomycin) supplemented with neurotrophic factors) as described before (Vierbuchen et al., 2010).
Phenotypic Characterization of iPS Cells
iPSC colonies were characterized after ∼21 days of reprogramming. Alkaline phosphatase activity was measured using an enzymatic assay for alkaline phosphatase (Alkaline Phosphatase Staining Kit II, Stemgent) according to the manufacturer’s instructions. Number of colonies formed in each condition was counted based on SSEA-4 positive colonies. Immunohistochemistry with SSEA-4 Antibody (DyLight 488 conjugate, Thermo Scientific) was performed according to the manufacturer’s protocol at 1:500 dilution. Cells were fixed with 4%PFA in PBS and permeabilized with 0.1% Tween-PBS before antibody incubation. The pluripotency staining was performed using the Pluripotent Stem Cell 4-Marker Immunocytochemistry Kit (Thermo Fisher Scientific) in addition with the NANOG rabbit (Thermo, PA1-097) antibody.
Pluripotency Teratoma Assay
The teratoma assay was done by EPO GmbH - Experimental Pharmacology & Oncology, Berlin, Germany. Briefly, in order to initiate the assay 1 x106 cells were suspended in 50 ul of PBS and thereafter mixed with 50 ul of Matrigel (Corning). These cell suspension was than subcutaneously transplanted into a NOG-mice and the tumor growth was documented on a weekly basis. After reaching the size of 1,5cm3 the tumor was extracted and pathologically analyzed.
Human ATAC-Seq
For ATAC-seq of human fibroblasts, 50.000 cells were harvested 48 h after siRNA transfection and the cell pellet was resuspended in transposase reaction mix as described previously (Buenrostro et al., 2013). Briefly, cell pellet was resuspended in the transposase reaction mix (25 μL 2× TD buffer, 2.5 μL transposase) using Nextera DNA Library Preparation Kit (Illumina) and the transposition reaction was carried out for 60 min at 37°C. The samples were purified using Zymo DNA Clean & Concentrator kit. Following purification, library fragments were amplified using NEBNext PCR master mix and previously published PCR primers (Buenrostro et al., 2013). Libraries were amplified for a total of 12 to 14 cycles and sequenced using paired-end-sequencing length of 75 nucleotides using NextSeq 500/550 High Output v2 kit (Illumina).
C. elegans Nuclei Isolation and ATAC-Seq
For C. elegans ATAC-seq, L1 animals were synchronized by harvesting embryos using sodium hypochlorite treatment and grown on RNAi plates until L4 stage (P0 RNAi as described above). Generally, we performed ATAC-seq without inducing che-1 overexpression in order to detect chromatin alterations that were caused by FACT depletion but not due to the activity of the reprogramming TF.
ATAC-seq upon RNAi against hmg-4 and spt-16 was performed in glp-4(bn2) mutant animals that lack a germline when grown at the non-permissive temperature (Beanan and Strome, 1992) to measure chromatin accessibility changes only in somatic cells. Synchronized L4 animals were washed 5 times in M9 buffer and collected on ice. Nuclei were isolated using a glass Dounce homogenizer with 50 strokes tight-fitting insert in buffer A (15 mM Tris–HCl pH7.5, 2 mM MgCl2, 340 mM sucrose, 0.2 mM spermine, 0.5 mM spermidine, 0.5 mM phenylmethanesulfonate [PMSF], 1mM DTT, 0.1% Trition X-100 and 0.25% NP-40 substitute) as described before (Ooi et al., 2010, Steiner et al., 2012). The debris were removed by spinning at 100×g for 5 min and nuclei were counted by Methylene blue staining. 100.000 nuclei per sample were pelleted by spinning at 1000×g for 10 min and proceeded immediately to transposition step of the ATACseq protocol as described above (Buenrostro et al., 2013). For ATAC-seq analysis of gonad DNA, 20 wild type gonads were dissected per replicate and nuclei were isolated as described above. Libraries were amplified for a total of 10 to 18 cycles and sequenced using paired-end-sequencing length of 75 nucleotides using NextSeq 500/550 High Output v2 kit (Illumina).
RNA-Seq Using C. elegans
For transcriptome analysis, RNA was isolated from HMG-4 and SPT-16-depleted animals using TRIzol (Life Technologies) and guanidinium thiocyanate-phenol-chloroform extraction. After adding chloroform to the sample containing TRIzol, phases were separated to an aquaous phase containing RNA, an interphase and an organic phase containing DNA and proteins. Guanidinium thiocyanate denatured proteins (including RNases) in the organic phase. RNA was purified from the aquaous phase using isopropanol. For reverse transcription GoScript Reverse Transcriptase (Promega) was used according to the manufacturers protocol. The preparation of libraries for whole-transcriptome sequencing was carried out using TruSeq RNA Library Prep Kit v2 (Illumina) according to the manufacturers instructions. Libraries were sequenced using single end sequencing length of a 100 nucleotides on a HiSeq4000 machine (Illumina). For transcriptome analysis of gonad RNA, 20 wild type gonads were dissected per replicate and RNA was isolated using Rneasy Micro Kit (Qiagen). PolyA-RNA enrichment was performed using oligo(dT)25 Dynabeads (Life Technologies) according to the manufacturers instructions. The libraries were prepared using NEXTflex Rapid Directional qRNA-Seq Kit (Bioo Scientific) according to the manufacturers instructions. The libraries were sequenced using paired end sequencing length of 75 nucleotides on a HiSeq4000 machine (Illumina).
RNA-Seq Using Human Cells
For transcriptome analysis in hiF-T cells, RNA was isolated from 80.000 cells treated with control, SSRP1 or SUPT16H siRNAs 48 h after transfection using RNeasy Plus Mini Kit (Qiagen). To remove ribosomal RNA, Ribo-Zero Gold rRNA Removal Kit (Illumina) was used as described in the manufacturers protocol. ERCC spike-in control mixes 1 and 2 (Thermo Fisher) were used for normalization control and the libraries were prepared using NEXTflex Rapid Directional qRNA-Seq Kit (Bioo Scientific) according to the manufacturers instructions. Libraries were sequenced using paired end sequencing length of 75 nucleotides on a HiSeq4000 machine (Illumina).
ChIP and ChIP-Seq
ChIP-Seq with Human Cells
For chromatin immunoprecipitation with sequencing (ChIP-seq), a batch of 25 million hiF-T cells were used per chromatin preparation. The cells were fixed with 1.1 % formaldehyde and iDeal ChIP-seq kit for Transcription Factors (Diagenode) was used for chromatin immunoprecipitation (ChIP) according to the manufactures instructions. The chromatin was sheared by sonication using the Bioruptor (Diagenode) for 20 cycles (30 sec on, 30 sec off) on high settings at 4°C. Go-ChIP-Grade anti-SSRP1mouse antibody (BioLegend) and anti-SPT16 (D7I2K) rabbit monoclonal antibody (Cell Signaling) were used for immunoprecipitation at 1:50 dilution. The libraries were prepared using NEXTflex® qRNA-Seq™ Kit v2 (Bioo Scientific) according to the manufacturers instructions. Libraries were sequenced using paired end sequencing length of 75 nucleotides on a HiSeq4000 machine (Illumina).
ChIP-Seq in C. elegans
M9 arrested L1 worms were grown on OP50/RNAi plates until L4/YA stage at RT. Animals were washed three times with M9 and fixed with 2% formaldehyde for 30 minutes followed by quenching with 0.125M glycine for 5 minutes. The samples were rinsed twice with PBS, and 100-200 ul of pellets were snap-frozen in liquid nitrogen and kept at -80°C. The pellets were washed once with 0.5 ml and resuspended in 1 ml FA Buffer (50nM HEPES/KOH pH 7.5, 1mM EDTA, 1% Triton X-100, 0.1 sodium deoxycholate, 150mM NaCl)+0.1% sarkosyl+protease inhibitor (Calbiochem) and then dounce-homogenized on ice with 30 strokes. The samples were sonicated using Bioruptor (Diagenode) with the setting of high power, 4°C, and 15 cycles, 30 sec on, 30 sec off. Soluble chromatin was isolated by centrifuging for 15 min at max speed at 4°C. The cellular debris was resuspended in 0.5 FA Buffer + 0.1% sarkosyl+protease inhibitor (Calbiochem) and sonicated again as described above. Isolated soluble chromatin was combined. The immunoprecipitation of HA- and AVI-tag proteins was performed overnight in a total volume of 600 μl with 4 μg of HA-antibody (ab9110, Abcam) and 80 μl of streptavidin coated dynamagnetic beads (Invitrogen), respectively, while 5% of samples were taken as input. Immunocomplexes with HA-tag were collected with Protein A-Sepharose beads (Sigma). The beads were washed with 1 ml of following buffers: twice with FA Buffer for 5 min, FA-1M NaCl for 5 min, FA-0.5M NaCl for 10 min, TEL Buffer (0,25M LiCL, 1% NP-40, 1% Sodium deoxycholate, 1mM EDTA, 10mM Tris-HCl pH 8.0) for 10 min, and twice with TE Buffer (pH8.0). DNA-protein complexes were eluted in 250 μl ChIP elution buffer (1%SDS, 250nM NaCl, 10 mM Tris pH8.0, 1mM EDTA) at 65°C for 30 min by shaking at 1400 rpm. The Inputs were treated for 5h with 20 μg of RNAse A (Invitrogen). The samples and inputs were treated with 10 μg of Proteinase K for 1h, and reverse cross-linked overnight at 65°C. DNA was purified with Qiagen MinElute PCR purification Kit (Qiagen).
The libraries were prepared using NEXTflex® qRNA-Seq™ Kit v2 (Bioo Scientific) according to the manufacturers instructions. Libraries were sequenced using paired end sequencing length of 75 nucleotides on a HiSeq4000 machine (Illumina).
Immunoprecipitation for Mass Spectrometry
Each immunoprecipitation was performed in triplicate. L4 staged wild type, hmg-3::3xHA and hmg-4::3xHA worms were collected and washed 4 times with M9 to get rid of bacteria. The worms were snap-frozen in liquid nitrogen and then cryo-fractured by using a pulverizer. In order to obtain a fine powder, worms were further ground using a mortar and pestle on dry ice. The worm powder was resuspended in 1.5x of lysis buffer (20mM Hepes pH 7.4, 150mM NaCl, 2mM MgCl2, 0.1% Tween20 and protease inhibitors), dounced with tight douncer 30 times and sonicated using a Bioruptor (Diagenode) (6 times 30 sec ON, 30 sec OFF; high settings at 4°C) followed by centrifugation at 16,000g at 4°C for 10 min. The supernatant was incubated with DNaseI (Thermo scientific) ON. Both control (N2) and IP (HMG-3::3xHA, HMG-4::3xHA) samples were incubated with HA antibody (ab9110, Abcam) for 30 min on a rotator at 4°C. Next, μMACS Protein A beads (Milteny Biotec) were added to samples as instructed in the kit, and samples were incubated for 30 min on ice rotating. Samples were diluted 5x with lysis buffer and μMACS columns (Milteny Biotec) were used to wash and elute the sample according to the manufacturers protocol. The proteins were eluted with elution buffer (100mM TrisCl pH 6.8, 4 % SDS, 20mM DTT) heated at 95°C. Eluted samples were prepared for mass spectrometry measurement by using SP3 (Hughes et al., 2014) before they were analyzed on a Q Exactive Plus (Thermo Scientific) connected to a Proxeon HPLC system (Thermo Scientific).
Quantification and Statistical Analysis
ATAC-Seq Analysis
Pre-processing
ATAC-seq reads were trimmed for adapters using flexbar v2.5 (-f i1.8 -u 10 -ae RIGHT -at 1.0) (Dodt et al., 2012) and mapped with bowtie2 v2.0.2 (Langmead and Salzberg, 2012) in default paired-end mode and restricting pair distances to 1500 (-X 1500 -- no-discordant) to version hg19 of the human genome or ce10 of the worm genome followed by removal of multimappers. PCR duplicates were removed using Picard Tools MarkDuplicates v1.90 (http://broadinstitute.github.io/picard) and reads were converted to .bed format using bedtools bamToBed v2.23 (Quinlan and Hall, 2010). Pairs were filtered out if they mapped to the same strand, to different chromosomes, or if the 5’end coordinate of the – strand read was less than or equal to the 5’end coordinate of the + strand read. Mapped pairs were split into single reads and converted to a 38-bp fragment reflecting the theoretical minimal spacing required for a transposition event by Tn5 transpososome (Adey et al., 2010) using bedtools slop on the read 5’ends (-l 15 -r 22;) (Quinlan and Hall, 2010). Replicates were concatenated after confirming high concordance. C. elegans datasets were further filtered for reads from the rDNA loci as well as those mapping to regions corresponding to transgenic reporter constructs existing in the strains and corresponding to sequences used in the RNAi vectors. See Table S1 for basic statistics.
Peak Calling and Differential Analysis
Peaks were called on concatenated processed bed files using JAMM peakcaller v1.0.7.5 (-f38 –b 100 –e auto (human) and –e 1.75 (worm)) (Ibrahim et al., 2015). The resulting “all” (for meta analyses described below) or “filtered” peaks output by JAMM for each condition (control, SSRP1 knockdown, and SUPT16H knockdown for human, rluc and hmg-3 knockdown, or rluc and hmg-4, and spt-16 knockdown for worms) were concatenated and then merged with bedtools merge v2.23 (Quinlan and Hall, 2010). Merged “all” peaks were further filtered for a minimum width of 25-bp and used for meta analyses as described below. Merged “filtered” peaks for human (totaling 106,967) and 25-bp width-filtered merged “all” peaks for worm (totaling 17,024 for the hmg-4/spt-16 experiment and 21,254 for the hmg-3 experiment) were then counted for the number of processed reads that intersected them from each replicate of each experimental condition using bedtools coverage v2.23 (-counts) (Quinlan and Hall, 2010). Count tables were then normalized (method = “TMM”) and subjected to differential analysis (exactTest) using edgeR (Robinson et al., 2010). Peaks were called as differential with a Benjamini-Hochberg adjusted p-value cutoff of 0.01 (adjust=”BH”).
Peak Annotations
“Filtered” human ATAC-seq peaks or 25-bp width-filtered merged “all” ATAC-seq peaks for worm were annotated as promoter-proximal if they located +/- 200bp from human gencode v19 annotated start sites, or +/- 500bp from worm annotated TSSs, using bedtools closest (Quinlan and Hall, 2010), otherwise they were considered promoter distal.
De Novo Motif Generation and Scanning
For the human motif generation, promoter distal peaks were summed for their edgeR normalized log2 fold changes for each condition, split into two groups according to summed changes greater (opening peaks) or less than (closing peaks) zero. The resulting summed changes were used as the ranking statistic for motif generation using the cERMIT program (parameters: seed_length=6, min_motif_length=6, max_motif_length=12, required_core_length=5, cluster_sim_threshold=0.8, hypegeom_p_val_cutoff=1.0e-30, num_random_runs=1000, max_gene_set_size_percentage_threshold=0.3, min_gene_set_size_percentage_threshold=0.01, degen_threshold=0.75, fraction_degen_pos_threshold=0.75, use_regression_scoring=no, bootstrap_motif_score=no, fast_mode=no; PMID: 20156354). The same analysis was also done with the log2 fold change ranks separate for each factor.
Worm motifs were generated the same way, except independent of peak annotation and log2 fold changes were used individually for each factor knockdown as the ranking statistic for cERMIT. Sequences were restricted to those with lengths <1000-bp and worm sequences for those >50-bp and <1000-bp prior to input into cERMIT. Sequence numbers put into cERMIT are as follows: human combined up 44068; human combined down 42720; SSRP1 up 45710, SSRP1 down 41087, SUPT16H up 42130, SUPT16H down 44667, hmg-3 down 10653, hmg-3 up 10486, hmg-4 down 8721, hmg-4 up 8221, spt-16 up 8616, spt-16 down 8326. cERMIT-generated motifs were converted to meme format and Tomtom (Gupta et al., 2007) was used in the default settings to match the generated motifs to the JASPAR CORE vertebrate 2016 database (Mathelier et al., 2016) for the human motifs or to a published database of protein binding microarray-generated motifs for C. elegans (Narasimhan et al., 2015). Known binding preferences matching generated motifs were scanned using Speakerscan (Megraw et al., 2009) with a 500bp sliding local first-order markov background window and the maximum score taken for each peak.
Fragment Length Analysis and Promoter ATAC-Seq Counts
Replicate-concatenated full-paired-end-fragments ATAC-seq .bed files were intersected with windows around annotated transcription start sites (see ChIP-seq section below) using bedtools intersect (Quinlan and Hall, 2010) and the distribution of resulting fragment sizes summarized and plotted using geom_density from the ggplot2 suite (Wilkinson, 2011). The same windows were counted for processed ATAC-seq reads (see above) and the distributions plotted using geom_violin from ggplot2 (Wilkinson, 2011).
Analysis of RNA-Seq
Human and Worm Gonad RNA-Seq
Unique molecular identifiers (UMIs) were extracted from .fastq files using UMI-tools (Smith et al., 2017) and reads trimmed using fastx_trimmer from the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads were then filtered for ERCC spike-in reads and rRNA by mapping to a custom index with Bowtie 1 (Langmead et al., 2009).Trimmed, filtered, reads were then mapped using STAR (Dobin et al., 2013) to the hg19 or ce10 version genome builds and GTF files from GENCODE for human (V19 including an additional pre-mRNA transcript for each gene (Mukherjee et al., 2017) or from Ensembl for worms. Mapped .bam files were then subjected to PCR deduplication using UMI-tools (Smith et al., 2017), followed by conversion to .fastq and remapping with STAR to generate final mapped files and normalized coverage tracks. Gene counts were quantified using RSEM (Li and Dewey, 2011) and, for human, imported for differential expression analysis by DESeq2 (Love et al., 2014) using tximport (Soneson et al., 2015) and the DESeqDataSetFromTximport function within DESeq2. Genes were considered differentially expressed if they passed the DESeq2’s independent filtering for both the supt16h knockdown and ssrp1 knockdown experiments and below the default FDR cutoff of 0.1 and one of the two experiments.
Whole Worm RNA-Seq
Multiplexed Illumina sequencing data was demultiplexed using Illumina bcl2fastq converter (version v2.17.1.14). Raw reads in fastq format were processed to get rid of low quality bases and possible adapter contamination using Trimmomatic (version 0.33) (Bolger et al., 2014) (settings: ILLUMINACLIP:TruSeq.fa:2:25:6 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36). The filtered reads were aligned to the C.elegans genome (ce10 genome build - WormBase WS220 released in October, 2010) using the splice-aware short read aligner STAR (version 2.4.2a) with the default settings (Dobin et al., 2013) except for setting “—outFilterMultimapNmax” argument to 1. The expression level of each gene was quantified using R/Bioconductor package quasR (Gaidatzis et al., 2015) using genome annotation data in GTF file format from the Ensembl database (version 70) (Yates et al., 2016) Differential expression analysis of the quantified expression levels of genes between different samples was done using the R/Bioconductor package DESeq2 (Love et al., 2014) Up/down regulated genes are detected based on the differential expression criteria of adjusted p-value of at least 0.1 and at least two-fold increase/decrease in expression levels in relation to the control samples.
ChIP-Seq Analysis
Human Ssrp1 and Supt16h, as well as worm HMG-3::HA, ChIP-seq raw fastq files were extracted for UMIs using UMI-tools (Smith et al., 2017) and then trimmed using fastx_trimmer from the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). Reads were then mapped using bowtie2 (Langmead and Salzberg, 2012) and mapped reads selected for unique mapping prior to PCR deduplication using UMI-tools (Smith et al., 2017). Uniquely mapped read pairs were then converted to .bed files reflecting coordinates of the full paired-end fragments using custom scripts and normalized bigwig files were generated on replicate-concatenated files using bamCoverage from the deepTools2 suite (Ramírez et al., 2016). Input normalized final bigwigs were then produced using bamCompare from the deepTools2 suite (Ramírez et al., 2016) setting the –ratio parameter to either “subtract” for the worm data or “ratio” for the human data. Annotated transcription start sites (TSS) were then quantified for ChIP signal by creating windows 500bp upstream and 1500bp downstream for human (250bp upstream and 1000bp downsteam for worm) and generating counts of full-paired-end-fragments from each replicate (the single input file for worms was split into two files to use as virtual replicates) using bedtools intersect (Quinlan and Hall, 2010). These count tables were then subjected to “differential” analysis using DESeq2 with default settings comparing ChIP counts versus input counts. DESeq2 normalized log2 “fold changes” were used as ChIP-seq signal in these annotated window regions and genes were classified to have “high” signal if their respective annotated TSS window DESeq2 fold-change was greater than zero with an FDR of < 0.1 and the gene passed the DESeq2 independent filtering from the accompanying RNA-seq data. Gene TSS windows were classified as ChIP-seq “Low” if they had an FDR > 0.1 and the gene passed the DESeq2 independent filtering from the accompanying RNA-seq data. The use of the RNA-seq filter to define these two classes is intended to have a more comparable set of “Low” genes that are at least basally expressed within the system used. For the worm data, this RNA detection requirement for ChIP High and ChIP Low classes was taken from the whole worm RNA-seq data when plotting whole worm ATAC-seq data or assessing the relationship beween ChIP-seq level and whole worm RNA-seq fold changes following FACT knockdown, or was taken from the gonad-specific RNA-seq (requiring an FPKM > 0 instead of DESeq2 filtering) for plotting gonad-specific ATAC-seq or assessing the relationship between ChIP-seq level and gene expression level.
Meta-Analyses
All average profile and heatmap meta-analysis plots were performed on normalized bigWig files described above using the computeMatrix followed by plotProfile or plotHeatmap tools from the deepTools suite (Ramírez et al., 2016).
Density Scatterplots
All density scatter plots were generated using ggplot2 geom_hex (bins=50) (Wilkinson, 2011).
Violin Plots
All violin summary plots were plotted using geom_violin (trim=TRUE, scale=”width”) from ggplot2 (Wilkinson, 2011).
Enrichment Tests
For FACT knockdown RNA-seq enrichment tests with reprogramming gene clusters from Cacchiarelli et al (Cacchiarelli et al., 2015), ENSEMBL gene ids were obtained from the published clusters and intersected with ENSEMBL gene ids of the union of differentially increased or decreased genes upon FACT knockdown (see RNA-seq section above). The numbers of these intersections were compared to the Cacchiarelli gene cluster intersections with all genes that passed the DESeq2 independent filtering for both Ssrp1 knockdown and Supt16h knockdown datasets after filtering for common ids with the entire Cacchiarelli dataset. The built-in R function phyper was used with parameter “lower.tail=FALSE” to obtain enrichment pvalues from the hypergeometric distribution.
For ATAC-seq peak enichements with the Cacchiarelli clusters, all ATAC-seq peaks were assigned to the closest annotated TSS and the resulting ENSEMBL gene ids were filtered to remove genes that that did not pass DESeq2’s independent filtering for both the Ssrp1 knockdown and Supt16h knockdown RNA-seq datasets (see RNA-seq analysis above). The unique union of genes assigned ATAC-seq peaks that were differentially increased or decreased for accessibility in either the Ssrp1 or Supt16h knockdowns was then intersected with the gene ids for each Cacchiarelli cluster and these numbers compared to the cluster intersections with the full set of RNA-seq-detected, ATAC-seq-peak-assigned genes after filtering for overlap with the entire set of Cacchiarelli ids. The pvalue from the hypergeometric distribution was calculated as described above.
Differential ATAC-seq and RNA-seq enrichments were tested as follows. Differentially regulated ATAC-seq peaks were assigned to the closest annotated TSS for each knockdown dataset and the resulting gene lists filtered to remove genes that had both increased-accessibility and decreased-accessibility peaks. For the human data, the union of increased-accessibility-peak-assigned genes and the union of decreased-accessibility-peak-assigned genes from the Ssrp1 knockdown and Supt16h knockdown datasets were generated. These two gene lists were then intersected with the union of up-regulated genes, the union of down-regulated genes, or the full set of genes that passed DESeq2’s independent filtering from both the Ssrp1 and Supt16h knockdown RNA-seq datasets. The numbers of these intersections were used to calculate pvalues from the hypergeometric distribution as described above. For the worm datasets, each ATAC-seq and RNA-seq dataset was treated independently (rather than taking the union of differential genes/peaks) due to the whole worm aspect of the data and the observed distinct tissue expression domains of the FACT complex components. Otherwise, the approach was identical to that for the human datasets.
For enrichments of differentially regulated ATAC-seq peaks from worm FACT knockdown experiments with available transcription factor (TF) datasets, reproducible peak calls using the IDR approach were downloaded from the modENCODE/modERN website for ce10. Intersections were counted between sets of differentially regulated ATAC-seq peaks with the TF peaks using bedtools intersect (Quinlan and Hall, 2010). These numbers were compared to the numbers of intersections for the same TF peaks with all ATAC-seq peaks and pvalues were obtained from the hypergeometric distribution as described above. For all enrichment tests, -log10 pvalues were plotted as heatmaps using the pheatmap package.
GO Analysis
GO analysis was performed using PANTHER overrepresentation test (Mi et al., 2013). The union of up- or down-regulated genes upon Ssrp1- or Supt16h-knockdown was compared to all genes detected in both experiments as a background and as output the PANTHER GO Slim ontologies.
Mass Spectrometry Data Processing
Label-free quantification (LFQ) was performed using Max Quant and the data was analyzed with an online tool http://projects.biotec.tu-dresden.de/msVolcano/ (Singh et al., 2016).
Other Statistical Analysis
Applied statistical analysis types used for phenotype scoring are indicated in the respective figure legends and Results section. Generally, the software application Prism (version 6, Graph Pad) has been used to calculate p-Values and determine standard deviations (SD) or standard error of the mean (SEM).
Data and Software Availability
The following link has been created to access to record GSE98758 containing all ChIP-seq, RNA-seq and ATAC-seq data sets:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=cnmnosmadlofpsz&acc=GSE98758
Additional Resources
C. elegans Strains Used in the Study
Information on strains including genotypes are provided in Table S7. All animals used for phenotype scoring were hermaphrodites.
Sequences
Oligonucleotide sequences used for gene editing (CRISPR/Cas9) and qPCRs, and smFISH probe sequences are provided in Table S8.
ENCODE/modENCODE/modERN Files Used in This Study
Human CEBP ChIP-seq: ENCFF377MTQ.bam
Human H2AFZ ChIP-seq: ENCFF255USS.bigWig
Human H3K27ac ChIP-seq: ENCFF858MGD.bigWig
Human H3K4me3 ChIP-seq: ENCFF837XME.bigWig
Worm ChIP-seq b0310.2_embryo: ENCFF230GNZ.bed
Worm ChIP-seq elt-1_l3: ENCFF217KEA.bed
Worm ChIP-seq elt-2_l3: ENCFF707HUO.bed
Worm ChIP-seq fkh-6_embryo: ENCFF671ZAU.bed
Worm ChIP-seq jun-1_l4: spp.optimal.JUN-1_OP234_WA_L4_IP_Rep0.tagAlign_VS_JUN-1_OP234_WA_L4_Input_Rep0.tagAlign.regionPeak
Worm ChIP-seq pha-4_l4: ENCFF209ONL.bed
Worm ChIP-seq skn-1_l4: ENCFF798LMX.bed
Worm ChIP-seq unc-55_l2: ENCFF346FAK.bed
Worm ChIP-seq ztf-16_embryo: ENCFF341MDZ.bed
Acknowledgments
We thank Oliver Hobert, Roger Pocock, Hannes Bülow, Luisa Cochella, and Claude Desplan for critical reading of the manuscript and comments. We thank Sergej Herzog, Alina El-Khalili, Philip Mertins, and Norman Krüger for technical advice and assistance. Also, we thank Uwe Ohler and Neelanjan Mukherjee for useful discussion and support with respect to bioinformatics. We thank members of the Tursun group for comments on the manuscript and Dr. Mikkelsen for providing hiF-T cells and CGC for worm strains (supported by the NIH). Scott Allen Lacadie is a BIH Delbrueck Fellow funded by the Stiftung Charité. This work was sponsored by the ERC-StG-2014-637530 and ERC CIG PCIG12-GA-2012-333922 and is supported by the Max Delbrueck Center for Molecular Medicine in the Helmholtz Association.
Author Contributions
Conceptualization, B.T. and E.K.; Methodology, S.D., E.K., S.A.L., and B.T.; Investigation, E.K., A.O., G.B, S.S., A.S., M.H., S.B., S.D., B.U., A.A., A.H., and, B.T; Validation, B.T., E.K., S.A.L., and S.D.; Writing – Original Draft, B.T. and E.K.; Writing – Review & Editing, B.T., E.K., S.A.L., B.U., A.A., D.V., and S.D.; Funding Acquisition, B.T.; Resources, B.T., S.A.L., and S.D.; Visualization, B.T., E.K., S.A.L., and S.D.; Supervision, B.T.; Project Administration, B.T.
Declaration of Interests
The authors declare no competing interests.
Published: August 2, 2018
Footnotes
Supplemental Information includes six figures and eight tables and can be found with this article online at https://doi.org/10.1016/j.devcel.2018.07.006.
Contributor Information
Sebastian Diecke, Email: sebastian.diecke@mdc-berlin.de.
Scott A. Lacadie, Email: scott.lacadie@mdc-berlin.de.
Baris Tursun, Email: baris.tursun@mdc-berlin.de.
Supplemental Information
References
- Adey A., Morrison H.G., Asan, Xun X., Kitzman J.O., Turner E.H., Stackhouse B., MacKenzie A.P., Caruccio N.C., Zhang X. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11:R119. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez Alvarado A., Yamanaka S. Rethinking differentiation: stem cells, regeneration, and plasticity. Cell. 2014;157:110–119. doi: 10.1016/j.cell.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere J.A., Cenik E.S., Jain N., Hess G.T., Lee C.H., Bassik M.C., Fire A.Z. Translation readthrough mitigation. Nature. 2016;534:719–723. doi: 10.1038/nature18308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N., Battaglioli E., Atouf F., Andres M.E., Chenoweth J., Anderson M.E., Burger C., Moniwa M., Davie J.R., Bowers W.J. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Beanan M.J., Strome S. Characterization of a germ-line proliferation mutation in C. elegans. Development. 1992;116:755–766. doi: 10.1242/dev.116.3.755. [DOI] [PubMed] [Google Scholar]
- Becker J.S., Nicetto D., Zaret K.S. H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet. 2016;32:29–41. doi: 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskaya R., Oh S., Bondarenko V.A., Orphanides G., Studitsky V.M., Reinberg D. FACT facilitates transcription-dependent nucleosome alteration. Science. 2003;301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- Bettinger J.C., Lee K., Rougvie A.E. Stage-specific accumulation of the terminal differentiation factor LIN-29 during Caenorhabditis elegans development. Development. 1996;122:2517–2527. doi: 10.1242/dev.122.8.2517. [DOI] [PubMed] [Google Scholar]
- Blau H.M., Baltimore D. Differentiation requires continuous regulation. J. Cell Biol. 1991;112:781–783. doi: 10.1083/jcb.112.5.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkent M., Bennett B.D., Lackford B., Bar-Nur O., Brumbaugh J., Wang L., Du Y., Fargo D.C., Apostolou E., Cheloufi S. A serial shRNA screen for roadblocks to reprogramming identifies the protein modifier SUMO2. Stem Cell Reports. 2016;6:704–716. doi: 10.1016/j.stemcr.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brivanlou A.H., Gage F.H., Jaenisch R., Jessell T., Melton D., Rossant J. Stem cells. Setting standards for human embryonic stem cells. Science. 2003;300:913–916. doi: 10.1126/science.1082940. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Wu B., Chang H.Y. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109 doi: 10.1002/0471142727.mb2129s109. 21.29.1-21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 2013;14:427–439. doi: 10.1038/nrg3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y., Faddah D.A., Cheng A.W., Itskovich E., Markoulaki S., Ganz K., Klemm S.L., van Oudenaarden A., Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a Late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D., Trapnell C., Ziller M.J., Soumillon M., Cesana M., Karnik R., Donaghey J., Smith Z.D., Ratanasirintrawoot S., Zhang X. Integrative analyses of human reprogramming reveal dynamic nature of induced pluripotency. Cell. 2015;162:412–424. doi: 10.1016/j.cell.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S., Hochedlinger K. Emerging roles of the histone chaperone CAF-1 in cellular plasticity. Curr. Opin. Genet. Dev. 2017;46:83–94. doi: 10.1016/j.gde.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S., Elling U., Hopfgartner B., Jung Y.L., Murn J., Ninova M., Hubmann M., Badeaux A.I., Euong Ang C.E., Tenen D. The histone chaperone CAF-1 safeguards somatic cell identity. Nature. 2015;528:218–224. doi: 10.1038/nature15749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Liu J., Yang J., Chen Y., Chen J., Ni S., Song H., Zeng L., Ding K., Pei D. BMPs functionally replace Klf4 and support efficient reprogramming of mouse fibroblasts by Oct4 alone. Cell Res. 2011;21:205–212. doi: 10.1038/cr.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J.A., Tapia-Ramírez J., Kim S., Toledo-Aral J.J., Zheng Y., Boutros M.C., Altshuller Y.M., Frohman M.A., Kraner S.D., Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Chronis C., Fiziev P., Papp B., Butz S., Bonora G., Sabri S., Ernst J., Plath K. Cooperative binding of transcription factors orchestrates reprogramming. Cell. 2017;168:442–459.e20. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano B., Collombet S., Jakobsen J.S., Wierer M., Sardina J.L., Lackner A., Stadhouders R., Segura-Morales C., Francesconi M., Limone F. C/EBPα creates elite cells for iPSC reprogramming by upregulating Klf4 and increasing the levels of Lsd1 and Brd4. Nat. Cell Biol. 2016;18:371–381. doi: 10.1038/ncb3326. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt M., Roehr J.T., Ahmed R., Dieterich C. FLEXBAR-flexible barcode and adapter processing for next-generation sequencing platforms. Biology (Basel) 2012;1:895–905. doi: 10.3390/biology1030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr J.S. Immunohistochemistry. WormBook. 2006:1–61. doi: 10.1895/wormbook.1.105.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duina A.A. Histone chaperones Spt6 and FACT: similarities and differences in modes of action at transcribed genes. Genet. Res. Int. 2011;2011:625210. doi: 10.4061/2011/625210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox P.M., Vought V.E., Hanazawa M., Lee M.H., Maine E.M., Schedl T. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development. 2011;138:2223–2234. doi: 10.1242/dev.059535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D., Lerch A., Hahne F., Stadler M.B. QuasR: quantification and annotation of short reads in R. Bioinformatics. 2015;31:1130–1132. doi: 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner A., MacQueen A.J., Villeneuve A.M. Methods for analyzing checkpoint responses in Caenorhabditis elegans. Methods Mol. Biol. 2004;280:257–274. doi: 10.1385/1-59259-788-2:257. [DOI] [PubMed] [Google Scholar]
- Gaydos L.J., Wang W., Strome S. Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science. 2014;345:1515–1518. doi: 10.1126/science.1255023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiev S., Boyle A.P., Jayasurya K., Ding X., Mukherjee S., Ohler U. Evidence-ranked motif identification. Genome Biol. 2010;11:R19. doi: 10.1186/gb-2010-11-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Guo C., Morris S.A. Engineering cell identity: establishing new gene regulatory and chromatin landscapes. Curr. Opin. Genet. Dev. 2017;46:50–57. doi: 10.1016/j.gde.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. Quantifying similarity between motifs. Genome Biol. 2007;8:R24. doi: 10.1186/gb-2007-8-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C.M., Strømme C.B., Huang H., Patel D.J., Groth A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017;18:141–158. doi: 10.1038/nrm.2016.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe B.D., Branda C.S., Krause M., Stern M.J., Fire A. MyoD and the specification of muscle and non-muscle fates during postembryonic development of the C. elegans mesoderm. Development. 1998;125:2479–2488. doi: 10.1242/dev.125.13.2479. [DOI] [PubMed] [Google Scholar]
- Hentze H., Soong P.L., Wang S.T., Phillips B.W., Putti T.C., Dunn N.R. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009;2:198–210. doi: 10.1016/j.scr.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Hobert O. Neurogenesis in the Nematode Caenorhabditis elegans. WormBook. 2010:1–24. doi: 10.1895/wormbook.1.12.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. The Neuronal Genome of Caenorhabditis elegans. WormBook. 2013:1–106. doi: 10.1895/wormbook.1.161.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C.S., Foehr S., Garfield D.A., Furlong E.E., Steinmetz L.M., Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014;10:757. doi: 10.15252/msb.20145625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M.M., Lacadie S.A., Ohler U. JAMM: a peak finder for joint analysis of NGS replicates. Bioinformatics. 2015;31:48–55. doi: 10.1093/bioinformatics/btu568. [DOI] [PubMed] [Google Scholar]
- Jamai A., Puglisi A., Strubin M. Histone chaperone Spt16 promotes redeposition of the original H3-H4 histones evicted by elongating RNA polymerase. Mol. Cell. 2009;35:377–383. doi: 10.1016/j.molcel.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Jiao J., Dang Y., Yang Y., Gao R., Zhang Y., Kou Z., Sun X.F., Gao S. Promoting reprogramming by FGF2 reveals that the extracellular matrix is a barrier for reprogramming fibroblasts to pluripotency. Stem Cells. 2013;31:729–740. doi: 10.1002/stem.1318. [DOI] [PubMed] [Google Scholar]
- Jin Y., Hoskins R., Horvitz H.R. Control of type-D GABAergic neuron differentiation by C. elegans UNC-30 homeodomain protein. Nature. 1994;372:780–783. doi: 10.1038/372780a0. [DOI] [PubMed] [Google Scholar]
- Jones A., Francis R., Schedl T. GLD-1, a cytoplasmic protein essential for oocyte differentiation, shows stage-and sex-specific expression during Caenorhabditis elegans germline development. Dev. Biol. 1996;180:165–183. doi: 10.1006/dbio.1996.0293. [DOI] [PubMed] [Google Scholar]
- Kamath R.S., Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kamath R.S., Fraser A.G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kelly W.G., Fire A. Chromatin silencing and the maintenance of a functional germline in Caenorhabditis elegans. Development. 1998;125:2451–2456. doi: 10.1242/dev.125.13.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly W.G., Schaner C.E., Dernburg A.F., Lee M.H., Kim S.K., Villeneuve A.M., Reinke V. X-chromosome silencing in the germline of C. elegans. Development. 2002;129:479–492. doi: 10.1242/dev.129.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolundzic E., Seelk S., Tursun B. Application of RNAi and heat-shock-induced transcription factor expression to reprogram germ cells to neurons in C. elegans. J. Vis. Exp. 2018 doi: 10.3791/56889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooreman N.G., Wu J.C. Tumorigenicity of pluripotent stem cells: biological insights from molecular imaging. J. R. Soc. Interface. 2010;7:S753–S763. doi: 10.1098/rsif.2010.0353.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudron M.M., Victorsen A., Gevirtzman L., Hillier L.W., Fisher W.W., Vafeados D., Kirkey M., Hammonds A.S., Gersch J., Ammouri H. The ModERN Resource: genome-wide binding profiles for hundreds of Drosophilaand Caenorhabditis elegans transcription factors. Genetics. 2018;208:937–949. doi: 10.1534/genetics.117.300657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A., Fornes O., Arenillas D.J., Chen C.Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2016;44:D110–D115. doi: 10.1093/nar/gkv1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough L., Connell Z., Petersen C., Formosa T. The abundant histone chaperones Spt6 and FACT collaborate to assemble, inspect, and maintain chromatin structure in Saccharomyces cerevisiae. Genetics. 2015;201:1031–1045. doi: 10.1534/genetics.115.180794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megraw M., Pereira F., Jensen S.T., Ohler U., Hatzigeorgiou A.G. A transcription factor affinity-based code for mammalian transcription initiation. Genome Res. 2009;19:644–656. doi: 10.1101/gr.085449.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Muruganujan A., Casagrande J.T., Thomas P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N., Calviello L., Hirsekorn A., de Pretis S., Pelizzola M., Ohler U. Integrative classification of human coding and noncoding genes through RNA metabolism profiles. Nat. Struct. Mol. Biol. 2017;24:86–96. doi: 10.1038/nsmb.3325. [DOI] [PubMed] [Google Scholar]
- Narasimhan K., Lambert S.A., Yang A.W.H., Riddell J., Mnaimneh S., Zheng H., Albu M., Najafabadi H.S., Reece-Hoyes J.S., Fuxman Bass J.I. Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities. Elife. 2015;4:1743. doi: 10.7554/eLife.06967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo A., Reddy P., Martinez-Redondo P., Platero-Luengo A., Hatanaka F., Hishida T., Li M., Lam D., Kurita M., Beyret E. In Vivo amelioration of age-associated Hallmarks by partial reprogramming. Cell. 2016;167 doi: 10.1016/j.cell.2016.11.052. 1719–1733.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder T.T., Kara N., Cherry A., Sinha A.U., Zhu N., Bernt K.M., Cahan P., Marcarci B.O., Unternaehrer J., Gupta P.B. Chromatin-modifying enzymes as modulators of reprogramming. Nature. 2012;483:598–602. doi: 10.1038/nature10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi S.L., Henikoff J.G., Henikoff S. A native chromatin purification system for epigenomic profiling in Caenorhabditis elegans. Nucleic Acids Res. 2010;38:e26. doi: 10.1093/nar/gkp1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G., LeRoy G., Chang C.H., Luse D.S., Reinberg D. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell. 1998;92:105–116. doi: 10.1016/s0092-8674(00)80903-4. [DOI] [PubMed] [Google Scholar]
- Orphanides G., Wu W.H., Lane W.S., Hampsey M., Reinberg D. The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature. 1999;400:284–288. doi: 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- Paix A., Wang Y., Smith H.E., Lee C.Y., Calidas D., Lu T., Smith J., Schmidt H., Krause M.W., Seydoux G. Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics. 2014;198:1347–1356. doi: 10.1534/genetics.114.170423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchem R.J., Ye J., Judson R.L., LaRussa M.F., Krishnakumar R., Blelloch A., Oldham M.C., Blelloch R. Two miRNA clusters reveal alternative paths in late-stage reprogramming. Cell Stem Cell. 2014;14:617–631. doi: 10.1016/j.stem.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H., Zhao R., West J.A., Yabuuchi A., Huo H., Ince T.A., Lerou P.H., Lensch M.W., Daley G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Patel T., Hobert O. Coordinated control of terminal differentiation and restriction of cellular plasticity. Elife. 2017;6 doi: 10.7554/eLife.24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel T., Tursun B., Rahe D.P., Hobert O. Removal of Polycomb repressive complex 2 makes C. elegans germ cells susceptible to direct conversion into specific somatic cell types. Cell Rep. 2012;2:1178–1186. doi: 10.1016/j.celrep.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper A.S.-R., Killian D.J., Hubbard E.J.A. Genetic analysis of Caenorhabditis elegans GLP-1 mutants suggests receptor interaction or competition. Genetics. 2003;163:115–132. doi: 10.1093/genetics/163.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo J.M., Anderssen E., Walsh R.M., Schwarz B.A., Nefzger C.M., Lim S.M., Borkent M., Apostolou E., Alaei S., Cloutier J. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Blaschke K., Wei G., Ohi Y., Blouin L., Qi Z., Yu J., Yeh R.F., Hebrok M., Ramalho-Santos M. Transcriptional analysis of pluripotency reveals the Hippo pathway as a barrier to reprogramming. Hum. Mol. Genet. 2012;21:2054–2067. doi: 10.1093/hmg/dds023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H., Diaz A., Blouin L., Lebbink R.J., Patena W., Tanbun P., LeProust E.M., McManus M.T., Song J.S., Ramalho-Santos M. Systematic identification of barriers to human iPSC generation. Cell. 2014;158:449–461. doi: 10.1016/j.cell.2014.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B.A., Jeronimo C., Robert F. Recent perspectives on the roles of histone chaperones in transcription regulation. Curr. Mol. Biol. Rep. 2017;3:1–10. [Google Scholar]
- Riddle M.R., Weintraub A., Nguyen K.C.Q., Hall D.H., Rothman J.H. Transdifferentiation and remodeling of post-embryonic C. elegans cells by a single transcription factor. Development. 2013;140:4844–4849. doi: 10.1242/dev.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehr J.T., Dieterich C., Reinert K. Flexbar 3.0 – SIMD and multicore parallelization. Bioinformatics. 2017;33:2941–2942. doi: 10.1093/bioinformatics/btx330. [DOI] [PubMed] [Google Scholar]
- Roopra A., Qazi R., Schoenike B., Daley T.J., Morrison J.F. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol. Cell. 2004;14:727–738. doi: 10.1016/j.molcel.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Ruan J., Li H., Chen Z., Coghlan A., Coin L.J.M., Guo Y., Hériché J.K., Hu Y., Kristiansen K., Li R. TreeFam: 2008 update. Nucleic Acids Res. 2008;36:D735–D740. doi: 10.1093/nar/gkm1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi-Tehrani P., Golipour A., David L., Sung H.K., Beyer T.A., Datti A., Woltjen K., Nagy A., Wrana J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Seelk S., Adrian-Kalchhauser I., Hargitai B., Hajduskova M., Gutnik S., Tursun B., Ciosk R. Increasing Notch signaling antagonizes PRC2-mediated silencing to promote reprograming of germ cells into neurons. Elife. 2016;5 doi: 10.7554/eLife.15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S., Hein M.Y., Stewart A.F. msVolcano: a flexible web application for visualizing quantitative proteomics data. Proteomics. 2016;16:2491–2494. doi: 10.1002/pmic.201600167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommermann E.M., Strohmaier K.R., Maduro M.F., Rothman J.H. Endoderm development in Caenorhabditis elegans: the synergistic action of ELT-2 and -7 mediates the specification→differentiation transition. Dev. Biol. 2010;347:154–166. doi: 10.1016/j.ydbio.2010.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A., Donahue G., Zaret K.S. Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanakis N., Carrera I., Hobert O. Regulatory logic of pan-neuronal gene expression in C. elegans. Neuron. 2015;87:733–750. doi: 10.1016/j.neuron.2015.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner F.A., Talbert P.B., Kasinathan S., Deal R.B., Henikoff S. Cell-type-specific nuclei purification from whole animals for genome-wide expression and chromatin profiling. Genome Res. 2012;22:766–777. doi: 10.1101/gr.131748.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tursun B., Patel T., Kratsios P., Hobert O. Direct conversion of C. elegans germ cells into specific neuron types. Science. 2011;331:304–308. doi: 10.1126/science.1199082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno M., Honjoh S., Matsuda M., Hoshikawa H., Kishimoto S., Yamamoto T., Ebisuya M., Yamamoto T., Matsumoto K., Nishida E. A fasting-responsive signaling pathway that extends Life span in C. elegans. Cell Rep. 2013;3:79–91. doi: 10.1016/j.celrep.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Südhof T.C., Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L. ggplot2: elegant graphics for data analysis by WICKHAM, H. Biometrics. 2011;67:678–679. [Google Scholar]
- Yang C.S., Lopez C.G., Rana T.M. Discovery of nonsteroidal anti-inflammatory drug and anticancer drug enhancing reprogramming and induced pluripotent stem cell generation. Stem Cells. 2011;29:1528–1536. doi: 10.1002/stem.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Chai L., Fowles T.C., Alipio Z., Xu D., Fink L.M., Ward D.C., Ma Y. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2008;105:19756–19761. doi: 10.1073/pnas.0809321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A., Akanni W., Amode M.R., Barrell D., Billis K., Carvalho-Silva D., Cummins C., Clapham P., Fitzgerald S., Gil L. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–D716. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S., Avery L., Baude E., Garbers D.L. Guanylyl cyclase expression in specific sensory neurons: a new family of chemosensory receptors. Proc. Natl. Acad. Sci. USA. 1997;94:3384–3387. doi: 10.1073/pnas.94.7.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.